

Abstract
COVID-19 exhibits extensive patient-to-patient heterogeneity. To link immune response variation to disease severity and outcome over time, we longitudinally assessed circulating proteins as well as 188 surface protein markers, transcriptome, and T cell receptor sequence simultaneously in single peripheral immune cells from COVID-19 patients. Conditional-independence network analysis revealed primary correlates of disease severity, including gene expression signatures of apoptosis in plasmacytoid dendritic cells and attenuated inflammation but increased fatty acid metabolism in CD56dimCD16hi NK cells linked positively to circulating interleukin (IL)-15. CD8+ T cell activation was apparent without signs of exhaustion. Although cellular inflammation was depressed in severe patients early after hospitalization, it became elevated by days 17–23 post symptom onset, suggestive of a late wave of inflammatory responses. Furthermore, circulating protein trajectories at this time were divergent between and predictive of recovery versus fatal outcomes. Our findings stress the importance of timing in the analysis, clinical monitoring, and therapeutic intervention of COVID-19.
Keywords: single cell multi-omics, COVID-19, time-resolved, exhaustion, CITE-seq, immune juncture, mixed-effect statistical modeling
Graphical abstract
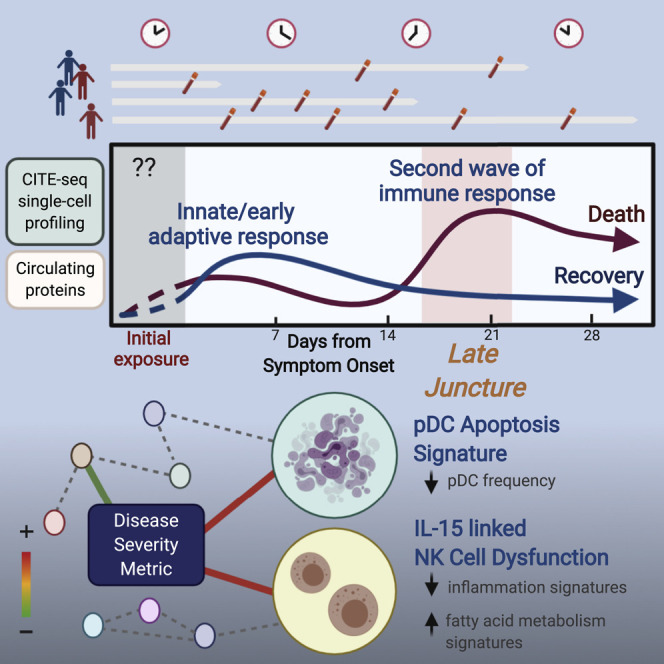
A quantitative disease severity metric is developed by integrating clinical data and circulating cytokines to capture finer shades of disease severity. Multimodal single-cell profiling and immune cell analyses of COVID-19 patients over time reveal a late wave of diverging inflammatory and host immune responses that predicts recovery versus fatality.
Introduction
COVID-19 is a potentially fatal disease caused by infection with the coronavirus SARS-CoV-2 (Zhou et al., 2020b), which has been fueling a global pandemic since December 2019 with more than 117 million confirmed cases and 2.6 million deaths to date (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). The clinical course of COVID-19 exhibits extensive heterogeneity: the infection can result in little to no symptoms, or mild disease with complete recovery, but also critical illness, hospitalization, and progression to acute respiratory distress syndrome (ARDS), tissue damage, and death (Wu and McGoogan, 2020). Variables such as age and host genetic variations (e.g., TLR7 in men [van der Made et al., 2020] or genes involved in type I interferon [IFN] production [Pairo-Castineira et al., 2020; Zhang et al., 2020c]), presence of autoantibodies (Bastard et al., 2020; Wang et al., 2020), and preexisting conditions, including obesity, diabetes mellitus, and cardiovascular disease have been identified as risk factors for severe disease and poor outcomes (CDC COVID-19 Response Team, 2020; Williamson et al., 2020). The research community has rapidly mobilized to investigate how human immune response variations can contribute to disease severity, progression, and outcome (for review, see Grigoryan and Pulendran, 2020). Understanding the mechanisms underlying immune response dynamics and differences across patients is critical for designing effective therapeutics, prognostic tools, and prevention strategies better tailored to individual patients.
The immune system protects the host but can also drive life-threatening inflammatory pathologies (Matheson and Lehner, 2020; Tay et al., 2020). Human immune responses to SARS-CoV-2 infection are highly dynamic; most of the data thus far have come from studies of hospitalized patients following symptom onset (Arunachalam et al., 2020; Laing et al., 2020; Lucas et al., 2020; Mathew et al., 2020; Rydyznski Moderbacher et al., 2020; Schulte-Schrepping et al., 2020; Su et al., 2020). COVID-19 patients are broadly characterized by elevation in neutrophils and monocytes concomitant with lymphopenia in blood, particularly with decreases in CD4+ and CD8+ T cells (Lucas et al., 2020; Mathew et al., 2020), including subsets such as γδ T cells (Laing et al., 2020). However, consistent with ongoing adaptive responses, increases in the frequency of activated and cycling CD4+ and CD8+ T cells as well as activated B cells and plasmablasts have been detected (Laing et al., 2020; Mathew et al., 2020). Accordingly, robust humoral responses have been observed with marked elevation of antibodies against SARS-CoV-2 proteins, such as the spike and nucleocapsid (Burbelo et al., 2020; Ju et al., 2020; Laing et al., 2020; Long et al., 2020; Zheng et al., 2020). However, lack of coordination among, and delay in, antigen-specific T and B cell responses tend to mark severe disease (Rydyznski Moderbacher et al., 2020). The level of proinflammatory cytokines such as interleukin (IL)-6, IL-8, IL-18, and tumor necrosis factor alpha (TNF-α) are often elevated in COVID-19 patients, and higher levels of IL-6 have been found to be predictive of poor outcomes in several studies (Liu et al., 2020; Mandel et al., 2020). TNF-α, interferon-γ-inducible protein 10 (IP-10 or CXCL-10), and IL-10 levels have also been shown to carry prognostic information (Abers et al., 2021; Del Valle et al., 2020; Laing et al., 2020; Lucas et al., 2020).
Innate immunity shows signs of dysregulation in COVID-19. Early reports noted the low levels of type I IFN activity detected in peripheral blood or lungs of COVID-19 patients (Acharya et al., 2020; Blanco-Melo et al., 2020; Hadjadj et al., 2020), suggesting a potential defect in viral defense. However, later reports found type I IFN transcriptional signatures (IFN-I signatures) in cells from bronchoalveolar lavage (BAL) fluids or peripheral blood of patients (Arunachalam et al., 2020; Liao et al., 2020; Schulte-Schrepping et al., 2020). The levels of both circulating IFN-α proteins and IFN-I signature in blood were negatively associated with the time post symptom onset and more mildly, with disease severity (Arunachalam et al., 2020; Hadjadj et al., 2020), suggesting that timing plays an important role in both the induction and experimental detection of type I IFN activities. In addition to the genetic evidence linking the type I IFN pathway to severe COVID-19 mentioned above, auto-antibodies against type I IFN have been found in some patients with life-threatening disease (Bastard et al., 2020; Wang et al., 2020), further implicating the importance of type I IFN response to protection against severe disease. Aside from infected epithelial cells, plasmacytoid dendritic cells (pDC) are another major producer of type I IFNs in response to viral infections. The frequency of pDCs in peripheral blood has been found to be lower in COVID-19 patients than healthy controls (HCs) and negatively correlated with disease severity (Arunachalam et al., 2020; Kuri-Cervantes et al., 2020; Laing et al., 2020), but substantial changes in pDC levels have not been found in BAL fluids or lung autopsies (Chua et al., 2020; Liao et al., 2020; Nienhold et al., 2020; Wauters et al., 2020). In addition, cell surface levels of HLA-DR have also been found to be lower in classical monocytes (Giamarellos-Bourboulis et al., 2020; Moratto et al., 2020; Schulte-Schrepping et al., 2020) and myeloid DCs (Arunachalam et al., 2020), suggesting that patients undergoing active inflammation and immune responses to SARS-CoV-2 have attenuated innate immune functions.
Given the importance of timing (Matheson and Lehner, 2020) and the involvement of diverse immune cells with evolving states in COVID-19, here we used multimodal single-cell profiling and computational approaches to longitudinally assess hospitalized COVID-19 patients. In addition to providing a time-resolved single-cell atlas of COVID-19 for further integration with other single-cell datasets (Schulte-Schrepping et al., 2020; Su et al., 2020; Unterman et al., 2020; Wilk et al., 2020; Yao et al., 2020; Zhang et al., 2020b), we revealed a network of dynamically evolving cell-type-specific signatures linked to disease severity. Strikingly, our analyses uncovered a late time window during which the host immune response undergoes divergences correlated with disease severity and predictive of fatal outcomes. Our results raise the prospect of more personalized intervention strategies and stress the importance of timing in the analysis and clinical monitoring of COVID-19.
Results
Study design and approach
Peripheral blood mononuclear cells (PBMCs) were obtained from 33 hospitalized patients from Brescia, Italy (Figures 1 A and S1 A) and 14 age- and gender-matched HCs (see STAR methods). The patients were classified as moderate (n = 3), severe (n = 5), and critical (n = 25 with 4 deceased during hospitalization) based on the National Institutes of Health guidelines (http://www.covid19treatmentguidelines.nih.gov/overview/clinical-presentation/) (Table S1). This distribution of disease severity is reflective of the early pandemic in Northern Italy when only the most severe patients were hospitalized due to capacity limitations. Longitudinal samples were obtained at up to four time points from each patient (time 0 [T0] to time 3 [T3]) that covered a range of times post symptom onset, thus providing a unique opportunity for linking immune state variations at T0 (at or near the time of hospital admission) to disease status and severity as well as reconstructing temporal immune response trajectories by pooling information across patients (Figure 1A).
Figure 1.

Study design and a multi-parameter disease severity metric (DSM)
(A) COVID-19 patient cohort overview and sample collection timeline. The distribution of sample collection timing since symptom onset (TSO) is summarized on the top while the sampling for each of the 33 donors (age and gender indicated in parentheses) is plotted below indicating individual T0 (1st) up to T3 (4th) time points. One patient (P098) with unknown TSO is depicted here by using his hospital admission date as the reference. Some samples were excluded from analysis after initial quality control as they did not have a sufficient number of cells (see STAR methods).
(B) Experimental design, analysis questions, and approach. Frozen PBMCs from the collected samples indicated in (A) and age-matched healthy controls were thawed and pooled, followed by combined surface protein/mRNA single-cell expression analysis (CITE-seq); data from individual samples were demultiplexed by a combination of SNP-based and “hashtag” antibody staining (see STAR methods). After automated clustering (plus additional targeted cell populations identified by manual gating) and cell subset identification based on surface protein profiles, pooled RNA data (“pseudobulk”) libraries were generated in silico for each sample per cell cluster/population and then used for downstream analysis using mixed effect statistical modeling to assess the effect of disease severity and TSO, while controlling for other variables such as TSO (when characterizing the effect of disease severity), age, and experimental batch.
(C) Heatmap of 18 clinical and serum protein measurements of patients after correction for days since hospital admission. Measurement values are standardized across subjects. Samples are divided into four groups based on unsupervised hierarchical clustering. Patients admitted to the intensive care unit (ICU) during their hospitalization are denoted with an asterisk (∗) next to their labels. None of the T0 samples were collected at the ICU.
(D) Parameter importance of the fitted coefficient values from partial least squares (PLS) ordinal regression models of disease severity. Parameters are ordered by their absolute median coefficient values from independent training iterations in leave-one-out cross validation. Error bars indicate SD of coefficient distribution across all iterations. CXCL9/MIG, monokine induced by γ interferon; IP-10, interferon-γ-inducible protein 10.
(E) Distribution of patient disease severity metric (DSM) groups based on the disease severity-outcome classification for the 30 patients with CITE-seq data (left panel, see STAR methods) and an independent set of 64 patients from Brescia (right panel). The DSM formula was trained using the CITE-seq cohort only. Significance of group difference is determined by two-tailed Wilcoxon test. ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
Figure S1.

Timeline of treatments relative to hospital admission date and DSM validation in an independent set of patients, related to Figure 1
(A) Timeline of treatments relative to hospital admission date. Dashed lines represent treatments with unknown start and/or end dates. Age and gender are listed next to each patient label. (B) Distribution of patient disease severity metric (DSM) grouped based on the WHO ordinal disease severity score of the CITE-seq cohort at the earliest time of PBMC sampling. The p value was computed using the Jonckheere trend test to assess the positive correlation between the WHO score and DSM. (C) Distribution of validation cohort disease severity metric (DSM) grouped by whether they were ever admitted to the ICU during the course of hospitalization for all patients (left) and only patients classified as Critical-Alive (right). Significance of difference between groups is determined by two-tailed Wilcoxon test.
Peripheral single immune cells were profiled by cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) (Stoeckius et al., 2017), simultaneously measuring (1) the expression of 188 surface protein markers (plus 4 isotype control antibodies) (Table S2), (2) the mRNA transcriptome, and (3) B and T cell receptor (BCR/TCR) sequences of the variable/diversity/joining (V(D)J) region. In addition to total PBMCs, we sorted non-naive B and T cells to increase their representation for T/B cell clonality and repertoire analysis. We integrated T cell clonality data here, while the corresponding B cell data will be reported separately. The data were analyzed at both the single cell and “pseudobulk” levels—the latter capturing the average gene expression profiles of cell clusters and states while mitigating single-cell mRNA measurement noise (Kotliarov et al., 2020; Soneson and Robinson, 2018). In addition to multimodal single-cell profiling, we have obtained and, in our analyses below, integrated blood cell count data and serum protein profiles (Abers et al., 2021) covering 63 circulating proteins including cytokines.
Our analyses focused on the gene expression program within cell clusters (capturing cell types and states). We built mixed effect statistical models to link these parameters to disease status (COVID-19 patients versus HCs), disease severity (after adjusting for the effects of timing; see below), and timing (time since symptom onset [TSO]) after accounting for age and experimental batch (Figure 1B). The goal was to first uncover cell type specific gene expression correlates of disease status and severity at T0 (near admission) followed by a dissection of how immune cell states evolve over time. Given the dynamic nature of the immune response to SARS-CoV-2, accounting for and disentangling the effects of timing when analyzing disease severity correlates were particularly important.
Empowering disease severity correlate analysis: development of a multi-parameter disease severity metric
Given the large proportion of patients classified as “critical” (or a World Health Organization [WHO] ordinal score of ≥5) in our cohort, we first sought to use established clinical, inflammatory, and cell count parameters to develop a quantitative disease severity score; such a metric would allow us to delineate finer shades of disease severity (e.g., breaking ties among patients in the same severity category) and thus better power downstream analyses. Based on the literature (Del Valle et al., 2020; Lucas et al., 2020; Mandel et al., 2020; Schett et al., 2020), we selected circulating proteins associated with inflammation (TNF-α/β, IL-6, and IL-18), IFN responses (IP-10 and CXCL9), immune responses (IFN-γ, IL-4, IL-13, and IL-17), and clinical markers known to be correlated with COVID-19 severity, namely C-reactive protein (CRP), neutrophil/lymphocyte ratio (NLR), lymphocyte and platelet counts, fibrinogen, D-dimer, lactate dehydrogenase (LDH), and pulse oximetry to fraction of inspired oxygen (SpO2/FiO2) ratio. Excluding three patients with very late initial sample time points (TSO ≥30 days), hierarchical clustering of the T0 patient samples using these parameters revealed several distinct patient profiles (Figure 1C), including (1) a profile of the most critical patients characterized by high levels of inflammatory markers but low in TNF-β and lymphocyte counts, (2) the moderate/severe patients with higher IL-17 and lymphocyte counts with either high or low levels of several cytokines, and (3) most of the critical-alive patients with a more mixed profile, consistent with the notion that a finer quantitative delineation of disease severity could be useful to characterize these patients.
Using ordinal partial least squares regression and leave-one-out cross validation within the CITE-seq cohort, we performed feature selection to identify parameters most predictive of the four severity-outcome categories (Figure 1D) (SpO2/FiO2 ratio was not used because it was used to define clinical disease severity). Those positively associated with severity include NLR, LDH, IP-10, D-dimer, IL-6, and IL-13, while lymphocyte count and TNF-β were correlated with less severe disease. These top parameters were then used along with the SpO2/FiO2 ratio to derive a severity score for each patient. As expected, this disease severity metric (DSM), was significantly associated with the severity-outcome categories within our CITE-seq cohort (Figure 1E) and also with the WHO ordinal score (Figure S1B). This was validated in an independent set of 64 patients from Brescia for whom DSM was computed using the same formula derived from our CITE-seq cohort only (Figure 1E). The DSM of the critical-alive patients in both the CITE-seq and the validation cohorts spanned a wide range, from levels comparable to the moderate and severe categories, to those of the critical-deceased patients (Figure 1E). As a further indication that DSM can delineate clinically relevant disease severity, DSM was significantly associated with intensive care unit (ICU) admission status in the validation cohort, even when assessed in the critical-alive patients only (Figure S1C). DSM thus provides a quantitative tool to order patients on a continuous severity scale; in our analyses below, we also used DSM to bin patients into discrete balanced groups of lower (DSM-low: DSM ≤ median DSM) versus higher disease severity (DSM-high: DSM > median DSM).
A multimodal time-resolved single immune cell atlas of COVID-19 patients and matching HCs
Given the interpretability of immune cell subsets identified using surface protein markers, we used the 188 surface protein profile to cluster ∼400,000 single cells and derived 30 coarse-resolution cell clusters (Table S3) spanning diverse innate and adaptive immune cell types and subsets (Figures 2A and 2B, small clusters not shown; Tables S2 and S3). Additional higher resolution clustering within each coarse level subset identified finer subpopulations (e.g., distinct memory subsets within the CD4+ memory T cell cluster) (Figure 2C). Manual gating was also used to obtain specific cell populations such as plasmablasts and circulating T follicular helper cells (cTfh). The relative frequency of the major cell populations obtained by CITE-seq is largely concordant with that obtained independently from 27-color flow cytometry (Figure S2 A). Furthermore, after accounting for TSO and age, we confirmed several previously reported associations between peripheral immune cell frequencies and disease status or severity near hospital admission (T0) (Figures S2B and S2C), including the negative correlation between severity (here captured by DSM) and the frequency of pDCs, CD8+ central memory T cells, and non-classical monocytes (Arunachalam et al., 2020; Grigoryan and Pulendran, 2020; Kuri-Cervantes et al., 2020; Laing et al., 2020; Mathew et al., 2020; Schulte-Schrepping et al., 2020).
Figure 2.

A multimodal single peripheral immune cell atlas of COVID-19
(A) Average DSB-normalized protein expression (see STAR methods) in each coarse-level cell cluster. Only proteins with a DSB value >3 in at least 1% of the cells are shown.
(B) UMAP visualization of single cells based on protein expression profiles for innate and adaptive groupings of cells labeled by the name of the corresponding coarse-level cluster.
(C) Average expression of selected surface protein markers in example finer-resolution CD4+ T cell clusters (CM, central memory; TM, transitional memory; EM.TE, terminal/effector memory).
(D–I) Transcript-based UMAP visualization of classical monocytes defined by surface proteins. Cells are colored according to donor class (D), disease severity group (only COVID-19 samples are shown) (E), days since symptom onset (only COVID-19 samples are shown) (F), surface marker CD163 expression (G), surface HLA-DR expression (H), and donor (I).
Figure S2.

Single immune cell atlas of COVID-19 reveals cell populations associated with COVID-19 disease status and severity, related to Figure 2
(A) Correlations of cell frequencies gated from CITE-seq and independently obtained 27-color flow cytometry data of the same samples. Shown are Pearson correlations and p values. Note that the cTfhs shown here were gated using the same strategy as flow data, i.e., based on CXCR5hiCD45RA-CD4+ T cells. Given the less-than-ideal staining of CXCR5 in both CITE-seq and flow cytometry, the cTfh being analyzed elsewhere in the paper were gated based on ICOS and PD1 in ICOShiPD1hi CD4+ T cell RNA-based cluster (see STAR methods). (B) Frequencies of immune cell clusters/subsets in HC, DSM-low (less severe disease; DSM at or below median of DSM) and DSM-high (more severe disease; DSM above median) groups at T0 (near hospitalization). Shown are the populations with the most significant changes in either COVID-19 versus HCs or correlation with DSM. P values shown are unadjusted p values based on linear models controlling for age, experimental batch and TSO (TSO only controlled for in DSM model, see STAR methods). P values are reported for assessing differences between COVID-19 versus HCs (green) and for correlation with DSM (red). Additional cell clusters meeting the p value cutoff are shown in (B). (CD4_Mem_CM: central memory CD4+ T cell cluster as a fraction of total CD4+ T cells; CD8_Mem_CM.TM: central/transitional memory CD8+ T cell cluster as a fraction of total CD8+ T cells; the frequency of γδT, mucosal associated invariant T cells (MAIT), cDC, pDC, Non-Classical Monocytes and Platelets are expressed as fractions of total PBMCs.) 104 populations were tested in total. (C) Heatmap showing cell frequencies of major cell clusters/subsets in individual subjects (columns), grouped by HC and DSM. Plasmablast and B cell subsets are expressed as fractions of CD19+ B cells; CD4+ T cell subsets are expressed as fractions of CD4+ T cells; CD8+ T cell subsets are expressed as fractions of CD8+ T cells; the CD163hi classical monocyte cluster is expressed as a fraction of total classical monocytes; others are shown as fractions of total PBMCs. Rows in the heatmap were scaled to have mean = 0 and variance = 1.
Due to the relatively lower number of cells obtained per sample by CITE-seq compared to flow cytometry, the frequency of rarer cell subsets showed weaker correlation between these two assays (Figure S2A). However, the power of CITE-seq lies in its ability to assess gene expression within cell subsets defined by surface proteins. As an example, a mRNA-only uniform manifold approximation and projection (UMAP) visualization of classical monocytes (identified by surface proteins only) shows separation of cells based on disease status, DSM, and to a lesser extent, TSO (Figures 2D–2F). Surface protein expression also differs across cells with distinct underlying RNA expression; for example, cells expressing CD163 or HLA-DR protein (Figures 2G and 2H) group together and are present in multiple donors (Figure 2I). Thus, these data (GEO: GSE161918) constitute a rich multimodal time-resolved single-cell dataset for exploring cell-type-specific transcriptomic and pathway signatures of disease severity and their dynamic trajectories.
Type I interferon signatures are negatively associated with disease severity and decrease over time
We systematically searched for transcriptional correlates of disease status (COVID-19 versus HCs), severity (DSM), and time (TSO) within individual cell clusters. Our goal was to first examine correlates at or close to hospital admission (T0) after accounting for TSO differences across patients (Figures 3A and 3B), followed by incorporation of longitudinal data within patients to reveal the effects of time (TSO) and the temporal evolution of cell-type-specific immune signatures in DSM-high and DSM-low patients separately (Figures S3 A and S3B). We fit the expression of each gene within individual cell clusters (Figure 2A, plus additional manually gated populations) via a model that incorporated the primary effect (COVID-19 versus HCs or DSM and TSO), age, and experimental batch, followed by gene set enrichment analysis (GSEA) (Subramanian et al., 2005) to uncover the biological processes and pathways involved.
Figure 3.

Cell-type-specific gene expression signatures of COVID-19 disease status and severity
(A) Gene set enrichment analysis (GSEA) result of COVID-19 versus HCs at T0. Selected gene sets (rows, see STAR methods) are grouped into functional/pathway categories. Differential expression moderated t statistics were computed from a linear model accounting for age, and experimental batch (TSO not accounted for as TSO does not exist for healthy individuals). Dot color denotes normalized gene set enrichment score (NES) and size denotes -log10(adjusted p value). p values were adjusted using the Benjamini-Hochberg method. The sign of the NES corresponds to increase/decrease of change in COVID-19 compared to HCs. Cell populations (columns) are grouped by lineage/type. See Table S4A for detailed results of all gene sets which passed the adjusted p value cutoff of 0.2.
(B) Similar to (A) but the enrichment analysis was performed based on association with DSM at T0, and the model controlled for TSO. The sign of the NES score denotes the sign of the association with DSM. See also Table S4B.
(C) Heatmap of type I IFN response in classical monocytes. Heatmap showing the scaled average mRNA expression (within the indicated cell cluster/population for a given subject) of shared leading-edge (LE) genes from the GSEA analysis of COVID-19 versus HCs (see A) and association with DSM (see B). Selected top LE genes are labeled by gene symbol and only the sample from the first time point for each patient (T0) is shown. For gene expression heatmaps, subjects are grouped by HC and DSM classes; also shown are the DSM value and severity-outcome classification of each patient (top of the heatmaps). Subjects with a small number of cells not included in the GSEA test are marked by a # (cell number <8).
(D) Per-sample gene set signature scores of the GO_Response to type I IFN gene set versus TSO (in days) in DSM-low and DS-high groups. Classical monocyte is shown as an example. Individual samples are shown as dots and longitudinal samples from the same individual are connected by gray lines. Trajectories (using LOESS smoothing, see STAR methods) were fitted to the groups separately with the shaded areas representing standard error. The median score of age- and gender-matched HCs is shown as a green dotted line. Gene set scores were calculated using the union of LE genes from both the timing (TSO) and disease severity (DSM) association models using gene set variation analysis (see STAR methods).
(E) Scatterplot comparing the effect size of association between TSO and the GO_Response to type I IFN gene set in the DSM-high (y axis) and DSM-low (x axis) patients. Each dot corresponds to a cell type/cluster. The effect size reflects the change of type I IFN signature enrichment with time (effect size <0 corresponds to the enrichment decreasing with time). Cell types are colored by their statistical significance (adjusted p value <0.05) of whether the association with time is significantly different between the two DSM groups (based on a model with a DSM-TSO interaction term, see STAR methods). The area framed by blue borders corresponds to cell types with more precipitous drop of IFN signature over time in the DSM-low than DSM-high groups.
(F) Heatmap of apoptosis/cell death signature in pDCs. Similar to (C), but instead of the shared LE genes, all LE genes from COVID-19 versus HCs and association with DSM are shown. Shared LE genes and genes in the REACTOME_Oxidative stress-induced senescence gene set are annotated on the right.
(G) Heatmap showing the sample-level pairwise Pearson correlations among serum IFN-α2a level, pDC frequency, the apoptosis signature score in pDCs, as well as the IFN-I and protein translation signature scores in classical monocyte, CD56dimCD16hi NK, and CD8 memory T cell clusters (∗p value <0.05). Selected scatterplots are shown, and the corresponding entry in the heatmap is indicated using bold borders. Samples were filtered to remove those with fewer than 8 cells or less than 40,000 unique molecular identifiers (UMI) in the pseudobulk library. Note that not all subjects had IFN-α2a measurements. Each dot indicates a subject and is colored by the severity-outcome category.
Figure S3.

Cell-type-specific gene expression signatures association with time since symptom onset and disease severity, related to Figure 3
(A and B) Similar to Figure 3B, but here showing GSEA results (of select gene sets) based on association with time since symptom onset (TSO) in DSM-low (A) and DSM-high (B) groups using all time points. See Tables S4C and S4D for detailed results of these selected gene sets and all sets that passed the adjusted P value cutoff of 0.2. Dot color denotes NES and size denotes -log10(adjusted P value). The sign of NES corresponds to the sign of correlation with TSO. Cell clusters/populations are grouped by lineage/type. (C) Similar to Figure 3C. Heatmap of translation/ribosomal gene signature in classical monocytes at T0. (D) Similar to Figure 3D. Time course of gene set signature scores of REACTOME_Translation and KEGG_Ribosome gene sets in DSM-low and DSM-high groups, respectively. Classical monocyte is shown as an example. (E and F) Similar to Figure 3F. Heatmap of apoptosis/cell death gene signature in pDCs of validation cohorts (Schulte-Schrepping et al., 2020) cohort 1 (E) and cohort 2 (F). The LE genes from the GSEA analysis of COVID-19 versus HCs and association with DSM in our Brescia CITE-seq cohort are shown (i.e., same genes as Figure 3F). Only the first time point (T0 sample) of each subject is shown. (G) GSEA results of Schulte-Schrepping et al. (2020) cohorts for pDC apoptosis/cell death signature identified in Brescia cohort. Given the limited pDC cell numbers in most samples, particularly in cohort 1, the enrichment was calculated without filtering based on cell number or library size cutoffs.
IFN-I and early viral response gene signatures from live influenza challenge (Woods et al., 2013) or yellow fever vaccination (Querec et al., 2009) (see STAR methods for signature compilation) were elevated in COVID-19 relative to HCs across myeloid and lymphoid cell lineages (Figure 3A; Table S4A). These signatures were negatively associated with DSM (Figures 3B and 3C; Table S4B): the DSM-high patients tended to have weaker IFN-I signatures than DSM-low patients even after accounting for TSO, although even some of the most critical patients had elevated IFN signatures relative to HCs (Figure 3C). IFN-I signatures decreased over time and the extent of the drop was more precipitous in DSM-low patients in most cell types, partly because those patients started off at a higher level (Figures 3D and 3E).
Translation and ribosome genes tended to be lower in most cell types with elevated IFN-I signatures (Figures 3A, 3G, and S3C; Table S4A, translation/ribosome signatures). Elevated type I IFNs are known to suppress protein translation to limit virus production (Ivashkiv and Donlin, 2014). Consistently, as IFN-I signatures decreased over time, translation increased (Figure S3D). Interestingly, even though IFN-I signatures were negatively associated with DSM in most cell types, translation signatures were not positively associated with DSM in the same cell types (Figure 3B; Table S4B), indicating that additional regulation was involved to tune protein translation in COVID-19 patients.
pDC apoptosis is associated with elevated disease severity and reduced pDC frequency
pDCs are major producers of type I IFNs and orchestrators of T and B cell responses upon viral infection (Swiecki and Colonna, 2015). It has been unclear why peripheral pDC frequencies were decreased in COVID-19 patients and negatively correlated with disease severity (Figures S2B and S2C) (Arunachalam et al., 2020; Kuri-Cervantes et al., 2020; Laing et al., 2020; Lucas et al., 2020); a better understanding of this observation could help explain the attenuated IFN-I signatures in the most severe patients (Figure 3B; Table S4B).
There is no evidence thus far that blood pDCs migrate into tissues after SARS-CoV-2 infection (Chua et al., 2020; Liao et al., 2020; Nienhold et al., 2020; Wauters et al., 2020); pDC infiltration was also not evident in bronchoscopy (Sánchez-Cerrillo et al., 2020). Alternatively, our analysis revealed that apoptotic gene signatures in pDCs, including BRCA2, CASP3, CASP8, BID, BAK1, and XBP1, were positively associated with disease status and severity (some of the most critical patients had few or no pDCs, so they were filtered out), as were oxidative stress-induced senescence genes such as FOS, HIST1H2AC, PHC3, MDM4, CBX6, and CDKN2D (Figures 3A, 3B, and 3F; Tables S4A and S4B). We further validated that the pDC apoptosis signature was significantly increased in COVID-19 patients relative to HCs using publicly available single-cell RNA sequencing (RNA-seq) data from two independent COVID-19 cohorts from Germany (Figures S3E–S3G) (Schulte-Schrepping et al., 2020), although the positive association with disease severity was not as apparent, likely due to weak statistical power given low cell numbers. The apoptotic gene signature score was also negatively correlated with pDC frequency (Pearson r = −0.72, p = 0.045), which was positively associated with IFN-I signatures in several cell populations (Figure 3G), suggesting that pDC apoptosis might have contributed to lower peripheral pDC numbers, which then led to the depressed IFN-I signatures in cells. We also confirmed that circulating IFN-α2a levels in serum were positively correlated with IFN-I signatures in cells (Figure 3G; CD8+ memory T cell shown; similar for other cell types). These analyses further confirmed the negative association between IFN-I and translation signatures within cell types discussed above (Figures 3A and 3G). Together, these results suggest that pDC apoptosis is a potential culprit of lower pDC frequencies and IFN-I signatures in severe COVID-19.
Conditional independence network analysis suggests IL-15-linked fatty acid metabolism and attenuated inflammation in CD56dimCD16hi NK cells as primary correlates of disease severity
Thus far, our analyses identified cell-type-specific signatures associated with DSM near hospital admission (T0) after accounting for TSO (Figure 3B; Table S4B), but many of them might not reflect primary correlates of disease severity and could have emerged because they correlated with ones that were. Thus, we next applied conditional independence network analysis to infer primary (or direct) correlates of DSM (see STAR methods). Nodes in the resultant network represent the gene expression state of a biological process in a cell population, except that DSM itself is one of the nodes; two nodes are connected if the correlation between them across patients is statistically independent of their correlations with other nodes in the network.
We found four nodes directly connected to DSM (Figure 4 A; the rest of the network is captured in Table S5). Oxidative stress-induced senescence in pDCs and fatty acid (FA) metabolism in CD56dimCD16hi NK cells were inferred as the primary positive correlates of DSM (Figure 4A; Tables S4B and S5). The senescence signal is reflective of the aforementioned pDC apoptosis signature (Figures 3B and 3G; Table S4B), e.g., the senescence and apoptosis signature scores in pDCs were significantly correlated across patients even though JUN is the only shared leading-edge gene in these two gene sets (Figure S4 A). Thus, this result further supports that pDC apoptosis was a potential driver of disease severity.
Figure 4.

Conditional independence network analysis points to IL-15-associated fatty acid metabolism and attenuated inflammation in CD56dimCD16hi NK cells as primary correlates of disease severity
(A) Disease severity network showing cell-type-specific gene set signatures directly connected with DSM (see STAR methods). Direct connections between the nodes to each other are also shown in a lighter shade. Edge value and width indicate Spearman correlation and its statistical significance, respectively, between DSM and the gene set signature score. In the legend: % leading-edge (LE) genes denotes the proportion of genes from the gene set that are in the LE based on gene set enrichment analysis. The top three LE genes based on effect size (association with DSM; see STAR methods) and other selected genes representative of the biology are shown for each gene set.
(B, C, G, and I) Scatterplots showing the correlation of circulating IL-15 level versus REACTOME_Fatty acid metabolism signature score (B) and DSM (C); REACTOME_Fatty acid metabolism score versus HALLMARK_TNFα signaling via NF-κB score and GO_Cellular response to IL-1 score (G); REACTOME_Fatty acid metabolism score versus HALLMARK_mTORC1 signaling score and normalized IFNG mRNA expression (I). Pearson correlation (R) and associated p value were computed using pairwise complete observations (see STAR methods). Each dot indicates a subject and is colored by the severity-outcome category.
(D) Heatmap of REACTOME_Fatty acid metabolism LE genes from the GSEA analysis of DSM association in CD56dimCD16hi NK cells at T0 (see Figure 3B). LE genes are grouped based on annotations obtained from Gene Ontology. Genes with an asterisk (∗) are those with both biosynthetic and catabolic annotations. Genes in red are those in the fatty acid oxidation set.
(E and F) Similar to (D), heatmaps of inflammation related gene sets in CD56dimCD16hi NK cells at T0: HALLMARK_TNFα signaling via NF-κB (E), GO_Cellular response to IL1 and KEGG_Chemokine signaling pathway (see A) (F). Heatmaps showing the scaled average mRNA expression (within the indicated cell cluster/population for a given subject) of LE genes from the GSEA analysis of DSM association in CD56dimCD16hi NK cells (see Figure 3B). Selected top LE genes are labeled by gene symbol. For all gene expression heatmaps (D–F), subjects are grouped by HC and DSM classes; also shown are the DSM value and severity-outcome classification of each patient (top of the heatmaps in D). Subjects with a small number of cells not included in the GSEA test are marked by a # (cell number <8).
(H) Average IFNG mRNA expression of CD56dimCD16hi NK cells; (D), (E), (F), and (H) are aligned column wise.
(J and K) Per-sample gene set signature scores of REACTOME_Fatty acid metabolism (J), HALLMARK_TNF-α signaling via NF-κB and GO_Cellular response to IL-1 (K) versus TSO (in days) in DSM-high (red) and DSM-low (blue) groups of CD56dimCD16hi NK cells. Individual samples are shown as dots and longitudinal samples from the same individual are connected by gray lines. Trajectories (using LOESS smoothing) were fitted to the groups separately with the shaded areas representing standard error. The median score of age- and gender-matched HCs is shown as a green dotted line. Gene set scores were calculated using the union of LE genes from both TSO association and DSM association models reflecting the change over time and severity differences at T0 (see STAR methods).
(L) Circulating IL-15 levels in DSM-low and DSM-high groups versus TSO.
See also Figures S4 and S5 and Table S5.
Figure S4.

Supporting data for dissecting primary gene expression signature correlates inferred by conditional independence network analysis, related to Figure 4
(A) Scatterplot of REACTOME_Oxidative stress-induced senescence signature score and GO_Apoptotic signaling signature score in pDCs (Pearson correlation and p value were computed using pairwise complete observations [samples filtered to those with at least 8 cells and 40,000 UMI detected in the pseudobulk library]). Each dot denotes a subject and is colored by the severity-outcome category. (B) Similar to (A), but between circulating IL-15 level and fatty acid metabolism signature score in CD56dimCD16hi NK cells after regressing out their associations with DSM. (C and D) Similar to Figure 4D. Heatmaps of REACTOME_Fatty acid metabolism in NK cells of two validation cohorts (Schulte-Schrepping et al., 2020) cohort 1 (C) and cohort 2 (D). The LE genes from the GSEA analysis of association with DSM in each cohort are shown. Only the first time point (T0 sample) of each subject is shown. (E) GSEA results of Schulte-Schrepping et al. (2020) cohorts for NK cell REACTOME_Fatty acid metabolism. (F) Similar to Figure 4G. Scatterplot of REACTOME_Fatty acid metabolism score and HALLMARK_TNFα signaling via NF-κB score in the validation cohorts (Schulte-Schrepping et al., 2020). (G and H) Similar to Figure 4E. Heatmaps of inflammation related gene sets in classical monocytes: HALLMARK_TNFα signaling via NF-κB (G) and HALLMARK_Inflammatory response (H).
The role of natural killer (NK) cell metabolism in COVID-19 is not known. However, CD56dimCD16hi NK cells are a cytolytic subset activated rapidly within hours of infection or after stimulation by cytokines such as IL-15 (Carson et al., 1994; De Maria et al., 2011). Strikingly, circulating levels of IL-15 were indeed positively correlated with the FA signature score in these NK cells (Figure 4B), and this association was DSM dependent: IL-15 was itself correlated with DSM (Figure 4C) and IL-15 and the FA signature were no longer correlated once their associations with DSM were statistically removed (Figure S4B). Both FA biosynthetic and catabolic/oxidative genes were elevated in the most severe (e.g., the critical-deceased subjects) versus the less severe patients, who tended to express these genes at lower levels than HCs (Figure 4D). By using single-cell RNA-seq data from two independent German COVID-19 cohorts (Schulte-Schrepping et al., 2020), we confirmed that this FA signature was indeed significantly associated with disease severity (Figures S4C–S4E).
Despite the positive association between IL-15 (a proinflammatory cytokine) and the FA signature, the FA signature was negatively correlated with inflammation related processes, including the nuclear factor κB (NF-κB) (Figures 4E and 4G; Pearson r = −0.55, p = 0.004) and IL-1 response signatures (see below) (Figures 4F and 4G; Table S4A), as well as with the mTORC1 signature (HALLMARK_mTORC1_signaling) (Pearson r = −0.63; p = 0.0008) and the IFNG transcript (Pearson r = −0.46; p = 0.02) (Figures 4H and 4I). This negative association between the FA and NF-κB related inflammation signature was also confirmed in the two independent German cohorts (Figure S4F) (Schulte-Schrepping et al., 2020). Thus, CD56dimCD16hi NK cells from the most critical patients were residing in a potentially dysfunctional state with attenuated inflammation (relative to less severe cases) and low IFNG transcription despite exposure to increased IL-15, as well as elevation in both FA biosynthesis and oxidation genes. Consistent with this notion, while CD56dimCD16hi NK cells are known to produce IFN-γ early (within hours) after cytokine stimulation, IFN-γ production decreases or stops by 16 h (De Maria et al., 2011); prolonged exposure of NK cells to inflammatory cytokines like IL-15 can actually lead to hyporesponsiveness to subsequent stimulation, partly via a loss in FA oxidation (Felices et al., 2018). Thus, here the increased FA oxidation genes may reflect a compensatory mechanism to counteract this dysfunctional metabolic state, although it is unclear why FA biosynthetic genes were also increased.
Consistently, the two primary negative correlates of DSM, chemokine signaling and IL-1 response signatures in the same subset of NK cells (Figures 4A and 4F), shared genes with the NF-κB and inflammation signatures, such as RELA, NFKB1B, STAT1, and IL8, although they also contained additional genes such as XCL1 and XCL2, which are chemokines that can be produced and secreted early after NK cell activation following an infection (Dorner et al., 2002). This inflammatory attenuation in the most severe patients was also found in other cell types including classical monocytes (Figures S4G and S4H), as reported earlier by others (Arunachalam et al., 2020; Schulte-Schrepping et al., 2020). Thus, the increased FA, together with the attenuated inflammatory and mTORC1 signatures in the most severe patients, might reflect an exhaustion-like NK cell state due to persistent exposure to inflammatory signals including IL-15.
Similar to the IFN-I signatures, the FA signature decreased over time, but here particularly in the DSM-high patients, because those had the most elevated expression of the FA genes near T0 (Figures 4J, S3A, and S3B). In contrast, the inflammation signatures increased, including the NF-κB (HALLMARK_TNFα_signaling_via_NF-κB) and IL-1 signatures (Figure 4K; Tables S4C and S4D), and the IFNG transcript (data not shown). Consistently, circulating IL-15 levels decreased particularly in the DSM-high patients (Figure 4L). Unlike the IFN-I signatures, however, both the FA and inflammation signatures deviated further from the HCs even though IL-15 dropped over time (Figures 4J-L). Thus, whereas exposure to IL-15 earlier in the disease course might have contributed to the dysregulated phenotypes in these NK cells, a reduction in IL-15 alone seemed insufficient to reset these cells; additional signals, such as those from other inflammatory cytokines, might have contributed as well (see below).
Exogenous corticosteroid use was associated with DSM (p = 0.001, F test from a linear model accounting for age, sex, ICU and intubation statuses, and TSO) and thus might have played a role in driving the inflammatory attenuation signature in the CD56dimCD16hi NK cells. However, DSM was not associated with a glucocorticoid transcriptional response signature derived from human immune cells (Franco et al., 2019) (Figure S5 A). Importantly, both this glucocorticoid transcriptional signature or another well-known marker of glucocorticoid or IL-10 exposure (TSC22D3/GILZ transcript [Berrebi et al., 2003; Cannarile et al., 2001]) was not negatively associated with the NF-κB, inflammatory, and mTORC1 signatures and also not positively correlated with the FA signature (Figures S5B and S5C). Although TSC22D3 mRNA level was trending higher in COVID-19 patients than HCs, it was similar between patients on or off corticosteroids (Figure S5D). Unexpectedly, the NF-κB and glucocorticoid signatures were positively correlated across patients (Figure S5B). These observations together suggest that the glucocorticoid response signature might be driven by endogenous glucocorticoids as a part of negative feedback regulation resulting from earlier inflammatory activation in COVID-19 patients (Jamieson et al., 2010; Newton et al., 2017). Thus, a “burnt-out”/exhausted, low inflammation, high FA gene expression state in CD56dimCD16hi NK cells could contribute to severe COVID-19 independent of exogenous corticosteroid use.
Figure S5.

Exogeneous corticosteroid treatment is not a major driver of cell-type-specific gene expression signatures associated with disease severity, related to Figure 4
(A) GSEA results for glucocorticoid response signature (see STAR methods on how the signature was derived) in DSM association model. Positive enrichment corresponds to higher level in the DSM-high samples. (B and C) Scatterplot showing the correlations between the indicated signature scores (computed using GSVA) and the glucocorticoid response signature score (B) or the TSC22D3 mRNA expression level (C) in CD56dimCD16hi NK cells. Pearson correlations and p values are reported for patients with corticosteroid use (orange; steroid use TRUE) and those without (cyan; steroid use FALSE) as well as for all samples (black). Each dot indicates a subject, shaped by steroid use status and colored by the severity-outcome category. (D) TSC22D3 mRNA expression levels of CD56dimCD16hi NK cells and classical monocytes in HC, no steroid-use and steroid-use COVID-19 patient groups. P values from testing for differences between the no steroid-use and steroid-use groups are shown. P values were obtained from an ANOVA test accounting for DSM, TSO, age and experimental batch.
Extensive single-cell and clonal expansion heterogeneity without signs of exhaustion in CD8+ T cells
T cell signatures were interestingly not implicated as primary correlates of disease severity. Consistent with an acute viral infection, signs of T cell activation and proliferation such as cell cycle and related metabolic signatures appeared in both CD4 + and CD8+ T cells in COVID-19 patients compared to HCs (Figures 3A and 3B). Examining CD8+ T cells further at the single-cell level including clonality information (utilizing our simultaneous TCR and gene expression measurements) revealed extensive heterogeneity (Figures S6 A and S6B), including subsets of activated cells with high clonality that were more abundant in COVID-19 (i.e., cluster 14 in Figures S6A–S6D). Similar patterns of heterogeneity were observed in CD4+ T cells, but most CD4+ subclusters were not expanded or less expanded compared to the CD8+ T cells (data not shown). Surprisingly, although clonality in all CD8+ memory T cells was unchanged between HCs and COVID-19 patients at T0, it was significantly higher in the DSM-high than the DSM-low patients after accounting for TSO (Figure S6E; p = 0.023 from linear model testing the effects of DSM while accounting for age, batch, and TSO), suggesting increased diversification (or depletion of clonal cells) in less severe patients relative to those with more severe disease.
Figure S6.

Single-cell and clonal expansion analysis in CD8+ T cells and exhaustion assessment in clonal CD8+ T cells, related to Results section “Extensive single-cell and clonal expansion heterogeneity without signs of exhaustion in CD8+ T cells
(A) Heatmap showing the gene expression profile of CD8+ T cell clusters identified based on single-cell mRNA expression of the leading-edge genes of selected pathways presented in Figure 3 (see text and STAR methods); only COVID-19 T0 samples are shown. Average scaled mRNA expression per cluster of genes (columns) in each of the CD8+ T cell clusters (rows) is shown. Gene memberships in selected gene sets are indicated by the color bars at the bottom of the heatmap. Total number of cells in each cluster is indicated on the right side of the heatmap. Bar plot shows the fraction of cells within each cluster that are defined as expanded (> 1 cell detected per CDR3 α-β pair). Two small clusters (< 32 cells each) are not shown. (B) Average expression of selected surface proteins in the clusters from (A). (C) Results of linear model accounting for age, and experimental batch, comparing the frequency of CD8+ T cell clusters from (A) between COVID-19 and HC samples. Shown is the difference in mean frequency between COVID-19 and HC (x axis) versus the -log10 (p value); horizontal line denotes an unadjusted p value of 0.05. 18 CD8 clusters were tested in linear model. (D) Fraction of overlap between RNA based clustering (from A) and surface protein based CD8+ naive and memory T cell cluster annotations (based on high resolution clustering). (E) CD8+ T cell clonality (median rarefied Simpson index, see STAR methods) in HC, DSM-low and -high groups. p values shown are from linear model adjusted for experimental batch, age, and TSO (green for assessing differences between COVID-19 versus HCs and red for correlation with DSM; TSO only adjusted for in the model assessing DSM association). (F) Coefficients from linear models of mean surface protein expression of canonical exhaustion markers fitted to COVID-19 patients and HCs. Positive coefficients (red bars) correspond to higher expression in COVID-19 versus HCs (above) or higher expression in DSM-high versus -low (below) and vice versa for negative coefficients (green bars). p values are indicated with red indicating significance at the 0.05 level (unadjusted). Among them, only CTLA4 was mildly significant but the association was in the opposite direction expected for exhaustion: its expression was lower in patients, particularly in the more severe. Similarly, insignificant changes or inconsistent directions of change were detected for the mRNA of these protein markers (data not shown). (G) Association of proportion of CD39+PD1+ cells with COVID-19 versus HCs and DSM in clonal CD8+ memory T cells using different cutoffs for CD39 and PD1 surface protein expression DSB counts (0.5, 1). p values are from Wilcoxon tests. Similarly, we also tested whether the frequency of cells co-expressing multiple exhaustion markers in (F) was different. Independent of the surface marker combination or protein expression cutoff used, we saw no signs of exhaustion or association with DSM (data not shown). (H) GSEA results for assessing enrichment for known exhaustion signatures in DE genes for expanded CD8+ T cells in COVID-19 versus HCs and DSM-high versus DSM-low comparisons. Positive enrichment corresponds to higher level in the first group. Both exhaustion gene sets were not enriched in the DSM-high versus DSM-low comparison, indicating that exhaustion was not associated with disease severity. The enrichment in the COVID versus HC comparisons could reflect cellular activation (“up” signature – see (I) below) and depression of translation genes in COVID-19 patients negatively associated with IFN-I signatures (“down” signature) – see also Figure 3A. (I) Gene set enrichment of Wherry et al. (2007) up and down genes in KEGG, GO BP, REACTOME, and Li BTM’s. Only top hits (ranked by adjusted p values from Fisher’s exact test) are shown. The “exhaustion up” genes appeared to reflect CD8+ T cell activation as they are enriched for NF-κB, JAK-STAT signaling, and IL-2 signaling genes. (J) GSEA result of Schulte-Schrepping et al. (2020) cohorts for exhaustion signatures of COVID-19 versus HCs and severe versus mild comparisons at T0. Differential expression moderated t statistics were computed from a linear model accounting for age, experimental batch and TSO (TSO only accounted for in the severe versus mild comparison). Dot color denotes normalized gene set enrichment score (NES) and size denotes -log10(adjusted P value). Dot shape indicates the significance of adjusted P values (adjusted P value < 0.05). Both cohorts 1 and 2 and both CD8+ T cell clusters in the original data and memory CD8+ T cell populations using transferred cell type labels from our CITE-seq data are shown. (K) Similar to (H). GSEA results of Schulte-Schrepping et al. (2020) cohorts for Wherry et al. (2007) exhaustion signatures of COVID-19 versus HCs and severe versus mild comparisons at T0. Positive enrichment corresponds to higher level in the first group. Only memory CD8 population based on transferred labels from our CITE-seq data is shown as an example.
Despite signs of activation and proliferation, we did not detect a cluster of “exhausted” CD8+ T cells associated with COVID-19 as suggested earlier (Laing et al., 2020; Zhang et al., 2020b), based on surface markers such as PD-1 (CD279) (Figures S6B and S6C). Furthermore, we did not detect association of exhaustion with disease status or severity by using surface markers or surface marker combinations (e.g., CD39+PD1+) (Gupta et al., 2015) in clonally expanded CD8+ memory T cells in our cohort (Figures S6F and S6G). Assessment using transcriptional signatures of exhaustion (Wherry et al., 2007) in our cohort and in two German cohorts (Schulte-Schrepping et al., 2020) revealed a more complex picture given that these signatures overlap with those of cellular activation and translation/ribosome (Figures S6H–S6K), which were detected earlier as disease status or severity correlates of COVID-19 (Figures 3A and 3B). Thus, transcriptional signatures alone are not sufficiently specific to indicate whether the cells were more exhausted or merely more activated. Although we uncovered an exhaustion related metabolic gene signature in CD56dimCD16hi NK cells as a primary disease severity correlate (Figure 4A), CD8+ T cells did not show signs of exhaustion beyond the transcriptional signatures expected of cellular activation or regulation by type I IFNs (Ivashkiv and Donlin, 2014), even in patients with critical disease.
Analysis of timing effects suggests a late immune response juncture
As shown earlier, although inflammatory signatures (e.g., HALLMARK_TNFα_signaling_via_NF-κB) were less elevated in the DSM-high patients than DSM-low patients early, they increased over time and stayed elevated compared to HCs as late as day 20 post symptom onset in cell subsets such as CD56dimCD16hi NK cells and classical monocytes (Figures 3B, 4K, S3A, and S3B; Tables S4C and S4D). Gene signatures of cytokine-mediated signaling pathways (e.g., IL-4/13 and IL-17 signaling) in classical monocytes also showed significant increases over time, particularly in the DSM-high patients (Figure S3B). Consistent with an earlier report (Lucas et al., 2020), the frequency of classical monocytes and its CD163hi subset increased in both patient groups over time and peaked around day 20 (Figure 5 A). The same was true for absolute monocyte and neutrophil counts in blood (Figure 5B). Thus, we next further examined the dynamics around day 20, particularly to assess differences between the DSM-high versus DSM-low patients.
Figure 5.

Analyses of timing effects suggest a late immune response juncture
(A) Time course of monocyte subset frequencies in DSM-low and DSM-high groups. Classical monocyte is expressed as fraction of total PBMCs; the CD163hi classical monocyte subset is expressed as a fraction of classical monocytes. The median cell frequency of age- and gender-matched HCs is shown as a green dotted line. Individual samples are shown as dots and longitudinal samples from the same individual are connected by gray lines. P values shown are from mixed effect linear models indicating the statistical significance of the timing effect (i.e., TSO) accounting for age and experimental batch in DSM-low and DSM-high groups, respectively. Trajectories (using LOESS smoothing) were fitted to DSM-low and DSM-high groups separately. TSO = days 17–23 period is highlighted with purple.
(B) Similar to (A) but showing the absolute blood neutrophil and monocyte counts. The two green dotted lines mark the approximate reference range of cell counts in healthy adults. The shaded areas around trajectories denote standard error.
(C) Effect size (normalized enrichment score from GSEA) comparison of the period before day 17 (TSO <day 17, green) and during the TSO = days 17–23 period (purple) for inflammatory related gene sets. Effect sizes correspond to differences between DSM-high versus DSM-low groups (e.g., effect size <0 corresponds to the gene signature being less enriched in the DSM-high group than DSM-low group). p values shown are FDR adjusted (via the Benjamini-Hochberg method) from the test reflecting the temporal changes in the difference between DSM-high and DSM-low groups from before the day 17 period to the days 17–23 time window (see STAR methods).
(D) Similar to Figure 3A, but showing GSEA results for assessing differences between the DSM-high versus DSM-low groups using only samples from days 17–23 since symptom onset. See Table S4E for detailed results of these selected gene sets and all sets that passed the adjusted p value cutoff of 0.2.
(E) Time course of gene set signature scores of inflammatory related gene sets in DSM-low and DSM-high patient groups in CD56dimCD16hi NK cells and classical monocytes. The gene set signature scores were calculated using the LE genes identified from the enrichment analysis shown in (C) above to highlight differences between the DSM-high versus DSM-low groups during the days 17–23 period. The median score of age- and gender-matched HCs is shown as a green dotted line. The shaded areas around trajectories denote standard error.
(F) Time course of serum protein levels from DSM-low and DSM-high patients, respectively. Similar to (E). Top 4 serum proteins significantly different between DSM-high versus DSM-low groups during the days 17–23 period are shown. See Table S6 for the full list of differentially expressed proteins.
We divided samples into three groups by TSO: before day 17 (27 samples), the week around day 20 (days 17–23, 16 samples), and after day 23 (5 samples), then fitted mixed effect models to evaluate changes between the first two periods in cell-type-specific gene expression differences between DSM-high and DSM-low patients (Figure S7 A; Table S4F; STAR methods). We detected several statistically significant “flips,” for example, inflammatory signatures in CD56dimCD16hi NK cells and classical monocytes were lower in DSM-high patients before day 17 but rose to be higher than the DSM-low patients by days 17–23 (Figure 5C). We then assessed differences between the DSM-high and DSM-low groups during days 17–23 (Figure 5D; Table S4E). In addition to CD56dimCD16hi NK cells and classical monocytes, the NF-κB gene signature was elevated in non-classical monocytes, other NK cell subsets, mucosal-associated invariant T (MAIT) cells, and memory and naive B cells in DSM-high compared to DSM-low patients (Figure 5D). The inflammation signature (i.e., the HALLMARK_Inflammatory_response gene set) behaved similarly (Figures 5D and 5E; Table S4E). Heightened signals of cellular activation and proliferation were also detected, such as cell-cycle signatures in CD8+ memory T cells (Figure 5D; Table S4E). We also assessed circulating proteins to reveal that several markers of inflammation (e.g., IL-6, soluble TNF receptors 1 and 2) were elevated in the DSM-high versus -low patients (Figure 5F; Table S7). Thus, this relative late period, or “juncture,” in the disease course (days 17–23) was characterized by increased inflammatory divergence associated with disease severity.
Figure S7.

Supporting data for critical juncture analysis, related to Figures 5 and 6
(A) Similar to Figure 3A, but here showing GSEA results for assessing the differences of delta between disease severity groups (DSM-high versus DSM-low) between the days 17-23 time window and the period before (TSO < day 17). See Table S4F for detailed results of these selected gene sets and the entire set that passed the adjusted p value cutoff of 0.2. (B) Time course of blood neutrophil and monocyte counts in recovered and deceased groups. Corresponding range of HCs are shown with green dotted lines. Longitudinal samples from the same individual are connected by gray lines. Trajectories were fitted to recovered and deceased groups separately with the shaded areas representing standard error. (C) Effect size comparison of DSM-high versus DSM-low (CITE-seq cohort) and deceased versus recovered (critical patients with distinct outcome subcohorts – see Figure 6A) at the days 17-23 period. Effect sizes were derived from the coefficients of the group comparison term in mixed-effect models. Proteins are colored by whether they were significantly different (with an unadjusted p value of less than or equal to 0.05) in the deceased versus recovered (“outcome”) and the DSM-high versus -low comparisons (“DSM”). See also Table S7 for the full list of DE proteins. (D) Similar to (C). Effect size comparison of Brescia deceased versus recovered and an independent US cohort (Yale cohort) (Lucas et al., 2020) deceased versus recovered patients (See STAR methods) for 38 overlapping circulating proteins/cytokines at the juncture period (TSO days 17-23). Proteins in red are those significantly different between the deceased and recovered patient groups in the Brescia cohort (unadjusted ANOVA p value < 0.05).
Divergences at the juncture predict fatal COVID-19
We next hypothesized that the immune statuses and trajectories at the juncture period are also divergent between the critically ill patients with distinct survival outcomes (Figure 6 A). Because our CITE-seq cohort only has a few patients with fatal outcomes, we started from the larger Brescia cohort with circulating protein measurements (175 patients; see also Abers et al. [2021]) and generated two age- and gender-matched sub-cohorts of critical patients with either recovery (n = 21) or fatal (n = 17) outcomes. ICU status, intubation, and corticosteroid use were not significantly different between these two groups during days 17–23 (Table S6).
Figure 6.

Divergence of deceased and recovered patients at the late juncture
(A) Approach for assessing and validating the late immune response juncture hypothesis by using serum protein profiles of critical ill patients with either recovery or deceased outcomes.
(B) Effect size plots of circulating serum proteins comparing the difference between critical deceased versus recovered patients before (days 7–16), during (days 17–23), and after (days 24–30) the juncture period. Mixed effect models were fitted to assess whether this difference between deceased and recovered groups changed significantly between (1) pre-juncture and juncture (top panel showing effect sizes in each period and its right bar plots showing the p value assessing the temporal difference), or (2) pre-juncture and after the juncture (bottom panel). The size of the circle denotes the statistical significance of the difference between deceased and recovered groups in the indicated period (p < 0.05 is marked by solid outlines). See also Table S7.
(C) Outcome prediction performance (recovered versus fatal) at (17–23 days; purple) or post (24–30 days; blue) juncture using leave-one-out cross-validation. Feature selection was performed using the same procedure that identified proteins shown in (B). Area under the curve (AUC) and permutation p values are shown.
(D) Similar to Figure 6F but showing serum protein levels of critical ill patients with recovered or deceased outcomes (see A). Trajectories (using LOESS smoothing) were fitted to the recovered versus deceased patient groups separately. Top proteins showing the largest temporal shifts in their differences between the deceased versus recovered patients in (B) are shown.
(E) Similar to (D) but for antibody measurements against SARS-CoV-2 spike and nucleocapsid proteins in critically ill patients with recovered or deceased outcomes. LU, light unit.
As seen above (Figure 5B), blood monocyte and neutrophil counts also peaked around day 20 in these patients (Figure S7B). We found 20 proteins significantly different between the deceased and recovered groups during the days 17–23 period (p < 0.05 based on a mixed effect model and the estimated false discovery rate [FDR] is 16%) (Table S7; STAR methods), including proinflammatory cytokines such as TNF-α, IL-6, and IFN-γ. These were consistent with the DSM-high versus -low comparison (Figure 5F; Table S7), with 45/63 proteins showing the same direction of change between the deceased versus recovered patients as that between DSM-high versus DSM-low groups (p = 0.0896, Fisher’s exact test; Figure S7C), suggesting that the inflammatory divergences between patients of high versus low disease severity were qualitatively similar to those between critical patients with distinct survival outcomes. Similarly, a majority of the differentially expressed (DE) proteins at the juncture had the same direction of change between the deceased and recovered individuals in an independent US cohort (Lucas et al., 2020) (Figure S7D).
We next reasoned that if, as suggested by the observations above, the days 17–23 window is an immune response juncture in the disease course, markers of inflammatory and other immune processes would not only differ between the two patient groups at the juncture but also exhibit temporal shifts during or after the juncture period relative to the period before the juncture—i.e., the juncture is a period of change and/or an inflection point orchestrating later changes. We thus searched for proteins that were DE between the patient groups during (days 17–23) or after the juncture (days 24–30, “post-juncture”) and also required that the differences between the deceased and recovered patients changed significantly over time between the pre-juncture (days 7–16) and juncture or between the pre- and post-juncture periods (Figure 6A). We found 12 proteins that satisfied these search criteria (Figure 6B) (all p < 0.05 and overall FDR = 14% based on a permutation test [i.e., ∼2 proteins are expected to be false positives]) and are predictive of fatal outcomes based on cross-validation machine learning analysis (Figure 6C).
These proteins revealed striking divergences between deceased and recovered patients around the juncture (Figures 6B and 6D). Several markers of tissue inflammation or damage started to increase more at the juncture in the deceased cohort, such as E-selectin, a marker of endothelial inflammation/leukocyte transmigration and lipocalin-2/NGAL, which is known to be associated with acute kidney injury and innate immune response to bacterial infections (Flo et al., 2004; Haase et al., 2009) (Figure 6D). Consistent with the IFN-I signatures above (Figures 3D, 3E, S3A, and S3B), IP-10, an IFN-stimulated protein product, was decreasing prior to the juncture with a steeper decline in the recovered cohort to reach lower levels than the deceased group by day 20 (Figure 6D). As observed earlier in the DSM-high versus DSM-low patients (Figure 5F), markers of inflammation and immune responses (e.g., IL-6, TNFRSF1B, and IL-17) were increasing at a higher rate or persisting (e.g., IL-18) at an elevated level in the deceased group (Figure 6D). However, antibody responses to both the SARS-CoV-2 spike and nucleocapsid appeared slower in the deceased compared to the recovered cohorts (Figure 6E), consistent with the delay and potential miscoordination of antigen-specific adaptive immune responses in the most severe patients (Rydyznski Moderbacher et al., 2020). Together, these findings further support our hypothesis that the days 17–23 period represents a critical juncture in the disease course characterized by a heightened wave of inflammatory responses in critical patients with fatal outcomes.
Discussion
Here, we integrated circulating cytokine and multimodal single-cell profiling and computational approaches to assess the cell surface protein phenotype, transcriptome, and T cell clonality of peripheral immune cells of COVID-19 patients over time. We revealed a network of cell-type-specific signatures linked to disease severity, dissected timing effects, and uncovered a late period during which the host immune response undergoes striking divergences between patients with distinct disease severity and outcomes. We validated key observations using public datasets in independent cohorts, including gene signatures of apoptosis in pDCs and FA metabolism and inflammatory attenuation in CD56dimCD16hi NK cells linked to disease severity.
Timing is a critical variable when assessing the host immune response to viral infections (Bernardes et al., 2020; Dunning et al., 2018; Huang et al., 2011; Zhang et al., 2020a). Live influenza challenge studies in humans and animal models have painted a prototypical immune response trajectory involving antiviral/IFN-I signatures in blood a few days after infection concomitant with symptom onset, which peak by days 5–7 and wane shortly after, followed by T cell activation, germinal center reaction, and plasmablast expansion during days ∼7–14, culminating in peak circulating antibody levels by ∼3–4 weeks after infection (Brandes et al., 2013; Huang et al., 2011; Pommerenke et al., 2012; Woods et al., 2013). In contrast, SARS-CoV-2 infections often have asymptomatic upper respiratory infections early (Matheson and Lehner, 2020), but more than 90% of cases displayed signs of pneumonitis a few days after symptom onset (Bernheim et al., 2020), suggesting that the patients we are studying here also had asymptomatic infections in the upper airway early (first ∼7–10 days after infection), followed by infection and inflammation in the lower airways and lung concomitant with symptom onset. Thus, at T0 (median = 11 days post symptom onset), most patients in our study may be in the mid to late phases of epithelial type I interferon production in response to the lung infection (Liao et al., 2020; Nienhold et al., 2020; Wauters et al., 2020). Consistent with recent reports (Arunachalam et al., 2020; Hadjadj et al., 2020; Schulte-Schrepping et al., 2020), we indeed detected increased IFN-I signatures in cells compared to matching HCs, including genes induced early in blood following influenza challenge or immunization with the yellow fever vaccine (Querec et al., 2009; Woods et al., 2013).
As observed earlier using bulk whole blood RNA expression analysis (Hadjadj et al., 2020), we found that IFN-I signatures were negatively associated with disease severity at T0 with single-cell resolution, even after accounting for timing effects. Timing contributed substantially to the variability of the IFN-I signature given its steep temporal decline, especially in the less severe patients. Concomitant with the elevation of the IFN-I signature were decreases in protein translation genes; the IFN-I and translation signatures tended to be negatively correlated within cell types, potentially reflective of the suppressive effect of type I IFNs on protein translation to limit viral replication (Ivashkiv and Donlin, 2014). However, whether a more prolonged suppression (e.g., with exogenous type I IFN use as an intervention) could negatively impact protein production in cells vital to the immune response (e.g., plasma cells producing antibodies) warrants further investigation.
In addition to infected epithelial cells, pDCs are another potential major source of type I IFNs. Consistent with earlier studies (Arunachalam et al., 2020; Kuri-Cervantes et al., 2020; Laing et al., 2020), we found that pDC frequencies were lower in COVID-19 patients than HCs and negatively associated with disease severity and IFN-I signatures in most immune cell subsets, suggesting that lower pDC levels might have contributed to the depressed IFN-I signatures in severe disease. Recent studies of lung autopsies and BAL fluids from COVID-19 patients did not indicate an association between pDC frequency and disease severity (Chua et al., 2020; Liao et al., 2020; Nienhold et al., 2020; Wauters et al., 2020); although pDCs can also migrate into secondary lymphoid organs via high endothelial venules (Diacovo et al., 2005; Randolph et al., 2008), data are lacking in SARS-CoV-2 infections. Thus, it is less likely that lower blood pDC frequency was solely due to pDC migration out of the periphery. Instead, we detected an apoptosis signature in COVID-19 pDCs, and the same signature was positively associated with disease severity and negatively associated with pDC frequency, thus providing a compelling hypothesis on why pDC frequencies were reduced in blood. Type I IFNs themselves can trigger apoptosis in pDCs based on animal studies (Swiecki et al., 2011), presumably as a negative feedback to modulate pDC numbers and limit the extent of type I IFN activation. Corticosteroids are also known to induce apoptosis and thus reduce the frequency of circulating pDCs in humans (Boor et al., 2006; Shodell and Siegal, 2001). Endogenous corticosteroids are induced by the hypothalamic-pituitary-adrenal system following viral infections to mediate negative feedforward regulation to partly limit the damaging effects of overt inflammation (Jamieson et al., 2010; Newton et al., 2017; Ruzek et al., 1997). Indeed, the more depressed IFN-I signatures in critically ill patients may be driven, at least in part, by the stronger negative regulation induced by more severe infections and disease. These interactions should be taken into consideration when designing therapies that target the type I IFN-pDC axis (Acharya et al., 2020; Hung et al., 2020).
Our disease severity network pointed to attenuated inflammation and an “exhaustion”-like gene expression state in CD56dimCD16hi NK cells as a primary positive correlate of disease severity. This FA signature was positively associated with circulating levels of IL-15, but paradoxically, negatively correlated with IFNG transcripts, NF-κB, mTORC1 signaling, and inflammatory gene expression signatures. Long-term exposures (on the timescale of days but not hours) to inflammatory activation are known to induce metabolic changes in NK cells, including the upregulation and utilization of both glycolysis and oxidative phosphorylation (OXPHOS) programs (O’Brien and Finlay, 2019). The role of FA metabolism is not well known, but prolonged exposure to proinflammatory cytokines like IL-15 can induce an “exhaustion”-like state linked to altered FA metabolism and hyporesponsiveness to future stimulations (Felices et al., 2018). Consistent with our data on the lowering of the mTORC1 signature and IFNG mRNA levels, decreased mTORC1 activity in NK cells is known to be associated with lower IFN-γ production (Nandagopal et al., 2014). The potential increase in lipid biosynthesis could also lead to lipid accumulation, which can downregulate mTORC1 signaling and thus NK cell effector functions (Michelet et al., 2018). Whereas the elevation in FA oxidation transcripts may reflect a compensatory mechanism to counteract the decreased FA oxidation capacity in NK cells persistently exposed to IL-15 (Felices et al., 2018), the concomitant increases in fatty acid biosynthesis and oxidation/catabolic genes may be a signature of dysfunction. Although not much is known about NK cells, CD8+ T cells are known to upregulate FA biosynthetic genes briefly early after viral infection, whereas FA oxidative genes appear later during the late effector/early memory phase to support memory cell functions, but not both simultaneously (Best et al., 2013; van der Windt et al., 2013). Overall, our findings are consistent with earlier observations of NK cells from COVID-19 patients, including increases in “adaptive” NK cells known to be associated with chronic activation as well as signs of exhaustion suggested by impaired cytolytic functions and inflammatory cytokine production, and elevation of surface markers or corresponding transcripts of NK cell exhaustion (Maucourant et al., 2020; Osman et al., 2020; Varchetta et al., 2020; Wilk et al., 2020).
The negative feedforward circuits regulating the delicate balance of type I IFN responses and pDC apoptosis discussed above might also play a role in and serve as cell “extrinsic” signals attenuating the inflammatory response in CD56dimCD16hi NK cells (and other cells such as classical monocytes) in severe disease. For example, endogenous corticosteroids can induce anti-inflammatory programs in monocytes (Franco et al., 2019), and so can IL-10 produced by monocytes themselves (or macrophages in tissues) and other cell types such as regulatory T cells following inflammatory activation (Mosser and Edwards, 2008; Neumann et al., 2020; Wynn et al., 2013; Xue et al., 2014). Indeed, circulating IL-10 levels are known to be elevated in COVID-19 patients (Lucas et al., 2020); here, IL-10 persisted at a high level particularly in non-survivors but dropped rapidly in survivors (data not shown). IL-10 and glucocorticoids can also increase CD163 expression on the surface of monocytes (Sulahian et al., 2000; Vallelian et al., 2010), thus could explain the rise of CD163hi classical monocytes in our patients over time. While exogenous corticosteroid, which is known to lower all-cause mortality in COVID-19 (WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group et al., 2020), was used by some patients in our cohort and its use correlated with DSM, our assessment using glucocorticoid gene expression signatures suggests that exogenous corticosteroid use did not play a role in shaping the inflammatory attenuation we observed. Our findings may also mirror some features of sepsis (López-Collazo et al., 2020): after the initial phase of hyperinflammation, a secondary phase of compensatory responses mediated by negative feedforward circuits can leave the patient immunosuppressed (Bellinvia et al., 2020; Bone, 1996; Rubio et al., 2019; Venet and Monneret, 2018).
Whether these attenuated inflammatory states were a consequence or cause of severe COVID-19 remains to be determined. Timing is again an important question. If attenuation happened early after the infection before or near symptom onset, it could negatively impact adaptive responses, which actually appeared largely intact in our patients. Consistent with previous reports (e.g., Laing et al., 2020; Mathew et al., 2020), we observed CD8+ T cell activation and proliferation. However, in contrast to some earlier reports (Laing et al., 2020; Zhang et al., 2020b), but consistent with a single-cell study of severe patients using mRNA expression of exhaustion markers (Wilk et al., 2020) and a recent report that SARS-Cov-2-specific CD8+PD1+ T cells are not exhausted (Rha et al., 2021), clear signs of CD8+ T cell exhaustion were not detected. Another report found increased PD-1- and TIM3-positive cells in COVID-19 patients versus controls but not across severity groups (Laing et al., 2020), thus suggesting that these might reflect T cell activation rather than exhaustion (Mathew et al., 2020). Together, our observations suggest that the attenuated antigen presentation and inflammatory activation in certain innate immune cells likely occurred later after symptom onset and did not severely impact the initial adaptive response to the infection.
Although the most severe patients had attenuated inflammation in CD56dimCD16hi NK cells and classical monocytes early, these signatures increased over time and were significantly more elevated than those in less severe patients by days 17–23. Circulating protein analysis confirmed that both severe (DSM-high) patients and non-survivors had elevated inflammatory and immune responses at this juncture compared to those with less severe disease (DSM-low) or critically ill survivors, respectively. This late period is distinct from known clinical junctures earlier in the disease course (e.g., the onset of ARDS and sepsis around days 10–12 post symptom onset) (Zhou et al., 2020a). However, the underlying driver of those earlier events might still be responsible for some of the divergences by days 17–23.
Secondary infections are potential triggers for this late wave of responses (Cox et al., 2020; Kim et al., 2020; Langford et al., 2020; Lansbury et al., 2020). Day 17 was the reported median onset of co-infections in a large retrospective cohort study (Zhou et al., 2020a), and we observed elevation in potential markers of bacterial infections (e.g., lipocalin-2/NGAL and lipopolysaccharide [LPS] binding protein [LBP]) around that time. The secondary infection rates reported thus far have been highly variable (from a few to tens of percent), and therefore more data are needed to further assess the prevalence and types of co-infections associated with severe COVID-19 and whether they are triggers of the late inflammatory responses we observed. Given the acute organ injury and tissue damage in severe disease, translocation of commensal bacterial products and other danger signals of tissue origin could also contribute to this late response wave (Estes et al., 2010; Marchetti et al., 2013). Indeed, the level of bacterial products (LPS and bacterial ribosomal DNA) in plasma has been reported to be associated with disease severity (Arunachalam et al., 2020).
Intriguingly, blood transcriptomic analyses of hospitalized, severe influenza patients have also revealed a second wave of responses (peaking around day 13 post symptom onset) involving a “bacterial” transcriptional signature that is associated significantly more with disease severity than with detectable bacterial co-infections (Dunning et al., 2018). Despite the timing differences and lack of information on the cellular origin of this late signature, these observations in influenza are qualitatively similar to ours in COVID-19. Thus, even without secondary infections, contributions from other factors including the lingering viral load and acute tissue injury in the critical patients may play important roles in driving the responses at the late juncture (Hirsch et al., 2020; Zheng et al., 2020; Zhou et al., 2020c). The lack of coordination among and delays in antigen-specific humoral and cellular immunity could be partly responsible for lingering viral loads (Rydyznski Moderbacher et al., 2020), which could also then trigger a second wave of inflammatory responses at the late juncture, particularly as the strength of the negative feedforward regulation from earlier innate responses waned.
Our observations highlight the importance of timing in designing therapeutics strategies for COVID-19. For example, although persistent IL-15 exposure could potentially lead to an exhaustion like phenotype in NK cells (Felices et al., 2018), an anti-IL-15 block followed by intermittent application of IL-15 (on-off-on) to revive and boost NK cell functions may help to ensure a productive but not overtly damaging response around the late juncture and thereafter. However, such strategies need to be guided by the immune and physiological profiles of the patient (e.g., whether there is an ongoing co-infection and the status of the patient around the critical juncture). Indeed, personalized intervention and clinical monitoring strategies hold promise to reduce the burden of COVID-19.
Limitations of study
The relatively small size of our cohort of mostly severe and critical patients, sparse sampling of very early time points (e.g., first week of symptom onset), and potential confounding factors such as therapeutic interventions in a natural history study are key limitations. Thus, further studies using larger patient cohorts with better longitudinal sampling will be helpful to further confirm and dissect our observations. Our longitudinal sampling within individual subjects was sparse; we thus borrowed information across subjects to infer trajectories and performed comparative analyses between patient groups. Denser sampling within individual subjects would be needed to dissect how earlier events may drive, for example, divergences at the late juncture. Although we have performed conditional independence analyses to disentangle correlative relationships, we still lack the ability to dissect causality, which would require interventional and animal model studies. Methodologically, our single-cell data do not directly measure parameters beyond gene and surface protein expression and variable lymphocyte receptor sequences. Thus, pathway activity and cellular parameters including metabolism were assessed using gene expression signatures as noted throughout. Direct measurements of those parameters would require further studies. See also STAR methods for limitations of antibody staining and surface protein quantification in CITE-seq.
Consortia
The members of the NIAID COVID Consortium are Michael S. Abers, Richard Apps, Marita Bosticardo, Pedro Milanez-Almeida, Matthew P. Mulè, Elana Shaw, and Yu Zhang.
The members of the COVID Clinicians are Francesco Castelli, Maria Lorenza Muiesan, Gabriele Tomasoni, Francesco Scolari, and Alessandra Tucci. Affiliations for all consortia members can be found in Data S1.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq-C Custom Human panel - All Ab’s (pouch) (see Table S2 for full antibody panel used for TotalSeq/CITEseq) | Biolegend | Cat# 99814 |
| TotalSeq-C0251 anti-human Hashtag 1, clones LNH-94; 2M2 | Biolegend | Cat# 394661; RRID:AB_2801031 |
| TotalSeq-C0252 anti-human Hashtag 2, clones LNH-94; 2M2 | Biolegend | Cat# 394663; RRID:AB_2801032 |
| TotalSeq-C0253 anti-human Hashtag 3, clones LNH-94; 2M2 | Biolegend | Cat# 394665; RRID:AB_2801033 |
| TotalSeq-C0255 anti-human Hashtag 5, clones LNH-94; 2M2 | Biolegend | Cat# 394669; RRID:AB_2801035 |
| Anti-human CD45 (APC/Cyanine7), clone 2D1 | Biolegend | Cat# 368516; RRID:AB_2566376 |
| Anti-human CD3 (AF488), clone SK7 | Biolegend | Cat# 344810; RRID:AB_10576234 |
| Anti-human CD19 (APC), clone SJ25C1 | Biolegend | Cat# 363006; RRID:AB_2564128 |
| Anti-human CCR7 (BV786), clone G043H7 | Biolegend | Cat# 353230; RRID:AB_2563630 |
| Anti-human CD95 (BV650), clone DX2 | Biolegend | Cat# 305642; RRID:AB_2632622 |
| Anti-human IgD (PerCP-Cy5.5), clone IA6-2 | Biolegend | Cat# 348208; RRID:AB_10641706 |
| Anti-human CD27 (PE/Cyanine7), clone M-T271 | Biolegend | Cat# 356412; RRID:AB_2562258 |
| Anti-human CD197 (BUV395), clone 150503 | BD Biosciences | CUSTOM |
| Anti-human CD16 (BUV496), clone 3G8 | BD Biosciences | Cat# 612944; RRID: AB_2870224 |
| Anti-human HLA-DR (BUV661), clone G46-6 | BD Biosciences | Cat# 612980; RRID:AB_2870252 |
| Anti-human CD196 (BUV737), clone 11A9 | BD Biosciences | Cat# 564377; RRID:AB_2738778 |
| Anti-human CD183 (BUV805), clone IC6/CXCR3 | BD Biosciences | Cat# 742048; RRID:AB_2871338 |
| Anti-human IgD (BV421), clone IA6-2 | BD Biosciences | Cat# 562518; RRID:AB_11153121 |
| Anti-human CD4 (eFluor450), clone SK3 | BD Biosciences | Cat# 560345; RRID:AB_1645572 |
| Anti-human CD127 (BV480), clone HIL-7R-M21 | BD Biosciences | Cat# 566101; RRID:AB_2869742 |
| Anti-human CD19 (BV570), clone HIB19 | BD Biosciences | CUSTOM |
| Anti-human CD194 (BV605), clone 1G1 | Biolegend | Cat# 359418; RRID:AB_2562483 |
| Anti-human CD123 (BV650), clone 7G3 | BD Biosciences | Cat# 563405; RRID:AB_2738185 |
| Anti-human CD25 (BV711), clone 2A3 | BD Biosciences | Cat# 563159; RRID:AB_2738037 |
| Anti-human CD14 (BV750), clone M5E2 | BD Biosciences | Cat# 746920; RRID:AB_2871712 |
| Anti-human CD27 (BV786), clone L128 | BD Biosciences | Cat# 563327; RRID:AB_2744353 |
| Anti-human CD45RA (BB515), clone H100 | BD Biosciences | Cat# 564552; RRID:AB_2738841 |
| Anti-human CD38 (PerCP-Cy5.5), clone HIT2 | BD Biosciences | Cat# 551400; RRID:AB_394184 |
| Anti-human CD24 (BB700), clone ML5 | BD Biosciences | Cat# 566524; RRID:AB_2744333 |
| Anti-human CD45 (BB790), clone HI30 | BD Biosciences | CUSTOM |
| Anti-human CD8 (PE), clone RPA-T8 | BD Biosciences | Cat# 555367; RRID:AB_395770 |
| Anti-human CD45RO (PE-Texas Red), clone UCHL1 | Beckman Coulter | Cat# IM2712U; RRID:AB_10639537 |
| Anti-human CD11c (PE-Cy5), clone B-Ly6 | BD Biosciences | Cat# 551077; RRID:AB_394034 |
| Anti-human CD20 (PE-Cy5.5), clone HI47 | ThermoFisher | Cat# MHCD2018; RRID:AB_10372042 |
| Anti-human CD185 (PE-Cy7), clone RF8B2 | Biolegend | Cat# 356924; RRID:AB_2562355 |
| Anti-human CCR10 (AlexaFluor 647), clone 314305 | R&D Systems | Cat# FAB3478A; RRID:AB_573043 |
| Anti-human CD56 (APC-R700), clone NCAM 16.2 | BD Biosciences | Cat# 565139; RRID:AB_2744429 |
| Anti-human CD3 (APC-H7), clone SK7 | BD Biosciences | Cat# 560176; RRID:AB_1645475 |
| Human TruStain FcX | Biolegend | Cat# 422302; RRID:AB_2818986 |
| Biological samples | ||
| Human PBMC and Serum samples (see donor characteristics Table S1. Clinical characteristics of the COVID-19 patients in this cohort, related to Figure 1, Table S7. Supporting data for circulating protein analysis in DSM-high versus -low (CITE-seq cohort) and deceased versus recovered critical patients (outcome cohort, see Figure 6G), related to Figures 6 and 7B) of COVID-19 individuals | ASST Spedali Civili Brescia (Italy); protocol NP 4000 – Studio CORONAlab | N/A |
| Healthy Donor Human PBMC samples | NIH protocol 99-CC-0168 | N/A |
| Healthy Donor Human PBMC samples | NIH protocol 18-I-0101 | N/A |
| Healthy Donor Human PBMC samples | NIH protocol 11-I-0187 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Live/Dead Blue | ThermoFisher | Cat# L23105 |
| Zombie Red Fixable viability dye | Biolegend | Cat# 423110 |
| DNase I | STEMCELL Technologies | Cat#07470 |
| Critical commercial assays | ||
| V-PLEX Human Cytokine 30-Plex Kit | Meso Scale Discovery | Cat# K15054D |
| S-PLEX Human IFN-α2a Kit | Meso Scale Discovery | Cat# K151P3S |
| Custom Multiplex bead assays | R&D Systems | Cat# LXSAHM |
| miRNAeasy micro kit | QIAGEN | Cat# 217084 |
| Truseq RNaseq library preparation kit | Illumina | Cat# 20020595 |
| Chromium Next GEM Single Cell 5ʹ Library & Gel Bead Kit v1.1 | 10x Genomics | Cat# 1000165 |
| Chromium Single Cell 5ʹ Library Construction Kit | 10x Genomics | Cat# 1000020 |
| Chromium Single Cell V(D)J Enrichment Kit, Human T Cell | 10x Genomics | Cat# 1000005 |
| NovaSeq S2 100 cycle sequencing kits | Illumina | Cat# 20012862 |
| NovaSeq S4 200 cycle sequencing kits | Illumina | Cat# 20027466 |
| NovaSeq S1 200 cycle sequencing kits | Illumina | Cat# 20012864 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE161918 |
| Schulte-Schrepping PBMC cohort 1 | Schulte-Schrepping et al., 2020 | https://beta.fastgenomics.org/datasets/detail-dataset-952687f71ef34322a850553c4a24e82e |
| Schulte-Schrepping PBMC cohort 2 | Schulte-Schrepping et al., 2020 | https://beta.fastgenomics.org/datasets/detail-dataset-7ae02f5553074bda92c14a8f0bce2d24 |
| Software and algorithms | ||
| Analysis Code (and full list of R packages used) | This paper | https://github.com/niaid/covid19-time-resolved |
| R (versions 3.5.2, 3.6.0, 3.6.1, 3.6.3) | The R Foundation | https://www.r-project.org |
| Seurat (versions 3.1.0, 3.1.4, 3.2.2) | Stuart et al., 2019 | https://cran.r-project.org/web/packages/Seurat/index.html |
| dsb (beta) | Mulè et al., 2020 | https://github.com/niaid/dsb |
| CytoML (1.12.0) | Finak et al., 2018 | https://www.bioconductor.org/packages/release/bioc/html/CytoML.html |
| flowWorkspace (3.34.1) | Finak and Jiang, 2020 | https://www.bioconductor.org/packages/release/bioc/html/flowWorkspace.html |
| Limma (versions 3.40.2, 3.42.2) | Ritchie et al., 2015 | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| Tidyverse (1.2.1, 1.3.0) | (Wickham, 2019) | https://www.tidyverse.org |
| ComplexHeatmap (2.2.0) | Gu et al., 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| edgeR (3.26.8, 3.28.1) | McCarthy et al., 2012 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FGSEA (1.10.1) | Sergushichev, 2016 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| lme4 (1.1-23) | Bates et al., 2015 | https://cran.r-project.org/web/packages/lme4/index.html |
| lmerTest | Kuznetsova et al., 2017 | https://cran.r-project.org/web/packages/lmerTest/index.html |
| plsRglm (1.2.5) | Bertrand and Maumy-Bertrand, 2019 | https://cran.r-project.org/web/packages/plsRglm/index.html |
| huge (1.3.4.1) | Zhao et al., 2012 | https://cran.r-project.org/web/packages/huge/index.html |
| Gsva (1.3.0) | Hänzelmann et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| Hmisc (4.4-1) | Harrell, 2019 | https://cran.r-project.org/web/packages/Hmisc/index.html |
| demuxlet (v2, from the ‘popscle’ software suite) | Kang et al., 2018 | https://github.com/statgen/popscle |
| CellRanger (3.1.0) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/3.1/ |
| Other | ||
| Hg19 human genome reference for CellRanger | 10x Genomics | https://cf.10xgenomics.com/supp/cell-exp/refdata-cellranger-hg19-1.2.0.tar.gz |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John S. Tsang (john.tsang@nih.gov).
Materials availability
This study did not generate new reagents.
Data and code availability
Raw and processed data from the single cell mRNA, surface protein, and TCR V(D)J sequencing and bulk RNaseq are available from the NCBI Gene Expression Omnibus, accession number GEO: GSE161918 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161918). Analysis code, extended patient and sample metadata, serum protein, and antibody data are available at: https://github.com/niaid/covid19-time-resolved. The single cell UMAPs in Figure 3B can be visualized interactively at: https://cellxgene.cziscience.com/collections/ed9185e3-5b82-40c7-9824-b2141590c7f0.
Experimental model and subject details
Patients
De-identified patient plasma and serum samples were obtained from discarded, clinically indicated collection of blood samples obtained from 382 patients admitted at ASST Spedali Civili Brescia, following positive nasopharyngeal swab and/or positive serology for SARS-CoV-2 infection. As described in the following sections and the main text, a subset of 60 unique patients, including 33 patients who had peripheral immune cells collected and 38 critically ill patients with deceased or recovery outcomes (for circulating protein/cytokine-based validation of the “critical juncture” concept in Figure 6) were analyzed in this study (i.e., 7 recovered and 4 deceased patients also had PBMCs collected). Ethical approval was obtained from the Comitato Etico Provinciale (NP 4000 – Studio CORONAlab), Brescia (Italy). Blood samples were processed to obtain serum and plasma. In addition, EDTA-blood samples were processed using Ficoll density gradient (GE Healthcare, Marlborough, MA) to obtain PBMCs, then washed twice in RPMI-1640; 5x106 PBMC were then resuspended in 1 mL of freezing medium consisting of RPMI plus 20% fetal calf serum and 10% DMSO, and then frozen at −80°C. Patient metadata is available in Tables S1 and S6 and deposited along with the sequencing data in GEO and code in Github listed above.
Healthy control (HC) samples
Blood samples of 14 age and gender matched HCs were collected at the NIH Clinical Center from individuals in Washington, DC metro area. These samples were obtained under NIH IRB-approved protocol 18-I-0101 and PBMCs were prepared using SepMate isolation tubes (STEMCELL Technologies, Vancouver, BC, Canada) and Ficoll density-gradient media, or under NIH IRB-approved protocol 99-CC-0168 and PBMCs were prepared using Ficoll-based density separation (without SepMate) according to manufacturer’s protocol. Cells were cryopreserved by standard methods and stored in liquid nitrogen until needed. One anonymized donor was excluded from further analysis, as we discovered an extremely expanded B cell phenotype consistent with lymphoma in this sample. Two additional healthy control samples were collected under NIH IRB-approved protocol 11-I-0187 and were used in preliminary testing to compare freezing of buffy coat samples with freezing of Ficoll-isolated PBMC (isolated as described for patients above); CITE-seq data was collected for both buffy and PBMC samples of each of these controls, but only the PBMC-derived data was used in our analysis. An additional leukopack-derived PBMC sample (anonymized and without demographic information, collected under NIH IRB-approved protocol 99-CC-0168 from the NIH Department of Transfusion Medicine) was used in each experimental batch as a technical control, but not used in later comparisons.
Method details
Circulating Protein/Cytokine Detection
Cytokine/biomarker analysis was performed on EDTA plasma or sera obtained from either patients or HCs. The cytokine data used in this paper was previously reported for the total Brescia cohort (Abers et al., 2021); the detection method is repeated here for completeness: Because of limited available volume, patient samples were analyzed as single determinations. Duplicate determinations of control samples and samples from HCs yielded coefficients of variation that were normally < 20%. Aliquots were stored in a −85°C freezer prior to analysis. Cytokines (IL-1β, IL-1α, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-12p40, IL-13, IL-15, IL-16, IL-17, IFN-γ, TNF-α, TNF-β, GM-CSF, VEGF, CCL-11/Eotaxin-1, CCL26/Eotaxin-3, CXCL10/IP-10, MCP-1/CCL2, MCP-4/CCL13, CCL22/MDC, MIP-1α/CCL3, MIP-1β/CCL4, CCL17/TARC) were measured using the V-PLEX Human Cytokine 30-Plex Kit (Meso Scale Discovery, Rockville, MD) an analyzed on a MESO QuickPlex SQ 120 reader (Meso Scale Discovery) according to the manufacturers specifications. Interferon-α2a was determined on a single analyte, ultra-sensitive S-PLEX IFN-α2a kit (Meso Scale Discovery) according to the manufacturer’s specifications. Additional cytokines (IL-1RA, IL-3, IL-18, IL-23, IL-33, G-CSF, M-CSF, CX3CL1/Fractalkine, TNFSF5 [sCD40 ligand], TNFSF6 [soluble Fas ligand], TNFSF14 [LIGHT], SCF [c-kit ligand]), and other soluble receptors/biomarkers (sTNFRSF1A [sTNF RI], sTNFRSF1B [sTNF RII], sST2 [sIL-33R], sCD25 [sIL-2R α], sCD54 [sICAM-1], sCD106 [sVCAM-1], sCD31 [sPECAM], sCD62L [sL-selectin], sCD62E [sE-selectin], RAGE [receptor for advanced glycation end products], sCD163 [macrophage scavenger receptor], sVEGFR1 [Flt-1], REG3A [regenerating islet-derived protein III-A], ferritin, S100A8, S100A9, MMP-9 [neutrophil gelatinase], lactoferrin, myeloperoxidase (MPO), lipocalin-2 [neutrophil gelatinase associated lipocalin, NGAL], LBP [LPS binding protein]) were measured on customized, magnetic bead-based, multiplex assay (R&D Systems, Minneapolis, MN) according to the manufacturers specifications for standards and dilutions. The magnetic beads were analyzed on Bio-Plex 3D instrumentation (Bio-Rad, Hercules, CA). Standard curves were analyzed using nonlinear curve fitting and unknowns were calculated based on the derived equation. Samples that exceeded the highest standards were reanalyzed more dilute until the values fell within the range of the known standards.
Luciferase immunoprecipitation system (LIPS) assays for measurement of SARS-CoV-2 antibodies
LIPS assays were used to detect antibody to the SARS-CoV-2 nucleocapsid and spike proteins as previously described (Burbelo et al., 2020). Briefly, plasmids expressing luciferase fused to either the SARS-CoV-2 nucleocapsid or spike protein were transfected into Cos-1 cells, and lysates were obtained. Heat-inactivated serum diluted 1:10 was added to the lysates, and protein A/G beads were added to capture antibody-antigen complexes. After washing the complexes, coelenterazine substrate was added and luciferase activity was quantified as light units (LUs) in a luminometer.
Sorting of non-naive B- and T cell populations
Pooled PBMC samples from different donors were washed with PBS and incubated with Zombie Red Fixable viability dye (1:1000 in PBS, BioLegend, San Diego, CA) for 20 minutes at 4°C protected from light. Then cells were washed with flow staining buffer (10% FBS in PBS) and Fc blocked (Human TruStain FcX, BioLegend) for 15 minutes on ice. The fluorescence-labeled antibody cocktail against human CD45 (APC/Cyanine7), CD3 (AF488), CD19 (APC), CCR7 (BV786), CD95 (BV650), IgD (PerCP-Cy5.5) and CD27(PE/Cyanine7; all antibodies obtained from Biolegend) were added at the end of blocking and incubated for 20 minutes at 4°C in the dark. Cells were washed and sorted on a BD Aria sorter (BD Biosciences, San Jose, CA) in Biosafety Level 3 (BSL3) lab. Non-naive B cell population were gated by CD45+CD19+IgD- or CD27+ and non-naive T cell population were gated by CD45+CD3+CCR7low or CD95+.
Single cell CITE-seq processing
Frozen PBMC samples were thawed, recovered and washed using RPMI media with 10% FBS and 10mg/mL DNase I (STEMCELL). Samples from different donors were pooled evenly before staining; single cells can be demultiplexed in silico using individual specific single nucleotide polymorphism (SNP) information during data analysis (see below). Multiple cell pools were prepared, such that cells from the same individual but different time points (Figure 1B) would be in different pools. PBMC pools were Fc blocked (Human TruStain FcX, BioLegend) and stained with Totalseq-C human ‘hashtag’ antibodies (BioLegend), washed with staining buffer (2% BSA in PBS). This round of hashtag staining allows the different pools to be identified in the analysis, which when combined with the SNP-based demultiplexing, allows full identification of each sample. A fraction of the combined cells was used for sorting non-naive T and B cells (see above). Separately for the unsorted and sorted cell fractions, hashtagged PBMC pools were combined and cells were stained with a cocktail of TotalSeq-C human lyophilized panel (BioLegend) of 192 surface proteins (Table S2). Then, cells were washed three times, resuspended in PBS, and counted before proceeding immediately to the single cell partition step. Note that the antibody concentrations used for CITE-seq were optimized by the manufacturer based on healthy PBMC samples, thus may not be optimal for COVID-19 samples. We have not independently verified the specificity of each of the antibodies, although most if not all have been used in flow/mass cytometry in the field and a subset has been titrated for CITE-seq in our previous work that also shows concordance with data from flow cytometry (Kotliarov et al., 2020). In addition, because we used staining conditions designed to retain optimal mRNA measurements, some surface markers that would stain better under different conditions (e.g., CD197/CCR7), were not detected well in our data; therefore, negative staining for certain markers should be interpreted with caution.
Single cell RNA sequencing
PBMC samples were mixed with the reverse transcription (RT) mix and partitioned into single cell Gel-Bead in Emulsion (GEM) using 10x 5′ Chromium Single Cell Immune Profiling Next GEM v1.1 chemistry (10x Genomics, Pleasanton, CA). The RT step was conducted in the Veriti Thermo Cycler (ThermoFisher Scientific, Waltham, MA). Single cell gene expression, cell surface protein, T cell receptor (TCR) and B cell receptor (BCR) libraries were prepared as instructed by 10x Genomics user guides (https://www.10xgenomics.com/resources/user-guides/). All libraries were quality controlled using Bioanalyzer (Agilent, Santa Clara, CA) and quantified using Qubit Fluorometric (ThermoFisher). 10x Genomics 5′ Single cell gene expression, cell surface protein tag, TCR and BCR libraries were pooled and sequenced on Illumina NovaSeq platform (Illumina, San Diego, CA) using the sequencing parameters recommended by the 10x Genomics 5′ v1.1 user guide. Sequencing saturation of the libraries ranged from approximately 60%–80% for the cDNA and 20%–40% for the surface protein tag libraries.
Flow cytometry
Flow cytometry was performed for a subset of 33 PBMC samples using a Cytek Aurora spectral cytometer (Cytek Biosciences, Fremont, CA) with a 27-color panel comprising viability dye and antibodies to CD3, CD4, CD8, CD11c, CD14, CD16, CD19, CD20, CD24, CD25, CD27, CD38, CD45, CD45RA, CD45RO, CD56, CD123, CD127, CCR4, CCR6, CCR7, CCR10, CXCR3, CXCR5, HLA-DR, and IgD (see Key Resources Table). The frequency of major populations was determined using Flowjo Software v10 (BD Biosciences) based on previously described manual gating strategies (Finak et al., 2016).
Bulk RNA sequencing and single cell sample demultiplexing
For each sample, a sample of 100,000-500,000 cells was processed in Trizol using the miRNAeasy micro kit (QIAGEN, Germantown, MD) and standard RNA sequencing libraries were generated using Illumina Truseq library preparation kits. These libraries were used to generate SNP calls for each donor. Sequencing results were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. The sequencing reads were adaptor and quality trimmed and then aligned to the human genome using the splice-aware STAR aligner and SNP calls were generated using the previously published protocol (Blay et al., 2019). We used the software package demuxlet (Kang et al., 2018) to then match single cells in the 10x RNaseq data to each donor and identify doublets. Because multiple samples from different time points for each donor may be collected and could not be demultiplexed by this method alone, we also used ‘hashtag’ antibodies (Biolegend) to uniquely label the different time points (Stoeckius et al., 2018).
Quantification and statistical analysis
Details on the statistical testing and values reported associated with each figure are reported in the figure legend or results section text.
Clinical Severity Classification
Clinical severity classification was assigned as follows:
Mild: modest symptoms, no pneumonia.
Moderate: Fever and respiratory symptoms plus radiological evidence of pneumonia. Use of low-flow oxygen is still part of this phenotype; O2 saturation is > 93% at rest.
Severe: oxygen saturation at rest 93% or lower, or respiratory rate > 30/min, or PaO2/FiO2 < 300; use of low-flow oxygen is still part of this phenotype.
Critical: any one of the following: Mechanical ventilation (CPAP, BiPAP, intubation, hi-flow oxygen), septic shock, organ damage requiring admission in the ICU.
Disease severity metric (DSM)
Quantitative disease severity scores, namely DSM, were derived for the cohort of patients with PBMC samples. The three subjects with initial PBMC sample collected at or later than 30 days since their symptom onset were excluded from this analysis (see Figure 1A). A total of 18 clinical/lab data and serum protein measurements were assembled from within 2 days of the earliest PBMC sample collection date for each of the remaining 30 subjects. These 18 measurements include TNF-α/β, IL-6, IL-18, IP-10, CXCL9, IFN-ϒ, IL-4, IL-13, IL-17, C-reactive protein (CRP), fibrinogen, D-dimer, lactate dehydrogenase (LDH), lymphocyte and platelet counts, neutrophil/lymphocyte ratio (NLR), and pulse oximetry to fraction of inspired oxygen (SpO2/FiO2) ratio. Missing values were imputed using an additive regression approach implemented in the R Hmisc package (Harrell, 2019). The measurements were normalized by accounting for difference in days between hospital admission and sample collection (i.e., by regressing out the “days since admission” variable using the removeBatchEffect function from limma). Since SpO2/FiO2 ratios were used in the initial clinical disease severity assessment, only the remaining 17 corrected measurements were employed as inputs to building machine learning models for predicting patient disease severity. We used an ordinal partial least-squares (PLS) regression approach with the response categories ordered from the least to most severe (i.e., Moderate-Alive, Severe-Alive, Critical-Alive, and Critical-Deceased). Classification performance was determined based on leave-one-out cross-validation where in each iteration one sample was reserved for testing and the rest were utilized for training of the model. Standardized model coefficients from the cross-validation models were evaluated for importance of each parameter. The first principal component of the top 8 most important parameters along with SpO2/FiO2 ratios were used as a continuous severity score, which we referred to as DSM. To validate our approach, DSM scores were also independently calculated using the same parameters for an additional 64 patients from the Brescia cohort with protein and clinical measurements collected < 30 days since symptom onset (Figure 1E). The modeling analyses were carried out with the R plsRglm package (Bertrand and Maumy-Bertrand, 2019) using proportional-odds logit models.
For several of the later analyses, we divided the 30 patients equally into DSM-high and DSM-low groups by using the median DSM value as the cutoff. Given the frequent use of this patient grouping, we assessed its robustness against the random number generator “noise” in the imputation step used above. We drew 200 random seeds for the imputation step to generate different DSM-high and -low groups. We then computed the rand-index (McShane et al., 2002), which measures the similarity between two different partitions of the subjects, between our observed DSM-high and -low partition and each of the random partitions. The average rand-index was 0.823, indicating that our disease severity grouping was robust to the random number generator “noise” in the imputation step. We have saved the seed used to generate the DSM-high and -low groups used in the paper (see Code Availability above).
Single cell data processing
CellRanger (10x Genomics) version 3.1.0 was used to map cDNA libraries to the hg19 genome reference and to count antibody tag features. Data were further processed using Seurat (v.3.1.0, 3.1.4, or 3.2.2) (Stuart et al., 2019) running in R v3.6.1. After filtering to single cells based on demuxlet output, we further demultiplexed the time points using the hashtag antibody staining using Seurat’s HTOdemux function. We removed cells with less than 200 or greater than 4,000 detected genes, greater than 30% mitochondrial reads, cell surface protein tag or mRNA counts greater than 15,000, or hashtag antibody counts greater than 5,000.
Denoised and Scaled by Background (DSB) normalization of CITE-seq protein data
We normalized the CITE-seq protein data using the DSB method (Mulè et al., 2020) [available at https://github.com/niaid/dsb], which removes technical noise associated with unbound antibody by rescaling each protein based on protein levels detected in empty droplets, then denoises each single cell by defining and regressing out the non-biological, technical component of the cell’s protein counts. The following parameters were used in the dsb normalization function: define.pseudocount = TRUE, pseudocount.use = 10, denoise_counts = TRUE. We set the use.isotype.control parameter to TRUE which models and regresses out a covariate corresponding to the technical component of the cell’s protein library by combining the per cell background and isotype control counts.
Manual gating of CITE-seq protein data
DSB normalized cell surface protein data were indexed and exported as csv file and gated in Flowjo Software v10 (BD Biosciences). Gated FCS files and gating files were imported and mapped to CITE-seq single cell data using R packages CytoML (Finak et al., 2018) and flowWorkspace R package version 4.2.0 (Finak and Jiang, 2020). Treg cells were gated on CD4+CD25hiCD127lo. Given the poor CXCR5 staining which could lead to contamination in the downstream differential expression analysis, cTfh cells were gated within CD4+ T cells following RNA based clustering as follows (He et al., 2013; King et al., 2008): Clustering was first performed on the scaled RNA data using variable features from Seurat’s FindVariableFeatures, ScaleData, RunPCA, FindNeighbors (first 15 PCs) and FindClusters (resolution 0.6) functions; ICOShiPD1hi clusters were further gated by using DSB value cutoffs of 4 and 0 for ICOS and PD1, respectively. The gated population exhibited higher expression of ICOS, MAF and SH2D1A (encoding SAP protein) mRNA (King et al., 2008; Xin et al., 2018) and was enriched for some reported human Tfh signatures (Locci et al., 2013; Weinstein et al., 2014) in comparison with other CD4+ T cell clusters. As a cautious note: given our gating scheme using ICOS and PD1, the cTfh cluster could overlap with some activated non-cTfh CD4+ T cells. CD71+ B cells were gated with a DSB value cutoff of 3 for CD71 within memory B cells. Plasmablasts were gated as CD19+CD20-CD38hi.
Automated clustering of CITE-seq protein data
DSB-normalized protein data, excluding the isotype control antibodies, were used to generate a Euclidean distance matrix computed for all single cells. The matrix was then used to build the shared nearest neighbor (SNN) graph followed by k-nearest neighbors clustering using the FindNeighbors and FindClusters functions, respectively, in Seurat (v3.1.0) using the Louvain algorithm. Two stages of clustering were performed: each of the three batches of single cells were first clustered separately using a resolution of 0.5. Major cell type clusters were manually identified within each batch and matched across batches. Cells of each major cell type were then merged across batches, and a second round of clustering (resolution = 1) was performed within each cell type, using DSB-normalized protein data that were batch-corrected using removeBatchEffect from the limma R package (Ritchie et al., 2015). The resulting clusters were manually annotated, and grouped into fine (e.g., CD4 central memory) and coarse levels (e.g., CD4 memory).
Cell frequency differential abundance
Cell frequencies expressed as percent of parent or percent of total were tested for association with COVID versus healthy and DSM through linear models using the limma package. All models controlled for age, batch, and days since symptom onset, with the exception of models comparing to healthy, where days since symptom onset was not controlled for as healthy individuals do not have a time since onset. Age was modeled as a continuous variable representing the age in years of the subject. The batch variable was modeled as factor variable representing the experimental batch (3 in total), and days since symptom onset was modeled as a continuous variable representing the number of days since self-reported symptom onset.
Cell frequency differential abundance: COVID-19 patients versus HC
Using the limma workflow as described in “Cell frequency differential abundance,” cell populations different between patient time point 0 (T0) samples and HC were identified with a model with the following formula in R: ∼covid_vs_healthy + age + batch, where covid_vs_healthy is a factor variable with two levels, COVID-19 and HC. P values were determined using the moderated T statistics associated with the covid_vs_healthy coefficient.
Cell frequency differential abundance: DSM
Using the limma workflow as described in “Cell frequency differential abundance,” cell populations associated with DSM in patient T0 samples were identified with a model with the following formula in R: ∼DSM + age + batch + days_since_onset, where DSM is the continuous measure of DSM. p values were determined using the moderated T statistics associated with the DSM coefficient.
Pseudobulk differential expression analysis
To create “pseudobulk” RNA libraries for differential expression analysis, all unsorted cells in a given sample were computationally “pooled” according to their automated cluster assignment by summing all counts for a given gene in a given cell-type in a given sample. In addition, this procedure was applied to specifically gated populations of interest (circulating T follicular helper (cTfh), Plasmablast, CD71 high B cells) from the sorted cells as these were not present at high enough quantities in the unsorted cells. Pseudobulk libraries made up by few cells and therefore likely not modeled properly by bulk differential expression methods were removed from analysis for each cell-type separately to remove samples that contained fewer than 8 cells and less than 40000 unique molecular identifier counts detected after pooling. Lowly expressed genes were removed for each cell type individually using the filterByExpr function from edgeR (McCarthy et al., 2012). Differentially expressed genes were identified using the limma voom (Law et al., 2014) workflow which models the log of the counts per million (cpm) of each gene. Scaling factors for library size normalization were calculated with the calcNormFactors function with method = “RLE.” In models utilizing repeated samples from the same subject, subject random effects were incorporated using the duplicateCorrelation function in limma. Models controlled for age, batch, and days since symptom onset, with the exception of models comparing to HC, where days since symptom onset was not controlled for as HC do not have a time since onset. Age was modeled as a continuous variable representing the age in years of the subject. The batch was modeled as factor variable representing the experimental batch (3 in total), and days since symptom onset was modeled as a continuous variable representing the number of days since self-reported symptom onset.
Visualizing gene expression in heatmaps
For heatmaps showing pseudo-bulk, sample level data, the log(cpm) for each sample for a given cell-type was calculated by pooling cells as described in “Pseudobulk differential expression analysis.” Library size normalization was performed without additional scaling factors. All single cell gene expression heatmaps show scaled log(counts per 10000) values computed using the logNormalize function in Seurat. Heatmaps were scaled to have mean = 0 and variance = 1 for each gene.
Gene set enrichment of differentially expressed (DE) genes
Enriched gene sets were identified using the pre-ranked gene-set enrichment analysis (GSEA) algorithm implemented in the FGSEA R package (Sergushichev, 2016). Genes were ranked using the moderated T statistics for the relevant coefficient from the limma voom model. Enrichment was assessed with a gene set list that included GO BP, KEGG, Reactome, MSIGDB’s Hallmark collection, Blood Transcriptomic Modules (Li et al., 2014), T cell exhaustion signatures (both upregulated and downregulated genes) from chronic murine LCMV infection (Wherry et al., 2007), the top genes that were consistently upregulated across two cohorts at early time points in response Yellow Fever vaccination (Querec et al., 2009), and lastly the union of the top 50 genes that allow for discrimination between symptomatic and asymptomatic infected inviduals in H1N1 and H3N2 infection in a human challenge study (Woods et al., 2013). Selected pathways shown in Figures 3A, 3B, and 5D and listed in Table S4 were manually curated to select gene sets relevant to immunology and often enriched in several cell-types across the various differential expression comparisons.
Gene set module scores
Specific module scores representing enriched pathway activities were calculated for relevant samples using leading edge genes identified by GSEA to enhance signal-to-noise ratio. Prior to score calculation, the pseudobulk gene counts were normalized with the varianceStabilizingTransformation function from DEseq2. The scores were generated using gene set variation analysis (GSVA) method implemented by the GSVA R package (Hänzelmann et al., 2013). DSM-high and DSM-low group trajectories (module scores versus TSO) were generated respectively using loess smoothing function with default span parameter (span = 0.75).
Differential expression: COVID-19 patients versus HCs
Using the pseudobulk limma voom workflow as described in “Pseudobulk differential expression analysis,” differentially expressed genes between patient T0 samples and HC were identified with a model with the following formula in R: ∼0 + covid_vs_healthy + age + batch, where covid_vs_healthy is a factor variable with two levels, COVID-19 and HC. The contrasts.fit function was then used to compare the estimated means between COVID-19 and HCs. Gene set enrichment analysis of the moderated T statistics was performed as described in “Gene set enrichment of DE genes”
Differential expression: DSM
Using the pseudobulk limma voom workflow as described in “Pseudobulk differential expression analysis,” genes associated with DSM in patient T0 samples were identified with a model with the following formula in R: ∼DSM + age + batch + days_since_onset, where DSM is the continuous measure of DSM. Gene set enrichment analysis of the moderated T statistics of the DSM term was performed as described in “Gene set enrichment of DE genes”
Glucocorticoid response gene signature
Genes induced/upregulated by glucocorticoid treatment were derived from Franco et al. (Franco et al., 2019) as part of a previous single cell RNaseq study of infant cord blood mononuclear cells (manuscript in preparation). Briefly, upregulated genes (between baseline and post glucocorticoid treatment) across all the tested cell types from Franco et al. were selected, filtered to only genes that were not also in the reactome ‘immune system’ pathway (to exclude immune genes), then further filtered to the top 50 most highly expressed based on average expression across all cells from our single cell RNA-seq dataset generated from infant cord blood (to filter out genes with lower average expression across diverse hematopoietic cell lineages).
RNA-based clustering of CD4 and CD8 cells
Single CD4+ and CD8+ T cells were separately clustered using mRNA expression profiles of genes found to be in the ‘leading edge’ of key gene sets significantly associated with COVID-19 versus HCs or DSM from the above GSEA analyses (see pathways shown in Figure S6A). Clustering was performed on the scaled RNA data after regressing out Batch and Donor variation using Seurat’s ScaleData function. PCA was performed using the leading-edge genes, and the shared nearest neighbors graph was constructed using the first 15 PCs using the Seurat’s FindNeighbors function. Louvain clustering was performed on the graph using a resolution of 1 using Seurat’s FindClusters function. Association of these clusters with COVID-19 patients versus HC and DSM was assessed using linear models as described in “Cell frequency differential abundance: COVID-19 patients vs. HC” and “Cell frequency differential abundance: COVID-19 patients vs. HC,” using all T0 samples and all HC samples. 18 CD8 clusters were tested.
TCR data processing
CellRanger (10x Genomics) version 3.1.0 was used to assemble V(D)J contigs. Cells were considered part of the same clone if they shared the same set of productive CDR3 nucleotide sequences (exact matches of alpha and beta chains). This was determined using the “raw_clonotype_id” of the filtered_contig_annotations.csv output from CellRanger (https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/algorithms/annotation).
Determination of clonal versus singleton T cells
Cells were removed from analysis as described in “Single cell data processing.” Cells were assigned to clones as described in “TCR data processing.” A cell was considered to be part of a non-singleton clone (i.e., clonal) if there were at least two cells from that clone in the same sample (Subject, time point combination). All other cells were deemed singleton clones.
Clonality in CD8 memory cells
Clonality in the CD8+ memory T cells in HC and patient T0 samples was determined as follows. Cells were assigned to clones as described in “TCR data processing.” Using only the sorted T cells labeled as CD8 Memory according to the automated clustering, samples with fewer than 100 cells were removed from further analysis. As measurements of clonality are biased by differing number of total cells in a sample, all samples were down-sampled to 100 cells and Simpson’s index (, where R = the total number of unique clones in the subsample and p i = the number of cells from clone i in a given subsample) was computed as a measure of clonality; Simpson’s index can be interpreted as the probability that two randomly selected cells are from the same clone. This process was repeated 1000 times with random subsamples of 100 cells per sample and the median Simpson’s index over the 1000 subsamples was used as a point estimate for a given sample. Differences in clonality (median Simpson’s index) between HC and COVID-19 patient and the association of clonality with DSM were then assessed with mixed effect models in the lme4 R package (Bates et al., 2015). Models were adjusted for age and days since symptom onset. In addition, batch was included as a random effect. Thus, the formulas in R were:
-
•
median_simpson_index ∼DSM + age + days_since_onset + (1|batch)
-
•
median_simpson_index ∼covid_vs_healthy + age + (1|batch)
The median_simpson_index is the median of the Simpson index over 1000 subsamples as described above; DSM is a continous measure of DSM; and covid_vs_healthy is factor variable with levels COVID19 and HC. The terms for age, batch and days_since_onset were coded as described in “Pseudobulk differential expression analysis.” Coefficients representing the association with DSM and the difference in means between COVID-19 patient and HC samples were tested for significance using the summary function from the lmerTest package (Kuznetsova et al., 2017).
Differential expression analysis of surface marker expression on clonal T cells
Cells were filtered to include only the sorted, clonal (singleton clones removed) CD8 memory T cells. Pseudobulk differential expression of canonical exhaustion surface markers (CD279, TIGIT, CD244, CD152, CD223, CD366) was tested using linear models through the limma package. To find CITE-seq surface protein markers differentially expressed between healthy and COVID, and associated with DSM, the mean DSB count per sample in the clonal CD8 memory cells was modeled using the traditional limma workflow. However, to model the RNA expression of the related genes, limma voom was used as previously described “Pseudobulk differential expression analysis.” The model comparing COVID to HC was identical to that described in “Differential expression: COVID vs. HC” and the model for DSM was identical to that described in “Differential expression: DSM.”
Enrichment of exhaustion signatures in DE genes
Using the same models of RNA expression in clonal CD8 memory cells as described in “Clonal T cell exhaustion marker differential expression” (COVID versus HC and DSM models), differential expression was assessed for all genes passing read depth filters using filterByExpr function from edgeR. Enrichment of exhaustion gene sets in differentially expressed genes in clonal (singleton clones removed) CD8 memory cells using the pre-ranked GSEA algorithm implemented in the FGSEA R package. Genes were ranked using the moderated T statistics for the relevant coefficient from the limma voom model.
Enrichment of other gene sets in Wherry exhaustion signatures
Enrichment of the Wherry exhaustion signatures (Wherry et al., 2007) in GO BP, Reactome, KEGG, MSIGDB Hallmark collection, and Blood Transcriptomic Modules (Li et al., 2014) was assessed using Fisher’s exact tests comparing the overlap of each exhaustion signature with each gene set. The union of all gene sets including the exhaustion signatures was used as the background. P values were adjusted with the Benjamini-Hochberg method (Benjamini and Hochberg, 1995).
Determining proportion of exhausted cells through surface marker co-expression
Cells were filtered to include only the sorted, clonal CD8 memory T cells. To evaluate whether exhaustion was associated with DSM and COVID-19 patients versus HC, we evaluated co-expression of CD39 and PD1 by using cutoffs of 0.5 and 1 DSB counts to determine positive cells.
Conditional independence network analysis
A severity network (Figure 4A) was created using a more expansive collection of annotated gene sets (similar to Figures 3A and 3B) with false discovery rate (FDR) ≤ 0.2 in the DSM gene set enrichment analysis (Table S4B) to infer “direct” correlations (see below) among biological processes in different cell subsets and their association with disease severity. Sample gene set scores generated using leading edge genes identified from the DSM differential expression GSEA were used for this analysis. Gene sets whose scores were not significantly correlated with DSM (Spearman’s correlation; p > 0.05) were removed. To distinguish between direct and indirect associations (“direct” are those that are correlated conditional on all other variables), an inverse correlation matrix of the remaining gene sets and DSM was derived using a regularized regression approach called Lasso (Meinshausen and Bühlmann, 2006) as implemented in the R Huge package (Zhao et al., 2012). Network nodes that are conditionally independent of each other would have zero coefficients in the inverse correlation matrix. By scanning the number of non-zeros in the matrix through a range of penalty values (λ) from 0 to 1, a parameter of 0.35 was selected as it represented an inflection point on the curve where the number of direct connections began to stabilize. Using DSM as the central node, a 2-level network was created with edges between nodes indicating possible direct and significant correlation (Table S5; Spearman’s correlation p value ≤ 0.05).
Differential abundance of cell populations: time since onset in DSM high and DSM low
Using the same limma workflow as described in “Cell frequency differential abundance,” an interaction model was fit using all COVID-19 samples with the formula in R: ∼DSM_group:days_since_onset + DSM_group + days_since_onset + age + batch, where DSM_group is a factor variable with levels DSM_high and DSM_low. The following contrasts were then used with the contrasts.fit function to find cell populations associated with days_since_onset in DSM high and DSM low respectively (DSM_low was reference level of DSM_group factor variable, and thus the days_since_onset term represents the slope for days_since_onset in the DSM_low group, and DSM_high:days_since_onset represents the difference in slopes between the two groups):
-
•
“days_onset_in_DSMhigh”: “DSM_high:days_since_onset + days_since_onset,”
-
•
“days_onset_in_DSMlow”: “days_since_onset”
Differential expression: critical juncture, DSM high versus low at days 17-23 and interaction model (DSM high-low X Juncture-pre-juncture) to assess temporal changes in the differences between DSM-high versus DSM-low patients during the days 17-23 period relative to the period before
Using the same pseudobulk limma voom workflow as described in “Pseudobulk differential expression analysis,” the DSM high group was contrasted with the DSM low group specifically at days 17-23 through the use of a contrast matrix and the contrasts.fit function in limma. The model was fit using all COVID-19 samples to improve variance estimates for individual genes and the following groups of samples were defined:
-
•
DSM_high_days < 17,
-
•
DSM_low_days < 17,
-
•
DSM_high_17 ≤ days ≤ 23,
-
•
DSM_low_17 ≤ days ≤ 23,
-
•
DSM_high_days > 23,
-
•
DSM_low_days > 23
The formula used in R was ∼0 + group + age + batch + days_since_onset, where group is a factor variable using the groups defined above. For the comparison of DSM high versus DSM low at the juncture, the contrast matrix specifically compared the DSM_high_17 ≤ days ≤ 23 group to the DSM_low_17 ≤ days ≤ 23 group. For the interaction model, the contrast matrix compared the difference in means between DSM high and low at the juncture versus before the juncture, i.e., (DSM_high_17 ≤ days ≤ 23 - DSM_low_17 ≤ days ≤ 23) - (DSM_high_days < 17-DSM_low_days < 17). Enriched gene sets for the contrast were identified as described in “Gene set enrichment of DE genes.”
Differential expression: time since onset in DSM high and DSM low
Using the same pseudobulk limma voom workflow as described in Pseudobulk differential expression analysis,” an interaction model was fit using the same approach described in “Differential abundance of cell populations: time since onset in DSM high and DSM low.” Enriched gene sets for the days_onset_in_DSMlow and days_onset_in_DSMhigh contrasts were identified as described in “Gene Set Enrichment of DE genes”
Assessing differences in circulating protein levels between DSM-high and -low patients at the late juncture (days 17-23)
For each of the 63 serum protein measurements, we tested for abundance difference between DSM-high and -low patients during the days 17-23 since symptom onset period. A total of 11 DSM-high and 10 DSM-low patients in our CITE-seq cohort had at least one measurement during this time interval. Protein levels were estimated by a mixed-effects model as follows:
ANOVA unadjusted p value of 0.05 or below for the dsm-group coefficient term was flagged as significant. Models with singular fits were not discarded for this purpose.
Analysis of circulating protein profiles of recovered versus deceased critical patients
All deceased patients (critical-deceased category) with longitudinal serum protein measurements (n = 17) in the larger Brescia cohort (see Patients above) were identified. Each of them was then matched with critical-alive subjects of the same sex and within ± 3 years in age (n = 21). We built two types of mixed-effect models to search for proteins exhibiting dynamic divergence around the days 17-23 juncture.
First, serum samples were divided into three time-interval groups based on TSO: days 7-16 (pre-juncture; 35 samples), days 17-23 (juncture; 38 samples), and days 24-30 (post-juncture; 28 samples) from symptom onset.
We then searched for proteins that 1) were differentially abundant between deceased and recovered patients at the juncture or post-juncture, and 2) the differences between deceased and recovered patients increased significantly at the juncture compared to the pre-juncture, or at the post-juncture compared to the pre-juncture. Identifying these proteins would help assess the extent of divergences around the juncture and the corresponding biological processes that shifted temporally at or after the juncture. We included the post-juncture period in our analysis because significant divergences after the juncture could be a result of what happened during the juncture.
For each of the 63 proteins, differential abundance at each interval was assessed by the model:
Model 1: Protein expression ∼patient group + sex + age + days from symptom onset to measurement + (1|subject)
ANOVA unadjusted p value of 0.05 or below for the patient group (deceased versus recovered) coefficient term was used to select proteins with significant differences for each interval (Table S7).
Next, significant changes between time intervals was assessed by the following model:
Model 2: Protein expression ∼patient group∗interval + sex + age + days from symptom onset to measurement + (1|subject)
where the group∗interval term was used to evaluate the temporal changes (between intervals) in the differences between the deceased and recovered groups. Proteins with an unadjusted p value of ≤ 0.05 from the resulting t statistics were selected (Table S7).
A subset of the 12 proteins that were 1) significantly different between the two patient groups at the juncture or post-juncture based on model 1, and 2) significantly shifted temporally based on model 2 are shown in Figure 6B with select protein trajectories shown in Figure 6D. False discovery rates (FDR) were estimated by running the same search procedures with 1000 permutations of the subject outcome labels. In total, 0.6% of permuted searches resulted in 12 or more proteins matching the search criteria (hits). All 12 proteins had an estimated p value of ≤ 0.05 individually based on this permutation test (Table S7). The average of number of protein hits was 1.73, leading to an overall FDR of 1.73/12 or 14.4% (i.e., ∼2 of the 12 proteins may be false positives).
We also assessed the concordance between the two models. The effect sizes estimated by these models were highly correlated for shared terms (Pearson’s correlation r = ∼0.9 among all proteins tested). However, two identified proteins in this group, Reg3α and CD25/IL-2Rα, had differences of more than 0.5 in the pre-juncture effects (i.e., differences between deceased and recovered patients) estimated by the post-juncture v. pre-juncture interval model versus the pre-juncture DE model. We thus did not focus on these two proteins in our data interpretation. Additionally, we also compared model 1 results at the juncture to results from the dsm-high and -low comparison (see previous section). Concordance of their effects had a p value of 0.0896 as assessed by Fisher’s exact test (Figure S7C). This mild concordance is not surprising given that the former is contrasting only very critical patients but with different outcomes, while the latter involves largely survivors with different disease severity.
To assess whether the above procedure can select proteins predictive of outcome using independent training and testing sets, using a leave-one-out cross-validation scheme we applied the above procedure to select proteins in training sets only and then the selected proteins were used as inputs to fit a logistic PLS model, which was then employed to compute a prediction score for each sample in the unseen patient. The prediction was carried out independently for the juncture (d17-23 TSO) and post-juncture (d24-30 TSO) periods. Based on these sample scores, classification performance was measured by the area under the receiver operating characteristic (ROC) curve (Figure 6C). Statistical significance of the prediction performance was evaluated by 1000 permutations of all subject labels to assess the ability of this procedure to generate predictive models. In each iteration of the permutation cross-validation, if no proteins were selected by the procedure, the unseen patient was assigned a score of 0.5 (middle point akin to best random guess) by default as no model could be constructed.
Time trajectories for the same cohort of recovered and deceased patients were created using all the measurements of the 12 temporally divergent proteins (Figure 6D). The same was done also for antibody measurements against SARS-CoV-2 spike and nucleocapsid proteins (Figure 6E) (Burbelo et al., 2020). Group trajectories were generated using the loess smoothing function in R (v3.6.2) with default span parameter (span = 0.75). For each subject, Individual measurements collected on the same day were averaged in the plots.
Assessing differences in circulating protein profiles between deceased and recovered patients at the juncture in an independent US cohort
Published serum protein measurement data from a US cohort of 113 COVID-19 patients (Lucas et al., 2020) were obtained to assess the differential abundance results observed in the Brescia cohort at the juncture. A sub-cohort of eight deceased/hospice (coded as 2 or 3 in the “Latest Outcome” column of the dataset) and four age- and sex-matched critical (4 or 5 in “Clinical Score” at any time point) patients with samples collected between days 17 and 23 since symptom onset was identified. Differential abundance analysis was carried out using the same procedures for the 38 overlapping proteins measured in our and the Lucas et al. (2020) studies. Concordance of statistically significant results found in the Brescia cohort (only 12 of 20 DE proteins at the juncture identified in our Brescia cohort can be examined) was assessed by Spearman’s correlation of effect sizes.
Validation of differentially expressed gene sets in external scRNA-seq data from Schulte-Schrepping et al. (2020)
Data from cohorts 1 and 2 of Schulte-Schrepping et al. (2020) were downloaded from fastgenomics at the following url: https://beta.fastgenomics.org/datasets/detail-dataset-952687f71ef34322a850553c4a24e82e, https://beta.fastgenomics.org/datasets/detail-dataset-7ae02f5553074bda92c14a8f0bce2d24. Using the pre-annotated clusters from the original publication, single cell gene expression data were pooled into pseudobulk libraries and differential expression models were run as described in “Pseudobulk differential expression analysis.” Associations with disease severity were determined using severity classifications present in the downloaded Seurat object metadata. Models controlled for TSO and age. Gene set enrichment was performed as described in “Gene set enrichment of differentially expressed genes” for select gene sets we wanted to validate. To assess exhaustion signatures in CD8+ T memory cells as close to ours as possible (Figures S6I and S6J), we utilized Seurat’s “TransferData” function to transfer cell cluster annotations from the single data of the Brescia cohort reported here to the data of Schulte-Schrepping et al. (2020). Note that since clonality/TCR sequencing data are not available in the Schulte-Schrepping et al. (2020) dataset, we could not assess clonally expanded cells only.
Data visualization and data table handling
The tidyverse suite of R packages was utilized for data table handling and data plotting (Wickham, 2019) [https://cran.r-project.org/web/packages/tidyverse/index.html]. ComplexHeatmap was used for heatmap visualizations (Gu et al., 2016).
Acknowledgments
We thank the NIAID OCICB (Office of Cyber Infrastructure and Computational Biology) and its Bioinformatics and Computational Biosciences (contract HHSN316201300006W/HHSN27200002 to MSC, Inc.) and Operations Engineering Branches for developing databases for patient and assay data, and we thank the NCI Advanced Biomedical Computational Science for data transformation support. We thank Helen Matthew, Sarah Webber, and Wade Green for logistical/regulatory assistance; the NCI CCR Sequencing Facility (CCR-SF) for sequencing support; the NIAID OCICB Locus team for high performance computing support; Iyadh Douagi, Thomas Moyer, and Melanie Cohen from the NIAID Flow Cytometry Section for supporting BSL3 cell sorting experiments; Philip Johnson (University of Maryland) for discussions regarding T cell clonality analysis; Rohit Farmer for code review; and Ronald Germain and Pamela Schwartzberg for comments on the manuscript. Illustrations in Figures 1B and 6A were created using BioRender.com. This research was supported by the Intramural Research Programs of the NIAID and NIDCR, Intramural Programs of the NIH supporting the NIH Center for Human Immunology, Regione Lombardia (project “Immune response in patients with COVID-19 and co-morbidities”), and federal funds from the National Cancer Institute, NIH (HHSN261200800001E). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author contributions
J.S.T. and L.D.N. conceived the study. A.J.M., C.L., and J.S.T. designed CITE-seq experiments with input from W.W.L., D.M., J.C., and S.M. C.L., J.C., and D.M. generated CITE-seq data with help from N.B. C.L., A.J.M., W.W.L., N.R., and J.S.T. designed data analysis strategies and approaches. C.L., A.J.M., W.W.L., and N.R. performed data analysis and generated figures with help from F.C. W.W.L. organized sample meta-data with help from the NIAID COVID Consortium. K.L.H. and B.A.S. generated flow cytometry data. NIAID COVID Consortium provided logistical and infrastructure and analysis support. L.I., A.S., E.Q.-R. and COVID Clinicians cared for patients, collected samples, and collected clinical data. O.M.D., L.I., L.F., A.S., E.Q.-R., M.S.L., H.C.S., C.R., and L.D.N. clinically characterized patients with help from NIAID COVID Consortium and COVID Clinicians. L.I., A.S., K.D., and N.B. processed and handled patient samples. R.S., L.F., N.B., T.-W.C., and S.M. contributed healthy control samples. D.L.F., D.B.K., M.S.L., and L.D.N. contributed circulating protein data. P.D.B. and J.I.C. contributed antibody data. C.L., A.J.M., W.W.L., N.R., and J.S.T. interpreted the data. J.S.T. drafted the manuscript. C.L., A.J.M., W.W.L., N.R., and J.S.T. finalized the manuscript with input from other authors. J.S.T. supervised the study.
Declaration of interests
The authors declare no competing interests.
Published: February 10, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.02.018.
Contributor Information
NIAID COVID Consortium:
Michael S. Abers, Richard Apps, Marita Bosticardo, Pedro Milanez-Almeida, Matthew P. Mulè, Elana Shaw, and Yu Zhang
COVID Clinicians:
Francesco Castelli, Maria Lorenza Muiesan, Gabriele Tomasoni, Francesco Scolari, and Alessandra Tucci
Supplemental information
Clusters were assigned based on surface marker expression. Results shown are for genes with log-fold change greater than 0.25 and expressed in at least 10% of the cells in either the cluster tested or all background cells, and with a p value < 0.01 (Wilcoxon test implemented in Seurat’s FindAllMarkers function).
Each tab corresponds to a different comparison. The corresponding figure in the main text is referenced in the caption at the top of each tab.
Each sample is annotated with the circumstances (ICU, intubation, and corticosteroid use) under which it was collected. Type II ANOVA test statistics for association between these circumstances and DSM at T0 time points and odd ratios statistics between deceased and recovered patients at juncture (d17-23 TSO) are also listed.
The coefficients/effect sizes and associated p values of each term in mixed-effect models are provided. All times are expressed as days since symptom onset.
References
- Abers M.S., Delmonte O.M., Ricotta E.E., Fintzi J., Fink D.L., de Jesus A.A.A., Zarember K.A., Alehashemi S., Oikonomou V., Desai J.V., et al. NIAID COVID-19 Consortium An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight. 2021;6:e144455. doi: 10.1172/jci.insight.144455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya D., Liu G., Gack M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020;20:397–398. doi: 10.1038/s41577-020-0346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.-H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., et al. HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D., Mächler M., Bolker B., Walker S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015;67:1–48. [Google Scholar]
- Bellinvia S., Edwards C.J., Schisano M., Banfi P., Fallico M., Murabito P. The unleashing of the immune system in COVID-19 and sepsis: the calm before the storm? Inflamm. Res. 2020;69:757–763. doi: 10.1007/s00011-020-01366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Bernardes J.P., Mishra N., Tran F., Bahmer T., Best L., Blase J.I., Bordoni D., Franzenburg J., Geisen U., Josephs-Spaulding J., et al. HCA Lung Biological Network. Deutsche COVID-19 Omics Initiative (DeCOI) Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity. 2020;53:1296–1314.e9. doi: 10.1016/j.immuni.2020.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim A., Mei X., Huang M., Yang Y., Fayad Z.A., Zhang N., Diao K., Lin B., Zhu X., Li K., et al. Chest CT Findings in Coronavirus Disease-19 (COVID-19): Relationship to Duration of Infection. Radiology. 2020;295:200463. doi: 10.1148/radiol.2020200463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrebi D., Bruscoli S., Cohen N., Foussat A., Migliorati G., Bouchet-Delbos L., Maillot M.-C., Portier A., Couderc J., Galanaud P., et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood. 2003;101:729–738. doi: 10.1182/blood-2002-02-0538. [DOI] [PubMed] [Google Scholar]
- Best J.A., Blair D.A., Knell J., Yang E., Mayya V., Doedens A., Dustin M.L., Goldrath A.W., Immunological Genome Project Consortium Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blay N., Casas E., Galván-Femenía I., Graffelman J., de Cid R., Vavouri T. Assessment of kinship detection using RNA-seq data. Nucleic Acids Res. 2019;47:e136. doi: 10.1093/nar/gkz776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone R.C. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit. Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Boor P.P.C., Metselaar H.J., Mancham S., Tilanus H.W., Kusters J.G., Kwekkeboom J. Prednisolone suppresses the function and promotes apoptosis of plasmacytoid dendritic cells. Am. J. Transplant. 2006;6:2332–2341. doi: 10.1111/j.1600-6143.2006.01476.x. [DOI] [PubMed] [Google Scholar]
- Brandes M., Klauschen F., Kuchen S., Germain R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo P.D., Riedo F.X., Morishima C., Rawlings S., Smith D., Das S., Strich J.R., Chertow D.S., Davey R.T., Cohen J.I. Sensitivity in Detection of Antibodies to Nucleocapsid and Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus 2 in Patients With Coronavirus Disease 2019. J. Infect. Dis. 2020;222:206–213. doi: 10.1093/infdis/jiaa273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarile L., Zollo O., D’Adamio F., Ayroldi E., Marchetti C., Tabilio A., Bruscoli S., Riccardi C. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001;8:201–203. doi: 10.1038/sj.cdd.4400798. [DOI] [PubMed] [Google Scholar]
- Carson W.E., Giri J.G., Lindemann M.J., Linett M.L., Ahdieh M., Paxton R., Anderson D., Eisenmann J., Grabstein K., Caligiuri M.A. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994;180:1395–1403. doi: 10.1084/jem.180.4.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC COVID-19 Response Team Preliminary Estimates of the Prevalence of Selected Underlying Health Conditions Among Patients with Coronavirus Disease 2019-United States, February 12-March 28, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020;69:382–386. doi: 10.15585/mmwr.mm6913e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- Cox M.J., Loman N., Bogaert D., O’Grady J. Co-infections: potentially lethal and unexplored in COVID-19. Lancet Microbe. 2020;1:e11. doi: 10.1016/S2666-5247(20)30009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maria A., Bozzano F., Cantoni C., Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc. Natl. Acad. Sci. USA. 2011;108:728–732. doi: 10.1073/pnas.1012356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle D.M., Kim-Schulze S., Huang H.-H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacovo T.G., Blasius A.L., Mak T.W., Cella M., Colonna M. Adhesive mechanisms governing interferon-producing cell recruitment into lymph nodes. J. Exp. Med. 2005;202:687–696. doi: 10.1084/jem.20051035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner B.G., Scheffold A., Rolph M.S., Hüser M.B., Kaufmann S.H.E., Radbruch A., Flesch I.E.A., Kroczek R.A. MIP-1α, MIP-1β, RANTES, and ATAC/lymphotactin function together with IFN-γ as type 1 cytokines. Proc. Natl. Acad. Sci. USA. 2002;99:6181–6186. doi: 10.1073/pnas.092141999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning J., Blankley S., Hoang L.T., Cox M., Graham C.M., James P.L., Bloom C.I., Chaussabel D., Banchereau J., Brett S.J., et al. MOSAIC Investigators Progression of whole-blood transcriptional signatures from interferon-induced to neutrophil-associated patterns in severe influenza. Nat. Immunol. 2018;19:625–635. doi: 10.1038/s41590-018-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes J.D., Harris L.D., Klatt N.R., Tabb B., Pittaluga S., Paiardini M., Barclay G.R., Smedley J., Pung R., Oliveira K.M., et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felices M., Lenvik A.J., McElmurry R., Chu S., Hinderlie P., Bendzick L., Geller M.A., Tolar J., Blazar B.R., Miller J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018;3:e96219. doi: 10.1172/jci.insight.96219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G., Jiang M. 2020. flowWorkspace: Infrastructure for representing and interacting with gated and ungated cytometry data sets. Release 3. Bioconductor version.https://rdrr.io/bioc/flowWorkspace/ [Google Scholar]
- Finak G., Langweiler M., Jaimes M., Malek M., Taghiyar J., Korin Y., Raddassi K., Devine L., Obermoser G., Pekalski M.L., et al. Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Sci. Rep. 2016;6:20686. doi: 10.1038/srep20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G., Jiang W., Gottardo R. CytoML for cross-platform cytometry data sharing. Cytometry A. 2018;93:1189–1196. doi: 10.1002/cyto.a.23663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo T.H., Smith K.D., Sato S., Rodriguez D.J., Holmes M.A., Strong R.K., Akira S., Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Franco L.M., Gadkari M., Howe K.N., Sun J., Kardava L., Kumar P., Kumari S., Hu Z., Fraser I.D.C., Moir S., et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med. 2019;216:384–406. doi: 10.1084/jem.20180595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.-E., Katsaounou P., et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan L., Pulendran B. The immunology of SARS-CoV-2 infections and vaccines. Semin. Immunol. 2020;50:101422. doi: 10.1016/j.smim.2020.101422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Gupta P.K., Godec J., Wolski D., Adland E., Yates K., Pauken K.E., Cosgrove C., Ledderose C., Junger W.G., Robson S.C., et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015;11:e1005177. doi: 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase M., Bellomo R., Devarajan P., Schlattmann P., Haase-Fielitz A., NGAL Meta-analysis Investigator Group Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am. J. Kidney Dis. 2009;54:1012–1024. doi: 10.1053/j.ajkd.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell F.E., Jr. 2019. Hmisc: Harrell Miscellaneous. R package version, 3(2) [Google Scholar]
- He J., Tsai L.M., Leong Y.A., Hu X., Ma C.S., Chevalier N., Sun X., Vandenberg K., Rockman S., Ding Y., et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013;39:770–781. doi: 10.1016/j.immuni.2013.09.007. [DOI] [PubMed] [Google Scholar]
- Hirsch J.S., Ng J.H., Ross D.W., Sharma P., Shah H.H., Barnett R.L., Hazzan A.D., Fishbane S., Jhaveri K.D., Abate M., et al. Northwell COVID-19 Research Consortium Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020;98:209–218. doi: 10.1016/j.kint.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Zaas A.K., Rao A., Dobigeon N., Woolf P.J., Veldman T., Øien N.C., McClain M.T., Varkey J.B., Nicholson B., et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011;7:e1002234. doi: 10.1371/journal.pgen.1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung I.F.-N., Lung K.-C., Tso E.Y.-K., Liu R., Chung T.W.-H., Chu M.-Y., Ng Y.-Y., Lo J., Chan J., Tam A.R., et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020;395:1695–1704. doi: 10.1016/S0140-6736(20)31042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson A.M., Yu S., Annicelli C.H., Medzhitov R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe. 2010;7:103–114. doi: 10.1016/j.chom.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B., Zhang Q., Ge J., Wang R., Sun J., Ge X., Yu J., Shan S., Zhou B., Song S., et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584:115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Quinn J., Pinsky B., Shah N.H., Brown I. Rates of Co-infection Between SARS-CoV-2 and Other Respiratory Pathogens. JAMA. 2020;323:2085–2086. doi: 10.1001/jama.2020.6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C., Tangye S.G., Mackay C.R. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- Kotliarov Y., Sparks R., Martins A.J., Mulè M.P., Lu Y., Goswami M., Kardava L., Banchereau R., Pascual V., Biancotto A., et al. Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat. Med. 2020;26:618–629. doi: 10.1038/s41591-020-0769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A., et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova A., Brockhoff P.B., Christensen R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017;82:1–26. [Google Scholar]
- Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I., et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- Langford B.J., So M., Raybardhan S., Leung V., Westwood D., MacFadden D.R., Soucy J.R., Daneman N. Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin. Microbiol. Infect. 2020;26:1622–1629. doi: 10.1016/j.cmi.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbury L., Lim B., Baskaran V., Lim W.S. Co-infections in people with COVID-19: a systematic review and meta-analysis. J. Infect. 2020;81:266–275. doi: 10.1016/j.jinf.2020.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Rouphael N., Duraisingham S., Romero-Steiner S., Presnell S., Davis C., Schmidt D.S., Johnson S.E., Milton A., Rajam G., et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014;15:195–204. doi: 10.1038/ni.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Liu F., Li L., Xu M., Wu J., Luo D., Zhu Y., Li B., Song X., Zhou X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020;127:104370. doi: 10.1016/j.jcv.2020.104370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locci M., Havenar-Daughton C., Landais E., Wu J., Kroenke M.A., Arlehamn C.L., Su L.F., Cubas R., Davis M.M., Sette A., et al. International AIDS Vaccine Initiative Protocol C Principal Investigators Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–769. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q.-X., Liu B.-Z., Deng H.-J., Wu G.-C., Deng K., Chen Y.-K., Liao P., Qiu J.-F., Lin Y., Cai X.-F., et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020;26:845–848. doi: 10.1038/s41591-020-0897-1. [DOI] [PubMed] [Google Scholar]
- López-Collazo E., Avendaño-Ortiz J., Martín-Quirós A., Aguirre L.A. Immune Response and COVID-19: A mirror image of Sepsis. Int. J. Biol. Sci. 2020;16:2479–2489. doi: 10.7150/ijbs.48400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel M., Harari G., Gurevich M., Achiron A. Cytokine prediction of mortality in COVID19 patients. Cytokine. 2020;134:155190. doi: 10.1016/j.cyto.2020.155190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti G., Tincati C., Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 2013;26:2–18. doi: 10.1128/CMR.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson N.J., Lehner P.J. How does SARS-CoV-2 cause COVID-19? Science. 2020;369:510–511. doi: 10.1126/science.abc6156. [DOI] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maucourant C., Filipovic I., Ponzetta A., Aleman S., Cornillet M., Hertwig L., Strunz B., Lentini A., Reinius B., Brownlie D., et al. Karolinska COVID-19 Study Group Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020;5:eabd6832. doi: 10.1126/sciimmunol.abd6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane L.M., Radmacher M.D., Freidlin B., Yu R., Li M.-C., Simon R. Methods for assessing reproducibility of clustering patterns observed in analyses of microarray data. Bioinformatics. 2002;18:1462–1469. doi: 10.1093/bioinformatics/18.11.1462. [DOI] [PubMed] [Google Scholar]
- Meinshausen N., Bühlmann P. High-Dimensional Graphs and Variable Selection with the Lasso. Ann. Stat. 2006;34:1436–1462. [Google Scholar]
- Michelet X., Dyck L., Hogan A., Loftus R.M., Duquette D., Wei K., Beyaz S., Tavakkoli A., Foley C., Donnelly R., et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018;19:1330–1340. doi: 10.1038/s41590-018-0251-7. [DOI] [PubMed] [Google Scholar]
- Moratto D., Chiarini M., Giustini V., Serana F., Magro P., Roccaro A.M., Imberti L., Castelli F., Notarangelo L.D., Quiros-Roldan E. Flow Cytometry Identifies Risk Factors and Dynamic Changes in Patients with COVID-19. J. Clin. Immunol. 2020;40:970–973. doi: 10.1007/s10875-020-00806-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulè M.P., Martins A.J., Tsang J.S. Normalizing and denoising protein expression data from droplet-based single cell profiling. bioRxiv. 2020 doi: 10.1101/2020.02.24.963603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandagopal N., Ali A.K., Komal A.K., Lee S.-H. The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front. Immunol. 2014;5:187. doi: 10.3389/fimmu.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J., Prezzemolo T., Vanderbeke L., Roca C.P., Gerbaux M., Janssens S., Willemsen M., Burton O., Mol P.V., Herck Y.V., et al. An open resource for T cell phenotype changes in COVID-19 identifies IL-10-producing regulatory T cells as characteristic of severe cases. medRxiv. 2020 doi: 10.1101/2020.05.31.20112979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R., Shah S., Altonsy M.O., Gerber A.N. Glucocorticoid and cytokine crosstalk: Feedback, feedforward, and co-regulatory interactions determine repression or resistance. J. Biol. Chem. 2017;292:7163–7172. doi: 10.1074/jbc.R117.777318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhold R., Ciani Y., Koelzer V.H., Tzankov A., Haslbauer J.D., Menter T., Schwab N., Henkel M., Frank A., Zsikla V., et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. medRxiv. 2020 doi: 10.1101/2020.06.17.20133637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien K.L., Finlay D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019;19:282–290. doi: 10.1038/s41577-019-0139-2. [DOI] [PubMed] [Google Scholar]
- Osman M., Faridi R.M., Sligl W., Shabani-Rad M.-T., Dharmani-Khan P., Parker A., Kalra A., Tripathi M.B., Storek J., Cohen Tervaert J.W., Khan F.M. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv. 2020;4:5035–5039. doi: 10.1182/bloodadvances.2020002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pairo-Castineira E., Clohisey S., Klaric L., Bretherick A.D., Rawlik K., Pasko D., Walker S., Parkinson N., Fourman M.H., Russell C.D., et al. GenOMICC Investigators. ISARICC Investigators. COVID-19 Human Genetics Initiative. 23andMe Investigators. BRACOVID Investigators. Gen-COVID Investigators Genetic mechanisms of critical illness in Covid-19. Nature. 2020 doi: 10.1038/s41586-020-03065-y. Published online December 11, 2020. [DOI] [Google Scholar]
- Pommerenke C., Wilk E., Srivastava B., Schulze A., Novoselova N., Geffers R., Schughart K. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS ONE. 2012;7:e41169. doi: 10.1371/journal.pone.0041169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querec T.D., Akondy R.S., Lee E.K., Cao W., Nakaya H.I., Teuwen D., Pirani A., Gernert K., Deng J., Marzolf B., et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2009;10:116–125. doi: 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph G.J., Ochando J., Partida-Sánchez S. Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- Rha M.-S., Jeong H.W., Ko J.-H., Choi S.J., Seo I.-H., Lee J.S., Sa M., Kim A.R., Joo E.-J., Ahn J.Y., et al. PD-1-Expressing SARS-CoV-2-Specific CD8+ T Cells Are Not Exhausted, but Functional in Patients with COVID-19. Immunity. 2021;54:44–52.e3. doi: 10.1016/j.immuni.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio I., Osuchowski M.F., Shankar-Hari M., Skirecki T., Winkler M.S., Lachmann G., La Rosée P., Monneret G., Venet F., Bauer M., et al. Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect. Dis. 2019;19:e422–e436. doi: 10.1016/S1473-3099(19)30567-5. [DOI] [PubMed] [Google Scholar]
- Ruzek M.C., Miller A.H., Opal S.M., Pearce B.D., Biron C.A. Characterization of early cytokine responses and an interleukin (IL)-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J. Exp. Med. 1997;185:1185–1192. doi: 10.1084/jem.185.7.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Cerrillo I., Landete P., Aldave B., Sánchez-Alonso S., Sánchez-Azofra A., Marcos-Jiménez A., Ávalos E., Alcaraz-Serna A., de Los Santos I., Mateu-Albero T., et al. REINMUN-COVID and EDEPIMIC groups COVID-19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J. Clin. Invest. 2020;130:6290–6300. doi: 10.1172/JCI140335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schett G., Sticherling M., Neurath M.F. COVID-19: risk for cytokine targeting in chronic inflammatory diseases? Nat. Rev. Immunol. 2020;20:271–272. doi: 10.1038/s41577-020-0312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 2016 doi: 10.1101/060012. [DOI] [Google Scholar]
- Shodell M., Siegal F.P. Corticosteroids depress IFN-α-producing plasmacytoid dendritic cells in human blood. J. Allergy Clin. Immunol. 2001;108:446–448. doi: 10.1067/mai.2001.117928. [DOI] [PubMed] [Google Scholar]
- Soneson C., Robinson M.D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods. 2018;15:255–261. doi: 10.1038/nmeth.4612. [DOI] [PubMed] [Google Scholar]
- Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., 3rd, Smibert P., Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., et al. ISB-Swedish COVID19 Biobanking Unit Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/j.cell.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulahian T.H., Högger P., Wahner A.E., Wardwell K., Goulding N.J., Sorg C., Droste A., Stehling M., Wallace P.K., Morganelli P.M., Guyre P.M. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. 2000;12:1312–1321. doi: 10.1006/cyto.2000.0720. [DOI] [PubMed] [Google Scholar]
- Swiecki M., Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015;15:471–485. doi: 10.1038/nri3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M., Wang Y., Vermi W., Gilfillan S., Schreiber R.D., Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 2011;208:2367–2374. doi: 10.1084/jem.20110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterman A., Sumida T.S., Nouri N., Yan X., Zhao A.Y., Gasque V., Schupp J.C., Asashima H., Liu Y., Cosme C., et al. Single-Cell Omics Reveals Dyssynchrony of the Innate and Adaptive Immune System in Progressive COVID-19. medRxiv. 2020 doi: 10.1101/2020.07.16.20153437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallelian F., Schaer C.A., Kaempfer T., Gehrig P., Duerst E., Schoedon G., Schaer D.J. Glucocorticoid treatment skews human monocyte differentiation into a hemoglobin-clearance phenotype with enhanced heme-iron recycling and antioxidant capacity. Blood. 2010;116:5347–5356. doi: 10.1182/blood-2010-04-277319. [DOI] [PubMed] [Google Scholar]
- van der Made C.I., Simons A., Schuurs-Hoeijmakers J., van den Heuvel G., Mantere T., Kersten S., van Deuren R.C., Steehouwer M., van Reijmersdal S.V., Jaeger M., et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA. 2020;324:663–673. doi: 10.1001/jama.2020.13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt G.J.W., O’Sullivan D., Everts B., Huang S.C.-C., Buck M.D., Curtis J.D., Chang C.-H., Smith A.M., Ai T., Faubert B., et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varchetta S., Mele D., Oliviero B., Mantovani S., Ludovisi S., Cerino A., Bruno R., Castelli A., Mosconi M., Vecchia M., et al. Unique immunological profile in patients with COVID-19. Cell. Mol. Immunol. 2020:1–9. doi: 10.1038/s41423-020-00557-9. Published online October 15, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venet F., Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 2018;14:121–137. doi: 10.1038/nrneph.2017.165. [DOI] [PubMed] [Google Scholar]
- Wang E.Y., Mao T., Klein J., Dai Y., Huck J.D., Liu F., Zheng N.S., Zhou T., Israelow B., Wong P., et al. Diverse Functional Autoantibodies in Patients with COVID-19. medRxiv. 2020 doi: 10.1101/2020.12.10.20247205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauters E., Mol P.V., Garg A.D., Jansen S., Herck Y.V., Vanderbeke L., Bassez A., Boeckx B., Malengier-Devlies B., Timmerman A., et al. Discriminating Mild from Critical COVID-19 by Innate and Adaptive Immune Single-cell Profiling of Bronchoalveolar Lavages. bioRxiv. 2020 doi: 10.1101/2020.07.09.196519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.S., Lezon-Geyda K., Maksimova Y., Craft S., Zhang Y., Su M., Schulz V.P., Craft J., Gallagher P.G. Global transcriptome analysis and enhancer landscape of human primary T follicular helper and T effector lymphocytes. Blood. 2014;124:3719–3729. doi: 10.1182/blood-2014-06-582700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E.J., Ha S.-J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group. Sterne J.A.C., Murthy S., Diaz J.V., Slutsky A.S., Villar J., Angus D.C., Annane D., Azevedo L.C.P., Berwanger O., et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: Meta-analysis. JAMA. 2020;324:1330–1341. doi: 10.1001/jama.2020.17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2019. tidyverse: Easily Install and Load the “Tidyverse.”.https://rdrr.io/cran/tidyverse/ [Google Scholar]
- Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson E.J., Walker A.J., Bhaskaran K., Bacon S., Bates C., Morton C.E., Curtis H.J., Mehrkar A., Evans D., Inglesby P., et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584:430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C.W., McClain M.T., Chen M., Zaas A.K., Nicholson B.P., Varkey J., Veldman T., Kingsmore S.F., Huang Y., Lambkin-Williams R., et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS ONE. 2013;8:e52198. doi: 10.1371/journal.pone.0052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., McGoogan J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- Wynn T.A., Chawla A., Pollard J.W. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin G., Zander R., Schauder D.M., Chen Y., Weinstein J.S., Drobyski W.R., Tarakanova V., Craft J., Cui W. Single-cell RNA sequencing unveils an IL-10-producing helper subset that sustains humoral immunity during persistent infection. Nat. Commun. 2018;9:5037. doi: 10.1038/s41467-018-07492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J., Schmidt S.V., Sander J., Draffehn A., Krebs W., Quester I., De Nardo D., Gohel T.D., Emde M., Schmidleithner L., et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C., Bora S.A., Parimon T., Zaman T., Friedman O.A., Palatinus J.A., Surapaneni N.S., Matusov Y.P., Chiang G.C., Kassar A.G., et al. Cell type-specific immune dysregulation in severely ill COVID-19 patients. medRxiv. 2020 doi: 10.1101/2020.07.23.20161182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Liu J., Yuan Y., Huang F., Ma R., Luo B., Xi Z., Pan T., Liu B., Zhang Y., et al. Two waves of pro-inflammatory factors are released during the influenza A virus (IAV)-driven pulmonary immunopathogenesis. PLoS Pathog. 2020;16:e1008334. doi: 10.1371/journal.ppat.1008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-Y., Wang X.-M., Xing X., Xu Z., Zhang C., Song J.-W., Fan X., Xia P., Fu J.-L., Wang S.-Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., et al. COVID-STORM Clinicians. COVID Clinicians. Imagine COVID Group. French COVID Cohort Study Group. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. NIAID-USUHS/TAGC COVID Immunity Group Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T., Liu H., Roeder K., Lafferty J., Wasserman L. The huge Package for High-dimensional Undirected Graph Estimation in R. J. Mach. Learn. Res. 2012;13:1059–1062. [PMC free article] [PubMed] [Google Scholar]
- Zheng S., Fan J., Yu F., Feng B., Lou B., Zou Q., Xie G., Lin S., Wang R., Yang X., et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ. 2020;369:m1443. doi: 10.1136/bmj.m1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Yang Y., Zhang X., Li Z., Liu X., Hu C., Chen C., Wang D., Peng Z. Clinical Course of 195 Critically Ill COVID-19 Patients: A Retrospective Multicenter Study. Shock. 2020;54:644–651. doi: 10.1097/SHK.0000000000001629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clusters were assigned based on surface marker expression. Results shown are for genes with log-fold change greater than 0.25 and expressed in at least 10% of the cells in either the cluster tested or all background cells, and with a p value < 0.01 (Wilcoxon test implemented in Seurat’s FindAllMarkers function).
Each tab corresponds to a different comparison. The corresponding figure in the main text is referenced in the caption at the top of each tab.
Each sample is annotated with the circumstances (ICU, intubation, and corticosteroid use) under which it was collected. Type II ANOVA test statistics for association between these circumstances and DSM at T0 time points and odd ratios statistics between deceased and recovered patients at juncture (d17-23 TSO) are also listed.
The coefficients/effect sizes and associated p values of each term in mixed-effect models are provided. All times are expressed as days since symptom onset.
Data Availability Statement
Raw and processed data from the single cell mRNA, surface protein, and TCR V(D)J sequencing and bulk RNaseq are available from the NCBI Gene Expression Omnibus, accession number GEO: GSE161918 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161918). Analysis code, extended patient and sample metadata, serum protein, and antibody data are available at: https://github.com/niaid/covid19-time-resolved. The single cell UMAPs in Figure 3B can be visualized interactively at: https://cellxgene.cziscience.com/collections/ed9185e3-5b82-40c7-9824-b2141590c7f0.