

Abstract
Aim: Deepening our understanding of the molecular mechanism of abdominal aortic aneurysm (AAA) progression will help set up novel avenues for therapeutic target identification. Our aim here was to unveil the mechanism function of STAT3 in AAA progression.
Methods: We investigated the functional role of STAT3 in AAA by evaluating vascular smooth muscle cell (VSMC) apoptosis and proliferation via terminal deoxynucleotidyl transferase dUTP nick end labeling, western blotting, 5-ethynyl-2′-deoxyuridine, and Cell Counting Kit-8 assays. The interplay of lncRNA-miRNA-mRNA was verified using the luciferase reporter assay and the RNA pull-down, RNA immunoprecipitation, and chromatin immunoprecipitation assays. Quantitative real-time polymerase chain reaction and western blot were utilized to quantitate the RNA and protein levels of the indicated molecules.
Results: Inhibition of STAT3 facilitated VSMC proliferation and repressed VSMC apoptosis. Moreover, It was demonstrated that small nucleolar RNA host gene 16 (SNHG16) sponged miR-106b-5p to release STAT3 from the inhibitory effect of miR-106b-5p. SNHG16 led to the upregulation of STAT3, and STAT3 was an upstream factor in the activation of SNHG16 transcription. Moreover, rescue experiments indicated that SNHG16 depended on STAT3 to regulate VSMC apoptosis and proliferation. In vivo assays showed that SNHG16 knockdown retarded the formation of AAA and upregulated STAT3 in vivo.
Conclusions: We identified that SNHG16/miR-106b-5p/STAT3 formed a complex circuitry for the deterioration of AAA via regulating VSMCs, suggesting a possible target for the pathogenesis of AAA.
Keywords: SNHG16, miR-106b-5p, STAT3, AAA, feedback loop
Introduction
Abdominal aortic aneurysm (AAA) is a focal bulge located in the abdominal aorta, with a 50% larger diameter compared to the proximal normal part (i.e., diameter of the aorta > 30 mm)1). AAA is a main contributing factor to sudden death among the elderly, with individuals above 75 years of age exhibiting a high risk of AAA2). Moreover, it has been revealed that hypercholesterolemia, hypertension, prior vascular disease(s), and family history are considered risk factors for AAA formation3). Depletion of vascular smooth muscle cells (VSMCs) has previously been substantiated to make a crucial contribution to AAA because it led to the elimination of cells that are conducive to connective tissue repair4). Basic studies regarding the molecular mechanism of AAA are relatively limited, and identifying new therapeutic targets is necessary.
Collective works elucidated that RNA transcripts of over 200 nucleotides belonging to long noncoding RNAs (lncRNAs) lack protein-coding potential5). In accordance with nucleotide length, transcripts whose length is between 20 and 24 nt are microRNAs (miRNAs), and the suppression or reduction of target mRNAs can be attributed to the binding of miRNAs to the 3′-untranslated region (3′-UTR) of mRNA6). lncRNAs and miRNAs are crucial modulators of gene expression and are responsible for the exceptional growth, mobility, and metastasis in the process of carcinogenesis7, 8). By functioning as a miRNA sponge, lncRNAs can mediate the availability and biological impact of miRNAs within disease onset and progression9, 10). lncRNA DANCR represses miR-149 expression as a ceRNA and is responsible for the malignant phenotypes of bladder cancer through MSI2 overexpression11). lncRNA AFAP1-AS1 serves as a ceRNA of miR-423-5p during the metastasis of nasopharyngeal carcinoma12). In Hirschsprung's disease, lncRNA FAL1 antagonizes the effect of miR-637 as a ceRNA on AKT1 inhibition13). Small nucleolar RNA host gene 16 (SNHG16) has been recognized as an oncogene in several kinds of cancer, such as cervical cancer14), pancreatic cancer15), and diffuse large B-cell lymphoma16). Recently, it has been proven that SNHG16 is a positive regulator of STAT3 in multiple types of cancer, such as bladder cancer and hepatocellular carcinoma17, 18). However, the relationship between SNHG16 and AAA has never been revealed, and whether SNHG16 regulates STAT3 in VSMCs to regulate AAA remains elusive.
In this work, our aim was to clearly define the biological function of SNHG16 and whether it could regulate STAT3 to affect the function of VSMCs in AAA.
Materials and Methods
VSMC Culture
Human VSMCs were purchased from the American Type Culture Collection (ATCC® PCS-100-012™, Manassas, VA, USA). For the VSMC culture, SmGM-2 Smooth Muscle Growth Medium-2 (Lonza Group, Basel, Switzerland) was the required medium for propagating VSMCs along with 10% fetal bovine serum, as per the manufacturer's instructions. VSMCs were maintained in a 5% CO2 incubator at 37°C.
VSMC Transfection
GenePharma (Shanghai, China) was responsible for the generation of short hairpin RNA (shRNA) against STAT3 and SNHG16 (sh-STAT3#1/2/3, sh-SNHG16#1/2/3) or controls (sh-NC). RiboBio Co., Ltd. (Guangzhou, China) provided the miR-106b-5p mimics, the NC mimics, the overexpression plasmids of STAT3 (pcDNA3.1/STAT3) and SNHG16 (pcDNA3.1/SNHG16 or pcDNA3.1/SNHG16 (Mut), without or with the miR-106b-5p site mutated), and the vector control. VSMC transfection was conducted using the above plasmids via a Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
VSMCs underwent total RNA extraction using a TRIzol reagent (Invitrogen) as per the manufacturer's instructions. TransScript® First-Strand cDNA Synthesis SuperMix (TransGen, Beijing, China) was used for total RNA reverse transcription. The quantitative realtime polymerase chain reaction (qRT-PCR) reaction was performed in an ABI Prism® 7500 Sequence Detection System (Life Technologies, Applied Biosystems, Foster City, CA, USA) using SYBR Green qPCR SuperMix (Life Technologies, Applied Biosystems). The results of STAT3 and SNHG16 were normalized to GAPDH, and their fold changes were measured using the 2−ΔΔCt method.
In vivo AAA model via Porcine Pancreatic Elastase Infusion in Mice
An in vivo model was established via porcine pancreatic elastase (PPE) infusion as per a former description19). C57BL/6 mice were bought from the Jackson Laboratory (Bar Harbor, ME, USA). Briefly, temporary ligatures were placed around the proximal and distal aorta, followed by an aortotomy at the bifurcation, following the infusion of 1.5 U/mL type I PPE (Sigma-Aldrich, St. Louis, MO, USA) contained in saline or the infusion of saline control into the aorta using an insertion catheter at 100 mmHg for 5 min. In order to knock down SNHG16 in vivo, at 0, 7, and 14 days after PPE infusion, the mice were intraperitoneally injected with LNA-anti-SNHG16 GapmeR (site-specific antisense oligonucleotides) or LNA-negative control-GapmeR from Exiqon (scr-GapmeR) (in 0.2 mL of phosphate-buffered saline [PBS], keeping about 3–6 s). Subsequently, the aortotomy underwent repair without lumen constriction. The aortic segments were harvested at 7, 14, or 28 days, including AAA (areas between the bifurcation and left renal artery) and suprarenal abdominal aorta (areas proximal to renal arteries up to the diaphragm). Thereafter, the samples were snap-frozen in liquid nitrogen and maintained at −80°C until further use.
Aortic Diameter Measurement by Ultrasound Imaging
At baseline and at 3, 7, 14, 21, and 28 days following aneurysm induction, the abdominal aortic diameter (AAD) was assessed via B-mode ultrasound imaging on the mice that were operated on following a previous description20). Assessments were carried out according to the random selection of each data set. In order to prevent recall bias, the operators were blinded. To limit operator variations, all assessments were collected by one observer, and the results were analyzed by another observer who was blinded to the treatment group.
Western Blot
VSMCs underwent protein extraction treatment using RIPA Lysis Buffer (Thermo Fisher Scientific, Rockford, IL, USA), followed by measuring the protein purity using a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). Extracts from the VSMCs were transferred to polyvinylidene fluoride (PVDF) membranes and blocked using non-fat powder milk. The PVDF membranes were then incubated at 4°C with anti-STAT3 (Abcam, Cambridge, UK) and anti-GAPDH (internal control; Abcam) overnight. Then, at room temperature, secondary antibodies were used for further incubation. An enhanced chemiluminescence (ECL) system was used to monitor the blot signals, and Image Lab™ software (Bio-Rad, Hercules, CA, USA) was used for density determination.
5-ethynyl-2′-Deoxyuridine Assay
Transfected VSMCs were kept at 37°C under treatment with 50 µmol L−1 5-ethynyl-2′-deoxyuridine (EdU) for 2 h. Cultured VSMCs were then fixed for 30 min with 4% paraformaldehyde (PFA), followed by staining with 1 × Apollo reaction cocktail for 30 min prior to incubation for 30 min with 100 µL of Hoechst 33342 at 5 µg mL−1. A fluorescence microscope was used to examine the percentage of EdU-positive VSMCs.
Cell Counting Kit-8 Assay
Referring to the manufacturer's guidelines for proliferation detection, a Cell Counting Kit-8 (CCK-8) (Beyotime) assay was conducted. In this assay, 96-well plates were used for VSMC cultivation for 24 h, and then the VSMCs were incubated for 1 h in a CCK-8 solution. VSMC proliferation was read using a Tecan Infinite M200 Multimode Microplate Reader (Tecan, Mechelen, Belgium) by measuring the absorbance at 450 nm.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Assay
After the VSMCs were transfected, they were rinsed with PBS and 4% PFA was used to fix them at room temperature for 30 min, followed by the incubation of the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) kit (Roche, Basel, Switzerland) with the VSMCs. Finally, under a microscope (TE200-U, Nikon, Tokyo, Japan), apoptotic cells were captured.
VSMC Cytoplasm/Nucleus Fraction Isolation
The SNHG16 sublocation in VSMCs was monitored using Nuclear and Cytoplasmic Extraction Reagents (Norgen, Belmont, CA, USA). The collected VSMCs were resuspended in a cell fraction buffer and incubated in ice for 10 min. Following centrifugation, the nuclear pellet and supernatant were used for RNA extraction by utilizing a cell disruption buffer, followed by qRT-PCR analysis of fractioned RNAs with GAPDH and U6 as cytoplasm/nucleus normalizers.
Luciferase Reporter Assay
Three fragment sequences or mutated ones of SNHG16 from the analysis of JASPAR were synthesized and ligated into the pGL3 - basic vector (OMEGA Engineering Inc., Norwalk, CT, USA), namely, E1-WT, E2-WUT, E3-WT, E1-MUT, E2-MUT, and E3-MUT. Meanwhile, full-length AC021078.1, SNHG16, and MALAT1 were also ligated into the pGL3 - basic vector to probe the relationship with miR-106b-5p, and the constructed vectors were cotransfected with miR-106b-5p mimics into VSMCs. Then, these vectors were transfected into VSMCs along with STAT3-overexpressing or STAT3-downregulating plasmids or their controls. Then, 48 h later, a Dual Luciferase Assay Kit (OMEGA Engineering Inc.) was utilized to assess the luciferase activity.
RNA Immunoprecipitation Assay
An RNA immunoprecipitation (RIP) assay was conducted using an EZ-Magna RIP Kit (Millipore, Billerica, MA, USA) as per the manufacturer's protocol. RIP magnetic beads were utilized for incubating the whole cell lysate of VSMCs, with anti-Argonaute-2 (anti-Ago2) and normal mouse IgG (Millipore) conjugated onto the beads. Following incubation in Proteinase K buffer, immunoprecipitated RNA by Ago2 was collected and the presence of certain targets was analyzed via qRT-PCR.
RNA Pull-Down Assay
Here, bio-miR-106b-5p sense and bio-miR-106b-5p antisense were produced using a Biotin RNA Labeling Mix (Roche Diagnostics, Indianapolis, IN, USA). VSMC lysates were first mixed with bio-miR-106b-5p sense and bio-miR-106b-5p antisense and then incubated for 60 min with streptavidin-agarose beads (Invitrogen) at 4 °C.qRT-PCR was utilized to purify RNA complex analysis.
Chromatin Immunoprecipitation Assay
A chromatin immunoprecipitation (ChIP) assay was performed by applying a STAT3 - specific antibody (Millipore), IgG antibody (Millipore), and EZ-ChIP-Chromatin Immunoprecipitation (Millipore) in VSMCs. Briefly, 1% formaldehyde was used to cross-link VSMCs, and 125 mM (final concentration) glycine was added to terminate the VSMCs. Clear nuclear lysates were mixed with STAT3 or IgG antibodies for immunoprecipitation. Following the purification of coprecipitated DNA, qRT-PCR was used to quantify the level of the target genes.
Statistical Analysis
All statistical analyses were performed using SPSS ver. 16.0 (SPSS Inc., Chicago, IL, USA), and statistical difference determination was performed using Student's t-test and one-way analysis of variance, which is dependent on group numbers. A p-value of < 0.05 was considered to indicate statistically significant differences. Data obtained from triplicate-performed experiments are expressed as the mean ± standard deviation.
Results
Depletion of STAT3 Induced VSMC Proliferation and Inhibited Apoptosis
It has been elucidated in prior work that STAT3 is evidently upregulated in AAA21). In order to identify the STAT3 function concerning AAA formation, we depleted STAT3 in VSMCs and investigated VSMC proliferation and apoptosis. qRT-PCR and western blot confirmed the depletion efficacy of STAT3 in VSMCs as a result of the lowered level of STAT3 (Fig. 1A). EdU and CCK-8 assays revealed that inhibiting STAT3 promoted the proliferation of VSMCs (Figs. 1B and 1C). On the contrary, VSMC apoptosis was retarded upon the decrease of STAT3, as shown by the TUNEL assay (Fig. 1D, Supplementary Figs. 3A–3B). Moreover, cleaved caspase-3 and Bax (proapoptotic proteins) were decreased and Bcl-2 (an antiapoptotic protein) was increased upon STAT3 knockdown, further indicating that STAT3 silence hindered the apoptosis of VSMCs (Fig. 1E, Supplementary Fig. 2A, and Supplementary Figs. 3C and 3D). Taken together, these data suggested that the knockdown of STAT3 might negatively affect AAA formation via inducing proliferation and retarding apoptosis in VSMCs.
Fig. 1.
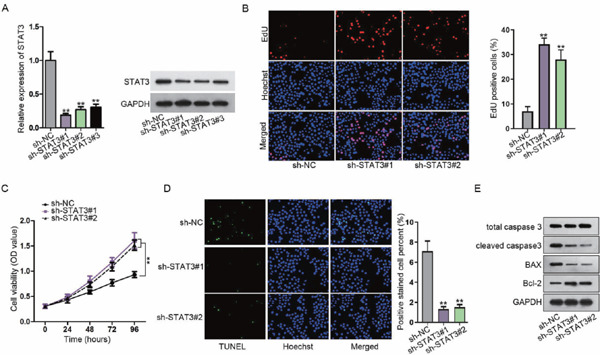
Depletion of STAT3 induces the proliferation and inhibits the apoptosis of vascular smooth muscle cells (VSMC)
(A) The STAT3 level was examined in VSMCs using qRT-PCR and western blotting. (B, C) VSMC proliferation was tested after STAT3 inhibition. (D, E) VSMC apoptosis in response to STAT3 suppression was assessed via detecting apoptosis-related proteins using TUNEL and western blotting. **P < 0.01.
Supplementary Fig. 3.
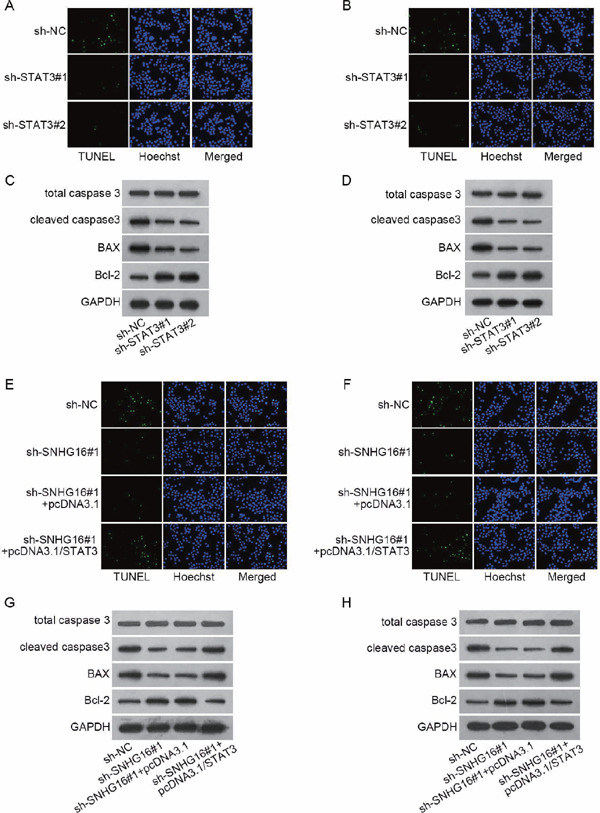
(A–H) Results of the repeated western blot and TUNEL staining in Figs. 1D, 1E, 4C, and 4D.
Supplementary Fig. 2.
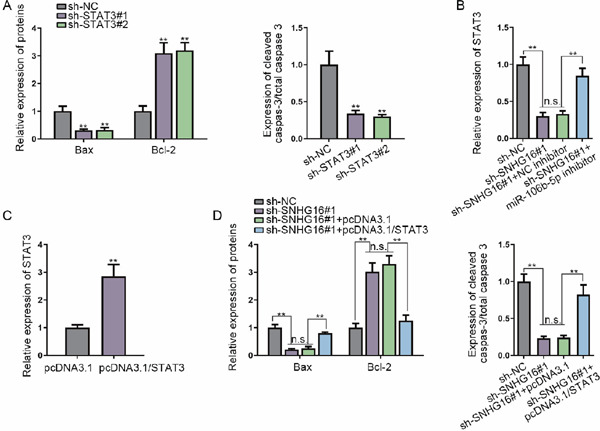
Western blot quantification
(A–D) Quantification of western blot bands in Figs. 1E (A), 2J (B), 3A (C), and 4D (D). **P < 0.01. n.s.: not significant.
STAT3 is a Target of miR-106b-5p and is Positively Regulated by SNHG16
Possible common miRNA response elements in STAT3 were searched through miRDB/miRWalK (http://mirdb.org/; http://mirwalk.umm.uni-heidelberg.de/). Both online tools uncovered 16 miRNAs, among which four were unexplored in terms of their association with STAT3 (Fig. 2A). Therefore, considerable attention was paid to these four miRNAs. qRT-PCR, consistent with western blotting, was used to assay the expression level of STAT3 in response to the upregulation of these miRNAs, and the results demonstrated that only miR-106b-5p mimics reduced STAT3 mRNA and protein expression (Fig. 2B). On the basis of the predicted binding sites between STAT3 and miR-106b-5p, a luciferase reporter assay was carried out. We found that miR-106b-5p mimics led to a decrease in the luciferase activity of STAT3-WT (Fig. 2C). Besides, RIP with an antibody against Ago2 confirmed the enrichment of STAT3 mRNA and miR-106b-5p in the RIP products in the Ago2 group (Fig. 2D). Starbase (http://starbase.sysu.edu. cn/) unmasked three lncRNAs with a high binding affinity, which could possibly serve as a ceRNA of miR-106b-5p. The luciferase reporter assay displayed decreased activity of SNHG16, rather than the other two lncRNAs, upon miR-106b-5p overexpression (Fig. 2E). Further, the luciferase reporter assay verified the binding of miR-106b-5p with SNHG16 (Fig. 2F). In order to verify whether SNHG16 could sponge miR-106b-5p to overexpress STAT3 in a ceRNA manner, the SNHG16 subcellular location was detected. The results suggested that SNHG16 was widely enriched in the cytoplasm in VSMCs (Fig. 2G). The RNA pull-down assay elucidated that STAT3 mRNA and SNHG16 were abundantly pulled down by biomiR-106b-5p sense (Fig. 2H). The impacts of SNHG16 silencing on STAT3 were probed, and SNHG16 was verified to be silenced under the transfection of sh-SNHG16#1/2/3 in VSMCs (Fig. 2I). As presented in Fig. 2J and Supplementary Fig. 2B, SNHG16 depletion caused a decrease in the STAT3 level, whereas downregulating miR-106b-5p counter-acted the inhibitory effect of SNHG16 depletion on STAT3 expression. In addition, we observed from the luciferase reporter assay that overexpression of SNHG16 restored the luciferase activity of the STAT3-WT reporter caused by the miR-106b-5p mimics, but the overexpression of SNHG16 (Mut) (with the miR-106b-5p site mutated) failed to reverse the effect of miR-106b-5p mimics, and the luciferase activity of the STAT3-Mut reporter presented no significant alterations (Supplementary Fig. 1A). Cotransfection of pcDNA3.1/SNHG16, rather than pcDNA3.1/SNHG16 (Mut), restored the mRNA and protein levels of STAT3 that were reduced by the miR-106b-5p mimics in VSMCs (Supplementary Figs. 1B and 1C). These results indicated that SNHG16 competed with STAT3 to bind with miR-106b-5p, which resulted in the upregulation of STAT3.
Fig. 2.
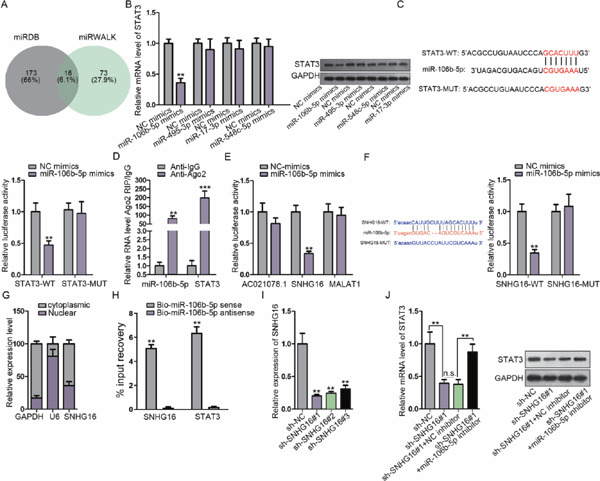
STAT3 is a target of miR-106b-5p and is positively regulated by SNHG16
(A) miRDB and miRWalK were used to predict miRNAs specifically binding to STAT3, among which four miRNAs were unexplored in terms of STAT3 interaction. (B) qRT-PCR combined with western blotting was used to determine the effects of these four miRNAs on STAT3. (C) The aim of the luciferase reporter assay was to discover the interplay between STAT3 and miR-106b-5p. (D) RIP confirmed the binding of miR-106b-5p to STAT3. (E) The top three lncRNAs with high miR-106b-5p binding scores were revealed via Starbase; the luciferase activity of the three lncRNA reporters was assayed using miR-106b-5p mimics. (F) The interaction of miR-106b-5p with SNHG16 was examined using the luciferase reporter assay. (G) qRT-PCR was used to quantify cytoplasmic and nuclear SNHG16 in VSMCs. (H) The RNA pull-down assay was used to probe the combination of miR-106b-5p with SNHG16 and STAT3. (I) Knockdown of SNHG16 was verified using qRT-PCR. (J) The mRNA and protein of STAT3 were determined after transfection with the indicated plasmids using qRT-PCR and western blotting. **P < 0.01, ***P < 0.001. n.s.: not significant.
Supplementary Fig. 1.
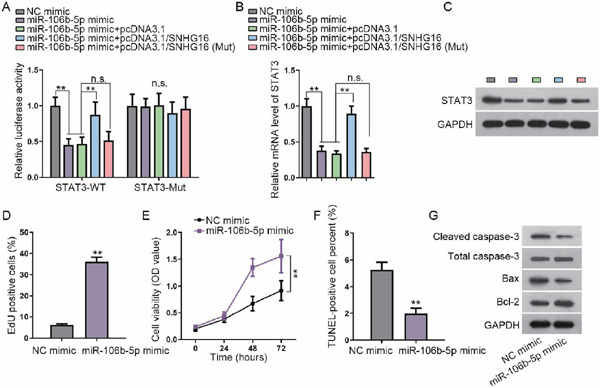
miR-106-5p is required for the regulation of SNHG16 on STAT3 and functions in VSMCs
(A) Luciferase activity of STAT3-WT and STAT3-Mut reporters in VSMCs under transfection of NC mimics, miR-106b-5p mimics, miR-106b-5p mimic+pcDNA3.1, miR-106b-5p mimics+pcDNA3.1/SNHG16, or miR-106b-5p mimics+pcDNA3.1/SNHG16 (Mut) (with the miR-106b-5p site mutated). (B, C) RT-qPCR and western blot results of the STAT3 level in VSMCs of the indicated group. (D, E) The proliferation of VSMCs under transfection of miR-106b-5p mimics versus NC mimics was evaluated using EdU and CCK-8 assays. (F) Quantification of TUNEL-positive VSMCs under miR-106b-5p overexpression versus control. (G) Western blot assay of cleaved caspase-3, total caspase-3, Bax, and Bcl-2 in VSMCs under miR-106b-5p overexpression versus control. **P < 0.01. n.s.: not significant.
Moreover, we tested the functional role of miR-106b-5p in VSMCs. The EdU and CCK-8 assays delineated that miR-106b-5p aggravated the proliferation of VSMCs (Supplementary Figs. 1D and 1E). The rate of apoptosis of VSMCs was reduced by the miR-106b-5p mimics in VSMCs (Supplementary Fig. 1F). In addition, miR-106b-5p overexpression reduced the levels of cleaved caspase-3 and Bax and induced the level of Bcl-2 in VSMCs (Supplementary Fig. 1G). These data indicated that miR-106b-5p might play a negative role in AAA by facilitating the proliferation and retarding the apoptosis of VSMCs. Overall, SNHG16 upregulated STAT3 via competitively binding to miR-106b-5p in VSMCs.
STAT3 is Required for SNHG16 Upregulation
Intriguingly, JASPAR (http://jaspar.genereg.net/) revealed that STAT3 is a transcription activator of SNHG16. qRT-PCR and western blotting were used to assess the overexpression of STAT3 in VSMCs (Fig. 3A, Supplementary Fig. 2C). qRT-PCR showed that any increase or decrease in STAT3 results in high or low expression of SNHG16, respectively, which is consistent with the western blot results (Fig. 3B). Three STAT3 binding sites on the SNHG16 promoter (score > 10) were obtained from the JASPAR tool (http://jaspar.genereg.net/). The STAT3 binding motif and predicted binding sequences on the SNHG16 promoter are shown in Fig. 3C. The luciferase activity of the E1 part of SNHG16 was elevated in the presence of pcDNA3.1/STAT3, and a decreased activity of E1 was found in response to STAT3 inhibition (Fig. 3D). The ChIP assay showed that the E1 part of SNHG16 bound to STAT3 with strong affinity (Fig. 3E). All of these findings showed that SNHG16 was upregulated by STAT3.
Fig. 3.
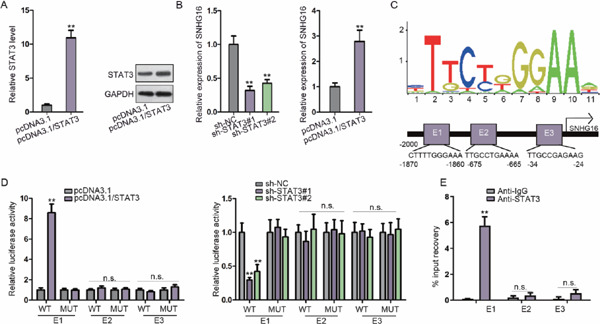
STAT3 is required for the upregulation of SNHG16
(A) The expression of STAT3 under pcDNA3.1/STAT3 transfection was assayed in VSMCs using qRT-PCR and western blotting. (B) The impacts of STAT3 elevation and suppression on SNHG16 were explored using qRT-PCR. (C) Potential binding sites of STAT3 within the promoter of SNHG16. (D) Luciferase activity of several reporters constructed from the SNHG16 promoter region in VSMCs cotransfected with STAT3-overexpressing or STAT3-downregulating plasmids. (E) ChIP was used to assay the enrichment of STAT3 on E1, E2, and E3 of the SNHG16 promoter region compared with IgG. **P < 0.01. n.s.: not significant.
STAT3 Mediates SNHG16-Regulated VSMC Proliferation and Apoptosis
In order to check the role of SNHG16-miR-106b-5p-STAT3 positive feedback loop in VSMC proliferation and apoptosis, rescue assays were designed. As shown in the data of EdU and CCK-8, SNHG16 depletion made a great contribution to VSMC proliferation. Subsequently, STAT3 overexpression rescued the facilitating impacts of SNHG16 depletion on VSMC proliferation (Figs. 4A and 4B). Conversely, SNHG16 depletion hampered VSMC apoptosis, whereas STAT3 overexpression mitigated the suppressive effects of SNHG16 knockdown on VSMC apoptosis (Figs. 4C and 4D, Supplementary Fig. 2D, and Supplementary Figs. 3E–3H). Altogether, SNHG16 knockdown induced proliferation and reduced apoptosis in VSMCs by regulating STAT3.
Fig. 4.
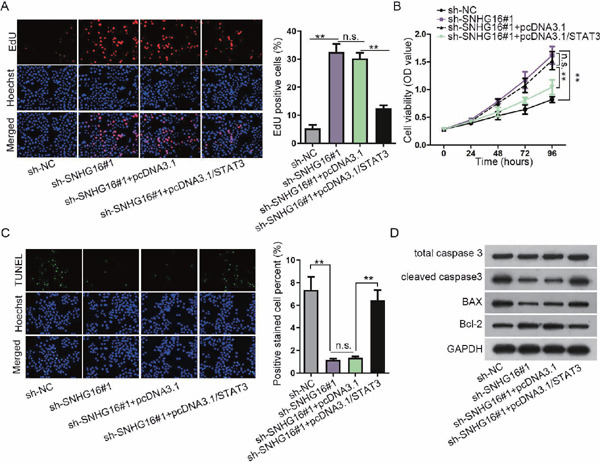
STAT3 mediates SNHG16-regulated VSMC proliferation and apoptosis
(A) EdU assay for the indicated VSMCs transfected with sh-SNHG16#1 or pcDNA3.1/STAT3 plasmids as required. (B) CCK-8 assay for VSMC viability following the transfection of sh-SNHG16#1 or pcDNA3.1/STAT3 plasmids as required. (C) TUNEL assay for VSMCs transfected with specific plasmids as indicated. (D) Western blot analysis of the apoptotic markers in VSMCs in response to several transfections. **P < 0.01. n.s.: not significant.
SNHG16 Knockdown Retards AAA Formation in vivo
Finally, we established a PPE-induced AAA in vivo model in mice to evaluate the effect of SNHG16 on AAA formation. The AAA mice induced by PPE were injected with LNA-anti-SNHG16 GapmeR or the negative control scr-GapmeR. By measuring the AAD, we observed that PPE-induced AAA growth in the mice and SNHG16 knockdown retarded PPE-induced AAA growth in mice (Fig. 5A). The levels of SNHG16 and STAT3 were increased, whereas the level of miR-106b-5p was decreased in PPE-induced AAA mice, and SNHG16 knockdown reversed the increase of SNHG16 and STAT3 in PPE-induced AAA mice (Fig. 5B). Western blot data verified that the levels of STAT3, cleaved caspase-3, and Bax increased, whereas the level of Bcl-2 decreased in PPE-induced AAA mice; such results were reversed by the knockdown of SNHG16 in AAA mice (Fig. 5C). Together, these data suggested that SNHG16 knockdown retarded AAA formation in vivo.
Fig. 5.
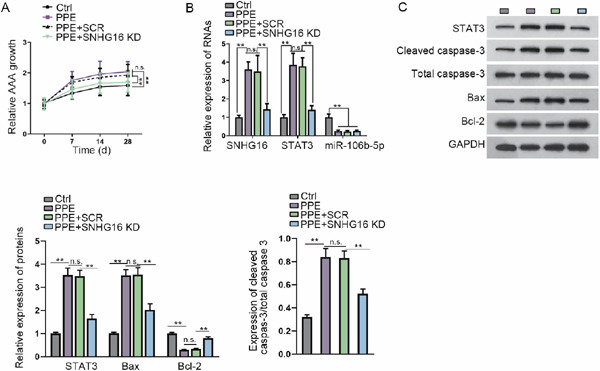
SNHG16 knockdown retards AAA formation in vivo
(A) Relative diameter of the aorta at the baseline and at 7, 14, and 28 days after aneurysm induction. AAD was assessed using B-mode ultrasound imaging. Data are expressed as growth fold change. In vivo SNHG16 knockdown was realized by site-specific antisense oligonucleotides (LNA-GapmeRs, SNHG16 KD) in order to limit aneurysm progression. (B) RT-qPCR results of the levels of SNHG16, STAT3 mRNA, and miR-106b-5p in the mice of each group. (C) Western blot results and quantification of STAT3, cleaved caspase-3, total caspase-3, Bax, and Bcl-2 in the mice of each group. **P < 0.01.
Discussion
Compelling evidence has shown that lncRNA and miRNA are crucial molecules in the initiation and development of multiple human diseases22, 23). For example, long noncoding RNA NNT-AS1 sponges miR-424 to overexpress E2F1 and promote tumorigenesis in gastric cancer24). Enhanced expression of BLACAT1 accelerates the development of hepatocellular carcinoma by competing against hsa-miR-485-5p25). However, a large number of lncRNAs are not well characterized in AAA.
As previously reported, SNHG16 contributes to gastric cancer progression and metastasis26) and is oncogenic in cervical cancer through miR-216A-5p/ZEB1 regulation27). SNHG16 also promotes the aggravation of glioma by sponging miR-4518 to upregulate the expression of PRMT5 28), and it also contributes to the development of bladder cancer29). Despite the widely reported tumor-promoting effect of SNHG16 in multiple diseases, the function of SNHG16 in AAA has never been studied. Therefore, our study is the first one evaluating the apoptosis and proliferation of VSMCs in response to SNHG16 knockdown. We showed that SNHG16 depletion exerts proliferation-promoting and antiapoptotic effects in VSMCs, indicating that SNHG16 might play a facilitating role in AAA via promoting VSMC deletion. As expected, we verified that the knockdown of SNHG16 retards PPE-induced AAA growth in mice and reduces the apoptosis of aortic SMCs in AAA mice.
STAT3 is extensively certified as a transcription driver and could lead to the activation of numerous diseases30, 31). For example, STAT3 contributes to the progression of pancreatic ductal adenocarcinoma32). Although several studies have shown that SNHG16 could positively regulate STAT3 17, 18), we are the first to validate that SNHG16 upregulates the expression of STAT3 in VSMCs. Moreover, STAT3 was for the first time found to be an activator of SNHG16 transcription. Previous work only described that the level of STAT3 increases in AAA, but no study investigated the function of STAT3 and the mechanism behind the regulation of STAT3 expression in AAA. Herein, we discovered that STAT3 knockdown leads to an increase in the proliferation and a decrease in the apoptosis of VSMCs. More importantly, STAT3 could act as a downstream target of lncRNA and also contribute to the upregulation of lncRNAs as a transcription factor, constituting a feedback loop33, 34). For instance, the positive feedback between lncRNA TNK2-AS1 and STAT3 enhances angiogenesis in non-small-cell lung cancer35). In this study, we were the first to demonstrate that miR-106b-5p is sponged by SNHG16 so that STAT3 is upregulated in VSMCs. Additionally, we validated that miR-106b-5p overexpression reduces the apoptosis and aggravates the proliferation of VSMCs. We also validated in vivo that SNHG16 knockdown leads to a decrease in STAT3 in AAA mice.
In conclusion, we have shown that SNHG16 facilitates the apoptosis and retards the proliferation of VSMCs to stimulate AAA formation via the miR-106b-5p/STAT3 feedback loop, indicating that targeting SNHG16 could be a potential approach for the management of AAA.
Acknowledgments
We deeply appreciate all members' support.
Conflicting Interests
None.
References
- 1). Kumar Y, Hooda K, Li S, Goyal P, Gupta N and Adeb M: Abdominal aortic aneurysm: pictorial review of common appearances and complications. Ann Transl Med, 2017; 5: 256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Ren H, Li F, Tian C, Nie H, Wang L, Li HH and Zheng Y: Inhibition of Proteasome Activity by Low-dose Bortezomib Attenuates Angiotensin II-induced Abdominal Aortic Aneurysm in Apo E(-/-) Mice. Sci Rep, 2015; 5: 15730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3). Senemaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB and Coscas R: Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol, 2017; 37: 401-410 [DOI] [PubMed] [Google Scholar]
- 4). Sachdeva J, Mahajan A, Cheng J, Baeten JT, Lilly B, Kuivaniemi H and Hans CP: Smooth muscle cell-specific Notch1 haploinsufficiency restricts the progression of abdominal aortic aneurysm by modulating CTGF expression. PloS one, 2017; 12: e0178538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Ponting CP, Oliver PL and Reik W: Evolution and functions of long noncoding RNAs. Cell, 2009; 136: 629-641 [DOI] [PubMed] [Google Scholar]
- 6). Stark A, Brennecke J, Bushati N, Russell RB and Cohen SM: Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3'UTR evolution. Cell, 2005; 123: 1133-1146 [DOI] [PubMed] [Google Scholar]
- 7). Schmitt AM and Chang HY: Long Noncoding RNAs in Cancer Pathways. Cancer Cell, 2016; 29: 452-463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Klinge CM: Non-coding RNAs: long non-coding RNAs and microRNAs in endocrine-related cancers. Endocr Relat Cancer, 2018; 25: R259-r282 [DOI] [PubMed] [Google Scholar]
- 9). Tay Y, Rinn J and Pandolfi PP: The multilayered complexity of ceRNA crosstalk and competition. Nature, 2014; 505: 344-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Smillie CL, Sirey T and Ponting CP: Complexities of post-transcriptional regulation and the modeling of ceRNA crosstalk. Crit Rev Biochem Mol Biol, 2018; 53: 231-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). Zhan Y, Chen Z, Li Y, He A, He S, Gong Y, Li X and Zhou L: Long non-coding RNA DANCR promotes malignant phenotypes of bladder cancer cells by modulating the miR-149/MSI2 axis as a ceRNA. J Exp Clin Cancer Res: CR, 2018; 37: 273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Lian Y, Xiong F, Yang L, Bo H, Gong Z, Wang Y, Wei F, Tang Y, Li X, Liao Q, Wang H, Zhou M, Xiang B, Wu X, Li Y, Li X, Chen X, Li G, Guo C, Zeng Z and Xiong W: Long noncoding RNA AFAP1-AS1 acts as a competing endogenous RNA of miR-423-5p to facilitate nasopharyngeal carcinoma metastasis through regulating the Rho/Rac pathway. J Exp Clin Cancer Res: CR, 2018; 37: 253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Li Y, Zhou L, Lu C, Shen Q, Su Y, Zhi Z, Wu F, Zhang H, Wen Z, Chen G, Li H, Xia Y and Tang W: Long non-coding RNA FAL1 functions as a ceRNA to antagonize the effect of miR-637 on the down-regulation of AKT1 in Hirschsprung's disease. Cell Prolif, 2018; 51: e12489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14). Tao L, Wang X and Zhou Q: Long noncoding RNA SNHG16 promotes the tumorigenicity of cervical cancer cells by recruiting transcriptional factor SPI1 to upregulate PARP9. Cell Biol Int, 2019; [DOI] [PubMed] [Google Scholar]
- 15). Yu Y, Dong JT, He B, Zou YF, Li XS, Xi CH and Yu Y: LncRNA SNHG16 induces the SREBP2 to promote lipogenesis and enhance the progression of pancreatic cancer. Future Oncol, 2019; [DOI] [PubMed] [Google Scholar]
- 16). Zhu Q, Li Y, Guo Y, Hu L, Xiao Z, Liu X, Wang J, Xu Q and Tong X: Long non-coding RNA SNHG16 promotes proliferation and inhibits apoptosis of diffuse large B-cell lymphoma cells by targeting miR-497-5p/PIM1 axis. J Cell Mol Med, 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Feng F, Chen A, Huang J, Xia Q, Chen Y and Jin X: Long noncoding RNA SNHG16 contributes to the development of bladder cancer via regulating miR-98/STAT3/Wnt/β-catenin pathway axis. J Cell Biochem, 2018; [DOI] [PubMed] [Google Scholar]
- 18). Lin Q, Zheng H, Xu J, Zhang F and Pan H: LncRNA SNHG16 aggravates tumorigenesis and development of hepatocellular carcinoma by sponging miR-4500 and targeting STAT3. J Cell Biochem, 2019; [DOI] [PubMed] [Google Scholar]
- 19). Azuma J, Asagami T, Dalman R and Tsao PS: Creation of murine experimental abdominal aortic aneurysms with elastase. J Vis Exp, 2009; 1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM and Tsao PS: Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest, 2012; 122: 497-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Fernandez-Garcia CE, Tarin C, Roldan-Montero R, Martinez-Lopez D, Torres-Fonseca M, Lindhot JS, Vega de Ceniga M, Egido J, Lopez-Andres N, Blanco-Colio LM and Martin-Ventura JL: Increased galectin-3 levels are associated with abdominal aortic aneurysm progression and inhibition of galectin-3 decreases elastase-induced AAA development. Clin Sci (London, England: 1979), 2017; 131: 2707-2719 [DOI] [PubMed] [Google Scholar]
- 22). Geisler S and Coller J: RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol, 2013; 14: 699-712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Gibb EA, Brown CJ and Lam WL: The functional role of long non-coding RNA in human carcinomas. Mol Cancer, 2011; 10: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Zai W, Chen W, Luan J, Fan J, Zhang X, Wu Z, Ding T, Ju D and Liu H: Dihydroquercetin ameliorated acetaminophen-induced hepatic cytotoxicity via activating JAK2/STAT3 pathway and autophagy. Appl Microbiol Biltechnol, 2018; 102: 1443-1453 [DOI] [PubMed] [Google Scholar]
- 25). Peng Y, Leng W, Duan S and Hong M: Long noncoding RNA BLACAT1 is overexpressed in hepatocellular carcinoma and its downregulation suppressed cancer cell development through endogenously competing against hsa-miR-485-5p. Biomed Pharmacother, 2019; 116: 109027 [DOI] [PubMed] [Google Scholar]
- 26). Lian D, Amin B, Du D and Yan W: Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark, 2017; 21: 151-160 [DOI] [PubMed] [Google Scholar]
- 27). Zhu H, Zeng Y, Zhou CC and Ye W: SNHG16/miR-216-5p/ZEB1 signal pathway contributes to the tumorigenesis of cervical cancer cells. Arch Biochem Biophys, 2018; 637: 1-8 [DOI] [PubMed] [Google Scholar]
- 28). Lu YF, Cai XL, Li ZZ, Lv J, Xiang YA, Chen JJ, Chen WJ, Sun WY, Liu XM and Chen JB: LncRNA SNHG16 Functions as an Oncogene by Sponging MiR-4518 and Up-Regulating PRMT5 Expression in Glioma. Cell Physiol Biochem, 2018; 45: 1975-1985 [DOI] [PubMed] [Google Scholar]
- 29). Feng F, Chen A, Huang J, Xia Q, Chen Y and Jin X: Long noncoding RNA SNHG16 contributes to the development of bladder cancer via regulating miR-98/STAT3/Wnt/beta-catenin pathway axis. J Cell Biochem, 2018; 119: 9408-9418 [DOI] [PubMed] [Google Scholar]
- 30). Yu H, Lee H, Herrmann A, Buettner R and Jove R: Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer, 2014; 14: 736-746 [DOI] [PubMed] [Google Scholar]
- 31). Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S, Zgodzinski W, Majewski M, Wallner G, Fang J, Huang E and Zou W: IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity, 2014; 40: 772-784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32). Fukuda A, Wang SC, Morris JPt, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ and Hebrok M: Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell, 2011; 19: 441-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Liu H, Li C, Yang J, Sun Y, Zhang S, Yang J, Yang L, Wang Y and Jiao B: Long noncoding RNA CASC9/miR-519d/STAT3 positive feedback loop facilitate the glioma tumourigenesis. J Cell Mol Med, 2018; 22: 6338-6344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Mo Y, He L, Lai Z, Wan Z, Chen Q, Pan S, Li L, Li D, Huang J, Xue F and Che S: LINC01287/miR-298/STAT3 feedback loop regulates growth and the epithelial-to-mesenchymal transition phenotype in hepatocellular carcinoma cells. J Exp Clin Cancer Res: CR, 2018; 37: 149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Wang Y, Han D, Pan L and Sun J: The positive feedback between lncRNA TNK2-AS1 and STAT3 enhances angiogenesis in non-small cell lung cancer. Biochem Biophys Res Commun, 2018; 507: 185-192 [DOI] [PubMed] [Google Scholar]