

In this study we provide for the first time an analysis of Bcl‐2 levels in various B‐cell subpopulations and antiapoptotic mRNA expression assays after CD40L and IL‐21 stimulation in naïve B cells of CVID patients. B cells from healthy donors presented an increase in Bcl‐2 intracellular expression in the transition from naïve to switched memory B cells. In contrast, this increase was reduced in CVID patients. We found reduced Bcl‐2 protein levels in memory B cells from CVID patients. We further explored the possible alteration of this crucial pro‐survival signalling pathway in CVID patients, by analysing the expression levels of mRNAs from antiapoptotic proteins in naïve B cells, mimicking T‐cell dependent activation in vitro with CD40L and IL‐21. BCL‐XL mRNA levels were decreased, together with reduced levels of AICDA, after naïve B‐cell activation in CVID patients. Our data suggested a molecular mechanism for a tendency toward apoptosis in B cells from CVID patients in relation to NFkB driven transcription. Lower Bcl‐2 protein levels in memory B cells could compromise their long‐term survival, and a possible less activity of NFkB in naïve B cells, munable to increase BCL‐XL mRNA levels, thus not promoting survival in the germinal centers.

Keywords: anti‐apoptotic proteins, B cells, common variable immunodeficiency
Summary
Common variable immunodeficiency (CVID) is a primary immunodeficiency characterized by hypogammaglobulinemia and different degrees of B cell compartment alteration. Memory B cell differentiation requires the orchestrated activation of several intracellular signaling pathways that lead to the activation of a number of factors, such as nuclear factor kappa B (NF‐κB) which, in turn, promote transcriptional programs required for long‐term survival. The aim of this study was to determine if disrupted B cell differentiation, survival and activation in B cells in CVID patients could be related to defects in intracellular signaling pathways. For this purpose, we selected intracellular readouts that reflected the strength of homeostatic signaling pathways in resting cells, as the protein expression levels of the Bcl‐2 family which transcription is promoted by NF‐κB. We found reduced Bcl‐2 protein levels in memory B cells from CVID patients. We further explored the possible alteration of this crucial prosurvival signaling pathway in CVID patients by analysing the expression levels of mRNAs from anti‐apoptotic proteins in naive B cells, mimicking T cell‐dependent activation in vitro with CD40L and interleukin (IL)‐21. BCL‐XL mRNA levels were decreased, together with reduced levels of AICDA, after naive B‐cell activation in CVID patients. The data suggested a molecular mechanism for this tendency towards apoptosis in B cells from CVID patients. Lower Bcl‐2 protein levels in memory B cells could compromise their long‐term survival, and a possible less activity of NF‐κB in naive B cells, may condition an inabilityto increase BCL‐XL mRNA levels, thus not promoting survival in the germinal centers.
Introduction
Common variable immunodeficiency (CVID) is the most frequent symptomatic primary immunodeficiency (PID), with an estimated prevalence of 1 : 100 000 to 1 : 10 000 of the population [1]. CVID is characterized by hypogammaglobulinemia and recurrent severe infections. Some patients, however, also present autoimmune complications, lymphoproliferative manifestations, granulomas, enteropathy and increased risk of malignancy [2, 3, 4]. A defining feature in all CVID patients is some degree of impairment in B cell differentiation into plasma cells (maturation). Different patterns of B cell alterations have been identified based on the stage in which B cell differentiation is halted [5]. Most CVID patients present normal numbers of peripheral B cells but with a poorly differentiated memory B cell compartment.
Despite significant efforts to identify the monogenic causes of CVID, fewer than 20% of CVID patients present a monogenic defect [6]. Several genes have been associated with CVID, including CD19 [7], CD81 [8], CD20 [9], CD21 [10], ICOS [11], TACI [12], BAFFR [13], LRBA [14], CTLA4 [15], PI3KCD [16], IKAROS [17], NFKB2 [18] and some others (TWEAK, CD27, IL‐21, IL‐21R, NFKB1, PRKCD, PIK3R1, VAV1, RAC2, BLK and IRF2BP2 [19, 20, 21]. Loss‐of‐function variants of nuclear factor kappa B subunit 1 (NF‐κB1) have recently been proposed as the most common monogenic cause of CVID in a European cohort [22]. The recent general consensus is that the pathogenesis of CVID is complex and multi‐factorial in most patients, rather than the consequence of a monogenic defect [6]. A plausible hypothesis for the pathogenesis of CVID is that of a polygenic or even epigenetic heterogeneous predisposing genetic background [6, 23], inducing several cumulative defects and leading to the deregulation of B cell differentiation.
B cells recognize a multitude of pathogens through the diversity of the Bcell receptor (BCR) and generate specific antibodies with diverse functional characteristics. This variability relies upon the variability, diversity and joining [V(D)J] recombination mechanism in the first stages of development in bone marrow and on the somatic hypermutation (SHM) and class‐switch recombination (CSR) processes in the germinal centers (GC) [24, 25].
B cell survival depends upon various signals through the BCR, B cell activating factor receptor (BAFFR) and cytokine receptors that trigger internal signal transduction pathways and ultimately activate several transcription factors that modulate the transcription of target genes [25, 26, 27, 28, 29, 30]. The nuclear factor of kappa B light chain (NF‐κB) family consists of various subunits: NF‐κB1 (p105/p50), NF‐κB2 (p100/p52), Rel A (p65), Rel B and c‐Rel, a key transcription factor that translocates to the nucleus and enhances the expression of survival proteins such as the Bcl‐2 family, among others [31, 32, 33, 34]. The Bcl‐2 anti‐apoptotic family consists of several integral membrane proteins located in the mitochondria, endoplasmic reticulum and nuclear membranes. Membrane B cell receptors, as well as BAFFR and CD40L bound by their ligands, induce intracellular signaling pathways that lead to NF‐κB activation and anti‐apoptotic gene expression [35, 36]. NF‐κB activity is, therefore, the result of the convergence of several signaling pathways.
The aim of this study was to determine whether disturbed B cell differentiation, survival and activation in CVID patients could be related to defects in intracellular signaling pathways. For this purpose, we selected intracellular readouts measurable by flow cytometry that reflected the strength of homeostatic signaling pathways in resting cells, as the protein expression levels of the Bcl‐2 family which transcription is promoted by NFκB. We further explored the possible alteration of this crucial prosurvival signaling pathway in CVID patients by analysing the expression levels of mRNAs from anti‐apoptotic proteins in naive B cells, mimicking an in‐vitro T cell‐dependent activation with CD40L and interleukin (IL)‐21.
Materials and methods
Samples
Heparinized peripheral blood samples from CVID patients (according to the criteria of the European Society for Immunodeficiencies) and healthy donors (HD) were collected at La Paz University Hospital after obtaining their informed consent. The study was approved by the hospital’s ethics committee (PI‐2833 and 2009/3348/I), and adhered to the principles set out in the Declaration of Helsinki. The clinical data were obtained from the clinical records updated during routine medical visits for the diagnosis and follow‐up at the outpatient clinic of the Clinical Immunology Department.
Routine enumeration of lymphocyte subpopulations, including the analysis of the B cell peripheral compartment, was performed on a fluorescence activated cell sorter (FACSCalibur) and FACS Canto flow cytometer (BD Biosciences, San Jose, CA, USA).
Intracellular proteins analysed by flow cytometry
We performed an intracellular staining protocol to evaluate Bcl‐2 expression in naive (CD19+IgD+CD27−), unswitched memory B cells (USm) CD19+IgD+CD27+ and switched memory B cells (Sm) (CD19+IgD−CD27+). Briefly, 300 μl of heparinized whole blood was lysed with FACS lysing solution (BD Biosciences) and permeabilized with BD FACS permeabilizing solution 2, according to the manufacturer’s recommendations. Samples were stained with intracellular Bcl‐2 fluorescein isothiocyanate (FITC) (clone Bcl‐2/100; BD Biosciences), CD19 peridinin chlorophyll‐cyanin 5.5 (PerCP‐Cy5.5) (clone SJ25C1; BD Biosciences), CD27 allophycocyanin (APC) (clone M‐T271, Miltenyi Biotec, San Diego, CA, USA), IgD phycoerythrin (PE) (Southern Biotech, Birmingham, AL, USA) and an isotype control mouse IgG1 FITC κ (BD Biosciences). Samples were analysed by flow cytometry using FACSCalibur and CellQuest software (BD Biosciences).
Intracellular and surface staining for Bcl‐xl expression was performed in 100 µl of whole blood, based on the manufacturer’s instructions and the original from Chow et al. [37]. Whole blood was stained with CD27 BV421 (clone M‐T271; BD Biosciences), then fixed with 10% formaldehyde and treated with 0·1% Triton X‐100. Cells were permeabilized with 50% methanol on ice. The cells were then stained with intracellular marker and the other surface markers: CD3 APC (clone UCHT1; BD Biosciences), CD19 phycoerythrin (PE)‐Cy7 (clone J3‐119; Beckman Coulter, Pasadena, CA, USA), IgD PE (Southern Biotech) and Bcl‐xl rabbit antibody (clone 54H6), conjugated with Alexa Fluor 488 (Cell Signaling, Danvers, MA, USA). The specific isotype control anti‐rabbit IgG for Bcl‐xl (Cell Signaling) was added at the same concentrations as the specific antibodies. The mean fluorescence intensity (MFI) values for each protein were obtained over the specific isotype controls. Samples were acquired in FACS Canto II (BD Biosciences). We performed the data analysis by employing FlowJo software LLC (Ashland, OR, USA).
Naive B cell purification and cell culture
Freshly obtained peripheral mononuclear cells (PBMCs) were isolated after performing Ficoll density gradient centrifugation. Naive B cells were isolated from PBMCs using the MACS naive B cell isolation kit II in an Automacs (Miltenyi Biotech). Naive B cell enrichment was assessed using CD19 PerCP‐Cy5.5, CD27 FITC, CD3 APC (BD Biosciences) and IgD PE (Southern Biotech). We resuspended 1 × 105 purified naive B cells in 200 µl RPMI‐1640 supplemented medium (10% fetal calf serum, 100 U/ml penicillium, 100 µg/ml streptomycin and 2 mM glutamine). Cells were activated for 4 days with 2 μg/ml of MegaCD40L soluble human recombinant (Enzo Life Sciences, Farmingdale, NY, USA) and 100 ng/ml of human recombinant IL‐21 (gibco, Life Technologies, Carlsbad, CA, USA); unstimulated samples were performed in parallel. Cell cultures were maintained for 4 days at 37°C in a 5% CO2 atmosphere. Cell activation was measured with the monoclonal antibody combinations CD19 PerCP‐Cy5.5, CD27 FITC, CD69 PE (clone L78) and CD86 APC (clone 2331 Fun‐1) (BD Biosciences). Responsiveness to stimulation was calculated as the fold increase of MFI in CD69 and CD86 in stimulated cells to the same markers in the unstimulated cells (fold change = MFI‐stimulated/MFI‐unstimulated). The analysis was performed by flow cytometry using FACS Canto II (BD Biosciences) and the FlowJo Software (LLC).
Molecular biology
BCL‐2, BCL‐XL and AICDA gene expression levels were detected by quantitative real‐time polymerase chain reaction (qPCR). Total RNA was extracted using the miRNeasy micro Kit (Qiagen, Valencia, CA, USA) and quantified in the Nanodrop 2000c (Thermo Scientific, Waltham, MA, USA). C‐DNA was synthesized from 50 ng of total RNA using the high‐capacity RNA‐to‐c DNA kit (Applied Biosystems, Foster City, CA, USA). We performed qPCR using the TaqMan gene expression master mix (Applied Biosystems) in an Applied Biosystems 7900HT fast real‐time PCR system, with the primer/probe real‐time PCR assays for BCL‐2 (Hs00608023_m1), BCL‐XL (Hs00236329_m1), AICDA (Hs00757808_m1) and 18 S (Hs 99999901_s1) (all from Applied Biosystems). PCR fragments were amplified for 2 min at 50°C, 10 min at 95°C followed by 50 cycles consisting of 15 sec at 95°C and 1 min at 60°C. Samples were amplified in triplicate, and target gene expression was normalized with 18S ribosomal RNA. We calculated the relative expression using the 2−ΔΔCt method and employed SDS Software and RQ Manager (Applied Biosystems) for the data analyses. We calculated the relative mRNA levels as the ratio of the relative gene expression in stimulated cells at day 4 compared with the relative gene expression in the unstimulated cells at day 4 of the culture, expressing the results as the fold increase.
Statistics
We performed the data analysis using GraphPad Prism version 6.0 software (San Diego, CA, USA). Statistical differences between CVID patients and controls were determined using the non‐parametric Mann–Whitney test. Differences were considered statistically significant for P‐values < 0·05, < 0·01 and < 0·001.
Results
Anti‐apoptotic protein levels
To determine whether unbalanced expression of Bcl‐2 family proteins could contribute to the disturbance in B cell compartment from CVID patients, we first analysed Bcl‐2 anti‐apoptotic protein levels in different B cell subsets, naive (CD19+IgD+CD27−), USm B cells and Sm B cells, from 43 CVID patients and 41 healthy donors (HD). Naive B cells from CVID patients presented similar levels of intracellular Bcl‐2 protein as healthy donors (expressed as mean MFI = 10·98 versus 10·92). However, USm B cells from CVID patients showed reduced Bcl‐2 expression compared with healthy controls (mean MFI = 17·31 versus 18·72) and significant Bcl‐2 protein levels in Sm B cells (mean MFI = 14·57 versus 18·11 in HD) (P = 0·0021) (Fig. 1a,b). B cells from healthy donors presented an increase in Bcl‐2 intracellular expression from naive to Sm B cells. In contrast, this increase was reduced in CVID patients (P < 0·0001) (Fig. 1c). The classification of patients according to B cell phenotype showed impaired Bcl‐2 expression in Sm B cells from CVID with a reduction in total memory B cells (P = 0·0567) (mean MFI = 15·53 versus 18·11 in HD, Fig. 2a). In CVID patients with a severe reduction in Sm B cell counts (< 2%), Bcl‐2 protein levels were lower in the unswitched (mean MFI = 16·59 versus 18·72 in HD; P = 0·0431) and in Sm cells (mean MFI = 14·61 versus 18·11 in HD; P = 0·0202) (Fig. 2b). CVID patients with expansion of CD21low B cells (> 10%) also presented lower Bcl‐2 levels in Sm B cells compared with the Sm B cells in healthy donors (mean MFI = 12·92 versus 18·11; P = 0·0100) (Fig. 2c).
Fig. 1.
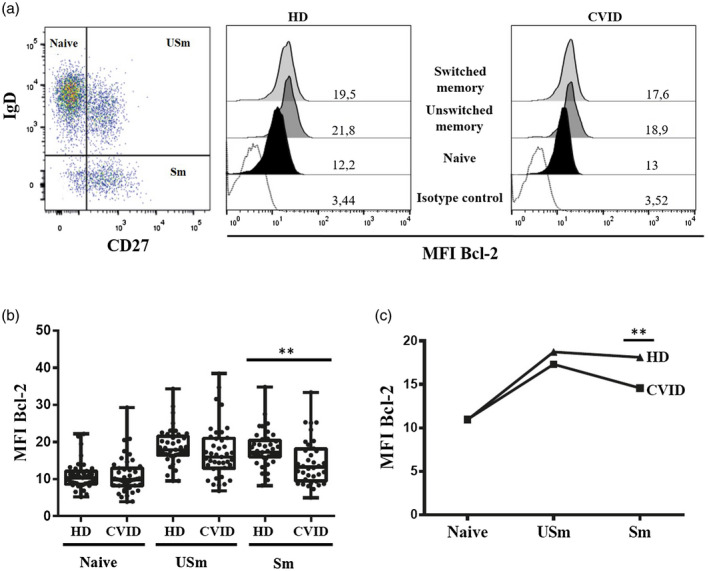
(a) Dot‐plots showing distribution of naive (CD19+IgD+CD27−), unswitched memory (USm) (CD19+IgD+CD27+) and switched memory (Sm) (CD19+IgD−CD27+) B cells. Histograms of representative healthy donor (HD) and common variable immunodeficiency (CVID) patient showing mean fluorescence intensity (MFI) of isotype control and MFI of Bcl‐2 in naive, USm and Sm B cells; (b) MFI of Bcl‐2 in naive, USm and Sm B cells. Box and whiskers represent mean and range. (c) Up‐regulation of Bcl‐2 protein expression in the transition from naive to Sm B cells. The P‐value is shown for the cases with a statistically significant difference using the Mann–Whitney test (*P < 0·05; **P < 0·01).
Fig. 2.
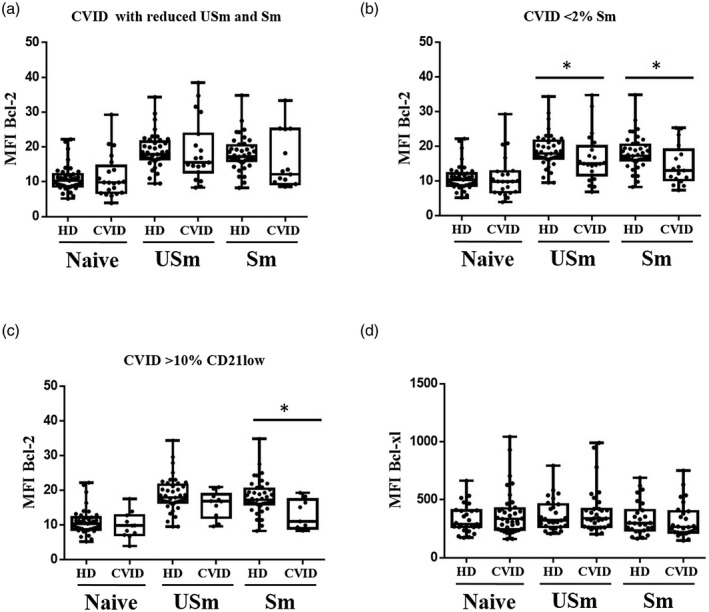
Mean fluorescence intensity (MFI) Bcl‐2 protein expression in naive, unswitched memory (USm) and switched memory (Sm) B cells according to B cell phenotype. (a) Common variable immunodeficiency (CVID) patients with reduced USm and Sm B cells. (b) CVID patients with severe reduction of Sm B cells (< 2%). (c) CVID patients with expansion of CD21low B cells (> 10%). (d) MFI Bcl‐xl protein expression in naive, USm and Sm B cells from HD and CVID patients. Box and whiskers represent mean with range. Significant P‐values using the Mann–Whitney test (*P < 0·05; **P < 0·01; ***P < 0·001).
To test whether the reduced Bcl‐2 protein levels were accompanied by other anti‐apoptotic proteins, we detected Bcl‐xl protein levels in B cell subsets from 37 CVID patients and 27 healthy donors. We detected no differences in MFI expression in naive, unSm memory B cells and Sm B cells between CVID patients and healthy donors (Fig. 2d).
Naive B cells from CVID presented reduced up‐regulation of anti‐apoptotic mRNA levels after activation with CD40L and IL‐21
To determine whether the reduced counts of Sm B cells together with the reduced Bcl‐2 protein levels were related to impaired B cell activation, we analysed the mRNA levels of Bcl‐2 and Bcl‐xl, two target genes of NF‐κB, upon naive B cell activation. Surface expression of the B cell activation proteins CD69 and CD86 were measured by flow cytometry as an internal control of proper naive B cell activation.
We examined the induction of target genes BCL‐2 and BCL‐XL together with AICDA, as a positive control of activation, in naive B cells after 4 days of in‐vitro stimulation with CD40L and IL‐21, mimicking a GC reaction (Fig. 3). CVID patients presented decreased BCL‐2 induction expressed as a fold change (mean = 1·9 versus 2·8) compared with healthy donors (although the difference was not statistically significant) (Fig. 3a). The relative expression of BCL‐XL induced after activation was diminished in CVID patients (mean = 4·72 versus 11·86 in HD; P = 0·0371) (Fig. 3b). AICDA was included in this study because AICDA was not expressed in naive B cells, but was induced after naive B cell activation during the GC reaction. We found that CVID patients and healthy donors expressed AICDA after naive B cell activation, but this increase was lower in CVID patients in comparison with healthy donors (P = 0·019) (Fig. 3c). The decreased up‐regulation of this mRNA did not appear to be related to a defective activation response of CVID naive B cells, due to similar induced levels of CD69 and CD86 after 4 days of stimulation with CD40L and IL‐21 in CVID patients and healthy controls (Fig. 4a,b).
Fig. 3.
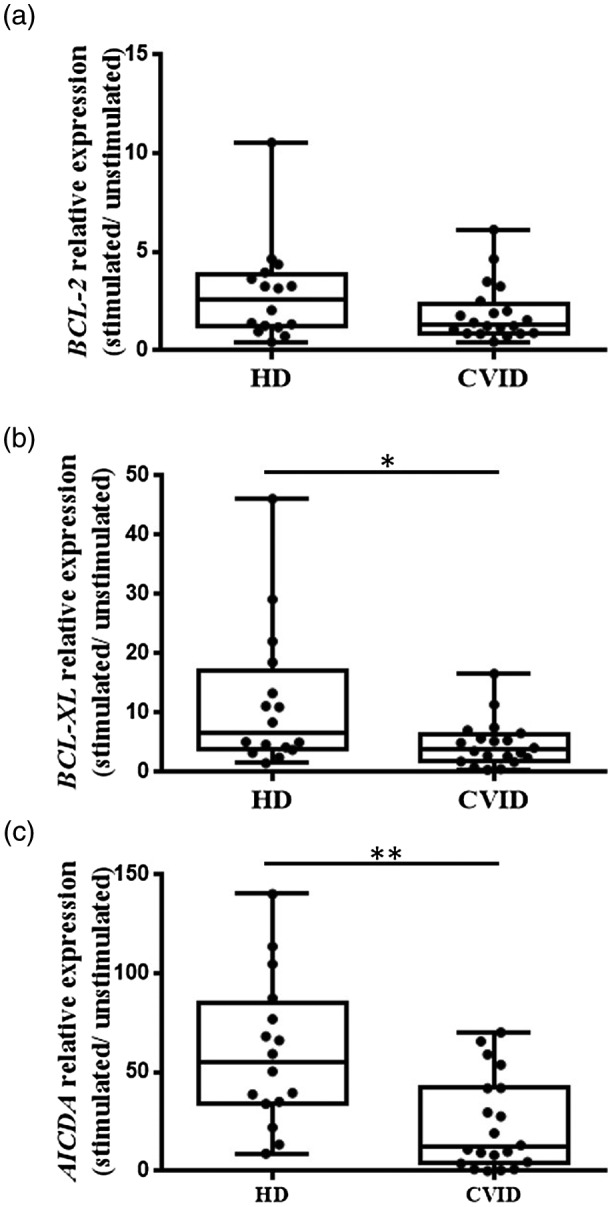
Relative gene expression (a) BCL‐2, (b) BCL‐XL and (c) AICDA in naive B cells from healthy donor (HD) and common variable immunodeficiency (CVID) patients after 4 days’ stimulation with CD40L and interleukin (IL)‐21. Relative mRNA levels were normalized to the expression of 18S ribosomal RNA and were calculated as the ratio of relative gene expression in stimulated cells at day 4 to the relative gene expression in unstimulated cells at day 4 of culture. Box and whiskers represent mean and range. Significant P‐values using the Mann–Whitney test (*P < 0·05; ** P < 0·01; ***P < 0·001).
Fig. 4.
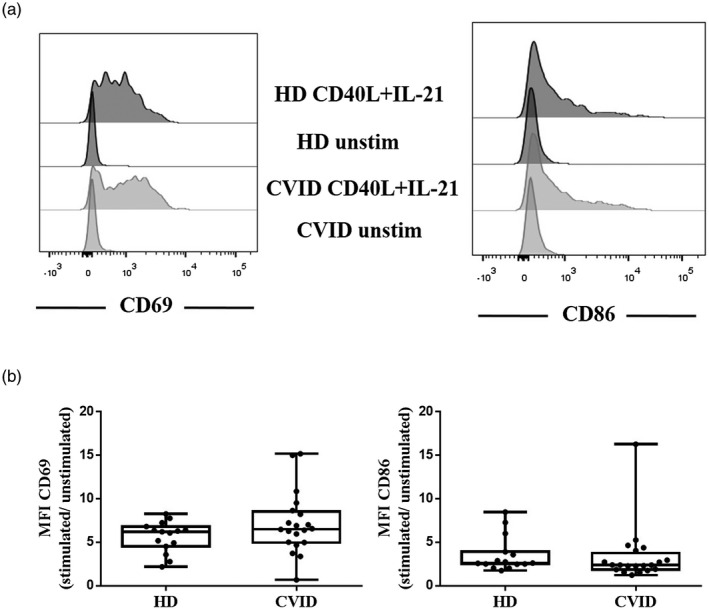
(a) Representative mean fluorescence intensity (MFI) in CD69 and CD86 activation markers expression in unstimulated and stimulated naive B cells with CD40L and interleukin (IL)‐21 from healthy donor (HD) and common variable immunodeficiency (CVID) patients. (b) No differences were detected in MFI expression in CD69 and CD86 in HD and CVID patients, represented as fold induction of naive B cells stimulated with CD40L and IL‐21 compared with unstimulated naive B cells. Box and whiskers represent mean and range.
Discussion
The pathogenesis of CVID is still poorly understood [1]. Adequate B cell activation allows the differentiation of naive B cells into memory B cells to enable a quick and efficient response during the immune reaction. The main differences between naive and memory B cells are the various gene expression profiles and the variety of stimuli they receive, including that from T cells [38]. The maintenance of circulating resting mature and memory B cells requires signaling via B cell antigen receptor (BCR), the tumor necrosis factor receptor (TNFR) family [via B cell activating factor receptor (BAFFR)] and cytokine receptors leading to the activation of key transcription factors, such as NF‐κB. The NF‐κB family consists of various subunits: NF‐κB1 (p105/p50), NF‐κB2 (p100/p52), Rel A (p65), Rel B and c‐Rel [31, 32]. Signaling through NF‐κB effectors eventually up‐regulates the expression of anti‐apoptotic members of the Bcl‐2 family, such as Bcl‐xl and Mcl‐1, with the reported increased of Bcl‐2 expression in memory B cells of particular relevance [35, 39].
Both anti‐apoptotic proteins Bcl‐2 and Bcl‐xl share functional homology, and their transcription is tightly regulated by NF‐κB activity but differs in the regulation of B cell survival in different subpopulations. Bcl‐2 has a relevant role for naive and memory B cell survival. In this study, we showed reduced Bcl‐2 protein levels in memory B cells from CVID patients, in particular in Sm B cells. In healthy donors, Bcl‐2 expression showed an increase in the transition from naive to Sm B cells, which was less evident in the B cells from CVID patients.
Whereas Bcl‐xl is weakly expressed in naive and memory B cells, Bcl‐xl is highly expressed in GC B cells where its regulator c‐Rel is constitutively expressed. Hence, the kinetics observed in our results barely showed changes in Bcl‐xl expression between the naive and Sm B cells [33]. To determine the signaling defect that prevents B cells from increasing the Bcl‐2 expression essential for progression to more differentiated stages, we activated naive B cells with CD40L and IL‐21. This stimulation leads to an activation and differentiation of naive B cells, mimicking the stimuli received during the in‐vivo GC reaction [40, 41]. B and T cell co‐operation activates both canonical and non‐canonical NF‐κB pathways for anti‐apoptotic BCL‐XL gene expression [34], given that c‐Rel (NF‐κB subunits) regulates the BCL‐XL promoter [33].
Activated naive B cells from CVID patients presented impaired up‐regulation of BCL‐2 and BCL‐XL mRNA levels. The increase in BCL‐XL levels appears to be crucial for GC B cells in vivo [33], precisely where BCL‐XL is highly expressed. Stimulated naive B cells from CVID patients showed impaired induction of BCL‐XL expression, which was even more diminished in the patients with reduced memory B cells. These results agree with those of Keller et al. [42], showing reduced induction of BCL‐XL after anti‐IgM stimulation in B cells from CVID patients. Recently, López‐Gómez et al. [43] also showed the B cell defect of CD27− B cells in CVID patients, analysing by flow cytometry the induction of Bcl‐2 and Bcl‐xl proteins after 20 h activation with anti‐BCR and anti‐CD40.
For the first time, to our knowledge, our results show alterations in Bcl‐2 protein expression in B cell subsets from CVID patients and reveal the complementary functions of the two anti‐apoptotic members of the Bcl‐2 family. Bcl‐2 has a relevant role in naive and memory B cells and Bcl‐xl in GC B cells. In the GC phase that we attempted to reproduce in vitro, the reduced BCL‐2 expression in this stage has been reported previously [33]. Bcl‐2 reduction can act as a tolerance mechanism to mediate apoptosis to eliminate possible autoreactive B cells that might be generated during the CSR and SHM process [44]. BCL‐2 has also been reported to be less inducible than BCL‐XL in memory B cells, even from healthy donors stimulated for 24 h with CD40L [45].
The discovery of NF‐κB signaling defects suggests a possible role for increased apoptosis as a contributor to the B cell defect in CVID. The inability to increase BCL‐XL mRNA levels would not favor survival in the GC maturation process, while expressing lower Bcl‐2 protein levels could compromise the long‐term survival of memory B cells. Other studies have recently described more related alterations in NF‐κB signaling in B cells from CVID patients. Keller et al. [42] identified impaired signaling in the canonical NF‐κB pathway in B cells from CVID patients, with diminished IκBα degradation and reduced p65 translocation, particularly in CVID patients with expanded CD21low B cells.
Deregulation in apoptosis in the CVID context has been previously reported, such as spontaneous apoptosis in B cells from CVID patients [46, 47], and the increased expression of cell death receptor FAS and TNF‐related apoptosis‐inducing ligand (TRAIL) in patients with low memory B cells [48]. T cells in CVID patients also showed higher levels of CD95, which was related to impaired survival and CD4+ lymphopenia [46, 49].
We also included AICDA in the study as a positive control, because AICDA is only expressed after naive B cell activation. The induction of AICDA expression could be mediated by both the canonical and alternative NF‐κB pathways [50]. Activated naive B cells from CVID patients and controls could increase AICDA expression, but the expression was dampened in CVID patients, particularly in those with reduced numbers of Sm B cells. Diminished AICDA induction together with low Sm B cells reveal the blockage in CSR and SHM present in CVID patients, with the reduced SHM rate observed in our CVID cohort [23].
Despite impaired BCL‐XL and AICDA induction, we found CD69 and CD86 expression levels comparable in the patients and controls after 4 days of activation of naive B cells with CD40L and IL‐21. The defects observed in vitro did not appear to be due to a lower responsiveness of the general activation in patients’ B cells. Previous studies have shown a defect in CD69 and CD86 induction; Lougaris et al. [51] observed reduced CD69 and CD86 expression in total PBMCs stimulated with cytosine– phosphate–guanosine (CpG). Groth et al. [52] also observed decreased expression in naive B cells from CVID patients stimulated with anti‐IgM and IL‐2. These results are only seemingly discordant with our findings if we consider that the subset of activated cells (total PBMCs), and the stimulation employed (CpG, anti IgM and IL‐2) was different from ours: naive B cells with CD40L and IL‐21, which more effectively reproduce the in‐vivo activation during the GC reaction and might generate a more powerful response [47].
In summary, having knowledge of the relevance of apoptosis in B cell survival, in this study we provide for the first time an analysis of Bcl‐2 levels in various B cell subpopulations and anti‐apoptotic mRNA expression assays after CD40L and IL‐21 stimulation in naive B cells of CVID patients. Our data suggest a molecular mechanism for this tendency to apoptosis, with potentially lower NF‐κB activation in the B cells of CVID patients (who might be unable to increase BCL‐XL mRNA levels, which does not favor survival in the GC maturation process) and memory B cells from CVID patients expressing lower Bcl‐2 protein levels (which could compromise their long‐term survival). Alterations in the induction of anti‐apoptotic proteins suggest defects in B cell signaling in CVID.
CVID is a complex disease with high variability in its clinical presentation and different penetrance, even when monogenic defects are present [21], which suggests that a polygenic heterogeneous predisposing genetic background [6], the involvement of epigenetic alterations [23, 53] and multiple cumulative defects could lead to the B cell deregulation present in CVID patients.
Disclosures
All authors state that they have no conflicts of interest.
Author contributions
E. L. and L. P. conceived the project. E. L. II, L. P. and J. T. designed the experiments. L. P. performed the experiments and O. P. provided technical assistance. L. P. and E. L. wrote the manuscript.
Acknowledgements
This study was supported by FIS PI16/01605 (Fondo de Investigación Sanitaria Instituto de Salud Carlos III, Madrid, Spain) and the Spanish Ministry of Economy and Competitiveness (MINECO; grant number SAF2009‐09899) and the European Regional Development Fund/European Social Fund FIS (FEDER/FSE). We would like to thank all the study participants. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Bonilla FA, Barlan I, Chapel H et al International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract [internet] 2016; 4:38–59. Available at: http://linkinghub.elsevier.com/retrieve/pii/S2213219815004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chapel H. Common variable immunodeficiency disorders (CVID) – diagnoses of exclusion, especially combined immune defects. J Allergy Clin Immunol Pract [internet] 2016; 4:1158–9. 10.1016/j.jaip.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 3. Chapel H, Lucas M, Lee M et al Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 2008; 112:277–86. [DOI] [PubMed] [Google Scholar]
- 4. Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet 2008; 372:489–502. [DOI] [PubMed] [Google Scholar]
- 5. Driessen GJ, Van Zelm MC, Van Hagen PM et al B‐cell replication history and somatic hypermutation status identify distinct pathophysiologic backgrounds in common variable immunodeficiency. Blood 2011; 118:6814–23. [DOI] [PubMed] [Google Scholar]
- 6. de Valles‐Ibanez G, Esteve‐Sole A, Piquer M et al Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol 2018; 9:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Zelm MC, Reisli I, Van Der Burg M et al An antibody‐deficiency syndrome due to mutations in the CD19 Gene. N Engl J Med 2006; 354:1901–12. [DOI] [PubMed] [Google Scholar]
- 8. Van Zelm MC, Smet J, Adams B et al CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest 2010; 120:1265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuijpers TW, Bende RJ, Baars PA et al CD20 deficiency in humans results in impaired T cell‐independent antibody responses. J Clin Invest 2010; 120:214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thiel J, Kimmig L, Salzer U et al Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol 2012; 129:801–810.e6. [DOI] [PubMed] [Google Scholar]
- 11. Grimbacher B, Hutloff A, Schlesier M et al Homozygous loss of ICOS is associated with adult‐onset common variable immunodeficiency. Nat Immunol 2003; 4:261–8. [DOI] [PubMed] [Google Scholar]
- 12. Salzer U, Chapel HM, Webster ADB et al Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet 2005; 37:820–8. [DOI] [PubMed] [Google Scholar]
- 13. Warnatz K, Salzer U, Rizzi M et al B‐cell activating factor receptor deficiency is associated with an adult‐onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA 2009; 106:13945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lopez‐Herrera G, Tampella G, Pan‐Hammarström Q et al Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 2012; 90:986–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schubert D, Bode C, Kenefeck R et al Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med [internet] 2014; 20:1410–6. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4668597&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elgizouli M, Lowe DM, Speckmann C et al Activating PI3Kδ mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol 2015; 183: 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuehn HS, Boisson B, Cunningham‐Rundles C et al Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med [internet] 2016; 374:1032–43. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26981933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu L, Hanson S, Gurugama P, Jones A, Clark B, Mohammed Ibrahim AA. Novel NFKB2 mutation in early‐onset CVID. J Clin Immunol 2014; 34:686–90. [DOI] [PubMed] [Google Scholar]
- 19. van Schouwenburg PA, Davenport EE, Kienzler AK et al Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol [internet] 2015; 160:301–14. 10.1016/j.clim.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenzweig S, Sarah Abraham R, Renee Forbes L et al Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016; 7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet [internet] 2016. Available at: http://jmg.bmj.com/lookup/doi/10.1136/jmedgenet‐2015‐103690 and http://www.ncbi.nlm.nih.gov/pubmed/27250108. [DOI] [PubMed] [Google Scholar]
- 22. Tuijnenburg P, Lango Allen H, Burns SO et al Loss‐of‐function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol 2018; 1:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. del Pino‐Molina L, Rodríguez‐Ubreva J, Torres Canizales J et al Impaired CpG demethylation in common variable immunodeficiency associates with B cell phenotype and proliferation rate. Front Immunol [internet] 2019; 10:1–11. Available at: https://www.frontiersin.org/article/10.3389/fimmu.2019.00878/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eibel H, Kraus H, Sic H, Kienzler A‐K, Rizzi M. B cell biology: an overview. Curr Allergy Asthma Rep [internet] 2014; 14:434 Available at: http://www.ncbi.nlm.nih.gov/pubmed/24633618. [DOI] [PubMed] [Google Scholar]
- 25. Berkowska MA, van der Burg M, van Dongen JJM, van Zelm MC. Checkpoints of B cell differentiation: visualizing Ig‐centric processes. Ann NY Acad Sci 2011; 1246:11–25. [DOI] [PubMed] [Google Scholar]
- 26. Harnett MM, Katz E, Ford CA. Differential signalling during B‐cell maturation. Immunol Lett 2005; 98:33–44. [DOI] [PubMed] [Google Scholar]
- 27. Reth M, Nielsen P. Signaling circuits in early B‐cell development, 1st edn. Adv Immunol [internet] 2014; 122:129–75. 10.1016/B978-0-12-800267-4.00004-3. [DOI] [PubMed] [Google Scholar]
- 28. Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol 2006; 18:263–275. [DOI] [PubMed] [Google Scholar]
- 29. Rowland SL, Leahy KF, Halverson R, Torres RM, Pelanda R. BAFF receptor signaling aids the differentiation of immature B cells into transitional b cells following tonic BCR signaling. J Immunol 2010; 185:4570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woo YJ, Yoon BY, Jhun JY et al Regulation of B cell activating factor (BAFF) receptor expression by NF‐ΚB signaling in rheumatoid arthritis B cells. Exp Mol Med 2011; 43:350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bonizzi G, Karin M. The two NF‐κB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25:280–8. [DOI] [PubMed] [Google Scholar]
- 32. De Silva NS, Anderson MM, Carette A et al Transcription factors of the alternative NF‐κB pathway are required for germinal center B‐cell development. Proc Natl Acad Sci USA [internet] 2016; 113:9063–8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27457956 and http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4987794 and http://www.pnas.org/lookup/doi/10.1073/pnas.1602728113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen C, Edelstein LC, Gélinas C, Ge L. The Rel/NF‐κB family directly activates expression of the apoptosis inhibitor Bcl‐x the Rel/NF‐κB family directly activates expression of the apoptosis inhibitor Bcl‐x L. Mol Cell Biol 2000; 20:2687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sen R. Control of B lymphocyte apoptosis by the transcription factor NF‐κB. Immunity 2006; 25:871–83. [DOI] [PubMed] [Google Scholar]
- 35. Opferman JT. Apoptosis in the development of the immune system. Cell Death Diff 2008; 15:234–42. [DOI] [PubMed] [Google Scholar]
- 36. Burlacu A. Regulation of apoptosis by Bcl‐2 family proteins. J Cell Mol Med [internet] 2003; 7:249–57. Available at: http://www.ncbi.nlm.nih.gov/pubmed/14594549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chow S, Hedley D, Grom P, Magari R, Jacobberger JW, Shankey TV. Whole blood fixation and permeabilization protocol with red blood cell lysis for flow cytometry of intracellular phosphorylated epitopes in leukocyte subpopulations. Cytom Part A 2005; 67:4–17. [DOI] [PubMed] [Google Scholar]
- 38. Davey AM, Pierce SK. Intrinsic differences in the initiation of B cell receptor signaling favor responses of human IgG(+) memory B cells over IgM(+) naive B cells. J Immunol [internet] 2012; 188:3332–41. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3311745&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol [lnternet] 2015; 15:149–59. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25677494. [DOI] [PubMed] [Google Scholar]
- 40. Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL‐21. J Immunol [internet] 2006; 177:5236–47. Available at: http://papers://91eab4a8-db2b-440e-8b23-89a059f56555/Paper/p1562. [DOI] [PubMed] [Google Scholar]
- 41. Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL‐21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp‐1 in human B cells. J Immunol [internet] 2013; 190:1827–36. Available at: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.1201678%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3563840&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keller B, Cseresnyes Z, Stumpf I et al Disturbed canonical nuclear factor of κ light chain signaling in B cells of patients with common variable immunodeficiency. J Allergy Clin Immunol 2017; 139:220–231.e8. [DOI] [PubMed] [Google Scholar]
- 43. López‐Gómez A, Clemente A, Cunill V, Pons J, Ferrer JM. IL‐21 and anti‐CD40 restore Bcl‐2 family protein imbalance in vitro in low‐survival CD27+ B cells from CVID patients. Cell Death Dis [internet] 2018; 9 10.1038/s41419-018-1191-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merino R, Ding L, Veis DJ, Korsmeyer SJ, Nuñez G. Developmental regulation of the Bcl‐2 protein and susceptibility to cell death in B lymphocytes. EMBO J 1994; 13:683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berard M, Casamayor‐Pallejà M, Billian G, Bella C, Mondière P, Defrance T. Activation sensitizes human memory B cells to B‐cell receptor‐induced apoptosis. Immunology 1999; 98:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iglesias J, Matamoros N, Raga S, Ferrer JM, Mila J. CD95 expression and function on lymphocyte subpopulations in common variable immunodeficiency (CVID); related to increased apoptosis. Clin Exp Immunol 1999; 117:138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Borte S, Pan‐Hammarström Q, Liu C et al Interleukin‐21 restores immunoglobulin production ex vivo in patients with common variable immunodeficiency and selective IgA deficiency. Blood 2009; 114:4089–98. [DOI] [PubMed] [Google Scholar]
- 48. Clemente A, Pons J, Lanio N, Matamoros N, Ferrer JM. CD27+ B cells from a subgroup of common variable immunodeficiency patients are less sensitive to apoptosis rescue regardless of interleukin‐21 signalling. Clin Exp Immunol 2013; 174:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saxon A, Keld B, Guo B, Hart NST, Lyon L. B cells from a distinct subset of patients with common variable CD38 expression, and undergo enhanced apoptosis. Clin Exp Immunol 1995; 95:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zan H, Casali P. Regulation of Aicda expression and AID activity. Autoimmunity [internet] 2013; 46:83–101. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3762583&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lougaris V, Baronio M, Vitali M et al B cell responses to CpG correlate with CXCL16 expression levels in common variable immunodeficiency. Sci World J 2012; 2012:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Groth C, Dräger R, Warnatz K et al Impaired up‐regulation of CD70 and CD86 in naive (CD27‐) B cells from patients with common variable immunodeficiency (CVID). Clin Exp Immunol 2002; 129:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rodríguez‐Cortez VC, Del Pino‐Molina L, Rodríguez‐Ubreva J et al Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naïve‐to‐memory B‐cell transition. Nat Commun 2015; 6:7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.