

Abstract
Background: Primary immunodeficiencies (PIDs) are a heterogeneous group of congenital disorders characterized by susceptibility to recurrent infections, allergy, malignancies and autoimmunity. The identification of disease-causing genetic defects is critically important for treatment options. In last decade, next-generation sequencing (NGS)-based methods has enabled the rapid genetic screening and the discovery of new genetic defects in PIDs. In this study, we investigated causative mutations in patients with PID by NGS.
Methods: We applied whole-exome sequencing in 8 PID patients. Detected mutations by NGS were validated by Sanger sequencing.
Results: We made a genetic diagnosis in 5 of 8 (63%) patients, including 3 novel disease-causing variants. The identified mutations were found in RAG1, RAG2, JAK3, RFXANK, and CYBA genes.
Conclusions: Our results show that whole-exome sequencing can facilitate the genetic diagnosis of the patients with PID.
Keywords: primary immunodeficiency, next-generation sequencing, whole-exome sequencing
Introduction
Primary immunodeficiencies (PIDs) are congenital disorders of immune system. The most common clinical symptoms in PID patients are high susceptibility to infections, inflammation, allergy, malignancies, and autoimmunity.1 The vast majority of PID diseases shows monogenic inheritance and almost 400 genetic defects have been identified so far.2 The overall prevalence of PIDs is ∼1:10,000. However, the high consanguinity and fertility rates lead to a higher prevalence in some countries such as Turkey.3,4
The use of cutting-edge technologies such as next-generation sequencing (NGS)-based methods has facilitated the genetic diagnosis in PID patients.5 These methods also provide the identification of new genetic defects at an increasing rate because next-generation technics allow high-throughput sequencing, including whole genome, whole exome, or a particular number of genes. In comparison with the conventional Sanger sequencing method, whole-exome or targeted sequencing are more cost-effective for genetic diagnosis.6
PID diseases present with a broad range of clinical phenotypes. The occurrence of severe infections in some PIDs such as severe combined immunodeficiency (SCID) can be very early in life. Hematopoietic stem cell transplantation (HSCT) is the only curative treatment for those patients.7 Thus, the rapid genetic diagnosis is highly important and can be life-saving. Moreover, the identification of causative gene defects allows the determination of disease prognosis and treatment options.8 Consequently, NGS may be considered the first option for genetic diagnosis in many PIDs, especially, there is no obvious candidate gene.
Materials and Methods
Patients
Eight PID patients were included in the study. The ages ranged between 4 months and 14 years. The patients were diagnosed as PID according to their clinical and laboratory findings in accordance with the fulfilling criteria of the European Society for Immunodeficiencies.9
The study was approved by the Ethics Committee of Istinye University. All the patient materials were obtained in accordance with the Declaration of Helsinki. Before the initiation of the study, informed consent forms were obtained from the patients or parents.
DNA isolation and sample preparation
Genomic DNA was isolated from peripheral blood using the DNeasy Blood and Tissue Kit (Qiagen). All samples were diluted and then measured on a Qubit 2.0 Fluorometer (Invitrogen/Life Technologies) for a total concentration of 200 ng/μL per each sample.
Whole-exome sequencing and data analysis
NGS library of the DNA samples were prepared by KAPA Library Preparation HTP/LTP Kit (Roche). The SeqCap® EZ Human Exome Probes v3.0 (Roche) was used for exome enrichment. Then we performed 100-bp paired-end-read sequencing on Illumina HiSeq2500 (Illumina, Inc.) platform running on HiSeq 2500 Control Software. The raw data in BCL format was converted to FASTQ by BCL2FASTQ (Illumina) software. The reads were aligned to human genome 19 (GRCh37/Hg19) as Sequence Alignment Map (Sam) format using Burrows-Wheeler Aligner tool. The Binary Alignment Map files were generated from SAM files by SamTools. Picard Tools (by Broad Institute, MIT) package was used for removing of intermediate files and the determination of duplicates. Variant calling was performed by GATK (Genome Analysis Toolkit; Broad Institute, MIT). Finally, we used VarSeq (Golden Helix, Inc.) software for the annotation of the variants. Variants found in 1,000 Genome Project, ExAC, and GnomAD databases with a minor allele frequency of more than 0.01 were excluded, and the other variants were filtered for rare nonsense, missense, and indel changes within the exons and splice-site regions.
Variant validation
All possible disease-causing variants obtained from NGS data were validated by Sanger sequencing according to standard methods as previously described.10
Results
Patient characteristics
A total of 8 PID patients aged between 4 months and 14 years without a genetic diagnosis were enrolled in the study. All patients had Turkish origin except 1 (patient 5 was from Iraq). The consanguinity rate was 63% (5/8). Three of them (patients 1, 2, and 3) were diagnosed as SCID (2 T− B− NK+ and 1 T− B+ NK−). The other patients had been diagnosed as the following: 3 with combined immunodeficiency (patients 4, 5, and 8), 1 possible Mendelian susceptibility to mycobacterial diseases (MSMD) (patient 6), and 1 chronic granulomatous disease (CGD) (patient 7). Patient characteristics are given in Table 1.
Table 1.
Patient Characteristics
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
|---|---|---|---|---|---|---|---|---|
| Age | 4 Months | 5 Months | 6 Months | 10 Months | 1 Year | 1 Months | 2 Years | 14 Years |
| Gender | M | F | F | M | M | M | F | M |
| Nationality | Turkish | Turkish | Turkish | Turkish | Iraqi | Turkish | Turkish | Turkish |
| Consanguinity | No | Yes | Yes | Yes | No | No | Yes | Yes |
| PID | T− B− NK+ SCID | T− B− NK+ SCID | T− B+ NK− SCID | MHC Class-2 | CID | MSMD? | CGD | Hyper IgE? |
| WBC (103/μL) | 2.4 | 2 | 19.2 | 10.5 | 4.2 | 12.4 | 18.9 | 5.2 |
| ALC (103/μL) | 0.9 | 0.8 | 1.5 | 3.8 | 3.7 | 1.9 | 5.4 | 2.1 |
| ANC (103/μL) | 1.5 | 0.9 | 17 | 6.2 | 0.3 | 9.8 | 13.4 | 2.5 |
| CD3 (%/count) | 1/0 | 0/0 | 1/0.15 | 54/2.1 | 73/2.7 | 89/1.7 | 29/1.6 | 63/1.3 |
| CD4 (%/count) | 0.6/0 | 0/0 | 0.5/0.25 | 40/1.5 | 50/1.9 | 81/1.5 | 17/0.9 | 25/0.5 |
| CD8 (%/count) | 0.0 | 0/0 | 0.2 | 13/0.5 | 21/0.8 | 7/0.1 | 11/0.6 | 26/0.5 |
| CD19 (%/count) | 2/0.01 | 1/0.08 | 92/1.4 | 25/1 | 3/0.1 | 7/0.1 | 61/3.3 | 20/0.4 |
| NK (%/count) | 91/0.8 | 89/0.7 | 0.2/0.02 | 12/0.5 | 13/0.5 | 4/0.07 | 4/0.2 | 10/0.2 |
| IgA (mg/dL) | 25 | 15 | 20 | <6 | 63 | 6 | 103 | 12/0.2 |
| IgG (mg/dL) | 25 | 10 | 37 | 25 | 1,502 | 182 | 65 | 311 |
| IgM (mg/dL) | 1,518 | 540 | 254 | 708 | 702 | 19 | 797 | 43 |
PID, primary immunodeficiency; SCID, severe combined immunodeficiency; CID, combined immunodeficiency; MSMD, Mendelian susceptibility to mycobacterial diseases; CGD, chronic granulomatous disease; WBC, white blood cell; ALC, absolute lymphocyte count; ANC, Absolute neutrophil count; Ig, immunoglobulin; NK, natural killer; MHC, major histocompatibility complex.
Patient 1 is a 4-month-old boy presented with skin rashes, eosinophilia, occipital lymphadenopathy, and severe pneumonia suggesting Omenn Syndrome. However, immunophenotyping revealed that there were quite low T and B cells in the circulation and the patient was diagnosed as T− B− NK+ SCID. He underwent HSCT from matched unrelated donor.
Patient 2, a 5-month-old girl, was admitted to our medical center with diarrhea and recurrent pneumonia. In complete blood count test, severe lymphopenia was detected. Flow cytometry analysis showed no T and B cells and T− B− NK+ SCID diagnosis was made. HSCT was performed from her haploidentical mother.
Patient 3 is another SCID patient (T− B+ NK−) suffered from recurrent fever and diarrhea. Then pneumothorax was developed. After SCID diagnosis, HSCT was applied from matched related donor.
Patient 4 was referred to us with severe pneumonia at the age of 3 months. Serum immunoglobulin levels were low and immunophenotyping revealed major histocompatibility complex (MHC) Class-2 deficiency (HLA-DR expression in CD20+ B cells was 0.02%) (Fig. 1).
FIG. 1.
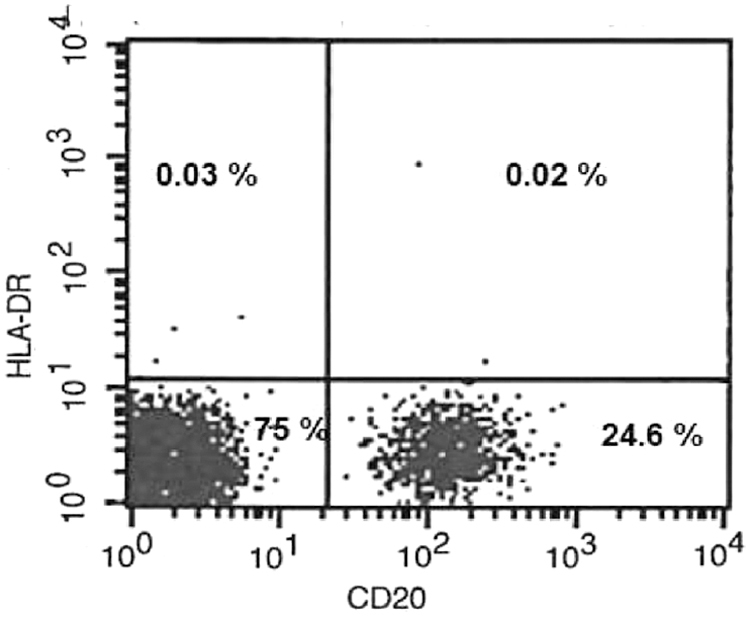
Flow cytometry plot of HLA-DR expression in the patient with MHC Class-2 deficiency. MHC, major histocompatibility complex.
Patient 5 presented with recurrent fever, hepatosplenomegaly, oral candidiasis and pancytopenia. His sibling died with similar clinical symptoms at the age of 5 months. In spleen biopsy specimen, germinal centers and B cells were not detected. Then, immunophenotyping revealed very low B cells (2%) in the circulation. In vitro T cell activation was significantly lower than age-matched healthy control. The patient has been following with possible combined immunodeficiency.
In patient 6, granulomatous lesions were observed in the first month of life. Then we detected Mycobacterium tuberculosis in liver biopsy and MSMD was considered first. There was no consanguinity between parents. The patient's maternal uncle died with similar symptoms in infancy.
Patient 7, a 2-year-old girl, was originally admitted to a local hospital due to skin abscess at the BCG vaccine inoculation site when she was 2 months. Then, deep abscesses were developed in arms and legs and granulomatous inflammation was detected. Nitro blue tetrazolium and dihydrorhodamine tests revealed CGD in the patient.
Patient 8, a 14-year-old boy, had multihospitalizations due to anemia, thrombocytopenia, and splenomegaly 5 years ago. Since he had autoimmunity symptoms, increased bacterial infections, failure to thrive, and mild mental retardation, hyperimmunoglobulin E (IgE) was considered first. However, serum IgE level was found to be normal, while the other Ig levels were low. The patient has been receiving intravenous immunoglobulin treatment.
Exome sequencing
The study, including the steps of sample preparation, sequencing, data analysis, and variant validation, was completed in 1 month. The average coverage of the sequenced regions with a minimum read depth of 30 × and a mapping quality score 30 was 94.8%. This ratio with 50 × and 100 × read depth were 89.3% and 84.6%, respectively.
Among the sequenced patients, we detected 6 variants considered having disease-causing genetic defects based on the respective patients' phenotype (Table 2). All detected variants in the patients and segregations in the parents were validated by Sanger sequencing. All mutations were found in known PID genes. However, 3 of them were novel mutations. Finally, the genetic diagnosis was made in 5 out of 8 patients (63%).
Table 2.
Likely Disease-Causing Variants Detected in the Patients
| Patient | Gene | Variant | Aa change | Type | Hom/Het | Sanger validation |
|---|---|---|---|---|---|---|
| 1 | RAG1 | c.1307C > A | p.Thr436Asn | Missense | Het | Yes |
| 1 | RAG1 | c.2005G > A | p.Glu669Lys | Missense | Het | Yes |
| 2 | RAG2 | c.581C > A | p.Ser194Ter | Nonsense | Hom | Yes |
| 3 | JAK3 | c.2134G > A | p.Gly712Ser | Missense | Hom | Yes |
| 4 | RFXANK | c.634C > T | p.Arg212Ter | Nonsense | Hom | Yes |
| 7 | CYBA | c.58 + 4_58 + 7delAGTG | Deletion | Hom | Yes |
Aa, amino acid; Hom, homozygous; Het, heterozygous.
In patient 1, T− B− NK+ SCID, we found 2 distinct RAG1 variants as compound heterozygous (c.1307C > A and c.2005G > A) with predicted damaging effects “damaging” and “disease-causing” (c.1307C > A: SIFT, score: 0 and Mutation Taster) and (c.2005G > A: SIFT, score: 0 and Mutation Taster). Combined Annotation Dependent Depletion (CADD) scores of these variants were 23.8 and 29.3, respectively. The first missense variant causes a threonine-to-asparagine change and it is located in a conserved region of the DNA-binding domain of the protein (Fig. 2b).11,12 This variant has not been published before. The second one leads an amino acid change (glutamic acid to lysine) in the core domain of RAG1 which is essential for V(D)J recombination.11,12
FIG. 2.

Mutation analyses of the patients. (a) Sanger sequencing results display the mutations in the patients and segregation pattern in the parents. (b) Domain architecture of the RAG1 and JAK3 proteins and locations of the missense mutations. Schematic representations of the truncated RAG2 protein compared with wild type one.
In patient 2, T− B− NK+ SCID, we found a novel, nonsense c.581C > A variant (CADD: 36) in RAG2 gene. The homozygous single-nucleotide variant leads a premature stop codon instead of serine at conserved amino acid position 194 (Fig. 2b).
In patient 3, T− B+ NK− SCID, homozygous c.2134G > A substitution resulted a glycine to serine change was found in JAK3 gene. The damaging effects of this variant were determined as “damaging” (SIFT, score:0) and “disease-causing” (Mutation Taster) with the CADD score 27. This new disease-causing variant is located in a conserved region and affects the Janus homology 2 (JH2) domain of the JAK3 protein (Fig. 2b). JH2 is a pseudokinase domain and essential for the regulation of kinase activity.13
We found a homozygous single-nucleotide change (c.634C > T) in RFXANK gene in the patient 4. The result of this variant was a premature stop codon in amino acid 212 instead of arginine. The same mutation published in the study of Wiszniewski et al.14 as a cause of MHC Class-2 deficiency before.
The last mutation found in our patients was a small deletion in donor splice side region of CYBA gene (c.58 + 4_58 + 7delAGTG). A very recent report15 showed this variant as a disease-causing mutation in a patient with CGD.
Sanger sequencing results of all mutations are given in Fig. 2a.
Discussion
PIDs are rare and heterogeneous diseases mainly present with susceptibility to infections. Different genetic defects may cause the similar clinical phenotypes and conversely, the same mutation may emerge with various clinical symptoms due to the type of mutations, environmental, or epigenetic factors.16 The use of next-generation-based sequencing is critical for genetically heterogeneous diseases such as PIDs, as these technologies provide a rapid and cost-effective genetic diagnosis.5 In this study, we investigated disease-causing variants in 8 PID patients by whole-exome sequencing. We made genetic diagnosis in 5 patients and identified 3 novel mutations.
The overall coverage of whole exome was adequately high with the rate of 94.8% and mapping quality score 30. We obtained proper sequencing data from all 8 patients and found the underlying genetic defects in 5 of them (63%). All causative mutations except 2 heterozygous RAG1 variants were found in the patients from consanguineous parents. Those mutations were autosomal recessive (AR) inherited as expected and found in RAG2, JAK3, RFXANK, and CYBA genes. These data support that in consanguineous populations, AR forms of PIDs are more frequent as in Turkey.17 A recent report18 studied in a highly consanguineous population showed that (consanguinity rate was 81%) more than 90% of the diagnosed patients had AR disorders. In another study that included patients from Iran,19 the consanguinity rate was 78% and the AR forms of PIDs were 63%. Compound heterozygous mutations were detected in 6% of the patients with AR PIDs in both studies. We identified compound heterozygous RAG1 mutations in 1 patient.
The novel mutations were found in RAG1, RAG2, and JAK3 genes. One of the heterozygous RAG1 mutations (c.1307C > A) found in patient 1 (T− B− NK+ SCID) effects the codon 436 in the DNA-binding region of the protein.12 A mutation located in the previous codon of the same domain (p.M435V) was reported in a patient with Omenn Syndrome in the study of Lee et al.20 Interestingly, our patient showed the clinical signs of Omenn syndrome, there was no T and B cells in the circulation. We suggest that the compound heterozygosity with the other RAG1 variant (c.2005G > A) causes the severe SCID phenotype in the patient. Another novel mutation (c.581C > A) leads a premature stop codon in RAG2 gene was found in patient 2 (T− B− NK+ SCID) who had a classical SCID phenotype. In patient 3 (T− B+ NK−), the missense JAK3 variant (c.2134G > A), which was located in pseudokinase domain, has been identified as a disease-causing mutation for the first time. Most of the JAK3 mutations in SCID patients have been reported in this region.21 All novel mutations were considered likely disease-causing genetic defects based on the respective patients' clinical phenotypes.
We also determined 2 identified mutation in RFXANK and CYBA genes.14,15 The patient who has c.634C > T mutation in RFXANK gene was diagnosed as MHC Class-2 deficiency. Finally, a small deletion in donor splice side region of CYBA gene (c.58 + 4_58 + 7delAGTG) was found in patient with CGD.
Three undiagnosed patients in our study presented with combined immunodeficiency and possible MSMD. Two of them were nonconsanguineous. Although the overall total coverage of our study was high, potentially disease-causing mutations in the patients may have been located in small regions with insufficient coverage. Another possible explanation is the genetic defects due to copy number variations (CNVs) not detected by our method or the genetic defects located in introns. In this case, the use of whole-genome sequencing may be useful for the identification of such causative variants.
NGS technology allows to analyze the whole genome, whole exome, or targeted coding regions. In last decade, this technology became the most useful approach for investigation of genetic defects in PID patients. Today whole-exome sequencing and targeted gene panels are in use for routine genetic diagnosis, as well. Although whole-genome sequencing provides more comprehensive evaluation such as the identification of CNVs and other genomic rearrangements, the data analysis is much more complex.22 Thus, whole-exome sequencing seems the most useful approach for PID diagnosis.5
There are lots of studies that showed the diagnostic yield of NGS in PIDs.19,23–32 Some of them included only specific disease populations such as SCID, combined immunodeficiency, common variable immunodeficiency, or primary antibody deficiency.19,23,26,28,32 The number of patients included in the studies reported as between 15 and 696. The percentage of genetic diagnosis for mixed PID groups ranged from 15% to 46%. This rate for specific PID populations ranged between 15% and 79%. In our study, the diagnostic rate was sufficiently high (63%). However, the limitation of our study is the low number of patients. Although the novel variants seem to be likely disease-causing mutations according the patients' clinical phenotypes, the functional effects of the mutations may need to be validated in a larger population. In addition, we did not perform CNV analysis in the study.
In conclusion, we made genetic diagnosis in 5 out of 8 PID patients by whole-exome sequencing and identified 3 novel mutations in known SCID genes. We suggest that our data will contribute to PID literature. Moreover, we showed that whole-exome sequencing is a useful strategy for rapid and accurate diagnosis of PID patients.
Acknowledgments
We thank “Can Sucak Candan Biseyler” Foundation (CSCBF) for their support and contributions during the study. CSCBF was founded in 2018 to honor Can Sucak who lost his life due to complications of PID. CSCBF supports research in the field of primary immunodeficiency and promotes awareness.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was partly supported by the “Can Sucak Candan Biseyler” Foundation.
References
- 1. Parvaneh N, Casanova JL, Notarangelo LD, et al. Primary immunodeficiencies: a rapidly evolving story. J Allergy Clin Immunol 2013; 131:314–323 [DOI] [PubMed] [Google Scholar]
- 2. Bousfiha A, Jeddane L, Picard C, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol 2018; 38:129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Notarangelo LD Primary immunodeficiencies. J Allergy Clin Immunol 2010; 125:S182–S194 [DOI] [PubMed] [Google Scholar]
- 4. Sanal O, Tezcan I. Thirty years of primary immunodeficiencies in Turkey. Ann N Y Acad Sci 2011; 1238:15–23 [DOI] [PubMed] [Google Scholar]
- 5. Meyts I, Bosch B, Bolze A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol 2016; 138:957–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chou J, Ohsumi TK, Geha RS. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiencies. Curr Opin Allergy Clin Immunol 2012; 12:623–628 [DOI] [PubMed] [Google Scholar]
- 7. Sponzilli I, Notarangelo LD. Severe combined immunodeficiency (SCID): from molecular basis to clinical management. Acta Biomed 2011; 82:5–13 [PubMed] [Google Scholar]
- 8. Latif AH, Tabassomi F, Abolhassani H, et al. Molecular diagnosis of primary immunodeficiency diseases in a developing country: Iran as an example. Expert Rev Clin Immunol 2014; 10:385–396 [DOI] [PubMed] [Google Scholar]
- 9. Seidel MG, Kindle G, Gathmann B, et al. The European Society for Immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract 2019; 7:1763–1770 [DOI] [PubMed] [Google Scholar]
- 10. Halacli SO, Ayvaz DC, Sun-Tan C, et al. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: a novel mutation. Clin Immunol 2015; 161:316–323 [DOI] [PubMed] [Google Scholar]
- 11. Matthews AG, Briggs CE, Yamanaka K, et al. Compound heterozygous mutation of Rag1 leading to Omenn syndrome. PLoS One 2015; 10:e0121489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Villa A, Sobacchi C, Notarangelo LD, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood 2001; 97:81–88 [DOI] [PubMed] [Google Scholar]
- 13. Chen M, Cheng A, Candotti F, et al. Complex effects of naturally occurring mutations in the JAK3 pseudokinase domain: evidence for interactions between the kinase and pseudokinase domains. Mol Cell Biol 2000; 20:947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wiszniewski W, Fondaneche MC, Louise-Plence P, et al. Novel mutations in the RFXANK gene: RFX complex containing in-vitro-generated RFXANK mutant binds the promoter without transactivating MHC II. Immunogenetics 2003; 54:747–755 [DOI] [PubMed] [Google Scholar]
- 15. Arts P, Simons A, AlZahrani MS, et al. Exome sequencing in routine diagnostics: a generic test for 254 patients with primary immunodeficiencies. Genome Med 2019; 11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fischer A Human primary immunodeficiency diseases. Immunity 2007; 27:835–845 [DOI] [PubMed] [Google Scholar]
- 17. Cipe FE, Dogu F, Aytekin C, et al. HLA-haploidentical transplantations for primary immunodeficiencies: a single-center experience. Pediatr Transplant 2012; 16:451–457 [DOI] [PubMed] [Google Scholar]
- 18. Al-Herz W, Chou J, Delmonte OM, et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol 2018; 9:3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abolhassani H, Chou J, Bainter W, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol 2018; 141:1450–1458 [DOI] [PubMed] [Google Scholar]
- 20. Lee YN, Frugoni F, Dobbs K, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol 2014; 133:1099–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberts JL, Lengi A, Brown SM, et al. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood 2004; 103:2009–2018 [DOI] [PubMed] [Google Scholar]
- 22. Rae W, Ward D, Mattocks C, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet 2018; 93:647–655 [DOI] [PubMed] [Google Scholar]
- 23. Abolhassani H, Aghamohammadi A, Fang M, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med 2019; 21:243–251 [DOI] [PubMed] [Google Scholar]
- 24. Al-Mousa H, Abouelhoda M, Monies DM, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol 2016; 137:1780–1787 [DOI] [PubMed] [Google Scholar]
- 25. Bisgin A, Boga I, Yilmaz M, et al. The utility of next-generation sequencing for primary immunodeficiency disorders: experience from a clinical diagnostic laboratory. Biomed Res Int 2018; 2018:9647253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Erman B, Bilic I, Hirschmugl T, et al. Investigation of genetic defects in severe combined immunodeficiency patients from Turkey by targeted sequencing. Scand J Immunol 2017; 85:227–234 [DOI] [PubMed] [Google Scholar]
- 27. Gallo V, Dotta L, Giardino G, et al. Diagnostics of primary immunodeficiencies through next-generation sequencing. Front Immunol 2016; 7:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maffucci P, Filion CA, Boisson B, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016; 7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moens LN, Falk-Sorqvist E, Asplund AC, et al. Diagnostics of primary immunodeficiency diseases: a sequencing capture approach. PLoS One 2014; 9:e114901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nijman IJ, van Montfrans JM, Hoogstraat M, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol 2014; 133:529–534 [DOI] [PubMed] [Google Scholar]
- 31. Stoddard JL, Niemela JE, Fleisher TA, et al. Targeted NGS: a cost-effective approach to molecular diagnosis of PIDs. Front Immunol 2014; 5:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu H, Zhang VW, Stray-Pedersen A, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol 2016; 138:1142–1151.e2. [DOI] [PubMed] [Google Scholar]