

Abstract
The use of kinase-directed precision medicine has been heavily pursued since the discovery and development of imatinib. Annually, it is estimated that around ~20 000 new cases of tropomyosin receptor kinase (TRK) cancers are diagnosed, with the majority of cases exhibiting a TRK genomic rearrangement. In this Perspective, we discuss current development and clinical applications for TRK precision medicine by providing the following: (1) the biological background and significance of the TRK kinase family, (2) a compilation of known TRK inhibitors and analysis of their cocrystal structures, (3) an overview of TRK clinical trials, and (4) future perspectives for drug discovery and development of TRK inhibitors.
Graphical Abstract
INTRODUCTION
The tropomyosin receptor kinase (TRK) family of enzymes are transmembrane receptor tyrosine kinases (RTKs) that regulate synaptic strength and plasticity in the mammalian nervous system.1–8 In this role, the TRK family regulates cell differentiation, proliferation, survival, and pain.9–23 There are three members of the TRK family: TRKA (encoded by NTRK1 gene), TRKB (NTRK2), and TRKC (NTRK3), which have all been implicated in the initiation and progression of malignancies.24–41 Similar to the BCR-ABL gene fusion product that drives chronic myelogenous leukemia (CML),42 NTRK rearrangements and fusion gene products have been observed in numerous tumor types, which has been comprehensively reviewed by Vaishnavi et al. and Amatu et al.43,44 Unlike CML, however, the incidence of NTRK fusion genes in each specific tumor type overall is rare. This generates profound difficulties for patient identification and adequate recruitment for clinical trials. For instance, NTRK2 gene fusions have been identified in 0.2% of lung adenocarcinoma,45 which represents approximately 184 patients of 92 138 diagnosed in 2010 in the USA.46 On the other end of the spectrum, NTRK3 fusion genes have been identified in virtually all secretory breast carcinomas and of mammary analogue secretory carcinomas (MASC), an extremely rare tumor of the salivary (in general, of the parotid) gland.47 In fact, the defining characteristic of MASC, when compared to other salivary carcinomas, is an NTRK gene fusion.47 In addition, NTRK fusions are found in about 50% of pediatric diffuse intrinsic pontine glioma and non-brainstem glioblastoma.48 In major cancer subgroups, NTRK fusions occur in 3.3% of lung cancers,45,49 2.2% of colorectal cancers,45,49–52 16.7% of thyroid cancers,45,53,54 2.5% of glioblastomas, and 7.1% of pediatric gliomas.43,55 Finally, similar to the receptor tyrosine kinase RET (rearranged during transfection), NTRK fusions (particularly ETV6-NTRK3) are common in post-Chernobyl radiation-induced papillary thyroid carcinomas.56,57 Thus, targeting TRK oncogenes is an attractive therapeutic approach for a diverse, frequently rare set of cancers.
The primary method to target TRK oncogenes is the use of small molecule kinase inhibitors. Because gene fusion products are the major oncogenes observed, other targeting strategies, such as antibody therapy, will not be effective since transmembrane tyrosine kinase fusions frequently lack an extracellular domain.58 In this case, the fusion proteins are only susceptible to small molecule inhibition.59–61 In general, small molecules are designed to target the adenosine triphosphate (ATP) binding site of TRK to block catalytic activity. This is based on the principle that protein kinases catalyze a phosphoryl transfer to a downstream substrate and only have micromolar affinity for ATP.62 Since turnover is rapid and kinase affinity for ATP is weak, small molecules can effectively bind and inhibit TRK despite a high endogenous concentration of ATP. Because of the druggability of the TRK enzyme class, a number of attempts to target TRKs have been completed. In this Perspective, we discuss current development and clinical application of TRK-targeted cancer therapy.
TRK Biology and Signaling.
The TRK oncogene was initially discovered in colon cancer in which the cytoskeletal protein tropomyosin was fused to an unknown catalytically active kinase domain.50,63 Further studies identified the kinase as a single-pass receptor tyrosine kinase expressed in the developing central nervous system and was given the name tropomyosin receptor kinase. In the extracellular region of TRK, there is a leucine-rich motif, two cysteine-rich domains, and two immunoglobulin-like domains and all are essential for ligand recognition and binding.64–66 Unlike typical RTKs, the TRK intracellular region is small and comprised of roughly 70 amino acids before and 15 amino acids after the kinase domain.64,65 In comparison to other RTKs, TRK is most similar to the insulin receptor and has been implicated in insulin signaling.67
The TRK family is comprised of three distinct isoforms: TRKA, TRKB, and TRKC. A major difference among all three isoforms is the ligand that activates the receptor. TRKA is activated by nerve growth factor (NGF),68–70 TRKB is activated by brain-derived neurotrophic factor (BDNF),69,70 while TRKC is primarily activated by neurotrophin 3 (NT3) (Figure 1).71,72 The kinase domains of TRKA, TRKB, and TRKC share between 72% and 78% sequence identity.73 When looking at the residues that interact directly with ATP in the active site,74 TRKA and TRKB share 95% sequence identity, while TRKB and TRKC are completely identical.73 In the TRK domain, the TRKA hinge is more structurally constrained compared to TRKB and TRKC, suggesting that the hinge environment differs between TRK isoforms.74 Further, the kinase insert domain of the TRK family is not structurally conserved, which is another key difference within the family.74
Figure 1.
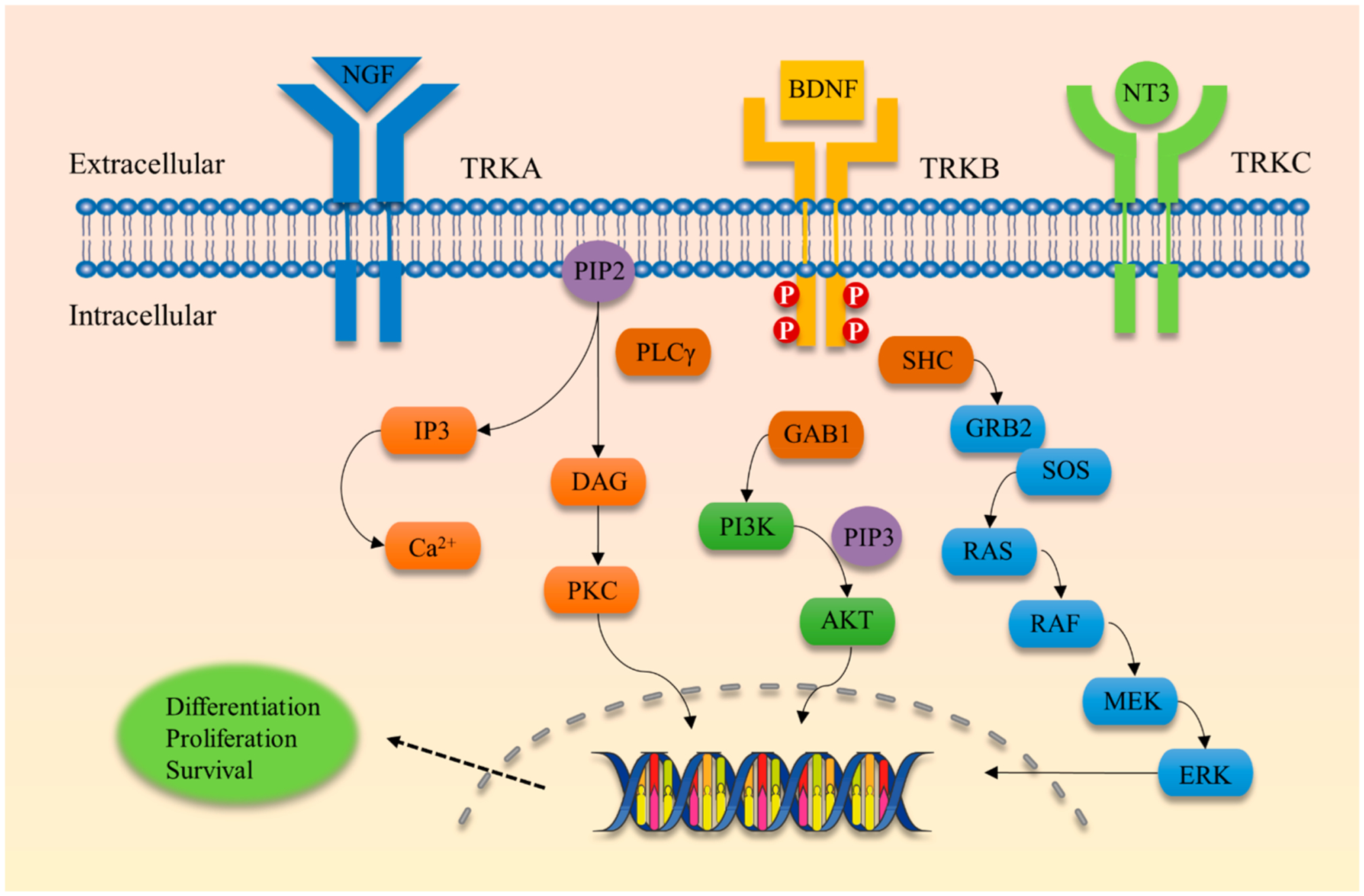
Schematic view of TRK receptor tyrosine kinases and major signal transduction pathways involved in cell differentiation, proliferation, and survival. TRKA is activated by nerve growth factor (NGF), TRKB is activated by brain-derived neurotrophic factor (BDNF), and TRKC is activated by neurotrophin-3 (NT3). RAS, rat sarcoma oncogene; RAF, rapidly accelerated fibrosarcoma oncogene; MEK, mitogen-activated protein kinase; ERK, extracellular-signal-regulated kinase; GRB2, growth factor receptor-bound protein 2; SHC, SRC homology 2 domain containing; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; AKT, v-AKT murine thymoma viral oncogene homologue; PLCγ, phospholipase C-γ; DAG, diacyl-glycerol; PKC, protein kinase C; IP3, inositol trisphosphate.
Similar to the RTK superfamily, TRKs dimerize in response to ligand binding (Figures 1 and 2).75 After ligand binding, TRKs autophosphorylate each monomer followed by rapid phosphorylation of the kinase activation loop.76–78 These phosphorylation events enhance catalytic activity of the kinase. To generate attachment sites for adapter proteins, the NPXY motif (Y490 in TRKA) in the juxtamembrane domain and the YLDIG motif (Y785 in TRKA) in the carboxy terminus are phosphorylated.79–82 These phosphorylation events create docking sites for SRC homology domain 2 (SH2) and phosphotyrosine binding domain (PTB) containing proteins, such as SHC and phospholipase C-γ (PLC-γ). After binding, SHC and PLC are activated through TRK-catalyzed phosphorylation.44,83
Figure 2.
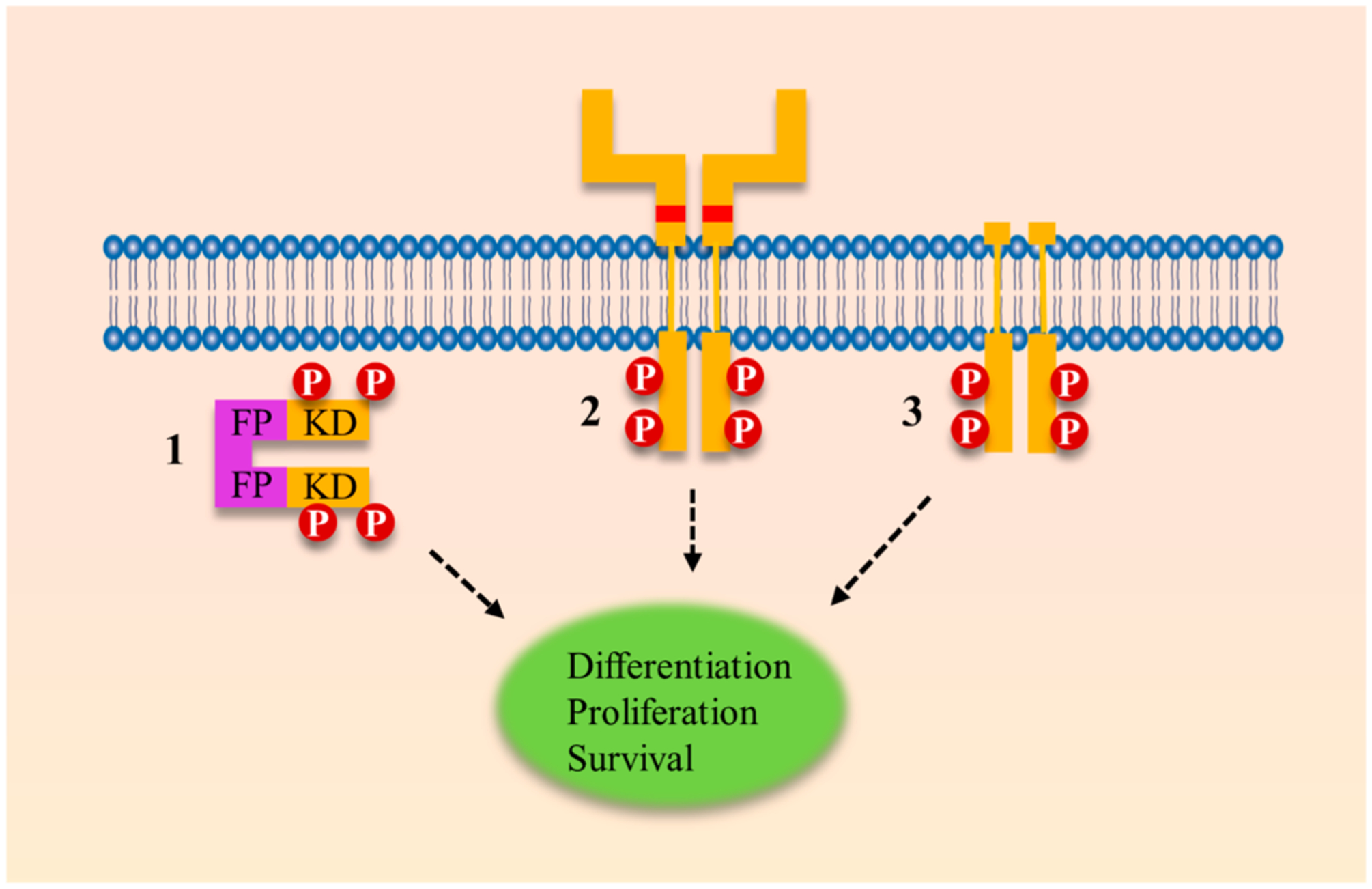
Ligand-independent signaling leading to constitutive TRK activation can occur through three distinct mechanisms: a genomic rearrangement event, extracellular point mutations, or truncations of the extracellular domain. (1) In a genomic rearrangement event, the kinase domain of TRK is fused to an unrelated protein, typically called the fusion partner. In this example, a cytoplasmic chimeric TRK fusion is shown without the transmembrane (TM) domain. Of note, TRK fusions can occur with or without the TM domain.102 (2) Point mutations can occur in the extracellular domain of TRK, generating a TRK oncogene with transforming abilities. Point mutations that are transformative under laboratory conditions have been identified at P203A and C345S.103,104 (3) Through in-frame deletions and alternative splicing events, the TRK kinase can present with a truncated extracellular domain. These aberrant gene products have been identified in AML and neuroblastoma.105–107 FP, fusion partner; KD, kinase domain.
SHC was the first adaptor protein identified to bind to the phosphorylated NPXY motif of TRK, which results in the activation of the AKT and RAS canonical pathways.80–82,84 After SHC is activated, a secondary adaptor protein, growth factor receptor-bound protein 2 (GRB2),85 is recruited and facilitates GTP-loading of RAS via the guanine nucleotide exchange factor, SOS.86 The activated GTP-bound form of RAS activates the MAP kinase cascade, which includes activation of RAF, MEK, and ERK.87 The ERK kinase translocates into the nuclear membrane activating transcription of target genes involved in cell growth, survival, and proliferation.88
Activation of the AKT pathway occurs via recruitment of SHC and GRB2 to the NPXY motif of TRK, which signals through the intermediary molecule GRB2-associated-binding protein 1 (GAB1). This simulates activity of phosphoinositide 3-kinase (PI3K) leading to phosphorylation of PI4,5 lipids at the 3′-position.89 On AKT, there is a conserved pleckstrin homology (PH) domain, which interacts with the 3′-phosphorylated lipids leading to AKT activation.90–93 AKT activation leads to increased expression of cell survival and proliferation genes that enhance prosurvival phenotypes mediated through TRK receptors.94–98
TRK Implication in Cancer.
Genetic mutations in the TRK family have been reported in many cancers, namely, carcinomas of the colon, thyroid, lung, ovary, breast (secretory breast carcinoma), salivary gland (mammary analogue secretory carcinoma), and pancreas, melanoma, spitzoid neoplasms, cholangiocarcinoma, stromal tumors (congenital fibrosarcoma, congenital mesoblastic nephroma, soft tissue sarcoma, gastrointestinal stromal tumor, inflammatory myofibroblastic tumor), brain tumors (pediatric glioma, astrocytoma, and glioblastoma), and leukemia (Table 1 and Figure 3).43,99,100,49,51,53,57,101 Within the TRK family, TRKA is the most commonly identified oncoprotein, which is found at a rate of approximately 7.4% across multiple tumor types.43 Following is TRKC and then TRKB, which are found at rates of 3.4% and 0.4%, respectively. The majority of TRKB mutations have a frequency of less than 0.5% and many TRKC mutations have a frequency of less than 1.0%.49
Table 1.
Oncogenic TRK Fusions Found across Multiple Tumor Typesa
| cancer site | estimated US cases per year | TRKA % | TRKB % | TRKC % |
|---|---|---|---|---|
| lung adenocarcinoma (NSCLC) | 92 13846 | 3.349 | 0.245 | |
| colorectal | 135 430108 | 1.549–52 | 0.745 | |
| intrahepatic cholangiocarcinoma | 2 970109,110 | 3.6111 | ||
| papillary thyroid cancer | 45 496108,112 | 12.353 | 14.554,113 | |
| glioblastoma | 11 376108,114 | 1.2555,115 | ||
| head and neck squamous cell carcinoma | 63 030116 | 0.245 | 0.2117 | |
| mammary analogue secretory carcinoma | 151108,118 | 100119 | ||
| Ph-like acute lymphoblastic leukemia | 1 192120,121 | 0.7122 | ||
| acute myeloid leukemia | 15 976121,123 | 0.012557,124 | ||
| skin cutaneous melanoma | 87 110121 | 0.345 | ||
| secretory breast carcinoma | 252121,125 | 92126 | ||
| estimated total cases per year | 10 917 | 310 | 8 325 | |
| estimated percent of cases per year | 55.8% | 1.6% | 42.6% |
Only positive studies are listed, and thus the actual prevalence may be lower than reported.
Figure 3.
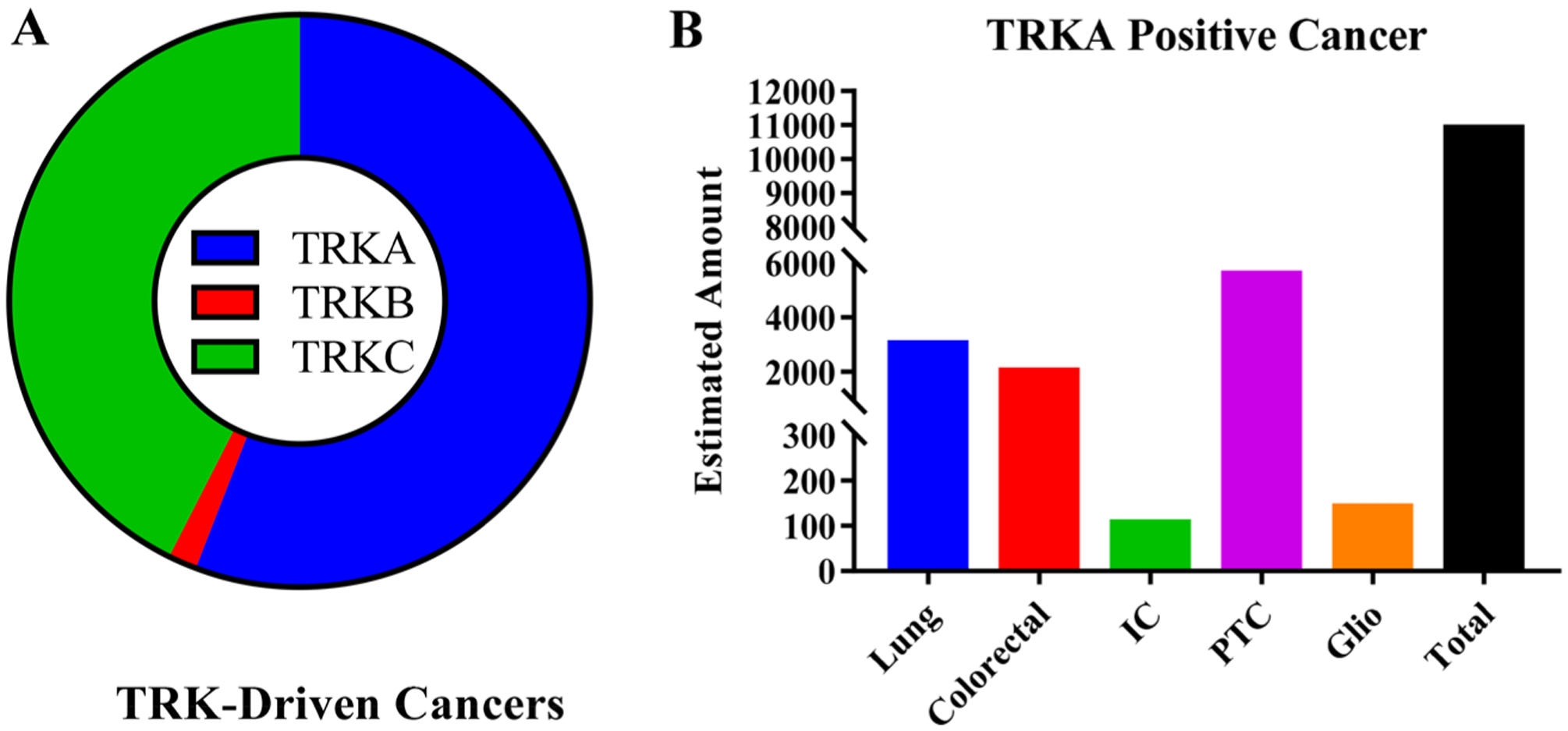
(A) Estimated contribution of TRKA/B/C mutations to all TRK-driven malignancies. Data is based on estimated incidence of cancers at major sites and contribution of TRK mutations at each site. It is estimated that a total of 19 552 cancers are diagnosed each year that have a TRK oncogene. Of the new cases, 55.8% (10 917) are TRKA, 42.6% are TRKC (8325), and 1.6% (310) are TRKB. Because of the limited sequencing data, the actual amount of TRK-driven tumors could be significantly greater or lower depending on robustness of sample size, sample selection, and data analysis. Also, tumors with a TRK mutation could be dependent on a separate pathway. (B) Estimated number of cancers positive for TRKA. Lung (lung adenocarcinoma), IC (intrahepatic cholangiocarcinoma), PTC (papillary thyroid cancer), and Glio (glioblastoma).
When an NTRK gene fusion occurs, the translocation event generates a hybrid oncogene composed of the active TRK kinase domain linked to an unrelated gene or fusion partner, triggering constitutive activation or overexpression of the TRK protein (Figure 2).44,53 The resulting aberrantly expressed novel oncogene can occur with or without the transmembrane domain.43,102 Certain mutations in the extracellular domain of TRKA, namely, P203A and C345S, have been identified as transforming under laboratory conditions but have yet to be identified in human tumor samples.103,104 On the other hand, in-frame deletions (ΔTRKA) and splice variants (TRKAIII) of NTRK1 have been functionally identified and characterized in human tumor samples.52,105–107 The ΔTRKA in-frame deletion, identified in acute myeloid leukemia (AML), contains a truncated extracellular domain that can transform both epithelial and fibroblast cells.107 The TRKAIII splice variant, identified in neuroblastoma, has deletions in exons 6, 7, and 9, which results in the loss of Ig-like C2-type I (IG-C2) and glycosylation sites in the extracellular domain (Figure 2).52,106 TRKA activating mutations from genomic rearrangements, point mutations, deletions, or splice variants compromise the ability to regulate the kinase domain. This suggests that a key attribute to the oncogenic potential of TRK is the loss of form or function of the extracellular domain.
In the tumor environment, TRK oncogenes stimulate uninhibited signaling through the RAS/RAF and PI3K/AKT pathways.49,127–129 The preferred signaling cascade is cell-specific, with dominance from the RAS/RAF pathway observed in both colorectal (KM-12) and lung (CUTO-3) cancers. In certain cell types, TRK oncogenic signaling also occurs through the PI3K/AKT and STAT3 signal transduction pathways,49 and in other cases the RAS/RAF and PI3K/AKT cascades are activated in concert.130 Because of multifaceted pathway activation, TRK oncogenes are potent and highly oncogenic by stimulating both antiapoptotic and pro-proliferative pathways.131 Further, TRK fusion oncogenes have been identified as important mediators to stimulate early tumor progression.132 Taken together, inhibition of TRK oncogenes can have chemotherapeutic and chemopreventive effects and, subsequently, has become a hotbed for therapeutic discovery efforts. In this Perspective, we summarize TRK inhibitors from peer-reviewed literature; compounds from patents have been previously reviewed by McCarthy and Bailey et al.133–135
TRK INHIBITORS
The following section represents a comprehensive overview of known TRK inhibitors and their corresponding discovery and development efforts. Because most TRK-activating mutations alter or eliminate the extracellular domain,59–61 antibodies directed at TRK or its ligands will not be effective as anticancer agents. Thus, all inhibitors reviewed are small-molecule kinase inhibitors that primarily target the TRK kinase domain. The inhibitors are at various stages of development from exploratory research and preclinical research to clinical trials. Inhibitors have been classified based on their interactions with TRK136 and are presented according to their binding modes. TRK inhibitors can be broken down into four, distinct subtypes based on ligand binding interactions. The classifications are (A) type I, (B) type II, (C) type III, and (D) type IV.137 (A) Type I TRK inhibitors are ATP competitive and bind to the ATP-binding site. The majority of TRK inhibitors under clinical investigation are type I. (B) Type II TRK inhibitors are ATP noncompetitive and exhibit noncompetitive or pseudocompetitive binding kinetics. Type II inhibitors bind at the ATP-binding site and also to an adjacent allosteric pocket. Only a subset of kinases permit such binding, including TRK, a binding mode that can be incorporated into compound design to increase selectivity. (C) Type III TRK inhibitors bind to the kinase domain outside of the ATP-binding site. Unlike type II inhibitors, type III inhibitors are true allosteric inhibitors and can exploit unique, kinase-specific functionality to help achieve selectivity among TRK isoforms. (D) Type IV TRK inhibitors bind to a region other than the kinase domain. The majority of kinases are expressed as multidomain proteins with a catalytic domain and a regulatory domain. Type IV inhibitors can interrupt key protein–protein interactions or ligand interactions that limit activity of the kinase domain.138 Because TRK genomic rearrangements often pair the TRK kinase domain to an unrelated gene, it is unclear how effective the anticancer properties will be for type IV TRK inhibitors. Type IV TRK inhibitors are not reviewed in this Perspective.
Type I TRK Inhibitors.
Type I TRK inhibitors exhibit vast diversity in compound architecture, which will alter activity and selectivity profiles of the compounds. This helps to better define chemical space that is relevant to TRK inhibition and suggests points of refinement for future drug discovery efforts. Wang et al. reported 4-aminopyrimidines with TRK inhibitory activity from a high throughput screening effort against TRKB (Figure 4A).139 The group discovered compound 1 with activity on TRKA and B (IC50 = 0.27 μM and 1.1 μM, respectively). To complete SAR studies on 1, the isoxazole was replaced with a phenyl group, and halogen substitution at the 4-position of phenyl was found to improve potency (1–1a and 1–1b, TRKA IC50 values were 0.10 μM and 0.36 μM, respectively).139 SAR studies at R2 indicated that the (S)-enantiomer is preferred over the (R)-enantiomer (1–1c vs 1–1d and 1–1e, TRKA IC50 values were 0.65 μM vs 0.10 μM and 0.006 μM, respectively). On the pyrimidine ring system, substituting bromine to fluorine or chlorine had little effect on TRKA activity (1–2a and 1–2b, TRKA IC50 values were 0.017 μM vs 0.004 μM, respectively).139 Replacement of cyclopropyl at R4 with isopropoxy is tolerated (1–2c, TRKA IC50 = 0.006 μM), but altering aliphatic length or bulk decreases potency likely from geometric constraints at the TRKA hinge region (1–2d and 1–2e, TRKA IC50 values were 0.081 μM and 3 μM, respectively). Further optimization and refinement strategies furnished orally bioavailable compound 1–3 (AZ23).139 Compound 1–3 was active against TRKA and B (IC50 = 0.002 μM and 0.008 μM, respectively) and exhibited anticancer activity by oral dosing in a TRKA-driven allograft model and TRK-expressing xenograft model of neuroblastoma.140 In 2012, a ring fusion study of this scaffold was reported by the same research group.141 Two different ring fusion strategies were employed to generate imidazo[4,5-b]pyridine and purine derivatives. Representative compounds of the two scaffolds, 1–4 and 1–5, exhibited potent TRK inhibition. Both compounds displayed IC50 values of 0.0005 μM against TRKA-dependent MCF10A cells (MCF10A-TRKA-Δ) and were also active in mice bearing 3T3-TRKA-Δ tumors.141
Figure 4.
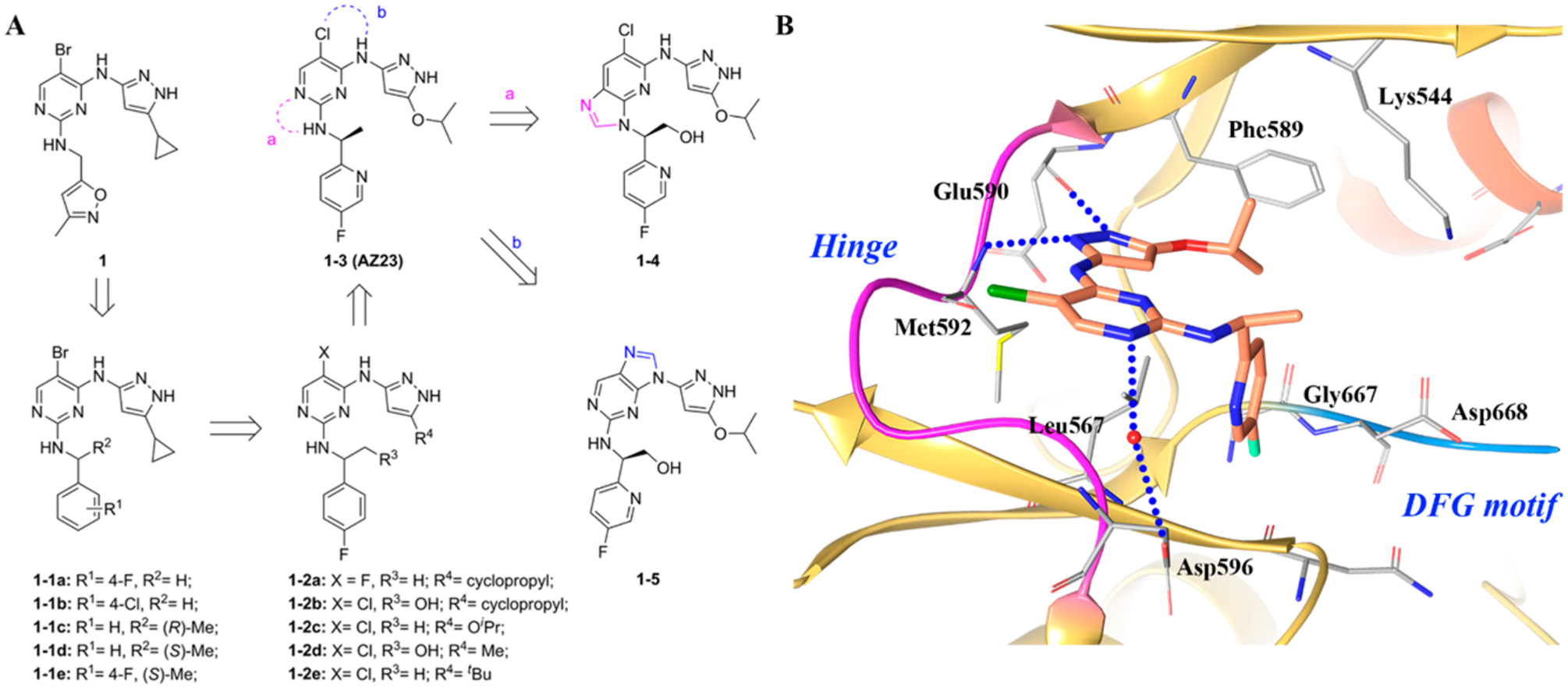
(A) Identification of compounds 1–3, 1–4, and 1–5 and (B) cocrystal structure of compound 1–3 in TRKA (PDB ID 4AOJ, 2.75 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; chlorine, green; fluorine, cyan.
In the cocrystal structure of compound 1–3 with TRKA (Figure 4B), TRKA is found in an inactive conformation with the C-helix pushed into a non-catalytically active orientation.141 Compound 1–3 forms two hydrogen bonds at the hinge region with the pyrazole moiety and interacts with the amide backbone of Glu590 and Met592. The isopropoxy group is oriented toward Phe589, which is the gatekeeper amino acid. The fluoropyridine ring engages in an interaction with Leu657, and the fluorine atom is in close proximity to Gly667.142 Numerous TRK kinase inhibitors exploit this region to increase selectivity and potency.141 Based on the crystal structure, compound 1–3 is a type I kinase inhibitor.
Further exploration of type I TRK inhibitors was reported by Brasca et al.143 The group initially identified compound 2 as a potent CDK-2 (cyclin dependent kinase) inhibitor (Figure 5A).143 Enhancement in selectivity, antiproliferative activity, and optimization of the physiochemical and pharmacokinetic properties led to the identification of compound 2–1 (milciclib, PHA-848125) (Figure 5A) with high potency (CDK-2 IC50 = 0.045 μM) and high bioavailability (F = 85%). Serendipitously, this compound exhibited near equal potency against TRKA (IC50 = 0.053 μM) with selectivity in the greater kinome (IC50 values >0.15 μM).143
Figure 5.
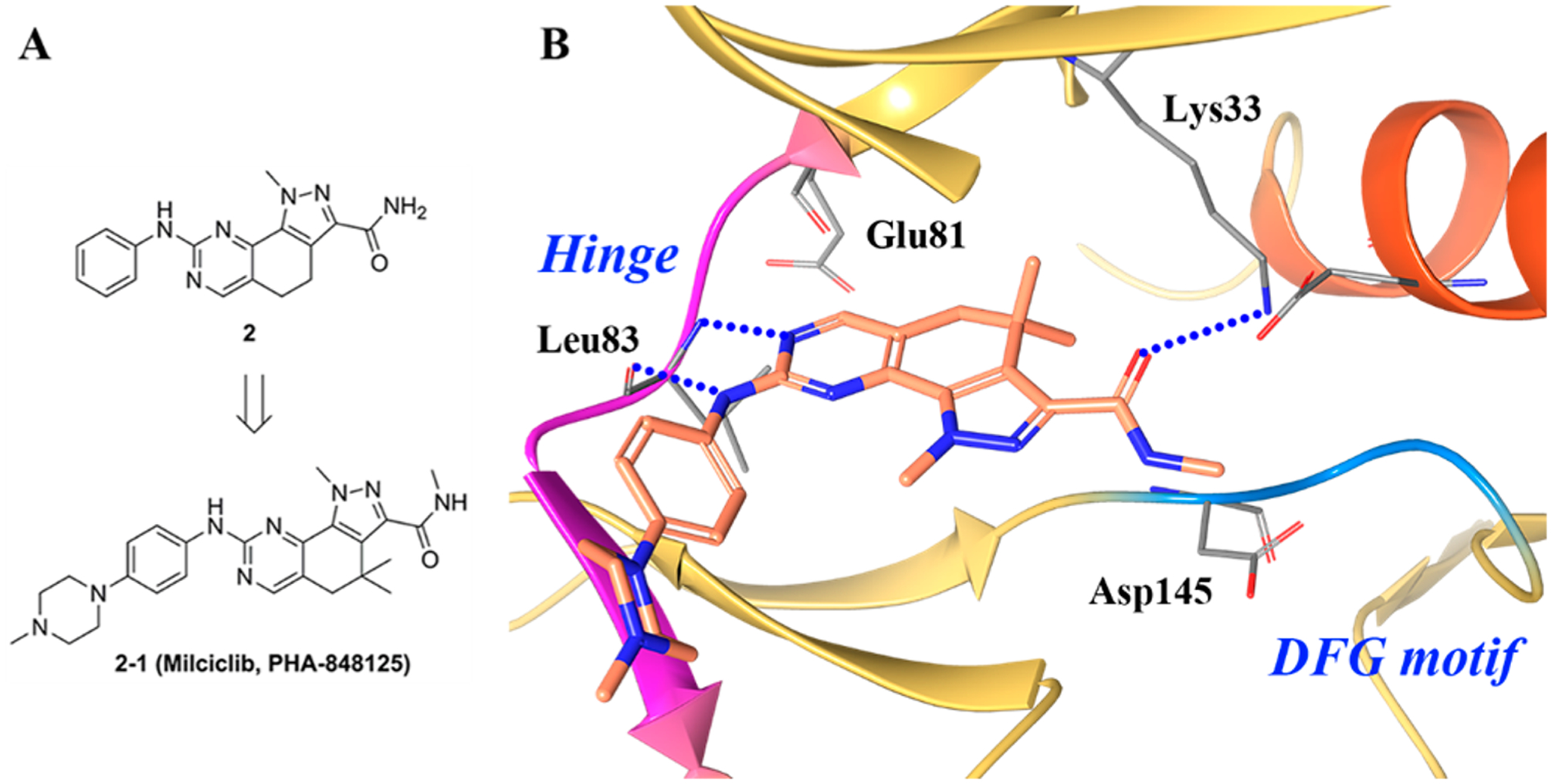
(A) Identification of 2–1 and (B) cocrystal structure of compound 2–1 in CDK-2/cyclin A (PDB ID 2WIH, 2.5 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red.
Figure 5B illustrates the crystal structure of compound 2–1 in complex with CDK2/cyclin A.143 The compound binds in the ATP pocket of the kinase with the pyrazoloquinazoline ring system occupying the adenine region, while the phenyl moiety points toward the solvent accessible region. Compound 2–1 creates two hydrogen bonds with the protein backbone of the hinge with residue Leu83. The amide carbonyl group of 2–1 is within hydrogen bonding distance of the conserved lysine (Lys33).143
Albanese et al. further evaluated the dual inhibitory activity of compound 2–1 against CDK-2 and TRKA.144,145 In vitro, 2–1 was able to inhibit NGF-induced phosphorylation of TRKA as well as downstream signaling in the DU-145 human prostate carcinoma line. In vivo, 2–1 inhibited tumor growth in a human prostate DU-145 xenograft model in a dose dependent manner. Because of the dual inhibitory properties of 2–1, the compound shifts the paradigm for precision medicine. Instead of generating a “magic-bullet” with high TRK kinase selectivity, Albanese et al. focused on validating and developing a “smart-bomb” with activity on the oncogene lesion (TRK) paired with cell cycle inhibition (CDK).146
In line with developing TRK inhibitors with dual activity, Fancelli et al. identified a series of 5-phenylacetyl 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole as Aurora kinase inhibitors (compound 3, Aurora-A IC50 = 0.027 μM) (Figure 6A).121 Further medicinal chemistry efforts lead to the discovery of compound 3–1b. Compound 3–1b was found to have dual activity on TRKA (IC50 = 0.03 μM) and Aurora A (IC50 = 0.013 μM)121 with weak ALK (anaplastic lymphoma kinase) inhibitory activity (IC50 = 0.091 μM).147 Akin to 2–1, compound 3–1b obtained dual activity on the oncogene lesion (TRKA) as well as cell cycle inhibition (Aurora A).147
Figure 6.
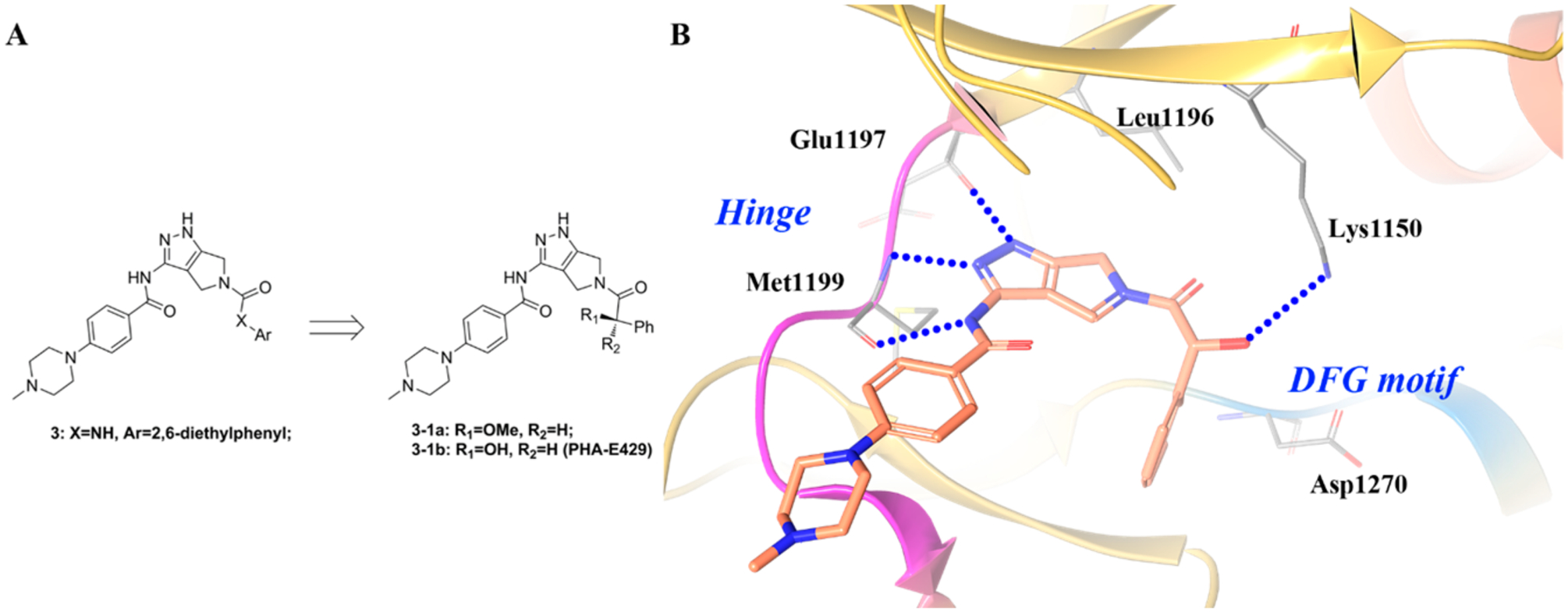
(A) Identification of compound 3–1b; (B) cocrystal structure of compound 3–1b with ALK (PDB ID: 2XBA, 1.95 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red.
Recently, Menichincheri et al. disclosed research efforts to generate compound 4–3 (entrectinib, RXDX-101) for ALK (IC50 = 0.012 μM) (Figure 7).148 To develop ALK inhibitors from compound 4 (ALK IC50 = 0.073 μM), the cocrystal complex of ALK with compound 3–1b was studied (see Figure 6B).148 Analysis of the complex suggested that a mono-substitution at the 2′-position would generate derivatives that occupy the ATP sugar pocket region while simultaneously displacing a water molecule. In addition, NH-R substituents at this position were able to stabilize the bioactive conformation through intramolecular hydrogen bonding. Based on this concept, different amines were attached to the 2′-position of the central phenyl ring.148
Figure 7.
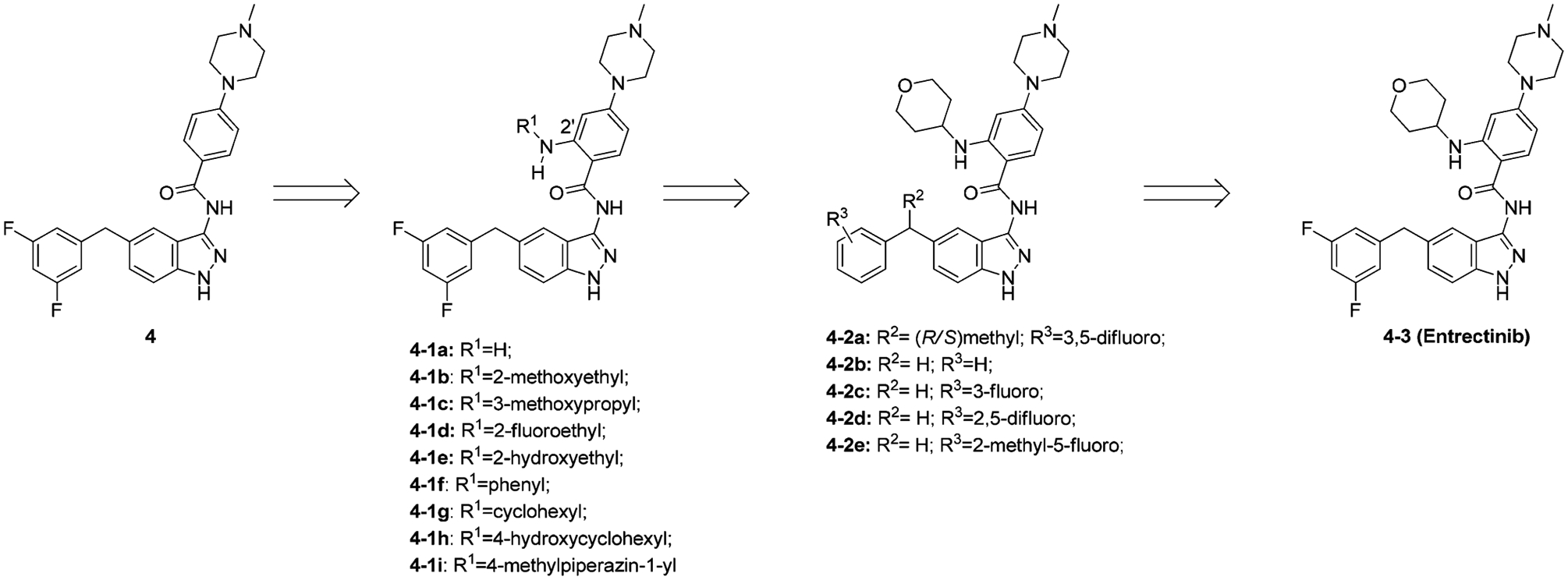
Structural optimization from compound 4 to 4–3.
The introduction of a primary amine at 2′ led to compound 4–1a with similar ALK potency (IC50 = 0.067 μM).148 This was consistent with modeling analysis as compound 4–1a lacked the substituent necessary to fill the sugar pocket. At R1, a methoxyethyl or methoxy propyl group generally led to a moderate loss in activity (4–1b and 4–1c, ALK IC50 = 0.21 μM and 0.135 μM, respectively) while a fluorine or a hydroxyl group increased activity (4–1d and 4–1e, ALK IC50 = 0.014 μM and 0.026 μM, respectively).148 In contrast, phenyl or cyclohexyl significantly decreased affinity because of size constraints of the sugar pocket (4–1f and 4–1g, ALK IC50 = 0.684 μM and 0.56 μM, respectively).148 Interestingly, introduction of a hydroxyl group furnished 4–1h of which the trans-isomer displayed 10-fold higher activity (ALK IC50= 0.01 μM) than the cis. When R1 is 4-amino-N-methylpiperidinyl (4–1i) or 4-aminotetrahydropyranyl (4–3), the resulting compounds exhibit excellent potency on ALK (IC50 = 0.015 and 0.012 μM, respectively). The latter of the two had better cellular activity (Cell IC50 = 0.438 μM vs 0.031 μM), which might be due to a difference in cell permeability.148 Introduction of a methyl group to R2 generated two enantiomers (4–2a), with one enantiomer being 3-fold more active than the other (ALK IC50= 0.059 μM and 0.019 μM, respectively, absolute configuration not determined).148 Due to labor-intensive chiral separation and stability of the benzhydryl stereogenic center further development of these compounds was terminated. Strong electron withdrawing groups on the phenyl ring are crucial for ALK activity. Removing the 3,5-difluoro substitution, changing the position of the fluorine, or replacing with methyl all decreased activity (compounds 4–2b to 4–2e, ALK IC50 = 0.106 μM, 0.03 μM, 0.038 μM, 0.181 μM, respectively).148
After initial development for ALK, compound 4–3 was subsequently found active on ROS1 (IC50 = 0.007 μM) and TRK (IC50 values for TRKA/B/C were 0.001 μM, 0.003 μM, and 0.005 μM, respectively).148,149 In antiproliferative studies, compound 4–3 was active against the colorectal cancer cell line KM-12 (IC50 = 0.0017 μM) and also induced tumor stabilization (>90% TGI) when administered po to mice bearing KM-12 xenografts.150 Compound 4–3 is currently in clinical trials for the treatment of patients with ALK-, ROS1-, and TRK-dependent tumors and is exhibiting remarkable signs of efficacy.148,149 In a Phase 2 clinical trial, 4–3 is being investigated for advanced or metastatic solid tumors that harbor TRKA/B/C, ROS1, or ALK gene rearrangements (NCT02568267).
The cocrystal structure of ALK with compound 4–3 was consistent with the above expectations (Figure 8A).148 A similar binding mode is observed in the cocrystal structure of 4–3 with TRKA (Figure 8B).151 The compound is anchored to the hinge through three hydrogen bonds between the aminoindazole moiety and the backbone of residues Glu590 and Met592 (Glu1197 and Met1199 in ALK). The difluorobenzyl-indazole core creates favorable contacts with Ala542 and Leu657 (Leu1256 and Val1130 in ALK) and the gatekeeper Phe589 (Leu1196 in ALK).148 Moreover, the 3,5-difluorobenzyl moiety is involved in multiple interactions with the backbone carbonyls of Asp668 from the DFG motif (Asn1254 and Gly1269 in ALK). The central phenyl ring makes a hydrophobic contact with Leu516 (Leu1122 in ALK), while the methylpiperazine is oriented toward the solvent front. In order to optimally fill the sugar pocket, the partially solvent-exposed tetrahydropyranyl moiety adopts a roughly orthogonal orientation with respect to the scaffold.148
Figure 8.
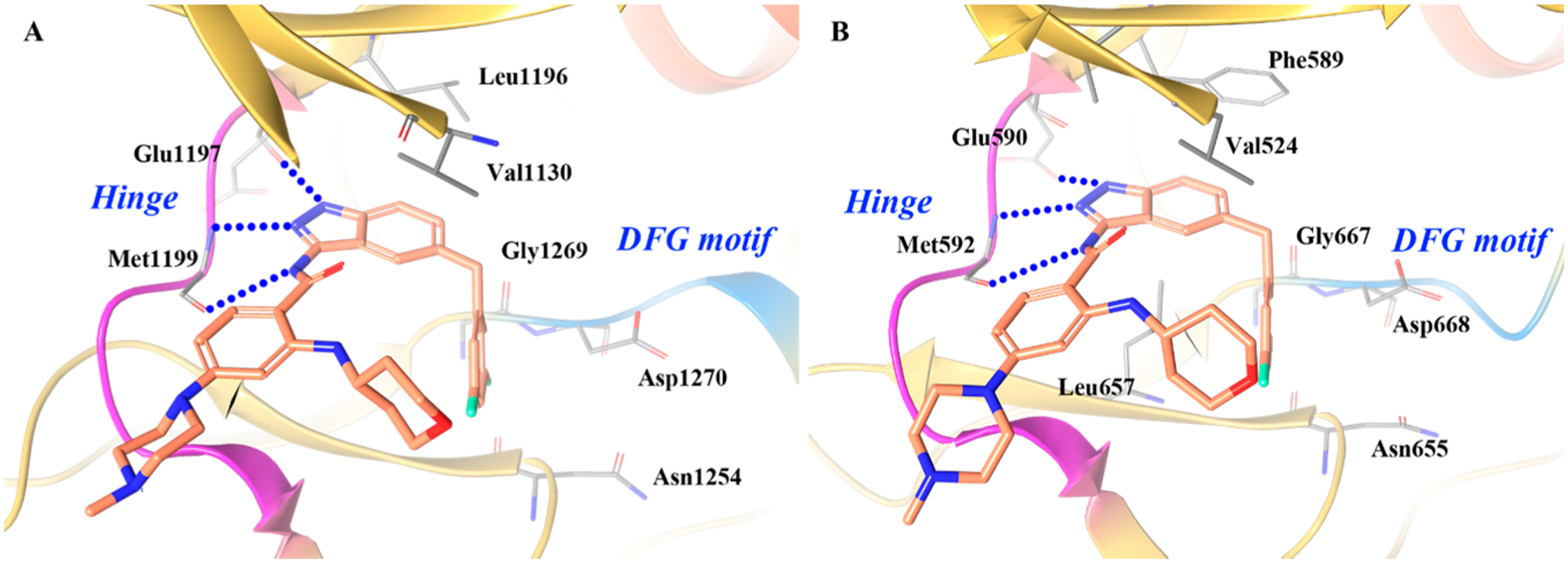
Cocrystal structure of compound 4–3 with (A) ALK (PDB ID 5FTO, 2.22 Å) and (B) TRKA (PDB ID 5KVT, 2.45 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
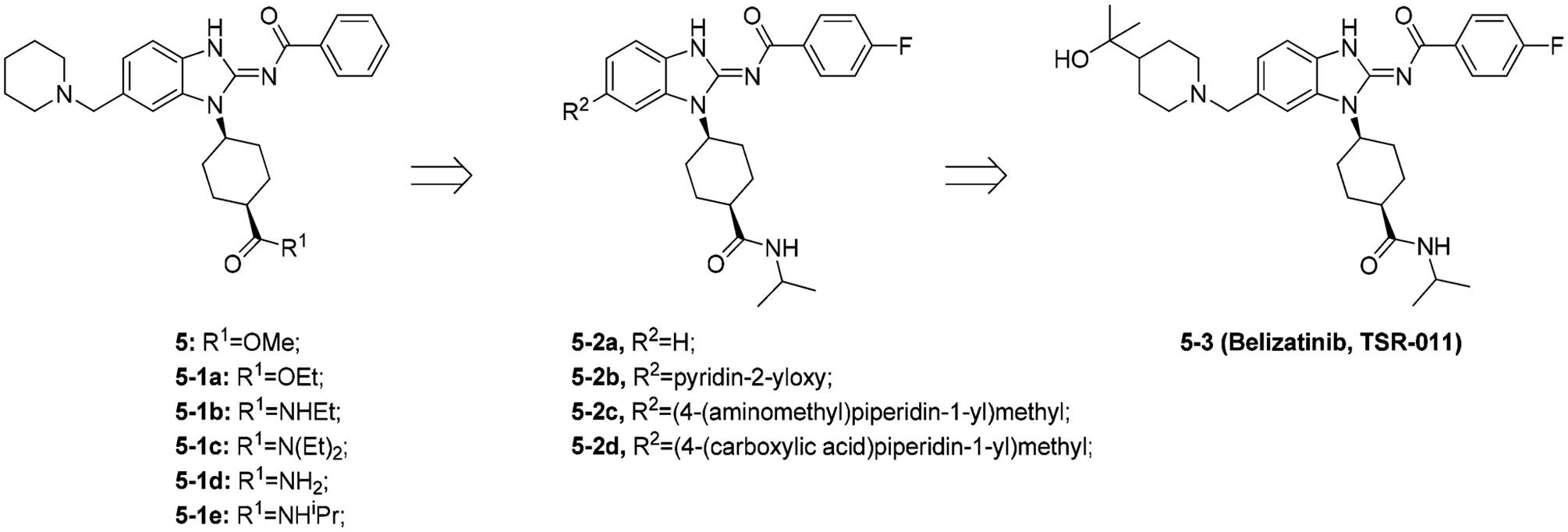
Another drug discovery campaign centered on ALK serendipitously uncovered 5–3 (belizatinib, TSR-011), a novel, potent TRK inhibitor (Figure 9).152 Through HTS (high-throughput screening), Lewis et al. identified the ALK inhibitor 5 (ALK kinase IC50 = 0.003 μM, pALK cell IC50 = 0.054 μM; 64-fold, 103-fold, and 15-fold selectivity over JAK2, SRC, and IGF1R, respectively),152 which was cocrystallized with ALK (Figure 10A).152 Through the benzoyl carbonyl oxygen and the benzimidazole NH, 5 was shown to bind to the hinge of ALK at Met1199, and the benzoyl ring was adjacent to the gatekeeper residue Leu1196. The cis-1,4 geometry of the cyclohexyl group pushes the methyl ester moiety into a hydrophobic pocket surrounded by Leu1256 and Gly1269. The piperidine moiety engages in van der Waals interactions with two backbone carbonyls on the N-terminal lobe (Gly1121 and Leu1122) in a shallow pocket on the surface of the protein at the entrance to the ATP binding pocket.152
Figure 9.
Structural optimization from compound 5 to 5–3.
Figure 10.
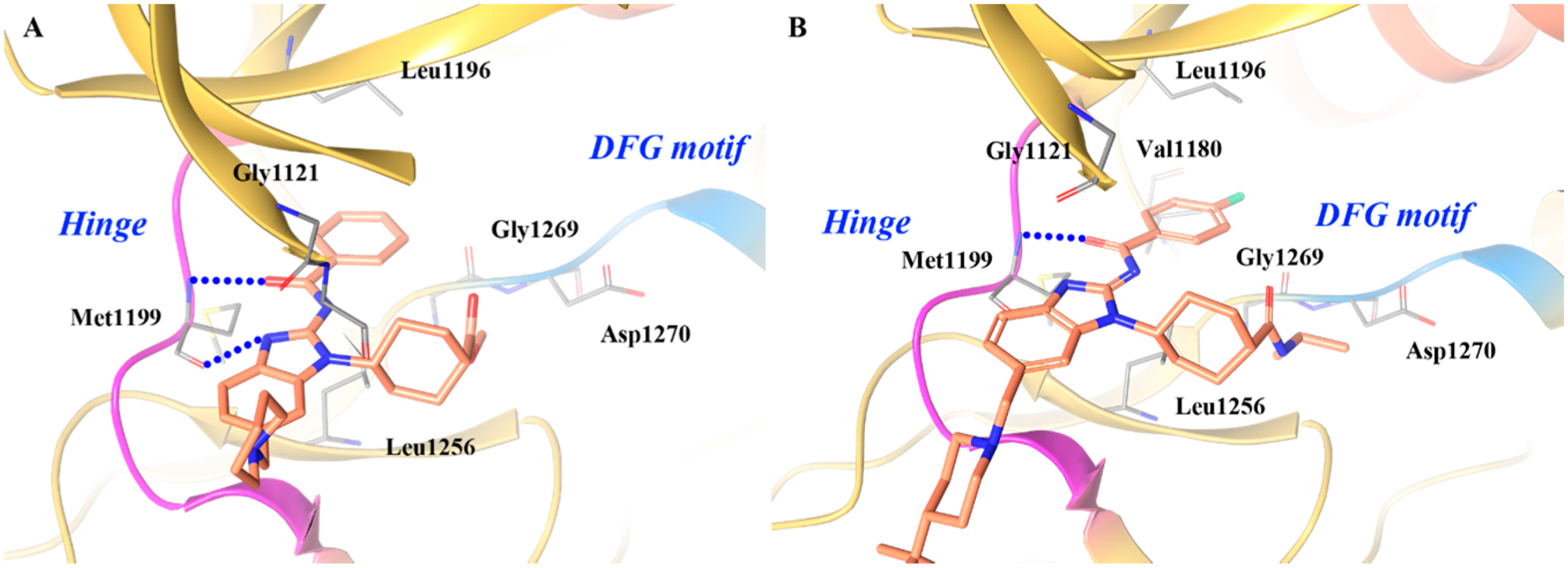
Co-crystal structure of (A) compound 5 in ALK (PDB ID 4FOC, 1.7 Å) and (B) compound 5–3 in ALK (PDB ID 4FOD, 2 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
To improve cell potency, selectivity, and metabolic stability, SAR optimization was completed. First, the methyl ester was replaced with different esters and amides (compounds 5–1a to 5–1e). The isopropyl amide compound 5–1e displayed the best combination of activity and selectivity (ALK kinase IC50 = 0.002 μM, pALK cell IC50 = 0.054 μM; 813-fold, 429-fold, and 36-fold selectivity over JAK2, SRC, and IGF1R, respectively).152 To improve metabolic stability, a wide variety of modifications were investigated at the R2 position. Figure 9 depicts some of these modifications, and the tertiary alcohol analogue 5–3 appeared optimal. Finally, different substitutions were introduced to the 4-fluoro phenyl ring; however, these derivatives either decreased selectivity or suffered from compromised cellular activity against ALK.152 When 5–3 was screened against a kinase panel, 5–3 was found to have potent pan-TRK activity (IC50 for TRKA/B/C < 0.003 μM).153 Currently, 5–3 is under a Phase 1/2 study in patients with advanced solid tumors and lymphomas (NCT02048488).
The cocrystal structure of 5–3 with ALK (Figure 10B) shows that 5–3 binds similarly to 5 (Figure 10A).152 Compound 5–3 pushes into the active site of the kinase in a pocket formed by Val1180 and Leu1256 and is in close proximity to the gatekeeper residue Leu1196. The iminobenzamide moiety adopts the exocyclic acylimine tautomer permitting key hydrogen bonding interactions with the hinge region residue Met1199. This tautomer is favored by extended π-delocalization into the pendant aryl ring, achieved by retaining a coplanar arrangement between benzamide and benzimidazole moieties.152
Isothiazole derivatives have been identified as TRK inhibitors (Figure 11). From a HTS campaign directed at TRKA, Lippa et al. identified compound 6 (TRKA IC50 = 0.007 μM).154 Initial SAR studies at R1 determined that substitution at the benzylic α-position remarkably influenced TRKA activity and VEGFR2 selectivity. Small aliphatic chains were well tolerated (6–1a and 6–1b, TRKA IC50 values were 0.003 μM and 0.004 μM, respectively), while larger substitutions significantly diminished activity (6–1d and 6–1e, TRKA IC50 values were 0.035 μM and 2.6 μM, respectively).154 The orientation of the R1 substitution also played a key role: the (R)-ethyl enantiomer 6–1b was >10-fold more active (TRKA IC50, 0.004 μM vs 0.052 μM) and was >100-fold more selective (VEGFR2 selectivity, 1300× vs 10×) than the (S)-enantiomer. SAR at R2 and R3 showed that substitution at these two positions is not necessary to maintain TRK activity (6–2a to 6–2d, TRKA IC50 values were 0.01 μM, 0.008 μM, 0.105 μM, and 0.111 μM, respectively).154 Further optimization identified that bicyclic moieties were important for TRK activity, especially the 7-membered ring system (compound 6–2e, TRKA IC50 < 0.001 μM), and consistent with previous data, the (R)-enantiomer was more active (6–3 vs 6–2f , TRKA IC50 values were <0.001 μM vs 0.091 μM). Compound 6–3 (Cpd5n) had picomolar kinase potency and single-digit nanomolar activity against TRK-driven cell growth.154
Figure 11.
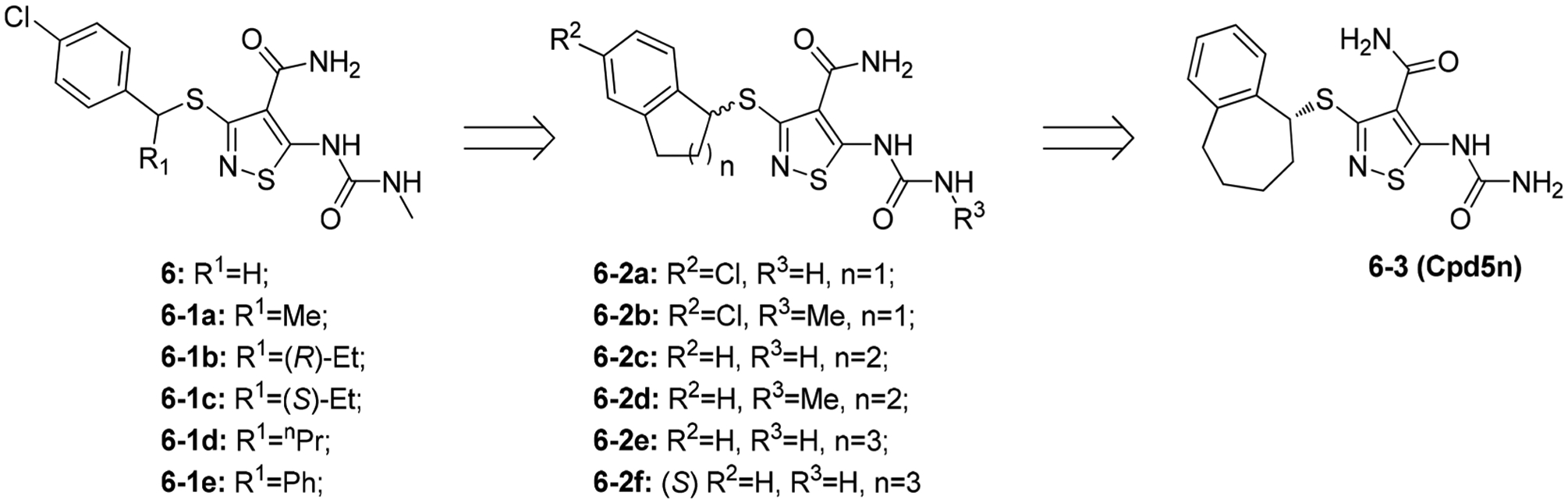
Structural optimization from compound 6 to 6–3.
The cocrystal complex of 6–3 in TRKB was obtained and is shown in Figure 12.73 The aliphatic portion of 6–3 is oriented toward the glycine-rich loop and exhibits clear type I binding.73 The glycine-rich loop forms a cage around the inhibitor and engages in strong hydrophobic interactions through Phe565. The urea and amide groups on the thiazole bind to the hinge of TRKB, forming three hydrogen bonds. The benzocycloheptene group is sandwiched between the gatekeeper Phe633 and glycine-rich loop Phe565 side chains, likely contributing to the stabilization of the TRKB DFG-in conformation.73 This also explains the potency differences between the two enantiomers as the S-enantiomer could still bind the hinge but would clash with Phe565 due to difference in stereochemistry of the benzo-cycloheptene. The affinity of 6–3 for TRKB is also enhanced by several water-bridged hydrogen bonds between the inhibitor and the kinase.73
Figure 12.
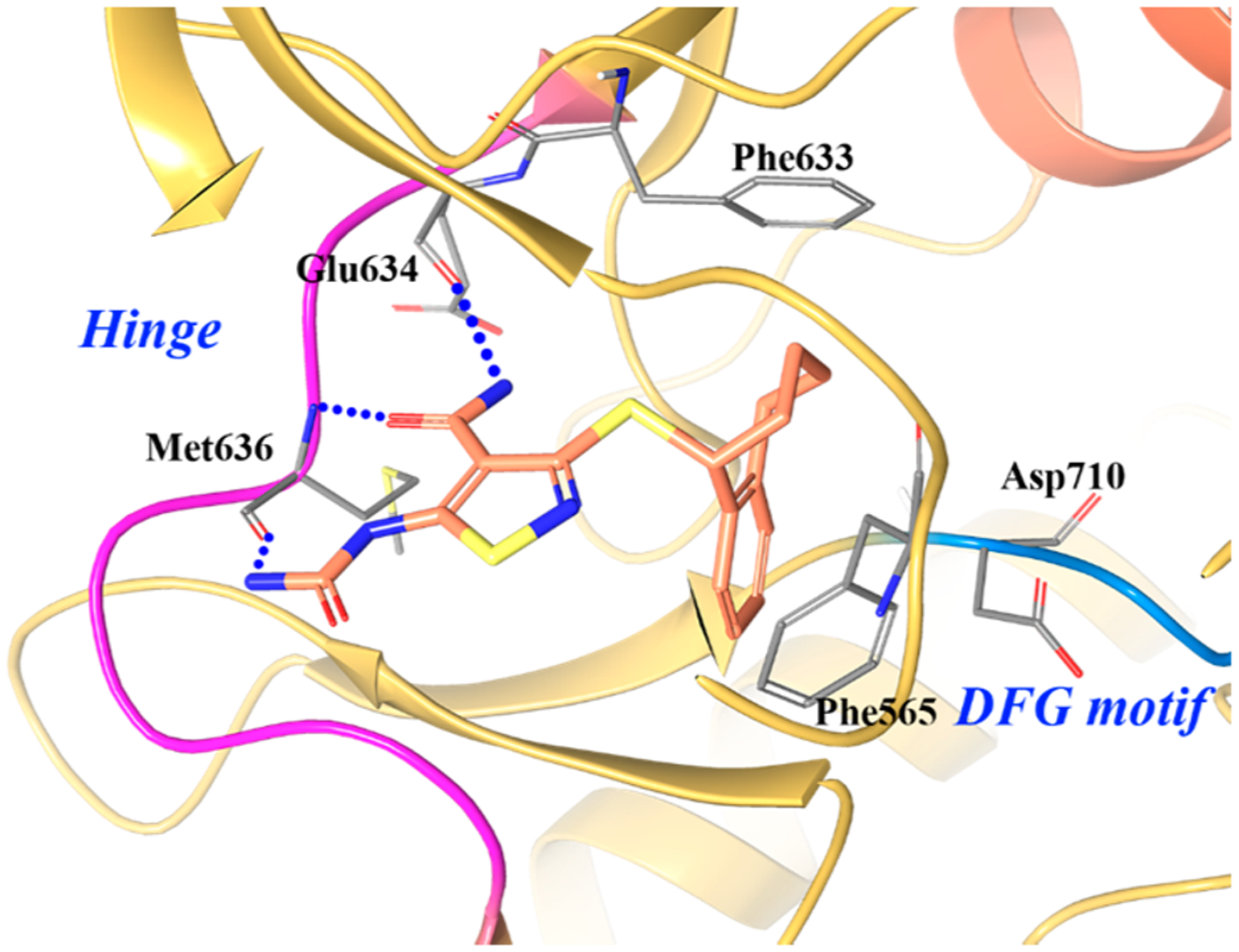
Co-crystal structure of compound 6–3 in TRKB (PDB ID 4AT3, 1.77 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; sulfur, yellow.
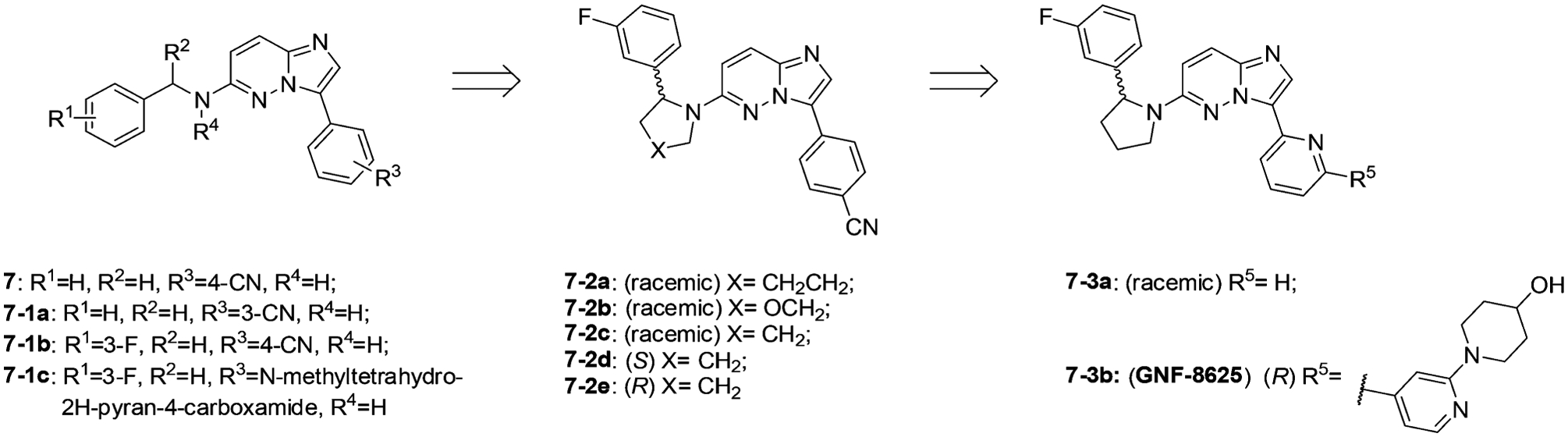
In another campaign to uncover novel TRK inhibitors, Choi et al. developed a series of substituted imidazopyridazine derivatives from compound 7 (TRKB IC50 = 0.083 μM) (Figure 13).155 The crystal complex of 7 with TRKC (Figure 14A) shows that the ligand binds to the inactivated (DFG-out) form of the kinase and the imidazopyridazine warhead engages in a hydrogen bond at the hinge with Met620. The benzonitrile ring is wedged between Phe617 and Phe698, and the phenyl ring sits under the glycine-rich loop and faces the solvent front of the active site.155
Figure 13.
Structural optimization from compound 7 to 7–3b.
Figure 14.
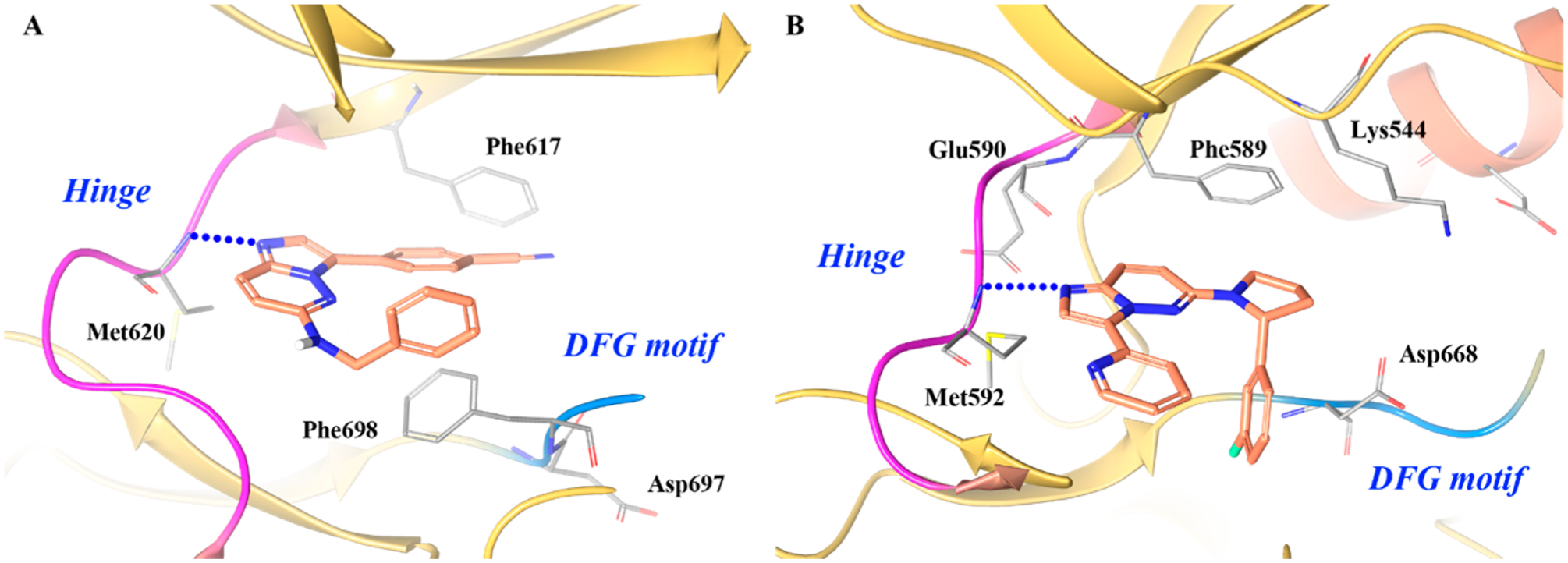
Co-crystal structure of (A) compound 7 in TRKC (PDB ID 4YMJ, 2 Å) and (B) compound 7–3a in TRKA (PDB ID 4YNE, 2.02 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
Preliminary medicinal chemistry efforts focused SAR studies at R1 to R4, with the goal of identifying compounds with improved potency and pharmacokinetic properties suitable for in vivo profiling. Nitrile substitutions in the phenyl group (R3) at the 3- or 4-position were equally tolerated (7 vs 7–1a, TRKA cell IC50 values = 0.85 μM and 0.74 μM). It was also observed that 3-substitution at the benzylamine phenyl ring (R1) led to a 10-fold increase in potency (7–1b, TRKA cell IC50 = 0.076 μM).155 Modifying the phenyl ring (R3) with 3-F substitution at R1 furnished many tolerated functional groups that could either increase solubility or further drive potency (i.e., compound 7–1c, TRKA cell IC50 = 0.006 μM). Methylation of the benzylic amine (R3) led to no significant change in potency indicating that the acidic proton on the benzylic amine does not hydrogen bond with TRK.155 Cyclizing the benzyl amine moiety with the adjacent phenyl ring was performed to rigidify the structure and reduce conformational entropy. The fused five-membered ring system was optimal for TRK inhibition and exhibited a preference for the R-enantiomer (7–2a to 7–2e, TRKA cell IC50 values = 0.17 μM, 0.075 μM, 0.068 μM, 3.13 μM, and 0.021 μM, respectively). Derivatization at R5 led to the discovery of the optimized compound 7–3b (GNF-8625).155 Compound 7–3b demonstrated potent antiproliferative activity against TRK transfected Ba/F3 and KM-12 cell lines (IC50 = 0.001 μM and 0.01 μM, respectively). In a KM-12-derived tumor xenograft, compound 7–3b demonstrated antitumor efficacy in a dose dependent manner, inducing 20% tumor regression at a dose of 50 mg/kg BID.155
Imidazopyridazines are frequently incorporated into kinase inhibitors as they readily form a hydrogen bond at the hinge with the C-6 carbon oriented toward the solvent.156 The warhead of 7–3a is based on imidazopyridazine and was expected to bind TRKA akin to 7. Surprisingly, the cocrystal structure of 7–3a with TRKA revealed an unexpected 180° rotation of the imidazopyridazine core at the hinge (Figure 14B).155 Only the (R)-enantiomer of 7–3a is active because the 3-F-phenyl is optimally positioned downward in the hydrophobic pocket. The moiety fills a pocket endogenously occupied by Phe669 and provides excellent shape complementary, likely contributing to increased affinity. Molecular modeling and crystallography studies indicated that two distinct binding modes exist (i.e., core flipping) with scaffold 7.155 Based on the different TRK cocrystal structures obtained with 7 and 7–3a, it was concluded that the preferred binding mode depends on C-6 substitution. If the C-6 substitution is modified to (R)-phenylpyrrolidine, the “flipped” orientation is preferred as the pyrrolidine anchors the phenyl group in the hydrophobic pocket.155 The phenomenon of “core flipping” could be employed to generate compounds with activity in multiple kinase families. Based on the crystal structure, compound 7–3a is a type I kinase inhibitor.
Compound 8 (larotrectinib, LOXO-101) is another selective pan-TRK inhibitor exhibiting low nanomolar cellular potency against the TRK family (IC50 ranging from 0.002 μM to 0.02 μM) with 100-fold selectivity over other kinases (Figure 15).49,157 The compound can induce cell-cycle arrest in the G1 phase and apoptosis in KM-12 cells.49 Compound 8 is currently in a Phase 2, open-label study for patients with advanced solid tumors harboring a fusion of TRKA, TRKB, or TRKC (NCT02576431).48 The medicinal chemistry effort leading to the discovery of this compound and its cocrystal complex with TRK have not been reported. However, because of the structure similarities between compounds 8 and 7–3b, it can be inferred that 8 exhibits similar SAR properties to that of 7–3b.
Figure 15.
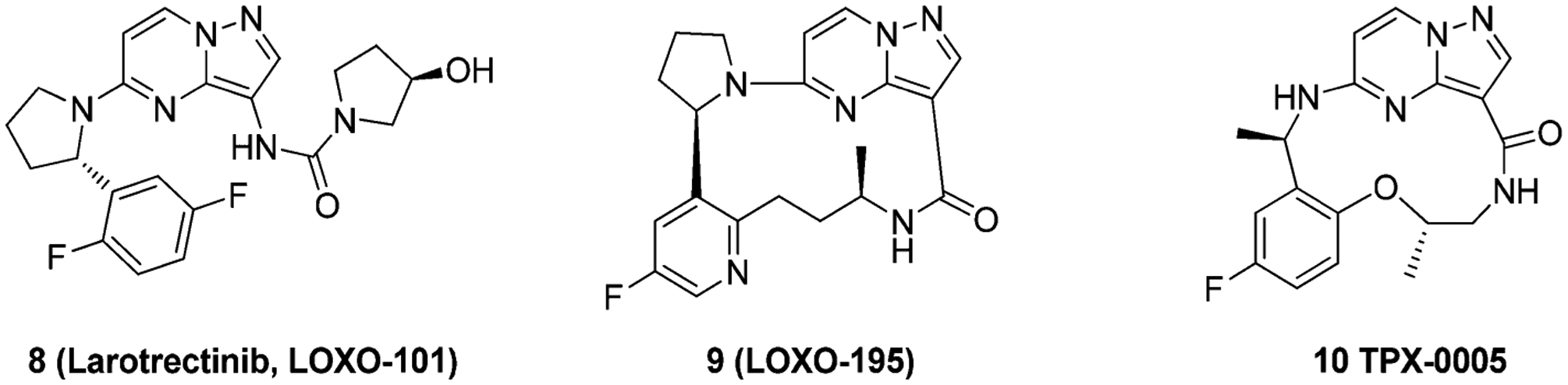
Chemical structures of compound 8 and second generation TRK inhibitors 9 and 10.
Second Generation Type I TRK Inhibitors.
All first generation TRK inhibitors exhibit profound, upfront efficacy but eventually lose effectiveness due to the development of secondary mutations in the TRK kinase domain. After treatment with 8 or 4–3, mutations in the TRK kinase domain have been reported158 that render further treatment ineffective.47,159 These mutations include TRKAG595R (and its paralogue TRKCG623R) in the solvent front of the active site, TRKAG667C (and its paralogue TRKCG696A) adjacent to the DFG motif, and TRKAF589L at the gatekeeper region.47,99,159–161 TRKAG595R and TRKAG667C are analogous to ALKG1202R and ALKG1269A, respectively.159 In TRKA, the native residues are important to accommodate 8 and 4–3, while the mutations create steric clashes with the inhibitors. In addition, TRKAG595R increases ATP affinity to the kinase.159 Structural modeling suggests that each mutation directly interferes with binding of 8, 4–3, and all other TRK tyrosine kinase inhibitors.47,160 Functional studies have subsequently confirmed that cancer cells harboring these mutations are cross-resistant to all TKIs with anti-TRK activity.162,163
To generate inhibitors that could overcome point mutations in TRK, compounds 9 (LOXO-195) and 24 (TPX-0005) were designed to limit the compound surface area in the active site (Figure 16). Both compounds were designed as fused macrocycles to reduce unnecessary interactions at either end of the binding pocket. Modeling suggested that TRKAG595R would introduce steric clashes between the arginine side chain and the hydroxypyrrolidine group of 8, and TRKAG667C would create steric clashes between the cysteine or alanine side chain and the difluorophenyl group of 8 (Figure 16A).159 To determine the impact of TRK kinase mutations on inhibitor activity, 8 and 9 were tested against TRKAG595R, TRKAG667C, and TRKCG623R. Compound 9 achieved low nanomolar inhibitory activity against all mutated kinase (IC50 values = 0.002 μM, 0.00098 μM, and 0.0023 μM); while compound 8 displayed significantly reduced inhibitory activity (IC50 values = 0.069 μM, 0.045 μM, and 0.048 μM).159 An innovative firstin-human clinical trial has demonstrated efficacy of compound 9 in 2 patients who had developed acquired resistance to 8 mediated by secondary TRKAG595R or TRKAG623R mutations.159 Compound 10, an analogue to 9, potently inhibited ROS1G2032R (IC50 = 0.0084 μM), TRKAG595R (IC50 = 0.0004 μM), TRKBG639R (IC50 = 0.0019 μM), and TRKCG623R (IC50 = 0.0004 μM).165 Compound 10 also exhibited activity in xenograft tumor models bearing WT and kinase domain mutations of ALK, ROS1, and TRKA.166,167
Figure 16.
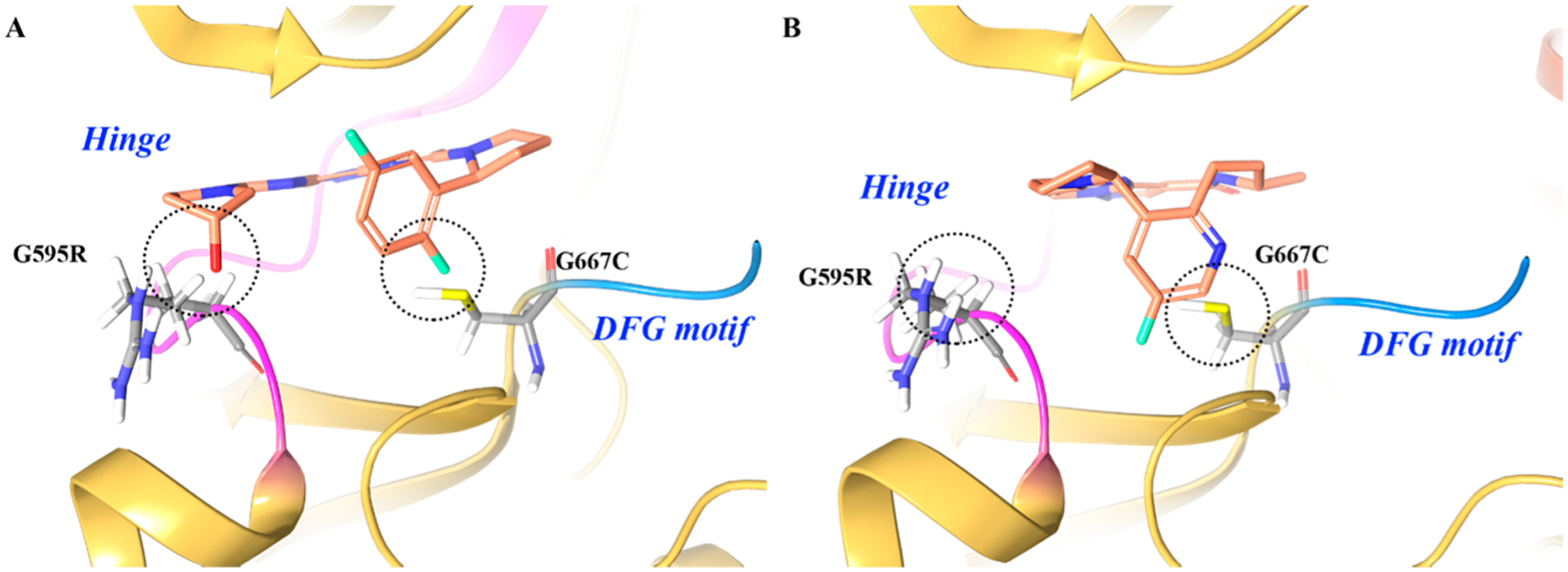
(A) Proposed binding and steric interactions of 8 with the TRKA double mutant G595R and G667C and (B) proposed binding of 9 to the TRKA double mutant for which steric interactions are not predicted between G595R and G667C. The kinase is depicted in yellow ribbons.164 Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
Other Type I TRK Inhibitors.
Several other TRK scaffolds have been reported that exhibit type I characteristics and are summarized in the following section (Figure 17). Aza-oxindole and oxindoles were reported as potent TRK inhibitors by Wood et al.168 The representative compound 11 exhibited pan-TRKA inhibitory activity (TRKA/B/C IC50 values = 0.030 μM, 0.007 μM, 0.005 μM, respectively) with selectivity in the greater kinome but also inhibited VEGFR2 (vascular endothelial growth factor receptor 2).168,169 Compound 11 shares similarities in structure to other known VEGFR2 inhibitors, such as sunitinib, which likely accounts for its VEGFR2 profile.168 Choe et al. reported a series of pyrrole[3,4-c]pyrazole TRKA inhibitors (Figure 17). The representative compound 12 inhibits TRKA with an IC50 of 0.019 μM. SAR studies indicated that the cyclopropyl and benzyl carbamate groups are essential for potency and removal of either resulted in a loss of activity.170 Carboni et al. developed compound 13 as a dual IGF-1R/IR inhibitor (IC50 = 0.0018 μM and 0.0017 μM, respectively). Kinase profiling showed that compound 13 was active on several other kinases, including TRKA/B (IC50 values = 0.007 μM and 0.004 μM, respectively).171 Cho et al. developed compound 14 as a c-MET inhibitor based on an aminopyridine core substituted with benzoxazole (IC50 = 0.08 μM).172 Compound 14 was later identified to have activity against the mutant c-METY1230D kinase (IC50 = 0.003 μM) and TRKA (IC50 = 0.039 μM).173 Stachel et al. reported a series of pyrido[3,2-d]pyrimidines as TRKA inhibitors; the representative compound 15 had a TRKA IC50 of 0.011 μM and moderate cell activity (IC50 = 1.688 μM).174
Figure 17.
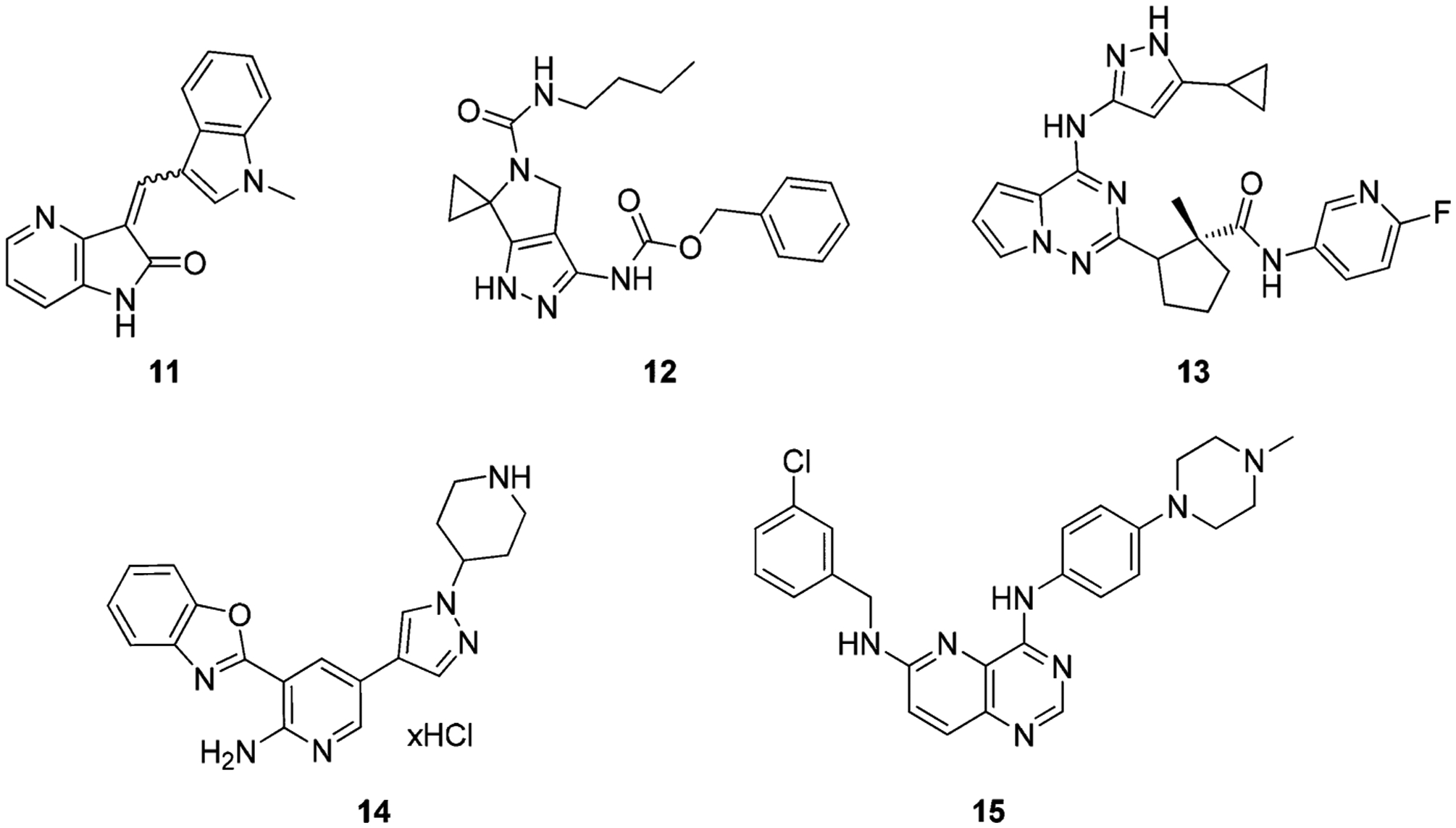
Chemical structures of other type I TRK inhibitors.
Radiolabeled Type I TRK Inhibitors.
Positron emission tomography (PET) is a noninvasive in vivo imaging technique that enables the visualization and quantification of the distribution of molecules labeled with positron-emitting isotopes. This method enables the evaluation of biochemical and physiological processes by monitoring the distribution and kinetics of a labeled molecule in vivo and has found broad application in clinical practice (personalized treatments), research, and pharmaceutical development.175–178
Because of the clinical importance of TRK in cancer as well as the central nervous system, Bernard-Gauthier et al. generated radiolabeled TRK ligands to assess and monitor TRKA/B/C levels in the brain. The TRK ligands were synthesized from known inhibitors using 11C- or 18F-labeled PET radiotracers suitable for in vivo imaging (compounds 16–20) (Figure 18).169,179–183 These compounds demonstrated comparable activity and selectivity profiles to the unlabeled precursor.181 Compound 16 displayed excellent TRK selectivity in a panel of brain and cancer relevant kinases and TRK-specific binding in vitro in rat brain and human neuroblastoma cryosections. The radiocompound also exhibited uniform regional brain distribution but was highly susceptible to CYP450 metabolism.169 Nevertheless, TRK radiocompounds can prove instrumental in studying TRK biology, diagnosing TRK-positive disease, and determining drug disposition of TRK inhibitors in real time.176,178 It is worth noting that compound 19 was the first TRK radiotracer clinical lead involved in a nonhuman primate imaging study;183 compound 20 was the first reported radiotracer used as an in vivo probe to explore endogenous kinase densities using PET neuroimaging in humans.182
Figure 18.
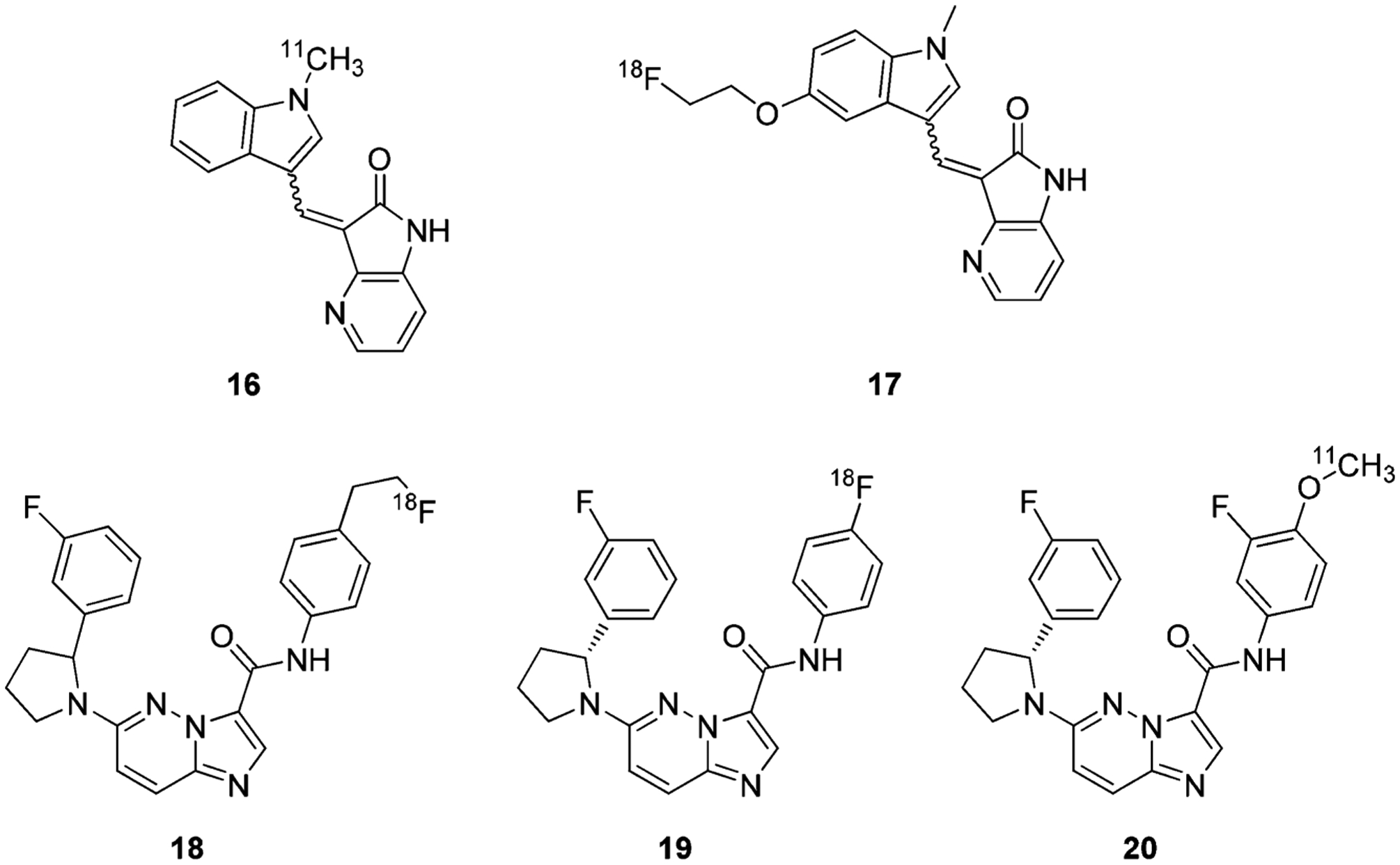
Chemical structures of representative radiolabeled type I TRK Inhibitors.
Type II TRK Inhibitors.
Type I TRK inhibitors are ATP competitive and typically possess pan-kinase activity. A method to increase kinase selectivity and potency is to generate inhibitors that bind in a type II fashion.184 Type II inhibitors are generally more active against tyrosine kinases, which can be exploited to increase selectivity and potency profiles.185 TRK inhibitors that display type II binding kinetics can be preferred depending on the biology being studied or the disease being treated. It is important to note that the most advanced TRK inhibitors in clinical trials are type I (8 and 4–3). These inhibitors are being evaluated for anticancer properties and may have inhibition characteristics optimal for a tumor environment. In other therapeutic areas, such as the central nervous system (CNS) or pain management, TRK inhibitors with a type II profile maybe preferred because, as a class, they are more selective than type I inhibitors.185 However, the typically larger molecular architecture of type II compounds can cause less favorable drug-like properties, leading to development challenges. In the following section, the development of type II TRK inhibitors is disclosed, and developmental efforts were primarily focused on optimizing for anticancer properties.
Albaugh et al. identified type II TRK inhibitors based on an oxindole-core (Figure 19).186 Oxindole 21 inhibited Ba/F3-Tel-TRKA/B/C with IC50 values less than 0.06 μM. SAR studies at the R1 position showed that the phenyl ring is critical for TRK specific inhibitory activity (21–1a vs 21–1b, TRKA cell IC50 2.3 μM vs 0.91 μM).186 To improve drug properties, several basic groups were introduced at the terminal phenyl ring and were well tolerated and increased potency but did not improve solubility. Substituting the aryl ring with 3-CF3 at R1 is crucial for TRK activity (21–1c and 21–1d, TRKA cell IC50 0.39 μM and 0.013 μM, respectively). Truncating the amide group to aniline resulted in a total loss of activity, indicating the amide is necessary for TRK activity (21–1e, TRKA cell IC50 > 10 μM).186 Replacement of the pyrrole with other groups or methylation of the pyrrole nitrogen yielded significantly less potent compounds suggesting that the acidic proton on pyrrole is involved in a key hydrogen bond at the hinge region. Substitution on the pyrrole showed that carbolic acid at position 4 is preferred for potency and selectivity, with or without methyl substitution (21–2a to 21–2c, TRKA cell IC50 0.014 μM, 0.01 μM, and 0.012 μM; selectivity 43-fold, 150-fold, and 108-fold, respectively).186 Replacement of the amide with urea generated compounds with increased potency and selectivity over VEGFR2 (21–3a, TRKA cell IC50= 0.004 μM, selectivity 130-fold).186 Introduction of a methyl substitution at R3 and a fluorine at R4 generated compound 21–3b (GNF-5837), which exhibits pan-TRK inhibition (TRKA/B/C IC50 values = 0.011 μM, 0.009 μM, and 0.007 μM, respectively) and about 300-fold selectivity over VEGFR2 (IC50 = 3.0 μM). In BALB/c mice and Sprague–Dawley rats, compound 21–3b demonstrated low drug clearance and moderate bioavailability. In mice bearing RIE xenografts expressing TRKA and NGF, compound 21–3b (100 mg/kg/day po) significantly inhibited tumor growth.186
Figure 19.
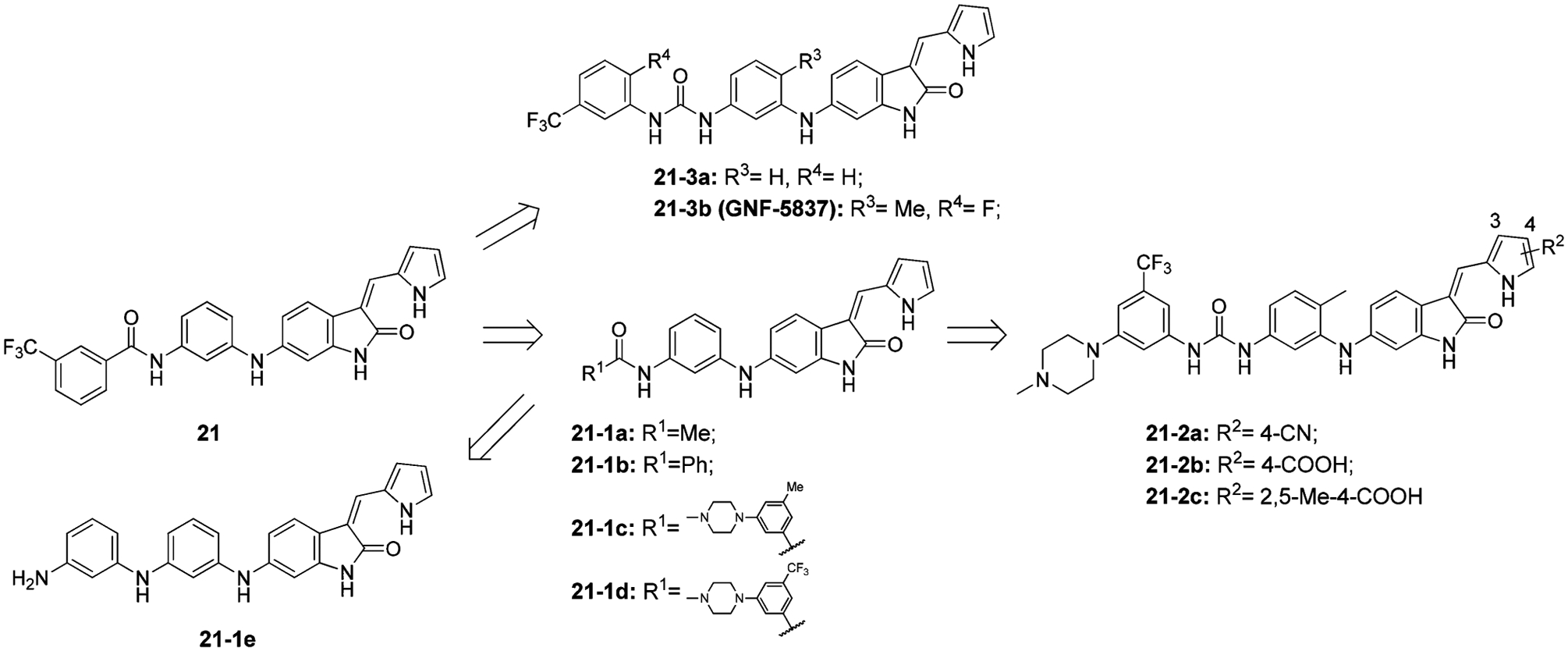
Structural optimization from compound 21 to 21–3b (GNF-5837).
In the cocrystal structure of 21–3a with TRKC (Figure 20), 21–3a is found to bind to the kinase in a type II manner.186 The oxindole core and pyrrole nitrogen form key hydrogen bonds with hinge residues Glu618, Tyr619, and Met620. The urea functionality interacts with Glu588 from the α-C-helix and Asp697 from the DFG motif. In addition, interactions occur at the phenyl bridge with Phe617 (gatekeeper) and a face to edge interaction with Phe698 from the DFG motif.186
Figure 20.
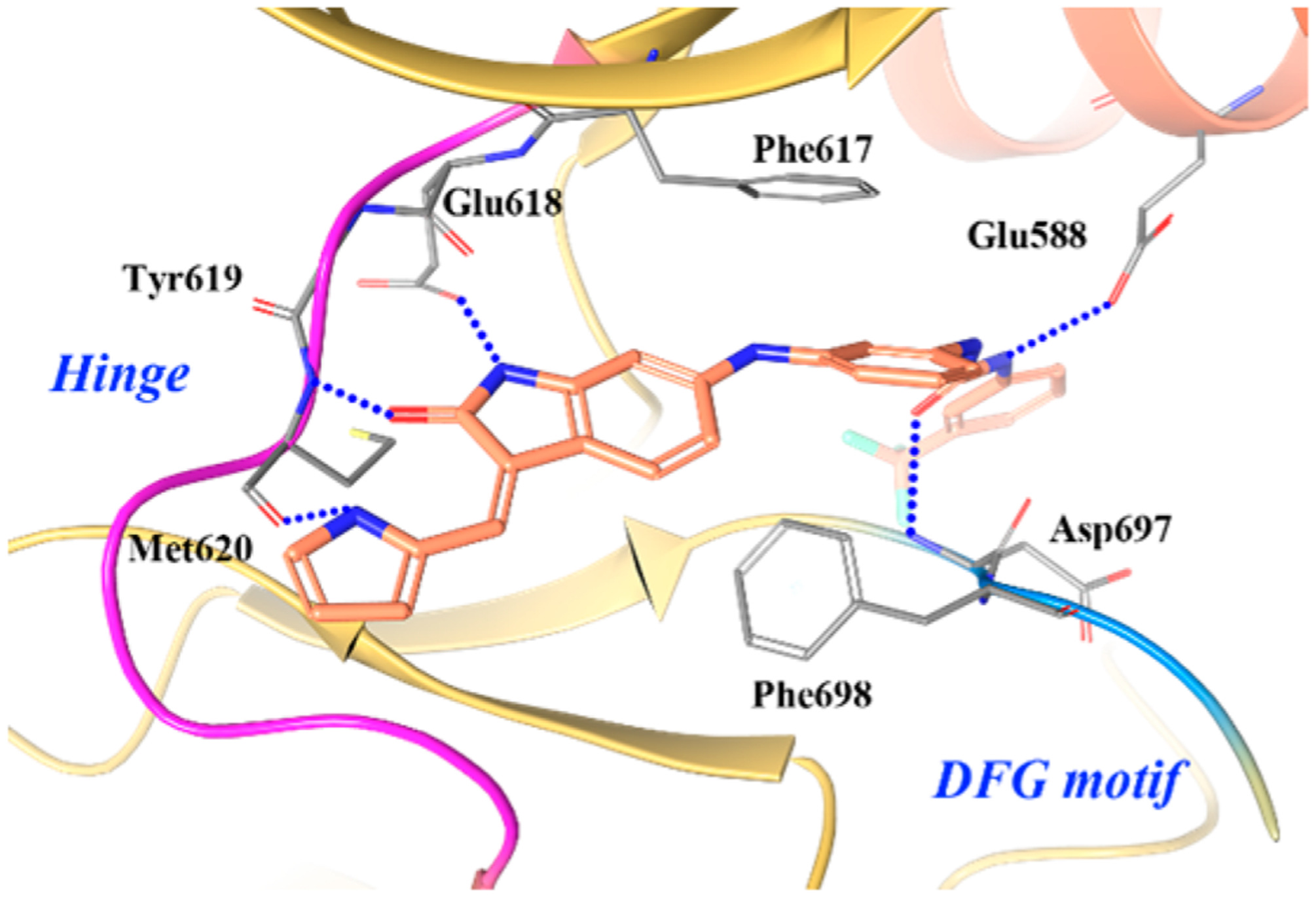
Co-crystal structure of compound 21–3a in TRKC (PDB ID 3V5Q, 2.2 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
Skerratt et al. reported a series of pyrrolopyrimidine derivatives as pan-TRK inhibitors (Figure 21A).187 By employing a high-throughput TRKA/B cell screening assay, compound 22 was identified as a pan-TRK inhibitor (TRKA/B/C IC50 values = 0.002 μM, 0.005 μM, and 0.004 μM, respectively) with strong kinome selectivity but poor water solubility (<0.3 μM).187 To improve solubility, the urea group was replaced with an amide and the 4-NH2 was removed to generate compound 22–1 (TRKA/B/C IC50 values = 0.012 μM, 0.004 μM, and 0.003 μM, respectively) with improved water solubility (~13.2 μM).187 It was determined that the scaffold was likely metabolized by aldehyde oxidase and inhibited hERG (human ether-à-go-go-related gene) potassium heart channels.188,189 In order to reduce metabolic liabilities and off target toxicities, an amino group was added at the 2-position furnishing compound 22–2 (TRKA/B/C IC50 values = 0.008 μM, 0.005 μM, and 0.004 μM, respectively; solubility = 30 μM).187 To further improve physical–chemical properties and aqueous solubility, a hydrophilic hydroxymethylene group was added to the N-iPr motif and the terminal ring was replaced with pyridine to obtain compound 22–3 (PF-06273340). Compound 22–3 was a potent pan-TRK inhibitor (TRKA/B/C IC50 values = 0.006 μM, 0.004 μM, and 0.003 μM, respectively; solubility = 131 μM) and exhibited selectivity over a large kinase panel.187 Currently, Phase 1 clinical trials are ongoing to examine the activity of different doses of 22–3 on a panel of evoked pain tests in healthy male subjects (NCT02260947).
Figure 21.
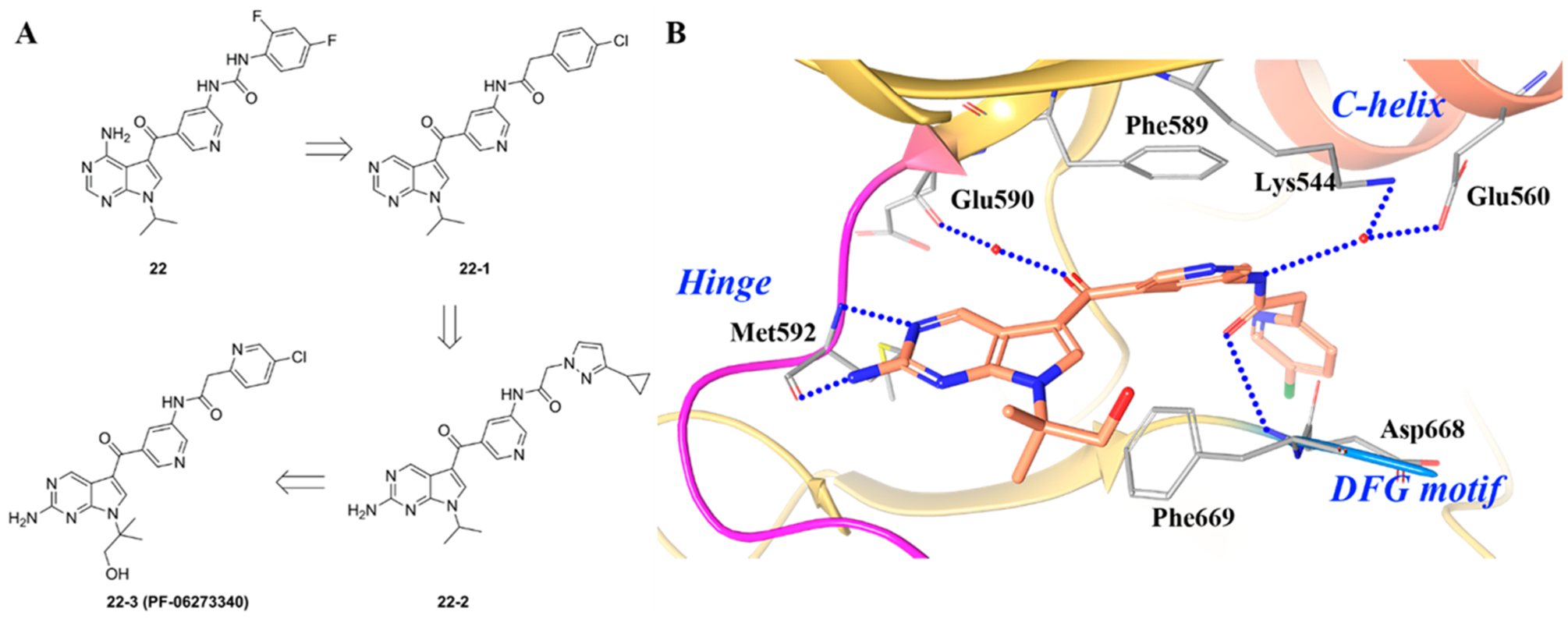
(A) Structural optimization from compound 22 to 22–3 and (B) cocrystal structure of compound 22–3 with TRKA (PDB ID 5JFX, 1.63 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; chlorine, green.
Figure 21B shows the cocrystal structure of 22–3 bound to TRKA and highlights key protein–ligand interactions.187 Compound 22–3 binds to the DFG-out form of TRK, with the 2-aminopyrrolopyrimidine forming hydrogen bonds at the hinge and the ketone binding to Glu590 through a water contact.187 Most kinase inhibitors engage in one or two interactions at the hinge, while 22–3 is found to engage in three. The central pyridine group of 22–3 engages in a double π–π stacking interaction with Phe589 (gatekeeper residue) and Phe669 (DFG motif). The carbonyl oxygen forms a hydrogen bond with the amide backbone of Asp668 (DFG motif) and the amide engages TRKA through a water contact between Lys544 and Glu560 from the c-helix. The lipophilic back pocket accommodates the chloropyridine group.187 Based on the binding characteristics of 22–3, the inhibitor can be considered type II.
Compound 23 (cabozantinib, XL184) is a multiple kinase inhibitor with activity against c-MET/VEGFR2 and was found to have a potent TRKB inhibitory activity (IC50 < 0.02 μM, Figure 22A).190 Compound 23 is currently under Phase 2 clinical trials in patients with RET fusion-gene non-small-cell lung cancer and those with other activating genes, such as ROS1, NTRK, MET, or AXL (NCT01639508). Compound 24 (sitravatinib, MGCD516) was developed by Patwardhan et al. as a potent multikinase inhibitor (Figure 22A) active against several kinases (IC50 values for AXL 0.0015 μM, VEGFR2 0.005 μM, FLT3 0.008 μM, c-KIT 0.006 μM, TRKA 0.005 μM, and TRKB 0.009 μM). Compound 24 blocked phosphorylation of several RTKs and induced potent anticancer effects in vitro; the compound was also active in vivo in MPNST (neurosarcoma) and LS141 (hybridoma) mouse xenograft models.191 Compound 24 is currently under Phase 1 clinical development for patients with advanced solid tumor malignancies (NCT02219711). Compound 25 (altiratinib, DCC2701) was reported by Smith et al. as a multitargeted kinase inhibitor with preferential activity on MET, TIE-2, and VEGFR2 (IC50 values = 0.0027 μM, 0.008 μM, and 0.0092 μM, respectively) (Figure 22A). Subsequently, 25 was identified to exhibit activity as a pan-TRK inhibitor (TRKA/B/C IC50 values = 0.00085 μM, 0.0046 μM, and 0.0083 μM, respectively) and FLT3 inhibitor (IC50 0.0093 μM).192 In cell antiproliferative assays, compound 25 exhibited IC50 values of 0.00069 μM in K562 cells, 0.0012 μM in SK-N-SH cells, and 0.0014 μM in KM-12 cells. Compound 25 inhibited tumor growth in the MET-amplified MKN-45 xenograft model in a dose-dependent manner. Further, compound 25 can actively penetrate the blood–brain barrier, indicating its potential for the treatment of brain cancers, brain metastases, and cancer pain. Compound 25 is currently under Phase 1 clinical development for patients with advanced solid tumors with NTRK or MET genomic alterations (NCT02228811).
Figure 22.
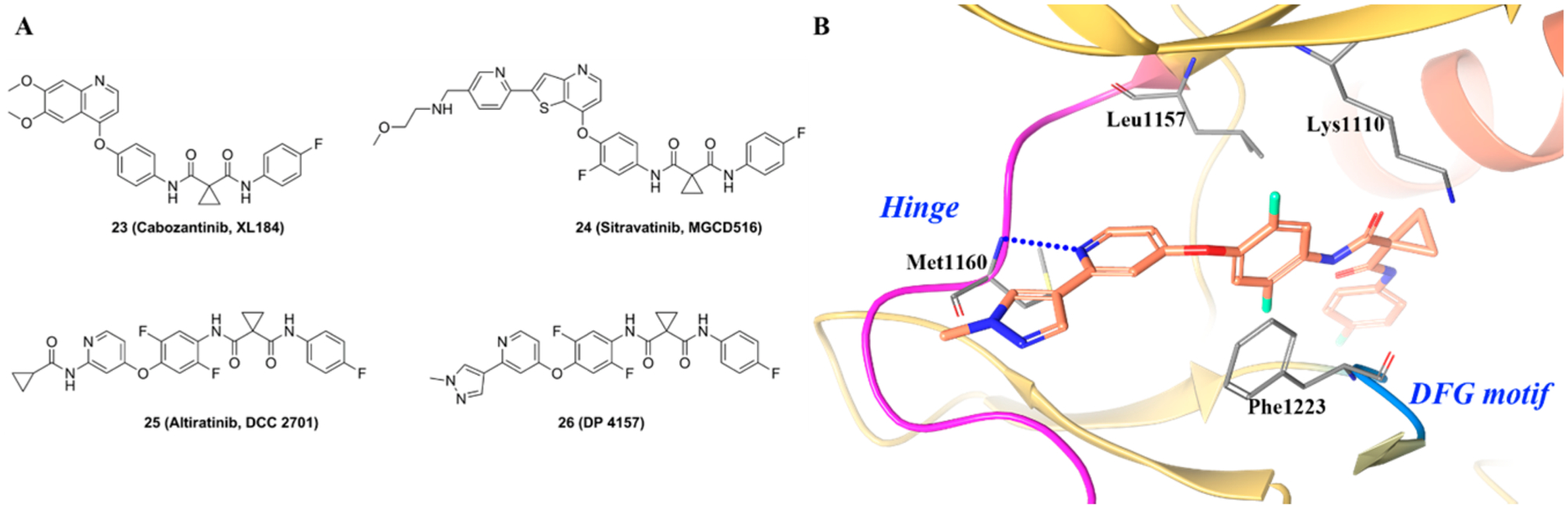
(A) Chemical structures of representative dicarboxamide TRKA inhibitors and (B) cocrystal structure of compound 26 in c-MET (PDB ID 5DG5, 2.6 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
Figure 22B illustrates the general type II binding mode of the dicarboxamide kinase inhibitor class.192 The pyridine of 26 (DP4157) binds to the hinge region and forms a hydrogen bond with Met1160. The central difluoro phenyl ring is in a hydrophobic pocket formed by Lys1110, Leu1157, and Phe1223 and engages in a π–π stacking interaction with Phe1223. The terminal para-fluoro phenyl ring binds in the allosteric pocket between the DFG motif and α-C-helix.192
Other Type II TRK Inhibitors.
El Zein et al. reported benzothiazole derivative 27 as multitargeted kinase inhibitors (Figure 23).193 Compound 27 was active on several kinases with IC50 values below 0.1 μM (including VEGFR2, ABL, TRKA, TRKB, and TIE2). Specifically, the IC50 values for TRKA and TRKB were 0.0038 μM and 0.0044 μM, respectively. In antiproliferative studies, compound 27 was active against the KM-12 cell line with an IC50 of 0.019 μM. Arcari et al. reported a series of 4-aminopyrrolopyrimidine derivatives as TIE-2 inhibitors.194 Compound 28 (Figure 23) was identified as a potent pan kinase inhibitor with activity against c-MET (IC50 = 0.0047 μM) and RON (IC50 = 0.0012 μM) and exhibited antitumor activities in multiple mouse xenograft models.157 Medicinal chemistry efforts led to the identification of compound 29 (Figure 23), which was found to be a TIE-2/TRKA dual inhibitor (IC50 for TIE-2 and TRKA were 0.0037 μM and 0.004 μM, respectively).194 A close analogue of 29, compound 30, progressed to clinical trials, but the Phase I multiple dose trials were terminated due to the development of significant CNS adverse events, including cognitive deficits, personality changes, and sleep disturbances.28,187 Compound 31 (Figure 23) is another compound reported by Stachel et al.174 with good TRK inhibitory activity (TRKA IC50 = 0.009 μM, cell IC50 = 0.007 μM). This compound was found to bind to the TRK kinase in the DFG-out conformation.195 On compound 31, the imidazole warhead binds to the hinge region, the central triazole ring forms an interaction with the gatekeeper residue, and the pyrazole occupies a selectivity pocket normally occupied by Phe669. This inhibitor is very unique because of the innovative triazole linker and imidazole warhead. Frett et al. described pyrazine-based derivatives as TRKA inhibitors (Figure 23).196 Compound 32 had a TRKA IC50 of 0.005 μM (Figure 23), and modeling studies determined the compound binds in a typical type II conformation. Compound 33 was initially reported by Conway et al. as a selective CSF-1R (colony stimulating factor 1 receptor) inhibitor (Figure 23) that completely inhibited CSF-1R in vitro at an IC50 of 0.06 μM but was inactive against 26 other kinases.197 Later, the same authors found compound 33 also inhibited TRKA with an IC50 of 0.88 μM.198 The radiolabeled analogue of 33 was reported by Bernard-Gauthier et al.181 Bertrand et al. reported that compound 33 bound to TRKB in a DFG-out conformation.73 Hong et al. reported a series of 3,5-disubstituted-7-azaindoles as TRK Inhibitors.199 Further modification uncovered compound 34 (Figure 23), which is a pan-TRK inhibitor with selectivity over 30 kinases. The activity of compound 34 against TRK isoforms was equal (pan-TRK IC50 = ~0.001 μM), but it was 100-fold more selective against other kinases.199 Amino pyrimidine derivatives were developed as TIE-2 (tunica interna endothelial-2) inhibitors with compound 35 (Figure 23) exhibiting TIE-2 inhibition (IC50 = 0.005 μM) with almost equal TRKA inhibitory activity (IC50 = 0.008 μM).200 Through screening of an in-house kinase library, Kim et al. found 2-amino-5-(thioaryl)thiazole to be a promising scaffold for TRK inhibition.108 Further SAR investigation led to the identification of compound 36 (Figure 23) (TRKA IC50 = 0.0006 μM), which was selective over CDK, MET, and IGF-1R (IC50 values = 0.54 μM, >1 μM, and 0.43 μM, respectively).
Figure 23.
Chemical structures of other Type II TRK inhibitors.
Pseudo-Type II TRK Inhibitors.
A quintessential property of type II inhibitors is the formation of a strong hydrogen bond at the hinge region.137 In certain inhibitors of TRK, however, the hydrogen bond at the hinge is abnormally weak with the majority of important interactions occurring in an adjacent allosteric pocket. This was first reported by Stachel et al. with the development of urea based compound 37 (Figure 24A) (TRKA kinase IC50 = 4.233 μM, cell IC50 = 0.549 μM).174 Structure optimization resulted in the identification of compound 37–1 (TRKA kinase IC50 = 0.011 μM, cell IC50 = 0.003 μM). Figure 24B illustrates the binding mode of 37 with TRKA.174 It is clear from this study that the compound bound to TRKA in a DFG-out conformation, which is a typical feature for amide- and urea-linked type II kinase inhibitors. However, 37 is structurally unlike any known type II TRK inhibitor because of its low molecular weight and simple architecture.174 At the ATP-binding site, the thiazole hetero-cycle forms a very weak hydrogen bond (3.45 Å) with the amide backbone of Met592. The benzylic ring is involved in hydrophobic interactions with the gatekeeper residue, Phe589. The para-trifluoromethoxyphenyl group occupies the hydrophobic pocket normally occupied by Phe669 in the DFG-in conformation. The amide carbonyl forms a hydrogen bond with Asp668 as is typical with most type II kinase inhibitors.137 A highly unique attribute to 37 is the cyclopropyl group, which occupies a hydrophobic cleft and is tucked against Val524. This is an uncommon trait, as most kinase inhibitors are unsubstituted at this position and hydrogen bond to a glutamic acid on the α-C-helix.174 One interpretation of the cyclopropyl SAR is that a critical hydrophobic mass is required to fill a small pocket near Val524 and Phe589. The placement of a hydrophobic group at this region is hypothesized to displace water, which would otherwise occupy the area.174 These interactions suggest the compound exhibits binding properties more typical of a type III inhibitor.
Figure 24.
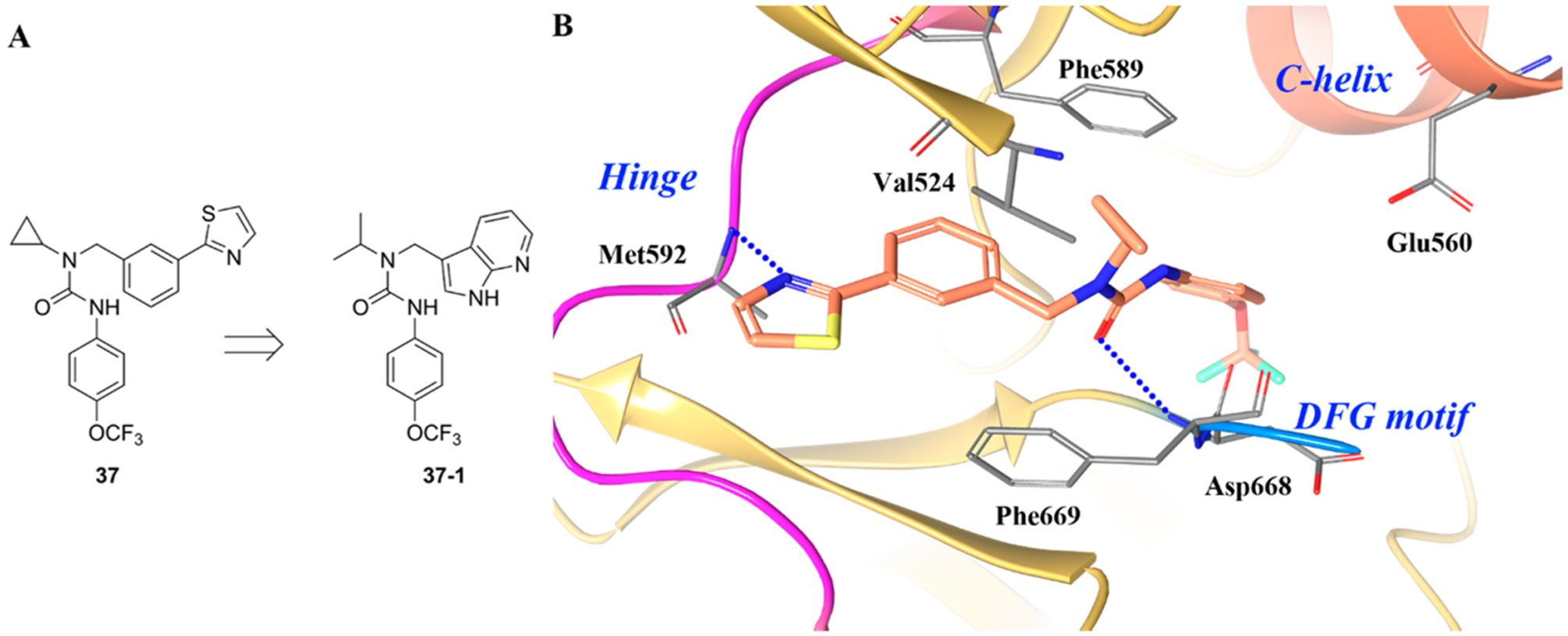
(A) Identification of compound 37–1 and (B) cocrystal structure of compound 37 and its binding mode in TRKA (PDB ID 4PMP, 1.8 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan; sulfur, yellow.
In the same manuscript by Stachel et al., the author discussed the development of compound 38 (TRKA kinase IC50 = 17.13 μM, cell IC50 = 1.162 μM) to compound 38–1 (TRKA kinase IC50 = 0.051 μM, cell IC50 = 0.029 μM) (Figure 25A).174 Compound 38 was shown to bind to the DFG-out conformation of TRKA (Figure 25B).174 The cocrystal structure revealed several unusual binding features. First, no direct interaction between the hinge backbone and the inhibitor was identified. Instead, the N5 nitrogen from the azaindole was shown to participate in a water-mediated hydrogen bond to the hinge region.174 The naphthalene moiety was buried in the hydrophobic cleft at the front of the hinge flanked by the activation loop residue Met671. One of the more unusual binding features is that the indole carboxylic acid interacts with two backbone amides in the activation loop. The unique interactions at the hinge and the activation loop regions anchor 38 to the ATP binding site. A third interaction was also evident between the carboxylic acid and Lys544 on the roof of the ATP binding pocket.174 Compound 38 exhibited phosphorylation-dependent binding, which was due to the unusual conformation between the indole carboxylic acid and the activation loop. In the cocrystal structure, Tyr676 was involved in a hydrogen-bond interaction with Asp596. Since Tyr676 is a known phosphorylation site on TRKA, phospho-Tyr676 would disrupt this interaction with Asp596, forcing the activation loop away from the active site and stabilizing the active conformation. As such, the phosphorylated form of the enzyme is not able to bind compound 38.174 Because of the highly unique binding of 38 to TRKA, the compound exhibits a profile more typical of type III.
Figure 25.
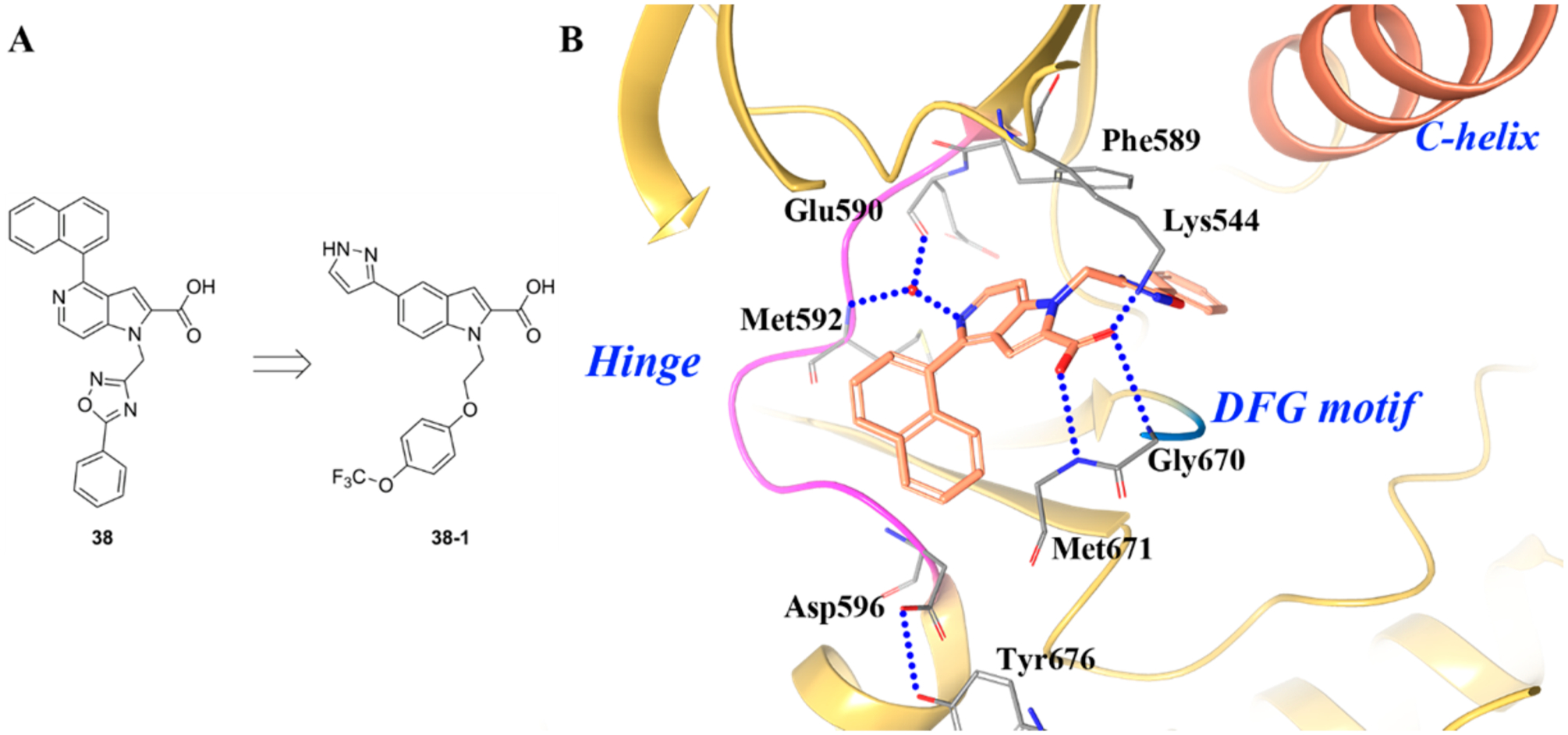
(A) Identification of compound 38–1 and (B) cocrystal structure of compound 38 and its binding mode in TRKA (PDB ID 4PMS, 2.8 Å). The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red.
Type III-JM TRK Inhibitors.
TRKA activation triggers intracellular signaling cascades that increase the sensitivity of nociceptors, thus leading to chronic sensitization and pain.201 However, TRKA/B/C share significant sequence homology at the kinase domain, which will make it extremely difficult to develop inhibitors selective for a single isoform. A method to circumvent selectivity issues is to develop allosteric inhibitors that do not bind to the highly conserved ATP active site. In an effort to identify TRKA specific inhibitors, Su et al. discovered compounds with novel scaffolds that bound to the juxtamembrane (JM) domain of TRKA.138 The JM domain is located between the kinase domain and the transmembrane domain, and there is only 36% identity between TRKA and TRKB and 40% identity between TRKA and TRKC.138 These findings provide the research community with a novel exploitable region to generate TRK selective small molecule inhibitors.
Compound 39 is a selective TRKA inhibitor (IC50 values for TRKA/B/C were 0.099 μM, >81 μM and 25 μM, respectively) (Figure 26A).138 In the cocrystal structure of 39 and TRKA (Figure 26B,C), compound 36 bound behind the DFG motif opposite of the kinase active site.138 The DFG motif was found in a DFG-out, inactive conformation, with Phe669 pointed toward the active site. Asp668, which coordinates to the phosphate groups on ATP, is away from the active site in the structure. The central urea of compound 39 makes two hydrogen bonds with Asp668. Asp668 is part of the DFG motif, and binding to compound 39 requires the DFG motif to be in the “out” conformation.138 The fluorine moiety of compound 39 occupies a relatively hydrophobic pocket formed primarily by aliphatic amino acids. The structure clearly reveals interactions at the JM region. Ile490, within the JM, sits on top of the fluorine moiety, aiding the formation of the hydrophobic pocket138 Similar to other TRKA cocrystal kinase structures, there is a shift in Phe646, which creates a pocket to accommodate the fluorine. The oxygen of the central urea in compound 39 forms a hydrogen bond with the amide nitrogen of Ile490. The phenyl moiety occupies a position between Lys544 of the β3 strand and Glu560 of the α-C-Helix.138
Figure 26.
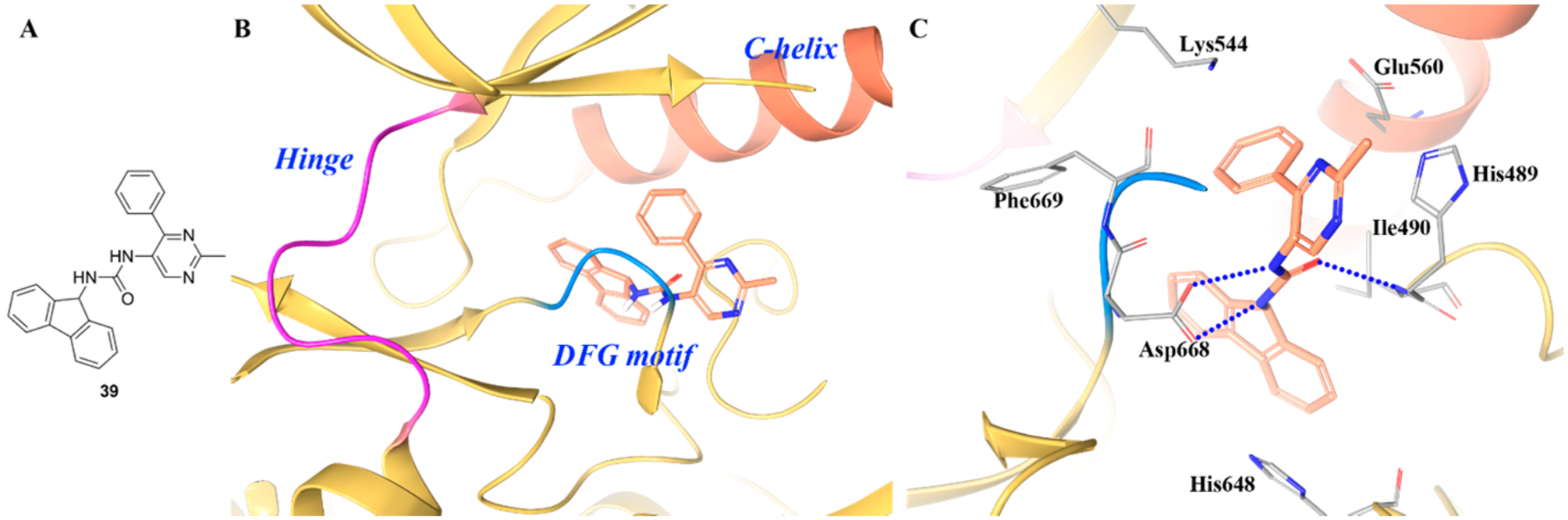
(A) Chemical structure of compound 39, (B) cocrystal structure of compound 39 in the allosteric pocket of TRKA (PDB ID 5KMI, 1.87 Å), and (C) interactions of compound 39 with surrounding residues. The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red.
Compound 40 is another selective TRKA inhibitor with interesting binding features (IC50 values for TRKA/B/C were 3.3 μM, >81.0 μM, and 27 μM, respectively) (Figure 27A).138 The cocrystal structure of compound 40 revealed the compound bound to the same pocket as 39, behind the DFG motif (Figure 27B,C). The trifluorophenyl moiety of 40 sits in the pocket occupied by the fluorine of compound 39. The central amide nitrogen is positioned close to the carboxylic acid of Asp668. The binding site is quite similar between the two structures except for the following differences: (1) for compound 39, Ile490 packs above the phenyl ring, but Leu486 packs above the phenyl ring for 40; (2) for 39, Phe646 was displaced by the bulky moiety in the hydrophobic pocket, but for 40, the smaller moiety accommodates Phe646 in a position closer to the active conformation.138
Figure 27.
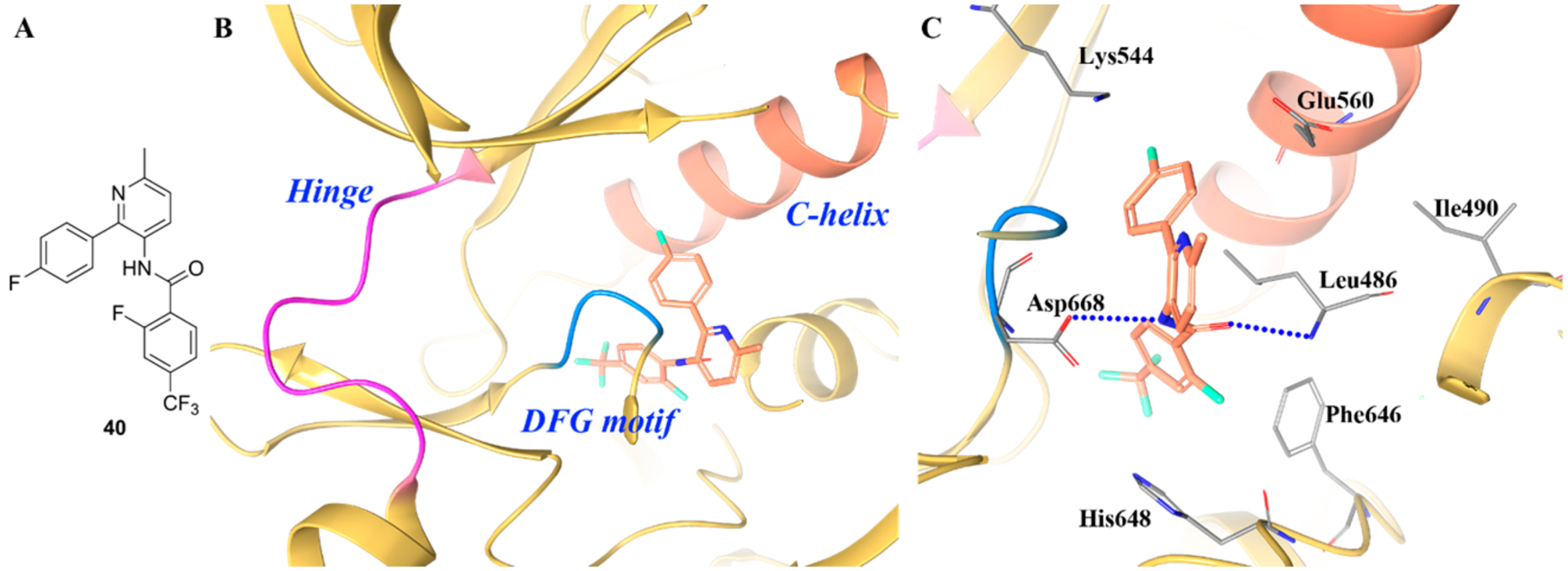
(A) Chemical structure of compound 40, (B) cocrystal structure of compound 40 in the allosteric pocket of TRKA (PDB ID 5KMK, 2.24 Å), and (C) interactions of compound 40 with surrounding residues. The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
A similar finding was also disclosed by Furuya et al.201 Compound 41 (Figure 28A) showed potent inhibitory activity against TRKA (IC50 = 0.0027 μM) but was selective against TRKB and TRKC (IC50 values were 1.3 μM and 2.5 μM, respectively). Figure 28B,C illustrates that compound 41 bound to the JM region of TRKA. The configuration of the kinase domain and A loop is very similar to that observed for Apo TRKA.201 Compound 41 binds to the deep pocket formed by the DFG region of the A loop, glycine-rich loop (G loop), C-helix, and JM region. This pocket is completely separate from the ATP site. There are four key interactions that 41 creates at the binding region: (1) Asp668 of the DFG motif forms two hydrogen bonds with the urea moiety; (2) His489 interacts with the pyrazole ring via π–π interactions and with the ethoxy group via CH–π interactions; (3) Ile490 forms a hydrogen bond with the urea moiety and interacts with the difluorobenzene group of 41 via CH–π interactions; (4) His648 interacts with the difluorobenzene moiety through a π–π stacking interaction.201 In addition to these, Leu486 is also involved in weak van der Waals interactions with the methyl and ethoxyl groups of 41. Interestingly, these amino acid residues are not conserved in TRKB nor TRKC but are integral for high binding affinity in TRKA.201 Further exploitation of these TRKA specific amino acids could furnish numerous allosteric scaffolds with high TRKA selectivity.
Figure 28.
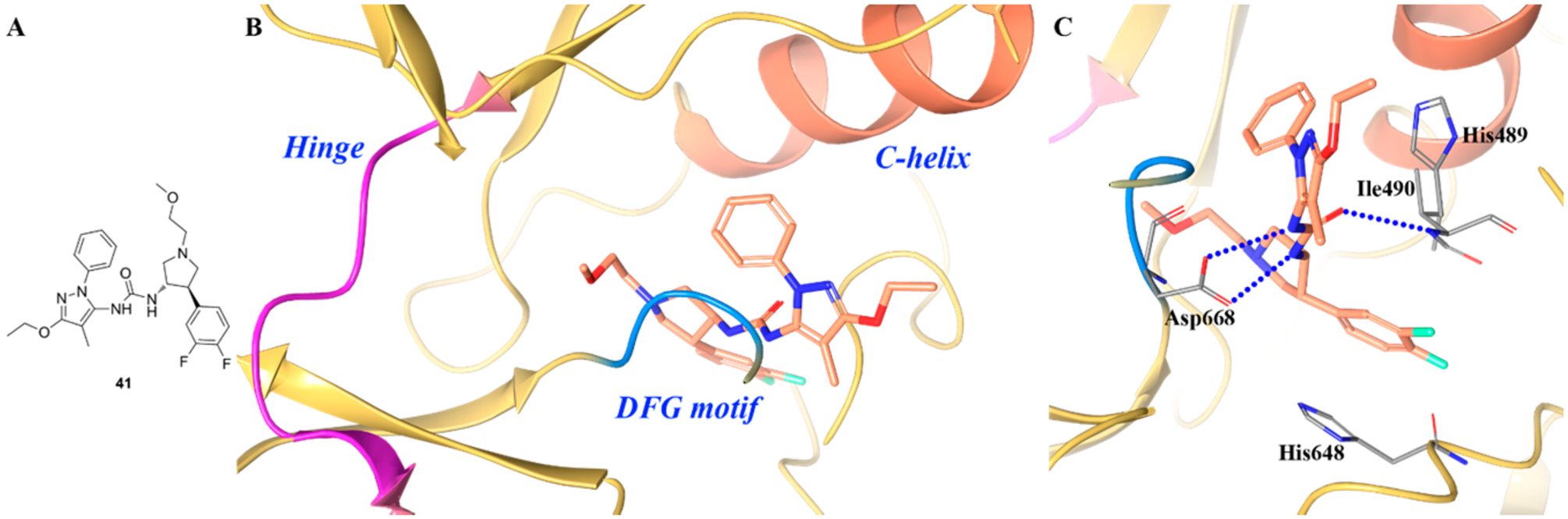
(A) Chemical structure of compound 41, (B) cocrystal structure of 41 bound in the allosteric pocket of TRKA (PDB ID 5H3Q, 2.1 Å), and (C) interactions of compound 41 with surrounding residues. The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; fluorine, cyan.
Recently, Bagal et al. reported the discovery of a series of N-phenyl-pyrazole based compounds as selective TRKA inhibitors (Figure 29A).202 The original hit 42 showed moderate TRKA inhibitory activity (TRKA cell IC50 = 3.3 μM) and >10-fold selectivity over TRKB/C (TRKB/C cell IC50 values >50 μM). By introducing an amino-pyridine ring to the dichlorophenyl ring and switching the methyl to an amide group, the resultant compound 43 demonstrated 66-fold improvement in TRKA potency (cell IC50 = 0.05 μM) and was >80-fold selective over TRKB/C (cell IC50 values were 14 μM and 4.1 μM, respectively) with improved drug-like properties.187 A variety of functional groups were evaluated on the pyridine and central phenyl ring, which led to the identification of 44 (TRKA/B/C IC50 values = 0.01 μM, 1.8 μM, and 0.7 μM, respectively) (Figure 29A). Compound 44 showed <15% inhibition against a panel of over 390 kinases at a concentration of 10 μM and was 72% orally bioavailable.202 Figure 29B,C illustrate the binding mode of 44 in the allosteric pocket of TRKA. The pyridyl nitrogen forms a hydrogen bond with the backbone of Asp668. The dichloro central phenyl ring occupies the lipophilic pocket surrounded by Leu486, Ile572, Phe646, and Ile666. The terminal carbonyl group interacts with the side chain of Arg673, and the tertiary alcohol is exposed to the solvent front.202
Figure 29.
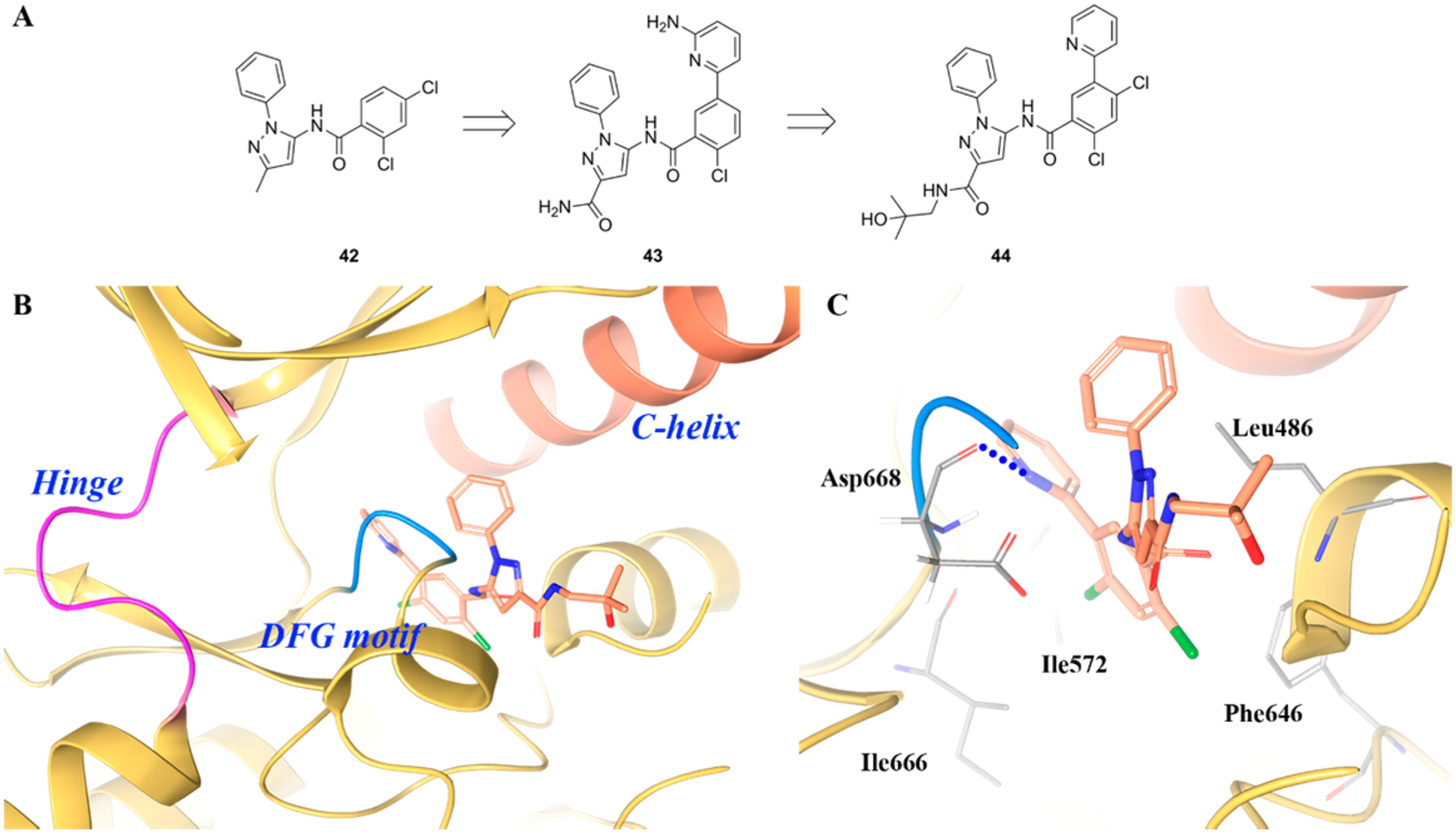
(A) Chemical structures of compounds 42, 43, and 44, (B) cocrystal structure of compound 44 bound to the allosteric pocket of TRKA (PDB ID 6D20, 1.94 Å), and (C) interactions of compound 44 with surrounding residues. The kinase is depicted in yellow ribbons, and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red; chlorine, green.
However, it is worth noting that two analogues of 39, compounds 45 and 46 (by switching the fluorine moiety to naphthalene or pyridine), were also reported by Su et al. (Figure 30A).138 These two compounds were active against TRKA and TRKC (45, TRKA/C IC50 values = 4.1 μM and 7.1 μM, respectively; 46, TRKA/C IC50 values = 4.0 μM and 2.3 μM, respectively). Interactions of compound 45 with the kinase domain are similar to 39, but the JM interactions are altered as His489 is shifted away from the compound (Figure 30B,C). Instead of binding to the allosteric pocket, 46 binds to the active site in an atypical type I conformation (Figure 30D). The urea carbonyl group forms a hydrogen bond with the hinge residue Met592, and the pyridine fragment has a weak π–π interaction with the DFG motif residue Phe669.138 In this case, it is still uncertain whether compounds with such scaffolds will exhibit TRKA selectivity. It could be expected that only a subset of type III inhibitors could be TRKA-selective because specific JM domain interactions are required for inhibition.133
Figure 30.
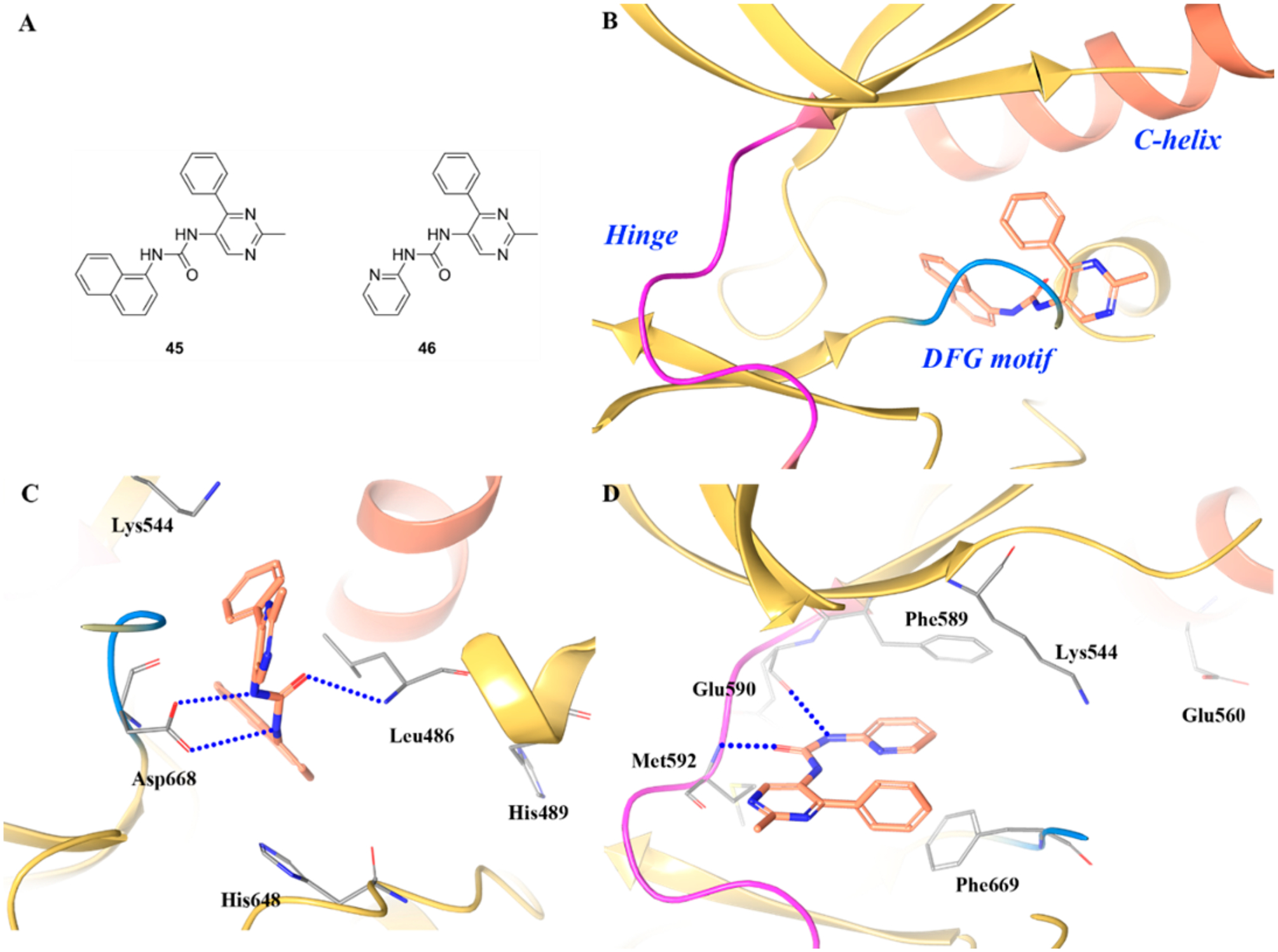
(A) Chemical structures of compounds 45 and 46, (B) cocrystal structure of compound 45 binding in the allosteric pocket of TRKA (PDB ID 5KMM, 2.1 Å), (C) interactions of compound 45 with surrounding residues, and (D) cocrystal structure of compound 46 in the active site of TRKA (PDB ID 5KMO, 2.7 Å). The kinase is depicted in yellow ribbons and the hydrogen bonds are illustrated with blue dashed lines. Compound atoms are colored as follows: carbon, orange; nitrogen, blue; oxygen, red.
Beyond compounds described above, there are three additional TRK inhibitors under clinical development with structures that have not been disclosed. These compounds include PLX7486 (NCT01804530), DS-6051B (NCT02279433), and F17752 (2013-003009-24).
CLINICAL EVALUATION OF TRK INHIBITORS
All TRK-directed compounds under clinical investigation are pan-TRK inhibitors (Table 2). This is likely due to the conservation of amino acids at the ATP binding domain of TRK isoforms. Pivotal TRK structural analysis completed by Bertrand et al. only identified a two amino acid difference between the ATP binding site of TRKA and that of TRKB.73 When they analyzed TRKB and TRKC, all ATP binding site residues were identical.73 Because all TRK inhibitors in clinical trials elicit activity by binding at the ATP binding site, they all possess pan-TRK activity. This activity profile is possibly beneficial because the inhibitors, in theory, can be utilized against a broad spectrum of tumors. However, this must be vetted cautiously, as dose-limiting toxicity can also increase based on inhibition profiles. An extensive clinical overview for the development of TRK inhibitors has been reviewed by Lange et al.203
Table 2.
Reported Drugs under Clinical Trials with Known Inhibitory Activity of TRK Kinases
| NCT identifier | phase | therapeutic agent | indication | status | patient selection | estimated completion |
|---|---|---|---|---|---|---|
| NCT03213704 | II | larotrectinib (8) | pediatric cancers | recruiting | NTRK gene fusions | 09/2024 |
| NCT02637687 | II | larotrectinib (8) | CNS cancers | recruiting | 06/2019 | |
| NCT02576431 | II | larotrectinib (8) | NTRK+ solid tumors | recruiting | NTRK gene fusions | 04/2018 |
| NCT02122913 | I | larotrectinib (8) | solid tumors | recruiting | NTRK gene fusions | 02/2017 |
| NCT03215511 | I/II | LOXO-195 (9) | TRK resistant tumors | recruiting | TRK-resistant lesions | 08/2019 |
| NCT02568267 | II | entrectinib (4–3) | solid tumors | recruiting | NTRK, ROS1, or ALK gene rearrangements | 10/2019 |
| NCT02097810 | I | entrectinib (4–3) | advanced/metastatic cancer | active | NTRK, ROS1, or ALK molecular alterations | 06/2019 |
| NCT02650401 | I | entrectinib (4–3) | pediatric and CNS cancers | recruiting | with or without NTRK, ROS1, or ALK gene fusions | 08/2018 |
| NCT01639508 | II | cabozantinib (23) | advanced NSCLC | recruiting | RET, ROS1, or NTRK fusions or increased MET or AXL activity | 07/2019 |
| NCT02048488 | I/IIa | belizatinib (5–3) | solid tumors and lymphomas | unknown | 09/2016a | |
| NCT02279433 | I | DS-6051b | advanced solid tumors | active | NTRK or ROS1 gene rearrangements | 11/2019 |
| NCT02675491 | I | DS-6051b | advanced solid tumors | active | NTRK or ROS1 gene rearrangements | 09/2018 |
| NCT02219711 | I | sitravatinib (24) | advanced cancers | recruiting | MET, NTRK, or DDR2 mutations, MET or KIT/PDGFRA/VEGFR2 gene amplification, MET, RET, AXL, NTRK1, or NTRK3 gene rearrangements, or CBL gene function loss mutations | 12/2018 |
| NCT01804530 | I | PLX7486 | advanced solid tumors | terminated | NTRK point or fusion mutations | 01/2018b |
| NCT02228811 | I | altiratinib (25) | advanced solid tumors | terminated | NTRK or MET gene alterations | 11/2017b |
Study has passed its completion date and status has not been verified in more than two years.
Actual primary completion date.
Larotrectinib (8, LOXO-101) is under clinical investigation to treat melanoma, glioblastoma, NSCLC, pancreatic and ovarian cancers, and pediatric cancers. Following a single oral dose, the unbound plasma concentration of larotrectinib is sufficient to inhibit TRKA/B/C signaling without substantial inhibition of other kinases.204 In a phase I study with larotrectinib, six patients with NTRK fusions with heavily pretreated advanced disease were enrolled (NCT02122913).205 Of the six patients, three had partial responses within 3–4 months and one patient had a near-complete response after 8 months.157 The drug was well tolerated, and the main side effects were fatigue, dizziness, and anemia. Larotrectinib was even well tolerated and efficacious in pediatric carcinomas harboring a TRK oncogene.206,207 Due to the overall positive results of the phase I study, Bayer and Loxo Oncology initiated a multicenter phase II basket trial to evaluate larotrectinib in patients with an NTRK fusion oncogene (NCT02637687 and NCT02576431). The overall response rate to treatment was 75%, with 13% exhibiting a complete response.99 At 1 year, 55% of the treated patients remained progression free. Based on the phase II results, larotrectinib has marked and durable antitumor efficacy in patients with NTRK fusions, regardless of age or tumor origin.99,208 Meeting future end points of clinical trials, such as median duration of response, progression-free survival, and overall survival, will further confirm the clinical utility of larotrectinib.
Entrectinib (4–3, RXDX-101) is a pan-kinase inhibitor under clinical development by Ignyta with activity against TRKA/B/C, ROS1, and ALK fusion oncogenes.149 In a phase 1 clinicaltrial, 119 patients were treated with entrectinib split between two subgroups: ALKA-372–001, 54 patients, and STARTRK-1, 65 patients (NCT02097810).209 Entrectinib was shown to be well tolerated and efficacious against NTRK1/2/3 and ALK gene fusions, including patients with primary or secondary CNS disease.209 The majority of treatment related toxicities were grades 1–2 and reversible with cessation or modification of treatment.209 In particular, responses were achieved regardless of histology, with responses observed in NSCLC, MASC, melanoma, glioneuronal tumor, colorectal cancer, and RCC.209 Response rates were highest in NTRK-rearranged tumors, which provided first-time, clinical “proof-of-concept” that NTRK-rearrangements are actionable drivers of tumor growth.209 Because of the success of phase 1 studies, a global, multicenter phase II basket study (STARTRK-2) is currently recruiting patients with NTRK-, ROS1-, and ALK-rearranged tumors (NCT02568267).210 These studies are being performed to confirm the results of the phase 1 study and to further establish the safety profile of entrectinib in a larger patient population. The clinical success of the phase 2 studies relies on the successful enrichment of patients with targetable gene fusions. Because of the rarity of NTRK-rearranged tumors and the success of the phase 1 trial, patients are being enrolled based on molecular, rather than histological, criteria.
Sitravatinib (24, MGCD516) is a multikinase inhibitor under clinical development by Mirati Therapeutics Inc. with activity against RTKs including MET, AXL, MER, and members of the VEGFR, PDGFR, DDR2, TRK, and Eph families.191 A phase 1/1b study is currently recruiting to evaluate dose and preliminary efficacy in patients harboring gene rearrangements involving MET, RET, AXL, or NTRK1/2/3 (NCT02219711).211 Based on preclinical data, sitravatinib is likely to be effective for metastatic soft-tissue sarcomas. Similar to entrectinib, the clinical success of sitravatinib will rest on the ability to appropriately enrich patients for targetable molecular lesions. Although blockade of multiple driver signaling pathways can be achieved with sitravatinib,191 clinical investigation must identify a therapeutic window to safely target NTRK rearrangements without breaching dose-limiting toxicity on other kinase pathways.
DS-6051b is under clinical development by Daiichi Sankyo, Inc., for advanced solid tumors harboring ROS1 or NTRK1/2/3 rearrangements. A phase 1, two-part clinical trial is underway to determine dose-limiting toxicities and tumor response for up to 2 years (NCT02279433). Results from a phase 1 trial in Japanese subjects have already been published with dose-limiting toxicities experienced at the highest dose of 800 mg QD (NCT02675491).212 Tolerability in American subjects was similar, with dose-limiting toxicities experienced at 800 and 1200 mg QD.213 Partial responses, based on tumor regression, were observed at or above the maximum tolerated dose warranting additional safety studies and better patient enrichment for a more durable, clinical response.
Cabozantinib (23, COMETRIQ) is a pan-kinase inhibitor developed by Exelixis with FDA approval to treat metastatic medullary thyroid cancer (mMTC) and renal cell carcinoma (RCC). Cabozantinib was first approved to treat mMTC in 2012, and later, in 2016, it was approved for RCC. Because of its pan-kinase profile demonstrating activity against c-Met, RET, ROS1, ALK, VEGFR2, and TRK receptors, cabozantinib is under phase II clinical investigation for patients harboring NTRK1 gene fusions in NSCLC (NCT01639508). In the same trial, the drug is being evaluated against patients with RET, ROS1, MET, and AXL oncogenes. Cabozantinib has been found to increase the risk of gastrointestinal perforations, fistulas, and hemorrhage and carries a black box warning for these side effects. In head-to-head studies, entrectinib exhibits stronger inhibition of NTRK fusion genes compared to cabozantinib, which suggests the clinical utility of entrectinib might be greater.161 Further, cabozantinib inhibits VEGFR2 at an IC50 of 0.000035 μM214 and other tyrosine kinases >0.001 μM, which positions cabozantinib as a highly potent, selective, and unique VEGFR2 inhibitor. The clinical utility of an inhibitor with strong VEGFR2 selectivity must be further evaluated for usefulness against TRK-driven disease. Because of the pan-kinase profile of cabozantinib, clinical response against TRK cancers can be robust, but this also must be weighed against the high propensity for DLTs. Phase II clinical investigation will further evaluate the usefulness of cabozantinib in safely treating TRK-driven cancers.
A major clinical consideration for TRK inhibitors is the propensity to select for drug resistant TRK mutations. For example, a metastatic colorectal cancer patient harboring a NTRK1 gene fusion displayed a marked response to entrectinib.160 However, after 4 months of treatment, entrectinib lost effectiveness in controlling tumor growth. By monitoring circulating tumor DNA (ctDNA), it was identified that the TRKA kinase domain underwent two different point mutations at G595R and G667C. These point mutations were also shown to limit the TRKA inhibitory activity of larotrectinib and belizatinib.160 To overcome TRK resistant mutations, LOXO-195 (9) was developed, which is a constrained analogue of larotrectinib. Due to conformational limitations, steric interactions with TRKA G667C and TRKC G623R (homologous to TRKA G595R) are limited and kinase inhibitory activity is maintained. These results have been translated into clinical trials where patients have responded to LOXO-195 after failing treatment with larotrectinib.159 Clinical trials for LOXO-195 are ongoing and are displaying the potential to extend durable remissions in patients with TRK-resistant lesions (NCT03215511).
CONCLUSION AND FUTURE PERSPECTIVE FOR TRK INHIBITORS
The use of kinase-directed precision medicine has been heavily pursued since the discovery and development of imatinib.42 Kinases have since emerged as one of the most intensely pursued drug targets in oncology research due to highly druggable active sites and their critical roles in cellular signaling. However, there has yet to be a clinically approved inhibitor for the TRK-receptor tyrosine kinase despite its intimate involvement in tumor pathology and disease. The vast majority of kinase inhibitors developed for TRK exhibit limited selectivity against any of the three isoforms (TRKA/B/C) and are active in the greater kinome. These types of inhibitors will have limited utility as tools to further study TRK biology but have exhibited excellent therapeutic potential in clinical trials, especially when patients treated with TRK inhibitors are selected based on their molecular pathology. To further study TRK biology, it will be beneficial to utilize highly selective inhibitors with activity specific for a single TRK isoform, such as 41 for TRKA. This will permit the inhibition of a single enzyme in the TRK family, which can help identify isoform importance and interplay in a variety of preclinical TRK models. It is important to note that TRK singaling, specifically via their corresponding growth factors,215 has been implicated in mediating and modulating the pain response. As such, beyond cancer, TRK inhibitors are being evaluated in clinical trials as agents to reduce neuropathic pain (NCT03346330).
The most clinically advanced TRK inhibitors include entrectinib, a pan-TRK, ALK, and ROS inhibitor, and larotrectinib, a specific pan-TRK inhibitor. Both larotrectinib and entrectinib have achieved orphan designation from regulatory authorities.48 A recent report of the clinical activity of entrectinib in 55 TRK mutant patients displayed an 80% overall response rate with long-lasting responses in 71% of patients (>12-months).100 An aspect that needs to be considered in the development of TRK inhibitors is, given their role in the nervous system, the possibility of adverse neuropsychiatric effects. One parameter that may influence this property is activity in the greater kinome, and another is the ability to cross the blood–brain barrier. Noteworthy, the most advanced TRK inhibitors under clinical development, such as larotrectinib and entrectinib, exhibited neurotoxicity. This is likely due to target specificity, although they are able to penetrate the blood–brain barrier and exert therapeutic effects against brain metastases.48 The identification of the clinical candidate PF-06273340 (22–3) provides a strategy to deliver peripherally restricted and orally bioavailable compounds, which was to design compounds in physicochemical space appropriate for absorption across the gastrointestinal epithelium while, simultaneously, engineering the compounds as substrates for blood–brain barrier efflux transporters.187,202
As observed with all kinase inhibitors, TRK resistance to precision medicine is a major therapeutic limitation for durable efficacy. To overcome resistance, LOXO-195 was developed and is exhibiting effectiveness to induce remissions in patients that have failed larotrectinib treatment. The exact duration of remission is being investigated in clinical trials, and the development of LOXO-195 is a hopeful addition for treating TRK-driven disease. Clinical studies have confirmed the importance of selecting patients based on molecular criteria, and this enrichment will become even more important when transitioning from first to second generation TRK inhibitors. Although all evaluation of TRK precision medicine is in the early stages of clinical development, significant breakthroughs have been uncovered for the treatment and management of TRK-driven tumors. With the completion of clinical studies incorporating larger patient populations, it is expected that TRK precision medicine will continue to exhibit efficacy. Larger clinical studies will also illuminate if selective (larotrectinib) or pan-kinase inhibitors (entrectinib) are more appropriate for treating TRK malignancies. With the advent of routine molecular profiling of tumors, patients will be treated with TRK precision medicine to induce a tailored response. This will translate to customized treatment profiles that will better target the unique pathology of a patient’s tumor.
ACKNOWLEDGMENTS
H.L. was supported by the grants (NIH 1R01CA194094-010 and 1R01CA197178-01A1).
ABBREVIATIONS USED
- AKT
v-AKT murine thymoma viral oncogene homologue
- ALIS
automated ligand identification system
- ALK
anaplastic lymphoma kinase
- AML
acute myeloid leukemia
- ATP
adenosine triphosphate
- BDNF
brain-derived neurotrophic factor
- CDK
cyclin-dependent kinase
- CML
chronic myelogenous leukemia
- DAG
diacyl-glycerol
- DDR2
discoidin domain-containing receptor 2
- ERK
extracellular-signal-regulated kinase
- FDA
food and drug administration
- FLT3
FMS-like tyrosine kinase 3
- Glio
glioblastoma
- GRB2
growth factor receptor-bound protein 2
- hERG
human ether-a-go-go-related gene
- IC
intrahepatic cholangiocarcinoma
- IGF1R
insulin-like growth factor 1 receptor
- IP3
inositol trisphosphate
- MASC
mammary analogue secretory carcinoma
- MEK
mitogen-activated protein kinase
- NGF
nerve growth factor
- NT3
neurotrophin-3
- PH
pleckstrin homology
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PLCγ
phospholipase C-γ
- PKC
protein kinase C
- PTC
papillary thyroid cancer
- PDGFR
platelet-derived growth factor receptor
- P-gp
P-glycoprotein
- QSAR
quantitative structure–activity relationship
- RAF
rapidly accelerated fibrosarcoma oncogene
- RAS
rat sarcoma oncogene
- RET
rearranged during transfection
- RIE
rat intestinal epithelial
- RON
recepteur d’origine nantais
- RTK
receptor tyrosine kinases
- SHC
Src homology 2 domain containing
- TIE-2
tunica interna endothelial-2
- TRK
tropomyosin receptor kinase
- VEGFR2
vascular endothelial growth factor receptor 2
Biographies
Wei Yan received his Ph.D. from East China University of Science and Technology, Shanghai, China, and worked jointly at Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Following, he worked at WuXi AppTec as a process chemistry scientist. He is currently a postdoctoral researcher at the University of Arkansas for Medical Sciences. His expertise is in medicinal chemistry and process chemistry for pilot plant manufacture of APIs.
Naga Rajiv Lakkaniga is currently a Ph.D. candidate in the Department of Pharmaceutical Sciences, University of Arkansas for Medical Sciences. He received a Bachelors in Pharmacy and Master of Pharmacy in Pharmaceutical Chemistry from Birla Institute of Technology and Science, Pilani, India. His current research is focused on discovering novel small molecule kinase inhibitors for anticancer therapy using synthetic medicinal chemistry and employing computational methods for lead optimization and conformational study of protein kinases.
Francesca Carlomagno is Professor of General Pathology at the University of Naples Federico II, Dept. Molecular Medicine and Medical Biotechnology. She is also associated with the Institute of Experimental Endocrinology and Oncology of the National Research Council (IEOS, CNR). She received her MD degree and Ph.D. from the University of Naples Federico II. She was a visiting fellow at NCI, NIH, and completed her postdoctoral training at Cambridge University and Federico II University. Her research is on the molecular biology of cancer with a specific interest in antineoplastic targeted therapy using small molecule kinase inhibitors. More recently in her lab, new projects related to alteration of DNA replication and iron metabolism are being conducted with the aim to identify new pathways relevant for cancer.
Massimo Santoro is Professor of General Pathology at the University of Naples Federico II, Dept. of Molecular Medicine and Medical Biotechnology. He graduated in Medicine and Surgery at the University of Naples Federico II and then pursued a Ph.D. degree in Molecular Pathology. He has been a fellow at NCI, NIH. His main interest has been molecular genetics of thyroid cancer. He participated in the identification of RET gene fusions in papillary thyroid cancer and activating point mutations in medullary thyroid cancer.
Neil Q. McDonald is currently a Senior Group Leader at the Francis Crick Institute. He is also Professor in Structural Biology at Birkbeck College, London. He received his Ph.D. degree from the University of London and did his postdoctoral training at Columbia University, NY. He previously worked at Cancer Research UK Laboratories in London where he focused on the structural biology of oncology targets. His current research interests are neurotrophic factor receptors and their downstream signaling pathways, in particular Rho-GTPase activated protein kinases.
Fengping Lv received his Ph.D. in Organic Chemistry from East China Normal University under the supervision of Professor Wenhao Hu. He completed his postdoctoral trainings at University of Arkansas for Medical Sciences in the field of drug discovery research of kinase inhibitors for cancer. He is currently a faulty member of Shaoxing University, Yuanpei College, and focused on translational targeted cancer drug development.
Naresh Gunaganti received his Ph.D. in Organic and Medicinal Chemistry from CSIR-Central Drug Research Institute, Jawaharlal Nehru University, New Delhi, India. Later, he joined the University of Arizona as a postdoctoral research associate under Dr. Hong-yu Li. He is currently working as a postdoctoral research associate at University of Arkansas for Medical Sciences in the area of kinase drug discovery. He is interested in developing irreversible inhibitors for various kinases using synthetic and medicinal chemistry tools.
Brendan Frett is an Assistant Professor of Pharmaceutical Sciences in the College of Pharmacy at the University of Arkansas for Medical Sciences. He received his Ph.D. degree from the University of Arizona, where he codiscovered a clinical candidate in IND studies. He has successfully transferred academic-based discoveries to pharmaceutical companies for clinical development. He is interested in pursuing translational research projects, where research completed in his laboratory can directly help patients.
Hong-yu Li is a Professor of Medicinal Chemistry at the University of Arkansas for Medical Sciences (UAMS). He is also an Arkansas Research Alliance (ARA) Scholar, the Helen Adams & ARA endowed chair in drug discovery, and codirector for the Therapeutics Science Program, Winthrop P Rockefeller Cancer Institute. He received his Ph.D. degree from the University of Tokyo and did postdoctoral training at Columbia University and Harvard University. He previously worked at Eli Lilly and the University of Arizona where he focused on oncology drug discovery. His current research interests are in chemical biology and drug discovery, especially for oncology related targets and phenotypes. In his lab at UAMS, a robust oncology pipeline is under development exploiting single agent polypharmacology and synergistic medicinal chemistry approaches.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Carter AR; Chen C; Schwartz PM; Segal RA Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J. Neurosci 2002, 22, 1316–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lai KO; Wong AS; Cheung MC; Xu P; Liang Z; Lok KC; Xie H; Palko ME; Yung WH; Tessarollo L; Cheung ZH; Ip NY TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat. Neurosci 2012, 15, 1506–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ren K; Dubner R Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol. Neurobiol 2007, 35, 224–235. [DOI] [PubMed] [Google Scholar]
- (4).Schuman EM Neurotrophin regulation of synaptic transmission. Curr. Opin. Neurobiol 1999, 9, 105–109. [DOI] [PubMed] [Google Scholar]
- (5).Kang H; Jia LZ; Suh KY; Tang L; Schuman EM Determinants of BDNF-induced hippocampal synaptic plasticity: role of the Trk B receptor and the kinetics of neurotrophin delivery. Learn. Mem 1996, 3, 188–196. [DOI] [PubMed] [Google Scholar]
- (6).McAllister AK; Katz LC; Lo DC Neurotrophins and synaptic plasticity. Annu. Rev. Neurosci 1999, 22, 295–318. [DOI] [PubMed] [Google Scholar]
- (7).Schinder AF; Poo M The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000, 23, 639–645. [DOI] [PubMed] [Google Scholar]
- (8).Huang EJ; Reichardt LF Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem 2003, 72, 609–642. [DOI] [PubMed] [Google Scholar]
- (9).Bartkowska K; Paquin A; Gauthier AS; Kaplan DR; Miller FD Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 2007, 134, 4369–4380. [DOI] [PubMed] [Google Scholar]
- (10).Islam O; Loo TX; Heese K Brain-derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK-B receptor, MAP kinase, AKT, and STAT-3 signaling pathways. Curr. Neurovascular Res 2009, 6, 42–53. [DOI] [PubMed] [Google Scholar]
- (11).Meakin SO; MacDonald JI; Gryz EA; Kubu CJ; Verdi JM The signaling adapter FRS-2 competes with Shc for binding to the nerve growth factor receptor TrkA. A model for discriminating proliferation and differentiation. J. Biol. Chem 1999, 274, 9861–9870. [DOI] [PubMed] [Google Scholar]
- (12).Kumar S; Kahn MA; Dinh L; de Vellis J NT-3-mediated TrkC receptor activation promotes proliferation and cell survival of rodent progenitor oligodendrocyte cells in vitro and in vivo. J. Neurosci. Res 1998, 54, 754–765. [DOI] [PubMed] [Google Scholar]
- (13).Matsumoto K; Wada RK; Yamashiro JM; Kaplan DR; Thiele CJ Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 1995, 55, 1798–1806. [PubMed] [Google Scholar]
- (14).Zhang YZ; Moheban DB; Conway BR; Bhattacharyya A; Segal RA Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J. Neurosci 2000, 20, 5671–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Barnabe-Heider F; Miller FD Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J. Neurosci 2003, 23, 5149–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Eggert A; Ikegaki N; Liu X; Chou TT; Lee VM; Trojanowski JQ; Brodeur GM Molecular dissection of TrkA signal transduction pathways mediating differentiation in human neuroblastoma cells. Oncogene 2000, 19, 2043–2051. [DOI] [PubMed] [Google Scholar]
- (17).DiCicco-Bloom E; Friedman WJ; Black IB NT-3 stimulates sympathetic neuroblast proliferation by promoting precursor survival. Neuron 1993, 11, 1101–1011. [DOI] [PubMed] [Google Scholar]
- (18).Glass DJ; Nye SH; Hantzopoulos P; Macchi MJ; Squinto SP; Goldfarb M; Yancopoulos GD Trkl3 mediates BDNF/NT-3-dependent survival and proliferation in fibroblasts lacking the low affinity NGF receptor. Cell 1991, 66, 405–413. [DOI] [PubMed] [Google Scholar]
- (19).Cohen RI; Marmur R; Norton WT; Mehler MF; Kessler JA Nerve growth factor and neurotrophin-3 differentially regulate the proliferation and survival of developing rat brain oligodendrocytes. J. Neurosci 1996, 16, 6433–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ibanez CF; Ebendal T; Barbany G; Murray-Rust J; Blundell TL; Persson H Disruption of the low affinity receptor-binding site in NGF allows neuronal survival and differentiation by binding to the trk gene product. Cell 1992, 69, 329–341. [DOI] [PubMed] [Google Scholar]
- (21).Lavenius E; Gestblom C; Johansson I; Nanberg E; Pahlman S Transfection of Trk-a into human neuroblastoma-cells restores their ability to differentiate in response to nerve growth-factor. Cell Growth Differ. 1995, 6, 727–736. [PubMed] [Google Scholar]
- (22).Chao MV Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci 2003, 4, 299–309. [DOI] [PubMed] [Google Scholar]
- (23).Bapat AA; Hostetter G; Von Hoff DD; Han H Perineural invasion and associated pain in pancreatic cancer. Nat. Rev. Cancer 2011, 11, 695–707. [DOI] [PubMed] [Google Scholar]
- (24).Miknyoczki SJ; Wan W; Chang H; Dobrzanski P; Ruggeri BA; Dionne CA; Buchkovich K The neurotrophin-trk receptor axes are critical for the growth and progression of human prostatic carcinoma and pancreatic ductal adenocarcinoma xenografts in nude mice. Clin. Cancer Res 2002, 8, 1924–1931. [PubMed] [Google Scholar]
- (25).Desmet CJ; Peeper DS The neurotrophic receptor TrkB: a drug target in anti-cancer therapy? Cell. Mol. Life Sci 2006, 63, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rubin JB; Segal RA Growth, survival and migration: the Trk to cancer. Cancer Treat Res. 2004, 115, 1–18. [DOI] [PubMed] [Google Scholar]
- (27).Brodeur GM; Minturn JE; Ho R; Simpson AM; Iyer R; Varela CR; Light JE; Kolla V; Evans AE Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res 2009, 15, 3244–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang T; Yu D; Lamb ML Trk kinase inhibitors as new treatments for cancer and pain. Expert Opin. Ther. Pat 2009, 19, 305–319. [DOI] [PubMed] [Google Scholar]
- (29).Weeraratna AT; Dalrymple SL; Lamb JC; Denmeade SR; Miknyoczki S; Dionne CA; Isaacs JT Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin. Cancer Res 2001, 7, 2237–2245. [PubMed] [Google Scholar]
- (30).Weeraratna AT; Arnold JT; George DJ; DeMarzo A; Isaacs JT Rational basis for Trk inhibition therapy for prostate cancer. Prostate 2000, 45, 140–148. [DOI] [PubMed] [Google Scholar]
- (31).Thiele CJ; Li Z; McKee AE On Trk–the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res 2009, 15, 5962–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Melck D; De Petrocellis L; Orlando P; Bisogno T; Laezza C; Bifulco M; Di Marzo V Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–126. [DOI] [PubMed] [Google Scholar]
- (33).Nakagawara A Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [DOI] [PubMed] [Google Scholar]
- (34).Sakamoto Y; Kitajima Y; Edakuni G; Sasatomi E; Mori M; Kitahara K; Miyazaki K Expression of Trk tyrosine kinase receptor is a biologic marker for cell proliferation and perineural invasion of human pancreatic ductal adenocarcinoma. Oncol. Rep 2001, 8, 477–484. [DOI] [PubMed] [Google Scholar]
- (35).Schneider MB; Standop J; Ulrich A; Wittel U; Friess H; Andren-Sandberg A; Pour PM Expression of nerve growth factors in pancreatic neural tissue and pancreatic cancer. J. Histochem. Cytochem 2001, 49, 1205–1210. [DOI] [PubMed] [Google Scholar]
- (36).McGregor LM; McCune BK; Graff JR; McDowell PR; Romans KE; Yancopoulos GD; Ball DW; Baylin SB; Nelkin BD Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 4540–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sclabas GM; Fujioka S; Schmidt C; Li Z; Frederick WA; Yang W; Yokoi K; Evans DB; Abbruzzese JL; Hess KR; Zhang W; Fidler IJ; Chiao PJ Overexpression of tropomysin-related kinase B in metastatic human pancreatic cancer cells. Clin. Cancer Res 2005, 11, 440–449. [PubMed] [Google Scholar]
- (38).Sugimoto T; Kuroda H; Horii Y; Moritake H; Tanaka T; Hattori S Signal transduction pathways through TRK-A and TRK-B receptors in human neuroblastoma cells. Jpn. J. Cancer Res 2001, 92, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Borrello MG; Bongarzone I; Plerotti MA; Luksch R; Gasparini M; Collini P; Pllotti S; Rlzzetti MG; Mondellini P; De Bernardi B; Di Martino D; Garaventa A; Brisigotti M; Tonini GP Trk and ret proto-oncogene expression in human neuroblastoma specimens: High frequency oftrk expression in non-advanced stages. Int. J. Cancer 1993, 54, 540–545. [DOI] [PubMed] [Google Scholar]
- (40).Okada Y; Eibl G; Guha S; Duffy JP; Reber HA; Hines OJ Nerve growth factor stimulates MMP-2 expression and activity and increases invasion by human pancreatic cancer cells. Clin. Exp. Metastasis 2004, 21, 285–292. [DOI] [PubMed] [Google Scholar]
- (41).Sapio MR; Posca D; Raggioli A; Guerra A; Marotta V; Deandrea M; Motta M; Limone PP; Troncone G; Caleo A; Rossi G; Fenzi G; Vitale M Detection of RET/PTC, TRK and BRAF mutations in preoperative diagnosis of thyroid nodules with indeterminate cytological findings. Clin. Endocrinol. (Oxford, U. K.) 2007, 66, 678–683. [DOI] [PubMed] [Google Scholar]
- (42).Druker BJ; Tamura S; Buchdunger E; Ohno S; Segal GM; Fanning S; Zimmermann J; Lydon NB Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med 1996, 2, 561–566. [DOI] [PubMed] [Google Scholar]
- (43).Vaishnavi A; Le AT; Doebele RC TRKing down an old oncogene in a new era of targeted therapy. Cancer Discovery 2015, 5, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Amatu A; Sartore-Bianchi A; Siena S NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Stransky N; Cerami E; Schalm S; Kim JL; Lengauer C The landscape of kinase fusions in cancer. Nat. Commun 2014, 5, 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dela Cruz CS; Tanoue LT; Matthay RA Lung cancer: epidemiology, etiology, and prevention. Clin. Chest Med 2011, 32, 605–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Drilon A; Li G; Dogan S; Gounder M; Shen R; Arcila M; Wang L; Hyman DM; Hechtman J; Wei G; Cam NR; Christiansen J; Luo D; Maneval EC; Bauer T; Patel M; Liu SV; Ou SH; Farago A; Shaw A; Shoemaker RF; Lim J; Hornby Z; Multani P; Ladanyi M; Berger M; Katabi N; Ghossein R; Ho AL What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann. Oncol 2016, 27, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Khotskaya YB; Holla VR; Farago AF; Mills Shaw KRM; Meric-Bernstam F; Hong DS Targeting TRK family proteins in cancer. Pharmacol. Ther 2017, 173, 58–66. [DOI] [PubMed] [Google Scholar]
- (49).Vaishnavi A; Capelletti M; Le AT; Kako S; Butaney M; Ercan D; Mahale S; Davies KD; Aisner DL; Pilling AB; Berge EM; Kim J; Sasaki H; Park S; Kryukov G; Garraway LA; Hammerman PS; Haas J; Andrews SW; Lipson D; Stephens PJ; Miller VA; Varella-Garcia M; Janne PA; Doebele RC Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med 2013, 19, 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Martin-Zanca D; Hughes SH; Barbacid M A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986, 319, 743–748. [DOI] [PubMed] [Google Scholar]
- (51).Ardini E; Bosotti R; Borgia AL; De Ponti C; Somaschini A; Cammarota R; Amboldi N; Raddrizzani L; Milani A; Magnaghi P; Ballinari D; Casero D; Gasparri F; Banfi P; Avanzi N; Saccardo MB; Alzani R; Bandiera T; Felder E; Donati D; Pesenti E; Sartore-Bianchi A; Gambacorta M; Pierotti MA; Siena S; Veronese S; Galvani A; Isacchi A The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol. Oncol 2014, 8, 1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tacconelli A; Farina AR; Cappabianca L; Gulino A; Mackay AR Alternative TrkAIII splicing: a potential regulated tumor-promoting switch and therapeutic target in neuroblastoma. Future Oncol. 2005, 1, 689–698. [DOI] [PubMed] [Google Scholar]
- (53).Greco A; Miranda C; Pierotti MA Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol. Cell. Endocrinol 2010, 321, 44–49. [DOI] [PubMed] [Google Scholar]
- (54).Kralik JM; Kranewitter W; Boesmueller H; Marschon R; Tschurtschenthaler G; Rumpold H; Wiesinger K; Erdel M; Petzer AL; Webersinke G Characterization of a newly identified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn. Pathol 2011, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Frattini V; Trifonov V; Chan JM; Castano A; Lia M; Abate F; Keir ST; Ji AX; Zoppoli P; Niola F; Danussi C; Dolgalev I; Porrati P; Pellegatta S; Heguy A; Gupta G; Pisapia DJ; Canoll P; Bruce JN; McLendon RE; Yan H; Aldape K; Finocchiaro G; Mikkelsen T; Prive GG; Bigner DD; Lasorella A; Rabadan R; Iavarone A The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet 2013, 45, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ricarte-Filho JC; Li S; Garcia-Rendueles ME; Montero-Conde C; Voza F; Knauf JA; Heguy A; Viale A; Bogdanova T; Thomas GA; Mason CE; Fagin JA Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J. Clin. Invest 2013, 123, 4935–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Leeman-Neill RJ; Kelly LM; Liu P; Brenner AV; Little MP; Bogdanova TI; Evdokimova VN; Hatch M; Zurnadzy LY; Nikiforova MN; Yue NJ; Zhang M; Mabuchi K; Tronko MD; Nikiforov YE ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer 2014, 120, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Segal RA Selectivity in neurotrophin signaling: theme and variations. Annu. Rev. Neurosci 2003, 26, 299–330. [DOI] [PubMed] [Google Scholar]
- (59).Greco A; Mariani C; Miranda C; Lupas A; Pagliardini S; Pomati M; Pierotti MA The DNA rearrangement that generates the Trk-T3 oncogene involves a nnovel gene on chromosome-3 whose product has a potential coiled-coil domain. Mol. Cell. Biol 1995, 15, 6118–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Mitra G; Martin-Zanca D; Barbacid M Identification and biochemical characterization of p70TRK, product of the human TRK oncogene. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 6707–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Greco A; Fusetti L; Miranda C; Villa R; Zanotti S; Pagliardini S; Pierotti MA Role of the TFG N-terminus and coiled-coil domain in the transforming activity of the thyroid TRK-T3 oncogene. Oncogene 1998, 16, 809–816. [DOI] [PubMed] [Google Scholar]
- (62).Gysin S; Salt M; Young A; McCormick F Therapeutic strategies for targeting ras proteins. Genes Cancer 2011, 2, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Martin-Zanca D; Oskam R; Mitra G; Copeland T; Barbacid M Molecular and biochemical characterization of the human trk proto-oncogene. Mol. Cell. Biol 1989, 9, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Ultsch MH; Wiesmann C; Simmons LC; Henrich J; Yang M; Reilly D; Bass SH; de Vos AM Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J. Mol. Biol 1999, 290, 149–159. [DOI] [PubMed] [Google Scholar]
- (65).Wiesmann C; Ultsch MH; Bass SH; de Vos AM Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999, 401, 184–188. [DOI] [PubMed] [Google Scholar]
- (66).Schneider R; Schweiger M A novel modular mosaic of cell adhesion motifs in the extracellular domains of the neurogenic trk and trkB tyrosine kinase receptors. Oncogene 1991, 6, 1807–1811. [PubMed] [Google Scholar]
- (67).Geetha T; Rege SD; Mathews SE; Meakin SO; White MF; Babu JR Nerve growth factor receptor TrkA, a new receptor in insulin signaling pathway in PC12 cells. J. Biol. Chem 2013, 288, 23807–23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kaplan DR; Hempstead BL; Martin-Zanca D; Chao MV; Parada LF The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science 1991, 252, 554–558. [DOI] [PubMed] [Google Scholar]
- (69).Squinto SP; Stitt TN; Aldrich TH; Davis S; Blanco SM; RadzieJewski C; Glass DJ; Masiakowski P; Furth ME; Valenzuela DM; Distefano PS; Yancopoulos GD TrkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell 1991, 65, 885–893. [DOI] [PubMed] [Google Scholar]
- (70).Klein R; Jing SQ; Nanduri V; O’Rourke E; Barbacid M The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [DOI] [PubMed] [Google Scholar]
- (71).Lamballe F; Klein R; Barbacid M TrkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell 1991, 66, 967–979. [DOI] [PubMed] [Google Scholar]
- (72).Yuen EC; Mobley WC Early BDNF, NT-3, and NT-4 signaling events. Exp. Neurol 1999, 159, 297–308. [DOI] [PubMed] [Google Scholar]
- (73).Bertrand T; Kothe M; Liu J; Dupuy A; Rak A; Berne PF; Davis S; Gladysheva T; Valtre C; Crenne JY; Mathieu M The crystal structures of trkA and trkB suggest key regions for achieving selective inhibition. J. Mol. Biol 2012, 423, 439–453. [DOI] [PubMed] [Google Scholar]
- (74).Cherry M; Williams DH Recent kinase and kinase inhibitor X-ray structures: mechanisms of inhibition and selectivity insights. Curr. Med. Chem 2004, 11, 663–673. [DOI] [PubMed] [Google Scholar]
- (75).Jing SQ; Tapley P; Barbacid M Nerve growth-factor mediates signal transduction through Trk homodimer receptors. Neuron 1992, 9, 1067–1079. [DOI] [PubMed] [Google Scholar]
- (76).Cunningham ME; Stephens RM; Kaplan DR; Greene LA Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. J. Biol. Chem 1997, 272, 10957–10967. [DOI] [PubMed] [Google Scholar]
- (77).Cunningham ME; Greene LA A function-structure model for NGF-activated TRK. EMBO J. 1998, 17, 7282–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Middlemas DS; Meisenhelder J; Hunter T Identification of Trkb autophosphorylation sites and evidence that phospholipase C-gamma-1 is a substrate of the Trkb receptor. J. Biol. Chem 1994, 269, 5458–5466. [PubMed] [Google Scholar]
- (79).Loeb DM; Stephens RM; Copeland T; Kaplan DR; Greene LA A Trk nerve growth factor (NGF) receptor point mutation affecting interaction with phospholipase C-gamma 1 abolishes NGF-promoted peripherin induction but not neurite outgrowth. J. Biol. Chem 1994, 269, 8901–8910. [PubMed] [Google Scholar]
- (80).Obermeier A; Lammers R; Wiesmuller KH; Jung G; Schlessinger J; Ullrich A Identification of Trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J. Biol. Chem 1993, 268, 22963–22966. [PubMed] [Google Scholar]
- (81).Obermeier A; Bradshaw RA; Seedorf K; Choidas A; Schlessinger J; Ullrich A Neuronal differentiation signals are controlled by nerve growth factor receptor/Trk binding sites for SHC and PLC gamma. EMBO J. 1994, 13, 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Stephens RM; Loeb DM; Copeland TD; Pawson T; Greene LA; Kaplan DR Trk receptors use redundant signal transduction pathways involving SHC and PLC-γ1 to mediate NGF responses. Neuron 1994, 12, 691–705. [DOI] [PubMed] [Google Scholar]
- (83).Patapoutian A; Reichardt LF Trk receptors: mediators of neurotrophin action. Curr. Opin. Neurobiol 2001, 11, 272–280. [DOI] [PubMed] [Google Scholar]
- (84).Songyang Z; Margolis B; Chaudhuri M; Shoelson SE; Cantley LC The phosphotyrosine interaction domain of SHC recognizes tyrosine-phosphorylated NPXY motif. J. Biol. Chem 1995, 270, 14863–14866. [DOI] [PubMed] [Google Scholar]
- (85).Lowenstein EJ; Daly RJ; Batzer AG; Li W; Margolis B; Lammers R; Ullrich A; Skolnik EY; Bar-Sagi D; Schlessinger J The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [DOI] [PubMed] [Google Scholar]
- (86).Bonfini L; Karlovich CA; Dasgupta C; Banerjee U The Son of sevenless gene product: a putative activator of Ras. Science 1992, 255, 603–606. [DOI] [PubMed] [Google Scholar]
- (87).Nishida E; Gotoh Y The map kinase sascade is essential for diverse signal transduction pathways. Trends Biochem. Sci 1993, 18, 128–131. [DOI] [PubMed] [Google Scholar]
- (88).Atwal JK; Massie B; Miller FD; Kaplan DR The TrkB-Shc site signals neuronal survival and local axon growth via MEK and PI3-kinase. Neuron 2000, 27, 265–277. [DOI] [PubMed] [Google Scholar]
- (89).Fishell G; Blazeski R; Godement P; Rivas R; Wang LC; Mason CA Optical microscopy. 3. Tracking fluorescently labeled neurons in developing brain. FASEB J. 1995, 9, 324–334. [DOI] [PubMed] [Google Scholar]
- (90).Datta K; Bellacosa A; Chan TO; Tsichlis PN Akt is a direct target of the phosphatidylinositol 3-kinase - Activation by growth factors, v-src and v-Ha-ras, in Sf9 and mammalian cells. J. Biol. Chem 1996, 271, 30835–30839. [DOI] [PubMed] [Google Scholar]
- (91).Hemmings BA Update: Signal transduction - PH domains - A universal membrane adapter. Science 1997, 275, 1899–1899. [DOI] [PubMed] [Google Scholar]
- (92).Alessi DR; James SR; Downes CP; Holmes AB; Gaffney PRJ; Reese CB; Cohen P Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol 1997, 7, 261–269. [DOI] [PubMed] [Google Scholar]
- (93).Andjelkovic M; Alessi DR; Meier R; Fernandez A; Lamb NJ; Frech M; Cron P; Cohen P; Lucocq JM; Hemmings BA Role of translocation in the activation and function of protein kinase B. J. Biol. Chem 1997, 272, 31515–31524. [DOI] [PubMed] [Google Scholar]
- (94).Burgering BM; Coffer PJ Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [DOI] [PubMed] [Google Scholar]
- (95).Dudek H; Datta SR; Franke TF; Birnbaum MJ; Yao R; Cooper GM; Segal RA; Kaplan DR; Greenberg ME Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [DOI] [PubMed] [Google Scholar]
- (96).Crowder RJ; Freeman RS Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci 1998, 18, 2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Yao R; Cooper GM Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [DOI] [PubMed] [Google Scholar]
- (98).Zirrgiebel U; Ohga Y; Carter B; Berninger B; Inagaki N; Thoenen H; Lindholm D Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: involvement of protein kinase C in neuronal survival. J. Neurochem 1995, 65, 2241–2250. [DOI] [PubMed] [Google Scholar]
- (99).Drilon A; Laetsch TW; Kummar S; DuBois SG; Lassen UN; Demetri GD; Nathenson M; Doebele RC; Farago AF; Pappo AS; Turpin B; Dowlati A; Brose MS; Mascarenhas L; Federman N; Berlin J; El-Deiry WS; Baik C; Deeken J; Boni V; Nagasubramanian R; Taylor M; Rudzinski ER; Meric-Bernstam F; Sohal DPS; Ma PC; Raez LE; Hechtman JF; Benayed R; Ladanyi M; Tuch BB; Ebata K; Cruickshank S; Ku NC; Cox MC; Hawkins DS; Hong DS; Hyman DM Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med 2018, 378, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Andre F Developing anticancer drugs in orphan molecular entities - a paradigm under construction. N. Engl. J. Med 2018, 378, 763–765. [DOI] [PubMed] [Google Scholar]
- (101).Harada T; Yatabe Y; Takeshita M; Koga T; Yano T; Wang Y; Giaccone G Role and relevance of TrkB mutations and expression in non-small cell lung cancer. Clin. Cancer Res 2011, 17, 2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Farago AF; Le LP; Zheng Z; Muzikansky A; Drilon A; Patel M; Bauer TM; Liu SV; Ou SH; Jackman D; Costa DB; Multani PS; Li GG; Hornby Z; Chow-Maneval E; Luo D; Lim JE; Iafrate AJ; Shaw AT Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J. Thorac. Oncol 2015, 10, 1670–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Coulier F; Kumar R; Ernst M; Klein R; Martin-Zanca D; Barbacid M Human trk oncogenes activated by point mutation, in-frame deletion, and duplication of the tyrosine kinase domain. Mol. Cell. Biol 1990, 10, 4202–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Arevalo JC; Conde B; Hempstead BI; Chao MV; Martin-Zanca D; Perez P A novel mutation within the extracellular domain of TrkA causes constitutive receptor activation. Oncogene 2001, 20, 1229–1234. [DOI] [PubMed] [Google Scholar]
- (105).Tomasson MH; Xiang Z; Walgren R; Zhao Y; Kasai Y; Miner T; Ries RE; Lubman O; Fremont DH; McLellan MD; Payton JE; Westervelt P; DiPersio JF; Link DC; Walter MJ; Graubert TA; Watson M; Baty J; Heath S; Shannon WD; Nagarajan R; Bloomfield CD; Mardis ER; Wilson RK; Ley TJ Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Tacconelli A; Farina AR; Cappabianca L; Desantis G; Tessitore A; Vetuschi A; Sferra R; Rucci N; Argenti B; Screpanti I; Gulino A; Mackay AR TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004, 6, 347–360. [DOI] [PubMed] [Google Scholar]
- (107).Reuther GW; Lambert QT; Caligiuri MA; Der CJ Identification and characterization of an activating TrkA deletion mutation in acute myeloid leukemia. Mol. Cell. Biol 2000, 20, 8655–8666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Kim SH; Tokarski JS; Leavitt KJ; Fink BE; Salvati ME; Moquin R; Obermeier MT; Trainor GL; Vite GG; Stadnick LK; Lippy JS; You D; Lorenzi MV; Chen P Identification of 2-amino-5-(thioaryl)thiazoles as inhibitors of nerve growth factor receptor TrkA. Bioorg. Med. Chem. Lett 2008, 18, 634–639. [DOI] [PubMed] [Google Scholar]
- (109).Gupta A; Dixon E Epidemiology and risk factors: intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr 2017, 6, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Selikoff IJ Epidemiology of gastrointestinal cancer. Environ. Health Perspect 1974, 9, 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Kim J; Lee Y; Cho HJ; Lee YE; An J; Cho GH; Ko YH; Joo KM; Nam DH NTRK1 fusion in glioblastoma multiforme. PLoS One 2014, 9, e91940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Sharma C An analysis of trends of incidence and cytohistological correlation of papillary carcinoma of the thyroid gland with evaluation of discordant cases. J. Cytol 2016, 33, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Rubin BP; Chen C-J; Morgan TW; Xiao S; Grier HE; Kozakewich HP; Perez-Atayde AR; Fletcher JA Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion. Am. J. Pathol 1998, 153, 1451–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Chien LN; Gittleman H; Ostrom QT; Hung KS; Sloan AE; Hsieh YC; Kruchko C; Rogers LR; Wang YF; Chiou HY; Barnholtz-Sloan JS Comparative brain and central nervous system tumor incidence and survival between the United States and Taiwan based on population-based registry. Front Public Health 2016, 4, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Wu G; Diaz AK; Paugh BS; Rankin SL; Ju B; Li Y; Zhu X; Qu C; Chen X; Zhang J; Easton J; Edmonson M; Ma X; Lu C; Nagahawatte P; Hedlund E; Rusch M; Pounds S; Lin T; Onar-Thomas A; Huether R; Kriwacki R; Parker M; Gupta P; Becksfort J; Wei L; Mulder HL; Boggs K; Vadodaria B; Yergeau D; Russell JC; Ochoa K; Fulton RS; Fulton LL; Jones C; Boop FA; Broniscer A; Wetmore C; Gajjar A; Ding L; Mardis ER; Wilson RK; Taylor MR; Downing JR; Ellison DW; Zhang J; Baker SJ The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet 2014, 46, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Mourad M; Jetmore T; Jategaonkar AA; Moubayed S; Moshier E; Urken ML Epidemiological trends of head and neck cancer in the United States: a SEER population study. J. Oral Maxillofac. Surg 2017, 75, 2562–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Pao W; Girard N New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [DOI] [PubMed] [Google Scholar]
- (118).Damjanov I; Skenderi F; Vranic S Mammary analogue secretory carcinoma (MASC) of the salivary gland: a new tumor entity. Bosnian J. Basic Med. Sci 2015, 16, 237–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Eguchi M; Eguchi-Ishimae M Absence of t(12;15) associated ETV6-NTRK3 fusion transcripts in pediatric acute leukemias. Med. Pediatr. Oncol 2001, 37, 417. [DOI] [PubMed] [Google Scholar]
- (120).Roberts KG; Gu Z; Payne-Turner D; McCastlain K; Harvey RC; Chen IM; Pei D; Iacobucci I; Valentine M; Pounds SB; Shi L; Li Y; Zhang J; Cheng C; Rambaldi A; Tosi M; Spinelli O; Radich JP; Minden MD; Rowe JM; Luger S; Litzow MR; Tallman MS; Wiernik PH; Bhatia R; Aldoss I; Kohlschmidt J; Mrozek K; Marcucci G; Bloomfield CD; Stock W; Kornblau S; Kantarjian HM; Konopleva M; Paietta E; Willman CL; Mullighan CG High frequency and poor outcome of philadelphia chromosome-like acute lymphoblastic leukemia in adults. J. Clin. Oncol 2017, 35, 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Fancelli D; Moll J; Varasi M; Bravo R; Artico R; Berta D; Bindi S; Cameron A; Candiani I; Cappella P; Carpinelli P; Croci W; Forte B; Giorgini ML; Klapwijk J; Marsiglio A; Pesenti E; Rocchetti M; Roletto F; Severino D; Soncini C; Storici P; Tonani R; Zugnoni P; Vianello P 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazoles: identification of a potent Aurora kinase inhibitor with a favorable antitumor kinase inhibition profile. J. Med. Chem 2006, 49, 7247–7251. [DOI] [PubMed] [Google Scholar]
- (122).Wiesner T; He J; Yelensky R; Esteve-Puig R; Botton T; Yeh I; Lipson D; Otto G; Brennan K; Murali R; Garrido M; Miller VA; Ross JS; Berger MF; Sparatta A; Palmedo G; Cerroni L; Busam KJ; Kutzner H; Cronin MT; Stephens PJ; Bastian BC Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun 2014, 5, 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Turbeville S; Francis KM; Behm I; Chiu GR; Sanchez H; Morrison B; Rowe JM Prevalence and incidence of acute myeloid leukemia may be higher than currently accepted estimates among the ≥ 65 year-old population in the United States. Blood 2014, 124, 958. [Google Scholar]
- (124).Knezevich S; McFadden D; Tao W; Lim J; Sorensen P A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet 1998, 18, 184–187. [DOI] [PubMed] [Google Scholar]
- (125).Lee SG; Jung SP; Lee HY; Kim S; Kim HY; Kim I; Bae JW Secretory breast carcinoma: A report of three cases and a review of the literature. Oncol. Lett 2014, 8, 683–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Russell JP; Powell DJ; Cunnane M; Greco A; Portella G; Santoro M; Fusco A; Rothstein JL The TRK-T1 fusion protein induces neoplastic transformation of thyroid epithelium. Oncogene 2000, 19, 5729–5735. [DOI] [PubMed] [Google Scholar]
- (127).Ranzi V; Meakin SO; Miranda C; Mondellini P; Pierotti MA; Greco A The signaling adapters fibroblast growth factor receptor substrate 2 and 3 are activated by the thyroid TRK oncoproteins. Endocrinology 2003, 144, 922–928. [DOI] [PubMed] [Google Scholar]
- (128).Miranda C; Greco A; Miele C; Pierotti MA; Van Obberghen E IRS-1 and IRS-2 are recruited by TrkA receptor and oncogenic TRK-T1. J. Cell. Physiol 2001, 186, 35–46. [DOI] [PubMed] [Google Scholar]
- (129).Roccato E; Miranda C; Ranzi V; Gishizki M; Pierotti MA; Greco A Biological activity of the thyroid TRK-T3 oncogene requires signalling through Shc. Br. J. Cancer 2002, 87, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Morrison KB; Tognon CE; Garnett MJ; Deal C; Sorensen PH ETV6-NTRK3 transformation requires insulin-like growth factor 1 receptor signaling and is associated with constitutive IRS-1 tyrosine phosphorylation. Oncogene 2002, 21, 5684–5695. [DOI] [PubMed] [Google Scholar]
- (131).Tognon C; Garnett M; Kenward E; Kay R; Morrison K; Sorensen PHB The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res 2001, 61, 8909–8916. [PubMed] [Google Scholar]
- (132).Edel MJ; Shvarts A; Medema JP; Bernards R An in vivo functional genetic screen reveals a role for the TRK-T3 oncogene in tumor progression. Oncogene 2004, 23, 4959–4965. [DOI] [PubMed] [Google Scholar]
- (133).Bailey JJ; Schirrmacher R; Farrell K; Bernard-Gauthier V Tropomyosin receptor kinase inhibitors: an updated patent review for 2010–2016-Part I. Expert Opin. Ther. Pat 2017, 27, 733–751. [DOI] [PubMed] [Google Scholar]
- (134).Bailey JJ; Schirrmacher R; Farrell K; Bernard-Gauthier V Tropomyosin receptor kinase inhibitors: an updated patent review for 2010–2016-Part II. Expert Opin. Ther. Pat 2017, 27, 831–849. [DOI] [PubMed] [Google Scholar]
- (135).McCarthy C; Walker E Tropomyosin receptor kinase inhibitors: a patent update 2009–2013. Expert Opin. Ther. Pat 2014, 24, 731–744. [DOI] [PubMed] [Google Scholar]
- (136).Cowan-Jacob SW; Jahnke W; Knapp S Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med. Chem 2014, 6, 541–561. [DOI] [PubMed] [Google Scholar]
- (137).Wu P; Nielsen TE; Clausen MH FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci 2015, 36, 422–439. [DOI] [PubMed] [Google Scholar]
- (138).Su HP; Rickert K; Burlein C; Narayan K; Bukhtiyarova M; Hurzy DM; Stump CA; Zhang X; Reid J; Krasowska-Zoladek A; Tummala S; Shipman JM; Kornienko M; Lemaire PA; Krosky D; Heller A; Achab A; Chamberlin C; Saradjian P; Sauvagnat B; Yang X; Ziebell MR; Nickbarg E; Sanders JM; Bilodeau MT; Carroll SS; Lumb KJ; Soisson SM; Henze DA; Cooke AJ Structural characterization of nonactive site, TrkA-selective kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E297–E306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Wang T; Lamb ML; Scott DA; Wang H; Block MH; Lyne PD; Lee JW; Davies AM; Zhang HJ; Zhu Y; Gu F; Han Y; Wang B; Mohr PJ; Kaus RJ; Josey JA; Hoffmann E; Thress K; Macintyre T; Wang H; Omer CA; Yu D Identification of 4-aminopyrazolylpyrimidines as potent inhibitors of Trk kinases. J. Med. Chem 2008, 51, 4672–4684. [DOI] [PubMed] [Google Scholar]
- (140).Thress K; Macintyre T; Wang H; Whitston D; Liu ZY; Hoffmann E; Wang T; Brown JL; Webster K; Omer C; Zage PE; Zeng L; Zweidler-McKay PA Identification and preclinical characterization of AZ-23, a novel, selective, and orally bioavailable inhibitor of the Trk kinase pathway. Mol. Cancer Ther 2009, 8, 1818–1827. [DOI] [PubMed] [Google Scholar]
- (141).Wang T; Lamb ML; Block MH; Davies AM; Han YX; Hoffmann E; Ioannidis S; Josey JA; Liu ZY; Lyne PD; MacIntyre T; Mohr PJ; Omer CA; Sjogren T; Thress K; Wang B; Wang HY; Yu DW; Zhang HJ Discovery of disubstituted imidazo 4,5-b pyridines and purines as potent TrkA inhibitors. ACS Med. Chem. Lett 2012, 3, 705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Carrera AC; Alexandrov K; Roberts TM The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Brasca MG; Amboldi N; Ballinari D; Cameron A; Casale E; Cervi G; Colombo M; Colotta F; Croci V; D’Alessio R; Fiorentini F; Isacchi A; Mercurio C; Moretti W; Panzeri A; Pastori W; Pevarello P; Quartieri F; Roletto F; Traquandi G; Vianello P; Vulpetti A; Ciomei M Identification of N,1,4,4-tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}−4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline-3-carboxamide (PHA-848125), a potent, orally available cyclin dependent kinase inhibitor. J. Med. Chem 2009, 52, 5152–5163. [DOI] [PubMed] [Google Scholar]
- (144).Albanese C; Alzani R; Amboldi N; Degrassi A; Festuccia C; Fiorentini F; Gravina G; Mercurio C; Pastori W; Brasca M; Pesenti E; Galvani A; Ciomei M Anti-tumour efficacy on glioma models of PHA-848125, a multi-kinase inhibitor able to cross the blood-brain barrier. Br. J. Pharmacol 2013, 169, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Albanese C; Alzani R; Amboldi N; Avanzi N; Ballinari D; Brasca MG; Festuccia C; Fiorentini F; Locatelli G; Pastori W; Patton V; Roletto F; Colotta F; Galvani A; Isacchi A; Moll J; Pesenti E; Mercurio C; Ciomei M Dual targeting of CDK and tropomyosin receptor kinase families by the oral inhibitor PHA-848125, an agent with broad-spectrum antitumor efficacy. Mol. Cancer Ther 2010, 9, 2243–2254. [DOI] [PubMed] [Google Scholar]
- (146).Gillies RJ; Verduzco D; Gatenby RA Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Bossi RT; Saccardo MB; Ardini E; Menichincheri M; Rusconi L; Magnaghi P; Orsini P; Avanzi N; Borgia AL; Nesi M; Bandiera T; Fogliatto G; Bertrand JA Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010, 49, 6813–6825. [DOI] [PubMed] [Google Scholar]
- (148).Menichincheri M; Ardini E; Magnaghi P; Avanzi N; Banfi P; Bossi R; Buffa L; Canevari G; Ceriani L; Colombo M; Corti L; Donati D; Fasolini M; Felder E; Fiorelli C; Fiorentini F; Galvani A; Isacchi A; Borgia AL; Marchionni C; Nesi M; Orrenius C; Panzeri A; Pesenti E; Rusconi L; Saccardo MB; Vanotti E; Perrone E; Orsini P Discovery of entrectinib: a new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros oncogene 1 kinase (ROS1), and pan-tropomyosine receptor kinases (Pan-TRKs) inhibitor. J. Med. Chem 2016, 59, 3392–3408. [DOI] [PubMed] [Google Scholar]
- (149).Ardini E; Menichincheri M; Banfi P; Bosotti R; De Ponti C; Pulci R; Ballinari D; Ciomei M; Texido G; Degrassi A; Avanzi N; Amboldi N; Saccardo MB; Casero D; Orsini P; Bandiera T; Mologni L; Anderson D; Wei G; Harris J; Vernier JM; Li G; Felder E; Donati D; Isacchi A; Pesenti E; Magnaghi P; Galvani A Entrectinib, a pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther 2016, 15, 628–639. [DOI] [PubMed] [Google Scholar]
- (150).Ardini E; Borgia AL; De Ponti C; Amboldi N; Ballinari D; Saccardo MB; Magnaghi P; Pesenti E; Isacchi A; Galvani A Identification and preclinical characterization of NMS-P626, a potent, selective and orally bioavailable TrkA inhibitor with anti-tumor activity in a TrkA-dependent colorectal cancer. Ejc Supplements 2010, 8, 39–40. [Google Scholar]
- (151).Jin L; Yan S; Wei G; Li G; Harris J; Vernier J-M The structure of TRKA kinase domain bound to the inhibitor Entrectinib. DOI: 10.2210/pdb5KVT/pdb , 10.2210/pdb5KVT/pdbhttps://www.rcsb.org/structure/5KVT, https://www.rcsb.org/structure/5KVT .
- (152).Lewis RT; Bode CM; Choquette DM; Potashman M; Romero K; Stellwagen JC; Teffera Y; Moore E; Whittington DA; Chen H; Epstein LF; Emkey R; Andrews PS; Yu VL; Saffran DC; Xu M; Drew A; Merkel P; Szilvassy S; Brake RL The discovery and optimization of a novel class of potent, selective, and orally bioavailable anaplastic lymphoma kinase (ALK) inhibitors with potential utility for the treatment of cancer. J. Med. Chem 2012, 55, 6523–6540. [DOI] [PubMed] [Google Scholar]
- (153).Sachdev J; Arkenau HT; Infante JR; Mita MM; Anthony SP; Natale RB; Ejadi S; Wilcoxen K; Kansra V; Laken H; Hughes L; Martell R; Weiss GJ 506 Phase (Ph) 1/2a study of TSR-011, a potent inhibitor of ALK and TRK, in advanced solid tumors including crizotinib-resistant ALK positive non-small cell lung cancer. Eur. J. Cancer 2014, 50, 165. [Google Scholar]
- (154).Lippa B; Morris J; Corbett M; Kwan TA; Noe MC; Snow SL; Gant TG; Mangiaracina M; Coffey HA; Foster B; Knauth EA; Wessel MD Discovery of novel isothiazole inhibitors of the TrkA kinase: Structure-activity relationship, computer modeling, optimization, and identification of highly potent antagonists. Bioorg. Med. Chem. Lett 2006, 16, 3444–3448. [DOI] [PubMed] [Google Scholar]
- (155).Choi HS; Rucker PV; Wang Z; Fan Y; Albaugh P; Chopiuk G; Gessier F; Sun F; Adrian F; Liu G; Hood T; Li N; Jia Y; Che J; McCormack S; Li A; Li J; Steffy A; Culazzo A; Tompkins C; Phung V; Kreusch A; Lu M; Hu B; Chaudhary A; Prashad M; Tuntland T; Liu B; Harris J; Seidel HM; Loren J; Molteni V (R)-2-Phenylpyrrolidine substituted imidazopyridazines: a new class of potent and selective pan-TRK inhibitors. ACS Med. Chem. Lett 2015, 6, 562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Sato Y; Onozaki Y; Sugimoto T; Kurihara H; Kamijo K; Kadowaki C; Tsujino T; Watanabe A; Otsuki S; Mitsuya M; Iida M; Haze K; Machida T; Nakatsuru Y; Komatani H; Kotani H; Iwasawa Y Imidazopyridine derivatives as potent and selective polo-like kinase (PLK) inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 4673–4678. [DOI] [PubMed] [Google Scholar]
- (157).Doebele RC; Davis LE; Vaishnavi A; Le AT; Estrada-Bernal A; Keysar S; Jimeno A; Varella-Garcia M; Aisner DL; Li Y; Stephens PJ; Morosini D; Tuch BB; Fernandes M; Nanda N; Low JA An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discovery 2015, 5, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Parikh AR; Corcoran RB Fast-TRKing drug development for rare molecular targets. Cancer Discovery 2017, 7, 934–936. [DOI] [PubMed] [Google Scholar]
- (159).Drilon A; Nagasubramanian R; Blake JF; Ku N; Tuch BB; Ebata K; Smith S; Lauriault V; Kolakowski GR; Brandhuber BJ; Larsen PD; Bouhana KS; Winski SL; Hamor R; Wu WI; Parker A; Morales TH; Sullivan FX; DeWolf WE; Wollenberg LA; Gordon PR; Douglas-Lindsay DN; Scaltriti M; Benayed R; Raj S; Hanusch B; Schram AM; Jonsson P; Berger MF; Hechtman JF; Taylor BS; Andrews S; Rothenberg SM; Hyman DM A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discovery 2017, 7, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Russo M; Misale S; Wei G; Siravegna G; Crisafulli G; Lazzari L; Corti G; Rospo G; Novara L; Mussolin B; Bartolini A; Cam N; Patel R; Yan SQ; Shoemaker R; Wild R; Di Nicolantonio F; Bianchi AS; Li G; Siena S; Bardelli A Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discovery 2016, 6, 36–44. [DOI] [PubMed] [Google Scholar]
- (161).Fuse MJ; Okada K; Oh-Hara T; Ogura H; Fujita N; Katayama R Mechanisms of resistance to NTRK inhibitors and therapeutic strategies in NTRK1-rearranged cancers. Mol. Cancer Ther 2017, 16, 2130–2143. [DOI] [PubMed] [Google Scholar]
- (162).Drilon A; De Braud FG; Siena S; Ou SI; Patel M; Ahn M; Lee J; Bauer TM; Farago AF; Liu SV; Reddinger N; Patel R; Luo D; Maneval EC; Multani PS; Doebele RC; Shaw AT Abstract CT007: Entrectinib, an oral pan-Trk, ROS1, and ALK inhibitor in TKI-naïve patients with advanced solid tumors harboring gene rearrangements: Updated phase I results. Cancer Res. 2016, 76, CT007. [Google Scholar]
- (163).Estrada-Bernal A; Le AT; Tuch B; Kutateladze T; Doebele RC Abstract C65: TRK kinase domain mutations that induce resistance to a pan-TRK inhibitor. Mol. Cancer Ther 2015, 14, C65. [Google Scholar]
- (164).The molecular docking was carried out using AutoDock4. The TRKA protein crystal structure was downloaded from the protein data bank (PDB ID 4AOJ). Amino acid mutations were carried out using Maestro 2018-2, and the figures were generated by Maestro 2018-2.
- (165).Cui JJ; Zhai D; Deng W; Rogers E; Huang Z; Whitten J; Li Y TPX-0005, a novel ALK/ROS1/TRK inhibitor, effectively inhibited a broad spectrum of mutations including solvent front ALK G1202R, ROS1 G2032R and TRKA G595R mutants. Eur. J. Cancer 2016, 69, S32–S32. [Google Scholar]
- (166).Cui JJ; Rogers E; Zhai DY; Deng W; Huang ZD; Whitten J; Li YS Ending the endless acquired tyrosine kinase resistance mutations - Design of TPX-0005, a multi-target ALK/ROS1/TRK inhibitor with broad spectrum activity against wild-type and mutants including ALK G1202R, ROS1 G2032R and TRKA G595R. Cancer Res. 2016, 76, 2133. [Google Scholar]
- (167).Cui JJ; Zhai D; Deng W; Rogers E; Huang Z; Whitten J; Li Y TPX-0005, a novel ALK/ROS1/TRK inhibitor, effectively inhibited a broad spectrum of mutations including solvent front ALK G1202R, ROS1 G2032R and TRKA G595R mutants. Eur. J. Cancer 2016, 69, S32. [Google Scholar]
- (168).Wood ER; Kuyper L; Petrov KG; Hunter RN; Harris PA; Lackey K Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and aza-oxindoles. Bioorg. Med. Chem. Lett 2004, 14, 953–957. [DOI] [PubMed] [Google Scholar]
- (169).Bernard-Gauthier V; Aliaga A; Aliaga A; Boudjemeline M; Hopewell R; Kostikov A; Rosa-Neto P; Thiel A; Schirrmacher R Syntheses and evaluation of carbon-11-and fluorine-18-radiolabeled pan-tropomyosin receptor kinase (Trk) inhibitors: exploration of the 4-aza-2-oxindole scaffold as Trk PET imaging agents. ACS Chem. Neurosci 2015, 6, 260–276. [DOI] [PubMed] [Google Scholar]
- (170).Choe H; Son YH; Byun BJ; Choi SU; Lee K Identification of pyrrole[3,4-c]pyrazoles as potent tropomyosin receptor kinase A (TrkA) inhibitors. Bull. Korean Chem. Soc 2016, 37, 1378–1380. [Google Scholar]
- (171).Carboni JM; Wittman M; Yang Z; Lee F; Greer A; Hurlburt W; Hillerman S; Cao C; Cantor GH; Dell-John J; Chen C; Discenza L; Menard K; Li A; Trainor G; Vyas D; Kramer R; Attar RM; Gottardis MM BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther 2009, 8, 3341–3349. [DOI] [PubMed] [Google Scholar]
- (172).Cho SY; Han SY; Ha JD; Ryu JW; Lee CO; Jung H; Kang NS; Kim HR; Koh JS; Lee J Discovery of aminopyridines substituted with benzoxazole as orally active c-Met kinase inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 4223–4227. [DOI] [PubMed] [Google Scholar]
- (173).Han SY; Lee CO; Ahn SH; Lee MO; Kang SY; Cha HJ; Cho SY; Ha JD; Ryu JW; Jung H; Kim HR; Koh JS; Lee J Evaluation of a multi-kinase inhibitor KRC-108 as an anti-tumor agent in vitro and in vivo. Invest. New Drugs 2012, 30, 518–523. [DOI] [PubMed] [Google Scholar]
- (174).Stachel SJ; Sanders JM; Henze DA; Rudd MT; Su HP; Li Y; Nanda KK; Egbertson MS; Manley PJ; Jones KL; Brnardic EJ; Green A; Grobler JA; Hanney B; Leitl M; Lai MT; Munshi V; Murphy D; Rickert K; Riley D; Krasowska-Zoladek A; Daley C; Zuck P; Kane SA; Bilodeau MT Maximizing diversity from a kinase screen: identification of novel and selective pan-Trk inhibitors for chronic pain. J. Med. Chem 2014, 57, 5800–5816. [DOI] [PubMed] [Google Scholar]
- (175).Rohren EM; Turkington TG; Coleman RE Clinical applications of PET in oncology. Radiology 2004, 231, 305–332. [DOI] [PubMed] [Google Scholar]
- (176).Rudin M; Weissleder R Molecular imaging in drug discovery and development. Nat. Rev. Drug Discovery 2003, 2, 123–131. [DOI] [PubMed] [Google Scholar]
- (177).Slobbe P; Poot AJ; Windhorst AD; van Dongen GA PET imaging with small-molecule tyrosine kinase inhibitors: TKI-PET. Drug Discovery Today 2012, 17, 1175–1187. [DOI] [PubMed] [Google Scholar]
- (178).Bernard-Gauthier V; Bailey JJ; Berke S; Schirrmacher R Recent advances in the development and application of radiolabeled kinase inhibitors for PET imaging. Molecules 2015, 20, 22000–22027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Bernard-Gauthier V; Bailey JJ; Aliaga A; Kostikov A; Rosa-Neto P; Wuest M; Brodeur GM; Bedell BJ; Wuest F; Schirrmacher R Development of subnanomolar radiofluorinated (2-pyrrolidin-1-yl)imidazo 1,2-b pyridazine pan-Trk inhibitors as candidate PET imaging probes. MedChemComm 2015, 6, 2184–2193. [Google Scholar]
- (180).Bernard-Gauthier V; Mahringer A; Vesnaver M; Fricker G; Schirrmacher R Design and synthesis of a fluorinated quinazoline-based type-II Trk inhibitor as a scaffold for PET radiotracer development. Bioorg. Med. Chem. Lett 2017, 27, 2771–2775. [DOI] [PubMed] [Google Scholar]
- (181).Bernard-Gauthier V; Schirrmacher R 5-(4-((4-[(18)F]-Fluorobenzyl)oxy)-3-methoxybenzyl)pyrimidine-2,4-diamine: a selective dual inhibitor for potential PET imaging of Trk/CSF-1R. Bioorg. Med. Chem. Lett 2014, 24, 4784–4790. [DOI] [PubMed] [Google Scholar]
- (182).Bernard-Gauthier V; Bailey JJ; Mossine AV; Lindner S; Vomacka L; Aliaga A; Shao X; Quesada CA; Sherman P; Mahringer A; Kostikov A; Grand’Maison M; Rosa-Neto P; Soucy JP; Thiel A; Kaplan DR; Fricker G; Wangler B; Bartenstein P; Schirrmacher R; Scott PJH A kinome-wide selective radiolabeled TrkB/C inhibitor for in vitro and in vivo neuroimaging: synthesis, preclinical evaluation, and first-in-human. J. Med. Chem 2017, 60, 6897–6910. [DOI] [PubMed] [Google Scholar]
- (183).Bernard-Gauthier V; Mossine AV; Mahringer A; Aliaga A; Bailey JJ; Shao X; Stauff J; Arteaga J; Sherman P; Grand’Maison M; Rochon PL; Wangler B; Wangler C; Bartenstein P; Kostikov A; Kaplan DR; Fricker G; Rosa-Neto P; Scott PJH; Schirrmacher R Identification of [(18)F]TRACK, a fluorine-18-labeled tropomyosin receptor kinase (Trk) inhibitor for PET imaging. J. Med. Chem 2018, 61, 1737–1743. [DOI] [PubMed] [Google Scholar]
- (184).Pargellis C; Tong L; Churchill L; Cirillo PF; Gilmore T; Graham AG; Grob PM; Hickey ER; Moss N; Pav S; Regan J Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol 2002, 9, 268–272. [DOI] [PubMed] [Google Scholar]
- (185).Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- (186).Albaugh P; Fan Y; Mi Y; Sun FX; Adrian F; Li NX; Jia Y; Sarkisova Y; Kreusch A; Hood T; Lu M; Liu GX; Huang SL; Liu ZS; Loren J; Tuntland T; Karanewsky DS; Seidel HM; Molteni V Discovery of GNF-5837, a selective TRK inhibitor with efficacy in rodent cancer tumor models. ACS Med. Chem. Lett 2012, 3, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Skerratt SE; Andrews M; Bagal SK; Bilsland J; Brown D; Bungay PJ; Cole S; Gibson KR; Jones R; Morao I; Nedderman A; Omoto K; Robinson C; Ryckmans T; Skinner K; Stupple P; Waldron G The discovery of a potent, selective, and peripherally restricted pan-Trk inhibitor (PF-06273340) for the treatment of pain. J. Med. Chem 2016, 59, 10084–10099. [DOI] [PubMed] [Google Scholar]
- (188).Pryde DC; Dalvie D; Hu Q; Jones P; Obach RS; Tran TD Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem 2010, 53, 8441–8460. [DOI] [PubMed] [Google Scholar]
- (189).Pryde DC; Tran TD; Jones P; Duckworth J; Howard M; Gardner I; Hyland R; Webster R; Wenham T; Bagal S; Omoto K; Schneider RP; Lin J Medicinal chemistry approaches to avoid aldehyde oxidase metabolism. Bioorg. Med. Chem. Lett 2012, 22, 2856–2860. [DOI] [PubMed] [Google Scholar]
- (190).Yakes FM; Chen J; Tan J; Yamaguchi K; Shi Y; Yu P; Qian F; Chu F; Bentzien F; Cancilla B; Orf J; You A; Laird AD; Engst S; Lee L; Lesch J; Chou YC; Joly AH Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther 2011, 10, 2298–2308. [DOI] [PubMed] [Google Scholar]
- (191).Patwardhan PP; Ivy KS; Musi E; de Stanchina E; Schwartz GK Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget 2016, 7, 4093–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Smith BD; Kaufman MD; Leary CB; Turner BA; Wise SC; Ahn YM; Booth RJ; Caldwell TM; Ensinger CL; Hood MM; Lu WP; Patt TW; Patt WC; Rutkoski TJ; Samarakoon T; Telikepalli H; Vogeti L; Vogeti S; Yates KM; Chun L; Stewart LJ; Clare M; Flynn DL Altiratinib inhibits tumor growth, invasion, angiogenesis, and microenvironment-mediated drug resistance via balanced inhibition of MET, TIE2, and VEGFR2. Mol. Cancer Ther 2015, 14, 2023–2034. [DOI] [PubMed] [Google Scholar]
- (193).El Zein N; D’Hondt S; Sariban E Crosstalks between the receptors tyrosine kinase EGFR and TrkA and the GPCR, FPR, in human monocytes are essential for receptors-mediated cell activation. Cell. Signalling 2010, 22, 1437–1447. [DOI] [PubMed] [Google Scholar]
- (194).Arcari JT; Beebe JS; Berliner MA; Bernardo V; Boehm M; Borzillo GV; Clark T; Cohen BD; Connell RD; Frost HN; Gordon DA; Hungerford WM; Kakar SM; Kanter A; Keene NF; Knauth EA; Lagreca SD; Lu Y; Martinez-Alsina L; Marx MA; Morris J; Patel NC; Savage D; Soderstrom CI; Thompson C; Tkalcevic G; Tom NJ; Vajdos FF; Valentine JJ; Vincent PW; Wessel MD; Chen JM Discovery and synthesis of novel 4-aminopyrrolopyrimidine Tie-2 kinase inhibitors for the treatment of solid tumors. Bioorg. Med. Chem. Lett 2013, 23, 3059–3063. [DOI] [PubMed] [Google Scholar]
- (195).Kufareva I; Abagyan R Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J. Med. Chem 2008, 51, 7921–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Frett B; McConnell N; Wang Y; Xu Z; Ambrose A; Li HY Identification of pyrazine-based TrkA inhibitors: design, synthesis, evaluation, and computational modeling studies. MedChemComm 2014, 5, 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Conway JG; McDonald B; Parham J; Keith B; Rusnak DW; Shaw E; Jansen M; Lin P; Payne A; Crosby RM; Johnson JH; Frick L; Lin MH; Depee S; Tadepalli S; Votta B; James I; Fuller K; Chambers TJ; Kull FC; Chamberlain SD; Hutchins JT Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 16078–16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Conway JG; Pink H; Bergquist ML; Han B; Depee S; Tadepalli S; Lin P; Crumrine RC; Binz J; Clark RL; Selph JL; Stimpson SA; Hutchins JT; Chamberlain SD; Brodie TA Effects of the cFMS kinase inhibitor 5-(3-methoxy-4-((4-methoxybenzyl)oxy)benzyl)pyrimidine-2,4-diamine (GW2580) in normal and arthritic rats. J. Pharmacol. Exp. Ther 2008, 326, 41–50. [DOI] [PubMed] [Google Scholar]
- (199).Hong S; Kim J; Seo JH; Jung KH; Hong SS; Hong S Design, synthesis, and evaluation of 3,5-disubstituted 7-azaindoles as Trk inhibitors with anticancer and antiangiogenic activities. J. Med. Chem 2012, 55, 5337–5349. [DOI] [PubMed] [Google Scholar]
- (200).Cee VJ; Albrecht BK; Geuns-Meyer S; Hughes P; Bellon S; Bready J; Caenepeel S; Chaffee SC; Coxon A; Emery M; Fretland J; Gallant P; Gu Y; Hodous BL; Hoffman D; Johnson RE; Kendall R; Kim JL; Long AM; McGowan D; Morrison M; Olivieri PR; Patel VF; Polverino A; Powers D; Rose P; Wang L; Zhao H Alkynylpyrimidine amide derivatives as potent, selective, and orally active inhibitors of Tie-2 kinase. J. Med. Chem 2007, 50, 627–640. [DOI] [PubMed] [Google Scholar]
- (201).Furuya N; Momose T; Katsuno K; Fushimi N; Muranaka H; Handa C; Ozawa T; Kinoshita T The juxtamembrane region of TrkA kinase is critical for inhibitor selectivity. Bioorg. Med. Chem. Lett 2017, 27, 1233–1236. [DOI] [PubMed] [Google Scholar]
- (202).Bagal SK; Omoto K; Blakemore DC; Bungay PJ; Bilsland JG; Clarke PJ; Corbett MS; Cronin CN; Cui JJ; Dias R; Flanagan NJ; Greasley SE; Grimley R; Johnson E; Fengas D; Kitching L; Kraus ML; McAlpine I; Nagata A; Waldron GJ; Warmus JS Discovery of allosteric, potent, subtype selective, and peripherally restricted TrkA kinase inhibitors. J. Med. Chem 2018, DOI: 10.1021/acs.jmedchem.8b00280. [DOI] [PubMed] [Google Scholar]
- (203).Lange AM; Lo HW Inhibiting TRK proteins in clinical cancer therapy. Cancers 2018, 10, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Burris HA; Shaw AT; Bauer TM; Farago AF; Doebele RC; Smith S; Nanda N; Cruickshank S; Low JA; Brose MS Abstract 4529: Pharmacokinetics (PK) of LOXO-101 during the first-in-human Phase I study in patients with advanced solid tumors: Interim update. Cancer Res. 2015, 75, 4529. [Google Scholar]
- (205).Hyman DM; Laetsch TW; Kummar S; DuBois SG; Farago AF; Pappo AS; Demetri GD; El-Deiry WS; Lassen UN; Dowlati A; Brose MS; Boni V; Turpin B; Nagasubramanian R; Cruickshank S; Cox MC; Ku NC; Hawkins DS; Hong DS; Drilon AE The efficacy of larotrectinib (LOXO-101), a selective tropomyosin receptor kinase (TRK) inhibitor, in adult and pediatric TRK fusion cancers. J. Clin. Oncol 2017, 35, LBA2501. [Google Scholar]
- (206).Laetsch TW; DuBois SG; Nagasubramanian R; Turpin B; Mascarenhas L; Federman N; Reynolds M; Smith S; Cruickshank S; Cox MC; Pappo AS; Hawkins DS A pediatric phase I study of larotrectinib, a highly selective inhibitor of the tropomyosin receptor kinase (TRK) family. J. Clin. Oncol 2017, 35, 10510. [Google Scholar]
- (207).Laetsch TW; DuBois SG; Mascarenhas L; Turpin B; Federman N; Albert CM; Nagasubramanian R; Davis JL; Rudzinski E; Feraco AM; Tuch BB; Ebata KT; Reynolds M; Smith S; Cruickshank S; Cox MC; Pappo AS; Hawkins DS Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Sidaway P Targeted therapy: Larotrectinib effective against TRK-fusion-positive cancers. Nat. Rev. Clin. Oncol 2018, 15, 264. [DOI] [PubMed] [Google Scholar]
- (209).Drilon A; Siena S; Ou SI; Patel M; Ahn MJ; Lee J; Bauer TM; Farago AF; Wheler JJ; Liu SV; Doebele R; Giannetta L; Cerea G; Marrapese G; Schirru M; Amatu A; Bencardino K; Palmeri L; Sartore-Bianchi A; Vanzulli A; Cresta S; Damian S; Duca M; Ardini E; Li G; Christiansen J; Kowalski K; Johnson AD; Patel R; Luo D; Chow-Maneval E; Hornby Z; Multani PS; Shaw AT; De Braud FG Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372–001 and STARTRK-1). Cancer Discovery 2017, 7, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Pishvaian MJ; Rolfo CD; Liu SV; Multani PS; Maneval EC; Garrido-Laguna I Clinical benefit of entrectinib for patients with metastatic pancreatic cancer who harbor NTRK and ROS1 fusions. J. Clin. Oncol 2018, 36, 521. [Google Scholar]
- (211).Werner T; Heist R; Carvajal R; Adkins D; Alva A; Goel S; Hong D; Bazhenova L; Saleh M; Siegel R; Kyriakopoulos C; Blakely C; Eaton K; Lauer R; Wang D; Schwartz G; Neuteboom S; Potvin D; Faltaos D; Chen I; Christensen J; Levisetti M; Chao R; Bauer T P2.06–001 A study of MGCD516, a receptor tyrosine kinase (RTK) inhibitor, in molecularly selected patients with NSCLC or other advanced solid tumors. J. Thorac. Oncol 2017, 12, S1068–S1069. [Google Scholar]
- (212).Takeda M; Seto T; Fujiwara Y; Yamamoto N; Nosaki K; Toyozawa R; Abe C; Shiga R; Nakamaru K; Nakagawa K 1362PSafety and clinical activity of DS-6051b, a ROS1/NTRK inhibitor, in Japanese patients with NSCLC harboring ROS1 fusion gene. Annals Oncol. 2017, 28, No. mdx380.063. [Google Scholar]
- (213).Papadopoulos KP; Borazanci E; Von Hoff D; Gandhi L; Patnaik A; Tachibana M; Zahir H; Gajee R; Goldberg T; Senaldi G; Ou S-H Abstract CT024: First-in-human phase 1 dose-escalation study of DS-6051b, an oral ROS1 and NTRK inhibitor, in subjects with advanced solid tumors. Cancer Res. 2016, 76, CT024. [Google Scholar]
- (214).You WK; Sennino B; Williamson CW; Falcon B; Hashizume H; Yao LC; Aftab DT; McDonald DM VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011, 71, 4758–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Pezet S; McMahon SB Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci 2006, 29, 507–538. [DOI] [PubMed] [Google Scholar]