

Abstract
Polycystic kidney disease (PKD) is a commonly inherited disorder characterized by cyst formation and fibrosis (Wilson, N Engl J Med 350:151–164, 2004) and is caused by mutations in cilia or cilia-related proteins, such as polycystin 1 or 2 (Oh and Katsanis, Development 139:443–448, 2012; Kotsis et al., Nephrol Dial Transplant 28:518–526, 2013). A major pathological feature of PKD is the development of interstitial inflammation and fibrosis with an associated accumulation of inflammatory cells (Grantham, N Engl J Med 359:1477–1485, 2008; Zeier et al., Kidney Int 42:1259–1265, 1992; Ibrahim, Sci World J 7:1757–1767, 2007). It is unclear whether inflammation is a driving force for cyst formation or a consequence of the pathology (Ta et al., Nephrology 18:317–330, 2013) as in some murine models cysts are present prior to the increase in inflammatory cells (Phillips et al., Kidney Blood Press Res 30:129–144, 2007; Takahashi et al., J Am Soc Nephrol JASN 1:980–989, 1991), while in other models the increase in inflammatory cells is present prior to or coincident with cyst initiation (Cowley et al., Kidney Int 43:522–534, 1993, Kidney Int 60:2087–2096, 2001). Additional support for inflammation as an important contributor to cystic kidney disease is the increased expression of many pro-inflammatory cytokines in murine models and human patients with cystic kidney disease (Karihaloo et al., J Am Soc Nephrol JASN 22:1809–1814, 2011; Swenson-Fields et al., Kidney Int, 2013; Li et al., Nat Med 14:863–868, 2008a). Based on these data, an emerging model in the field is that disruption of primary cilia on tubule epithelial cells leads to abnormal cytokine cross talk between the epithelium and the inflammatory cells contributing to cyst growth and fibrosis (Ta et al., Nephrology 18:317–330, 2013). These cytokines are produced by interstitial fibroblasts, inflammatory cells, and tubule epithelial cells and activate multiple pathways including the JAK-STAT and NF-κB signaling (Qin et al., J Am Soc Nephrol JASN 23:1309–1318, 2012; Park et al., Am J Nephrol 32:169–178, 2010; Bhunia et al., Cell 109:157–168, 2002). Indeed, inflammatory cells are responsible for producing several of the pro-fibrotic growth factors observed in PKD patients with fibrosis (Nakamura et al., Am J Nephrol 20:32–36, 2000; Wilson et al., J Cell Physiol 150:360–369, 1992; Song et al., Hum Mol Genet 18:2328–2343, 2009; Schieren et al., Nephrol Dial Transplant 21:1816–1824, 2006). These growth factors trigger epithelial cell proliferation and myofibroblast activation that stimulate the production of extracellular matrix (ECM) genes including collagen types 1 and 3 and fibronectin, leading to reduced glomerular function with approximately 50% of ADPKD patients progressing to end-stage renal disease (ESRD). Therefore, treatments designed to reduce inflammation and slow the rate of fibrosis are becoming important targets that hold promise to improve patient life span and quality of life. In fact, recent studies in several PKD mouse models indicate that depletion of macrophages reduces cyst severity. In this chapter, we review the potential mechanisms of interstitial inflammation in PKD with a focus on ADPKD and discuss the role of interstitial inflammation in progression to fibrosis and ESRD.
12.1. Introduction
Polycystic kidney diseases (PKD) are a group of genetically inheritable disorders that are characterized by the formation of cysts in the kidney and other organs (Wilson 2004). Two forms of autosomal PKD exist, depending on their underlying genetic mutation. Autosomal dominant PKD (ADPKD) affects approximately 1 in 1000 people and is caused by a mutation in either PKD1 or PKD2, which encode the polycystin-1 (PC1) or polycystin-2 (PC2) protein, respectively (Wilson 2004). Autosomal recessive PKD (ARPKD) affects 1 in 20,000 individuals and is caused by a mutation in PKHD1, which encodes the protein fibrocystin/polyductin (Wilson 2004). For both forms of the disease, the affected proteins localize to the primary cilia although both proteins are also found in regions outside of the primary cilium (Wilson 2004; Oh and Katsanis 2012). While the function of primary cilia in the kidney remains uncertain, it has been proposed that they are involved in mechanosensation to detect fluid flow through the tubule lumen, in regulation of cell proliferation and oriented cell divisions, as well as in cell-to-cell and cell-to-matrix signaling (Yoder 2007; Yoder et al. 2002; Zimmerman and Yoder 2015; Kotsis et al. 2013; Basten and Giles 2013). In ADPKD, cyst growth is associated with changes in epithelial cell proliferation and differentiation, fluid secretion, and basement membrane abnormalities (Wilson 2004). Expansion of cysts leads to obstruction and compression of surrounding nephrons that can significantly reduce kidney function (Grantham et al. 2011). During late stages of disease progression, cyst formation is accompanied by ECM deposition and fibrosis, further reducing glomerular filtration and eventually leading to ESRD (Wilson 2004; Wilson and Goilav 2007; Wilson and Falkenstein 1995). Importantly, disruption of primary cilia or the polycystins using a variety of mouse models causes cyst formation in the kidney and liver along with associated fibrosis in both organs (Yoder et al. 1995; Ma et al. 2013). Recently, in vivo studies using conditional cilia and polycystin mutant mice showed that the rate of cyst formation depends on the timing of gene deletion. Disruption of cilia or polycystin proteins prior to postnatal day 12–14 caused rapid cyst formation. In contrast, deletion of these proteins after postnatal day 14 caused slow focal cyst formation (Davenport et al. 2007; Piontek et al. 2007; Lin et al. 2003). However, the protracted rate of cyst formation in the adult-induced mutants can be greatly exacerbated by injury (Sharma et al. 2013; Takakura et al. 2009; Patel et al. 2008). Expression profiling data indicated there is an increased response to injury with marked elevation of innate immune response genes in cilia or polycystin mutant samples, leading to the idea that inflammatory cells are involved in PKD progression (Mrug et al. 2008). This is further supported by data showing reduced cystic pathology and improved renal function in mouse PKD models in which macrophages were depleted (Karihaloo et al. 2011; Swenson-Fields et al. 2013). The involvement of other inflammatory cell types in PKD is less understood. Inflammatory cells can serve as potent producers of pro-inflammatory, pro-mitotic, and pro-fibrotic cytokines; therefore, understanding the involvement of inflammatory cells in cyst formation and fibrosis and their relationship with signaling pathways regulated by the primary cilia located on the epithelium will provide innovative insights into the mechanism of disease progression. More important, this understanding will open new targets for therapeutic intervention to slow the progression of cystic kidney disorders in humans.
12.2. Inflammation in the Pathogenesis of PKD
12.2.1. Immune Cells in PKD
Accumulation of inflammatory cells in the renal interstitial space is becoming appreciated as a hallmark of human and animal models of PKD (Fig. 12.1a, b) (Ta et al. 2013; Grantham 2008). One of the most studied inflammatory cells is the macrophage. Macrophages are involved in innate immunity and in tissue development, repair, and homeostasis (Ginhoux et al. 2015; Wynn et al. 2013). In kidney, they express surface markers including F4/80, CD11b, and Ly6c in mice and CD14 and CD16 in humans, which are used to identify different subtypes of macrophage (Nelson et al. 2012). An interesting feature of macrophages is their ability to become polarized and express either pro-inflammatory or anti-inflammatory cytokines in response to signals found in the tissue microenvironment. This paradigm is supported by data showing two distinct responses to treatment with cytokines in vitro (Gordon and Taylor 2005; Mantovani et al. 2002). Macrophages stimulated with LPS or interferon-gamma demonstrate a pro-inflammatory Th1-like phenotype with potent antimicrobial and antitumor activity. This macrophage is referred to as the classically activated or M1-like macrophage. M1 macrophages are characterized by expression of iNOS, TNF-α, and IL-1β amongst others and have a pro-inflammatory function (Sica and Mantovani 2012; Benoit et al. 2008). On the other hand, naı¨ve macrophages treated with interleukin-4 or 13 produce an anti-inflammatory response and are referred to as alternatively activated or M2-like macrophages (Adams and Hamilton 1984). These M2 macrophages express arginase 1 and IL-10 and have anti-inflammatory and wound healing functions (Sica and Mantovani 2012; Benoit et al. 2008). However, several in vivo studies demonstrated that macrophages cannot be clearly defined into either M1- or M2-like states, but have a wide degree of plasticity depending on their environment (Helm et al. 2014; Kratochvill et al. 2015; Stables et al. 2011; Xue et al. 2014; Ginhoux and Jung 2014; Lavin et al. 2014). It is now believed that macrophages which are present in vivo display a range of phenotypes that fall somewhere in between the M1 and M2 spectrum and that they are able to rapidly switch these phenotypes based on external cues. Intriguingly, evidence suggests that M1 macrophages can transition to an M2-like polarization following phagocytosis of necrotic or apoptotic cells (Huen and Cantley 2015; Fadok et al. 1998) or when cultured with renal epithelium demonstrating the importance of the tissue microenvironment on macrophage polarization (Swenson-Fields et al. 2013).
Fig. 12.1.
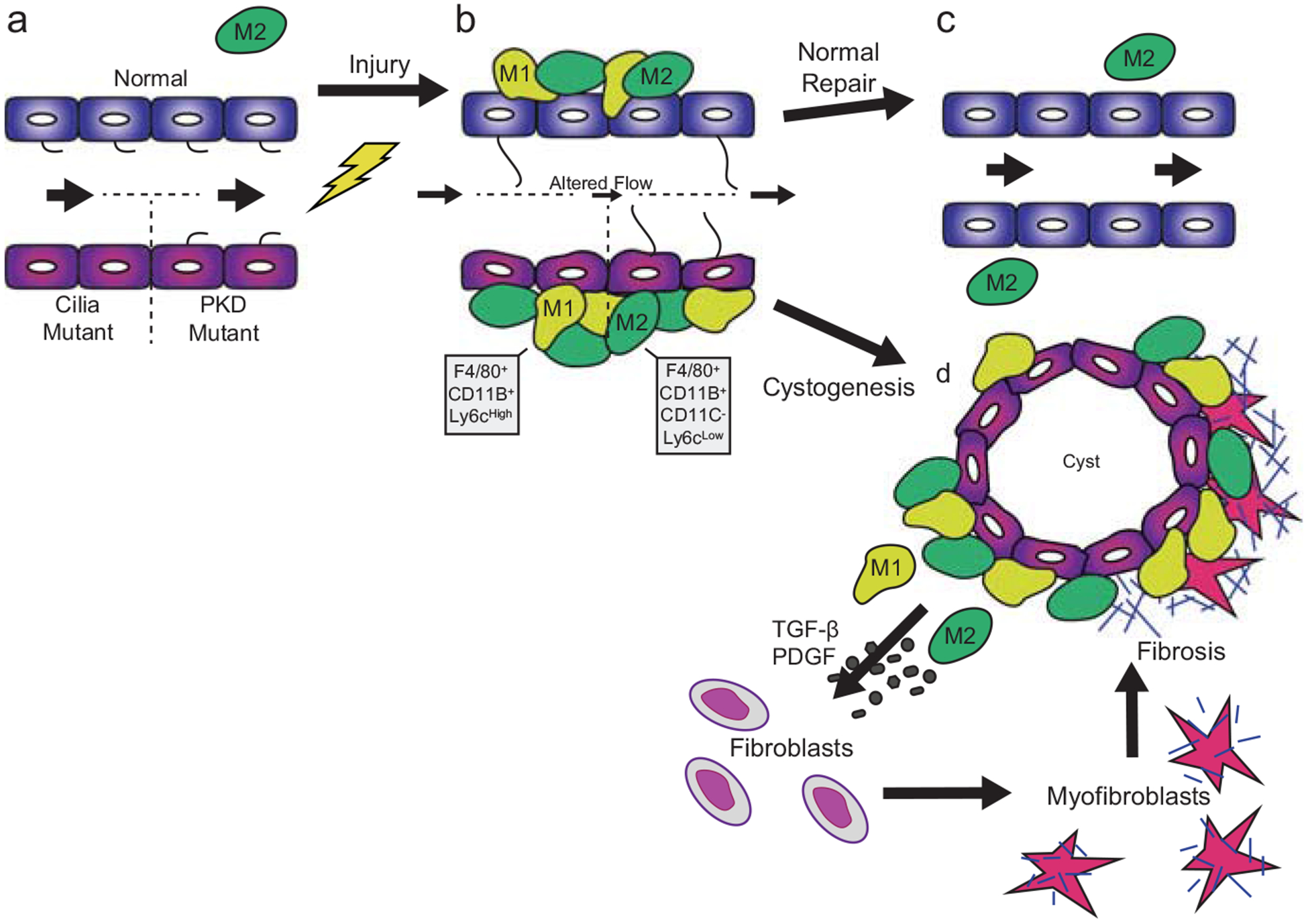
Proposed model focused on the involvement of macrophages following renal injury under normal conditions and in the presence of ciliary or polycystin dysfunction. (a) During steady state, primary cilia protrude into the lumen of renal tubules. Macrophages (M2 like) are present in the interstitial space surrounding the renal tubules. (b) Following injury, there is increased accumulation of macrophage populations (M1-like) or (M2-like) in the interstitial space of the kidney. Furthermore, cilia length initially increases following injury followed by cilia regression during stages of rapid epithelial cell proliferation. (c) During the repair phase in the normal mouse kidney, the injured tubules proliferate, downregulate inflammatory cell accumulation, reform primary cilia, and fully repair the injured epithelium. (d) However, in the cilia or PKD mutant mouse kidney, there is an increased accumulation of macrophage populations (M1 and M2) which fail to properly resolve following injury. The persistent increase in macrophage number leads to enhanced secretion of pro-inflammatory and pro-fibrotic cytokines which causes transition of fibroblasts to a myofibroblast phenotype. These myofibroblasts produce large amounts of extracellular matrix proteins leading to deposition of collagen into the extracellular matrix and fibrosis in the PKD kidney
The origin of macrophages in specific tissues is an area of intense debate. The old model regarding macrophage origin suggests that all macrophages present in tissue can be maintained from a constant supply of bone marrow-derived circulating monocytes (van Furth and Cohn 1968; Katz et al. 1979). However, the new model of macrophage origin has demonstrated that there are two distinct populations of macrophages that can be distinguished by their progenitors, developmental history, and renewal mechanisms (Schulz et al. 2012). One subtype of macrophage is derived from bone marrow progenitor cells and is referred to as monocytes or infiltrating macrophages (in mice F4/80lo and Cd11bhi) once they reach their destination organ (Schulz et al. 2012; Merad et al. 2002; Ginhoux et al. 2010). In contrast, tissue-resident macrophages (in mice F4/80hi and Cd11blo) develop in the yolk sac, enter tissues early during development, and originate from embryonic progenitors (Schulz et al. 2012). The yolk sac-derived macrophages persist in some adult tissues (e.g., epidermis, liver) independent of bone marrow-derived hematopoietic stem cells (HSCs) (Schulz et al. 2012; Ginhoux et al. 2010). They can self-proliferate in response to growth factors such as CSF-1 and other proliferative cytokines (Ginhoux and Jung 2014). In normal kidney, about half of the resident macrophages are derived from the yolk sac and maintained through self-renewal while the remainders are derived from the hematopoietic lineage (Schulz et al. 2012). Resident renal macrophages have important developmental roles influencing ureteric bud branching, producing cytokines, and monitoring surrounding cells for abnormal cell death (Rae et al. 2007; Pollard 2009). The second distinct population of renal macrophage, the infiltrating macrophage, rapidly accumulates in the kidney in response to injury (e.g., following renal ischemia–reperfusion injury; IRI), produces large amounts of pro-inflammatory cytokines, and is associated with worsened kidney injury and fibrosis. Bone marrow-derived infiltrating macrophages can be further classified based on expression of the surface marker Ly6c (Ly6chi, Ly6cint, Ly6clo) (Clements et al. 2015; Lin et al. 2009). Following IR injury, Ly6chi infiltrating macrophages present in the kidney and produce pro-inflammatory cytokines (Clements et al. 2015). Several days after infiltration, these Ly6chi macrophages downregulate expression of Ly6c (Ly6clo) associated with kidney repair and production of extracellular matrix genes (Lin et al. 2009). Despite functional similarities to M1 and M2 macrophages, gene expression analysis comparing Ly6chi, Ly6cint, and Ly6clo macrophages to M1- and M2-like macrophages indicates that these populations do not fit into the canonical M1/M2 classification (Clements et al. 2015).
Both infiltrating and resident macrophage numbers increase in PKD. While data indicate that macrophages impact the rate of cyst formation based on depletion studies in mice, it is not clear whether inflammation is important for cyst initiation or is simply a consequence of the pathology (Ta et al. 2013). In the Lewis polycystic kidney (lpk) and pck rats, data indicate cysts are present prior to the increase in inflammatory cells (Phillips et al. 2007; Takahashi et al. 1991); however, in Han: SPRD rats inflammatory cells are present prior to or coincident with cyst initiation (Cowley et al. 1993, 2001). Furthermore, using a heterozygous ADPKD mouse model exposed to IRI, two groups demonstrated an increased number of neutrophils, macrophages, and pro-inflammatory cytokines prior to cyst formation (Bastos et al. 2009; Prasad et al. 2009). The functional importance of macrophages in cystic disease progression was demonstrated by depletion of macrophages using liposomal clodronate (LC), a phagocytic poison that leads to macrophage death. Treatment of mice with LC improved cystic indices and renal function compared to controls, suggesting that macrophages do have a role in promoting cyst formation (Karihaloo et al. 2011; Swenson-Fields et al. 2013).
The overlap between resident and infiltrating macrophage populations in mice and humans remains uncertain as many of the macrophage surface markers are not shared between species. In human PKD, CD68-positive macrophages, which resemble M2-like or resident macrophages in mice, are present in the renal interstitium of ADPKD patients at both early and advanced stages of kidney disease (Zeier et al. 1992). More recently, HAM56 (a marker for human macrophages)-positive macrophages were shown to be distributed throughout the interstitium of kidneys from ARPKD and ADPKD patients including sites adjacent to cysts. However, the ratio of M2-like macrophages out of total macrophages was similar between control and PKD kidneys (Swenson-Fields et al. 2013), suggesting that the markers used in this study were not specific for human M2-like macrophages or that the ratio of M2-like macrophages was unaltered in the cystic kidneys. Interestingly, epithelial cells derived from control and cystic human kidneys produced soluble factors which induced polarization of macrophages to an M2-like phenotype in vitro (Swenson-Fields et al. 2013). However, marker characterization studies of the M2-like macrophages found within ADPKD kidneys have not revealed the origins of these macrophages (yolk sac or bone marrow derived). The M2-like macrophage response in mice was first noted in a rapidly progressive model of ARPKD, the Cys1-cpk mouse (Mrug et al. 2008); however, when these macrophages enter the kidney remains controversial. In contrast, CD-68 (ED-1)-positive macrophages have been detected in heterozygous Han:SPRD-Cy rats when cystic dilatations were initiating, suggesting a possible causal rather than a responsive role for macrophages in cyst development (Cowley et al. 2001). Subsequent studies using the PKD1fl/fl; Pkhd1-Cre mouse also demonstrated the presence of alternatively activated macrophages based on expression of surface markers (F4/80+, CD45+, CD11c−) and low levels of Ly6C (Ly6Clo) (Karihaloo et al. 2011).
The involvement of other immune cells such as T lymphocytes, mast cells, and neutrophils has been reported in human PKD patients and rodent models (Zeier et al. 1992; Ibrahim 2007; Takahashi et al. 1991; Vogler et al. 1999; Kaspareit-Rittinghausen et al. 1989; McPherson et al. 2004; Bernhardt et al. 2007). Currently, there is significant debate about the presence of dendritic cells in the kidney. Several groups have shown that dendritic cells are present in the kidney based on their surface expression of CD11b, F4/80, and CD11c (Li et al. 2008b; Hochheiser et al. 2013; Hull et al. 2015). However, CD11c has also been reported to be expressed by tissue-resident macrophages, raising the possibility that resident macrophages and dendritic cells may be overlapping populations (Cao et al. 2015). In addition to macrophages, the number of CD4-positive T cells is increased in the interstitial space of ADPKD patients and in animal models of PKD (Zeier et al. 1992; Takahashi et al. 1991; Vogler et al. 1999; Kaspareit-Rittinghausen et al. 1989). Furthermore, it is believed that mast cells may be involved in PKD progression through production of pro-inflammatory molecules including chymase and AngII (McPherson et al. 2004; Nadel 1991; Dell’Italia and Husain 2002). Finally, an elevated number of neutrophils have been reported in human and canine cystic kidney disease models (Bernhardt et al. 2007; O’Leary et al. 1999). However, conclusions about the contribution of neutrophils to disease pathogenesis are limited as the markers used for neutrophils in those studies have subsequently been shown to be non-neutrophil specific. While a wide variety of immune cells are increased in cystic models, their importance in cyst formation remains to be determined. Future studies should be directed at better defining these populations and their respective roles in cyst formation and fibrosis.
12.2.2. Dysregulation of Chemokines and Cytokines in PKD
Chemokines are important for immune cell infiltration, activation, and polarization and in regulating immune cell behavior (Fig. 12.2b, c). Therefore, determining the involvement of chemokines and cytokines in mouse models of PKD will provide insight into their role in modulating immune cells and inflammation associated with cystic disease.
Fig. 12.2.
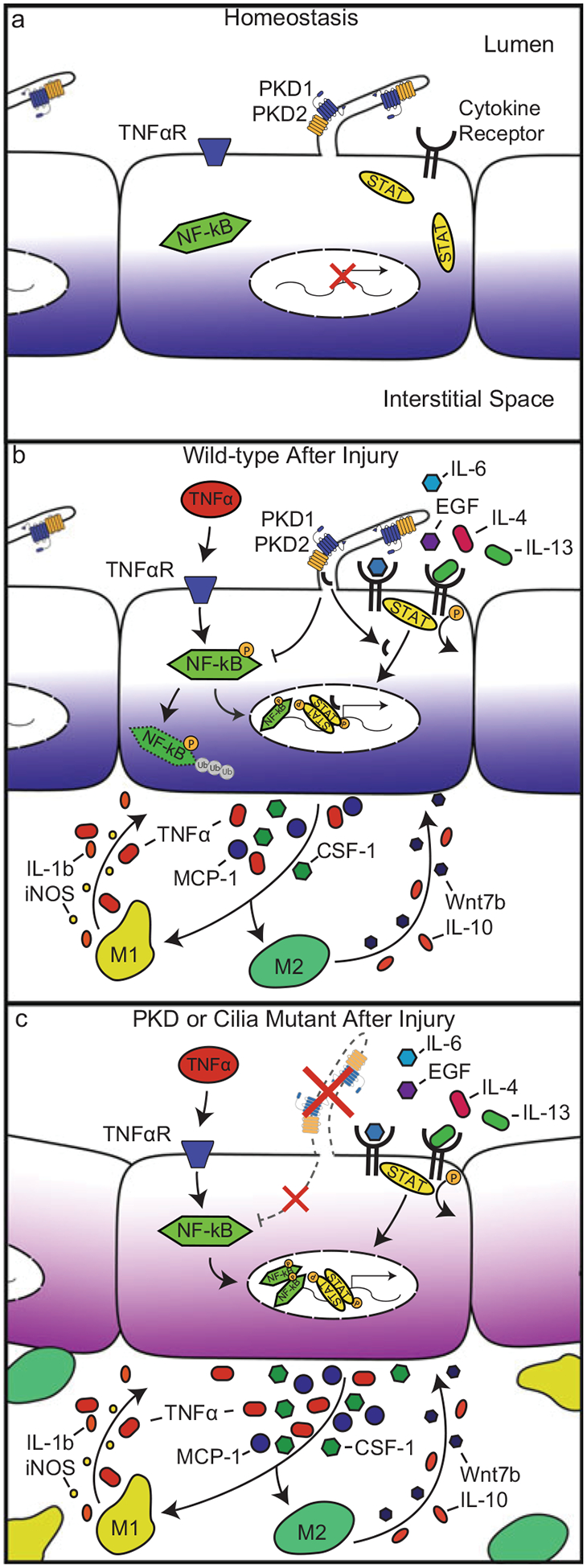
Inflammatory signaling pathways activated under normal or injured conditions in control, cilia, or PKD mutant epithelial cells. (a) During homeostasis, NF-κB and JAK-STAT signaling pathways are not activated. (b) In injured wild-type epithelial cells, PC1 undergoes C-terminal tail (CTT) proteolytic cleavage. The CTT of PC1 translocates into the nucleus where it serves as a co-activator of the STAT signaling pathway. The NF-κB pathway is also activated through ligands, such as TNF-α, binding to their cognate receptors on the cell surface. Collectively, activation of the NF-κB and JAK-STAT signaling pathways induces transient activation of pro-inflammatory genes such as MCP-1, TNF-α, and CSF-1. These cytokines rapidly recruit macrophages to the site of injury leading to production of pro-inflammatory (IL-1β, TNF-α, iNOS) and anti-inflammatory cytokines (IL-10, Wnt7b) that feed back to the injured epithelium leading to tissue repair. (c) In injured PKD or cilia mutant epithelial cells, there is persistently increased NF-κB and JAK-STAT pathway activation. The persistent activation of NF-κB and JAK-STAT signaling leads to enhanced production of pro-inflammatory, pro-fibrotic, and chemoattractant cytokines and increased macrophage accumulation. Accumulated macrophages produce cystogenic cytokines such as TNF-α or Wnt7b that may be responsible for driving cyst formation
One of the best studied chemokines is MCP-1 (Ccl2), which binds to its cognate receptor, CCR2. CCR2 is a member of the CC chemokine family of G-protein coupled receptors and is typically expressed on the surface of monocytes or T cells (Li et al. 2008b; Bruhl et al. 2004). CCR2 mediates cell infiltration in renal inflammatory states such as diabetic nephropathy, glomerulonephritis, and ischemia–reperfusion injury. More important, MCP-1 is markedly elevated in the cyst fluid of ADPKD patients, and increased levels of MCP-1 are associated with worsened renal function and cystic disease outcome (Zheng et al. 2003). The increased expression of MCP-1 in mouse and rat models of PKD appears to parallel that observed in humans. In Han:SPRD rats, MCP-1 mRNA was elevated in homozygous mutant rats compared to wild-type controls (Cowley et al. 2001). Interestingly, heterozygous Han:SPRD cy/+ males displayed higher MCP-1 levels compared to females, which correlates with observations in humans that males often experience higher MCP-1 concentration in urine and serum than females (Cowley et al. 2001). In vitro models suggest that MCP-1 is produced by tubule epithelial cells as PKD1−/− tubule cells have significantly higher expression of MCP-1 compared to control cells. However, in a mouse adriamycin nephropathy model, data suggest that renal F4/80+ CD11c+ mononuclear phagocytes in kidney are also a major source of MCP-1 (Cao et al. 2015). Based on the increased MCP-1 expression across multiple cystic models and human patients, MCP-1 has been proposed as a biomarker for PKD. However, there is no evidence demonstrating a direct causative role for MCP-1 in disease progression as inhibition of MCP-1 signaling using Bindarit did not affect cyst formation (Zoja et al. 2015). Nevertheless, Bindarit treatment did reduce the renal accumulation of ED-1-positive inflammatory cells, suggesting that CCR2-mediated inflammatory cell infiltrates are not a critical component of PKD.
In addition to MCP-1, expression of several other chemokines/cytokines is increased in cystic kidney disease models. This includes osteopontin, IL-1beta, CCL6, CX3CL1, colony-stimulating factor 3 (CSF3), CXCL1, CSF1, CCL28, IL33, IL8, IL6, and IL17D and CXCL8 that function in the recruitment of myeloid-derived cells (macrophages, mast cells, neutrophils) into the cystic kidney. The importance of these chemokines in disease progression still needs to be thoroughly evaluated.
In contrast to the above cytokines, TNF-α appears to have a significant role in cyst formation (Fig. 12.2b, c). In human ADPKD cyst fluid, levels of TNF-α were dramatically increased compared to controls (Li et al. 2008a; Gardner et al. 1991). Using the cpk mouse model, Nakamura et al. demonstrated that cystic mice had increased expression of TNF-α mRNA compared to wild-type controls (Nakamura et al. 1993). Furthermore, TNF-α expression increased with age and cyst severity, suggesting that TNF-α accumulation is associated with cyst progression (Li et al. 2008a; Nakamura et al. 1993). Addition of TNF-α to cultured PKD2+/− and wild-type embryonic kidney explants induced cystogenesis that could be blocked by the TNF-α inhibitor etanercept (Li et al. 2008a). Inhibition of TNF-α-converting enzyme (TACE) led to a significant reduction in cystic index, serum creatinine, and BUN levels in the bpk mouse model of PKD (Dell et al. 2001). Taken together, these data implicate TNF-α as a key contributor to cyst formation.
12.2.3. Inflammatory Pathways in PKD
Recent studies in PKD disease models demonstrate that disruption of ciliary proteins specifically in the tubule epithelium causes cyst formation that is associated with an altered inflammatory response (Ta et al. 2013). Importantly, immune cells do not possess cilia, raising the possibility that cilia on tubule epithelium have a role in regulating inflammatory responses and that dysregulation of signals between the epithelium and inflammatory cells contributes to cyst development (Fig. 12.2) (Qin et al. 2012; Dinsmore and Reiter 2016; Wann and Knight 2012; Wann et al. 2014; Low et al. 2006). The signaling pathway involved and the potential role for either PC1 or PC2 in the inflammatory response are unknown although mice heterozygous for PC1 or PC2 show enhanced inflammation following renal injury compared to their wild-type littermates (Bastos et al. 2009; Prasad et al. 2009). The global gene analysis of human PKD1 kidneys found that many inflammatory pathways are activated in PKD including the JAK-STAT pathway and the NF-κB signaling pathway (Song et al. 2009). NF-κB is an important inflammatory pathway activated in response to a variety of cytokines including TNF-α, IL-1β, IL6, and MCP-1 (Pahl 1999). Following ligand binding, phosphorylation of the NF-κB inhibitor, IκB, results in its degradation and subsequent activation and nuclear translocation of NF-κB. Data from several studies support a role for NF-κB in cyst formation (Qin et al. 2012). PKD1 knockout epithelial cells have increased NF-κB-induced luciferase reporter activity and enhanced phosphorylation of the NF-κB subunit P65 compared to control cells (Qin et al. 2012). Furthermore, Park et al. demonstrated there is an increase of NF-κB protein and that it is phosphorylated in epithelial cells from PKD2 knockout mice, but not in the control mice (Park et al. 2010). Collectively, these data indicate that upregulation of NF-κB pathway activity occurs in animal models of PKD. Wnt7b may function downstream of NF-κB as studies by Qin et al. showed that increased NF-κB signaling results in overexpression of Wnt7b, which is also present in PKD1 mutant mice. Importantly, inhibition of NF-κb or Wnt7b results in a significant decrease in cyst severity in the PKD1 mutant mouse and in organ culture models (Qin et al. 2012).
The JAK-STAT pathway is activated during inflammation and is believed to contribute to cyst formation as demonstrated by increased STAT3 activity in cyst-lining epithelial cells of PKD mutant animals (Fig. 12.2c) (Weimbs and Talbot 2013). In vitro and In vivo studies have shown that PC1 and PC2 are required for JAK1/2 activation and that the c-terminal tail of PC1 regulates STAT3 activation through the src tyrosine kinase (Talbot et al. 2014; Bhunia et al. 2002). The JAK-STAT pathway is also regulated by the suppressor of cytokine signaling (SOCS-1) which limits the inflammatory activity of macrophages. SOCS-1 knockout leads to the development of renal cysts in mice, but it is unknown whether this effect is mediated by inflammation or other contributions of the JAK-STAT signaling pathway (Metcalf et al. 2002).
12.3. Fibrosis in PKD
12.3.1. Extracellular Matrix in PKD
The hallmark of tubule interstitial fibrosis is the accumulation of extracellular matrix (ECM) proteins such as collagen I and III (Zeisberg and Neilson 2010). Similar to many renal disorders, abnormalities in ECM accumulation were also found in kidneys from PKD patients and from orthologous animal models of PKD (Wilson et al. 1992; Wilson and Falkenstein 1995; Wilson and Sherwood 1991; Geng et al. 1999; Wilson and Burrow 1999; Guay-Woodford 2003; Wilson 2008). While ECM accumulation was often described in end-stage kidney disease (ESRD), abnormal ECM composition and increased basement membrane thickness were also observed in early-stage, pre-dialysis ADPKD patients (Wilson et al. 1992). This suggests that tissue remodeling and ECM deposition may be an essential component of cyst progression rather than simply being the result of cyst formation and inflammation. The ADPKD-associated changes include abnormalities in ECM production, composition and turnover, and differences in content of collagen types I, III, and basement membrane collagen type IV, fibronectin, and heparin sulfate proteoglycan (HSPG) (Wilson et al. 1992). These abnormalities were most commonly noted in regions surrounding cysts and interstitial fibroblasts (Norman 2011). While many factors may regulate the activity of pro-fibrotic pathways, recent studies in animal models indicate that polycystins may control production of ECM more directly and that inadequate regulation of this process is responsible for PKD-associated vascular abnormalities (Liu et al. 2014). Importantly, upregulation of the major pro-fibrotic growth factor, transforming growth factor β (TGF-β), was observed in PKD1 haploinsufficient mice and loss of PKD1 leads to increased responsiveness of the cystic epithelium and fibroblasts to TGF-β (Wilson et al. 1992; Song et al. 2009; Hassane et al. 2010). These observations may explain enhanced Smad2/3 nuclear localization observed in PKD patients and animal models (Hassane et al. 2010). Similarly, loss of polycystins is associated with notochord collagen overexpression in zebrafish that is thought to contribute to their body axis curvature defects (Mangos et al. 2010). Together, these data suggest that the PKD-associated changes in ECM composition may be a direct consequence of polycystin mutation.
12.3.2. Dysregulation of ECM Turnover in PKD
Development of fibrosis in PKD and other fibrotic models is dependent on both the amount of ECM that is produced and the extent of matrix turnover which occurs during disease progression (Norman 2011; Catania et al. 2007). Among the many factors that influence ECM turnover, matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) are perhaps the most extensively studied in PKD (Nakamura et al. 2000; Hassane et al. 2010; Catania et al. 2007; Schaefer et al. 1996; Obermuller et al. 2001; Rankin et al. 1996). Despite the clear evidence that MMPs are dysregulated in PKD, the importance and cause of these changes remains controversial. Schaefer and colleagues showed that MMP2 was present in proximal tubule cells from normal rat kidney but significantly decreased in the cystic Han:SPRD rat (Schaefer et al. 1996). This group also demonstrated that TIMP-1 and TIMP-2 mRNA levels were increased in the mutant rat (Schaefer et al. 1996). Paradoxically, MMP14 mRNA was increased in cyst-lining epithelia and distal tubules in the Han:SPRD rat (Obermuller et al. 2001). Inhibition of MMP activity with batimastat reduced cyst number and kidney weight suggesting a detrimental role for MMPs in cyst progression (Obermuller et al. 2001). Likewise, conditioned media from kidney tubule cells isolated from the cpk mouse contained elevated amounts of MMP-2, MMP-3, MMP-9, and TIMP1 (Rankin et al. 1996). The increased expression of MMPs in PKD models was recently confirmed by Hassane et al. who showed increased expression of MMP-2 using a PKD1 mouse model of cyst formation (Hassane et al. 2010). In human ADPKD, patient serum contains increased levels of multiple MMPs, TIMPs, and plasminogen activator inhibitors (PAI) in both pre-dialysis and end-stage ADPKD kidneys (Nakamura et al. 2000). Data demonstrating an important role of the plasminogen activators and inhibitors in PKD are limited, although increased expression of PAI-1 has been reported in both mouse and human PKD samples (Norman 2011; Eddy 2009). It is believed that PAI are pro-fibrotic due to their ability to recruit macrophage and myofibroblasts to the tubulointerstitial area rather than their inhibitory effects on the serine proteases tPA and uPA (Norman 2011; Eddy 2009). Together, these data suggest that MMP expression is dysregulated in PKD and may lead to cyst progression by promoting ECM turnover and allowing expansion of cystic epithelial cells.
The functional consequences of changes in ECM-degrading enzymes in PKD are not fully understood. However, they likely include changes in cellular function and immune regulation. One mechanism of immune regulation may occur through breakdown of collagen I into a PGP fragment. PGP is a key regulator of inflammatory neutrophil accumulation, a process that is central to the pathogenesis of chronic obstructive pulmonary disease (Snelgrove et al. 2010). Since one of two MMPs that participate in generation of PGP is increased in ADPKD (MMP-9), PGP or other ECM collagen fragments may exert important immunoregulatory functions in PKD kidneys.
The interactions of cells with ECM or its breakdown products are mediated by matrix receptors including integrins and syndecans (Geiger et al. 2001). Following binding of ECM to these receptors, focal adhesion complexes form allowing for intracellular signaling to occur through a variety of pathways (Ehrhardt et al. 2002; Wozniak et al. 2004). To date, it has been demonstrated that several ECM receptor components are increased in PKD patients including various integrins, syndecan-4, as well as the integrin-associated focal adhesion kinase (Geng et al. 1999; Wilson and Burrow 1999; Zeltner et al. 2008; Wallace et al. 2014; Joly et al. 2003). Mitigation of renal cyst formation in response to disruption of these pathways (e.g., by genetic targeting of integrin in mice) points to functional relevance of the cell–ECM interactions in the pathogenesis of PKD (Lee et al. 2015). Of note, a hypomorphic mutation in the laminin alpha5 gene was sufficient to drive renal cyst formation (Shannon et al. 2006).
12.3.3. Fibrotic Cells in PKD
Myofibroblasts are specialized cells that can exert large contractile forces, mediate wound healing, and substantially contribute to the expansion of ECM and development of renal tubulointerstitial fibrosis (Fig. 12.1d) (Qi et al. 2006). A hallmark of myofibroblasts is expression of cytoskeletal alpha smooth muscle actin (αSMA) which is incorporated into stress fibers. While αSMA expressing interstitial cells were found in focal areas during early-stage ADPKD, they were found in abundance in end-stage ADPKD kidneys (Hassane et al. 2010). αSMA-positive cells were also found in kidneys from PKD1-deficient mice (Hassane et al. 2010) and end-stage PKD kidneys from Han:SPRD-Cy rats (Song et al. 2009; Schieren et al. 2006). Cells expressing high amounts of αSMA are potent producers of ECM proteins including type I and III collagen. Production of excess ECM proteins leads to interstitial fibrosis, tubule and nephron loss, a decline in glomerular filtration rate, and eventual end-stage renal disease. Although the origin of renal PKD myofibroblasts remains unknown, it appears that these cells can differentiate from several different precursors, including resident interstitial fibroblasts, or pericytes (Kuo et al. 1997). Such transformation may be regulated by TGF-β or perhaps by PKD-related alterations in calcium flux (Follonier Castella et al. 2010). Targeting of αSMA-positive cells for anti-fibrotic therapy to help reduce scarring and retain renal function is an attractive idea, although, to date, no such treatment exists.
The origin of myofibroblasts in the kidney of PKD mouse models remains unknown; however, in a UUO model of renal fibrosis, it was shown that a majority of myofibroblasts in the kidney were derived through proliferation of resident fibroblasts although bone marrow-derived fibrocytes also contributed (~35%) to the myofibroblast population (LeBleu et al. 2013). Using fate mapping studies, this group concluded the vascular pericytes likely do not contribute to renal fibrosis. In contrast, Humphreys and colleagues determined that resident pericytes were major contributors to renal myofibroblasts following UUO-induced injury (Humphreys et al. 2010). While the exact reasoning for the discrepancy is unknown, both groups showed that epithelial-to-mesenchymal transition (EMT) was a minor contributor to the SMA+ myofibroblast population following UUO-induced renal injury. Interestingly, EMT may be enhanced in PKD through factors such as TGF-β, platelet-derived growth factor (PDGF), FGF-1, and Insulin-like growth factor (IGF) I and II, all of which are upregulated in cystic disease (Norman 2011; Kuo et al. 1997). Togawa et al. showed that expression of E-cadherin and beta-catenin, components important for epithelial adhesion, was significantly attenuated in tubules from PCK rats (Togawa et al. 2011). Furthermore, cystic epithelial cells found in fibrotic regions showed increased mesenchymal markers including vimentin and fibronectin (Togawa et al. 2011). Gene expression analysis from human ADPKD samples showed significant alterations in the expression of genes related to smooth muscle which led the authors to speculate on a possible EMT in these patients (Schieren et al. 2006). However, a recent study concluded that EMT provides only a minor contribution to the SMA-positive population in PKD kidneys using a PKD1 mouse model (Hassane et al. 2010). Despite evidence that EMT plays an important role in the formation of myofibroblasts in the kidney (Zeisberg and Neilson 2010), the extent of EMT and its importance for the pathogenesis of cysts and fibrosis in PKD patients is unknown.
The involvement of macrophages in promoting accumulation and degradation of extracellular matrix proteins is well established. Macrophages secrete growth factors and cytokines that induce myofibroblast activation and extracellular matrix production leading to fibrosis (Fig. 12.1d) (Vernon et al. 2010). The most well-defined macrophage-secreted growth factor is TGF-β, which drives extracellular matrix production and fibrosis through binding to the TGF-β receptor present on myofibroblasts (Wynn and Barron 2010). In addition to TGF-β, macrophages are major producers of PDGF and drive extracellular matrix production and fibrosis in a bleomycin model of idiopathic pulmonary fibrosis (Bonner 2004). Macrophages also produce other pro-fibrotic growth factors including galectin-3, insulin-like growth factor-I, IL-4, and IL-13, factors that are potent inducers of extracellular matrix production and fibrosis (Wynn and Barron 2010). Specifically, galectin-3 is critical for activation of renal fibroblasts and knockout of galectin-3 reduced fibrosis following unilateral ureteric obstruction (UUO) (Henderson et al. 2008). Despite extensive studies demonstrating that macrophages promote fibrosis, other data indicate that macrophages produce several matrix-degrading proteases including MMP1, MMP2, MMP8, MMP9, and MMP13 and TIMPs (Wynn and Barron 2010). For example, overexpression of MM9 in alveolar macrophages significantly reduced fibrotic lesions and hydroxyproline content compared to controls (Cabrera et al. 2007). Furthermore, adoptive transfer of bone marrow-derived macrophages into leukocyte-depleted mice significantly attenuated fibrosis following UUO-induced injury (Nishida et al. 2005). Collectively, these data suggest that different macrophage subtypes are responsible for promoting or regressing fibrosis through production of matrix promoting or degrading factors although this concept has not been extensively studied in PKD models.
12.3.4. Fibrotic Cytokines and Signaling Pathways
A number of intracellular signaling pathways are activated in cyst-lining epithelial cells in ADPKD, many of which are also activated in fibrosis (Norman 2011). TGF-β, a major fibrogenic cytokine, is highly expressed in cystic epithelia in human, rat, and mouse models of PKD. Studies in rodent models suggest that increased TGF-β expression and signaling correlate with later stages of the disease and that TGF-β is important in cyst progression and fibrosis rather than cyst initiation (Hassane et al. 2010). Activin A is a cytokine that belongs to the TGF-β family of growth factors. During development, activin A is produced by the ureteric bud (UB) and negatively regulates its branching; furthermore, activin A is also involved in kidney regeneration following injury, suggesting an important role for this cytokine during kidney formation/regeneration (Maeshima et al. 2003; Ritvos et al. 1995). More important, inhibition of activin A signaling slows the progression of PKD (Leonhard et al. 2016). Additionally, activin A expression is increased in a PKD1 mouse, model raising the possibility that altered activin A expression has a role in renal cystic disease (Hassane et al. 2010). Its expression is also associated with conditions wherein loss of cilia or polycystins causes rapid cyst formation (e.g., after injury or in juvenile ages). These studies indicate that activin A is a critical cytokine in cyst progression and ESRD since it is involved in epithelial regeneration and is a member of the TGF-β family of pro-fibrotic growth factors capable of inducing ECM gene expression in renal cells (Yamashita et al. 2004).
In addition to TGF-β, several other pro-fibrotic growth factors were associated with ECM production in PKD including epithelial growth factor (EGF), fibroblast growth factor-1 (FGF-1), and hepatocyte growth factor (HGF). Levels of these pro-fibrotic cytokines increased in PKD over time and their highest levels were often found in end-stage or near-end-stage PKD kidneys (Kuo et al. 1997; Du and Wilson 1995). Furthermore, overexpression of the bone morphogenic protein receptor (ALK-3) or knockout of Bmp-7 triggers cyst formation in mice (Hu et al. 2003; Jena et al. 1997). Importantly, in complementary studies, BMP-7 treatment attenuated renal cystic disease progression in a jck mouse model of PKD (Leonhard et al. 2016). Similarly, EGF is another important regulator of cystic epithelial cell proliferation and inhibition of this pathway is being targeted for therapeutic development (Du and Wilson 1995). It could be presumed that targeting multiple pro-fibrotic pathways may be an effective means of intervention for patients progressing toward ESRD. Overall, upregulation of multiple pro-fibrotic growth factor components suggests that these cytokines work in conjunction to increase extracellular matrix during PKD progression. However, the specific pathways involved in fibrosis associated with ADPKD remain to be defined.
12.3.5. Anti-Fibrotic Therapies in PKD
Slowing cyst expansion and the development of fibrosis is key to prolonging patient life span and improving palliative care. Although progress has been made (Boor et al. 2007; Fine and Norman 2008; Vilayur and Harris 2009; Albaqumi et al. 2008; Grgic et al. 2009), development of effective anti-fibrotic strategies in PKD patients is very limited. Therefore, it is imperative that we better understand the mechanisms underlying the initiation and progression of fibrosis. Identifying key molecular mechanisms that are impacted during the development of cysts and how they ultimately contribute to ESRD should provide novel targets for anti-fibrotic therapies.
References
- Adams DO, Hamilton TA (1984) The cell biology of macrophage activation. Annu Rev Immunol 2:283–318 [DOI] [PubMed] [Google Scholar]
- Albaqumi M, Srivastava S, Li Z, Zhdnova O, Wulff H, Itani O, Wallace DP, Skolnik EY (2008) KCa3.1 potassium channels are critical for cAMP-dependent chloride secretion and cyst growth in autosomal-dominant polycystic kidney disease. Kidney Int 74:740–749 [DOI] [PubMed] [Google Scholar]
- Basten SG, Giles RH (2013) Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia 2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos AP, Piontek K, Silva AM, Martini D, Menezes LF, Fonseca JM, Fonseca II, Germino GG, Onuchic LF (2009) Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J Am Soc Nephrol JASN 20:2389–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M, Desnues B, Mege JL (2008) Macrophage polarization in bacterial infections. J Immunol 181:3733–3739 [DOI] [PubMed] [Google Scholar]
- Bernhardt WM, Wiesener MS, Weidemann A, Schmitt R, Weichert W, Lechler P, Campean V, Ong AC, Willam C, Gretz N, Eckardt KU (2007) Involvement of hypoxia-inducible transcription factors in polycystic kidney disease. Am J Pathol 170:830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, Germino FJ, Germino GG (2002) PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 109:157–168 [DOI] [PubMed] [Google Scholar]
- Bonner JC (2004) Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15:255–273 [DOI] [PubMed] [Google Scholar]
- Boor P, Sebekova K, Ostendorf T, Floege J (2007) Treatment targets in renal fibrosis. Nephrol Dial Transplant 22:3391–3407 [DOI] [PubMed] [Google Scholar]
- Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, Schlondorff D, Mack M (2004) Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol 172:890–898 [DOI] [PubMed] [Google Scholar]
- Cabrera S, Gaxiola M, Arreola JL, Ramirez R, Jara P, D’Armiento J, Richards T, Selman M, Pardo A (2007) Overexpression of MMP9 in macrophages attenuates pulmonary fibrosis induced by bleomycin. Int J Biochem Cell Biol 39:2324–2338 [DOI] [PubMed] [Google Scholar]
- Cao Q, Wang Y, Wang XM, Lu J, Lee VW, Ye Q, Nguyen H, Zheng G, Zhao Y, Alexander SI, Harris DC (2015) Renal F4/80+ CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J Am Soc Nephrol JASN 26:349–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania JM, Chen G, Parrish AR (2007) Role of matrix metalloproteinases in renal pathophysiologies. Am J Physiol Renal Physiol 292:F905–F911 [DOI] [PubMed] [Google Scholar]
- Clements M, Gershenovich M, Chaber C, Campos-Rivera J, Du P, Zhang M, Ledbetter S, Zuk A (2015) Differential Ly6C expression after renal ischemia-reperfusion identifies unique macrophage populations. J Am Soc Nephrol JASN 27:159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley BD Jr, Gudapaty S, Kraybill AL, Barash BD, Harding MA, Calvet JP, Gattone VH 2nd (1993) Autosomal-dominant polycystic kidney disease in the rat. Kidney Int 43:522–534 [DOI] [PubMed] [Google Scholar]
- Cowley BD Jr, Ricardo SD, Nagao S, Diamond JR (2001) Increased renal expression of monocyte chemoattractant protein-1 and osteopontin in ADPKD in rats. Kidney Int 60:2087–2096 [DOI] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17:1586–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell KM, Nemo R, Sweeney WE Jr, Levin JI, Frost P, Avner ED (2001) A novel inhibitor of tumor necrosis factor-alpha converting enzyme ameliorates polycystic kidney disease. Kidney Int 60:1240–1248 [DOI] [PubMed] [Google Scholar]
- Dell’Italia LJ, Husain A (2002) Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol 17:374–379 [DOI] [PubMed] [Google Scholar]
- Dinsmore C, Reiter JF (2016) Endothelial primary cilia inhibit atherosclerosis. EMBO Rep 17:156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wilson PD (1995) Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Phys 269:C487–C495 [DOI] [PubMed] [Google Scholar]
- Eddy AA (2009) Serine proteases, inhibitors and receptors in renal fibrosis. Thromb Haemost 101:656–664 [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt A, Ehrhardt GR, Guo X, Schrader JW (2002) Ras and relatives—job sharing and networking keep an old family together. Exp Hematol 30:1089–1106 [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101:890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine LG, Norman JT (2008) Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int 74:867–872 [DOI] [PubMed] [Google Scholar]
- Follonier Castella L, Gabbiani G, McCulloch CA, Hinz B (2010) Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res 316:2390–2401 [DOI] [PubMed] [Google Scholar]
- Gardner KD Jr, Burnside JS, Elzinga LW, Locksley RM (1991) Cytokines in fluids from polycystic kidneys. Kidney Int 39:718–724 [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM (2001) Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol 2:793–805 [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Jung S (2014) Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14:392–404 [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK (2015) New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17:34–40 [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev Immunol 5:953–964 [DOI] [PubMed] [Google Scholar]
- Grantham JJ (2008) Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 359:1477–1485 [DOI] [PubMed] [Google Scholar]
- Grantham JJ, Mulamalla S, Swenson-Fields KI (2011) Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 7:556–566 [DOI] [PubMed] [Google Scholar]
- Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J, Muller A, Kaistha A, Schmidt C, Raman G, Wulff H, Strutz F, Grone HJ, Kohler R, Hoyer J (2009) Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc Natl Acad Sci USA 106:14518–14523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guay-Woodford LM (2003) Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 285:F1034–F1049 [DOI] [PubMed] [Google Scholar]
- Hassane S, Leonhard WN, van der Wal A, Hawinkels LJ, Lantinga-van Leeuwen IS, ten Dijke P, Breuning MH, de Heer E, Peters DJ (2010) Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol 222:21–31 [DOI] [PubMed] [Google Scholar]
- Helm O, Held-Feindt J, Grage-Griebenow E, Reiling N, Ungefroren H, Vogel I, Kruger U, Becker T, Ebsen M, Rocken C, Kabelitz D, Schafer H, Sebens S (2014) Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer 135:843–861 [DOI] [PubMed] [Google Scholar]
- Henderson NC, Mackinnon AC, Farnworth SL, Kipari T, Haslett C, Iredale JP, Liu FT, Hughes J, Sethi T (2008) Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 172:288–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochheiser K, Heuser C, Krause TA, Teteris S, Ilias A, Weisheit C, Hoss F, Tittel AP, Knolle PA, Panzer U, Engel DR, Tharaux PL, Kurts C (2013) Exclusive CX3CR1 dependence of kidney DCs impacts glomerulonephritis progression. J Clin Invest 123:4242–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Piscione TD, Rosenblum ND (2003) Elevated SMAD1/beta-catenin molecular complexes and renal medullary cystic dysplasia in ALK3 transgenic mice. Development 130:2753–2766 [DOI] [PubMed] [Google Scholar]
- Huen SC, Cantley LG (2015) Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol 30:199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull TD, Kamal AI, Boddu R, Bolisetty S, Guo L, Tisher CC, Rangarajan S, Chen B, Curtis LM, George JF, Agarwal A (2015) Heme oxygenase-1 regulates myeloid cell trafficking in AKI. J Am Soc Nephrol JASN 26:2139–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim S (2007) Increased apoptosis and proliferative capacity are early events in cyst formation in autosomal-dominant, polycystic kidney disease. Sci World J 7:1757–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena N, Martin-Seisdedos C, McCue P, Croce CM (1997) BMP7 null mutation in mice: developmental defects in skeleton, kidney, and eye. Exp Cell Res 230:28–37 [DOI] [PubMed] [Google Scholar]
- Joly D, Morel V, Hummel A, Ruello A, Nusbaum P, Patey N, Noel LH, Rousselle P, Knebelmann B (2003) Beta4 integrin and laminin 5 are aberrantly expressed in polycystic kidney disease: role in increased cell adhesion and migration. Am J Pathol 163:1791–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, Somlo S, Cantley LG (2011) Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol JASN 22:1809–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspareit-Rittinghausen J, Rapp K, Deerberg F, Wcislo A, Messow C (1989) Hereditary polycystic kidney disease associated with osteorenal syndrome in rats. Vet Pathol 26:195–201 [DOI] [PubMed] [Google Scholar]
- Katz SI, Tamaki K, Sachs DH (1979) Epidermal Langerhans cells are derived from cells originating in bone marrow. Nature 282:324–326 [DOI] [PubMed] [Google Scholar]
- Kotsis F, Boehlke C, Kuehn EW (2013) The ciliary flow sensor and polycystic kidney disease. Nephrol Dial Transplant 28:518–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, McEvoy J, Roussel MF, Dyer MA, Qualls JE, Murray PJ (2015) TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep 12:1902–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo NT, Norman JT, Wilson PD (1997) Acidic FGF regulation of hyperproliferation of fibroblasts in human autosomal dominant polycystic kidney disease. Biochem Mol Med 61:178–191 [DOI] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I (2014) Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R (2013) Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19:1047–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Boctor S, Barisoni LM, Gusella GL (2015) Inactivation of integrin-beta1 prevents the development of polycystic kidney disease after the loss of polycystin-1. J Am Soc Nephrol JASN 26:888–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard WN, Kunnen SJ, Plugge AJ, Pasternack A, Jianu SB, Veraar K, El Bouazzaoui F, Hoogaars WM, Ten Dijke P, Breuning MH, De Heer E, Ritvos O, Peters DJ (2016) Inhibition of activin signaling slows progression of polycystic kidney disease. J Am Soc Nephrol JASN 27:3589–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Huang L, Sung SS, Vergis AL, Rosin DL, Rose CE Jr, Lobo PI, Okusa MD (2008b) The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int 74:1526–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Magenheimer BS, Xia S, Johnson T, Wallace DP, Calvet JP, Li R (2008a) A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat Med 14:863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Castano AP, Nowlin BT, Lupher ML Jr, Duffield JS (2009) Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol 183:6733–6743 [DOI] [PubMed] [Google Scholar]
- Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100:5286–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Wang CJ, Judge DP, Halushka MK, Ni J, Habashi JP, Moslehi J, Bedja D, Gabrielson KL, Xu H, Qian F, Huso D, Dietz HC, Germino GG, Watnick T (2014) A Pkd1-Fbn1 genetic interaction implicates TGF-beta signaling in the pathogenesis of vascular complications in autosomal dominant polycystic kidney disease. J Am Soc Nephrol JASN 25:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T (2006) Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell 10:57–69 [DOI] [PubMed] [Google Scholar]
- Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S (2013) Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45:1004–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima A, Yamashita S, Maeshima K, Kojima I, Nojima Y (2003) Activin a produced by ureteric bud is a differentiation factor for metanephric mesenchyme. J Am Soc Nephrol JASN 14:1523–1534 [DOI] [PubMed] [Google Scholar]
- Mangos S, Lam PY, Zhao A, Liu Y, Mudumana S, Vasilyev A, Liu A, Drummond IA (2010) The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech 3:354–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555 [DOI] [PubMed] [Google Scholar]
- McPherson EA, Luo Z, Brown RA, LeBard LS, Corless CC, Speth RC, Bagby SP (2004) Chymase-like angiotensin II-generating activity in end-stage human autosomal dominant polycystic kidney disease. J Am Soc Nephrol JASN 15:493–500 [DOI] [PubMed] [Google Scholar]
- Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, Weissman IL, Cyster JG, Engleman EG (2002) Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol 3:1135–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS (2002) Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc Natl Acad Sci USA 99:943–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrug M, Zhou J, Woo Y, Cui X, Szalai AJ, Novak J, Churchill GA, Guay-Woodford LM (2008) Overexpression of innate immune response genes in a model of recessive polycystic kidney disease. Kidney Int 73:63–76 [DOI] [PubMed] [Google Scholar]
- Nadel JA (1991) Biology of mast cell tryptase and chymase. Ann N Y Acad Sci 629:319–331 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ebihara I, Fukui M, Osada S, Tomino Y, Masaki T, Goto K, Furuichi Y, Koide H (1993) Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol JASN 4:1064–1072 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ushiyama C, Suzuki S, Ebihara I, Shimada N, Koide H (2000) Elevation of serum levels of metalloproteinase-1, tissue inhibitor of metalloproteinase-1 and type IV collagen, and plasma levels of metalloproteinase-9 in polycystic kidney disease. Am J Nephrol 20:32–36 [DOI] [PubMed] [Google Scholar]
- Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J (2012) The renal mononuclear phagocytic system. J Am Soc Nephrol JASN 23:194–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Okumura Y, Fujimoto S, Shiraishi I, Itoi T, Hamaoka K (2005) Adoptive transfer of macrophages ameliorates renal fibrosis in mice. Biochem Biophys Res Commun 332:11–16 [DOI] [PubMed] [Google Scholar]
- Norman J (2011) Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim Biophys Acta 1812:1327–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary CA, Mackay BM, Malik R, Edmondston JE, Robinson WF, Huxtable CR (1999) Polycystic kidney disease in bull terriers: an autosomal dominant inherited disorder. Aust Vet J 77:361–366 [DOI] [PubMed] [Google Scholar]
- Obermuller N, Morente N, Kranzlin B, Gretz N, Witzgall R (2001) A possible role for metalloproteinases in renal cyst development. Am J Physiol Renal Physiol 280:F540–F550 [DOI] [PubMed] [Google Scholar]
- Oh EC, Katsanis N (2012) Cilia in vertebrate development and disease. Development 139:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866 [DOI] [PubMed] [Google Scholar]
- Park EY, Seo MJ, Park JH (2010) Effects of specific genes activating RAGE on polycystic kidney disease. Am J Nephrol 32:169–178 [DOI] [PubMed] [Google Scholar]
- Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P (2008) Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 17:1578–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JK, Hopwood D, Loxley RA, Ghatora K, Coombes JD, Tan YS, Harrison JL, McKitrick DJ, Holobotvskyy V, Arnolda LF, Rangan GK (2007) Temporal relationship between renal cyst development, hypertension and cardiac hypertrophy in a new rat model of autosomal recessive polycystic kidney disease. Kidney Blood Press Res 30:129–144 [DOI] [PubMed] [Google Scholar]
- Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG (2007) A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13:1490–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JW (2009) Trophic macrophages in development and disease. Nat Rev Immunol 9:259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, McDaid JP, Tam FW, Haylor JL, Ong AC (2009) Pkd2 dosage influences cellular repair responses following ischemia-reperfusion injury. Am J Pathol 175:1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Chen X, Poronnik P, Pollock CA (2006) The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int J Biochem Cell Biol 38:1–5 [DOI] [PubMed] [Google Scholar]
- Qin S, Taglienti M, Cai L, Zhou J, Kreidberg JA (2012) c-Met and NF-kappaB-dependent overexpression of Wnt7a and −7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol JASN 23:1309–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae F, Woods K, Sasmono T, Campanale N, Taylor D, Ovchinnikov DA, Grimmond SM, Hume DA, Ricardo SD, Little MH (2007) Characterisation and trophic functions of murine embryonic macrophages based upon the use of a Csf1r-EGFP transgene reporter. Dev Biol 308:232–246 [DOI] [PubMed] [Google Scholar]
- Rankin CA, Suzuki K, Itoh Y, Ziemer DM, Grantham JJ, Calvet JP, Nagase H (1996) Matrix metalloproteinases and TIMPS in cultured C57BL/6J-cpk kidney tubules. Kidney Int 50:835–844 [DOI] [PubMed] [Google Scholar]
- Ritvos O, Tuuri T, Eramaa M, Sainio K, Hilden K, Saxen L, Gilbert SF (1995) Activin disrupts epithelial branching morphogenesis in developing glandular organs of the mouse. Mech Dev 50:229–245 [DOI] [PubMed] [Google Scholar]
- Schaefer L, Han X, Gretz N, Hafner C, Meier K, Matzkies F, Schaefer RM (1996) Tubular gelatinase A (MMP-2) and its tissue inhibitors in polycystic kidney disease in the Han:SPRD rat. Kidney Int 49:75–81 [DOI] [PubMed] [Google Scholar]
- Schieren G, Rumberger B, Klein M, Kreutz C, Wilpert J, Geyer M, Faller D, Timmer J, Quack I, Rump LC, Walz G, Donauer J (2006) Gene profiling of polycystic kidneys. Nephrol Dial Transplant 21:1816–1824 [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90 [DOI] [PubMed] [Google Scholar]
- Shannon MB, Patton BL, Harvey SJ, Miner JH (2006) A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease. J Am Soc Nephrol JASN 17:1913–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Malarkey EB, Berbari NF, O’Connor AK, Vanden Heuvel GB, Mrug M, Yoder BK (2013) Proximal tubule proliferation is insufficient to induce rapid cyst formation after cilia disruption. J Am Soc Nephrol JASN 24:456–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, Shastry S, Rowe SM, Shim YM, Hussell T, Blalock JE (2010) A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science 330:90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y (2009) Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18:2328–2343 [DOI] [PubMed] [Google Scholar]
- Stables MJ, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, Farrow S, Gilroy DW (2011) Transcriptomic analyses of murine resolution-phase macrophages. Blood 118:e192–e208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, Wallace DP, Fields TA (2013) Macrophages promote polycystic kidney disease progression. Kidney Int 83:855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta MH, Harris DC, Rangan GK (2013) Role of interstitial inflammation in the pathogenesis of polycystic kidney disease. Nephrology 18:317–330 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Calvet JP, Dittemore-Hoover D, Yoshida K, Grantham JJ, Gattone VH 2nd (1991) A hereditary model of slowly progressive polycystic kidney disease in the mouse. J Am Soc Nephrol JASN 1:980–989 [DOI] [PubMed] [Google Scholar]
- Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, Zhou J (2009) Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 18:2523–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JJ, Song X, Wang X, Rinschen MM, Doerr N, LaRiviere WB, Schermer B, Pei YP, Torres VE, Weimbs T (2014) The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol JASN 25:1737–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togawa H, Nakanishi K, Mukaiyama H, Hama T, Shima Y, Sako M, Miyajima M, Nozu K, Nishii K, Nagao S, Takahashi H, Iijima K, Yoshikawa N (2011) Epithelial-to-mesenchymal transition in cyst lining epithelial cells in an orthologous PCK rat model of autosomal-recessive polycystic kidney disease. Am J Physiol Renal Physiol 300:F511–F520 [DOI] [PubMed] [Google Scholar]
- van Furth R, Cohn ZA (1968) The origin and kinetics of mononuclear phagocytes. J Exp Med 128:415–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon MA, Mylonas KJ, Hughes J (2010) Macrophages and renal fibrosis. Semin Nephrol 30:302–317 [DOI] [PubMed] [Google Scholar]
- Vilayur E, Harris DC (2009) Emerging therapies for chronic kidney disease: what is their role? Nat Rev Nephrol 5:375–383 [DOI] [PubMed] [Google Scholar]
- Vogler C, Homan S, Pung A, Thorpe C, Barker J, Birkenmeier EH, Upadhya P (1999) Clinical and pathologic findings in two new allelic murine models of polycystic kidney disease. J Am Soc Nephrol JASN 10:2534–2539 [DOI] [PubMed] [Google Scholar]
- Wallace DP, White C, Savinkova L, Nivens E, Reif GA, Pinto CS, Raman A, Parnell SC, Conway SJ, Fields TA (2014) Periostin promotes renal cyst growth and interstitial fibrosis in polycystic kidney disease. Kidney Int 85:845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wann AK, Knight MM (2012) Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell Mol Life Sci 69:2967–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wann AK, Chapple JP, Knight MM (2014) The primary cilium influences interleukin-1beta-induced NFkappaB signalling by regulating IKK activity. Cell Signal 26:1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs T, Talbot JJ (2013) STAT3 signaling in polycystic kidney disease. Drug Discov Today Dis Mech 10:e113–e118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PD (2004) Polycystic kidney disease. N Engl J Med 350:151–164 [DOI] [PubMed] [Google Scholar]
- Wilson PD (2008) Mouse models of polycystic kidney disease. Curr Top Dev Biol 84:311–350 [DOI] [PubMed] [Google Scholar]
- Wilson PD, Burrow CR (1999) Cystic diseases of the kidney: role of adhesion molecules in normal and abnormal tubulogenesis. Exp Nephrol 7:114–124 [DOI] [PubMed] [Google Scholar]
- Wilson PD, Falkenstein D (1995) The pathology of human renal cystic disease. Curr Top Pathol 88:1–50 [DOI] [PubMed] [Google Scholar]
- Wilson PD, Goilav B (2007) Cystic disease of the kidney. Annu Rev Pathol 2:341–368 [DOI] [PubMed] [Google Scholar]
- Wilson PD, Sherwood AC (1991) Tubulocystic epithelium. Kidney Int 39:450–463 [DOI] [PubMed] [Google Scholar]
- Wilson PD, Geng L, Li X, Burrow CR (1999) The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest J Tech Methods Pathol 79:1311–1323 [PubMed] [Google Scholar]
- Wilson PD, Hreniuk D, Gabow PA (1992) Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia. J Cell Physiol 150:360–369 [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ (2004) Focal adhesion regulation of cell behavior. Biochim Biophys Acta 1692:103–119 [DOI] [PubMed] [Google Scholar]
- Wynn TA, Barron L (2010) Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 30:245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496:445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL (2014) Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40:274–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Maeshima A, Kojima I, Nojima Y (2004) Activin A is a potent activator of renal interstitial fibroblasts. J Am Soc Nephrol JASN 15:91–101 [DOI] [PubMed] [Google Scholar]
- Yoder BK (2007) Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol JASN 18:1381–1388 [DOI] [PubMed] [Google Scholar]
- Yoder BK, Hou X, Guay-Woodford LM (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol JASN 13:2508–2516 [DOI] [PubMed] [Google Scholar]
- Yoder BK, Richards WG, Sweeney WE, Wilkinson JE, Avener ED, Woychik RP (1995) Insertional mutagenesis and molecular analysis of a new gene associated with polycystic kidney disease. Proc Assoc Am Physicians 107:314–323 [PubMed] [Google Scholar]
- Zeier M, Fehrenbach P, Geberth S, Mohring K, Waldherr R, Ritz E (1992) Renal histology in polycystic kidney disease with incipient and advanced renal failure. Kidney Int 42:1259–1265 [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Neilson EG (2010) Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol JASN 21:1819–1834 [DOI] [PubMed] [Google Scholar]
- Zeltner R, Hilgers KF, Schmieder RE, Porst M, Schulze BD, Hartner A (2008) A promoter polymorphism of the alpha 8 integrin gene and the progression of autosomal-dominant polycystic kidney disease. Nephron Clin Pract 108:c169–c175 [DOI] [PubMed] [Google Scholar]
- Zheng D, Wolfe M, Cowley BD Jr, Wallace DP, Yamaguchi T, Grantham JJ (2003) Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease. J Am Soc Nephrol JASN 14:2588–2595 [DOI] [PubMed] [Google Scholar]
- Zimmerman K, Yoder BK (2015) SnapShot: sensing and signaling by cilia. Cell 161(692–692): e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoja C, Corna D, Locatelli M, Rottoli D, Pezzotta A, Morigi M, Zanchi C, Buelli S, Guglielmotti A, Perico N, Remuzzi A, Remuzzi G (2015) Effects of MCP-1 inhibition by bindarit therapy in a rat model of polycystic kidney disease. Nephron 129:52–61 [DOI] [PubMed] [Google Scholar]