

Abstract
For the last century we have relied on model organisms to help understand fundamental biological processes. Now, with advancements in genome sequencing, assembly and annotation, non-model organisms may be studied with the same advanced bioanalytical toolkit as model organisms. Proteomics is one such technique, which classically relies on predicted protein sequences to catalog and measure complex proteomes across tissues and biofluids. Applying proteomics to non-model organisms can advance and accelerate biomimicry studies, biomedical advancements, veterinary medicine, agricultural research, behavioral ecology, and food safety. In this post-model organism era we can study almost any species, meaning that many non-model organisms are in fact important emerging model organisms. Herein we focus specifically on eukaryotic organisms and discuss steps to generating sequence databases, analyzing proteomic data with or without a database, interpreting results, and future research opportunities. Proteomics is more accessible than ever before, and will continue to rapidly advance in the coming years, enabling critical research and discoveries in non-model organisms that were hitherto impossible.
Keywords: comparative biology, biomimicry, proteomics, genomics, non-model
Graphical Abstract
Introduction
Beginning with fruit flies at the turn of the 20th century, model organisms have enabled biological discoveries by bringing testable yet complex biological systems to the lab. These organisms, including mice, rats, frogs, zebra fish, roundworms, yeast, maize, Arabidopsis, and Escherichia coli, are easily maintained in the lab, have well established methods and molecular tools, can be manipulated at the molecular level, and have a vast body of associated literature1–2. Over the last 100 years, these model organisms have been crucial to revealing fundamental biological truths and developing the theoretical framework for molecular biology. But as cutting-edge biomolecular analysis in non-model organisms is becoming more routine, we are on the cusp of a new frontier3–4. Over billions of years, natural selection has favored countless adaptations to allow organisms to inhabit and thrive in all corners of the earth. Through the study of non-model organisms, scientists are revealing insights into the molecular basis of a vast array of human conditions. Naturally occurring adaptations and susceptibilities inform the evolutionary and genetic underpinning of chronic and genetic disease in humans, from cancer to neurodegeneration (expertly reviewed by Stenvinkel et al.5–6). For instance, elephants and naked mole rats illuminate molecular mechanisms at play in cancer resistance7–9, deer antlers hold clues to organ regeneration10–11, diving mammals inform ischemia reperfusion injury resistance12–16, hibernating arctic squirrels and grizzly bears teach lessons about neuroprotection and metabolism17–19, and naked mole rats and bats possess secrets to longevity20–23. Plants are also treasure troves of information including how compartmentalization in plants avoids senescence24–25. This field of study, biomimetics, seeks to understand these adaptations and provide insight into chronic disease in humans, and is made possible by non-model organism research.
In addition to biomimetics, non-model organisms are important in other key areas, including agricultural research, veterinary medicine, behavioral ecology, and food safety. Emerging plant diseases in high value agricultural crops, such as citrus greening disease26–28 and cotton blue disease29–33, are forcing rapid development of bioanalytical capabilities to study non-model plant species, plant pathogens and insect vectors of plant pathogens. Knowledge of these virus-interacting host proteins paves the way for genome editing approaches to develop durable resistance. Non-model organism research is also critical in veterinary medicine for industries such as zoos and aquariums or pets ($3 billion and $18 billion annual, respectively34–35), which rely on bioanalytical measurements for development and validation of new techniques translated from human medicine. In cases such as bats, genomic and proteomic analysis is being harnessed to better understand longevity, as well as their innate immunity and ability to serve as disease reservoirs36–38. Maybe less apparent is the applicability of proteomics to behavioral ecology (expertly reviewed by Valcu and Kempenaers39), such as eusociality in the naked mole-rat40. Lastly, with growing concerns of food authenticity, proteomic analysis in non-model organisms offers promising avenues of tracking and validation in aquaculture and agriculture41–45. These topics are economically critical and rely on our ability to rapidly develop cutting edge analytical capabilities in non-model organisms. Classical model organisms are vitally important, but there is an enormous promise of untold discoveries and applications by studying non-model organisms (Figure 1).
Figure 1. General overview of proteomics in non-model organisms.
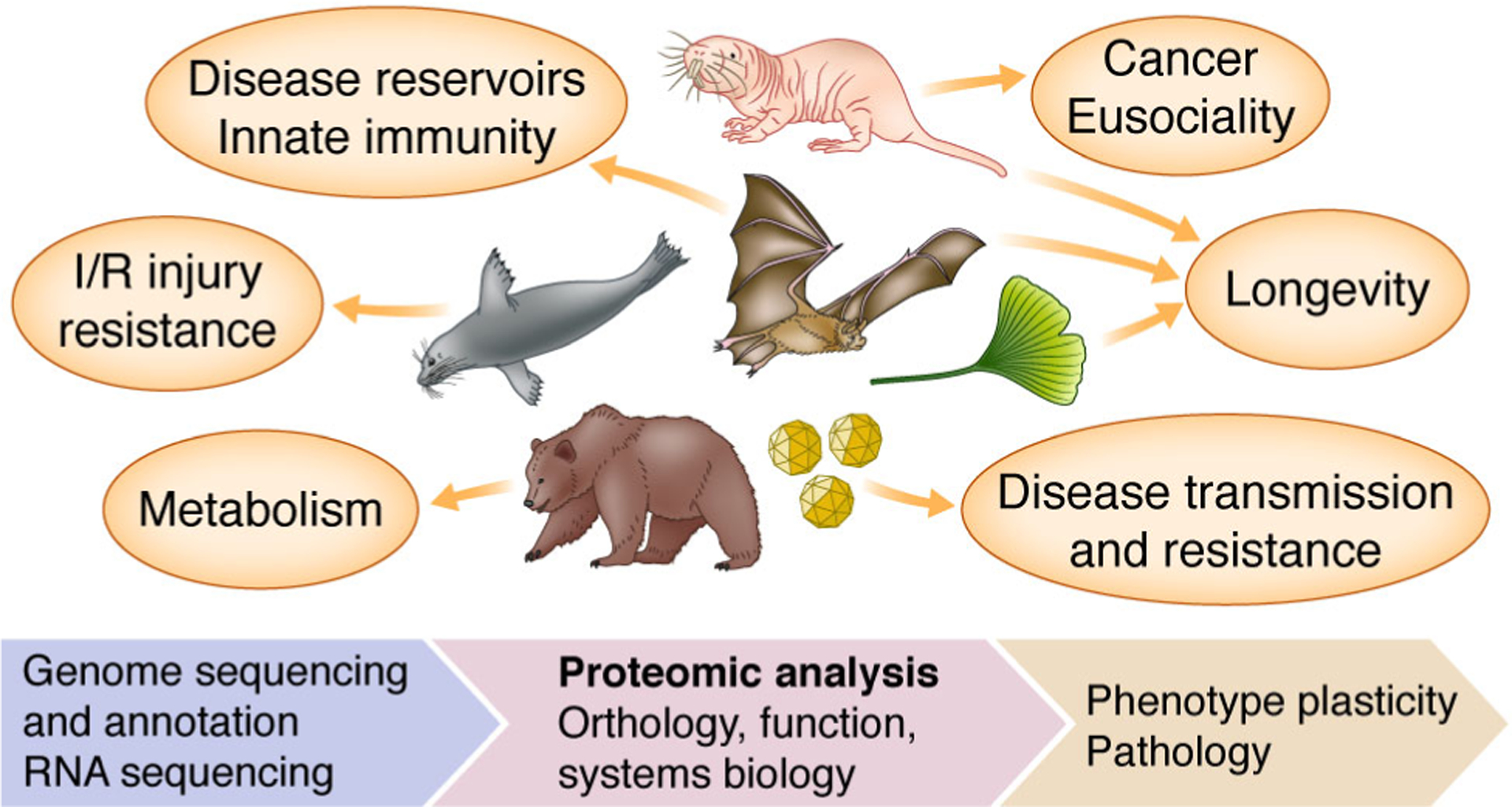
Some notable examples of non-model proteomics are shown (clockwise from top): naked mole-rat, bat, ginkgo, viruses, hibernating mammals, and diving mammals. I/R refers to ischemia reperfusion.
Given the current SARS-CoV-2 pandemic, it is important to specifically highlight the importance and potential of non-model organism research when studying zoonotic viruses. Although predicting zoonosis includes many non-molecular factors46–47, it has been shown that sequence homology between host receptors (e.g., ACE2 in SARS and SARS-CoV-2 infection) can predict zoonotic potential and species tropism48–50. For instance, SARS-CoV-2 entry potential correlates with ACE2 receptor homology between reservoirs and hosts50. Current studies of SARS-CoV-2 utilizing structural information such as receptor binding domain51 and glycosylation of the viral spike protein52 will continue to improve predictive power even further53–54. Undoubtedly proteomic analysis of these non-model reservoirs can provide empirical evidence of structural and glycosylation predictions on vital molecular targets, which is made possible only by directly studying the non-model organism. Furthermore, determining host-virus interactions by protein-protein interaction (PPI) proteomic analysis is critical to understanding infection, such as with Ebola virus55, Zika virus56 and SARS-CoV-257, but similar PPI studies in viral reservoirs themselves could be equally enlightening for innate and adaptive immunity as well as zoonotic spillover potential. Broad proteomic studies across hundreds of reservoir and host species will improve our understanding of viral infection, zoonotic prediction, and highlight possible treatments.
Proteomics in non-model systems is driven by genomics
Currently, the most common method of proteomic analysis is shotgun proteomics (also referred to as bottom-up proteomics). By digesting proteins to peptides, proteomics researchers use mass spectrometers and database search algorithms to identify and quantify near complete proteomes (such as recent studies in human58–59 and yeast60). Traditional data-dependent acquisition (DDA) or newer techniques such as data-independent acquisition (DIA) or BoxCar61 may be used if the search space can be defined using protein sequences and appropriate post-translational modifications. When using shotgun proteomics to study a non-model organism that may not have an annotated genome available, the search space is the major hurdle62–63. If an annotated genome is available, any limitation that applies to proteomics in model organisms will also apply to a non-model (e.g., studying glycosylation with DIA data is difficult regardless). Knowledge of current solutions and best practices in non-model organisms is limiting widespread adoption. For starters, in the past, genome sequencing and annotation could prove an insurmountable or very expensive problem to overcome, but this is no longer the case.
In recent years, post-2015, there has been an explosion of genome sequencing as the cost to generate accurate highly-contiguous genome assemblies has continued to drop64. There are ongoing large sequencing efforts (e.g., Earth BioGenome Project, Vertebrate Genomes Project, DNA Zoo, Zoonomia Project, Bat1K Project, Bird 10 000 Genomes Project) that are beginning to hit their stride and soon will be releasing hundreds of genomes per year to achieve many thousands of genomes by completion. Of the roughly 5400 mammal species, currently 430 have genomes, while of the roughly 400 000 plant species, there are 630 plant genomes, and of the nearly 1 million named species of insects less than 500 genomes are complete65 (Figure 2). When a genome is not available, especially in the case of mammals the most affordable path to de novo sequencing is to use short-read sequencing with proximity ligation techniques (such as Hi-C or Chicago). The resulting genome assemblies are accurate for transcriptomic and proteomic studies (e.g., proteomic analysis using a Chicago-based mammalian assembly66). In cases of highly heterogeneous and repetitive genomes (e.g., Mollusca), additional and more expensive techniques may be required such as long-read sequencing based on Oxford Nanopore Technologies or PacBio, as well as optical mapping. But in the case of many species, a de novo assembly can be generated in the matter of months for minimal cost and expertise. Once completed, quality measures including contiguity (i.e., scaffold and contig N50) and completeness (i.e., BUSCO67) can be evaluated, but since there are no universal quality thresholds, we recommend comparing quality metrics of phylogenetically similar published assemblies. Alternatively, proteogenomics represents experimental validation of a genome assembly using proteomics66, 68–69. Peptide identification can help validate polymorphisms and predicted alleles while top-down proteomic methods can confirm predicted isoforms. Future development of genome annotation pipelines may include ways to integrate mass spectrometry-based proteomic data.
Figure 2. Distribution of species with proteomic data sets (P), genome assemblies (G), and genome annotations (A) across phylogenetic clades.
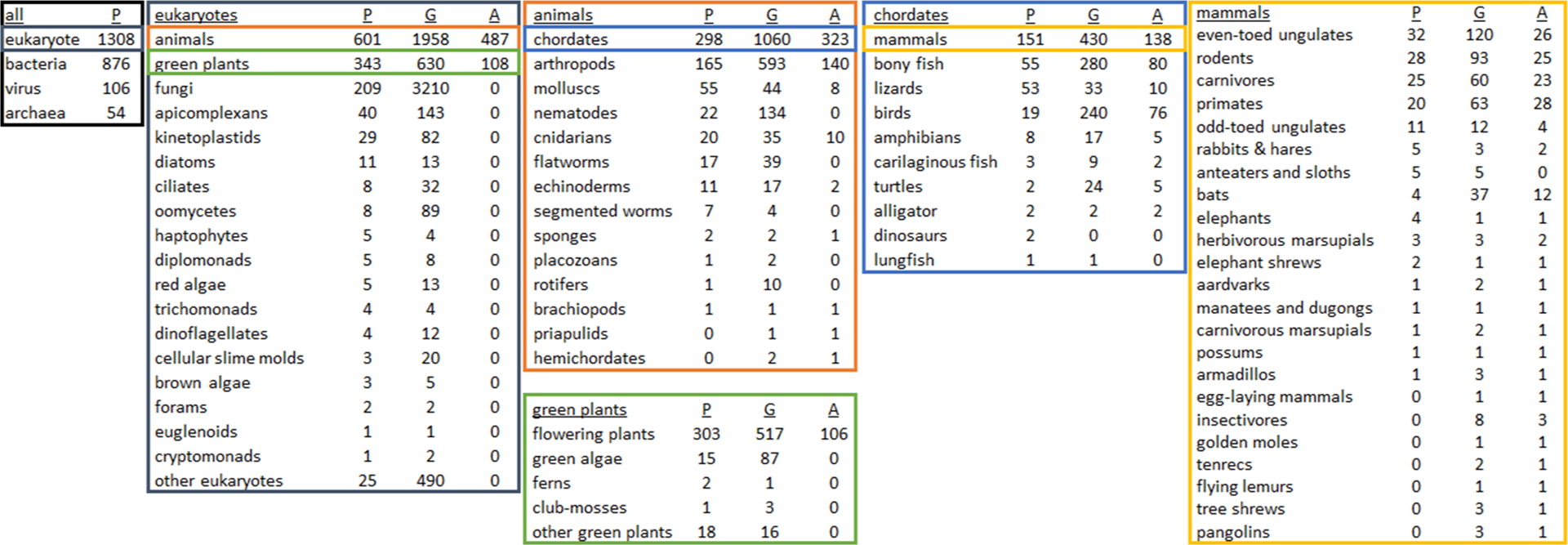
This series of charts was created by cross-referencing taxon IDs of genome assemblies on Genbank and genome annotations on RefSeq (valid as of 12 March 2020) with published proteomic data sets on the Proteomics Identifications Database (PRIDE; as of 17 January 2020) in order to emphasize clades lacking proteomic analysis, or clades that should be focused on for genomic sequencing and annotation. Specific to Eukaryotes, there are approximately 1308 species with published proteomic results, 6718 with available genomes and 595 species with complete genomic annotations. Note that these charts do not show the total number of species per clade (e.g., 950 000 named insect species65), or the number of proteomic data sets per species (e.g., 68 % of the 12 660 PRIDE data sets are human and mouse). For this cataloging, we focused only on genome and annotation resources at NCBI and acknowledge that for non-animal non-flowering plants, there are other more appropriate repositories such as the Joint Genome Institute. Terms in tables are accepted common or scientific NCBI designations, except in a few cases under ‘mammals’ (the full mammal table with taxon IDs is available in Supplemental Table S1), and that these numbers may not be exact due to self-reporting on PRIDE and matching taxon identifiers between sources (note that the two dinosaur proteomic data sets are from Brachylophosaurus canadensis and Tyrannosaurus rex).
Genome sequencing and availability does not seem to be the main roadblock to proteomics in non-model species: the current bottleneck is genome annotation. Complete and accurate genome annotation is key to proteomic workflows. Broadly there are two paths to annotation: in-house annotation or making the data publicly available for annotation by NCBI or Ensembl. In the case of in-house annotation, publicly available and often free, high-performance computing (HPC) resources such as XSEDE (Extreme Science and Engineering Discovery Environment; including JetStream), ACI-REF (Advanced Cyberinfrastructure Research and Education Facilitators) Network, NCI (National Computational Infrastructure) Australia, PRACE (Partnership for Advanced Computing in Europe) and the ELIXIR (the European life-sciences Infrastructure for biological Information) network, have enabled novice bioinformaticians without a local HPC to annotate genome assemblies using pipelines like MAKER (see tutorials70–71). Although still an iterative task, this is made possible by having thousands of processing hours on a virtual cluster. The second path to genome annotation is using free public resources like NCBI RefSeq or Ensembl. Specifically, the RefSeq project maintains and curates new and updated genome annotations72–73. RefSeq has published guidelines on how genomes are selected for annotation74, related to assembly contiguity, secondary RNA-seq data, and need by researchers. Once a high-quality genome assembly has been made public on DDBJ (DNA Data Bank of Japan), ENA (European Nucleotide Archive) or GenBank, it may be annotated and publicly released by RefSeq in a matter of weeks. But of the hundreds of genomes on GenBank, only a fraction have been annotated (Figures 2 and 3). What may not be evident is that a researcher not associated with the original genome project but with access to RNA-seq data may upload new sequence read archives (SRA), which can help begin or improve annotations of already existing high-quality genome assemblies. This is also an excellent way to demonstrate interest and improve annotation as more secondary gene evidence and transcript diversity typically results in more complete and accurate annotations. For instance, multiple tissue single-stranded RNA sequencing data were used by RefSeq to annotate the California sea lion genome75. The current annotation system is ripe for democratization and team science approaches, especially for pre-existing publicly available genome assemblies.
Figure 3. Distribution of mammals with proteomic data sets or genome annotations.
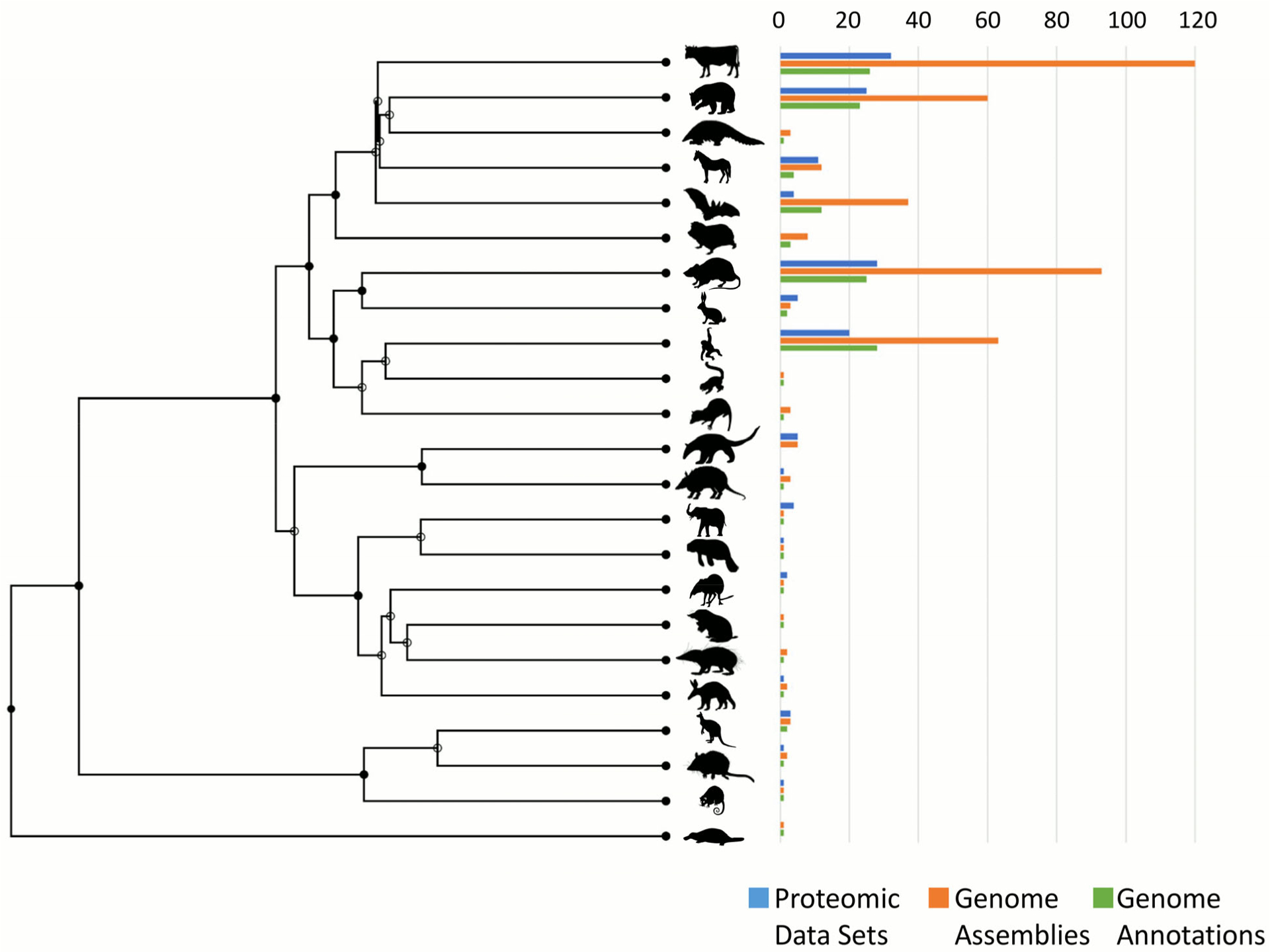
Using a time tree (http://www.timetree.org), which incorporates fossil evidence76, information regarding the 23 groups of mammals with proteomic data sets on the Proteomics Identifications Database (PRIDE; as of 17 January 2020), genome assemblies on Genbank and genome annotations on RefSeq (valid as of 12 March 2020) was plotted. This is per taxa and does not include the number of proteomic data sets, genome assemblies or genome annotations within taxa. The order, from top to bottom is: even-toed ungulates, carnivores, pangolins, odd-toed ungulates, bats, insectivores, rodents, rabbits and hares, primates, flying lemurs, tree shrews, anteaters and sloths, armadillos, elephants, manatees and dugongs, elephant shrews, golden moles, tenrecs, aardvarks, herbivorous marsupials, carnivorous marsupials, possums, egg-laying mammals.
Following in-house or RefSeq genome annotation as described above, there will be a sequence database (a collection of protein sequences, typically as a FASTA, XML or GFF file) that can be used to search proteomic data. There are secondary resources such as UniProtKB that may import and evaluate annotations from RefSeq and Ensembl, but it is important to note that UniProtKB does not import all available high-quality annotations. Moreover, whereas RefSeq groups genome annotations by release or by user submission, the provenance of protein assignments from other resources may be difficult to determine. For these reasons, retrieving species-specific proteomes from UniProtKB, RefSeq and Ensembl will yield different protein databases, often based on different genome assemblies, annotation pipelines, and curation. Since interpretations of proteomic data in non-model organisms are only as useful as the functional annotation of the proteome, a careful manual inspection of automated annotations may be required before proceeding with biological and functional experiments, as even RefSeq annotations may not be perfect due to issues with homology-based assignments, likely because orthologs in well-annotated species may not always exist77. When proteins of interest are unknown or hypothetical, manual single protein curation is required using tools like UniRule78. In insects, community and student-led annotation efforts have helped identify families of well-conserved genes and proteins79–81, but species-specific genes and genes under positive selection are more difficult to annotate and require detailed, tissue-specific proteomic investigation82–85. As more annotations come online, and with continued curation by the community, pipeline generated genome annotations will continue to improve.
There is a wealth of public genome assemblies, even multiple genomes from the same species, challenging the concept of a reference genome. In this pan-genome era, researchers are realizing that one individual cannot capture the genomic (and therefore proteomic) diversity of a population or species. In other words, more than a single human, aphid or African elephant reference genome may be required to describe the genome or proteome. Moreover, even with reference genomes there will be numerous annotation versions as gene predictions improve, more evidence is acquired, assemblies are improved, or more genomes are completed. The reference human annotation is on its tenth release, and less obvious species like the cow are on their seventh release. For protein databases to evolve and effectively capitalize on pan-genomic data, a new framework will need to be developed.
Analyzing proteomic data from non-model organisms
Given the global resources devoted to genome annotation and how these data products are maintained and propagated throughout data hubs, when possible the greatest community benefits will come from applying resources to genome annotation (described above), as opposed to one-off transcriptomes. Yet there are still occasions where it is not possible due to resources, time, or simply availability of quality samples for genomic sequencing. If samples can be acquired with RNA stability in mind, it should be possible to generate predicted protein sequences using RNA sequencing (such as86; Figure 4A). In cases such as biofluids, choosing a relevant tissue to sequence may be difficult since protein provenance is unknown. For instance, due to its proximity to all organs the complete blood proteome is translated in many tissues, not the blood itself87. Another consideration is that when using short-read RNA sequencing, de novo assembly is required, which can be computationally costly and may not provide sequence coverage of protein isoforms or very large proteins (e.g., human titin isoform N2BA mRNA is over 100 000 residues in length). To overcome these issues, full-length RNA sequencing can be employed88, negating the need for transcriptome assembly since the direct transcript evidence can define the search space. As the capability and accessibility of full-length RNA sequencing improves, this could become a viable parallel approach to support mass spectrometry-based proteomics.
Figure 4. Different approaches to analyzing proteomic data without an annotated genome.
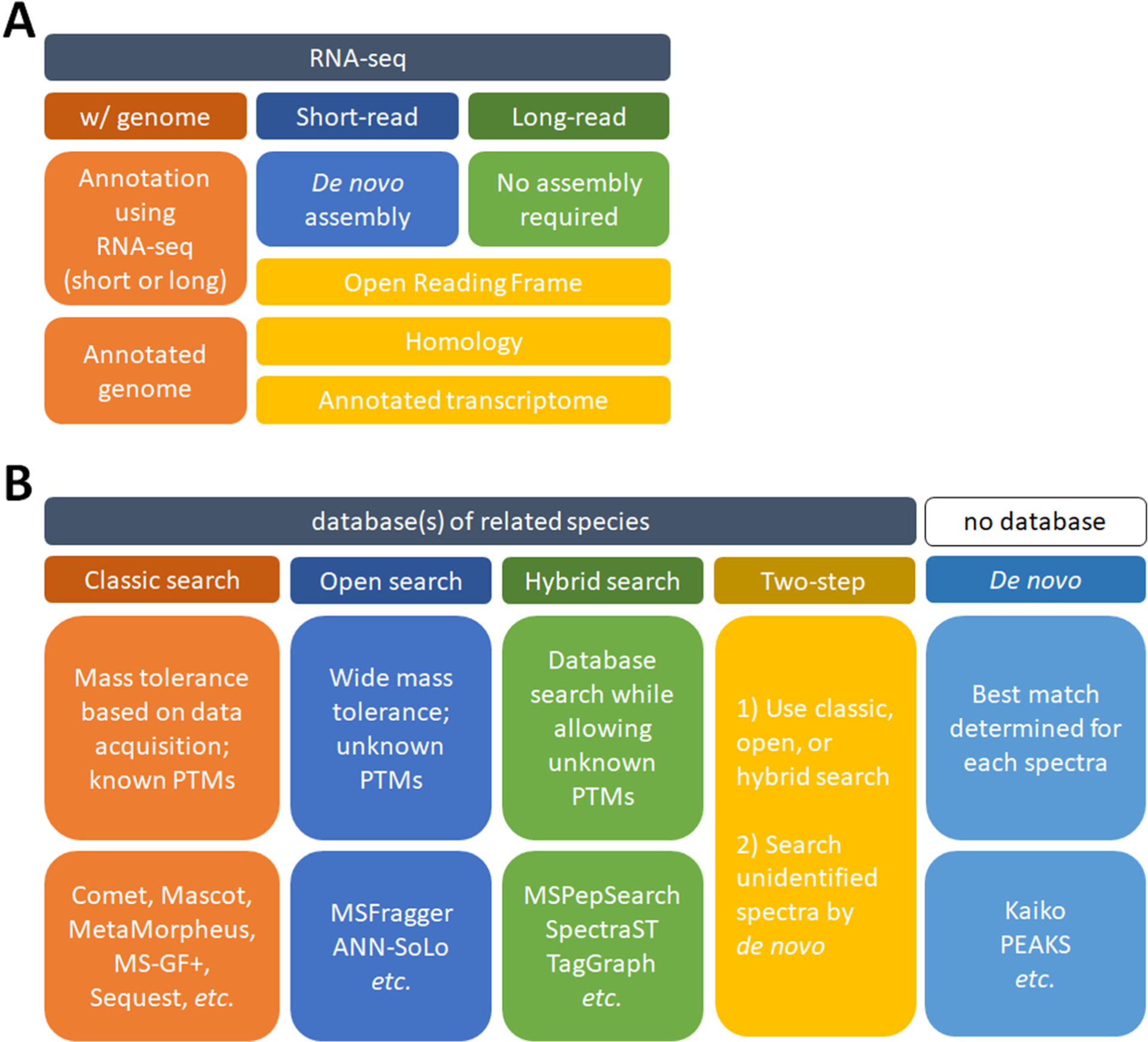
A. Using nucleic acid sequencing to generate an annotated genome or transcriptome. These can be used to analyze shotgun proteomic data. B. Searching shotgun proteomic data without prior/relevant nucleic acid sequencing. The software listed is not exhaustive and there are many suitable alternatives.
Proteomic analysis of a non-model organism in the absence of any additional nucleic acid sequencing is also possible (Figure 4B). Homology searching is when the database of a closely related species is used for conventional database searching. In this case, only tryptic peptides with complete homology will be identified. Depending on the clade this may work better than others (e.g., using the Weddell seal and Pacific walrus annotations to search California sea lion proteomic data provided valid conclusions89). Recently an approach was developed that can assist in selecting species by evaluating how well a homologous sequence database can explain an unknown proteome63. Other search approaches can involve using wide mass tolerances such that single amino acid variants (SAAVs) can be tolerated between the known and unknown species. This is accomplished by employing an open search with a wide mass tolerance, and searching using a closely related species with a tool like MSFragger90 or ANN-SoLo91. Similarly, utilizing a hybrid spectral library approach may work with closely related species92. Also, TagGraph93 should likewise be applicable with an unknown proteome by combining database and de novo searching to identify unknown post-translational modifications (PTMs). Another approach is to take the remaining unmatched spectra from any of these approaches and perform de novo searching with tools such as Kaiko94, PEAKS95, or those packaged within DeNovoGUI96. Using de novo proteomic techniques in conjunction with database searching, or as a stand-alone search, is a viable alternative when database searching of shotgun proteomic data isn’t possible. Although the accuracy of de novo is still improving, recent and ongoing advancements continue to make de novo a valid approach. Finally, it is important to note that employing these search approaches may be useful even in well studied organisms where we have complete confidence in the validity of sequence databases. Until we appreciate and account for the genetic diversity within a species, there will always remain the possibility that the reference sequence database is incomplete or inaccurate.
In the given suggestions, it is assumed that the source of the proteome is known. In some cases there may be a primary or secondary unidentified species. In the case of unknown disease agents, we can use proteomics where other methods fail due to antibody reactivity, RNA degradation or unknown PCR probes. This can present an enormous search space (such as the nearly 12 million proteins listed on UniProtKB for Fungi), but one that is not insurmountable given the numerous databases available, though high computational costs and significant false-discovery concerns exist. Conversely, we may be tasked with identifying non-model organisms within model systems. For instance, when studying the gut proteome there is the microbiome and host, but there are also remnants of ingested food. In an effort to correctly define search space and accurately identify mass spectra, we must account for numerous unknowns. In these cases, multiple search steps, greater reliance on known spectral libraries, or de novo is required.
Technical capabilities, hurdles, and future advancements
The discussion and solutions presented so far have focused on current approaches to define and evaluate the search space for shotgun proteomic analysis in non-model organisms, but there are other proteomic approaches worth mentioning. In contrast to digesting proteins prior to analysis, intact proteins can be analyzed using mass spectrometry by top-down proteomics97–99. Although top-down is not yet routinely used in non-model organisms, it would face similar hurdles as those in humans, but given accurate protein sequences top-down non-model proteomics is possible. Protein arrays, composed of antibodies, lectins, or aptamers, are another possible approach to proteomics in non-model organisms. Though these arrays are typically geared towards human proteins, such as the SOMAscan, PETAL100, or Olink platforms, they may be used in other species with varying success when there is enough cross-reactivity between conserved epitopes101–102. It seems likely that arrays will be developed for animal-models if the market exists. There is a paucity of commercial antibodies against proteins from non-model organisms, therefore targeted mass spectrometry-based proteomics is a convenient alternative and a tremendously valuable tool to aid researchers interested in quantification of specific proteins in non-model organisms31, 103–104. Overall, it seems only a matter of time before other proteomic techniques become routine in non-model organism studies.
Once protein identifications have been made, data interpretation in non-model organisms presents its own unique challenges. A typical workflow will result in a list of differentially abundant proteins, or proteins that are drivers of some difference, and this list is used for downstream analysis. Depending on the source of the non-model organism sequence database, the protein identification may be meaningful (e.g., alpha-2 microglobulin) or it may simply be a generic locus or transcript ID which is unsuitable for biological interpretation. One possible first step is to convert the identifications to a known model-species by using BLAST to assign protein sequences to their model-species equivalent (a notable implementation is the PAW BLASTer105). When determining orthology, the conversion may still require manual inspection due to the high degree of homology between certain proteins (e.g., human alpha-2 microglobulin and pregnancy zone protein are 72% identical). Once the protein identifications have been converted to a model-species, a typical downstream analysis is gene set enrichment or pathways analysis. The underpinning biology and literature inform these lists, which means results from a non-model organism may be irrelevant or at least filled with caveats. Currently, the Molecular Signatures Database (MSigDB v7.0) includes five species (the vast majority being human and mouse), the Reactome pathway database and WebGestalt have 15 and 12 species, respectively, while the Plant Reactome has 97 plant species106 and the OmicsDB::Pathogens has 11 species107. But the further away, phylogenetically speaking, you move from the few well-studied model organisms, the less applicable the results may be. One alternative is to use co-expression or stabilized regression analysis108, which can utilize abundance patterns to identify novel pathways or gene sets. Although these approaches are far from perfect, they do allow conditional conclusions to be reached and generation of testable hypotheses.
Proteomics provides unique insights into biology
Given the hurdles described, an apparent question is why use proteomics to study non-model organisms. Studying biology at the molecular level is not mutually exclusive to nucleic acid sequencing techniques, but there are cases where proteomics provides more relevant and actionable insights as compared to gene or transcript studies. Since the abundance of the majority of proteins does not correlate with transcript or gene abundance109–115, proteomic analysis can provide a clearer understanding of the plasticity of phenotypes. Moreover, there are specific cases where proteomics can provide more relevant and actionable insights, such as biofluids (blood, urine, cerebrospinal fluid, insect hemolymph, etc.) where the mRNA in the fluid is not related to the proteome of the biofluid. Instead, the proteome of a biofluid is reflected by organs proximal to the fluid and any pathology causing flux between the fluid and organ(s). Also, it is important to note that genes do not change with age, but protein abundance, turnover, localization, modifications and interactions do change116–117. In other words, phenotypes may arise from unique combinations of protein interactions and protein abundance, which will not be captured from genomic information alone. In addition to examining protein abundance differences, proteomic analysis can capture measurements of functionally important endogenous peptides118–119, PTMs120 and proteoforms121. In the case of proteoforms especially, abundance is highly dynamic, such as histone H4 with over 40 proteoforms that may vary over two orders of magnitude122. It is not to say that other techniques cannot answer these questions, but it is important to note where proteomics is uniquely capable.
The complexity of the proteome is further magnified when one considers that different proteoforms can interact to form functional protein complexes with unknown roles in cellular homeostasis and pathogensis (e.g., tumor suppressor protein PTEN123). Only proteomic methods can provide quantitative measurements of protein complex formation in cells at proteome-wide scales29, 124–132, improving our understanding of the evolutionary origins of protein interactions, protein co-localization, and functional protein interaction networks133. Genetic and proteomic diversity also extends to an organism’s microbiota134. This proteomic diversity is critical to understanding biological phenotypes, including cancer135 and transmission of viruses134, 136. Metaproteomic techniques can also provide insights into host-symbiont functional compartmentalization of proteomes, predictions on resident microbiota in eukaryotic host tissues and knowledge on co-evolutionary mechanisms regulating symbiosis in non-model organisms is crucial to our understanding of eukaryotic biology137–139. It is evident that proteomics can provide an additional modality to improve nucleic acid based analyses, while also providing unique insight into the molecular landscape responsible for a given phenotype or pathology.
Future Outlook
There is great potential in applying proteomics in non-model organisms and the technical hurdles are not formidable. This is best exemplified by a recent large scale proteomic study that cataloged 340 000 proteins from 100 species140. In the future, proteomic studies of non-model organisms will undoubtedly become more commonplace. As it becomes possible to define the molecular landscape in species across the tree of life, the next step is finding the non-model organism experts. But this doesn’t need to be for an exotic species in a distant land. In the shallow waters along the eastern seaboard is a euryhaline stingray that can achieve and maintain an extraordinarily high renal transtubular osmotic gradient141 and regenerate glomeruli142. Understanding the elasmobranch kidney could provide insight into chronic disease in humans. Exciting topics like this are all around us if we just look, explore and experiment. Proteomics researchers must work with biologists in these adjacent fields who can direct experimental design of naturally occurring phenotypes to empower comparative studies, and may be unaware of the power of proteomics or its accessibility via cores and collaborations. Empowering these scientists with advanced bioanalytical capabilities is a major goal of proteomics in non-model organisms. There is great potential to address pressing issues such as food supply security, help facilitate commerce, protect human and animal health, and use these techniques in biomimetics to help accelerate biomedical breakthroughs. The post-model organism era opens the door to new applications of maturing technology, hopefully leading to new fundamental biological truths unattainable using classic model organisms.
Supplementary Material
Supplementary Table S1. Specific NCBI taxon IDs and their designation for 23 mammal groups used in Figures 2 and 3.
Acknowledgements
The graphics in Figure 1 were created specifically for this manuscript by Debbie Maizels. Funding for MH by USDA ARS Project # 8062-22410-006-00-D. The authors wish to thank Juan Antonio Vizcaino (European Bioinformatics Institute) for providing current information on data sets at PRIDE, and Magnus Palmblad (Leiden University Medical Center) and Michael G. Janech (College of Charleston) for critical feedback. Identification of certain commercial equipment, instruments, software or materials does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
Footnotes
Competing Interests: The authors declare that they have no competing financial interests.
Supporting Information:
The following supporting information is available free of charge at ACS website http://pubs.acs.org/
References
- 1.Muller B; Grossniklaus U, Model organisms--A historical perspective. Journal of proteomics 2010, 73 (11), 2054–63. [DOI] [PubMed] [Google Scholar]
- 2.Tang B; Wang Y; Zhu J; Zhao W, Web resources for model organism studies. Genomics, proteomics & bioinformatics 2015, 13 (1), 64–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell JJ; Theriot JA; Sood P; Marshall WF; Landweber LF; Fritz-Laylin L; Polka JK; Oliferenko S; Gerbich T; Gladfelter A; Umen J; Bezanilla M; Lancaster MA; He S; Gibson MC; Goldstein B; Tanaka EM; Hu CK; Brunet A, Non-model model organisms. BMC biology 2017, 15 (1), 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldstein B; King N, The Future of Cell Biology: Emerging Model Organisms. Trends in cell biology 2016, 26 (11), 818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stenvinkel P; Painer J; Johnson RJ; Natterson-Horowitz B, Biomimetics - Nature’s roadmap to insights and solutions for burden of lifestyle diseases. Journal of internal medicine 2020, 287 (3), 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenvinkel P; Painer J; Kuro OM; Lanaspa M; Arnold W; Ruf T; Shiels PG; Johnson RJ, Novel treatment strategies for chronic kidney disease: insights from the animal kingdom. Nature reviews. Nephrology 2018, 14 (4), 265–284. [DOI] [PubMed] [Google Scholar]
- 7.Callier V, Core Concept: Solving Peto’s Paradox to better understand cancer. Proceedings of the National Academy of Sciences of the United States of America 2019, 116 (6), 1825–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vazquez JM; Sulak M; Chigurupati S; Lynch VJ, A Zombie LIF Gene in Elephants Is Upregulated by TP53 to Induce Apoptosis in Response to DNA Damage. Cell reports 2018, 24 (7), 1765–1776. [DOI] [PubMed] [Google Scholar]
- 9.Seluanov A; Gladyshev VN; Vijg J; Gorbunova V, Mechanisms of cancer resistance in long-lived mammals. Nature reviews. Cancer 2018, 18 (7), 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C; Zhao H; Liu Z; McMahon C, Deer antler--a novel model for studying organ regeneration in mammals. The international journal of biochemistry & cell biology 2014, 56, 111–22. [DOI] [PubMed] [Google Scholar]
- 11.Dong Z; Haines S; Coates D, Proteomic Profiling of Stem Cell Tissues during Regeneration of Deer Antler: A Model of Mammalian Organ Regeneration. Journal of Proteome Research 2020, 19 (4), 1760–1775. [DOI] [PubMed] [Google Scholar]
- 12.Zapol WM; Liggins GC; Schneider RC; Qvist J; Snider MT; Creasy RK; Hochachka PW, Regional blood flow during simulated diving in the conscious Weddell seal. Journal of applied physiology: respiratory, environmental and exercise physiology 1979, 47 (5), 968–73. [DOI] [PubMed] [Google Scholar]
- 13.Halasz NA; Elsner R; Garvie RS; Grotke GT, Renal recovery from ischemia: a comparative study of harbor seal and dog kidneys. The American journal of physiology 1974, 227 (6), 1331–5. [DOI] [PubMed] [Google Scholar]
- 14.Vazquez-Medina JP; Zenteno-Savin T; Elsner R; Ortiz RM, Coping with physiological oxidative stress: a review of antioxidant strategies in seals. Journal of comparative physiology. B, Biochemical, systemic, and environmental physiology 2012, 182 (6), 741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elsner R; Oyasaeter S; Almaas R; Saugstad OD, Diving seals, ischemia-reperfusion and oxygen radicals. Comparative biochemistry and physiology. Part A, Molecular & integrative physiology 1998, 119 (4), 975–80. [DOI] [PubMed] [Google Scholar]
- 16.Allen KN; Vazquez-Medina JP, Natural Tolerance to Ischemia and Hypoxemia in Diving Mammals: A Review. Frontiers in physiology 2019, 10, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drew KL; Rice ME; Kuhn TB; Smith MA, Neuroprotective adaptations in hibernation: therapeutic implications for ischemia-reperfusion, traumatic brain injury and neurodegenerative diseases. Free radical biology & medicine 2001, 31 (5), 563–73. [DOI] [PubMed] [Google Scholar]
- 18.Grabek KR; Cooke TF; Epperson LE; Spees KK; Cabral GF; Sutton SC; Merriman DK; Martin SL; Bustamante CD, Genetic variation drives seasonal onset of hibernation in the 13-lined ground squirrel. Communications biology 2019, 2, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jansen HT; Trojahn S; Saxton MW; Quackenbush CR; Evans Hutzenbiler BD; Nelson OL; Cornejo OE; Robbins CT; Kelley JL, Hibernation induces widespread transcriptional remodeling in metabolic tissues of the grizzly bear. Communications biology 2019, 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Austad SN, Methusaleh’s Zoo: how nature provides us with clues for extending human health span. Journal of comparative pathology 2010, 142 Suppl 1, S10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson GS; Adams DM, Recurrent evolution of extreme longevity in bats. Biology letters 2019, 15 (4), 20180860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Podlutsky AJ; Khritankov AM; Ovodov ND; Austad SN, A new field record for bat longevity. The journals of gerontology. Series A, Biological sciences and medical sciences 2005, 60 (11), 1366–8. [DOI] [PubMed] [Google Scholar]
- 23.Johnson AA; Shokhirev MN; Shoshitaishvili B, Revamping the evolutionary theories of aging. Ageing research reviews 2019, 55, 100947. [DOI] [PubMed] [Google Scholar]
- 24.Bernard C; Compagnoni A; Salguero-Gómez R, Testing Finch’s hypothesis: The role of organismal modularity on the escape from actuarial senescence. Functional Ecology 2020, 34 (1), 88–106. [Google Scholar]
- 25.Wang L; Cui J; Jin B; Zhao J; Xu H; Lu Z; Li W; Li X; Li L; Liang E; Rao X; Wang S; Fu C; Cao F; Dixon RA; Lin J, Multifeature analyses of vascular cambial cells reveal longevity mechanisms in old Ginkgo biloba trees. Proceedings of the National Academy of Sciences of the United States of America 2020, 117 (4), 2201–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chin EL; Ramsey JS; Mishchuk DO; Saha S; Foster E; Chavez JD; Howe K; Zhong X; Polek M; Godfrey KE; Mueller LA; Bruce JE; Heck M; Slupsky CM, Longitudinal Transcriptomic, Proteomic, and Metabolomic Analyses of Citrus sinensis (L.) Osbeck Graft-Inoculated with “Candidatus Liberibacter asiaticus”. J Proteome Res 2020, 19 (2), 719–732. [DOI] [PubMed] [Google Scholar]
- 27.Ramsey JS; Chin EL; Chavez JD; Saha S; Mischuk D; Mahoney J; Mohr J; Robison FM; Mitrovic E; Xu Y; Strickler SR; Fernandez N; Zhong X; Polek M; Godfrey KE; Giovannoni JJ; Mueller LA; Slupsky CM; Bruce JE; Heck M, Longitudinal Transcriptomic, Proteomic, and Metabolomic Analysis of Citrus limon Response to Graft Inoculation by Candidatus Liberibacter asiaticus. J Proteome Res 2020, 19 (6), 2247–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruse A; Fleites LA; Heck M, Lessons from One Fastidious Bacterium to Another: What Can We Learn about Liberibacter Species from Xylella fastidiosa. Insects 2019, 10 (9), 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeBlasio SL; Chavez JD; Alexander MM; Ramsey J; Eng JK; Mahoney J; Gray SM; Bruce JE; Cilia M, Visualization of Host-Polerovirus Interaction Topologies Using Protein Interaction Reporter Technology. J Virol 2015, 90 (4), 1973–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBlasio SL; Johnson R; Mahoney J; Karasev A; Gray S; Cilia M, Insights into the polerovirus-plant interactome revealed by co-immunoprecipitation and mass spectrometry. Molecular Plant-Microbe Interactions 2015, 28 (4), 467–481. [DOI] [PubMed] [Google Scholar]
- 31.DeBlasio SL; Johnson R; Sweeney MM; Karasev A; Gray SM; MacCoss MJ; Cilia M, Potato leafroll virus structural proteins manipulate overlapping, yet distinct protein interaction networks during infection. Proteomics 2015, 15 (12), 2098–112. [DOI] [PubMed] [Google Scholar]
- 32.DeBlasio SL; Johnson RS; MacCoss MJ; Gray SM; Cilia M, Model System-Guided Protein Interaction Mapping for Virus Isolated from Phloem Tissue. J Proteome Res 2016, 15 (12), 4601–4611. [DOI] [PubMed] [Google Scholar]
- 33.DeBlasio SL; Xu Y; Johnson RS; Rebelo AR; MacCoss MJ; Gray SM; Heck M, The Interaction Dynamics of Two Potato Leafroll Virus Movement Proteins Affects Their Localization to the Outer Membranes of Mitochondria and Plastids. Viruses 2018, 10 (11), 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.IBISWorld Zoos & Aquariums in the US industry statistics. https://www.ibisworld.com/united-states/market-research-reports/zoos-aquariums-industry/ (accessed 27 March).
- 35.APPA Pet Industry Market Size & Ownership Statistics. https://www.americanpetproducts.org/press_industrytrends.asp (accessed 27 March).
- 36.Jebb D; Huang Z; Pippel M; Hughes GM; Lavrichenko K; Devanna P; Winkler S; Jermiin LS; Skirmuntt EC; Katzourakis A; Burkitt-Gray L; Ray DA; Sullivan KAM; Roscito JG; Kirilenko BM; Dávalos LM; Corthals AP; Power ML; Jones G; Ransome RD; Dechmann D; Locatelli AG; Puechmaille SJ; Fedrigo O; Jarvis ED; Springer MS; Hiller M; Vernes SC; Myers EW; Teeling EC, Six new reference-quality bat genomes illuminate the molecular basis and evolution of bat adaptations. bioRxiv 2019, 836874. [Google Scholar]
- 37.Pavlovich SS; Lovett SP; Koroleva G; Guito JC; Arnold CE; Nagle ER; Kulcsar K; Lee A; Thibaud-Nissen F; Hume AJ; Muhlberger E; Uebelhoer LS; Towner JS; Rabadan R; Sanchez-Lockhart M; Kepler TB; Palacios G, The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell 2018, 173 (5), 1098–1110.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Z; Whelan CV; Foley NM; Jebb D; Touzalin F; Petit EJ; Puechmaille SJ; Teeling EC, Longitudinal comparative transcriptomics reveals unique mechanisms underlying extended healthspan in bats. Nature ecology & evolution 2019, 3 (7), 1110–1120. [DOI] [PubMed] [Google Scholar]
- 39.Valcu C-M; Kempenaers B, Proteomics in behavioral ecology. Behavioral Ecology 2014, 26 (1), 1–15. [Google Scholar]
- 40.Mulugeta E; Marion-Poll L; Gentien D; Ganswindt SB; Ganswindt A; Bennett NC; Blackburn EH; Faulkes CG; Heard E, Molecular insights into the pathways underlying naked mole-rat eusociality. bioRxiv 2017, 209932. [Google Scholar]
- 41.Paredi G; Sentandreu MA; Mozzarelli A; Fadda S; Hollung K; de Almeida AM, Muscle and meat: new horizons and applications for proteomics on a farm to fork perspective. Journal of proteomics 2013, 88, 58–82. [DOI] [PubMed] [Google Scholar]
- 42.Ortea I; O’Connor G; Maquet A, Review on proteomics for food authentication. Journal of proteomics 2016, 147, 212–225. [DOI] [PubMed] [Google Scholar]
- 43.Piras C; Roncada P; Rodrigues PM; Bonizzi L; Soggiu A, Proteomics in food: Quality, safety, microbes, and allergens. Proteomics 2016, 16 (5), 799–815. [DOI] [PubMed] [Google Scholar]
- 44.Wulff T; Nielsen ME; Deelder AM; Jessen F; Palmblad M, Authentication of fish products by large-scale comparison of tandem mass spectra. J Proteome Res 2013, 12 (11), 5253–9. [DOI] [PubMed] [Google Scholar]
- 45.Righetti PG; Fasoli E; D’Amato A; Boschetti E, Chapter 6 - Making Progress in Plant Proteomics for Improved Food Safety In Comprehensive Analytical Chemistry, García-Cañas V; Cifuentes A; Simó C, Eds. Elsevier: 2014; Vol. 64, pp 131–155. [Google Scholar]
- 46.Becker DJ; Albery GF; Sjodin AR; Poisot T; Dallas TA; Eskew EA; Farrell MJ; Guth S; Han BA; Simmons NB; Carlson CJ, Predicting wildlife hosts of betacoronaviruses for SARS-CoV-2 sampling prioritization. bioRxiv 2020, 2020.05.22.111344. [Google Scholar]
- 47.Letko M; Seifert SN; Olival KJ; Plowright RK; Munster VJ, Bat-borne virus diversity, spillover and emergence. Nature reviews. Microbiology 2020, 18, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li F, Receptor recognition and cross-species infections of SARS coronavirus. Antiviral research 2013, 100 (1), 246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li W; Zhang C; Sui J; Kuhn JH; Moore MJ; Luo S; Wong SK; Huang IC; Xu K; Vasilieva N; Murakami A; He Y; Marasco WA; Guan Y; Choe H; Farzan M, Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. The EMBO journal 2005, 24 (8), 1634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao X; Chen D; Szabla R; Zheng M; Li G; Du P; Zheng S; Li X; Song C; Li R; Guo J-T; Junop M; Zeng H; Lin H, Broad and differential animal ACE2 receptor usage by SARS-CoV-2. bioRxiv 2020, 2020.04.19.048710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lan J; Ge J; Yu J; Shan S; Zhou H; Fan S; Zhang Q; Shi X; Wang Q; Zhang L; Wang X, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581 (7807), 215–220. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe Y; Berndsen ZT; Raghwani J; Seabright GE; Allen JD; Pybus OG; McLellan JS; Wilson IA; Bowden TA; Ward AB; Crispin M, Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nature communications 2020, 11 (1), 2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pach S; Nguyen TN; Trimpert J; Kunec D; Osterrieder N; Wolber G, ACE2-Variants Indicate Potential SARS-CoV-2-Susceptibility in Animals: An Extensive Molecular Dynamics Study. bioRxiv 2020, 2020.05.14.092767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Damas J; Hughes GM; Keough KC; Painter CA; Persky NS; Corbo M; Hiller M; Koepfli K-P; Pfenning AR; Zhao H; Genereux DP; Swofford R; Pollard KS; Ryder OA; Nweeia MT; Lindblad-Toh K; Teeling EC; Karlsson EK; Lewin HA, Broad Host Range of SARS-CoV-2 Predicted by Comparative and Structural Analysis of ACE2 in Vertebrates. bioRxiv 2020, 2020.04.16.045302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Batra J; Hultquist JF; Liu D; Shtanko O; Von Dollen J; Satkamp L; Jang GM; Luthra P; Schwarz TM; Small GI; Arnett E; Anantpadma M; Reyes A; Leung DW; Kaake R; Haas P; Schmidt CB; Schlesinger LS; LaCount DJ; Davey RA; Amarasinghe GK; Basler CF; Krogan NJ, Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175 (7), 1917–1930.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah PS; Link N; Jang GM; Sharp PP; Zhu T; Swaney DL; Johnson JR; Von Dollen J; Ramage HR; Satkamp L; Newton B; Hüttenhain R; Petit MJ; Baum T; Everitt A; Laufman O; Tassetto M; Shales M; Stevenson E; Iglesias GN; Shokat L; Tripathi S; Balasubramaniam V; Webb LG; Aguirre S; Willsey AJ; Garcia-Sastre A; Pollard KS; Cherry S; Gamarnik AV; Marazzi I; Taunton J; Fernandez-Sesma A; Bellen HJ; Andino R; Krogan NJ, Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175 (7), 1931–1945.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gordon DE; Jang GM; Bouhaddou M; Xu J; Obernier K; White KM; O’Meara MJ; Rezelj VV; Guo JZ; Swaney DL; Tummino TA; Huettenhain R; Kaake RM; Richards AL; Tutuncuoglu B; Foussard H; Batra J; Haas K; Modak M; Kim M; Haas P; Polacco BJ; Braberg H; Fabius JM; Eckhardt M; Soucheray M; Bennett MJ; Cakir M; McGregor MJ; Li Q; Meyer B; Roesch F; Vallet T; Mac Kain A; Miorin L; Moreno E; Naing ZZC; Zhou Y; Peng S; Shi Y; Zhang Z; Shen W; Kirby IT; Melnyk JE; Chorba JS; Lou K; Dai SA; Barrio-Hernandez I; Memon D; Hernandez-Armenta C; Lyu J; Mathy CJP; Perica T; Pilla KB; Ganesan SJ; Saltzberg DJ; Rakesh R; Liu X; Rosenthal SB; Calviello L; Venkataramanan S; Liboy-Lugo J; Lin Y; Huang XP; Liu Y; Wankowicz SA; Bohn M; Safari M; Ugur FS; Koh C; Savar NS; Tran QD; Shengjuler D; Fletcher SJ; O’Neal MC; Cai Y; Chang JCJ; Broadhurst DJ; Klippsten S; Sharp PP; Wenzell NA; Kuzuoglu D; Wang HY; Trenker R; Young JM; Cavero DA; Hiatt J; Roth TL; Rathore U; Subramanian A; Noack J; Hubert M; Stroud RM; Frankel AD; Rosenberg OS; Verba KA; Agard DA; Ott M; Emerman M; Jura N; von Zastrow M; Verdin E; Ashworth A; Schwartz O; d’Enfert C; Mukherjee S; Jacobson M; Malik HS; Fujimori DG; Ideker T; Craik CS; Floor SN; Fraser JS; Gross JD; Sali A; Roth BL; Ruggero D; Taunton J; Kortemme T; Beltrao P; Vignuzzi M; García-Sastre A; Shokat KM; Shoichet BK; Krogan NJ, A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muntel J; Gandhi T; Verbeke L; Bernhardt OM; Treiber T; Bruderer R; Reiter L, Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Molecular omics 2019, 15 (5), 348–360. [DOI] [PubMed] [Google Scholar]
- 59.Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sorensen KD; Hoyer S; Orntoft TF; Andersen CL; Nielsen ML; Olsen JV, An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell systems 2017, 4 (6), 587–599.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richards AL; Hebert AS; Ulbrich A; Bailey DJ; Coughlin EE; Westphall MS; Coon JJ, One-hour proteome analysis in yeast. Nature protocols 2015, 10 (5), 701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meier F; Geyer PE; Virreira Winter S; Cox J; Mann M, BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nature methods 2018, 15 (6), 440–448. [DOI] [PubMed] [Google Scholar]
- 62.Cilia M; Tamborindeguy C; Rolland M; Howe K; Thannhauser TW; Gray S, Tangible benefits of the aphid Acyrthosiphon pisum genome sequencing for aphid proteomics: Enhancements in protein identification and data validation for homology-based proteomics. J Insect Physiol 2011, 57 (1), 179–90. [DOI] [PubMed] [Google Scholar]
- 63.Johnson RS; Searle BC; Nunn BL; Gilmore JM; Phillips M; Amemiya CT; Heck M; MacCoss MJ, Assessing Protein Sequence Database Suitability Using De Novo Sequencing. Molecular & cellular proteomics: MCP 2020, 19 (1), 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wetterstrand K DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). www.genome.gov/sequencingcostsdata (accessed 10 March).
- 65.Vaughan TA; Ryan JM; Czaplewski NJ, Mammalogy. 6th ed.; Jones & Bartlett Learning: U.S.A., 2013; p 756. [Google Scholar]
- 66.Neely BA; Ellisor DL; Davis WC, Proteomics as a metrological tool to evaluate genome annotation accuracy following de novo genome assembly: a case study using the Atlantic bottlenose dolphin (Tursiops truncatus). bioRxiv 2018, 254250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simão FA; Waterhouse RM; Ioannidis P; Kriventseva EV; Zdobnov EM, BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics (Oxford, England) 2015, 31 (19), 3210–2. [DOI] [PubMed] [Google Scholar]
- 68.Volkening JD; Bailey DJ; Rose CM; Grimsrud PA; Howes-Podoll M; Venkateshwaran M; Westphall MS; Ane JM; Coon JJ; Sussman MR, A proteogenomic survey of the Medicago truncatula genome. Mol Cell Proteomics 2012, 11 (10), 933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carolan JC; Caragea D; Reardon KT; Mutti NS; Dittmer N; Pappan K; Cui F; Castaneto M; Poulain J; Dossat C; Tagu D; Reese JC; Reeck GR; Wilkinson TL; Edwards OR, Predicted effector molecules in the salivary secretome of the pea aphid (Acyrthosiphon pisum): a dual transcriptomic/proteomic approach. J Proteome Res 2011, 10 (4), 1505–18. [DOI] [PubMed] [Google Scholar]
- 70.Campbell MS; Holt C; Moore B; Yandell M, Genome Annotation and Curation Using MAKER and MAKER-P. Current protocols in bioinformatics 2014, 48, 4.11.1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MAKER 2.31.9 with CCTOOLS Jetstream Tutorial. https://cyverse.atlassian.net/wiki/spaces/TUT/pages/258736333/MAKER+2.31.9+with+CCTOOLS+Jetstream+Tutorial (accessed 14 August).
- 72.Thibaud-Nissen F; Souvorov A; Murphy T; DiCuccio M; Kitts P, Eukaryotic Genome Annotation Pipeline. 2nd ed.; National Center for Biotechnology Information (US): Bethesda (MD), 2013. https://www.ncbi.nlm.nih.gov/books/NBK169439/ (accessed 27 March 2020). [Google Scholar]
- 73.O’Leary NA; Wright MW; Brister JR; Ciufo S; Haddad D; McVeigh R; Rajput B; Robbertse B; Smith-White B; Ako-Adjei D; Astashyn A; Badretdin A; Bao Y; Blinkova O; Brover V; Chetvernin V; Choi J; Cox E; Ermolaeva O; Farrell CM; Goldfarb T; Gupta T; Haft D; Hatcher E; Hlavina W; Joardar VS; Kodali VK; Li W; Maglott D; Masterson P; McGarvey KM; Murphy MR; O’Neill K; Pujar S; Rangwala SH; Rausch D; Riddick LD; Schoch C; Shkeda A; Storz SS; Sun H; Thibaud-Nissen F; Tolstoy I; Tully RE; Vatsan AR; Wallin C; Webb D; Wu W; Landrum MJ; Kimchi A; Tatusova T; DiCuccio M; Kitts P; Murphy TD; Pruitt KD, Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic acids research 2016, 44 (D1), D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.NCBI Eukaryotic Genome Annotation Policy On Which Genomes Are Annotated https://www.ncbi.nlm.nih.gov/genome/annotation_euk/policy/ (accessed 27 March).
- 75.NCBI Zalophus californianus Annotation Release 100. https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Zalophus_californianus/100/.
- 76.Hedges SB; Marin J; Suleski M; Paymer M; Kumar S, Tree of life reveals clock-like speciation and diversification. Molecular biology and evolution 2015, 32 (4), 835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hosseinzadeh S; Ramsey J; Mann M; Bennett L; Hunter WB; Shams-Bakhsh M; Hall DG; Heck M, Color morphology of Diaphorina citri influences interactions with its bacterial endosymbionts and ‘Candidatus Liberibacter asiaticus’. PLoS One 2019, 14 (5), e0216599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.MacDougall A; Volynkin V; Saidi R; Poggioli D; Zellner H; Hatton-Ellis E; Joshi V; O’Donovan C; Orchard S; Auchincloss AH; Baratin D; Bolleman J; Coudert E; de Castro E; Hulo C; Masson P; Pedruzzi I; Rivoire C; Arighi C; Wang Q; Chen C; Huang H; Garavelli J; Vinayaka CR; Yeh LS; Natale DA; Laiho K; Martin M, UniRule: a unified rule resource for automatic annotation in the UniProt Knowledgebase. Bioinformatics (Oxford, England) 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen W; Hasegawa DK; Kaur N; Kliot A; Pinheiro PV; Luan J; Stensmyr MC; Zheng Y; Liu W; Sun H; Xu Y; Luo Y; Kruse A; Yang X; Kontsedalov S; Lebedev G; Fisher TW; Nelson DR; Hunter WB; Brown JK; Jander G; Cilia M; Douglas AE; Ghanim M; Simmons AM; Wintermantel WM; Ling KS; Fei Z, The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol 2016, 14 (1), 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.International Aphid Genomics C, Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol 2010, 8 (2), e1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saha S; Hosmani PS; Villalobos-Ayala K; Miller S; Shippy T; Flores M; Rosendale A; Cordola C; Bell T; Mann H; DeAvila G; DeAvila D; Moore Z; Buller K; Ciolkevich K; Nandyal S; Mahoney R; Van Voorhis J; Dunlevy M; Farrow D; Hunter D; Morgan T; Shore K; Guzman V; Izsak A; Dixon DE; Cridge A; Cano L; Cao X; Jiang H; Leng N; Johnson S; Cantarel BL; Richards S; English A; Shatters RG; Childers C; Chen MJ; Hunter W; Cilia M; Mueller LA; Munoz-Torres M; Nelson D; Poelchau MF; Benoit JB; Wiersma-Koch H; D’Elia T; Brown SJ, Improved annotation of the insect vector of citrus greening disease: biocuration by a diverse genomics community. Database (Oxford) 2017, 2017, baz035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carolan JC; Fitzroy CI; Ashton PD; Douglas AE; Wilkinson TL, The secreted salivary proteome of the pea aphid Acyrthosiphon pisum characterised by mass spectrometry. Proteomics 2009, 9 (9), 2457–67. [DOI] [PubMed] [Google Scholar]
- 83.Bos JI; Prince D; Pitino M; Maffei ME; Win J; Hogenhout SA, A functional genomics approach identifies candidate effectors from the aphid species Myzus persicae (green peach aphid). PLoS Genet 2010, 6 (11), e1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mathers TC; Chen Y; Kaithakottil G; Legeai F; Mugford ST; Baa-Puyoulet P; Bretaudeau A; Clavijo B; Colella S; Collin O; Dalmay T; Derrien T; Feng H; Gabaldon T; Jordan A; Julca I; Kettles GJ; Kowitwanich K; Lavenier D; Lenzi P; Lopez-Gomollon S; Loska D; Mapleson D; Maumus F; Moxon S; Price DR; Sugio A; van Munster M; Uzest M; Waite D; Jander G; Tagu D; Wilson AC; van Oosterhout C; Swarbreck D; Hogenhout SA, Rapid transcriptional plasticity of duplicated gene clusters enables a clonally reproducing aphid to colonise diverse plant species. Genome Biol 2017, 18 (1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kruse A; Ramsey JS; Johnson R; Hall DG; MacCoss MJ; Heck M, Candidatus Liberibacter asiaticus Minimally Alters Expression of Immunity and Metabolism Proteins in Hemolymph of Diaphorina citri, the Insect Vector of Huanglongbing. J Proteome Res 2018, 17 (9), 2995–3011. [DOI] [PubMed] [Google Scholar]
- 86.Guo Q; Li D; Zhai Y; Gu Z, CCPRD: A Novel Analytical Framework for the Comprehensive Proteomic Reference Database Construction of NonModel Organisms. ACS Omega 2020, 5 (25), 15370–15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Uhlén M; Karlsson MJ; Hober A; Svensson AS; Scheffel J; Kotol D; Zhong W; Tebani A; Strandberg L; Edfors F; Sjöstedt E; Mulder J; Mardinoglu A; Berling A; Ekblad S; Dannemeyer M; Kanje S; Rockberg J; Lundqvist M; Malm M; Volk AL; Nilsson P; Månberg A; Dodig-Crnkovic T; Pin E; Zwahlen M; Oksvold P; von Feilitzen K; Häussler RS; Hong MG; Lindskog C; Ponten F; Katona B; Vuu J; Lindström E; Nielsen J; Robinson J; Ayoglu B; Mahdessian D; Sullivan D; Thul P; Danielsson F; Stadler C; Lundberg E; Bergström G; Gummesson A; Voldborg BG; Tegel H; Hober S; Forsström B; Schwenk JM; Fagerberg L; Sivertsson Å, The human secretome. Science signaling 2019, 12 (609). [DOI] [PubMed] [Google Scholar]
- 88.Amarasinghe SL; Su S; Dong X; Zappia L; Ritchie ME; Gouil Q, Opportunities and challenges in long-read sequencing data analysis. Genome biology 2020, 21 (1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Neely BA; Prager KC; Bland AM; Fontaine C; Gulland FM; Janech MG, Proteomic Analysis of Urine from California Sea Lions (Zalophus californianus): A Resource for Urinary Biomarker Discovery. J Proteome Res 2018, 17 (9), 3281–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI, MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nature methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bittremieux W; Laukens K; Noble WS, Extremely Fast and Accurate Open Modification Spectral Library Searching of High-Resolution Mass Spectra Using Feature Hashing and Graphics Processing Units. J Proteome Res 2019, 18 (10), 3792–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Burke MC; Mirokhin YA; Tchekhovskoi DV; Markey SP; Heidbrink Thompson J; Larkin C; Stein SE, The Hybrid Search: A Mass Spectral Library Search Method for Discovery of Modifications in Proteomics. J Proteome Res 2017, 16 (5), 1924–1935. [DOI] [PubMed] [Google Scholar]
- 93.Devabhaktuni A; Lin S; Zhang L; Swaminathan K; Gonzalez CG; Olsson N; Pearlman SM; Rawson K; Elias JE, TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nature biotechnology 2019, 37 (4), 469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee J-Y; Mitchell HD; Burnet MC; Jenson SC; Merkley ED; Shukla AK; Nakayasu ES; Payne SH, Proteomics of natural bacterial isolates powered by deep learning-based de novo identification. bioRxiv 2018, 428334. [Google Scholar]
- 95.Ma B; Zhang K; Hendrie C; Liang C; Li M; Doherty-Kirby A; Lajoie G, PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid communications in mass spectrometry: RCM 2003, 17 (20), 2337–42. [DOI] [PubMed] [Google Scholar]
- 96.Muth T; Weilnbock L; Rapp E; Huber CG; Martens L; Vaudel M; Barsnes H, DeNovoGUI: an open source graphical user interface for de novo sequencing of tandem mass spectra. J Proteome Res 2014, 13 (2), 1143–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kafader JO; Melani RD; Durbin KR; Ikwuagwu B; Early BP; Fellers RT; Beu SC; Zabrouskov V; Makarov AA; Maze JT; Shinholt DL; Yip PF; Tullman-Ercek D; Senko MW; Compton PD; Kelleher NL, Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nature methods 2020, 17, 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rose RJ; Damoc E; Denisov E; Makarov A; Heck AJ, High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nature methods 2012, 9 (11), 1084–6. [DOI] [PubMed] [Google Scholar]
- 99.Snijder J; Schuller JM; Wiegard A; Lossl P; Schmelling N; Axmann IM; Plitzko JM; Forster F; Heck AJ, Structures of the cyanobacterial circadian oscillator frozen in a fully assembled state. Science (New York, N.Y.) 2017, 355 (6330), 1181–1184. [DOI] [PubMed] [Google Scholar]
- 100.Wang Z; Li Y; Hou B; Pronobis MI; Wang M; Wang Y; Cheng G; Weng W; Wang Y; Tang Y; Xu X; Pan R; Lin F; Wang N; Chen Z; Wang S; Ma LZ; Li Y; Huang D; Jiang L; Wang Z; Zeng W; Zhang Y; Du X; Lin Y; Li Z; Xia Q; Geng J; Dai H; Yu Y; Zhao XD; Yuan Z; Yan J; Nie Q; Zhang X; Wang K; Chen F; Zhang Q; Zhu Y; Zheng S; Poss KD; Tao SC; Meng X, An array of 60,000 antibodies for proteome-scale antibody generation and target discovery. Sci Adv 2020, 6 (11), eaax2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Loffredo FS; Steinhauser ML; Jay SM; Gannon J; Pancoast JR; Yalamanchi P; Sinha M; Dall’Osso C; Khong D; Shadrach JL; Miller CM; Singer BS; Stewart A; Psychogios N; Gerszten RE; Hartigan AJ; Kim MJ; Serwold T; Wagers AJ; Lee RT, Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013, 153 (4), 828–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Löfgren M; Svala E; Lindahl A; Skiöldebrand E; Ekman S, Time-dependent changes in gene expression induced in vitro by interleukin-1β in equine articular cartilage. Research in veterinary science 2018, 118, 466–476. [DOI] [PubMed] [Google Scholar]
- 103.Neely B; Carlin K; Arthur J; McFee W; Janech M, Ratiometric Measurements of Adiponectin by Mass Spectrometry in Bottlenose Dolphins (Tursiops truncatus) with Iron Overload Reveal an Association with Insulin Resistance and Glucagon. Frontiers in endocrinology 2013, 4 (132), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pinheiro PV; Ghanim M; Alexander M; Rebelo AR; Santos RS; Orsburn BC; Gray S; Cilia M, Host Plants Indirectly Influence Plant Virus Transmission by Altering Gut Cysteine Protease Activity of Aphid Vectors. Molecular & cellular proteomics: MCP 2017, 16 (4 suppl 1), S230–s243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilmarth P PAW_BLAST. https://github.com/pwilmart/PAW_BLAST.
- 106.Naithani S; Preece J; D’Eustachio P; Gupta P; Amarasinghe V; Dharmawardhana PD; Wu G; Fabregat A; Elser JL; Weiser J; Keays M; Fuentes AM; Petryszak R; Stein LD; Ware D; Jaiswal P, Plant Reactome: a resource for plant pathways and comparative analysis. Nucleic acids research 2017, 45 (D1), D1029–d1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hansen BO; Olsson S, OmicsDB::Pathogens - A database for exploring functional networks of plant pathogens. bioRxiv 2020, 2020.03.18.979971. [Google Scholar]
- 108.Pfister N; Williams EG; Peters J; Aebersold R; Bühlmann P, Stabilizing Variable Selection and Regression. arXiv preprint arXiv:1911.01850 2019. [Google Scholar]
- 109.Gygi SP; Rochon Y; Franza BR; Aebersold R, Correlation between protein and mRNA abundance in yeast. Molecular and cellular biology 1999, 19 (3), 1720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Foss EJ; Radulovic D; Shaffer SA; Ruderfer DM; Bedalov A; Goodlett DR; Kruglyak L, Genetic basis of proteome variation in yeast. Nature genetics 2007, 39 (11), 1369–75. [DOI] [PubMed] [Google Scholar]
- 111.Edfors F; Danielsson F; Hallstrom BM; Kall L; Lundberg E; Ponten F; Forsstrom B; Uhlen M, Gene-specific correlation of RNA and protein levels in human cells and tissues. Molecular systems biology 2016, 12 (10), 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu Y; Beyer A; Aebersold R, On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165 (3), 535–50. [DOI] [PubMed] [Google Scholar]
- 113.Nusinow DP; Szpyt J; Ghandi M; Rose CM; McDonald ER 3rd; Kalocsay M; Jane-Valbuena J; Gelfand E; Schweppe DK; Jedrychowski M; Golji J; Porter DA; Rejtar T; Wang YK; Kryukov GV; Stegmeier F; Erickson BK; Garraway LA; Sellers WR; Gygi SP, Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180 (2), 387–402.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salovska B; Zhu H; Gandhi T; Frank M; Li W; Rosenberger G; Wu C; Germain PL; Zhou H; Hodny Z; Reiter L; Liu Y, Isoform-resolved correlation analysis between mRNA abundance regulation and protein level degradation. Molecular systems biology 2020, 16 (3), e9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buccitelli C; Selbach M, mRNAs, proteins and the emerging principles of gene expression control. Nature reviews. Genetics 2020, Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 116.Lehallier B; Gate D; Schaum N; Nanasi T; Lee SE; Yousef H; Moran Losada P; Berdnik D; Keller A; Verghese J; Sathyan S; Franceschi C; Milman S; Barzilai N; Wyss-Coray T, Undulating changes in human plasma proteome profiles across the lifespan. Nature medicine 2019, 25 (12), 1843–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kelmer Sacramento E; Kirkpatrick JM; Mazzetto M; Baumgart M; Bartolome A; Di Sanzo S; Caterino C; Sanguanini M; Papaevgeniou N; Lefaki M; Childs D; Bagnoli S; Terzibasi Tozzini E; Di Fraia D; Romanov N; Sudmant PH; Huber W; Chondrogianni N; Vendruscolo M; Cellerino A; Ori A, Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Molecular systems biology 2020, 16 (6), e9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boschiero C; Dai X; Lundquist PK; Roy S; de Bang T; Zhang S; Zhuang Z; Torres-Jerez I; Udvardi MK; Scheible WR; Zhao PX, MtSSPdb: the Medicago truncatula Small Secreted Peptide Database. Plant Physiol 2020, 183 (1), 399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fleites LA; Johnson R; Kruse AR; Nachman RJ; Hall DG; MacCoss M; Heck ML, Peptidomics Approaches for the Identification of Bioactive Molecules from Diaphorina citri. J Proteome Res 2020, 19 (4), 1392–1408. [DOI] [PubMed] [Google Scholar]
- 120.Zheng Y; Sweet SM; Popovic R; Martinez-Garcia E; Tipton JD; Thomas PM; Licht JD; Kelleher NL, Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc Natl Acad Sci U S A 2012, 109 (34), 13549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smith LM; Kelleher NL; Consortium for Top Down, P., Proteoform: a single term describing protein complexity. Nat Methods 2013, 10 (3), 186–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zheng Y; Huang X; Kelleher NL, Epiproteomics: quantitative analysis of histone marks and codes by mass spectrometry. Curr Opin Chem Biol 2016, 33, 142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Malaney P; Uversky VN; Dave V, PTEN proteoforms in biology and disease. Cell Mol Life Sci 2017, 74 (15), 2783–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cristea IM; Williams R; Chait BT; Rout MP, Fluorescent proteins as proteomic probes. Mol Cell Proteomics 2005, 4 (12), 1933–41. [DOI] [PubMed] [Google Scholar]
- 125.Cristea IM; Carroll JW; Rout MP; Rice CM; Chait BT; MacDonald MR, Tracking and elucidating alphavirus-host protein interactions. J Biol Chem 2006, 281 (40), 30269–78. [DOI] [PubMed] [Google Scholar]
- 126.Moorman NJ; Sharon-Friling R; Shenk T; Cristea IM, A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol Cell Proteomics 2010, 9 (5), 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cristea IM; Chait BT Affinity purification of protein complexes Cold Spring Harb Protoc [Online], 2011. http://www.ncbi.nlm.nih.gov/pubmed/21536765. [DOI] [PMC free article] [PubMed]
- 128.Miteva YV; Budayeva HG; Cristea IM, Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal Chem 2013, 85 (2), 749–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weisbrod CR; Chavez JD; Eng JK; Yang L; Zheng C; Bruce JE, In Vivo Protein Interaction Network Identified with a Novel Real-Time Cross-Linked Peptide Identification Strategy. J Proteome Res 2013, 12 (4), 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zheng C; Weisbrod CR; Chavez JD; Eng JK; Sharma V; Wu X; Bruce JE, XLink-DB: Database and Software Tools for Storing and Visualizing Protein Interaction Topology Data. J Proteome Res 2013, 12 (4), 1989–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chavez JD; Eng JK; Schweppe DK; Cilia M; Rivera K; Zhong X; Wu X; Allen T; Khurgel M; Kumar A; Lampropoulos A; Larsson M; Maity S; Morozov Y; Pathmasiri W; Perez-Neut M; Pineyro-Ruiz C; Polina E; Post S; Rider M; Tokmina-Roszyk D; Tyson K; Vieira Parrine Sant’Ana D; Bruce JE, A General Method for Targeted Quantitative Cross-Linking Mass Spectrometry. PLoS One 2016, 11 (12), e0167547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ramsey JS; Chavez JD; Johnson R; Hosseinzadeh S; Mahoney JE; Mohr JP; Robison F; Zhong X; Hall DG; MacCoss M; Bruce J; Cilia M, Protein interaction networks at the host-microbe interface in Diaphorina citri, the insect vector of the citrus greening pathogen. R Soc Open Sci 2017, 4 (2), 160545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kuriyan J; Eisenberg D, The origin of protein interactions and allostery in colocalization. Nature 2007, 450 (7172), 983–90. [DOI] [PubMed] [Google Scholar]
- 134.Cilia M; Tamborindeguy C; Fish T; Howe K; Thannhauser TW; Gray S, Genetics coupled to quantitative intact proteomics links heritable aphid and endosymbiont protein expression to circulative polerovirus transmission. J Virol 2011, 85 (5), 2148–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mertins P; Mani DR; Ruggles KV; Gillette MA; Clauser KR; Wang P; Wang X; Qiao JW; Cao S; Petralia F; Kawaler E; Mundt F; Krug K; Tu Z; Lei JT; Gatza ML; Wilkerson M; Perou CM; Yellapantula V; Huang KL; Lin C; McLellan MD; Yan P; Davies SR; Townsend RR; Skates SJ; Wang J; Zhang B; Kinsinger CR; Mesri M; Rodriguez H; Ding L; Paulovich AG; Fenyo D; Ellis MJ; Carr SA; Nci C, Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534 (7605), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cilia M; Howe K; Fish T; Smith D; Mahoney J; Tamborindeguy C; Burd J; Thannhauser TW; Gray S, Biomarker discovery from the top down: Protein biomarkers for efficient virus transmission by insects (Homoptera: Aphididae) discovered by coupling genetics and 2-D DIGE. Proteomics 2011, 11 (12), 2440–58. [DOI] [PubMed] [Google Scholar]
- 137.Poliakov A; Russell CW; Ponnala L; Hoops HJ; Sun Q; Douglas AE; van Wijk KJ, Large-scale label-free quantitative proteomics of the pea aphid-Buchnera symbiosis. Mol Cell Proteomics 2011, 10 (6), M110 007039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ramsey JS; Johnson RS; Hoki JS; Kruse A; Mahoney J; Hilf ME; Hunter WB; Hall DG; Schroeder FC; MacCoss MJ; Cilia M, Metabolic Interplay between the Asian Citrus Psyllid and Its Profftella Symbiont: An Achilles’ Heel of the Citrus Greening Insect Vector. PLoS One 2015, 10 (11), e0140826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kruse A; Fattah-Hosseini S; Saha S; Johnson R; Warwick E; Sturgeon K; Mueller L; MacCoss MJ; Shatters RG Jr.; Cilia Heck M, Combining ‘omics and microscopy to visualize interactions between the Asian citrus psyllid vector and the Huanglongbing pathogen Candidatus Liberibacter asiaticus in the insect gut. PLoS One 2017, 12 (6), e0179531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Müller JB; Geyer PE; Colaço AR; Treit PV; Strauss MT; Oroshi M; Doll S; Virreira Winter S; Bader JM; Köhler N; Theis F; Santos A; Mann M, The proteome landscape of the kingdoms of life. Nature 2020, 582, 592–596. [DOI] [PubMed] [Google Scholar]
- 141.Janech MG; Fitzgibbon WR; Chen R; Nowak MW; Miller DH; Paul RV; Ploth DW, Molecular and functional characterization of a urea transporter from the kidney of the Atlantic stingray. American journal of physiology. Renal physiology 2003, 284 (5), F996–f1005. [DOI] [PubMed] [Google Scholar]
- 142.Elger M; Hentschel H; Litteral J; Wellner M; Kirsch T; Luft FC; Haller H, Nephrogenesis is induced by partial nephrectomy in the elasmobranch Leucoraja erinacea. Journal of the American Society of Nephrology: JASN 2003, 14 (6), 1506–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Specific NCBI taxon IDs and their designation for 23 mammal groups used in Figures 2 and 3.