

Different mechanisms play a role in the anti‐tumour effect of mAbs and both target engagement with the Fab arm as well as Fc‐mediated effector functions contribute to the efficacy of treatment. As Ig isotypes differ in their ability to bind to FcRs on immune cells as well as in their ability to activate complement, they differ in the immune responses they activate. Therefore, the choice of antibody isotype for therapeutic mAbs is dictated by its intended mechanism of action. Here, we discuss the current knowledge of the therapeutic effector functions of different isotypes and Fc‐engineering strategies to improve mAbs application.

Keywords: cancer, effector functions, Fc tail, isotype, mAbs
Summary
The clinical application of monoclonal antibodies (mAbs) has revolutionized the field of cancer therapy, as it has enabled the successful treatment of previously untreatable types of cancer. Different mechanisms play a role in the anti‐tumour effect of mAbs. These include blocking of tumour‐specific growth factor receptors or of immune modulatory molecules as well as complement and cell‐mediated tumour cell lysis. Thus, for many mAbs, Fc‐mediated effector functions critically contribute to the efficacy of treatment. As immunoglobulin (Ig) isotypes differ in their ability to bind to Fc receptors on immune cells as well as in their ability to activate complement, they differ in the immune responses they activate. Therefore, the choice of antibody isotype for therapeutic mAbs is dictated by its intended mechanism of action. Considering that clinical efficacy of many mAbs is currently achieved only in subsets of patients, optimal isotype selection and Fc optimization during antibody development may represent an important step towards improved patient outcome. Here, we discuss the current knowledge of the therapeutic effector functions of different isotypes and Fc‐engineering strategies to improve mAbs application.
Introduction
Monoclonal antibodies (mAbs) have become an increasingly important class of drugs, with a global market comprising a total of 93 mAbs with marketing approval [1], with cancer being their most prevalent target disease [2]. Significant breakthroughs made in the areas of hybridoma technology, phage display and recombinant antibody production have enabled the development of a large variety of specific mAbs of any isotype.
In the case of cancer therapy, most therapeutic mAbs have been designed to interfere with the biological function of their target molecules and Fab arm specificity is, therefore, extremely important for mAb therapeutic efficacy. In addition, the Fc tail, which dictates downstream effector functions of an antibody, also plays an important role. Therefore, the final outcome of the binding of an antibody to its target is also influenced by the chosen isotype (Fig. 1, Table 1). Moreover, Fc‐ or glyco‐engineering of the chosen isotype can be used to further optimize its effector functions and half‐life. In this review we focus on optimal isotype selection for three mAb types receiving much clinical attention, which are according to their mechanism of action: (a) tumour antigen‐targeting, (b) immune checkpoint inhibiting and (c) tumour necrosis factor receptor (TNFR) family targeting agonistic mAbs.
Fig. 1.
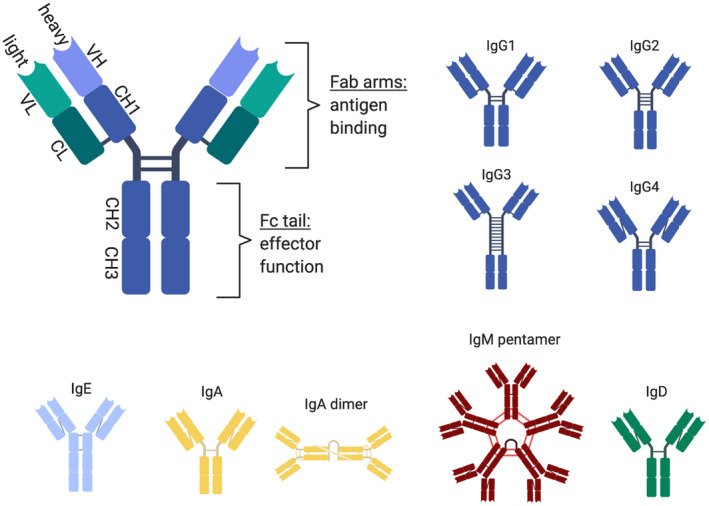
Antibody structure and isotypes. Human antibodies can be classified into five main isotypes – immunoglobulin (Ig)G, IgA, IgM, IgE and IgD, with IgG and IgA being further divided into the subclasses IgG1, IgG2, IgG3, IgG4, and IgA1 and IgA2, respectively. Overall structural organization of an antibody molecule is similar for all isotypes. It consists of two heavy and two light chains joined by disulphide bonds. Both the heavy and light chains have a highly diverse variable domain (VH and VL, respectively) and one or more constant domains (CH1 CH2, CH3 and CL, respectively). The constant domains of heavy chain are identical for all antibodies of the same isotype/subclass. Antibodies can also be divided into two functional subunits: (1) Fab arm, responsible for the specific binding to the antigen, (2) Fc tail, responsible for the activation of antibody effector functions [complement‐dependent cytotoxicity (CDC), antibody‐dependent cell‐mediated phagocytosis (ADCP), antibody‐dependent cell‐mediated cytotoxicity (ADCC), antigen cross‐presentation] through interaction with the complement system and binding to Fc receptors present on immune and other cells. A graphical overview of different isotypes and subclasses is shown.
Table 1.
Immunoglobulin (Ig) isotypes mediate their Fc effect via different receptors and activate different immune cells
| Antibody isotype | IgG1, IgG3* | IgE | IgA1, IgA2** | IgM | |||
|---|---|---|---|---|---|---|---|
| Main effector Fc receptor/ C1q | FcγRIIIa | FcγRIIa | C1q | FcεRI | FcεRII | FcαRI | C1q |
| Activated immune cells [3, 4, 5] | Macrophages, monocytes, NK cells | Macrophages, monocytes, eosinophils, neutrophils, DCs | – | Mast cells, granulocytes, macrophages, monocytes, DCs | Eosinophils, neutrophils, macrophages, monocytes, DCs | Eosinophils, neutrophils, macrophages***, monocytes, DCs*** | ‐ |
Tumour antigen‐targeting mAbs
Mechanism of action of tumour antigen‐targeting mAbs
The first generation of mAbs approved for clinical application – and still the most common group of mAbs in cancer therapy – consisted of mAbs directly targeting tumour antigens. These tumour antigens are, to a greater or lesser extent, important for the growth, survival and invasiveness of the tumour. The interference with tumour cell signalling pathways affects cell proliferation and leads to tumour cell death [e.g. anti‐human epidermal growth factor receptor 2 (HER2), anti‐epidermal growth factor receptor (EGFR)] [8, 9]. However, several observations in humans and mice suggest that Fc‐mediated activation of immune cells is an important additional mechanism of action of many of these mAbs [9, 10] (Fig. 2). Tumour cell‐bound antibodies can bind with their Fc tail to activating FcRs present on effector cells, such as natural killer (NK) cells, macrophages or neutrophils, which then mediate tumour cell lysis [10]. This can occur via release of cytotoxic mediators [antibody‐dependent cell‐mediated cytotoxicity (ADCC)] or via phagocytosis of tumour cells [antibody‐dependent cell‐mediated phagocytosis (ADCP)]. In addition, with their Fc tail, antibodies can activate the complement cascade through binding of C1q which can result in tumour cell lysis via several different mechanisms [11]. These include the formation of the membrane attack complex (MAC), that directly induces the lysis of target cells [complement‐dependent cytotoxicity (CDC)], or the attraction of immune cells through the chemoattractive activity of the complement components C3a and C5a. Furthermore, the opsonization by C3b and C4b marks the target cells for complement‐dependent cell‐mediated cytotoxicity (CDCC) by NK cells, macrophages/monocytes and granulocytes, or for complement‐dependent cell‐mediated phagocytosis (CDCP) by myeloid cells [11]. Antibody‐mediated cell death also leads to the release of tumour antigens and the formation of immune complexes (IC) which facilitate the initiation of anti‐tumour T cell responses, sustaining the tumour control and rejection. During this process, binding to FcγRs and activation of complement have been shown to play a critical role in the uptake of IC and cross‐presentation of IC‐derived tumour antigens by dendritic cells (DCs) in vivo [12, 13].
Fig. 2.
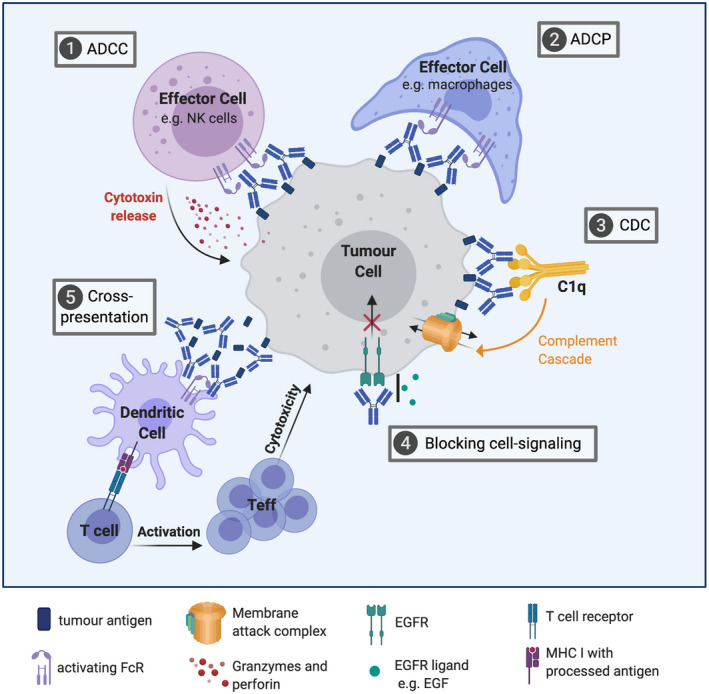
Mechanisms of action of tumour antigen‐targeting antibodies. Tumour antigen‐targeting (tumour‐depleting) antibodies mediate tumour cell‐killing via different mechanisms: (1, 2) activation of immune effector cells [antibody‐dependent cell‐mediated cytotoxicity (ADCC), antibody‐dependent cell‐mediated phagocytosis (ADCP)], (3) initiation of complement cascade [complement‐dependent cytotoxicity (CDC)], (4) blocking important signalling pathways in tumour cells and (5) formation of immune complexes inducing enhanced tumour antigen cross‐presentation by dendritic cells (DCs), leading to adaptive immune response.
In conclusion, in addition to blocking important signalling pathways in tumour cells with their Fab arm, tumour‐targeting antibodies furthermore deliver their effect through Fc‐mediated ADCC, ADCP and CDC. Therefore, an antibody isotype with the highest capacity to induce these effects should show the highest clinical efficacy. We will discuss different strategies to improve immunoglobulin (Ig)G Fc‐effector functions, as well as the potential use of alternative isotypes such as IgE and IgA (Fig. 4a).
Fig. 4.
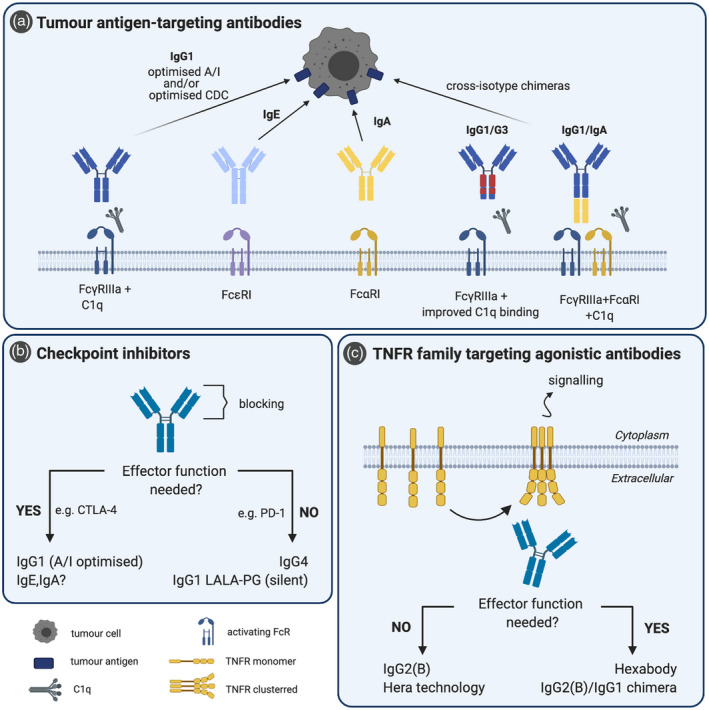
Optimal immunoglobulin (Ig) isotypes for therapeutic monoclonal antibodies (mAbs). (a) Tumour antigen‐targeting antibodies are mainly of IgG1 isotype, which can be further improved by optimizing their A/I ratio and complement activation through Fc‐ and glyco‐engineering. Recently, interest in IgE, IgA and cross‐isotype chimeras is rising, as they can offer alternative immune responses against tumour cells and the chimeras combine the advantages of two different isotypes. (b) For checkpoint inhibitors, a functional Fc tail may be either beneficial [anti‐cytotoxic T lymphocyte antigen (CTLA‐4)] or detrimental [anti‐programmed cell death 1 (PD‐1]. If Fc‐effector function is needed, isotype selection is similar to depleting antibodies (a). If Fc‐mediated effects are unwanted, optimal isotypes are IgG4 (poor Fc effector function) or IgG1 with abrogated FcR and C1q binding, for instance via LALA‐PG mutation [86]. (c) Agonistic antibodies target different receptors of the tumour necrosis factor receptor (TNFR) family, which require receptor clustering for initiation of their signalling cascade. Different strategies for achieving receptor clustering are available. In addition, Fc‐mediated cell depletion plays an important role in some cases. Prior to selecting the optimal isotype, the need for a functional Fc tail should be considered.
Optimizing IgG effector function
IgG Fc‐effector functions are mediated via complement and FcγRs which are either activating (FcγRI, FcγRIIa/IIc, FcγRIIIa, FcγRIIIb [14]) or inhibitory (FcγRIIb) [15]. As most effector cells co‐express both activating and inhibitory FcγRs, the outcome of IgG binding is a result of the relative binding affinity, receptor availability and signalling capacity. The relative affinity of an antibody for its receptors is defined as the activating‐to‐inhibitory (A/I) ratio [16] (Fig. 3). The concept of the A/I ratio is based on observations in mice which show a high A/I for mIgG2a, a low A/I for mIgG1 and an intermediate A/I for mIgG2b [16]. As a consequence, therapeutic antibodies of the mIgG2a subclass have been shown to clear the tumours more efficiently in many in‐vivo model systems [17]. Although the differences in the A/I ratio are less pronounced between human IgG subclasses, they also differ in their capability to induce immune responses due to their different FcR binding profile [18, 19]. IgG1 and IgG3 bind to all FcRs, but show higher affinity for the activating ones. Thus, they are defined as highly activating with strong Fc‐effector function. Conversely, IgG4 binds with similar affinities to most activating FcRs and inhibitory FcγRIIb and is considered as poorly activating. Finally, IgG2 shows overall poor binding to most FcRs, with the exception of the high‐affinity H131 FcγRIIa allele (as discussed later), and has limited Fc‐effector function. Therefore, IgG1 and IgG3 are capable of exerting potent effector functions desirable for depleting antibodies, whereas IgG2 and IgG4 are preferred when Fc‐mediated cell depletion is to be avoided.
Fig. 3. T.
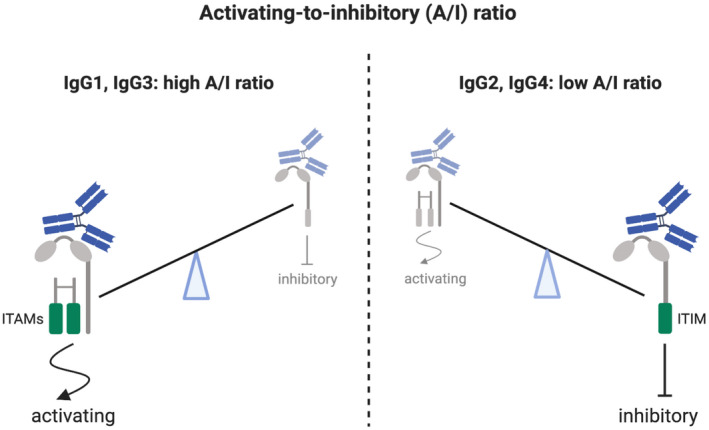
A/I ratio dictates the outcome of Fc‐effector function of immunoglobulin (Ig)G antibodies. FcγRs can be either inhibitory (FcγRIIb) with immunoreceptor tyrosine‐based inhibition motif (ITIM) or activating (FcγRI, FcγRIIa/IIc, FcγRIIIa, FcγRIIIb) [15]. Activating FcγRI and IIIa are associated with the common FcR gamma chain dimer containing two immunoreceptor tyrosine‐based activating motifs (ITAMs); FcγRIIa and IIc are not associated with the gamma chain, but contain their own ITAM motif, whereas FcγRIIIb does not contain an ITAM motif and is not always considered as an activating receptor [15]. Expressed mainly on neutrophils, FcγRIIIb has been shown to favour phagocytosis (ADCP) in co‐operation with FcγRIIa but, conversely, it has a negative impact on neutrophilic antibody‐dependent cell‐mediated cytotoxicity (ADCC) by acting as a decoy receptor for IgG, thus competing with FcγRIIa for antibody binding [14]. Cross‐linking of an activating receptor with the inhibiting receptor results in down‐regulation of the activating signal. Therefore, all activating FcγRs are counterbalanced by one inhibiting receptor FcγRIIb. Differential affinity of IgG for FcRs is defined as activating‐to‐inhibitory (A/I) ratio.
Although less relevant for effector functions, an additional IgG receptor is the neonatal Fc receptor (FcRn) which mediates IgG transport through the placenta as well as IgG cellular recycling, providing IgG with a relatively long serum half‐life and thus favourable pharmacokinetic properties [20]. FcRn also binds albumin with similar effects [20] which can be exploited for mAb engineering, as will be discussed later.
Currently, most of the clinically approved tumour‐targeting mAbs are of IgG1 isotype, which was shown to be superior to other isotypes and subclasses in inducing ADCC by mononuclear cells as well as CDC in vitro [21]. IgG1 achieves most of its Fc‐effector functions via FcγRIIIa present on macrophages and NK cells (ADCC, ADCP), as well as via complement activation [22]. Furthermore, IgG1 shows favourable biopharmaceutical characteristics with regard to production and purification. However, independently of their specificity, all tumour‐targeting IgGs used hitherto in the clinic displayed a therapeutic effect only in a subset of patients. Therefore, over the years different strategies have been explored to further optimize tumour‐targeting mAbs, many of which focused upon improving Fc‐mediated functions.
Improving the A/I ratio
One of the common approaches to improve the IgG Fc‐effector functions is to optimize the A/I ratio by increasing affinity for the activating FcγRs, on one hand, and decreasing binding to the inhibitory FcγRIIb on the other hand. One approach to improve the A/I ratio was successfully achieved by glycoengineering. The most relevant modification is afucosylation of N297 glycan, which significantly increased the affinity for FcγRIIIa improving the ADCC effect in vitro [23], which was mirrored by improved in‐vivo anti‐tumour responses in mouse models [24]. Two afucosylated mAbs have already received marketing approval [mogamulizumab (anti‐CCR4) [25], obinutuzumab (anti‐CD20)] [26], and several others are currently in clinical trials [27]. Obinituzumab was found to be superior to rituximab (non‐glycoengineered anti‐CD20) in terms of complete response rate and progression‐free survival in various clinical settings [28, 29, 30]. Another commonly used strategy to improve the A/I ratio is the introduction of point mutations in the Fc tail [22]. The most promising mAb in this group is margetuximab, an anti‐HER2 antibody featuring five point‐mutations in its Fc tail resulting in improved binding to FcγRIIIa and FcγRIIa, as well as a decreased FcγRIIb binding [31]. These modifications translated into improved ADCC in vitro [31], enhanced anti‐tumour activity in mice [31] and higher response rate and progression‐free survival in HER2‐positive metastatic breast cancer when compared to its non‐Fc‐engineered analogue trastuzumab [32]. In conclusion, glyco‐ and Fc‐ engineered IgG1 mAbs with an optimized A/I ratio appear superior to non‐engineered IgG1, due most probably to enhanced ADCC.
Optimizing CDC
CDC has been recognized as an important mechanism of action for some therapeutic mAbs, such as anti‐CD20 [33, 34, 35]. Thus, strategies to optimize Fc‐mediated complement activation are currently being developed.
Intrinsically, due to its naturally occurring pentameric and hexameric forms, IgM demonstrates the highest capacity for complement activation. However, IgM has received little attention in therapeutic mAbs development, and only a few tumour‐targeting IgM mAbs have been evaluated in clinical trials [36], most prominently with PAT‐SM6 receiving orphan drug designation by the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) for multiple myeloma [37, 38].
Among IgG subclasses, IgG1 and IgG3 are good complement activators, with IgG3 appearing to be the more potent isotype. Nevertheless, although IgG3 intrinsic problems, such as a short in‐vivo half‐life, have successfully been addressed [39], specific manufacturing issues make it a less attractive candidate for drug development. A way to combine the advantages of both IgG1 (favourable manufacturing characteristics) and IgG3 (enhanced CDC) was achieved through the construction of IgG1/IgG3 chimeric antibodies [40]. The optimal construct, called 113F, combined the CH1 and the hinge of IgG1 with the CH2 of IgG3 and a CH3 which was partly of IgG3 and partly of IgG1 origin. The non‐fucosylated version of this chimeric antibody showed enhanced CDC and ADCC comparable to non‐fucosylated IgG1, in addition to preserved protein A binding, important for the purification process. The improved efficacy of this chimeric construct was confirmed in cynomolgus monkeys, where an anti‐CD20 113F antibody construct showed greater B cell depletion if compared to IgG1 (both antibodies were non‐fucosylated for improved ADCC). This study indicates that the combination of optimized complement activation and A/I ratio represents a promising strategy for the improvement of tumour‐depleting antibodies.
Other strategies for enhanced complement activation include the introduction of point mutations to improve IgG1 binding to C1q [22], a key component for the initiation of the complement cascade. Importantly, mutations that potentiate CDC can be combined with ADCP‐ and ADCC‐enhancing mutations in a single IgG1 [41], thus broadening the effector function of these antibodies. Finally, the mutations that favour IgG hexamer formation also significantly enhance C1q fixation and thus CDC [42, 43]. However, currently it remains to be seen whether or not these Fc mutations that enhance CDC in in‐vitro and ex‐vivo studies translate into improved clinical efficacy.
Use of alternative Ig isotypes
IgE
Several epidemiological studies have suggested a protective effect of some allergies and IgE antibodies against specific types of tumours [44, 45], providing a rationale for exploring the potential use of mAbs of the IgE isotype as anti‐tumour agents. IgE can mediate its Fc‐effector function via two activating receptors: the high‐affinity FcεRI and the low‐affinity FcεRII. While predominantly expressed by mast cells (MC) and basophils, FcεRI expression can also be found on eosinophils, dendritic cells (DCs) and myeloid cells, although 10–100‐fold lower than on fully matured and activated MC [46]. Compared to the IgG class, IgE offers several advantages that can be of interest for cancer therapy. For instance, it shows two orders of magnitude higher affinity for its receptor FcεRI than IgG for its high‐affinity receptor FcγRI [47]. Because of such a high FcɛRI affinity, IgE is locally retained on the cells expressing FcɛRI and has excellent bioavailability in tissues, which is of great importance for treatment of solid tumours. In addition, IgE lacks inhibitory Fc receptors that could cause immunosuppression such as FcγRIIb in the case of IgG [47]. Consequently, the use of IgE antibodies in cancer therapy has been tested both in in‐vitro and in‐vivo mouse models, using transgenic hFcɛRI mice [48] as well as rats.
Side‐to‐side studies demonstrated that an IgE mAb targeting tumour‐associated antigen was superior to its IgG1 counterpart in terms of survival and reduction of tumour growth [48, 49, 50]. Furthermore, it was found that the main anti‐tumour effector function of IgE antibodies was mediated by myeloid cells [50, 51], and in‐vitro experimental data showed that monocytes can mediate IgE tumour killing via both ADCC through FcɛRI as well as ADCP through FcɛRII [52]. Remarkably, the IgE antibodies both recruited tumour‐associated macrophages (TAM) for ADCC and ADCP, but also differentiated them towards activated M1‐like phenotype characterized by up‐regulation of a tumour necrosis factor (TNF)α/monocyte chemoattractant protein‐1 (MCP)‐1/interleukin (IL)‐10 cytokine signature, suggesting a potential role of IgE in tumour microenvironment (TME) modification [53]. Furthermore, IgE has been shown to facilitate DC cross‐presentation of IgE IC‐derived antigens, further supporting the anti‐tumour effect by inducing a T cell‐based anti‐tumour response [54, 55, 56].
An intrinsic concern regarding IgE therapy is the risk of inducing potentially life‐threatening anaphylaxis triggered by degranulation of MC or basophils. Although tumour antigens released into the blood as monomers are not expected to induce cross‐linking of FcɛR‐bound IgE required for degranulation [54], circulating tumour cells expressing multiple copies of targeted antigen would have the potential to induce degranulation. However, no signs of anaphylaxis were found in preclinical models and safety data were satisfactory in both rodents and monkeys [54, 57, 58], supporting the initiation of the first clinical trial using a tumour‐targeting anti‐folate receptor alpha IgE mAb MOv18 (NCT02546921). Interim Phase I data from 24 patients support the safety and potential efficacy of MOv18 IgE [59]. Potential to develop systemic allergic toxicity was evaluated by pretreatment skin prick test and ex‐vivo basophil activation test (BAT). Readily manageable urticaria was the most common side effect. Only one patient experienced anaphylaxis – the only patient with a positive BAT test – indicating that the BAT test might be an important tool to exclude patients with potential risk of anaphylaxis. In addition, although the primary objective was to evaluate the safety and low doses of MOv18 IgE were used, anti‐tumour effect was observed in one patient.
In conclusion, the experimental data suggest that IgE might be an attractive Ig isotype to improve the clinical efficacy of tumour‐depleting mAbs. Upcoming results of the MOv18 IgE clinical trial will be a major step forward in evaluating IgE‐based therapy in human settings.
IgA
Another promising Ig isotype for tumour‐depleting mAbs is IgA, which mediates its effector functions through FcαRI. The FcαRI induces activating signals when IgA is encountered as an immune complex; however, it induces inhibitory signals upon monomeric binding [3, 60]. FcαRI is highly expressed on polymorphonuclear cells (PMNs), making neutrophils the most relevant cell type for IgA mAb therapy. Neutrophils represent the most abundant cytotoxic cell type in humans. They are armed with a variety of potent cell destruction mechanisms, including the release of cytotoxic molecules, induction of apoptosis and necrosis. Furthermore, they are well established for their recruitment of other immune cells and phagocytosis [60, 61]. Importantly, it has been shown that, compared to cross‐linking of FcγR, FcαRI cross‐linking is far more efficient in the activation of neutrophils [61, 62, 63].
However, in‐vivo studies are still scarce, due largely to the fact that mice lack a FcαR homologue. Creation of a transgenic human FcαR mouse strain[64] allowed in‐vivo studies in which the anti‐tumour effect of IgA antibodies was demonstrated [65, 66]. Surprisingly, macrophages were shown to be the crucial effector population for anti‐EGFR IgA in vivo, leaving the role of neutrophils unclear. Unfortunately, the transgenic hFcαR mouse only partially resolved the lack of a useful model, as human IgA has a very short half‐life in mice. Therefore, hIgA mouse pharmacokinetics and exposure were enhanced by attaching an albumin‐binding domain to improve FcRn binding and thus the recycling of the antibody [67]. Furthermore, the clearance by the asialoglycoprotein receptor in the liver could be reduced by Fc‐engineering [68]. In both cases, increased IgA half‐life translated into improved anti‐tumour efficacy in mouse models. These strategies may direct more extensive exploration of IgA‐based cancer therapies in murine models and might be considered for extending the relatively short serum half‐life of IgA mAbs in humans.
Similar to IgG1/IgG3 chimeras, attempts to construct IgG1/IgA chimeras have been made with the intention of combining the advantages of the two different isotypes [69, 70]. Binding to FcRn, FcγRs and C1q with the IgG1 part as well as to FcαR with the IgA part, has been successfully achieved [70]. It provided the IgG1/IgA chimera with an extended half‐life, the capability to activate macrophages and complement and initiate recruitment of neutrophils, respectively, resulting in an overall improved cytotoxicity [70]. Thus, the combined effector functions of such a chimeric isotype mAb construct may further improve the clinical efficacy of tumour‐targeting mAbs.
Isotypes and patient‐tailored medicine
With so many different strategies to improve the downstream effector functions of tumour‐targeting antibodies, the question arises as to which approach to follow. It is tempting to speculate that personalized medicine approach may give an answer, by taking into consideration patient‐related factors and tumour intrinsic characteristics. For example, two FcγR polymorphisms that affect the binding of IgG antibodies have been described: H131R in FcγRIIa and V158F in FcγRIIIa. The R131 variant shows lower affinity for IgG2 while the F158 variant shows lower affinity for IgG1 and IgG3. The clinical implication of these variants has not yet been fully resolved, with some studies finding a negative correlation with therapeutic efficacy while others do not [71, 72]. Nevertheless, if larger and better‐designed studies confirm the negative correlation between lower‐affinity FcRs variants and response to IgG antibody treatment, these patients may benefit more from IgE, IgA or mAbs optimized for complement activation, given that these are proven to be effective anti‐tumour treatments in the future. With regard to complement optimized mAbs, it may further be important to consider tumour microenvironment (TME) factors, such as pH, that can affect CDC [73] or the expression level of complement regulatory proteins, which allow complement evasion by cancer cells [43]. Furthermore, it has been shown that C‐reactive protein (CRP) shares its binding site on FcγRs (I and II) and FcαRI with IgG and IgA, respectively, whereas it can also bind to C1q [74, 75]. Whether CRP can act as a competitive inhibitor for FcRs and complement binding of those antibodies in vivo has not yet been studied, but it could have important implications. For instance, patients suffering from chronic inflammatory and neurodegenerative diseases, such as atherosclerosis, type 2 diabetes mellitus or Parkinson’s disease, have chronically elevated CRP levels [76] which may interfere with antibody treatment. Thus, for those patients, IgE‐based antibody treatment might be an attractive choice. In conclusion, antibody engineering offers a wide range of opportunities to improve effector functions of mAbs, but patient‐related factors should also be taken into consideration for optimal isotype selection. This multi‐level approach could result in a more effective personalized treatment.
Antibodies targeting immunological checkpoint proteins
A recently identified class of mAbs for cancer therapy are the so‐called checkpoint inhibitors. These antibodies do not target the tumour directly but enhance anti‐tumour immune responses by targeting immunological checkpoint proteins, such as programmed cell death 1 (PD‐1) or cytotoxic T lymphocyte antigen 4 (CTLA‐4), or their ligands, such as programmed cell death ligand 1 (PD‐L1). These checkpoint proteins are expressed on activated T cells and limit excessive T cell responses. As a means of immune resistance, the ligands of PD‐1 are often expressed by tumour cells [77] as well as by myeloid cells infiltrating the tumour microenvironment (TME) [78, 79]. Checkpoint blockade leads to enhanced T cell activation [77, 80] and, consequently, the clinical introduction of checkpoint inhibitors led to a tremendous improvement of cancer therapy for several different types of cancers.
In theory, checkpoint blocking antibodies do not require Fc‐mediated effects, as their main effector function is expected to be derived from blocking the receptor–ligand interaction (Fab‐mediated). However, it was found that a functional Fc tail contributed to the therapeutic efficacy of anti‐CTLA‐4 checkpoint inhibitors in mouse models [81, 82]. These studies revealed that whereas both effector T cell (Teff) and regulatory T cell (Treg) populations were increased in lymph nodes after therapy within tumours, specifically the Treg but not the Teff population was decreased. This decrease was only observed with anti‐CTLA‐4 of the IgG2a isotype (the isotype with the highest A/I ratio in mouse) and appeared mFcγRIV‐dependent. The underlying mechanism was found to be caused by a selectively high abundance of macrophages expressing high levels of FcγRIV in tumours but not in lymph nodes [81]. Furthermore, Tregs express much higher levels of CTLA‐4 than Teff cells and were, therefore, preferentially depleted [83]. These findings point to the importance of the TME for therapeutic mAb efficacy.
There are indications that human anti‐CTLA‐4 mAbs show the same effect. A recent study confirmed the importance of Treg depletion for human anti‐CTLA‐4 antibody in a hFcγR mouse model [84]. In addition, in advanced melanoma patients with a high neoepitope burden the authors found a positive correlation between the presence of the high‐affinity V158 FcγRIIIa allele and increased response to the CTLA‐4 targeting antibody ipilimumab, providing further clinical evidence for the importance of Fc‐mediated function. These findings may be relevant to explain why only some patients respond to anti‐CTLA‐4 therapy and provide further rationale to optimize CTLA‐4 mAbs by improving their A/I ratio [85] or switching to IgA or IgE isotypes, given that the microenvironmental requirements for selective tumour Treg depletion are met.
Similarly, it was shown that the binding of anti‐PD‐L1 mAb to activating FcγRs enhances its therapeutic efficacy in mouse models, due to Fc‐mediated depletion of immunosuppressive myeloid cell subsets in the TME [78]. However, although another study confirmed that Fc‐mediated depletion of myeloid cells in the TME contributes to the therapeutic effect of anti‐PD‐L1 antibodies, this effect was found to be dependent upon the mouse genetic background, as it occurred in CT26 tumours transplanted in BALB/c but not MC38 tumours in C57BL/6 mice [79]. The depleted myeloid cell subset was the one with the highest PD‐L1 expression, whereas PD‐L1 expression on the tumour cells did not contribute to the therapeutic effect of the anti‐PD‐L1 antibody [79]. Currently, there are three clinically approved anti‐PD‐L1 mAbs, two of which have a mutated Fc tail with abrogated FcγR binding (atezolizumab, durvalumab) and one is a wild‐type IgG1 (avelumab). As hundreds of clinical trials with these antibodies are currently ongoing, future results might help to resolve the question whether a functional Fc tail improves clinical efficacy of PD‐L1 targeting antibodies in humans. If so, a further Fc‐effector function optimization might be an appealing step forward.
In contrast to anti‐CTLA‐4 and anti‐PD‐L1, a functional Fc tail compromised the activity of anti‐PD‐1 mAbs in vivo. The underlying mechanism of this detrimental effect was the depletion of tumour‐infiltrating CD8+ T cells, which are characterized by high PD‐1 expression[78]. Not surprisingly, two clinically approved anti‐PD‐1 mAbs are of the IgG4 subclass with poor Fc effector functions. However, as IgG4 still binds to activating FcγRs to some extent, it would be interesting to compare its therapeutic efficacy with that of a mutated mAb with completely abolished FcγR binding [86]. Similarly, antibodies targeting CD47, a ‘don’t eat me signal’ often up‐regulated by tumour cells to avoid elimination by myeloid cells as part of CD47/SIRP‐α checkpoint pathway, do not require Fc‐effector function either [87].
In conclusion, these findings strongly suggest that the cellular composition of the TME as well as the relative expression of the target molecule on different immune cell populations can greatly affect the outcome of checkpoint‐blocking mAb therapy. These factors dictate the need for Fc‐mediated mechanisms for an optimal therapeutic effect and, thus, the isotype selection for checkpoint inhibitors. (Fig. 4b).
Tumour necrosis factor receptor family targeting agonistic antibodies
Recently, the Fc tail of agonistic mAbs that target specific members of the TNFR family has been shown to play a critical role in their therapeutic efficacy. This class of mAbs is designed to either activate death receptors such as DR4, DR5 and FAS on cancer cells in order to induce cell death, or to activate co‐stimulatory receptors such as CD40, 41BB, OX40, GITR and CD27 on immune cells in order to improve anti‐tumour immune responses.
TNFRs require trimerization in order to initiate their associated signalling cascade [88]. Therefore, bivalent engagement of these receptors with Fab arms is usually not sufficient for their activation, and additional cross‐linking is required. For these antibodies, the interaction with FcγRs functions as an effective scaffold for clustering. Specifically, it has been shown that FcγRIIb represents a dominant scaffold for antibody‐mediated TNFR cross‐linking and activation of downstream signalling because of its relatively high expression [89, 90]. Consequently, agonistic antibody activity was found to be highly dependent upon successful FcγRIIb engagement in mice [91, 92], and Fc‐engineered antibodies with improved FcγRIIb binding showed stronger anti‐tumour activity [93, 94]. However, the expression of FcgRIIb is dynamic and can be down‐regulated by particular cytokines [95], rendering the success of FcγRIIb‐mediated cross‐linking for receptor clustering unpredictable. In addition, effective FcR‐engagement by agonistic antibodies was found to be associated with serious hepatotoxicity [96, 97, 98], which could potentially be explained by the high expression of FcγRIIb on certain subsets of liver cells [99]. Therefore, new strategies have been explored to improve the agonistic activity of these mAbs independently of FcγR engagement. One of these strategies is the use of hIgG2(B). This compact and highly agonistic conformation of hIgG2 [100] is a consequence of a unique disulphide bonds rearrangement in the hinge region [101]. Compared to hIgG2(A), whose Fab arms are not linked to the hinge via disulphide bonds, hIgG2(B) presents with two disulphide bonds between each Fab arm and hinge, making them more rigid and potentially able to pack TNFR molecules closer together [102]. In line with this finding, the agonistic effect of anti‐CD40 hIgG2 antibodies was demonstrated to be FcγR‐independent both in vitro and in vivo. Importantly, it is possible to lock hIgG2 in B conformation via a specific cysteine mutation in CH1 region which allows its recombinant production [100]. Thus, the use of hIgG2(B) is a viable strategy for improving the FcγR‐independent agonistic activity of mAbs targeting TNFR family members [103]. Furthermore, isotype switching from hIgG1 to hIgG2 was sufficient to convert an immunosuppressive anti‐CD40 antagonistic antibody into a potent agonist with anti‐tumour activity [104]. These findings constitute one of the most striking examples of how the choice of the isotype can completely change the activity of a mAb.
Another approach to improve the agonistic activity of TNFR family targeting mAbs, independently of FcγR engagement, is the recently developed HERA platform. HERA is an artificial chimeric molecule which, instead of Fab‐arms, has two trimeric TNFR binding domains, fused to an IgG1 Fc backbone with abrogated FcγR binding. The resulting hexavalent molecule is capable of exerting its agonistic activity without FcγR‐mediated cross‐linking. So far, two HERA molecules targeting CD27 and CD40 have shown promising anti‐tumour activity, without significant toxicological signs in preclinical mouse models [105, 106]. These findings suggest that agonistic HERA molecules may offer improved safety combined with unaltered efficacy and thus an advantageous clinical profile.
The strategies described to improve agonistic activity in a FcγR‐independent manner could have an additional advantage, as they prevent unwanted depletion of immune cells expressing the target molecule. However, experiments in mice suggest that the therapeutic effect of some TNFR family targeting agonistic antibodies (such as GITR [107], anti‐OX40 [108] or anti‐4‐1BB) [109] also involved Treg depletion, suggesting that, analogous to anti‐CTLA‐4, a functional Fc tail might be advantageous. Similarly, some Fc‐mediated downstream effector functions may be useful for agonistic mAbs targeting death receptors on cancer cells, as Fc‐mediated cytotoxicity and ADCP would act as an additional tumour cell‐depleting mechanism and might facilitate cross‐presentation inducing an adaptive anti‐tumour response.
A few solutions have been proposed to combine the divergent properties, as mentioned above, in a single Ig molecule. For instance, a pentameric IgM antibody with high complement activation capacity has been used to successfully induce DR5 clustering via multi‐valent interaction, inducing tumour regression in preclinical models [110]. An alternative approach which takes advantage of Ig multimerization, but avoids IgM manufacturing issues, is the so‐called HexaBody technology. It is based on a single point mutation (E430G) in the Fc domain of IgG1 that enhances Fc–Fc interactions upon binding to membrane‐bound targets [111]. Consequently, these antibodies have a strong tendency to form hexamers on the target cell, ultimately leading to both high agonistic activity and improved CDC [112]. A combination of different HexaBodies targeting two different epitopes on DR5 is currently in early clinical testing (NCT03576131). Given the enhanced complement activation of HexaBodies, this antibody form could furthermore be attractive whenever tumour cell lysis is intended, such as for classical tumour antigen‐targeting antibodies (e.g. anti‐CD20), suggestive for the design of an entirely novel type of tumour antigen‐targeting antibodies.
In addition to HexaBodies, a highly agonistic anti‐4‐1BB recombinant Ig with potent Fc‐effector function was achieved by combining human IgG2 CH1 and hinge locked in B conformation, with murine IgG2a CH2 and CH3 (the IgG subclass with the highest A/I ratio in the mouse) [109]. In mice, tumour treatment with this chimeric construct induced both Teff stimulation in lymph nodes (strong 4‐1BB agonism) and Fc‐mediated Treg depletion within tumours, leading to an increased intratumoral Teff/Treg ratio and enhanced survival compared to a wild‐type mIgG2a construct [109]. By analogy with the mouse example, a chimera of hIgG2(B) and hIgG1 might be applicable in humans.
In conclusion, important breakthroughs have been made in the design of TNFR agonistic antibodies by making their activity FcγR‐independent. It is precisely the FcγR‐independency that may overcome initial problems seen in the clinic, such as severe toxicity and modest efficacy. However, the contribution of Fc‐mediated cell depletion to the therapeutic efficacy represents an important consideration for the optimal design of a specific agonistic antibody (Fig. 4c).
Conclusion
The introduction of mAbs into the clinic has fundamentally changed cancer therapy. Nevertheless, it has increasingly become apparent that mAbs mediate their effects via a multitude of different mechanisms of action. Since the selection of the correct Ig isotype was recognized as crucial, much effort has been put into understanding the Fc‐mediated effects of different antibody isotypes as well as into Fc‐modifications for further improvement of mAbs efficacy. Consequently, several strategies have been developed in order to optimize Fc‐mediated effector functions, opening entirely novel opportunities to improve mAbs‐based cancer therapy. Furthermore, by considering patient‐related factors, such as their immune status, characteristics of the TME or FcγR polymorphism, the isotype selection may either allow for the development of antibodies that are active in a wider range of patients or may allow for the selective use of antibodies tailored towards the individual’s needs. Such considerations may lead us one step further to patient‐tailored medicine and more effective mAb treatment in the future.
Disclosures
The authors have no competing interests.
Acknowledgements
We are grateful to Keri Pallister (University of Edinburgh) for her help in the figure design. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement no. 765394 (N. V.).
Contributor Information
J. S. Verbeek, Email: j.s.verbeek@toin.ac.jp.
D. M. W. Zaiss, Email: Dietmar.Zaiss@ed.ac.uk.
Data Availability Statement
Data availability are not applicable to this review article.
References
- 1. Antibody therapeutics approved or in regulatory review in the EU or US. The Antibody Society. Available at: https://www.antibodysociety.org/resources/approved‐antibodies/ (accessed 14 April 2020).
- 2. Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol 2019; 37:9–16. 10.1016/j.tibtech.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 3. Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcαRI; its function and potential as target in disease. Immunol Rev 2015; 268:123–38. [DOI] [PubMed] [Google Scholar]
- 4. Bournazos S, Ravetch JV. Fcγ receptor function and the design of vaccination strategies. Immunity 2017; 47:224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jensen‐Jarolim E, Bax HJ, Bianchini R et al AllergoOncology – the impact of allergy in oncology: EAACI position paper. Allergy 2017; 72:866–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rösner T, Kahle S, Montenegro F et al Immune effector functions of human IgG2 antibodies against EGFR. Mol Cancer Ther 2019; 18:75–88. [DOI] [PubMed] [Google Scholar]
- 7. Steffen U, Koeleman CA, Sokolova MV et al IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat Commun 2020; 11:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hudis CA. Trastuzumab – mechanism of action and use in clinical practice. N Engl J Med 2007; 357:39–51. [DOI] [PubMed] [Google Scholar]
- 9. Weiner GJ. Monoclonal antibody mechanisms of action in cancer. Immunol Res 2007; 39:271–8. [DOI] [PubMed] [Google Scholar]
- 10. Chenoweth AM, Wines BD, Anania JC, Mark Hogarth P. Harnessing the immune system via FcγR function in immune therapy: a pathway to next‐gen mAbs. Immunol Cell Biol 2020. 10.1111/imcb.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bordron A, Bagacean C, Tempescul A et al Complement system: a neglected pathway in immunotherapy. Clin Rev Allergy Immunol 2020; 58:155–71. [DOI] [PubMed] [Google Scholar]
- 12. Ho NI, Camps MGM, de Haas EFE, Trouw LA, Verbeek JS, Ossendorp F. C1q‐dependent dendritic cell cross‐presentation of in vivo‐formed antigen–antibody complexes. J Immunol 2017; 198:4235–43. [DOI] [PubMed] [Google Scholar]
- 13. DiLillo DJ, Ravetch JV. Differential Fc‐receptor engagement drives an anti‐tumor vaccinal effect. Cell 2015; 161:1035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Treffers LW, van Houdt M, Bruggeman CW et al FcγRIIIb restricts antibody‐dependent destruction of cancer cells by human neutrophils. Front Immunol 2019; 9:3124 10.3389/fimmu.2018.03124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol Rev 2015; 268:25–51. [DOI] [PubMed] [Google Scholar]
- 16. Nimmerjahn F. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science 2005; 310:1510–2. [DOI] [PubMed] [Google Scholar]
- 17. Nimmerjahn F, Ravetch JV. Fcγ Receptors: old friends and new family members. Immunity 2006; 24:19–28. [DOI] [PubMed] [Google Scholar]
- 18. Bruhns P, Iannascoli B, England P et al Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113:3716–25. [DOI] [PubMed] [Google Scholar]
- 19. Hogarth PM, Pietersz GA. Fc receptor‐targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov 2012; 11:311–31. [DOI] [PubMed] [Google Scholar]
- 20. Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The neonatal Fc receptor (FcRn). A Misnomer? Front Immunol 2019; 10:1540 10.3389/fimmu.2019.01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brüggemann M, Williams GT, Bindon CI et al Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med 1987; 166:1351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half‐life. Front Immunol 2019; 10:1296 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shields RL, Lai J, Keck R et al Lack of fucose on human IgG1 N‐linked oligosaccharide improves binding to human Fcgamma RIII and antibody‐dependent cellular toxicity. J Biol Chem 2002; 277:26733–40. [DOI] [PubMed] [Google Scholar]
- 24. Junttila TT, Parsons K, Olsson C et al Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2‐amplified breast cancer. Cancer Res 2010; 70:4481–9. [DOI] [PubMed] [Google Scholar]
- 25. Beck A, Reichert JM. Marketing approval of mogamulizumab. mAbs 2012; 4:419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goede V, Fischer K, Engelke A et al Obinutuzumab as frontline treatment of chronic lymphocytic leukemia: updated results of the CLL11 study. Leukemia 2015; 29:1602–4. [DOI] [PubMed] [Google Scholar]
- 27. Pereira NA, Chan KF, Lin PC, The SZ. ‘Less‐is‐more’ in therapeutic antibodies: afucosylated anti‐cancer antibodies with enhanced antibody‐dependent cellular cytotoxicity. mAbs 2018; 10:693–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sehn LH, Goy A, Offner FC et al Randomized Phase II trial comparing obinutuzumab (GA101) with rituximab in patients with relapsed CD20+ indolent B‐cell non‐Hodgkin lymphoma: final analysis of the GAUSS study. J Clin Oncol Off J Am Soc Clin Oncol 2015; 33:3467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Townsend W, Buske C, Cartron G et al Comparison of efficacy and safety with obinutuzumab plus chemotherapy versus rituximab plus chemotherapy in patients with previously untreated follicular lymphoma: updated results from the phase III Gallium Study. J Clin Oncol 2020; 38:8023. [Google Scholar]
- 30. Valentin Goede VG, Fischer Kirsten, Dyer MJS et al Overall survival benefit of obinutuzumab over rituximab when combined with chlorambucil in patients with chronic lymphocytic leukemia and comorbidites: final survival analysis of the CLL11 study. Vol. 43 European Hematology Library, 2018:562–3. [Google Scholar]
- 31. Nordstrom JL, Gorlatov S, Zhang W et al Anti‐tumor activity and toxicokinetics analysis of MGAH22, an anti‐HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res 2011; 13:R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rugo HS, Im S‐A, Wright GLS et al SOPHIA primary analysis: a phase 3 (P3) study of margetuximab (M) + chemotherapy (C) versus trastuzumab (T) + C in patients (pts) with HER2+ metastatic (met) breast cancer (MBC) after prior anti‐HER2 therapies (Tx). J Clin Oncol 2019; 37 10.1200/JCO.2019.37.15_suppl.1000. [DOI] [Google Scholar]
- 33. Rogers LM, Veeramani S, Weiner GJ. Complement in Monoclonal Antibody Therapy of Cancer. Immunol Res 2014; 59:203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Di Gaetano N, Cittera E, Nota R et al Complement activation determines the therapeutic activity of rituximab in vivo . J Immunol 2003; 171:1581–7. [DOI] [PubMed] [Google Scholar]
- 35. Sato F, Ito A, Ishida T et al A complement‐dependent cytotoxicity‐enhancing anti‐CD20 antibody mediating potent antitumor activity in the humanized NOD/Shi‐scid, IL‐2Rγ(null) mouse lymphoma model. Cancer Immunol Immunother CII 2010; 59:1791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goulet DR, Atkins WM. Considerations for the design of antibody‐based therapeutics. J Pharm Sci 2019. 10.1016/j.xphs.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rasche L, Menoret E, Dubljevic V et al A GRP78‐directed monoclonal antibody recaptures response in refractory multiple myeloma with extramedullary involvement. Clin Cancer Res 2016; 22:4341–9. [DOI] [PubMed] [Google Scholar]
- 38. Rasche L, Duell J, Morgner C et al The natural human IgM antibody PAT‐SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLOS ONE 2013; 8:e63414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stapleton NM, Andersen JT, Stemerding AM et al Competition for FcRn‐mediated transport gives rise to short half‐life of human IgG3 and offers therapeutic potential. Nat Commun 2011; 2:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Natsume A, In M, Takamura H et al Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res 2008; 68:3863–72. [DOI] [PubMed] [Google Scholar]
- 41. Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. mAbs 2010; 2:181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diebolder CA, Beurskens FJ, de Jong RN et al Complement is activated by IgG hexamers assembled at the cell surface. Science 2014; 343:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldberg BS, Ackerman ME. Antibody‐mediated complement activation in pathology and protection. Immunol Cell Biol 2020; 98:305–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Josephs DH, Spicer JF, Corrigan CJ, Gould HJ, Karagiannis SN. Epidemiological associations of allergy, IgE and cancer. Clin Exp Allergy 2013; 43:1110–23. [DOI] [PubMed] [Google Scholar]
- 45. Ferastraoaru D, Rosenstreich D. IgE deficiency is associated with high rates of new malignancies: results of a longitudinal cohort study. J Allergy Clin Immunol Pract 2020; 8:413–5. [DOI] [PubMed] [Google Scholar]
- 46. Josephs DH, Spicer JF, Karagiannis P, Gould HJ, Karagiannis SN. IgE immunotherapy: a novel concept with promise for the treatment of cancer. mAbs 2014; 6:54–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leoh LS, Daniels‐Wells TR, Penichet ML. IgE immunotherapy against cancer In: Lafaille JJ, Curotto de Lafaille MA, eds. IgE Antibodies: Generation and Function, vol. 388 Basel, Switzerland: Springer International Publishing, 2015:109–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Daniels TR, Martínez‐Maza O, Penichet ML. Animal models for IgE‐meditated cancer immunotherapy. Cancer Immunol Immunother 2012; 61:1535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gould HJ, Mackay GA, Karagiannis SN et al Comparison of IgE and IgG antibody‐dependent cytotoxicity in vitro and in a SCID mouse xenograft model of ovarian carcinoma. Eur J Immunol 1999; 29:3527–37. [DOI] [PubMed] [Google Scholar]
- 50. Josephs DH, Bax HJ, Dodev T et al Anti‐folate receptor‐α IgE but not IgG recruits macrophages to attack tumors via TNFα/MCP‐1 signaling. Cancer Res 2017; 77:1127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karagiannis SN, Wang Q, East N et al Activity of human monocytes in IgE antibody‐dependent surveillance and killing of ovarian tumor cells. Eur J Immunol 2003; 33:1030–40. [DOI] [PubMed] [Google Scholar]
- 52. Karagiannis SN, Bracher MG, Hunt J et al IgE‐antibody‐dependent immunotherapy of solid tumors: cytotoxic and phagocytic mechanisms of eradication of ovarian cancer cells. J Immunol 2007; 179:2832–43. [DOI] [PubMed] [Google Scholar]
- 53. Pellizzari G, Bax HJ, Josephs DH et al Harnessing therapeutic IgE antibodies to re‐educate macrophages against cancer. Trends Mol Med 2020; 26:615–26. [DOI] [PubMed] [Google Scholar]
- 54. Daniels TR, Leuchter RK, Quintero R et al Targeting HER2/neu with a fully human IgE to harness the allergic reaction against cancer cells. Cancer Immunol Immunother CII 2012; 61:991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Daniels‐Wells TR, Helguera G, Leuchter RK et al A novel IgE antibody targeting the prostate‐specific antigen as a potential prostate cancer therapy. BMC Cancer 2013; 13:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Platzer B, Elpek KG, Cremasco V et al IgE/FcεRI‐mediated antigen cross‐presentation by dendritic cells enhances anti‐tumor immune responses. Cell Rep 2015; 10:1487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Josephs DH, Nakamura M, Bax HJ et al An immunologically relevant rodent model demonstrates safety of therapy using a tumour‐specific IgE. Allergy 2018; 73:2328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams IP, Crescioli S, Sow HS et al In vivo safety profile of a CSPG4‐directed IgE antibody in an immunocompetent rat model. mAbs 2019; 12:1685349 10.1080/19420862.2019.1685349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spicer J, Basu B, Montes A et al Abstract CT141: Phase 1 trial of MOv18, a first‐in‐class IgE antibody therapy for cancer. Cancer Res 2020; 80 10.1158/1538-7445.AM2020-CT141. [DOI] [Google Scholar]
- 60. Leusen JHW. IgA as therapeutic antibody. Mol Immunol 2015; 68:35–9. [DOI] [PubMed] [Google Scholar]
- 61. Heemskerk N, van Egmond M. Monoclonal antibody‐mediated killing of tumour cells by neutrophils. Eur J Clin Invest 2018; 48:e12962 10.1111/eci.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Valerius T, Stockmeyer B, van Spriel AB et al FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood 1997; 90:4485–92. [PubMed] [Google Scholar]
- 63. Brandsma AM, Bondza S, Evers M et al Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front Immunol 2019; 10:704 10.3389/fimmu.2019.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Egmond M, van Vuuren AJ, Morton HC et al Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood 1999; 93:4387–94. [PubMed] [Google Scholar]
- 65. Pascal V, Laffleur B, Debin A et al Anti‐CD20 IgA can protect mice against lymphoma development: evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica 2012; 97:1686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Boross P, Lohse S, Nederend M et al IgA EGFR antibodies mediate tumour killing in vivo . EMBO Mol Med 2013; 5:1213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meyer S, Nederend M, Jansen JHM et al Improved in vivo anti‐tumor effects of IgA‐Her2 antibodies through half‐life extension and serum exposure enhancement by FcRn targeting. mAbs 2015; 8:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lohse S, Meyer S, Meulenbroek LAPM et al An anti‐EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo . Cancer Res 2016; 76:403–17. [DOI] [PubMed] [Google Scholar]
- 69. Kelton W, Mehta N, Charab W et al IgGA: A ‘cross‐isotype’ engineered human Fc antibody domain that displays both IgG‐like and IgA‐like effector functions. Chem Biol 2014; 21:1603–9. [DOI] [PubMed] [Google Scholar]
- 70. Borrok MJ, Luheshi NM, Beyaz N et al Enhancement of antibody‐dependent cell‐mediated cytotoxicity by endowing IgG with FcαRI (CD89) binding. mAbs 2015; 7:743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol 2013; 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu F, Ding H, Jin X et al FCGR3A 158V/F polymorphism and response to frontline R‐CHOP therapy in diffuse large B‐cell lymphoma. DNA Cell Biol 2014; 33:616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dantas E, Erra Díaz F, Pereyra Gerber P et al Low pH impairs complement‐dependent cytotoxicity against IgG‐coated target cells. Oncotarget 2016; 7:74203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lu J, Marjon KD, Mold C, Marnell L, Du Clos TW, Sun P. Pentraxins and IgA share a binding hot‐spot on FcαRI. Protein Sci Publ Protein Soc 2014; 23:378–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lu J, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc receptor‐Mediated immune responses. Front Immunol 2018; 9:2607 10.3389/fimmu.2018.02607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Luan Y, Yao Y. The clinical significance and potential role of C‐reactive protein in chronic inflammatory and neurodegenerative diseases. Front Immunol 2018; 9:1302 10.3389/fimmu.2018.01302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol 2016; 34:539–73. 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 78. Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs modulate the anti‐tumor activity of antibodies targeting the PD‐1/PD‐L1 axis. Cancer Cell 2015; 28:285–95. [DOI] [PubMed] [Google Scholar]
- 79. Sow HS, Benonisson H, Breukel C et al FcγR interaction is not required for effective anti‐PD‐L1 immunotherapy but can add additional benefit depending on the tumor model. Int J Cancer 2019; 144:345–54. [DOI] [PubMed] [Google Scholar]
- 80. Walunas TL, Lenschow DJ, Bakker CY et al CTLA‐4 can function as a negative regulator of T cell activation. Immunity. 1994; 1:405–13. [DOI] [PubMed] [Google Scholar]
- 81. Simpson TR, Li F, Montalvo‐Ortiz W et al Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti‐CTLA‐4 therapy against melanoma. J Exp Med 2013; 210:1695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Selby MJ, Engelhardt JJ, Quigley M et al Anti‐CTLA‐4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 2013; 1:32–42. [DOI] [PubMed] [Google Scholar]
- 83. Furness AJS, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol 2014; 35:290–8. [DOI] [PubMed] [Google Scholar]
- 84. Arce Vargas F, Furness AJS, Litchfield K et al Fc effector function contributes to the activity of human anti‐CTLA‐4 antibodies. Cancer Cell 2018; 33:649–663.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Korman AJ, Engelhardt J, Loffredo J et al Abstract SY09‐01: next‐generation anti‐CTLA‐4 antibodies. Cancer Res 2017; 77 10.1158/1538-7445.AM2017-SY09-01. [DOI] [Google Scholar]
- 86. Lo M, Kim HS, Tong RK et al Effector‐attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem 2017; 292:3900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chao MP, Takimoto CH, Feng DD et al Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol 2020; 9:1380 10.3389/fonc.2019.01380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wajant H. Principles of antibody‐mediated TNF receptor activation. Cell Death Diff 2015; 22:1727–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. White AL, Chan HTC, Roghanian A et al Interaction with FcγRIIB is critical for the agonistic activity of anti‐CD40 monoclonal antibody. J Immunol 2011; 187:1754–63. [DOI] [PubMed] [Google Scholar]
- 90. Wilson NS, Yang B, Yang A et al An Fcγ receptor‐dependent mechanism drives antibody‐mediated target‐receptor signaling in cancer cells. Cancer Cell 2011; 19:101–13. [DOI] [PubMed] [Google Scholar]
- 91. Li F, Ravetch JV. Antitumor activities of agonistic anti‐TNFR antibodies require differential FcγRIIB coengagement in vivo . Proc Natl Acad Sci 2013; 110:19501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Waight JD, Gombos RB, Wilson NS. Harnessing co‐stimulatory TNF receptors for cancer immunotherapy: current approaches and future opportunities. Hum Antibodies 2017; 25:87–109. [DOI] [PubMed] [Google Scholar]
- 93. Li F, Ravetch JV. Inhibitory Fcγ receptor engagement drives adjuvant and anti‐tumor activities of agonistic CD40 antibodies. Science 2011; 333:1030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic activity of agonistic, human anti‐CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell 2016; 29:820–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Verbeek JS, Hirose S, Nishimura H. The complex association of FcγRIIb with autoimmune susceptibility. Front Immunol 2019; 10:2061 10.3389/fimmu.2019.02061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4‐1BB: mechanistic rationale, clinical results, and future strategies. Blood 2018; 131:49–57. [DOI] [PubMed] [Google Scholar]
- 97. Byrne KT, Leisenring NH, Bajor DL, Vonderheide RH. CSF‐1R‐dependent lethal hepatotoxicity when agonistic CD40 antibody is given before but not after chemotherapy. J Immunol 2016; 197:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Compte M, Harwood SL, Muñoz IG et al A tumor‐targeted trimeric 4‐1BB‐agonistic antibody induces potent anti‐tumor immunity without systemic toxicity. Nat Commun 2018; 9:4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ganesan LP, Kim J, Wu Y et al FcγRIIb on liver sinusoidal endothelium clears small immune complexes. J Immunol 2012; 189:4981–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. White AL, Chan HTC, French RR et al Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 2015; 27:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Martinez T, Guo A, Allen MJ et al Disulfide connectivity of human immunoglobulin G2 structural isoforms. Biochemistry 2008; 47:7496–508. [DOI] [PubMed] [Google Scholar]
- 102. Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood 2016; 127:1097–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yu X, Chan HTC, Orr CM et al Complex interplay between epitope specificity and isotype dictates the biological activity of anti‐human CD40 antibodies. Cancer Cell 2018; 33:664–75.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yu X, Chan HTC, Fisher H et al Isotype switching converts anti‐CD40 antagonism to agonism to elicit potent antitumor activity. Cancer Cell 2020. 10.1016/j.ccell.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Merz C, Sykora J, Marschall V et al The hexavalent CD40 agonist HERA‐CD40L induces T‐cell‐mediated antitumor immune response through activation of antigen‐presenting cells. J Immunother 2018; 41:385–98. [DOI] [PubMed] [Google Scholar]
- 106. Thiemann M, Richards DM, Heinonen K et al A single‐chain‐based hexavalent CD27 agonist enhances T cell activation and induces anti‐tumor immunity. Front Oncol 2018; 8:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bulliard Y, Jolicoeur R, Windman M et al Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor‐targeting antibodies. J Exp Med 2013; 210:1685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol 2014; 92:475–80. [DOI] [PubMed] [Google Scholar]
- 109. Buchan SL, Dou L, Remer M et al Antibodies to costimulatory receptor 4‐1BB enhance anti‐tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 2018; 49:958–70.e7. [DOI] [PubMed] [Google Scholar]
- 110. Wang B, Wang L, Kothambawala T et al IGM‐8444 as a potent agonistic death receptor 5 (DR5) IgM antibody: induction of tumor cytotoxicity, combination with chemotherapy and in vitro safety profile. J Clin Oncol 2020; 38 10.1200/JCO.2020.38.15_suppl.3595. [DOI] [Google Scholar]
- 111. de Jong RN, Beurskens FJ, Verploegen S et al A novel platform for the potentiation of therapeutic antibodies based on antigen‐dependent formation of IgG hexamers at the cell surface. PLOS Biol 2016; 14:e1002344 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Overdijk MB, Strumane K, Buijsse AO et al Abstract 2391: DR5 agonist activity of HexaBody®‐DR5/DR5 (GEN1029) is potentiated by C1q and independent of Fc‐gamma receptor binding in preclinical tumor models. Cancer Res 2019; 79:2391. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data availability are not applicable to this review article.