

Abstract
Purpose:
Genomic and transcriptomic alterations during metastasis are considered to affect clinical outcome of colorectal cancers (CRCs), but detailed clinical implications of metastatic alterations are not fully uncovered. We aimed to investigate the effect of metastatic evolution on in vivo treatment outcome, and identify genomic and transcriptomic alterations associated with drug responsiveness.
Experimental Design:
We developed and analyzed patient-derived xenograft (PDX) models from 35 CRC patients including five patients with multiple organ metastases (MOMs). We performed whole-exome, DNA methylation and RNA sequencing for patient and PDX tumors. With samples from patients with MOMs, we conducted phylogenetic and subclonal analysis and in vivo drug efficacy test on the corresponding PDX models.
Results:
Phylogenetic analysis using mutation, expression and DNA methylation data in patients with MOMs showed that mutational alterations were closely connected with transcriptomic and epigenomic changes during the tumor evolution. Subclonal analysis revealed that initial primary tumors with larger number of subclones exhibited more dynamic changes in subclonal architecture according to metastasis, and loco-regional and distant metastases occurred in a parallel or independent fashion. The PDX models from MOMs demonstrated therapeutic heterogeneity for targeted treatment, due to subclonal acquisition of additional mutations or transcriptomic activation of bypass signaling pathway during tumor evolution.
Conclusions:
This study demonstrated in vivo therapeutic heterogeneity of CRCs using PDX models, and suggests that acquired subclonal alterations in mutations or gene expression profiles during tumor metastatic processes can be associated with the development of drug resistance and therapeutic heterogeneity of CRCs.
Keywords: colorectal cancer, metastasis, tumor evolution, patient-derived xenograft, therapeutic heterogeneity
Introduction
Tumor metastasis is a major cause of cancer-related deaths, and, to date, appropriate treatment modalities are very limited in most types of cancers (1,2). To develop the strategies for preventing or targeting tumor metastasis, understanding the nature and biology of tumor metastasis is inevitable. Genomic instable nature of cancer cells can result in genetic diversity of cells within a given tumor (intra-tumoral heterogeneity (ITH)) (3), and genetic and epigenetic alterations during metastasis process has been suggested to contribute to tumor progression, treatment resistance and survival outcome (4–7). Recent advances of high-throughput genomic analysis technology provided landscape of genomic alterations specific to metastasized tumors (8), and comparative studies between primary and matched metastasized tumors have described the evolutionary characteristics of metastatic tumors (9–11). However, detailed clinical implications of genetic and epigenetic alterations during metastasis remain limited for most cancer types.
Patient-derived xenografts (PDXs) are cancer models that are established by the transfer of patient tumor tissues into immunodeficient mice. PDXs retain several key characteristics of patients’ tumors including histology, genomic alterations and transcriptomic signatures (12–14). Previous studies have also suggested that PDX models exhibit drug responsiveness comparable to that of the primary tumors in the patients (15–17), and overcome many limitations of conventional in vitro cell line models and cell line xenograft models (13,18). Most critically, PDX tumors have the capacity to retain the ITH of the original patient tumors and therefore can be applied to the study of clonal selection and tumor evolution (13,19).
Colorectal cancer (CRC) is the third most prevalent cancer and the fourth common cause of cancer-related death worldwide (20), and a cancer type reported to exhibit significant ITH (21,22). In this study, we examined the clonal dynamics and tumor evolution of CRC during metastasis by analyzing the genome, transcriptome and epigenome of patient and PDX tumors from CRC patients with multiple organ metastases (MOMs), and experimentally demonstrated that acquired genomic and transcriptomic heterogeneities during tumor evolution can be associated with the heterogeneity of drug responsiveness.
Materials and Methods
CRC patient sample collection and generation of PDX models
CRC tissue samples from primary and metastasis sites and blood samples were obtained from individuals who underwent colectomies at Gil Medical Center from 2014 to 2015. All samples were obtained with informed consent at the Gil Medical Center, and the study was approved by the institutional review board in accordance with the Declaration of Helsinki. For PDX models, surgically resected tissues were minced into pieces approximately ~2 mm in size and injected subcutaneously in the flanks of 6-week-old NOD/SCID/IL-2γ-receptor null (NSG) female mice (The Jackson laboratory). When a tumor volume reached > 700~1000 mm3, the mouse was sacrificed and tumor tissues were extracted and cryopreserved in liquid nitrogen and stored at −80 °C for generating future PDX passages. Mice were cared for according to institutional guidelines of the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (No. 14–0016-C0A0).
Exome sequencing processing and variant calling
DNA reads were aligned to the merged references for human GRCh19 and mouse mm10 genome versions using BWA-MEM (23). Sorting and marking duplicate reads were performed using Picard tools. Additional processing was conducted to remove mouse sequences using an in-house bioinformatics pipeline, followed by the IndelRealignment, BaseQualityScoreRecalibration, PrintReads commands as Best Practices recommendations of the Genome Analysis Tool Kit (GATK) (24).
After processing the BAM files, Mutect (25) and IndelGenotyper were employed for somatic mutation calling, followed by ANNOVAR for functional annotations. Variants with at least 8 read depths, or 4 alternate allele depths and a minimum genotype quality of 20 were maintained. For somatic indels, a strand bias Phred-scale P-value of greater than 20, by Fisher’s exact test, was discarded. Common variants found in dbSNP142 were also removed. Finally, exonic and splicing variants were selected based on the RefSeq database and having population frequencies lower than 0.01, based on The Exome Aggregation Consortium, 1000 Genomes Project and NHLBI ESP6500.
The pairwise distance of SNPs were calculated based on 4,784 rare non-silent somatic mutations in tumor tissues from the patient #21. The maximum composite likelihood substitution model, which is an accurate alignment-free estimator of the number of substitutions per site based on the lengths of exact matches between pairs of sequences, were applied using MEGA (version 7.0) program (26).
Copy number alteration analysis using WES data
Copy-number alterations (CNAs) were identified in WES data based on the RPKM (Read Per Kilobase per Million mapped reads) value of exonic regions from CONIFER (27). The logarithm of germline blood and sample data were used for further CNA analysis. Somatic CNAs were segmented and called with the Bioconductor package ‘DNAcopy’.
To identify CNAs emerging during the generation of PDXs, we compared copy number profiles between patient and derived PDX tumors. First, a comparative value was obtained for each probe region by subtracting the log2-transformed copy number value of patient tumor from that of PDX tumor. The circular binary segmentation analysis was then applied to relative copy number data with the ‘DNAcopy’ R package. If |segment value| > 0.3 and size > 5 Mb, segment calls were retained. Second, segment calls from relative data and each sample were summarized to gene-level calls. A discordant CNA call between the patient and PDX tumor was considered an actual PDX-specific CNA, and “false” discordant CNA calls, which is called in both samples but showed a high relative value because of the high purity of the PDX tumor, were eliminated. The number of discordant CNA calls was divided by the total number of genes (excluding genes with a neutral copy number call in both datasets).
Calculation of heterogeneity score and analysis of clonal architecture
The subclonal genomic structure of the patient and PDX tumors was inferred using mutant allele fractions and copy number data from WES. Based on the Bayesian clustering method, SciClone (28) and PyClone (29) estimated the clonal population structure of each sample. In particular, PyClone was used for the estimation of cellular prevalence of each cluster of each sample with respect to normal cell contamination and copy number changes. PyClone was run for 10,000 iterations with a burn in period of 1,000 iterations using a beta binomial parameter of 1,000. We plotted clusters with at least 5 mutations and selected the representative genes for each cluster if mutated genes were included in our cancer gene list or showed high mutation frequencies in cBioPortal CRC studies (http://www.cbioportal.org). Of the primary patient-PDX tumor pairs, the number of input mutations in two patients (Patient #1 and 2) was too small or large to obtain the results in PyClone analysis.
RNA-seq data processing and expression analysis
RNA-seq reads from each WTS experiments were aligned to the same reference as for WES using STAR aligner (30). The following procedures were performed as the Best Practices workflow for RNA-seq using GATK. Gene expression levels were quantified using deduplicated bam files by FPKM (fragments per kilobase of exon per million mapped reads) using HTSeq-count (31) based on the Homo sapiens GRCh37 Ensemble v65. FPKM values were normalized and log transformed with edgeR. We also used pairwise average-linkage clustering for Pearson distance measurement of common clustering patterns. The DESeq2 algorithm (32) was used to detect metastatic tumor-specific gene expression patterns when compared to the primary tumor of each patient.
Methyl-capture sequencing
Methyl-capture sequencing was performed using the SureSelectXT Human Methyl-Seq Target Enrichment System, which utilized a capture-then-bisulfite-convert approach, as directed by the manufacturer. Genomic DNA (3 μg) of PDX tumors was sheared to a median fragment size of 150–200 bp using a Corvaris E210. Libraries were generated and hybridized using SureSelectXT Methyl-Seq Kit (Agilent). Bisulfite conversion was performed using EZ DNA Methylation Gold Kit (Zymo Research). The enriched and bisulfite-converted libraries were indexed by PCR amplification as directed in the protocol and purified using AMPure XP beads (Beckman Coulter). After the quality and quantity of the library sample was assessed by Qubits dsDNA HS Assay Kit (Thermo Fisher Scientific) and the Bioanalyzer DNA 1000 Kit (Agilent), the libraries were sequenced with HiSeq instruments.
The raw sequence data were trimmed for potential adapter sequences using trimgalore and aligned to combined reference using Bismark program with bowtie1. Deduplication and extraction of methylation was also performed with Bismark. Since the capture-then-bisulfite-convert approach captures the top strand of the DNA, only reads that aligned to the original top strand were considered for calling cytosine methylation. Only CpGs with at least 7X coverage were kept for comparisons. The methylation ratio was the number of methylated Cs divided by the total of methylated and unmethylated Cs. Average methylation level of each CpG island and CpG shores were calculated annotated against RefSeq gene database. For 13,199 genes, expression of each gene was correlated with multiple methylation probes per gene and the region that is most anticorrelated with expression data was selected for a given gene.
Phylogenetic analysis using sequencing data
Phylogenetic trees were constructed separately using (i) point mutations, (ii) gene expression and (iii) methylation from each patient and corresponding PDX models with multiple metastatic tumors using the neighbor joining method of the MEGA7 program (26). Rare functional mutations were used for WES phylogeny tree analysis.
Generation of cancer-related gene list
The list of cancer-related genes used to select important mutations was based on a total of 608 genes found in the Cancer Gene Census of The Sanger Institute (33), TARGET by The Broad Institute and Vogelstein’s cancer genes (34). The TARGET database v3 was used to select for druggable gene targets.
Single sample gene set enrichment analysis projection on a collection of hallmark gene sets
An unsupervised gene enrichment method that calculates a separate enrichment score (ES) to gene set for each individual sample, independent of sample phenotype labeling, single sample gene set enrichment analysis (ssGSEA), was performed on expression and methylation data. We scored signatures of hallmark processes of the Molecular Signatures Database (MSigDB) through the ssGSEA Projection module of GenePattern, ES was obtained and further analyzed by transforming into a z-score by subtracting the mean of the ES’s assigned to all other gene-sets and by dividing the result to their standard deviation. A positive ES denotes a significant overlap of the signature gene set with groups of genes at the top of the ranked list, whereas a negative ES denotes a significant overlap of the signature gene set with groups of genes at the bottom of the ranked list.
Histological analysis and immunohistochemistry
After resection, tumor tissue samples were fixed in 10% neutral buffered formalin and embedded in paraffin. Hematoxylin-Eosin staining (H&E) was performed according to standard protocols. For immunohistochemistry, tumor tissue samples obtained from patients and PDX mice were stained using antibodies including CEA (Cell Marque), CK7 (Dako), CK20 (Dako), Ki-67 (Dako), E-cadherin (Zymed), Vimentin (Zymed), CD3 (Dako), and CD31 (Cell Marque). Formalin fixed and paraffin embedded tissue sections of tumor samples were immunohistochemically stained for expression of those antibodies using a BenchMArk ULTRA automatic immunostaining device (Ventana Medical Systems) with the OptiView DAB IHC Detection Kit (Ventana Medical Systems) according to the manufacturer’s instructions.
In vivo pharmacological studies
For PDX mice, drug treatments began after tumors reached approximately 200 mm3. For standard treatments, mice were randomly divided into two treatment groups, consisting of 5 mice in each group: 1. vehicle only, 2. 5-FU (ApexBio, 50 mg/kg, weekly) + oxaliplatin (ApexBio, 5 mg/kg, weekly). For targeted treatments, mice were randomly divided into four treatment groups consisting of 5 mice in each group: 1. vehicle only, 2. lapatinib (ChemieTek, 30 mg/kg, twice a day), 3. trametinib (ApexBio, 2 mg/kg, daily), and 4. BYL719 (ChemieTek, 25 mg/kg, daily). The vehicle for 5-FU and oxaliplatin was 5% (w/v) glucose in water (Sigma). The vehicle for lapatinib was 0.5% (v/v) methylcellulose (Sigma) and 0.5% (v/v) Tween 80 (Sigma) in phosphate-buffered saline (PBS). The vehicle for trametinib was 0.5% (v/v) hydroxypropyl methylcellulose (HPMC, Sigma) and 0.2% (v/v) Tween 80 in PBS. The vehicle for BYL719 was 10% (v/v) ethanol (Merck), 30% (v/v) polyethylene glycol (Sigma) and 60% (v/v) Phosal 50 PG (Lipoid). 5-FU and oxaliplatin was administered via intraperitoneal injection while lapatinib, trametinib and BYL719 were all administered via oral injection for 21 – 26 days.
Data availability
The data in this study have been submitted to the National Center for Biotechnology Information (NCBI) BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA400542.
Results
Generation of genomically defined PDXs from CRC patients
We generated a PDX cohort of CRCs and analyzed the mutational profiles of the original tumors and the derived PDX models. We implanted a total of 95 tumor tissues from 42 CRC patients (detailed clinical information in Supplementary Table S1), and successfully established 72 serially-transplantable PDXs from 35 patients (Fig. 1A; Supplementary Table S2). Anatomical location of primary tumors, tumor stage, MSI status and organ sites of metastatic tumors were not associated with PDX engraftment rates (Supplementary Table S3).
Figure 1.
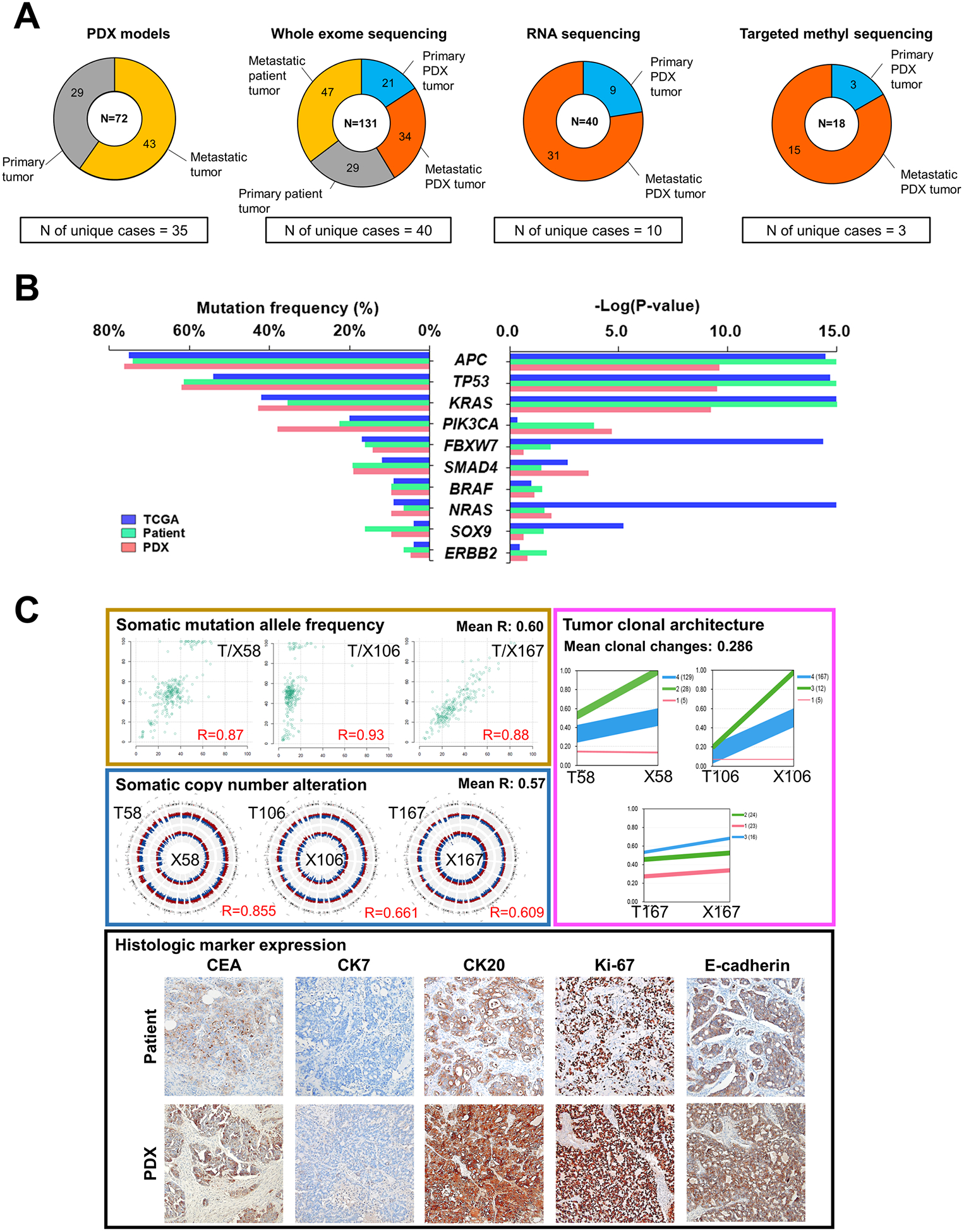
Genomic characterization of PDX tumors of CRCs. A, Summary of sample composition for PDX generation, whole exome sequencing, RNA sequencing and DNA targeted methyl-capture sequencing. B, Distribution of the mutation frequencies of the top 10 significantly-mutated cancer genes from the TCGA cohort (n = 224; blue), primary tumors of our patient cohort (n = 29; green) and primary tumors of our PDX cohort (n = 18; pink). Left panel represents the frequency of mutations from three cohorts, calculated by considering only primary tumors. Right panel shows P-values of each gene estimated by the MutSig algorithm. C, Panels with individual examples for comparison between matched patient and PDX tumors. Left upper panel contains scatterplots of somatic mutation allele frequencies of matched samples, and left middle panel represents genomic distribution of somatic copy number alterations (red region, amplification; blue region, deletion) in matched patient (outer circle) and PDX (inner circle) tumors. Right upper panel shows tumor clonal architecture of matched patient and PDX tumors, estimated by PyClone algorithm. Line widths indicate the number of mutations in each cluster (numbers in brackets next to each cluster). Lower panel is the representative microscopic images of immunohistological analysis in matched patient and PDX tumors (100X). Immunohistochemical staining were performed for carcinoembryonic antigen (CEA), cytokeratin 7 (CK7), CK20, Ki-67, and E-cadherin.
Given that a genomically well-defined PDX cohort is a valuable tool for assessing drug efficacy and understanding drug resistance mechanisms, we analyzed the genomic profiles of the original patient and PDX tumors using whole exome sequencing (WES). We sequenced 76 of the original tumor samples from 29 patients (29 primary and 47 metastatic tumors; Fig. 1A; Supplementary Table S2), and 55 PDX samples from 24 patients (21 primary and 34 metastatic tumors; Fig. 1A; Supplementary Table S2). The most frequently mutated cancer genes in our PDX tumors paralleled those seen in CRCs in The Cancer Genome Atlas (TCGA) database (35) (Fig. 1B); with the exception of an increased frequency of PIK3CA mutations in our PDX tumors. This suggests that overall, our CRC tumors and derived PDX models retain mutation profiles similar to that of other CRC cohorts. In addition, we performed RNA sequencing for 40 PDX samples from 10 patients (9 primary and 31 metastatic tumors) and DNA methyl-capture sequencing for 18 PDX samples from 3 patients with MOMs (3 primary and 15 metastatic tumors; Fig. 1A; Supplementary Table S2).
In mutational analysis, the allele frequencies (AFs) of mutations were correlated between these patient and PDX tumors (average Pearson correlation coefficient = 0.60), but AF usually increased in PDX tumors consistent with the notion that tumor cells are enriched in PDX samples (Fig. 1C; Supplementary Fig. S1A). Somatic copy number alterations (SCNAs) profiling using WES data showed that SCNAs were also generally correlated between patient tumors and corresponding PDX tumors (average Pearson correlation coefficient = 0.57) (Fig. 1C; Supplementary Fig. S1B). We also estimated the clonal architecture using PyClone (29) and found that each tumor had 2 to 6 major subclones, which were well conserved and enriched in PDX tumors (average changes in clonal prevalence: 0.286) (Fig. 1C; Supplementary Fig. S1C; Supplementary Table S4).
In histological analyses, the PDX tumors retained comparable histologic architecture of the primary patient tumors, and cancer cells exhibited similar expression patterns with respect to carcinoembryonic antigen (CEA), cytokeratin 7 (CK7), CK20, Ki-67 and E-cadherin staining (Fig. 1C; Supplementary Fig. S2). When evaluating the tumor microenvironment, stromal vimentin expressions of the primary patient tumors were retained in half of the derived PDX tumors (Fig. 1C; Supplementary Fig. S2), and vessels of human origin (CD31 positive cells) were replaced with mouse vessels (Fig. 1C; Supplementary Fig. S2). In addition, CD3+ lymphocytes were enriched in PDX tissues, suggesting that human lymphocytes are expanding in immunocompromised states of early PDX passages (Supplementary Fig. S2). Taken together, our observations showed that our PDX models retain genomic and histologic characteristics of the primary patient tumors, but lose some characteristics associated with tumor microenvironment.
Analysis of genomic alterations during tumor metastasis
In our CRC cohort, we have 5 patients with MOMs, and successfully generated PDX models from the primary tumors of these 5 patients as well as 25 of their metastases (patient #10, #14, #21, #34 and #35 in Supplementary Table S2). In data analysis from WES on the original patient samples and WES/RNA-seq/methyl-capture sequencing of the derived PDX samples (unfortunately the RNA quality from the patient samples precluded RNA-seq), phylogenetic analyses revealed branched evolution for the primary tumors and derived PDXs for all five patients (Fig. 2), consistent with the notion that the evolutionary processes were well conserved in the PDX models. Furthermore, the evolutionary processes, determined by somatic mutations, mRNA expressions and DNA methylation, exhibited similar patterns in the primary patient tumors and the derived PDX samples (Fig. 2), indicating that genomic changes during the cancer evolution process were closely connected with transcriptomic and epigenomic changes.
Figure 2.
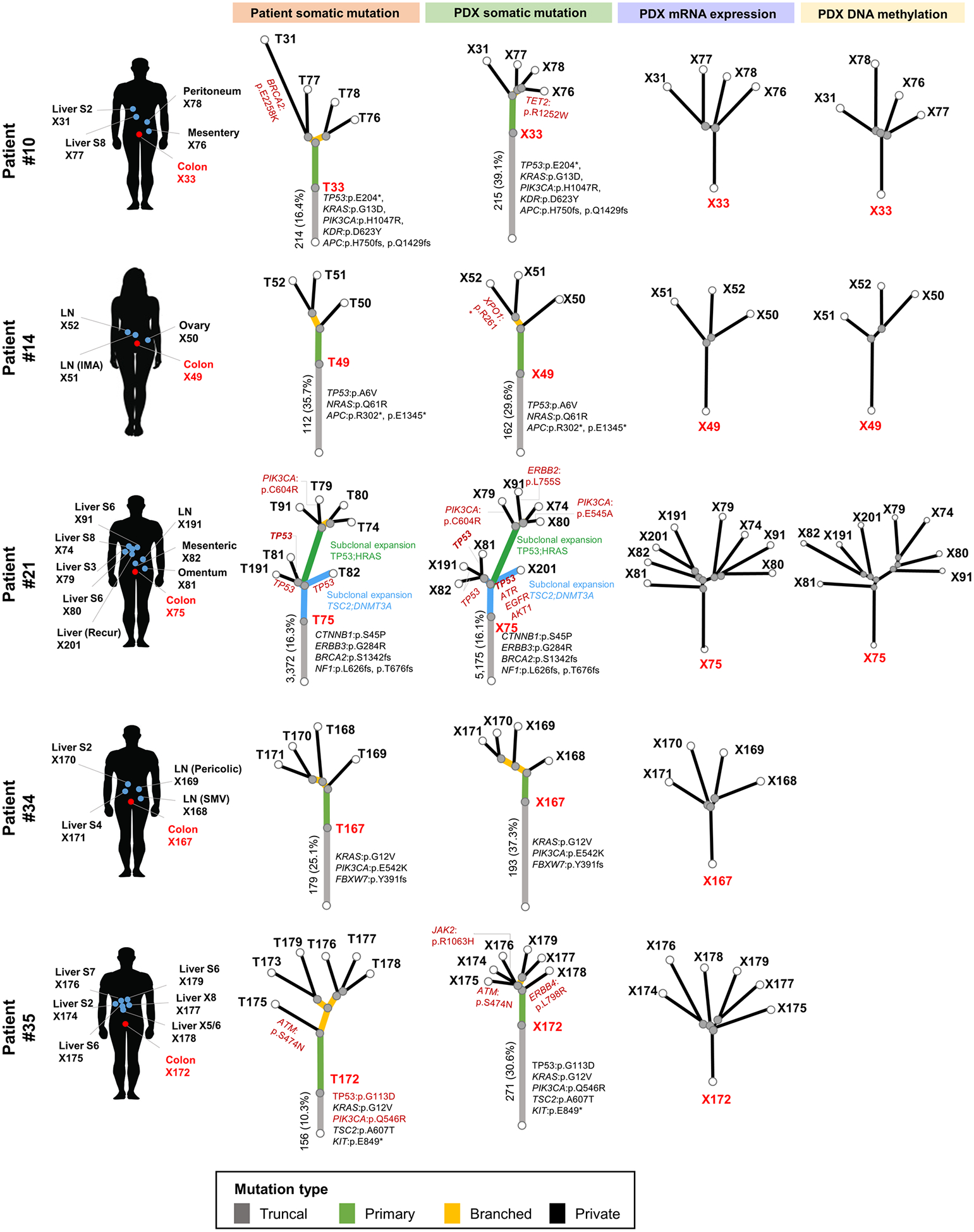
Branched evolutionary processes of tumors from CRC patients with multiple organ metastasis. Phylogenetic trees were reconstructed by neighbor-joining method using patient somatic mutations from patient whole exome sequencing analysis (left; Patient somatic mutation), PDX somatic mutations from PDX WES analysis (middle left; PDX somatic mutation), PDX mRNA expression from RNA-seq analysis (middle right; PDX mRNA expression), and PDX DNA methylation analysis (left; PDX DNA methylation). Colors of each line indicate the truncal mutations (gray), mutations found only in primary tumors (green), branched mutations (yellow) and private mutations (black). Representative genes with mutations in each evolutionary process are indicated next to the branch where the mutation occurred. Angles between branches were chosen only for convenience of display.
In tumors with MOMs, the somatic mutations were divided into three categories: truncal (mutations present in all tumor regions), branched (mutations present in at least two, but not all regions) and private mutations (mutations present in one region). In the 5 patients analyzed, the average rates of truncal, branched and private mutations were 30.5% (range 16.1 – 39.1%), 16.3% (range 9.6 – 25.9%) and 53.2% (range 36.8 – 63.7%), respectively (Supplementary Table S5). Of the 608 cancer-related genes with known importance (the selection criteria are described in Materials and Methods), mutations in KRAS (3/5), PIK3CA (3/5), TP53 (2/5) and APC (2/5) were detected as truncal mutations in two or more patients (Fig. 2), suggesting that mutations of these genes play a critical role in tumor initiation and early development of CRCs.
Comparison of genetic changes between metastasis and PDX generation
PDX models provide unique tumor microenvironment different to human, raising the possibility that increased genomic instability occurs. To estimate the de novo genetic changes during PDX generation in mice, we compared the changes of mutations and copy number alterations during PDX generations with those during tumor evolution. In patient #21, primary tumor (T75) was metastasized to liver (T74, T79, T80, and T91) and regional lymph nodes (T81, T82, and T191), and we generated four different PDX models by engrafting T75 (Fig. 3A). Analysis of WES data showed that estimated differences between primary tumor and PDXs (range 0.07 – 0.12, mean: 0.09) were lower than those between primary and metastasized tumors, especially tumors metastasized to liver (range 0.20 – 0.23, mean: 0.22; Fig. 3B and 3C). These data suggest that genetic alterations during the PDX generation were not significantly augmented compared to the metastasis within the patient.
Figure 3.
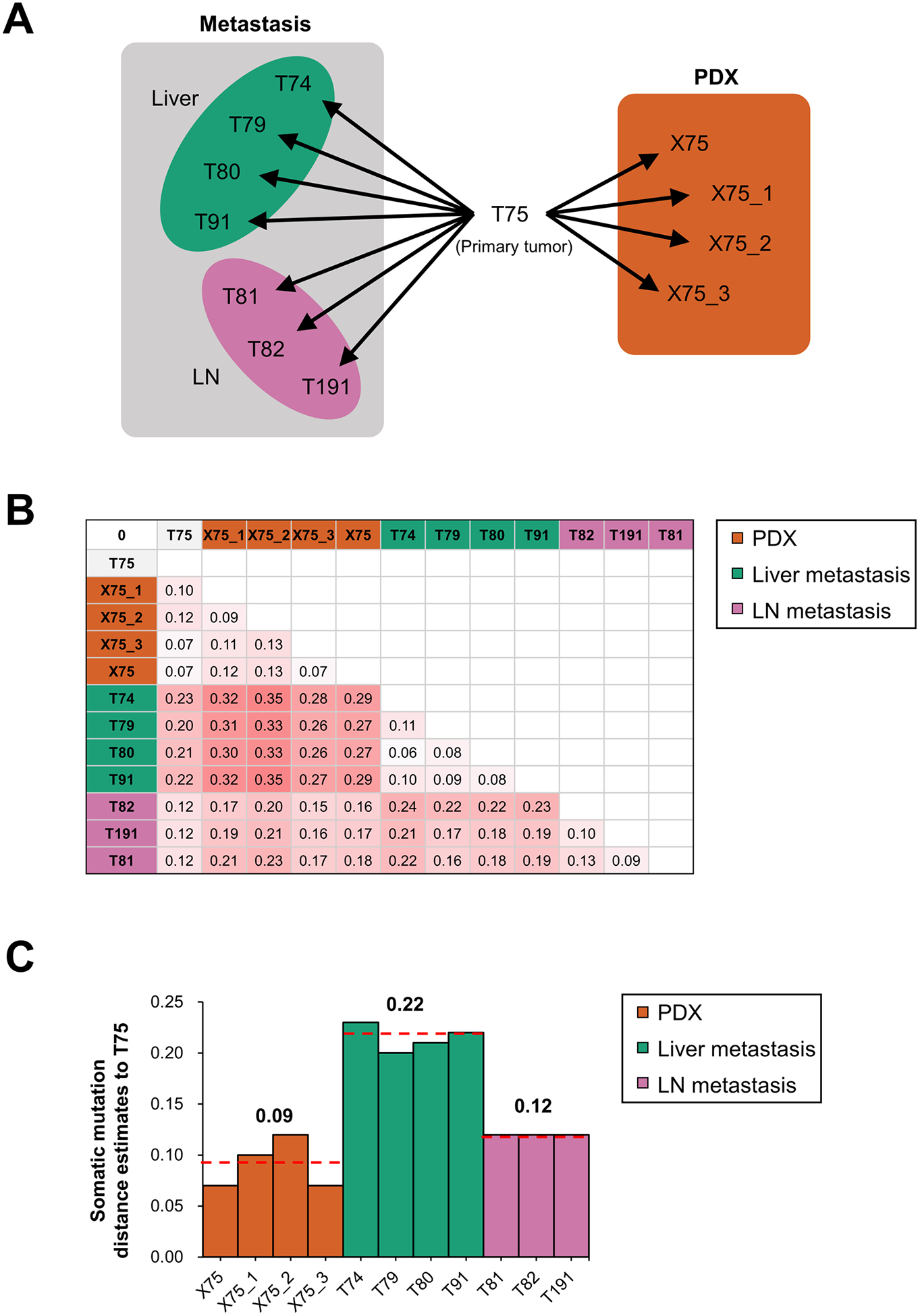
Genomic distances compared to primary tumor in patient metastatic tumors and PDXs. A, Schematic presentation of analyzed tumors from patient #21 and primary tumor-derived PDXs. From primary tumor (T75), 4 tumors were metastasized to liver (T74, T79, T80 and T91), 3 tumors metastasized to regional lymph nodes (T81, T82 and T191), and 4 tumors were generated as PDXs. B, Estimates of evolutionary divergence in metastatic and PDX tumors revealed by maximum composite of somatic mutations. C, Somatic mutation distance estimates of patient metastatic tumors and PDXs compared to primary tumor.
Clonal architecture dynamics during tumor metastasis
Next, we analyzed the clonal architectures of the PDX tumors from 5 patients with MOMs, because clonal architecture of patient tumors was conserved in PDX tumors (Fig. 1C; Supplementary Fig. S1C). The SciClone analysis, modeling both clonal and subclonal mutation clusters (28), revealed that the mutations in the tumor samples from each individual could be organized into 2 to 8 clusters (Fig. 4). All metastasized tumors showed multiple subclones, and these subclones were usually found in primary tumors (Fig. 4), suggesting that metastasis occurs via polyclonal seeding, not a single cell. Patients having primary tumors with a small number of subclones (2 – 3 subclones; patient #10 and #35) showed little change in subclonal architecture during metastasis (Fig. 4). However, patients having primary tumors with a large number of subclones (5 – 7 subclone; patient #14, 21 and 34) exhibited dynamic changes in subclonal architecture during metastasis (Fig. 4), and these changes were largely dependent on the organs where the cancer had metastasized (Fig. 4). Tumors metastasized to lymph nodes showed similar subclonal architecture with primary tumors, but tumors metastasized to ovary or liver showed enrichment of specific subclones showing little ratios in primary tumors (Fig. 4). For example, C4 subclone in patient #14 was enriched in tumors metastasized to ovary, and C3 suclone in patient #21 and C4 subclone in patient #34 were significantly enriched in tumors metastasized to liver (Fig. 4), suggesting that loco-regional metastasis to lymph nodes and distant metastasis to liver or ovary probably occurs in a parallel or independent fashion and distant organs provide unique tumor microenvironment for distinct clonal selection.
Figure 4.
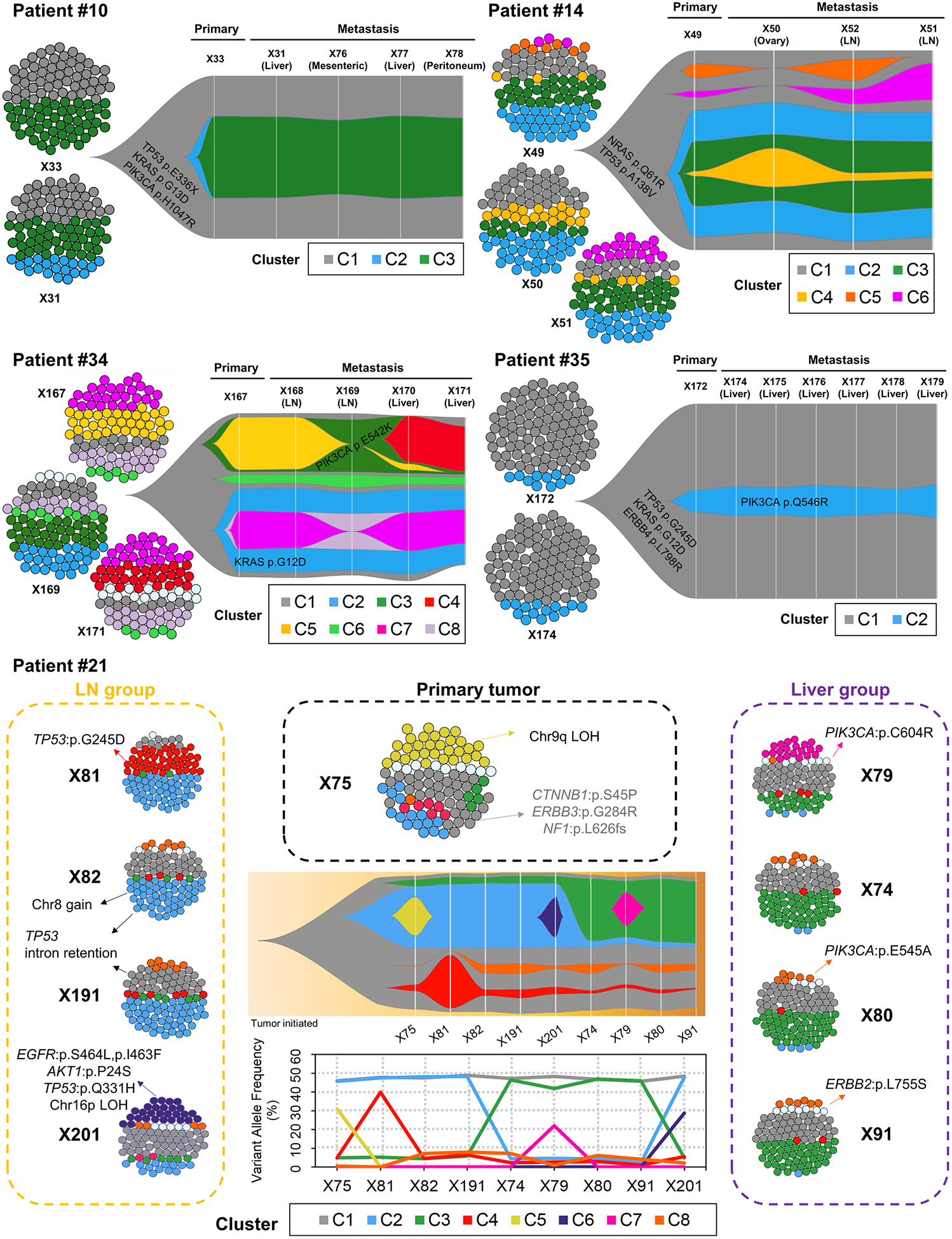
Clonal architecture and clonal dynamics of tumors of CRC patients with multiple organ metastasis by SciClone analysis. In each patient, cell cluster figures represent inferred composition of subclone clusters in primary and metastasized tumors, and fish plots represents inferred schematic of clonal evolution, showing percentage of cells belonging to each primary or metastasized tumor. Each color depicted each subclone cluster (C1 to C8). In patient #21, middle lower graph shows variant allele frequency of each subclone cluster in each primary or metastasis tumor.
Transcriptomic and epigenomic changes during tumor metastasis
We also analyzed the RNA-seq data of PDX tumors from patients with MOMs. Clustering analysis and principal component analysis (PCA) analysis showed that samples from the same patient were more adjacently clustered compared to metastases to the same organ (Supplementary Fig. S3). When we investigated enriched hallmark gene sets (36) among the metastasized tumors using single sample gene set enrichment analysis (ssGSEA), gene sets of ‘UV_response’, ‘DNA_repair’, ‘Apoptosis’, ‘Myc_targets’ and ‘Protein secretion’ were enriched in metastasized tumors in 4 or more patients (Fig. 5). Interestingly, gene sets of ‘Epithelial_mesenchymal_transition’ and ‘Hypoxia’, which were known to promote metastasis (37,38), were depleted in metastasized tumors in 5 or more patients (Fig. 5). It is probable that metastasized tumors are exposed to more oxygen compared to primary tumor sites, but detailed investigation was needed in further studies.
Figure 5.
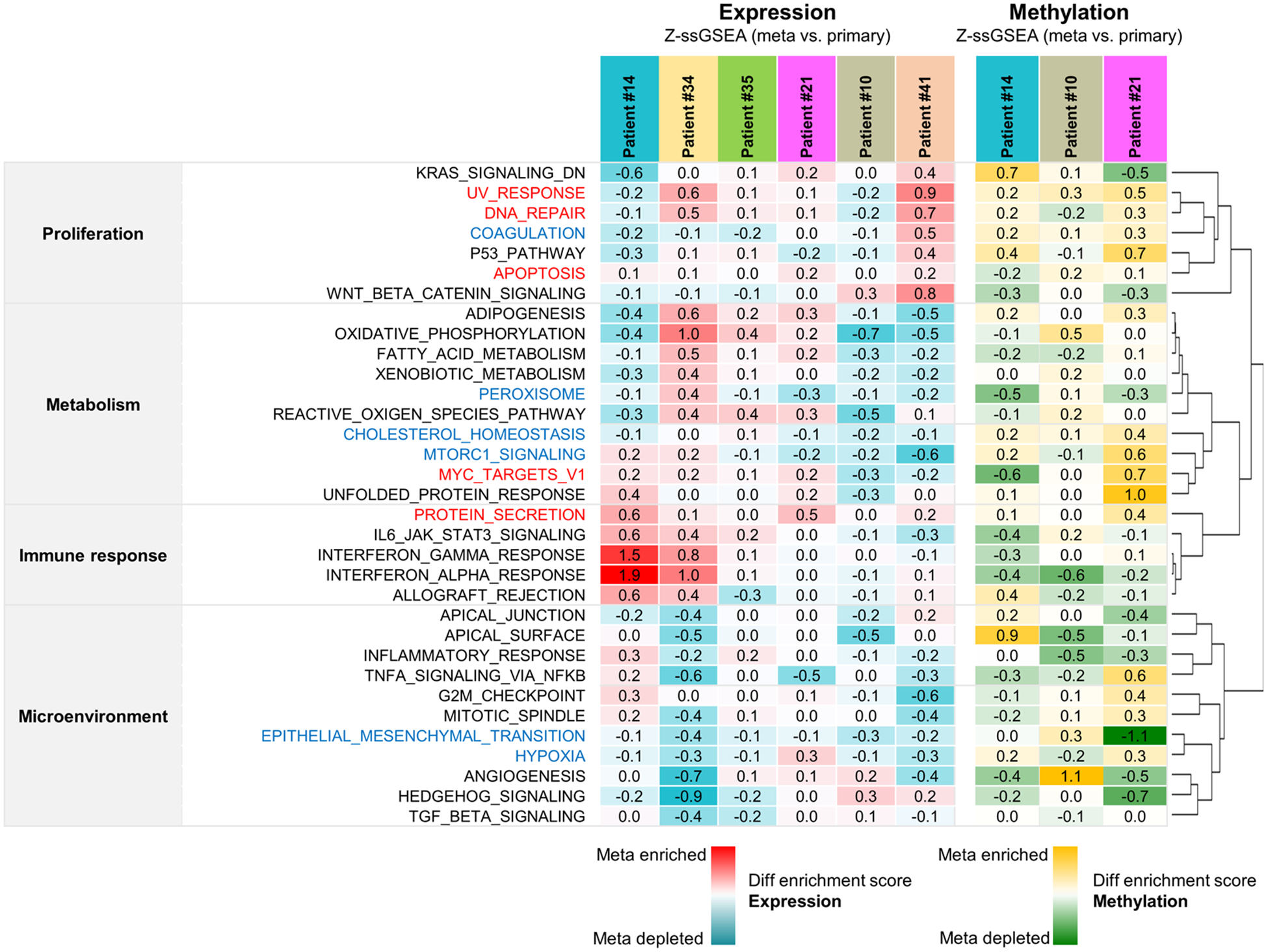
Metastasis-specific altered cancer hallmark gene sets from transcriptome and DNA methylation analysis in PDX tumors of CRC patients with multiple organ metastasis. Clustered heatmaps represent gene set activity scores calculated relative to primary tumors, which show a comparison of hallmark gene set activities between primary and metastasized tumors, from RNA sequencing (left panel) and DNA methylation sequencing (right panel). In expression analysis, red indicates that a hallmark is more active relative to primary tumor and blue indicates that a hallmark is less active. In methylation analysis, yellow indicates that a hallmark is more active relative to primary tumor and green indicates that a hallmark is less active.
We also performed ssGSEA using DNA methyl-capture sequencing data and investigated hallmark gene sets enriched in metastasized tumors. However, there is little gene set enriched or depleted in metastasized tumors of all tested 3 patients with MOMs (Fig. 5). The promoter methylation in gene set of ‘Cholesterol_homeostasis’ was enriched in metastasized tumors of all 3 patients, and the mRNA expression levels of this gene set were inversely correlated with methylation status (Fig. 5). Although cholesterol was reported to promote metastasis (39), gene set of ‘Cholesterol_homeostasis’ was down-regulated in metastasized tumors, and detailed molecular mechanisms need to be investigated.
Tumor heterogeneity analysis of CRC patient with MSI-H status
Among the five patients with MOMs, we focused on one patient exhibiting the most divergent mutational profiles due to high levels of microsatellite instability (patient #21). This case showed prominent differences in mutation and expression profiles between two distinct groups of tumors; primary/LN group and liver-metastasized group (Fig. 4; Supplementary Fig. S4).
In subclone analysis, the C1 subclone was found in all tumors (Fig. 4), and possesses several truncal mutations including CTNNB1, ERBB3, NF1, BRCA2, SYK mutations (Fig. 4; Supplementary Fig. S4A). In addition, copy neutral loss of heterozygosity (cnLOH) in chromosome 3p, which contains CTNNB1 gene, were also found as truncal alterations (Supplementary Fig. S4B). The differences between primary/LN group and liver-metastasized groups were predominantly based on the different prevalence of two subclones, C2 and C3 (Fig. 4). In the primary/LN group, the most enriched subclone was a C2 subclone, which included mutations such as TSC2 and MSH6 mutations (Fig. 4; Supplementary Fig. S4A). Interestingly, convergent bi-allelic inactivation of TP53 gene was observed in metastatic tumors in LN group (Supplementary Fig. S5). On the other hand, the liver-metastasized group showed enrichment of the C3 subclone, which harbored the TP53 R273C mutation (Fig. 4; Supplementary Fig. S4A). Comparison of copy number (CN) values for chromosome 7 (chr7) between the two groups showed that primary/LN group showed significant increase in CN alterations on chr7 (Supplementary Fig. S4B; P = 0.012). Notably, copy number changes for EGFR showed significant differences between the primary/LN group compared to the primary/liver group (P = 0.004), and the pathway-level expression analysis also showed that EGFR signaling in cancer was upregulated in the primary/LN group (Supplementary Fig. S4C). In addition, expression of genes in ‘Signaling by Wnt’ increased in primary/liver group (Supplementary Fig. S4C), which was compatible with decrease of promoter methylation in ‘Wnt_beta_catenin_signaling’ pathway (Supplementary Fig. S4D).
For the X201 samples (which were derived from recurrence in the liver after adjuvant chemotherapy with FOLFIRI and cetuximab), we identified a subclone that was not evident in the pre-chemotherapy samples (C6; Fig. 4). In these treatment-resistant subclones, potential driver mutations included mutations of EGFR S464L, was previously reported to be related to the cetuximab resistance (40).
Association of genomic and transcriptomic signatures with therapeutic heterogeneity as delineated by PDX models
Next, we investigated the responsiveness of targeted treatment based on genomic profiling in the PDX models from patient #21 (Supplementary Table S6). The ERBB3 G284R mutation was reported to activate the ERBB2 signaling pathway, which can be targeted by the ERBB2 inhibitor, lapatinib (41). NF1 mutations result in activation of the Ras-MEK-ERK signaling pathway, which can be targeted by a MEK inhibitor, trametinib (42), and PIK3CA E545A mutations, found in X80, can be targeted by PI3K inhibitors such as BYL719 (43,44) (Supplementary Table S6). When the normal growth curves of these PDX tumors were examined, the tumor with a PIK3CA E545A activation mutation (X80) showed the fastest growth (Supplementary Fig. S6).
The combination treatment of 5-florouricil and oxaliplatin (5-FU/Oxa), which is a standard non-targeted treatment for CRC, showed a significant tumor inhibitory effect in three out of the five samples tested (Fig. 6A; Supplementary Fig. S7A). Lapatinib treatment resulted in significant tumor inhibition in three samples (X75, X79, X80) (Fig. 6B; Supplementary Fig. S7B). However, one sample with ERBB2 L755S mutation (X91) was refractory to lapatinib treatment (Fig. 6B; Supplementary Fig. S7B), which was explained by the presence of the lapatinib-resistant mutation in ERBB2 gene (ERBB2 L755S; Supplementary Table S6) (45). We validated this mutation with droplet digital PCR (ddPCR), and found the ERBB2 L755S mutation to be a private mutation (i.e., found in only one sample; Supplementary Fig. S8). Trametinib treatments showed effective inhibition on tumor growth in four sample out of 5 samples (Fig. 6C; Supplementary Fig. S7B). Unexpectedly, BYL719 was effective only in the primary tumor (Fig. 6D; Supplementary Fig. S7B), which was not anticipated by its PIK3CA mutation status.
Figure 6.
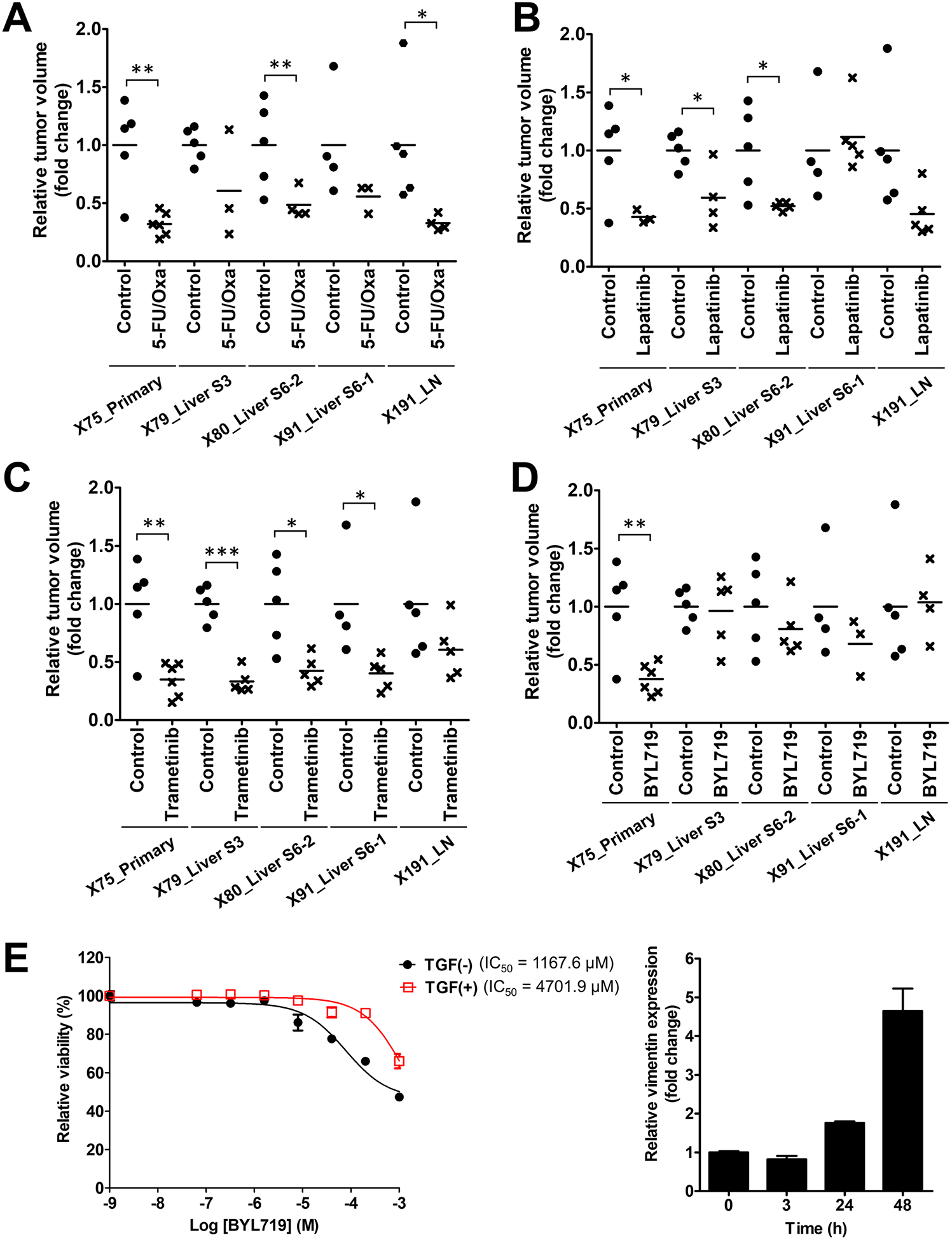
Therapeutic heterogeneity of tumors from one CRC patient with multiple organ metastasis. A-D, Relative tumor volume of the primary and metastasis PDX tumors at the endpoint of drug treatment. Relative tumor volume was calculated as fold changes based on the mean of the vehicle-treated (control) groups for each PDX tumor. Mice were treated with 5-FU (50 mg/kg/week) + oxaliplatin (5 mg/kg/week) (5-FU/Oxa; A), lapatinib (30 mg/kg, twice a day; B), trametinib (2 mg/kg/day; C), and BYL719 (25 mg/kg/day; D). Asterisks indicate statistically significant differences (*, p < 0.05; **, p < 0.01; ***, p < 0.001) between control and drug-treated groups. E, TGFβ1 effect on sensitivity to a PI3K inhibitor, BYL719, in HEK293 cells. TGFβ1 (5 ng/ml) was treated 3 h before BYL719 treatment, and WST assays were used to examine the cytotoxic effect after 72 h treatment of BYL719 in each concentration. IC50 values for BYL719 are given. Right graph shows the effect of TGFβ1 treatment on the expression of vimentin, which is a known TGFβ1-responsive gene.
To understand the factor(s) associated with BYL719 responsiveness, we compared genomic and transcriptomic differences between responsive and non-responsive tumors. In mutational profiling, mutations in TP53 (R273C and A159V) were detected only in metastatic tumors (Supplementary Table S6). However, overexpression of mutant TP53 did not appear to significantly contribute to resistance to BYL719, as compared to the wild-type (Supplementary Fig. S9). From expression profiling, the TGFβ signaling pathway was enriched in the metastatic tumors (Supplementary Fig. S10), and treatment of TGFβ1 increased the IC50 for BYL719 (Fig. 6E), suggesting that activation of TGFβ signaling pathway is associated with resistance to BYL719. Taken together, in vivo experiments using our PDX models verified that subtle differences in the genome and transcriptome could largely affect the therapeutic responsiveness.
Discussion
During metastasis, genetic and epigenetic alterations are generated due to the genomic instability of cancer cells, and are dynamically reshaped via evolutionary process and adaptation to environmental changes. The tumor evolution process is generally delineated as a branched evolution model, based on phylogenetic reconstructive analyses (10,11,46–48). The branched evolution model emphasizes the importance of targeting truncal alterations. However, resistance for targeted drug may also occur due to subclonal alterations, at the level of gene mutations or gene expression as shown in our data (Fig. 6).
It has been ambiguous whether the subclonal alterations associated with drug resistance exist as minor subclones in the original tumor or emerge during the evolutionary process. The Big Bang model of tumor growth suggests that a single expansion produces diverse subclones where clonal and subclonal alterations can develop early in cancer growth (49). A study using a complex DNA barcode labelling system showed that, during the development of drug resistance caused by ITH, the majority of resistant clones were part of pre-existing subpopulations (50). Our analysis of clonal architecture of metastatic tumors also demonstrated that most subclones in metastatic tumors were found in primary tumors (Fig. 4), suggesting that genomic divergence during metastasis occurs usually by evolutionary reshaping of pre-existing subclones. However, our ddPCR results also supports the notion that some drug-resistant alterations could appear in the middle of evolutionary process from de novo alterations (Supplementary Fig. S8), and the branched or private alterations play critical roles in the development of drug resistance. In our study, one metastatic tumor with the ERBB2 L755S mutation showed the resistance to lapatinib, and transcriptional activation of the TGFβ signaling pathway increases the drug resistance to BYL719 (Fig. 6). Therefore, for the treatment of cancer patients with MOMs, genomic and transcriptomic heterogeneities should be considered for pertinent treatment, especially in CRC patients having primary tumors with a large number of subclones, because subclonal architecture changes dynamically in these patients.
Our analyses provide several applicable points for the targeted treatment of CRC patients with MOMs. Considering the heterogeneous nature of primary and metastatic tumors, targeting the truncal alterations would be the best way for the treatment of patients with MOMs, because all primary and metastatic tumors possess the truncal alterations. In our study, targeting NF1 mutations, which were truncal alterations, with trametinib reduced tumor growth in all tested samples (Fig. 6C). However, at the same time, the incidence of resistance due to subclonal alterations needs to be carefully examined. Because multiple biopsies are usually difficult to perform, one alternative way is to analyze the genomic profiles of circulating tumor cells or cell-free DNA, which represent the summation of genomic alterations from all cancer cells. Detection of resistance-related alterations in liquid biopsy can guide to treat the patients with other targeted drugs.
Detailed analysis of subclonal architecture of tumors from CRC patients with MOMs suggested some implications about mode of tumor metastasis. Several subclones were found simultaneously in primary and metastatic tumors (Fig. 4), which support polyclonal metastasis rather than metastasis via a single cell dissemination. And, initial number of subclones in primary tumors is associated with diversity in subclonal architecture according to metastasis, and primary tumors harboring more subclones showed more diverse subclonal architecture among metastatic tumors, probably due to higher chance of clonal selection in metastasized tumor microenvironment. In addition, loco-regional metastasis to lymph nodes and distant metastasis to liver or ovary exhibited distinctive subclonal architectures, suggesting that loco-regional and distant metastasis probably occurs in parallel or independent ways.
PDX models retain several characteristics of patient tumors and showed drug responsiveness compatible with the patient’s original tumors (15–17). Our comparison of the patient’s primary tumors with the derived PDX tumors indicated that the derived PDXs appear to maintain the characteristics of the patient original tumor on the basis of mutational profile and signatures, SCNAs, tumor clonal architectures and histology (Fig. 1). PDX models also retained the characteristics related with evolutionary processes in terms of somatic mutation, gene expression and DNA methylation (Fig. 2). In addition, genetic changes during PDX generation were similar or lower than genetic changes during metastasis processes in patients (Fig. 3). However, PDX models showed some changes in its tumor microenvironment, such as a decrease in extracellular proteins and vessels of human origin (Supplementary Fig. S2), which was probably due to the replacement of these cells with mouse stromal cells. In addition, analysis of the mutation allele frequencies as well as clonal architecture in the PDX models exhibited an enrichment of cancer cells having driver mutations or drug responsiveness-associated mutations, compared to the patient samples. For example, the prevalence of PIK3CA mutations in our PDX cohort was higher than our patient cohort (Fig. 1B), which suggests that the subclones with PIK3CA mutations (pre-exist or de novo) had growth advantages in the PDX environment (Supplementary Fig. S6). Therefore, PDX models show accordant prediction of drug responsiveness in patients by augmenting the portion of subclones with driver mutations or drug responsiveness-determining mutations.
Our study demonstrated that development of genomic and transcriptomic alterations during the metastatic process plays critical roles in determining drug responsiveness, and, as a result, are highly associated with clinical outcome of patients. Furthermore, PDX models are valuable tools for studying the correlation between tumor evolution and therapeutic heterogeneity. More studies of tumor heterogeneity during metastasis with respect to therapeutic effectiveness would pave the ways for prediction and improvement of clinical outcome in CRC patients.
Supplementary Material
Translational Relevance.
Colorectal cancers (CRCs) exhibit various genomic and transcriptomic changes during metastasis because of genomic instable nature of cancer cells. However, detailed clinical implications of metastasis-associated alterations have not been fully understood. Herein, we developed and analyzed patient-derived xenograft models from patients with multiple organ metastases. Phylogenetic analysis showed that mutational alterations were closely connected with transcriptomic and epigenomic changes during the tumors’ metastatic process. Primary tumors with heterogeneous subclonal architecture showed more dynamic subclonal changes during metastasis, and subclonal acquisition of additional mutations or transcriptomic activation of bypass signaling pathway were responsible for the development of treatment resistance. Especially, gain of ERBB2 L755S mutation and activation of TGFβ signaling pathway were associated with the resistance to an ERBB2 inhibitor, lapatinib and a PI3K inhibitor, BYL719, respectively. Thus, therapeutic heterogeneity from genome and transcriptome dynamics during metastasis should be considered for the treatment of CRC patients with metastasis.
Acknowledgments
This work was supported by Gil Hospital Research Grant (grant No. GCU-2015-5092); the Korean Healthcare Technology R&D project through the Korean Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (grant No. HI13C2148); the International Research & Development Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (grant No. 2015K1A4A3047851); the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (grant No. 2017R1C1B2002183); the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant No. 2017R1D1A1B03035453); and the National Cancer Institute of the NIH (grant No. P30CA034196). C.L. is a distinguished Ewha Womans University Professor supported in part by the Ewha Womans University Research grant of 2018.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer 2006;6:449–58. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Targeting metastasis. Nat Rev Cancer 2016;16:201–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015;27:15–26. [DOI] [PubMed] [Google Scholar]
- 4.Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol 2014;12:e1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee AJX, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 2011;71:1858–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012;486:395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010;467:1109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science 2016;352:169–75. [DOI] [PubMed] [Google Scholar]
- 10.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014;346:251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol 2014;15:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin Cancer Res 2012;18:5160–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho SY, Kang W, Han JY, Min S, Kang J, Lee A, et al. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol Cells 2016;39:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9:338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther 2011;10:1311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosfjord E, Lucas J, Li G, Gerber HP. Advances in patient-derived tumor xenografts: from target identification to predicting clinical response rates in oncology. Biochem Pharmacol 2014;91:135–43. [DOI] [PubMed] [Google Scholar]
- 18.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 2014;4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015;518:422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–86. [DOI] [PubMed] [Google Scholar]
- 21.Naxerova K, Brachtel E, Salk JJ, Seese AM, Power K, Abbasi B, et al. Hypermutable DNA chronicles the evolution of human colon cancer. Proc Natl Acad Sci U S A 2014;111:E1889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013:13033997. [Google Scholar]
- 24.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016;33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 2012;22:1525–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol 2014;10:e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 2014;11:396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith BN, Bhowmick NA. Role of EMT in metastasis and therapy resistance. J Clin Med 2016;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006;440:1222–6. [DOI] [PubMed] [Google Scholar]
- 39.Nelson ER, Chang CY, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends Endocrinol Metab 2014;25:649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arena S, Bellosillo B, Siravegna G, Martinez A, Canadas I, Lazzari L, et al. Emergence of multiple egfr extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res 2015;21:2157–66. [DOI] [PubMed] [Google Scholar]
- 41.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 2013;23:603–17. [DOI] [PubMed] [Google Scholar]
- 42.Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, et al. A Genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 2013;3:350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A 2005;102:802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 2013;10:143–53. [DOI] [PubMed] [Google Scholar]
- 45.Kancha RK, von Bubnoff N, Bartosch N, Peschel C, Engh RA, Duyster J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PloS One 2011;6:e26760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014;46:225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014;346:256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015;47:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhang HEC, Ruddy DA, Radhakrishna VK, Caushi JX, Zhao R, Hims MM, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med 2015;21:440–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data in this study have been submitted to the National Center for Biotechnology Information (NCBI) BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA400542.