

Abstract
The discovery of new natural products that have some combination of unprecedented chemical structures, biological activities of therapeutic interest for urgent medical needs, and new molecular targets provides the fuel that sustains the vitality of natural products chemistry research. Unfortunately, finding these important new compounds is neither routine or trivial and a major challenge is finding effective discovery paradigms. This review presents examples that illustrate the effectiveness of a chemical genetics approach to marine natural product (MNP) discovery that intertwines compound discovery, molecular target identification, and phenotypic response/biological activity. The examples include MNPs that have complex unprecedented structures, new or understudied molecular targets, and potent biological activities of therapeutic interest. A variety of methods to identify molecular targets are also featured.
Graphical Abstract

I). Introduction
Discovery of a new natural product with an unprecedented chemical structure and potent bioactivity relevant to treating an unmet human medical need, mediated by interacting with a new molecular target, generates a wave of excitement and scientific activity among natural product chemists and in the associated fields of chemical synthesis, biosynthesis, genetics, cell biology, drug discovery and oftentimes ecology.1 The challenge is how to find these iconic biologically active natural products that capture scientist’s imaginations and drive multiple fields forward. Many creative and effective approaches to finding new natural product structures have emerged in recent years. These include genome mining,2,3 metabolomics,4 molecular networking,5 chemical prospecting using NMR,6 a genomisotopic approach,7 and MALDI imaging.8 One drawback to these structure-focussed methods is that they usually do not immediately associate biological activity with the newly discovered compounds, consigning the discovery of any biological activity they might have to the hit or miss proposition of subsequent unfocussed bioassay screens. Reversing the process and using either whole-cell phenotypic assays or in vitro enzyme modulation assays conducted on crude extract libraries as the first screen in the search for new natural products guarantees that the responsible natural products will have bioactivities of interest. This approach has been used for decades and it has proven to be highly effective. For example, NCI pioneered the use of panels of cancer cell-line-based cytotoxicity assays of crude extracts to not only find potent and cancer type-selective new anticancer drug leads but also to identify their putative molecular targets and mechanisms of action (MOA) by using the NCI COMPARE technology.9,10 New cell-based methodologies developed to aid bioactive natural product and/or molecular target/MOA discovery11 feature the use of image-based phenotypic screening (MorphoBase),12 proteomic profiling (ChemProteoBase),12 and gene expression profiles (Fusion)13 to identify MOAs, and the simultaneous use of image-based phenotypic screening and mass spectrometry-based metabolomics (Compound Activity Mapping)14 to profile crude extracts for hits with a combination of new chemical structures and interesting MOAs. The pharmaceutical industry has made extensive use of bioactive compound discovery with pure molecular targets in their robotic high throughput drug screening (HTS) programs as a way to be certain that pure synthetic compound hits would have biological activities of interest. This HTS approach has also been applied to natural product crude extracts but with mixed success because the timelines for identifying the active compounds in crude extracts is unpredictable and not always compatible with industrial drug discovery metrics.15
Chemical genetics (or chemical genomics) has emerged in the last two decades as a specialized field of scientific enquiry that seeks to establish the important relationship between a chemical structure, a desired cellular phenotypic response, and a cellular molecular target (Figure 1).16–19 Forward Chemical Genetics screens compounds for their ability to elicit a desired phenotypic response in cells or whole animals such as zebrafish and then sets out to identify the molecular target. Reverse Chemical Genetics starts by screening compounds for modulation of a pure molecular target of interest in an in vitro assay and then examines the active hits in a cellular or whole animal context to see if the hit molecule will elicit the desired phenotypic response. Both approaches have advantages and limitations. The big challenge of the Forward Chemical Genetics approach is identifying the molecular target. Methodology for target identification is improving rapidly, but this process is still not routine.11–14,20 A real strength of Forward Chemical Genetics is that it is completely unbiased in terms of molecular target so it is a good way to find potential new targets for treating diseases modeled by the phenotypic response selected in the screening assay. Another strength of phenotypic cell-based assays is that they provide important initial information about toxicity of hits and cellular permeability. A strength of Reverse Chemical Genetics screens is that they provide immediate information about the potency of modulation of a validated molecular target and counter screens against other related targets can evaluate limited target selectivity. A weakness of Reverse Chemical Genetics screens is that there is no information about cellular permeability, on-target engagement in cells, and off-target toxicity in cells.
Figure 1.

Marine Natural Products (red and blue) and their cellular molecular targets (pink) discovered using forward chemical genetics (red) and reverse chemical genetics (blue) approaches. PS: phosphatidylserine; PARP: poly ADP ribose polymerase; AR NTD: androgen receptor N-terminus domain; PKC: protein kinase C; PP2A: protein phosphatase 2A; Chk1: checkpoint kinase 1; SHIP1: Src homology 2-containing inositol 5-phosphatases; MRSA PK: methicillin resistant Staphylococcus aureus pyruvate kinase; SETD8: histone-lysine N-methyltransferase; IDO: indoleamine 2,3-dioxygenase; HPA: human pancreatic amylase.
As outlined above, most high-profile natural product discoveries feature unprecedented chemical structures, potent bioactivity relevant to treating an unmet human medical need, and a new molecular target. The combined chemical structure, phenotypic response, and molecular target focus of chemical genetics makes it an ideal platform for the discovery of new high profile bioactive natural products. The bioactivity screening component of both forward and reverse chemical genetics has been used effectively by natural product chemists as a discovery paradigm for decades and is not new. The emphasis on identification of molecular targets is more recent, but there are now many natural product discovery papers that also include target identification.11–14,21 Therefore, much current natural product discovery research is using a chemical genetics approach even if it is not explicitly described as such in publications. In order to discover new bioactive natural products that might be useful cell biology tools and or drug leads by using a chemical genetics approach, it is necessary to have a crude extract library that is rich in chemical diversity and structural novelty and bioactivity screens that are robust and highly predictive of effectiveness in a disease-treatment animal model.
This review will present some examples of bioactive natural product discovery from our research program that has involved using both forward and reverse chemical genetics approaches and multiple molecular targets (Figure 1). Our focus has been on marine invertebrate and marine microbial culture crude extracts, the most prolific sources of new marine natural products (MNPs) reported in the literature since the 1970s.22 The examples will illustrate the first natural product modulators of new and under exploited molecular targets and illustrate several different methods that have been used to identify or validate new molecular targets. An absolutely essential element of our discovery efforts has been collaborations with a committed group of biologists who are world leaders in their particular areas of human disease, have an interest in finding chemical research tools, drug leads, and new drug targets for these diseases, have developed biological assays to find such compounds and molecular targets, and have the patience to screen extract libraries and support assay-guided isolation of hits from crude extracts.
II). Marine Natural Products (MNPs) Discovered Using Reverse Chemical Genetics Screens
A). MNP Binds to Target and Phenotype Data Exists
i). SHIP1 Activators
The phosphoinositide 3-kinase (PI3K) signaling pathway plays a central role in regulating cellular survival, adhesion, movement, proliferation, differentiation, and end cell activation. Aberrant dysregulated activation of the PI3K pathway leads to inflammatory/immune disorders and cancer and as a consequence there have been extensive efforts in the Pharma industry to find selective PI3K inhibitors to control PI3K signaling.23 PI3K becomes activated in response to binding between extracellular ligands and their membrane bound receptors and in response it phosphorylates phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to generate the important second messenger phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3 or PIP3). PIP3 stimulates signaling by interacting with other downstream kinases such as Akt that becomes phosphorylated during active signaling and acts as a useful On/Off biomarker for the pathway. The normal levels of PIP3 in cells is very low, but it is rapidly synthesized from PI-4,5-P2 in response to extracellular stimuli. Under normal conditions, the concentration of PIP3 is balanced by the relative rates of formation via PI3K phosphorylation of PI-4,5-P2 and destruction via hydrolysis back to PI-4,5-P2 by the tumor suppressor phosphatase and tensin homolog (PTEN) or hydrolysis to PI-3,4-P2 by Src homology 2-containing inositol 5-phosphatases (SHIP1, sSHIP, and SHIP2).
Our biological collaborators proposed that activating the phosphatase SHIP1 rather than inhibiting the kinase PI3K should be a promising new approach to inhibiting the aberrant PI3K signalling that leads to inflammation and blood cancers.24 SHIP1 is only found in hematopoietic (blood) cells and this restricted cell-type distribution should limit the effects of a highly selective SHIP1 agonist to target cells. Activation of a phosphatase was also expected to act via an allosteric effect rather than by binding at the catalytically active site. Most kinase inhibitors are competitive inhibitors of ATP binding and there is a high degree of homology at ATP binding sites in different kinases making it challenging to find selective PI3K inhibitors. It is not expected that there would be high degree of homology at allosteric activation sites on phosphatases, increasing the chances of finding a selective drug candidate acting by inhibition of PI3K signalling by allosteric activation of SHIP1. SHP099 discovered in a screen of a synthetic chemical library is a potent and selective allosteric inhibitor of the tyrosine phosphatase SHP2 that has subsequently been reported, supporting the concept of allosteric modulation of a phosphatase as a drug discovery paradigm.25 However, at the outset of our work there were no literature reports of activation of any phosphatase as a therapeutic approach to inhibiting a signaling pathway.
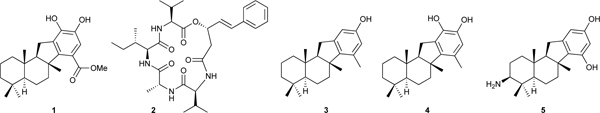
Our crude extract library was screened for SHIP1 activators using recombinant SHIP1 in a colorimetric kinetic assay that measured the rate of conversion of inositol-1,3,4,5-tetrakisphosphate to inositol-1,3,4-trisphosphate.24 The meroterpenoid pelorol (1) isolated from the sponge Dactylospongia elegans collected in Papua New Guinea (PNG)24 and a new cyclic depsipeptide turnagainolide B (2)26 isolated from laboratory cultures of a marine Bacillus sp. isolate from British Columbia (BC) marine sediments were found to be potent SHIP1 activators. Pelorol (1) was unknown when we isolated it, but while we were examining its biology and filing patent applications it was reported by several other groups.27,28 We have synthesized many SHIP1 activating analogs of pelorol (1) including MN100 (3), AQX16A (4), and AQX151 (5) that have been used as tools to validate SHIP1 as a new therapeutic target.29,30 Our collaborator Krystal and his co-workers made a SHIP1 (−/−) knockout mouse line that provided access to SHIP1 (−/−) macrophages and SHIP1(−/−) mast cells as biological tools.29
A number of cell-based assays were used to show that pelorol analogs exhibited selectivity for SHIP1 activation and generated phenotypic responses consistent with inhibiting stimulated activation of PI3K signaling.29 The synthetic pelorol analog 16A (4) activated recombinant SHIP1 in vitro as strongly as pelorol (1) but did not activate the closely related phosphatase SHIP2, demonstrating good selectivity. Measurement of the cellular lipids PIP3 and PI-3,4-P2 in macrophages that had been stimulated with lipopolysaccharide (LPS) in the presence or absence of AQX16A (4) showed that LPS induced a 3–5 fold increase in PIP3 levels, but this increase was abolished in the presence of AQX16A (4) and there was a corresponding increase in the PIP3 hydrolysis product PI-3,4-P2. AQX16A (4) was also shown to inhibit TNFα production and Akt phosphorylation in macrophages stimulated by LPS in a dose dependent manner in SHIP1(+/+) macrophages but not in SHIP1(−/−) macrophages, which are the expected phenotypic responses for inhibition of PI3K signaling via activation of SHIP1. It was shown using truncated SHIP1 protein analogs that activation of SHIP1 by MN-100 (3) required the C2 domain, the binding site of PI-3,4-P2 the endogenous allosteric activator of SHIP1, confirming that MN100 was also working as an allosteric activator that binds to the endogenous allosteric activator site. AQX151 (5) showed potent anti-inflammatory activity with a clear dose response (IC50 0.1 μM) via oral administration in a standard passive cutaneous mouse model of inflammation.30 The biological activity data obtained for pelorol (1) and its synthetic analogs described in part above provided proof of principle confirmation that SHIP1 is a viable new anti-inflammatory drug target and that allosteric activation of a phosphatase is a promising approach to controlling signaling pathways stimulated by dysregulated activation of kinases. Pelorol (1), synthetic analogs of pelorol, and turnagainolide B (2), the first known SHIP1 activators, represent interesting cell biology tools and antiinflammatory and anticancer drug leads.
ii). MRSA PK inhibitors
Infection with methicillin-resistant Staphylococcus aureus (MRSA) is a major health problem in hospitals, long-term care facilities, and community medicine settings worldwide.31,32,33 New antibiotics employing alternate strategies to prevent drug resistance are urgently needed. Our collaborators Reiner et al. explored the MRSA protein interactome and identified the hub protein MRSA pyruvate kinase (MRSA PK) as a promising new antibiotic drug target.34,35 Highly connected hub proteins are considered to be associated with cellular essentiality and the removal of a hub protein from a protein interaction network (PIN) is likely to lead to lethality.36 Therefore, it is thought that hub protein antibacterial targets may be less likely to develop resistance than conventional targets because their central network positions should make them less tolerant of mutations. The majority of antibiotics already in clinical use hit targets outside of the hub-core of the protein interaction network (PIN), so targeting a hub protein represents a largely unexplored approach to overcoming antibiotic resistance.
MRSA PK was found to be one of the most highly connected proteins in the MRSA PIN and it has not yet been exploited as an antibiotic target.34 In addition to being a hub protein, MRSA PK has a number of other attributes that make it an attractive new antibacterial target. First, it is the product of a single-copy gene and it has been shown to be essential for S. aureus viability by PK antisense and gene disruption experiments. Second, a high level of MRSA PK enzymatic activity is observed during the exponential phase of S. aureus growth, which is consistent with its essential role in bacterial replication. Third, there are significant differences in the protein sequences and structures between MRSA PK and the corresponding human isozymes creating opportunities for selective inhibition of the bacterial target, a requirement for a useful antibiotic.
MRSA PK is a tetramer that is allosterically activated by AMP and ribose-5-phosphate, which promotes the formation of the active R state from the inactive T state by inducing a symmetrical 6º rocking motion of the rigid A and C domain cores of the monomeric units relative to each other at their contact points in the tetramer (Figurer 2).34,35 There is notable sequence divergence and unique amino acid deletion differences between MRSA PK and human PK isoforms in the A and C monomer domains that are responsible for the A-A large interface and C-C small interface binding regions in the tetramers, suggesting potential for selectivity.
Figure 2.

Structure of S. aureus PK in complex with compound 6. A, tetrameric structure of PK illustrating the ligand density at the small interface. Position of active and effector sites are for reference only. B, interface binding pocket. Refined ligand position shown with ligand omit map (green). Anomalous map calculated from data collected at bromine edge (purple) indicates the position of the ligand bromine atoms. Residues lining the binding pocket from chain A are labeled. Hydrogen bonds between Ser362 and indole nitrogens are marked in red. C and D, comparison of S. aureus (C) and representative human PK structure (PDB 1ZJH) (D) illustrating closer packing of helix 340–350 and differences in the primary structure of this helix and the interface binding pocket, which contribute to selectivity. This research was originally published in the Journal of Biological Chemistry. R. Zoraghi et al. Methicillin-resistant Staphylococcus aureus (MRSA) Pyruvate Kinase as a Target for Bis-indole Alkaloids with Antibacterial Activities. J. Biol. Chem. 2011; 286, 44716–44725. © the American Society for Biochemistry and Molecular Biology.
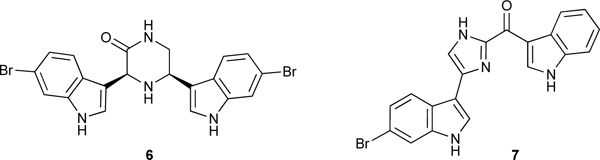
Our crude extract library was screened against recombinant MRSA and human PK’s enzymatic activity. Extracts of the sponge Topsentia pachastrelloides collected off the coast of South Africa yielded the known compounds cis-3,4-dihydrohamacanthin B (6) and bromodeoxytopsentin D (7) as potent (IC50’s 16–60 nM vs MRSA PK) and selective (166 – 600 fold compared with human isoforms) MRSA PK inhibitors that also showed in vitro inhibition of MRSA strains with MIC’s in the range of 6 – 12 μM.37
The apo- and cis-3,4-dihydrohamacanthin B (6)-bound MRSA PK structures were solved by x-ray diffraction analysis (Figure 2).37 The structure with bound 6 showed a small but significant conformational change in the relative orientation of the monomers in the tetrameric structure compared with the apo protein. Single molecules of the ligand 6 were found bound at each of the two small C-C interface domains in the symmetrical tetrameric structure, rather than at the catalytic or allosteric effector sites, suggesting a noncompetitive allosteric mechanism of inhibition. It appears that binding of 6 at the C-C interface prevents the conformational change from the inactive T state to the active R state required for PK enzyme activity. Comparison of the MRSA PK crystal structure with the structures of PKs from higher eukaryotes revealed a much tighter packing in the small C-C interface in the eukaryotic PKs that significantly alters the binding pocket occupied by cis-3,4-dihydrohamacanthin B (6) in MRSA PK, making it smaller and much less accessible to ligand binding in the eukaryotic versions. In addition, changes in amino acid residues in the eukaryotic PK C regions compared with the MRSA PK result in loss of potential hydrogen bonding interactions between Ser362 and ligand indole nitrogen atoms. Taken together, these differences in PK structure provide in part an explanation for the selectivity of 6 for inhibition of MRSA PK compared with human PK isoforms and offer opportunities for further refinement of the ligand binding. A number of investigators have used the lead structures 6 and 7 to guide the synthesis of MRSA PK inhibitory antibiotics containing the 6-bromoindole structural motif.38,39
iii). SETD8 Inhibitors
Histone methyl transferases catalyze the transfer of methyl groups from the co-factor S-adenosylmethionine (SAM) to the side chain amino groups of lysine residues in the histone proteins that make up the core of nucleosomes, the basic building blocks of chromatin. Methylation of histone lysine residues plays an important role in epigenetic regulation of gene expression.40 SETD8, a monomethyltransferase that monomethylates the side chain amino group of lysine 20 of histone H4 (H4K20), is overexpressed in various cancers and may be involved in carcinogenesis.41,42 Finding selective histone methyl transfer inhibitors that bind at the SAM site has emerged as an attractive frontier for anticancer drug development.43 At the outset of this project, there were no literature reports of SETD8 inhibitors that targeted the SAM binding site. A screen of our crude extract and pure MNP libraries by our collaborators Arrowsmith et al. identified the highly hydroxylated polyketide nahuoic acid A (8) as a SETD8 inhibitor.43,44 Nahuoic acid A (8) and the closely related congeners B to E (9-12) are produced in culture by a Streptomyces sp. obtained from a marine sediment collected in PNG. The carbon skeleton of nahuoic acid A (8) had not been previously encountered in polyketide natural products.
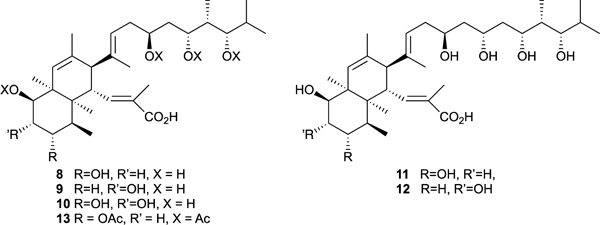
Nahuoic acid A (8) inhibited SETD8 in vitro with an IC50 of 6.5 μM and a kinetic analysis of the inhibition showed that 8 is a competitive inhibitor of SAM binding with a Ki value of 2 μM but not a competitive inhibitor of peptide substrate binding.44 It had no significant inhibitory effect on the activity of other protein methyltransferases such as G9a, EHMT1, SETD7, SUV39H2, SUV420H1, SUV420H2, DOT1L,PRMT3, and PRMT5 and MLL complexes demonstrating strong target selectivity. A loss of SETD8 activity in osteosarcoma U2OS cells is known to trigger cell proliferation defects.45,46 Exposure to nahuoic acid A (8) and the peracetate prodrug 13 resulted in a significant decrease in U2OS cell proliferation with modest potency (8: IC50 65 μM; 13: IC50 39 μM).44 Immunoblotting was used to examine if nahuoic acid A (8) was inhibiting SETD8 inside the U2OS cells. As a control, it was shown that siRNA depletion of SETD8 led to a decrease in both K4K20me1 and H4K20 me3 levels. Treatment of U2OS cells with nahuoic acid A (8) at its IC50 value also led to a significant decrease in the levels of both K4K20me1 and H4K20 me3 levels but did not decrease the levels of other methylated histone lysine marks. U2OS cells exposed to nahuoic acid at its proliferation IC50 concentration displayed an abnormal accumulation at the entry of S phase that mirrored the effect of RNAi depletion of SETD8 in the U2OS cells. Therefore, it was concluded the observed cellular toxicity of nahuoic acid A (8) in U2OS cells was linked to its ability to permeate the cells and selectively inhibit SETD8. The novel chemical structure, SAM competitive binding to SETD8, the in vitro histone methyl transferase selectivity, and the osteosarcoma U2OS proliferation inhibition phenotype involving selective SETD8 inhibition and abnormal accumulation in S phase are exactly the desired attributes of a drug lead and chemical biology tool for the potential new anticancer drug target SETD8. However, the lack of required potency of nahuoic acid A (8) will limit its usefulness as a cell biology tool or drug candidate. This challenge has been recognized by the synthetic chemistry community who are developing methodology to access the nahuoic acid pharmacophore and that effort will hopefully lead to more potent and, therefore, more useful synthetic analogs.47,48
B). NP Binds to Target – No Cell-based Phenotype Data
i). IDO inhibitors
It has been proposed that indoleamine-2,3-dioxygenase (IDO) plays a central role in the immune escape that is a characteristic of both mammalian fetal development and solid tumor cancer progression.49,50,51 IDO catalyzes the conversion of tryptophan into N-formylkynurenine in the first and rate limiting step in the catabolism of this essential amino acid. Munn et al. were the first to demonstrate a link between IDO and immune escape when they found that treatment of pregnant mice with the weak IDO inhibitor 1-methyl tryptophan (14) removed the toleragenic state protecting fetal tissue from the maternal immune system.52 T cells are very sensitive to local tryptophan concentrations. In environments where tryptophan has been depleted by IDO, killer T cells cannot be activated by antigens and they undergo G1 cell cycle arrest leading to apoptosis and immunosuppression. IDO is overexpressed in many tumor cell types and it has been found that increased expression of IDO in tumor cells is correlated with poor prognosis for survival in patients with solid tumor cancers.51,52,53 In an early study, Muller et al found that administration of the IDO inhibitor 14 together with paclitaxel led to regression of tumors in mouse models that did not respond to paclitaxel alone.55 At the outset of our IDO inhibitor discovery efforts, all of the interest, including a clinical trial, was focussed on 14, which is not very water soluble or potent, so there was clearly a need for better IDO inhibitors that could be used as cell biology tools to validate the target and as drug leads.
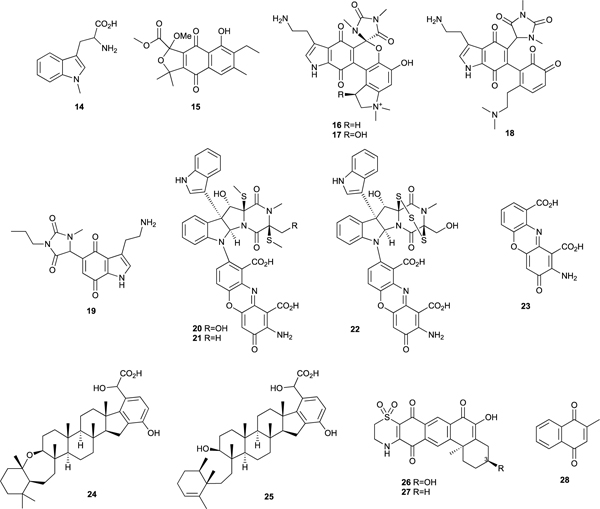
A screen of our crude extract library for inhibitors of IDO uncovered a number of potent hits. One of the first IDO inhibitory natural products discovered in this program was annulin C (15) obtained from the marine hydroid Garveia annulata collected in BC coastal waters.56 It had a Ki for IDO inhibition of 140 nM making it significantly more potent than 14 (Ki 34 μM). Exiguamine A (16) was subsequently isolated from the marine sponge Neopetrosia exigua collected in PNG.57 It has a complex hexacyclic alkaloid skeleton without precedent among known natural products that contains both the indole and quinone structural elements found in 14 and 15. Exiguamine A (16) had a Ki of 41 nM for inhibition of IDO making it the most potent inhibitor known at the time of its discovery. We subsequently identified the hydroxylated analog exiguamine B (17) from the same sponge extract and Trauner synthesized exiguamines A (16) and B (17) as well as the putative biosynthetic intermediate 18 and showed that 17 and 18 had Ki’s of ≈ 80 nM for IDO inhibition.58 We synthesized the simplified pharmacophore analog 19 that is much more accessible than the natural products and still has a potent Ki of ≈ 200 nM.59
Plectosphaeroic acids A (20), B (21), and C (22), produced in culture by the fungus Plectosphaerella cucumerina collected from marine sediments in BC coastal waters were found to inhibit IDO with IC50’s ≈ 2 μM.60 It was found that cinnabarinic acid (23), a substructure of these complex alkaloids, was the active IDO inhibitory pharmacophore. The meroterpenoids halicloic acids A (24) and B (25) isolated from the marine sponge Haliclona sp. collected in the Philippines showed modest IDO inhibition, with IC50 values of ≈ 10 μM.61 Xestosaprol A (26), another new meroterpenoid isolated from the sponge Xestospongia vansoesti, showed inhibition of IDO with an IC50 of 4 μM.62 We prepared a synthetic analog 27, that differs from 26 only by the absence of the C-3 hydroxyl substituent. 27 had an IC50 for IDO inhibition of 110 nM, 40-fold lower than the inspirational natural product 26, making it one of the most potent IDO inhibitors known.62
There is no cell-based disease model phenotypic assay for measuring the immunosuppressive properties of IDO inhibitors. The only disease model that exists is a very complex mouse cancer model. Based on our discovery of the annulins as potent in vitro IDO inhibitors, Muller and Pendergast tested a series of commercially available naphthoquinones for IDO inhibition and they discovered that menadione (28) (vitamin K3) showed good in vitro activity.63 They also showed, that like 14, the combination of 28 and paclitaxel regressed mouse tumors that did not respond to paclitaxel or 28 alone and they used an IDO knockout mouse line to show that IDO was essential for this activity. These mouse studies on an annulin analog provided phenotypic support for the relevance of the in vitro activity of an IDO inhibitor discovered in a reverse chemical genetics screen.
ii). Human Pancreatic Amylase Inhibitor
Diabetes is a serious disease caused by the inability to produce insulin or effectively respond to the insulin being produced. It currently affects an estimated 425 million people worldwide. Type II diabetes, that accounts for 90% of all diabetes cases, results from pancreatic β-cell impairment and a gradual loss of cellular responsiveness to insulin. Drugs such as metformin and acarbose that lower blood glucose levels independent of insulin by controlling the influx of dietary glucose into the blood stream are the preferred therapeutic intervention.64 Starch is the major source of glucose in human diets. The digestion of starch starts with salivary amylases that hydrolyze insoluble starch polymers into shorter oligomers. Pancreatic α-amylase in the small intestine cleaves starch into the disaccharide maltose and the trisaccharide maltotriose. This mixture then passes to the brush border of the small intestine where it is processed into glucose by brush border glucosidases and the resulting glucose is transferred to the blood stream. The commonly used therapeutics acarbose and miglitol are non-selective inhibitors of both the amylases that break down starch polymers and the brush border glucosidases. They prevent starch breakdown as desired, but they also prevent the hydrolysis of dietary sugars such as sucrose. These drugs have side effects including diarrhea, hepatotoxicity, bloating, and flatulence, which lead to poor compliance. It is thought that displacement of dietary di- and trisaccharides to the lower gut leads to osmotic-induced diarrhea and fermentation that causes flatulence and bloating.
Our collaborators Withers and Brayer proposed that highly selective human pancreatic amylase (HPA) inhibitors would minimize or eliminate the aforementioned side effects.65 Inhibition of only HPA and not the brush border glucosidases would prevent conversion of starch to glucose lowering blood sugar levels as desired, but it would allow brush border glucosidase catalyzed hydrolysis of dietary disaccharides such as sucrose to glucose that would enter the blood stream. The net result would be transport of intact starch but not disaccharides to the lower gut. Starch is not fermented in the lower gut and it does not contribute to osmotic-induced diarrhea, so there would be none of the diarrhea, bloating, or flatulence caused by accumulation of disaccharides when the brush border glucosidases are inhibited.

A screen of our crude extract library for potent and selective -HPA inhibitors identified an extract from the Caribbean sea anemone Stichodactyla helianthus as the most promising hit.66 The active component in the extract was found to be the peptide helianthamide (29) whose structure was elucidated by x-ray diffraction analysis of recombinant helianthamide bound to porcine pancreatic amylase (Figure 3). Helianthamide is a novel linear 44 amino acid peptide composed of a four-stranded antiparallel beta sheet with three disulfide bonds in a 1–5, 2–4, 3–6 topology. A BLAST search revealed no peptide sequence-based homologs of helianthamide. However, helianthamide (29) is related to the mammalian and avian antimicrobial peptides in the β-defensin family67 that are characterized by a cationic and amphipathic nature, a 1–5, 2–4, 3–6 disulfide topology, an antiiparallel β-sheet core, and toxic and antibacterial bioactivities. ShI, a β-defensin-like toxin, has also been isolated from S. helianthus.68
Figure 3.

Crystal structure of helianthamide (29) (blue and red spheres) bound to the active cleft of porcine pancreatic amylase. This research was originally published in ACS Central Science. C. Tysoe et al. Potent human α-amylase inhibition by the β-defensin-like protein helianthamide. ACS Cent. Sci. 2016, 2, 154–161. © the American Chemical Society.
Helianthamide (29) inhibited HPA with a remarkable Ki of 10 picomolar, making it the most potent HPA inhibitor known to date. Cationic charge on the surface of the β-defensins is required for their toxic and antimicrobial activities. Helinathamide (29) is unique in this family because it lacks significant surface charge and its potent HPA inhibition instead depends on its highly lipophilic surface. It showed excellent selectivity for inhibition of HPA versus a panel of closely related glycosidases and an E. coli expression system was developed that makes it easy to manufacture in high yields. The disulfide linkages provide impressive resistance to acidic hydrolysis. Control of glucose blood levels by inhibiting HPA only requires that a therapeutic agent acts topically in the intestinal tract so there is no need for systemic uptake. Helianthamide’s resistance to acid hydrolysis should make it stable at the low pHs found in the gut and the inherently low bioavailability of proteins should prevent systemic uptake and any associated side effects. These properties make helianthamide (29) a promising candidate for an orally delivered drug to control postprandial blood glucose levels.
III). Marine Natural Products Discovered Using Forward Chemical Genetic Screens and Target Identification Methodologies
i). Cell Membrane Phosphatidylserine – In Vitro Cytotoxicity and Anti-HIV Activity
Papuamide B (30) is a complex cyclic depsipeptide, isolated independently at NCI and UBC from the sponges Theonella mirabilis and T. swhinhoei collected in PNG, that has in vitro cytotoxic activity against a panel of human cancer cell lines (average IC50 ≈ 75 nM), anti-HIV activity (EC50 4 nM), and antifungal activity against Saccharomyces cerevisiae (MIC < 1 μg/mL).69 Our collaborator Boone and co-workers used yeast haploid deletion methodology to show that papuamide B (30) binds selectively to phosphatidylserine on the yeast cell membrane surface leading to destabilization of the membrane integrity and its antifungal, cytotoxic, and anti-HIV activities.70 Exoplasmic leaflet membrane PS represents a previously unrecognised non-protein cytotoxic and anti-HIV molecular target.

The composition of phospholipids in eukaryotic cell membranes is highly asymmetric. Phosphatidylcholine (PC) and sphingolipids are found in the exoplasmic leaflet and phosphatidylserine (PS), phosphatidylethnaolamine (PE), phosphatidylinositol (PI), and phosphoinositides are predominantly found in the cytosolic leaflet.71,72 In model membranes, phospholipid transfer between faces of the bilayer is not spontaneous. Therefore, the asymmetric distribution of phospholipids in cell membranes is catalyzed by the phospholipid translocases flippase, floppase, and scramblase. When papuamide B (30) binds phosphatidylserine on the outer surface of a cell membrane, it disrupts the cell membrane stability leading to cytotoxicty, making it a specific sensor for exoplasmic leaflet pools of PS and a useful tool for studying the phospholipid translocases. This useful sensor property created a significant demand for papuamides A and B (30) as cell biology research tools,71,72 a demand that has been met by UBC for several years through an at-cost supply of the natural products.
ii). PARP3 and PSRP16 Inhibitors – del508-CFTR Correction
Cystic fibrosis (CF) is a common lethal genetic disease that affects primarily Caucasians with an estimated incidence of 1 in 2500/3500 people in the USA. Deletion of phenylalanine at position 508 (F508del) in the CF transmembrane conductance regulator (CFTR), the primary chloride channel in the membrane of epithelial cells, is the major cause of CF.73 F508del-CFTR misfolds and is recognized and destroyed by the protein quality control machinery in the endoplasmic reticulum, so it never traffics to the cell surface.74,75 Diminished chloride channel function leads to dry viscous mucus in airways that prevents clearance of pathogenic bacteria, resulting in chronic infections causing fibrosis and destruction of normal lung function. However, if F508del-CFTR reaches the cell surface it functions as a normal chloride channel and therapies called ‘correctors’, that partially rescue and traffic the F508del-CFTR ER retention, may be beneficial. It has been estimated that therapeutic benefits could be gained if only a fraction of the endoplasmic reticulum retained F508del-CFTR equal to 10 to 25% of wild type expression could be rescued. Our collaborator, Thomas developed a cell-based assay to screen for ‘correctors’ that was used to interrogate our marine natural product crude extract and pure compound libraries for hits with ‘corrector’ activity.76
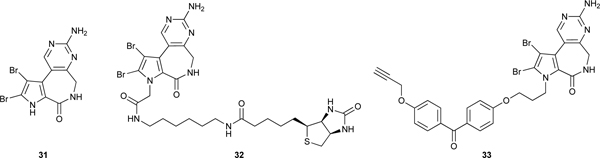
Latonduine A (31),77 a novel tricyclic alkaloid isolated from the marine sponge Stylissa carteri collected in Indonesia, was found to correct F508del-CFTR trafficking activity up to 45% of wild type CFTR surface expression with an EC50 of ≈ 1nM in the cell-based assay.76 31 also produced correction equal to 25% of normal CFTR expression in CFBE410- cells, which are a human epithelial cell line derived from a F508del-CFTR patient. A standard mouse salivary secretion assay comparing wild type mice and F508del-CFTR mice was used to evaluate the in vivo corrector activity of latonduine A (31). F508del-CFTR and wild type mice were treated either with a control or fed with 50 mg/kg latonduine A (31) by oral gavage once daily for 2 days before stimulation of salivary production by co-injection of atropine and isoprenaline into their cheeks. Latonduine A treated CF mice saliva production was 9% of wild type mice production, demonstrating that latonduine A administered PO corrects F508del-CFTR in whole animals at a level deemed to be therapeutically useful.
Two Streptavidin pull down experiments were used with CFBE41o- cell lysates to identify the molecular target of latonduine A (31).76 One of them used the biotinylated probe BIOLAT (32) and the other one used the Photoaffinity/Click chemistry probe PHOTOLAT (33). In both cases the identity of bound proteins was determined by SDS-PAGE and trypsinization followed by mass spectrometry. In several replicate experiments, the highly abundant human enzyme PARP-1 was the only protein identified in all replicates and no other protein was found in even two replicates. This result was confirmed by using BIOLAT (32) and WESTERN blots with an anti-PARP-1 antibody. Competition pull down experiments using BIOLAT (32) and recombinant PARP-1 in the presence of known active site inhibitors of PARP-1 demonstrated that latonduine binds directly to PARP-1 in the catalytic site, competitively with NAD. The same experiment was used to show that latonduine A also binds to the active sites of PARPs −2, −3, −4 and −5a. Measurement of latonduine A’s ability to inhibit the enzymatic activity of the PARP family showed inhibitory activities for PARP’s −1, −2, −4 and −5a with EC50 values in the single digit or double digit micromolar range. The one exception in this group was PARP-3, which had an extremely potent EC50 of 400 pM. siRNA knockdown of PARP-3 was found to significantly reduce the amount of latonduine A (31) needed to elicit optimal correction of F508del-CFTR, with the EC50 of latonduine A (31) dropping from 8 nM to 200 pM. The high cellular concentration of PARP-1 and the low cellular concentration of PARP-3 was thought to explain why the cell lysate Streptavidin pull down experiments only identified PARP-1 even though PARP-3 is a much more potently inhibited by latonduine A (31). A subsequent SAR study, that examined additional PARP family members as targets of synthetic latonduine A analogs, revealed that latonduine A (31) was also a potent inhibitor of PARP-16, and siRNA knockdown experiments showed that simultaneous inhibition of both PARP-3 and PARP-16 is required for the full F508del-CFTR ‘corrector’ activity of latonduine A (31).78 The use of an unbiased cell-based phenotypic assay to discover the F508del-CFTR ‘corrector’ activity of latonduine A (31), and the subsequent identification of its mechanism of action involving the polypharmacological inhibition of PARP-3 and PARP-16, identified a novel F508del-CFTR ‘corrector’ pharmacophore drug lead that acts via potent simultaneous inhibition of two PARP enzymes that were not previously associated with CFTR trafficking defects.
iii). Androgen Receptor N-Terminal Domain Antagonists – mCRPC Drug Leads
Prostate cancer (PC) is the second leading cause of cancer death in American men after lung cancer.79 Localized PC is treated with surgery, radiation, or active surveillance. Roughly 20–30% of patients receiving treatments for localized PC will have recurrence of their disease signaled by an increase in levels of prostate-specific antigen (PSA). Most PC tumor growth is driven by the androgen receptor (AR), which is a ligand-activated transcription factor comprised of a ligand binding domain (LBD), a hinge region (HR), a DNA binding domain (DBD), and a N-terminal domain (NTD).80 Patients with recurrent disease are treated with androgen deprivation therapy (ADT) to reduce the levels of the androgens testosterone and 5α-dihydrotestosterone (DHT) in their bodies, thereby reducing activation of the AR by reducing its ligand titer. ADT includes surgical castration, chemical castration, the use of the CYP17 inhibitors to prevent the synthesis of androgens,81 and administration of LBD binding antiandrogens such as enzalutamide.82 ADT effectively reduces serum levels of PSA and reduces tumor burden leading to a significant period of progression-free disease.
All current ADTs that target the AR LBD eventually fail in patients with advanced disease leading to metastatic castration-resistant prostate cancer (mCRPC), characterized by a rise in PSA levels and tumor burden.83,84,85 Most men with mCRPC die from their cancer within 2–3 years of reaching this stage. PSA is an androgen-regulated gene that is dependent on AR activation, so the rising PSA in mCRPC suggests continued AR transcriptional activity in the absence of testicular androgens.85 AR splice variants that are missing the LBD and are constitutively active are an important source of androgen-independent AR transcriptional activity in mCRPC. Transcriptional activity of the AR splice variants and the full-length AR, with or without androgens, requires binding of co-activating proteins to the activity function 1 (AF1) region of the androgen receptor N-terminal domain (AR NTD). Interleukin 6 (IL6) and protein kinase A (PKA) signal transduction pathways converge on the AF1 region of the AR NTD and are also known to drive transcriptional activity of full-length AR in the absence of androgens and may contribute to CRPC tumor growth. Our collaborator Sadar was the first to propose that small molecules able to bind to the AF1 region of the AR NTD in splice variant and full-length ARs should prevent binding of co-activating proteins and therefore block transcriptional activation resulting from all of the most common mechanisms of ADT resistance that lead to mCRPC.86
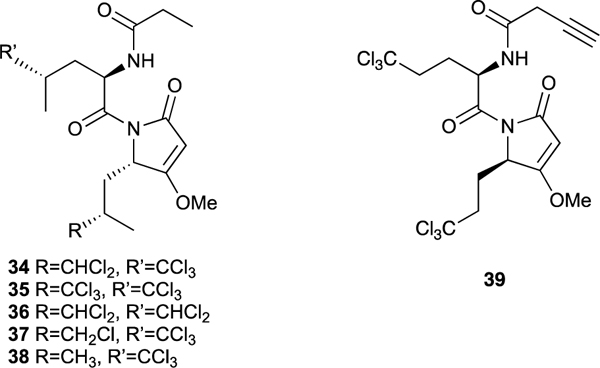
Sadar used LNCaP human PC cells expressing a functional full-length AR that drives proliferation and gene expression in response to androgens and have AR-driven luciferase (Luc) reporter genes such as PSA-luciferase to screen our library of marine sponge extracts for the presence of compounds able to block AR transcriptional activity in vitro. Extracts of a Dysidea sp. marine sponge collected in Indonesia showed strong transcriptional inhibition activity in the screen and bioassay guided fractionation identified the sintokamides A (34) to E (38) as the active constituents.87 The sintokamides are highly modified Leu-Leu dipeptides featuring mono-, di-, or trichlorination of a side chain methyl group in one or both of the two leucine-derived fragments, acetate extension of the C-terminus carboxylic acid followed by cyclization and methylation to give a methyltetramic acid moiety, and an N-terminus propionamide cap.
A subsequent assay involving transcriptional activation of PC cells stimulated by binding of IL6 or PKA to the AR NTD demonstrated that sintokamide A (34) was able to inhibit transcriptional activity of PC cells by blocking the AR NTD.88 It represented the first known AR NTD antagonist. Sintokamide A (34) did not inhibit transcription in the closely related progesterone and glucocorticoid receptors and it showed selective inhibition of AR receptor driven proliferation of LNCaP cells compared to PC3 and DU145 cells that do not contain androgen receptor. The synthetic Click chemistry probe 39 was used in a Streptavidin pull-down experiment to show that the sintokamides slowly and in very low yield form a covalent bond to the AF1 region of the AR NTD. Consistent with the findings described above, sintokamide A (34) inhibited the in vitro growth of enzalutamide-resistant LNCaP95 cells and expression of the UBE2C gene that are driven by splice variant transcription and it caused regression of CRPC tumors in mouse xenografts and reduced expression of PSA in the tumors. The data collected on sintokamide A (34) provided preliminary in vitro and in vivo validation of the AR NTD as a new molecular target for drugs to treat mCRPC and the sintokamide scaffold represents a marine natural product pharmacophore inspiration for the development of a new class of PC-selective anticancer drugs that block the transcriptional activity of this target. Sintokamide A (34) was part of the core technology of the startup biotech company ESSA Pharma (NASDAQ EPIX) that is evaluating AR NTD antagonists in human clinical trials as a new class of drugs for treatment of mCRPC.89
iv). PKC Activators - HIV Latency Reversal Agents
The World Health Organization estimates that globally there were 38 million people living with HIV and 770,000 deaths from HIV in 2018. Highly active antiretroviral therapy (HAART) has dramatically improved the life expectancy and quality of life for people infected with HIV, but a sterilizing cure has remained elusive, so lifelong administration of HAART is required in order to realize the full therapeutic benefits.90,91,92 HAART reduces plasma virus loads to undetectable levels and limits onward transmission but it does not eliminate latent viral reservoirs that can lead to reinfection if HAART is stopped. Integrated and transcriptionally silenced HIV-1 provirus in resting CD4+ T cells represents a major reservoir of latent infection that can become activated after prolonged periods of dormancy. The quest for a sterilizing cure for HIV has focussed on trying to clear these latent reservoirs.
A potential latent reservoir clearing strategy referred to as “Shock and Kill” involves de-repressing HIV-1 provirus transcription with small molecule drugs and then killing the released replicating virus with HAART. Upon activation, the reservoir CD4+ T cells express antigens on their cell surfaces which are recognized by CD8+ T cells that kill the activated CD4+ T cells. Several natural products, operating by a variety of mechanisms of action, are known to have Latency Reversal Agent (LRA) properties in vitro. These include bryostatin (40),93 prostratin (41),94 and synthetic analogs of these two compounds that work by activating protein kinase C (PKC) leading to translocation of the transcription factor NF-kb to the nucleus where it activates HIV-1 gene transcription. Clinical trials involving known LRAs have thus far failed to reduce latent viral reservoirs in HIV-1 infected patients and it has been suggested that combinations of LRA agents acting by different mechanisms and/or having greater potency are required to realize a sterilizing cure via this approach.
Our collaborator Tietjens used a Jurkat helper T cell line, which encodes an integrated non-infectious HIV-1 provirus containing a GFP reporter sequence, to screen our crude extract and pure marine natural product libraries for LRA hits.95 Sestertepenoids belonging to the alotaketal96 and ansellone97 families, that were discovered by our group from extracts of the sponges Hamigera sp collected in PNG and Phorbas sp. collected in BC and represent the first examples of the new alotane and ansellane sesterterpenoid carbon skeletons, showed strong LRA properties. Ansellone A (42) showed potency and efficacy almost identical to the positive control prostratin (41), while alotaketal C (43) was significantly more potent and more efficacious (i.e. larger amount of viral reactivation) than prostratin (41) in the cell-based screening assay.98 It has been observed that LRAs acting through different mechanisms show synergistic effects when added in combination, but LRAs with the same MOA show only additive or weaker effects. Tietjen showed strong synergistic effects with combinations of alotaketal C (43) and TNFα (a proinflammatory cytokine) and panobinostat (an HDAC inhibitor), but no synergistic effects with the PKC activator prostratin (41), suggesting that alotaketal C (43) is a PKC activator. Combination of alotaketal C (43) and various PKC inhibitors eliminated the LRA activity of alotaketal C (43). Taken together, these results strongly suggest that alotaketal C (43) is a PKC activator and similar results were found for ansellone A (42). Alotaketal C (43) and ansellone A (42) are the first sesterterpenoids known to be agonists of PKC and able to activate latent HIV-1 reservoirs. They are interesting lead structures for the development of a new LRA drug scaffold.

v). PP2 Inhibitor and Microtubule Targeting Agents
Our collaborator Roberge developed a cell-based assay to detect cells arrested in mitosis.99 The assay uses human breast cancer MCF7 cells and an antibody TG3 that detects a specific phosphoepitope on the nuclear protein nucleolin that only exists when cells are in mitosis. This was the first cell-based assay that could be used in a 96 well plate format to screen large numbers of crude extracts directly for antimitotic agents and by using microscopy to examine the microtubules in the arrested cells to immediately identify tubulin as the molecular target and further distinguish microtubule depolymerizing agents from microtubule stabilizing agents. Using this assay, we discovered the new microtubule stabilizing agents caribaeosidie (44)99,100 and ceratamine A (45)101,102 and the tubulin depolymerizing agent rhizoxin seco acid 46.99 Spirastrellolides A (47)-G isolated from the Caribbean marine sponge Spirastrella coccinea were also discovered using this assay.103–106 The spirastrellolides consist of a 47 carbon linear polyketide backbone incorporated into a highly functionalized 38-membered lactone containing a tetrahydropyran and two spiro bispyran substructures embedded in the macrocycle and a side chain terminating in a carboxylic acid which was converted to the methyl ester (48) to aid the isolation. The methyl ester of spirastrellolide A (48) exhibited potent mitotic arrest. However, it did not affect tubulin polymerization, but rather had the unusual property of being able to drive cells from S phase directly into mitosis before causing mitotic arrest. Premature entry into mitosis and mitotic arrest are biological properties typical of the Ser/Thr phosphatase inhibitors fostriecin, okadaic acid, and calyculin A that are potent protein phosphatase 2A inhibitors, suggesting a ‘candidate molecular target’ for the spirastrellolides. Therefore, spirastrellolide A methyl ester (48) was screened against a panel of protein phosphatases and it was found to inhibit the activity of protein phosphatase 2A potently (IC50 = 1 nM), PP1 much less potently (IC50 = 50 nM) and PP2C not at all, confirming PP2A as its cellular target.
vi). ChK1 inhibitor
Normal cells respond to DNA damage by activating cell cycle checkpoints that delay the transition from G1 to S phase and from G2 to M phase while DNA is repaired.107 Most cancer cells have an inoperative G1 checkpoint due to inactivation of the p53 tumor suppressor gene but a functioning G2 checkpoint. With this in mind, it was proposed that treatment with radiotherapy or DNA-damaging chemotherapeutic agents in combination with drugs that inhibit the G2 checkpoint might promote the selective killing of tumors bearing p53 mutations by driving cells into mitosis with lethally damaged DNA, thereby providing therapeutic benefit. Roberge used a modification of his cell-based antimitotic assay employing human breast carcinoma MCF-7 cells in which p53 is inactivated to screen our extract library for G2 checkpoint inhibitors. The assay identified the novel alkaloid isogranulatimide (49) isolated from the Brazilian ascidian Didemnum granulatum as a potent G2 checkpoint inhibitor.108 Evaluation of a panel of natural and synthetic isogranulatimide analogues showed that the imide nitrogen and a basic nitrogen at position 14 or 15 in the imidazole ring were important for checkpoint inhibition.108 Isogranulatimide (49) shows structural resemblance to the aglycon of UCN-01 (50), a potent inhibitor of the checkpoint kinase Chk1 (IC50, 0.007 μmol/L). This structural similarity suggested that Chk1 was a ‘candidate molecular target’ for isogranulatimide (49). In vitro kinase inhibition assays confirmed that isogranulatimide (49) inhibited Chk1 (IC50, 0.1 μmol/L) and glycogen synthase kinase-3β (IC50, 0.5 μmol/L), but not 13 additional protein kinases tested.109 A crystal structure of the Chk1 catalytic domain complexed with isogranulatimide (49) revealed that like UCN-01 (50), isogranulatimide (49) binds in the ATP-binding pocket of Chk1.

Conclusions and Outlook
Interest in the discovery of new MNPs with unprecedented chemical structures and potent biological activities continues unabated.110 Most of the motivation comes from the recognition that MNPs are important lead compounds for drug development. The recent approval for clinical use of the anticancer drugs Yondelis, Halavan, Adcertis, and Aplidin provides proof that this motivation to find marine natural product drug leads is well founded. Chemical biology has advanced to a stage where it is now unlikely that any new drug would be developed if its cellular target and mechanism of action was not well understood, and first in class drug candidates with new targets and new chemical scaffolds are more highly valued than me too drugs.13,16,111 Therefore, MNP discovery programs that incorporate cellular target identification and identify lead compounds for new molecular targets will have the greatest chance of having a natural product discovery progress to preclinical drug development or become a useful chemical biology tool.
The aim of this review is to illustrate that bioactivity driven MNP discovery is still highly effective, particularly if it focusses on new molecular targets and/or phenotypic responses that are relevant to satisfying unmet medical needs. Reverse chemical genetics discovery screens provide the most direct path to link target identification and phenotypic response as illustrated above for SHIP1 activators, MRSA PK inhibitors, and SETD8 inhibitors. However, there is often no corresponding cell-based assay to confirm that reverse chemical genetics hits will elicit a desired phenotypic response and complex animal models are required to demonstrate the desired phenotypic response as illustrated for the HPA and IDO inhibitors. Cell-based phenotypic assays are attractive because they can identify new molecular targets, they provide immediate information about cellular permeability and cytotoxicity, and they work well with crude natural product extracts that often cause problems in reverse chemical genetics screens due to the presence of pigments. The challenge with phenotypic assays is identifying the molecular target and several approaches are illustrated in this review. Phosphotidylserine was identified as the cytotoxic/anti-HIV target of papuamide B (30) using yeast haploinsufficiency methodology and PARPs 3 and 16 were identified as the del508-CFTR corrector targets of latonduine A (31) using a combination of Streptavidin pull-down experiments with biotinylated and photoaffinity/Click chemistry probe analogs of latonduine (32, 33) followed by screening against a panel of pure PARP enzymes. Engineered cells that screen for a very focussed phenotype can be used to look for hits against a particular molecular target. The AR NTD antagonist sintokamide A (34) was discovered by using a LNCaP PC cell transcriptional inhibition screen with a PSA-luciferase reporter. Follow up assays and Streptavidin pull-down experiments showed that sintokamide A (34) binds to the AF1 region of the AR NTD. The HIV-1 LRAs alotaketal C (43) and ansellone A (42) were discovered using a Jurkat helper T cell line that was engineered to contain transcriptionally silenced HIV-1 provirus, again with a luciferase reporter for transcriptional activity. Alotaketal C (43) and ansellone A (42) were shown to be PKC activators by looking for synergism with several compounds that hit well known LRA ‘candidate targets’. The spirastrellolides (eg., 47) and isogranulatimide (49) were both discovered by screening with human breast cancer MCF-7 cells and looking specifically for an antimitotic phenotype with a TG3 antibody. Molecular targets of both compounds were subsequently identified by screening against putative ‘candidate targets’ identified by comparisons of their phenotypes or chemical structures with known compounds. An x-ray diffraction structure of a bioactive MNP bound to a putative protein molecular target as illustrated above for cis-3,4-dihydrohamacanthin B (6) bound to MRSA PK and helianthamide (29) bound to PPA provides definitive molecular target engagement evidence and as a bonus serves as a template to guide analog design and synthesis.
An important take home message from the work reviewed herein is that screening for new types of bioactivities using novel forward or reverse chemical genetics assays leads to the discovery of interesting new MNP chemistry and the associated chemical biology. The MNPs discovered have potent activities with IC50/EC50/Ki values ranging from low single digit micromolar to double digit picomolar values and they represent alkaloid, non-ribosomal peptide, terpenoid, meroterpenoid, and polyketide biosynthetic origins. In most of the examples described above, the screening assays were new, the MNPs were either the first known ligand or the first known natural product ligand for a particular protein target, and in most cases the MNPs had unprecedented chemical scaffolds. Bioactivity driven natural product discovery is not new but the increased emphasis on MOA and target identification is recent. Continued interest in this approach to natural product discovery is evidenced by the development and application of the Morphobase,12 ChemProteoBase,12 Fusion,13 and Compound Activity Mapping14 platforms and increasing numbers of publications describing bioactive MNP discoveries and their molecular targets.21,112–114 Combining the objectives of bioactive natural product discovery and molecular target identification under a chemical genetics umbrella is a very effective discovery paradigm that guarantees newly discovered natural products will garner attention as drug development candidates and/or cell biology research tools.
Acknowledgements
The authors thank the following biological collaborators: Alice Mui, Christopher Ong, Gerry Krystal, Neil Reiner, Natalie Strynadka, Peter Brown, Cheryl Arrowsmith, Masoud Vedadi, Eric Julien, Grant Mauk, Steve Withers, Gary Brayer, Theresa Allen, Charlie Boone, David Thomas, Graeme Carlile, Marianne Sadar, Ian Tietjen, and Michel Roberge.
Footnotes
Conflicts of interest
D.E. Williams and R.J. Andersen own shares in ESSA Pharma and R.J. Andersen is a consultant to ESSA Pharma.
This review presents examples that illustrate the effectiveness of using a chemical genetics approach for the discovery of biologically active marine natural products and their molecular targets.
References
- 1.For example see: Martens E, Demain AL, J. Antibiot, 2011, 64,705–710. [DOI] [PubMed] [Google Scholar]
- 2.For example see: Ziemert N, Alanjary M and Weber T, Nat. Prod. Rep, 2016, 33, 988–1005. [DOI] [PubMed] [Google Scholar]
- 3.For example see: Heslfrich EJN, Reiter S and Piel Jorn, Curr. Opin. Biotech, 2014, 29, 107–115. [DOI] [PubMed] [Google Scholar]
- 4.For example see: Dona AC, Kyriakides M, Scott F, Shephard EA, Varshavi D, Veselkov K, Everett JR, Computational and Structural Biotechnology Journal, 2016, 14, 135–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For example see: Duncan KR, Crusemann Max, Lechner A, Sarkar A, Li J, Ziemert N, Wang M, Bandeira N, Moore BS, Dorrestein PC and Jensen Paul R., Chem. Biol, 2015, 22, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schroeder FC, Gibson DM, Churchill ACL, Sojikul P, Eursthorn EJ, Kransoff SB, Clardy J, Angew. Chem. Int. Ed, 2007, 46, 901–904. [DOI] [PubMed] [Google Scholar]
- 7.Gross H, Stockwell VO, Henkels MD, Novak-Thompson B, Loper J, Gerwick WH, Chem. Biol, 2007, 14, 53–63. [DOI] [PubMed] [Google Scholar]
- 8.Spraker JE, Luu GT and Sanchez LM, Nat. Prop. Rep, 2019, DOI: 10.1039/c9np00038k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chabner BA, J. Natl. Cancer I, 2016, 108, dvj388. [DOI] [PubMed] [Google Scholar]
- 10.Monks A, Scudiero DA, Johnson GS, Paull KD, Sausville EA, Anti-Cancer Drug Des., 1997, 12, 533–541. [PubMed] [Google Scholar]
- 11.Kurita KL and Linington RG, J. Nat. Prod 2015, 78, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muroi M, Futamura Y, and Osada Nat H. Prod. Rep 2016, 33, 621–625. [DOI] [PubMed] [Google Scholar]
- 13.Potts MB, Kim HS, Fisher KW, Hu Y, Carrasco YP, Bulut GB, Ou YH, Herrera-Herrera ML, Cubillos F, Mendiratta S, Xiao G, Hofree M, Ideker T, Xie Y, Huang LJ-S, Lewis RE, MacMillan JB, and White MA, Sci. Signal 2013, 6, ra90–ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurita KL, Glassey E, and Linington RG PNAS 2015, 112, 11999–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koehn FE and Carter GT, Nat. Rev. Drug Discov, 2005, 4, 206–220. [DOI] [PubMed] [Google Scholar]
- 16.Cacace E, Kritikos G and Typas A, Curr. Opin. Syst. Biol, 2017, 4, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong F, Cheung AK and Huang SMA, Annu. Rev. Pharmacol. Toxicol, 2012, 52, 57–78. [DOI] [PubMed] [Google Scholar]
- 18.Burkard ME, Jallepalli PV, Biochim. Biophys. Acta, 2010, 1806, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roemer T, Davies J, Giaever G, and Nislow C, Nat. Chem. Biol, 2012, 8, 46–56. [DOI] [PubMed] [Google Scholar]
- 20.Schenone M, Danick V, Wagner BK, and Clemons PA, Nat. Chem. Biol 2013, 9, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang X, Luo D, Yan J-L, Rezaei MA, Salvador-Reyes LA, Gunasekera SP, Li C, Ye T, Paul VJ, and Luesch H, Org. Lett, 2019, 21, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blunt J, Copp BR, Keyzers RA, Munro MHG, and Prinsep MR, Nat. Prod. Rep 2015, 31, 160–258. [DOI] [PubMed] [Google Scholar]
- 23.Janne PA, Gray N and Settleman J, Nat. Rev. Drug Discovery, 2009, 8, 709–723. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Williams DE, Mui A, Ong C, Krystal G, van Soest R and Andersen RJ, Org. Lett, 2005, 7, 1073–1076. [DOI] [PubMed] [Google Scholar]
- 25.Fortanet JG, Chen CH-T, Chen Y-NP, Chen Z, Deng Z, Firestone B, Fekkes P, Fodor M, Fortin PD, Fridrich C, Grunenfelder D, Ho S, Kang ZB, Karki R, Kato M, Keen N, LaBonte LR, Larrow J, Lenoir F, Liu G, Liu S, Lombardo F, Majumdar D, Meyer MJ, Palermo M, Perez L, Pu M, Ramsey T, Sellers WR, Shultz MD, Stams T, Towler C, Wang P, Williams SL, Zhang J-H and LaMarche MJ J. Med. Chem 2016, 59, 7773–7782. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Carr G, Zhang Y, Williams DE, Amlani A, Bottriell H, Mui AL-F and Andersen RJ, J. Nat. Prod, 2011, 74, 1093–1099. [DOI] [PubMed] [Google Scholar]
- 27.Goclik E, Konig GM, Wright AD and Kaminsky R, J. Nat. Prod, 2000, 63, 1150–1152. [DOI] [PubMed] [Google Scholar]
- 28.Kwak JH, Schmitz FJ and Kelly M J. Nat. Prod, 2000, 63, 1153–1156. [DOI] [PubMed] [Google Scholar]
- 29.Ong CJ, Ming-Lum A, Nodwell M, Ghanipour A, Yang L, Williams DE, Kim J, Demirjian L, Qasimi P, Ruschmann J, Cao L-P, Ma K, Chung SW, Duronio V, Andersen RJ, Krystal G and Mui AL-F, Blood, 2007, 110, 1942–1949. [DOI] [PubMed] [Google Scholar]
- 30.Meimetis LG, Nodwell M, Yang L, Wang X, Wu J, Harwig C, Stenton GR, Mackenzie LF, MacRury T, Patrick BO, Ming-Lum A, Ong CJ, Krystal G, Mui AL-F and Andersen RJ, Eur. J. Org. Chem, 2012, 2012, 5195–5207. [Google Scholar]
- 31.Cuddy SM, Plastic Surgical Nursing, 2008, 28, 168–169. [DOI] [PubMed] [Google Scholar]
- 32.Kurosu M, Siricilla S and Mitachi K, Expert Opin. Drug Dis, 2013, 8, 1095–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein E, Smith DL and Laxminarayan R, Emerg. Infect. Dis, 2007, 13, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cherkasov A, Hsing M, Zoraghi R, Foster L, H See R, Stoynov N, Jiang J, Kaur S, Liap T, Jackson L, Gong H, Swayze RD, Amandoron E, Hormozdiari F, Dao P, Sahinalp C, Santos-Filho O, Axerio-Cilies P, Byler K, McMaster WR, Brunham RC, Finlay BB and Reiner NE, J. Proteome Res, 2011, 10, 1139–1150. [DOI] [PubMed] [Google Scholar]
- 35.Axerio-Cilies P, See RH, Zoraghi R, Worrall L, Lian T, Stoynov N, Jiang J, Kaur S, Jackson L, Gong H, Swayze RD, Amandoron E, Kumar NS, Moreau A, Hsing M, Strynadka NC, McMaster WR, Finlay BB, Foster LJ, Young RN and Reiner NE, ACS Chem. Biol, 2012, 7, 350–359. [DOI] [PubMed] [Google Scholar]
- 36.Park K and Kim D, Proteomics, 2009, 9, 5143–5154. [DOI] [PubMed] [Google Scholar]
- 37.Zoraghi R, Worrall L, See RH, Strangman W, Popplewell WL, Gong H, Samaai T, Swayze RD, Kaur S, Vuckovic M, Finlay BB, Brunham RC, McMaster WR, Davies-Coleman MT, Strynadka NC, Andersen RJ and Neil E Reiner, J. Biol. Chem, 2011, 286, 44716–44725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veale CGL, Zoraghi R, Young RM, Morrison JP, Pretheeban M, Lobb KA, Reiner NE, Andersen RJ and Davies-Coleman MT, J. Nat. Prod, 2015, 78, 355–362. [DOI] [PubMed] [Google Scholar]
- 39.Labriere C, Gong H, Finlay BB, Reiner NE and Young RN, Eur. J. Med. Chem, 2017, 125, 1–13. [DOI] [PubMed] [Google Scholar]
- 40.Arrowsmith CH, Bountra C, Fish PV, Lee K and Schapira M, Nat. Rev. Drug Discov, 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- 41.Yang F, Sun L, Li Q, Han X, Lei L, Zhang H and Shang Y, EMBO J., 2012, 31, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takawa M, Cho H-S, Hayami S, Toyokawa G, Kogure M, Yamane Y, Iwai Y, Maejima K, Ueda K, Masuda A, Dohmae H, Field HI, Tsunoda T, Kobayashi T, Akasu T, Sugiyama M, Ohnuma S, Atomi Y, Ponder BAJ, Nakamura Y and Hamamoto R, Cancer Res., 2012, 72, 3217–3227. [DOI] [PubMed] [Google Scholar]
- 43.Williams DE, Dalisay DS, Li F, Amphlett J, Maneerat W, Chavez MAG, Wang YA, Matainaho T, Yu W, Brown PJ, Arrowsmith CH, Vedadi M and Andersen RJ, Org. Lett, 2013, 15, 414–417. [DOI] [PubMed] [Google Scholar]
- 44.Williams DE, Izard F, Arnould S, Dalisay DS, Tantapakul C, Maneerat W, Matainaho T, Julien E and Andersen RJ, J. Org. Chem, 2016, 81, 1324–1332. [DOI] [PubMed] [Google Scholar]
- 45.Tardat M, Murr R, Herceg Z, Sardet C and Julien EJ, Cell Biol., 2007, 179, 1413–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jørgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, Helleday T, Helin K and Sørensen CSJ, Cell Biol., 2007, 179, 1337–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Q, Deng Y and Amos B AB Smith III, J. Am. Chem. Soc, 2017, 139, 13668–13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guillade L, González-Pérez AB and de Lera ÁR, Org. Biomol. Chem, 2017, 15, 7430–7438. [DOI] [PubMed] [Google Scholar]
- 49.Muller AJ and Prendergast GC, Cancer Res., 2005, 65, 8065–8068. [DOI] [PubMed] [Google Scholar]
- 50.Muller AJ, Malachhowski WP and Prendergast GC, Expert Opin. Ther. Tar, 2005, 9, 831–849. [DOI] [PubMed] [Google Scholar]
- 51.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T and van den Eynde B, Nat. Med, 2003, 9, 1269–1274. [DOI] [PubMed] [Google Scholar]
- 52.Munn MD, Zhou amnd J M. Attwood T, Science, 1998, 281, 1191–1193. [DOI] [PubMed] [Google Scholar]
- 53.Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, Ishii N, Yanaihara N, Yamada K, Takikawa O, Kawaguchi R, Isonishi S, Tanaka T and Urashima M, Clin. Cancer Res, 2005, 11, 6030–6039. [DOI] [PubMed] [Google Scholar]
- 54.Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Orbist P, Winkler C, Werner ER, Werner-Felmayer G, Weiss HG, Gobel G, Margreiter R, Konigsrainer A, Fuchs D and Amberger A, Clin. Cancer Res, 2006, 12, 1144–1151. [DOI] [PubMed] [Google Scholar]
- 55.Muller AJ, Duhadaway JB, Donover PS, Sutanto-Ward E and Prendergast GC, Nat. Med, 2005, 11, 312–319. [DOI] [PubMed] [Google Scholar]
- 56.Pereira A, Vottero E, Roberge M, Mauk AG and Raymond J Andersen, J. Nat. Prod,2006, 69, 1496–1499. [DOI] [PubMed] [Google Scholar]
- 57.Brastianos HC, Vottero E, Patrick BO, Soest RV, Matainaho T, Grant Mauk A and Andersen RJ, J. Am. Chem. Soc, 2006, 128, 16046–16047. [DOI] [PubMed] [Google Scholar]
- 58.Volgraf M, Lumb J-P, Brastianos HC, Carr G, Chung MKW, Munzel M, Mauk AG, Andersen RJ and Trauner D, Nat. Chem. Biol, 2008, 4 (9), 535–537. [DOI] [PubMed] [Google Scholar]
- 59.Carr G, Chung MKW, Mauk AG and Andersen RJ, J. Med. Chem, 2008, 51 (9), 2634–2637. [DOI] [PubMed] [Google Scholar]
- 60.Carr G, Tay W, Bottriell H, Andersen SK, Mauk AG and Andersen RJ, Org. Lett, 2009, 11, 2996–2999. [DOI] [PubMed] [Google Scholar]
- 61.Williams DE, Steinø A, de Voogd NJ, Mauk AG and Andersen RJ, J. Nat. Prod, 2012, 75, 1451–1458. [DOI] [PubMed] [Google Scholar]
- 62.Centko RM, Steinø A, Rosell FI, Patrick BO, de Voogd NJ, Mauk AG and Andersen RJ, Org. Lett, 2014, 16, 6480–6483. [DOI] [PubMed] [Google Scholar]
- 63.Kumar S, Malachowski WP, DuHadaway JB, LaLonde JM, Carroll PJ, Jaller D, Metz R, Prendergast GC and Muller AJ, J. Med. Chem, 2008, 51, 1706–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scott LJ and Spencer CM, Drugs, 2000, 59, 521–549. [DOI] [PubMed] [Google Scholar]
- 65.Tarling CA, Woods K, Zhang R, Brastianos HC, Brayer CD, Andersen RJ and Withers SG, Chem. Bio. Chem, 2008, 9, 433–438. [DOI] [PubMed] [Google Scholar]
- 66.Tysoe C, Williams LK, Keyzers R, Nguyen NT, Tarling C, Wicki J, Goddard-Borger ED, Aguda AH, Perry S, Foster LJ, Andersen RJ, Brayer GD and Withers SG, ACS Cent. Sci, 2016, 2, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ganz T, Nat. Rev. Immunol 2003, 3, 710–720. [DOI] [PubMed] [Google Scholar]
- 68.Kem WR, Parten B, Pennington MW, Price DA and Dunn BM, Biochemistry 1989, 28, 3483–3489. [DOI] [PubMed] [Google Scholar]
- 69.Ford PW, Gustafson KR, McKee TC, Shigematsu N, Maurizi LK, Pannell LK, Williams DE, de Silva ED, Lassota P, Allen TM, V Soest R, Andersen RJ and Michael R Boyd, J. Am. Chem. Soc, 1999, 121, 5899–5909. [Google Scholar]
- 70.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, C-H Ho J Wang, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ and Boone C, Cell, 2006, 126, 611–625. [DOI] [PubMed] [Google Scholar]
- 71.Chen S, Wang J, Muthusamy B-P, Liu K, Zare S, Andersen RJ and Graham TR, Traffic, 2006, 7, 1503–1517. [DOI] [PubMed] [Google Scholar]
- 72.Mioka T, Fujimura-Kamada K, Mizugaki N, Kishimoto T, Sano T, Nunome H, Williams DE, Andersen RJ and Tanaka K. Molecular Biology of the Cell, 2018, 29, 1203–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, L Chou J, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC, Science, 1989, 245, 1066–1073. [DOI] [PubMed] [Google Scholar]
- 74.Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR and Grinstein S, J. Biol. Chem, 1993, 268, 21592–21598. [PubMed] [Google Scholar]
- 75.Hwang TC and Sheppard DN, J. Physiol, 2009, 587, 2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carlile GW, Keyzers RA, Teske KA, Robert R, Williams DE, Linington RG, Gray CA, Centko RM, Yan L, Anjos SM, Sampson HM, Zhang D, Liao J, Hanrahan JW, Andersen RJ and Thomas DY, Chem. Biol, 2012, 19, 1288–1299. [DOI] [PubMed] [Google Scholar]
- 77.Linington RG, Williams DE, Tahir A, van Soest R and Andersen RJ, Org. Lett, 2003, 5, 2735–2738. [DOI] [PubMed] [Google Scholar]
- 78.Carlile GW, Robert R, Matthes E, Yang Q, Solari R, Hatley R, Edge CM, Hanrahan JW, Andersen R, Thomas DY and Birault V, Mol. Pharmacol, 2016, 90, 65–79. [DOI] [PubMed] [Google Scholar]
- 79.American Cancer Society. https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html (Accessed June 8, 2019).
- 80.Debes JD, Tindall DJ, Cancer Lett., 2002, 187, 1–7. [DOI] [PubMed] [Google Scholar]
- 81.Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PFA, Sternberg CN, Miller K, Logothetis CJ, Shore ND, Small EJ, Carles J, Flaig TW, Taplin M-E, Higano CS, de Souza P, de Bobo JS, Griffin TW, De Porre P, Yu MK, Park YC, Li J, Kheoh T, Naini V, Molina A, Rathkopf DE, Lancet Oncol., 2015, 16, 152–60. [DOI] [PubMed] [Google Scholar]
- 82.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, David ID, de Bono JS, N. Engl. J. Med, 2014, 371, 424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Feldman BJ, Feldman D, Nat. Rev. Cancer, 2001, 1, 34–45. [DOI] [PubMed] [Google Scholar]
- 84.Katsogiannou M, Ziouziou H, Karaki S, Andrieu C, Henry de Villeneuve M, Rocchi P, Cancer Treat. Rev, 2015, 41, 588–97. [DOI] [PubMed] [Google Scholar]
- 85.Antonarakis ES, Armstrong AJ, Dehm SM, Luo J, Prostate Cancer P. D, 2016, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sadar MD, Cancer Res., 2011, 71, 1208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sadar MD, Williams DE, Mawji NR, Patrick BO, Wikanta T, Chasanah E, Irianto HE, Van Soest R and Andersen RJ, Org. Lett, 2008, 10, 4947–4950. [DOI] [PubMed] [Google Scholar]
- 88.Banuelos CA, Tavakoli I, Tien AH, Caley DP, Mawji NR, Li Z, Wang J, Chi Yang Y, Imamura Y, Yan L, Wen JG, Andersen RJ and Sadar MD, J. Biol. Chem, 2016, 291, 22231–22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Obst JK, Wang J. Jian K, Williams DE, Tien AH, Mawji N, Tam T, Yang YC, Andersen RJ, Chi KN, Momtgomery B and Sadar MD ACS Pharmacol. Transl. Sci 2019. DOI: 10.1021/acsptsci.9b00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chun TW, Moir S and Fauci AS, Nat. Immunol, 2015, 16, 584–589. [DOI] [PubMed] [Google Scholar]
- 91.Archin NM, Sung JM, Garrido C, Soriano-Sarabia N and Margolis DM, Nat. Rev. Microbiol, 2014, 12, 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cary DC, Fujinaga K and Peterlin BM, J. Clin. Invest, 2016, 126,448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rasmussen TA, Lewin SR, Curr. Opin. HIV AIDS, 2016, 11, 394–401. [DOI] [PubMed] [Google Scholar]
- 94.Xing S, Siliciano RF, Drug Discov. Today, 2013, 18, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Richard K, Williams DE, de Silva ED, Brockman MA, Brumme ZL, Andersen RJ and Tietjen I, Viruses, 2018, 10, 348; DOI: 10.3390/v10070348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Daoust J, Fontana A, Merchant CE, de Voogd NJ, Patrick BO, Kieffer TJ and Andersen RJ, Org. Lett, 2010, 12, 3208–3211. [DOI] [PubMed] [Google Scholar]
- 97.Daoust J, Chen M, Wang M, Williams DE, Chavez MAG, Wang YA, Merchant CE, Fontana A, Kieffer TJ and Andersen RJ, J. Org. Chem, 2013, 78, 8267–8273. [DOI] [PubMed] [Google Scholar]
- 98.Wang M, Tietjen I, Chen M, Williams DE, Daoust J, Brockman MA and Andersen RJ, J. Org. Chem, 2016, 81, 11324–11334. [DOI] [PubMed] [Google Scholar]
- 99.Roberge M, Cinel B, Anderson HJ, Lim L, Jiang X, Xu L, Bigg CM, Kelly MT and Raymond J Andersen, Cancer Res., 2000, 60, 5052–5058. [PubMed] [Google Scholar]
- 100.Cinel B, Roberge M, Behrisch H, van Ofwegen L, Castro CB and Andersen RJ, Org. Lett, 2000, 2, 257–260. [DOI] [PubMed] [Google Scholar]
- 101.Manzo E, van Soest R, Matainaho L, Roberge M and Andersen RJ, Org. Lett, 2003, 5, 4591–4594. [DOI] [PubMed] [Google Scholar]
- 102.Karjala G, Chan Q, Manzo E, Andersen RJ and Roberge M, Cancer Res., 2005, 65, 3040–3043. [DOI] [PubMed] [Google Scholar]
- 103.Williams DE, Roberge M, Van Soest R and Andersen RJ, J. Am. Chem. Soc, 2003, 125, 5296–5297. [DOI] [PubMed] [Google Scholar]
- 104.Williams DE, Lapawa M, Feng X, Tarling T, Roberge M and Andersen RJ, Org. Lett, 2004, 6, 2607–2610. [DOI] [PubMed] [Google Scholar]
- 105.Warabi K, Williams DE, Patrick BO, Roberge M and Andersen RJ, J. Am. Chem. Soc, 2007, 129, 508–509. [DOI] [PubMed] [Google Scholar]
- 106.Williams DE, Keysers RA, Warabi K, DesJardin K, Riffell JL, Roberge M and Andersen RJ, J. Org. Chem, 2007, 75, 9842–9845. [DOI] [PubMed] [Google Scholar]
- 107.Anderson HJ, Andersen R,J. and Roberge M, Prog. Cell Cycle Res, 2003, 5, 423–430. [PubMed] [Google Scholar]
- 108.Roberge M, Berlinck RGS, Xu L, Anderson HJ, Lim LY, Curman D, Stringer CM, Friend SH, Davies P, Vincent I, Haggarty SJ, Kelly MT, Britton R, Piers E and Andersen RJ, Cancer Res., 1998, 58, 5701–5706. [PubMed] [Google Scholar]
- 109.Jiang X, Zhao B, Britton R, Lim LY, Leong D, Sanghera JS, B-B S Zhou, Piers E, Andersen RJ and Roberge M, Mol. Cancer. Ther, 2004, 3, 1221–1227. [PubMed] [Google Scholar]
- 110.Blunt J, Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR, Nat. Prod. Rep 2018, 35, 8–53. [DOI] [PubMed] [Google Scholar]
- 111.Chan JNY, Nislow C and Emili A Trends in Pharmacological Sciences, 2009, 31,82–88 [DOI] [PubMed] [Google Scholar]
- 112.Ochoa JL, Bray WM, Lokey RS, and Linington RG, J. Nat. Prod 2015, 78, 2242–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou X, Liang Z, Li K, Feng W, Tian Y, Luo X, Chen Y, Zhan Z, Zhang T, Liao S, Liu S, Liu Y, Fenical W, and Tang L, J. Med. Chem 2019, 62, 7058–7069. [DOI] [PubMed] [Google Scholar]
- 114.Hammons JC, Trzoss L, Jimenez PC, Hirata AS, Costa-Lotufo LV, La Clair JJ, and Fenical W, ACS Med. Chem. Lett 2019, 10, 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]