

Abstract
ClpL is a member of the HSP100 family of the AAA+ chaperone that is widely present in Gram-positive but surprisingly absent in Gram-negative bacteria. ClpL is involved in various cellular processes including stress tolerance response, long-term survival, virulence, and antibiotic resistance. ClpL is poorly characterized and its molecular mechanisms of chaperone activity are largely unclear. Here, we biochemically characterized the ClpL protein from Streptococcus mutans, a dental pathogen, to understand its biological functions. ClpL harbors five domains: N-domain, two nucleotide binding domains (NBD-1 and NBD-2), M-domain, and C-domain. NBD-1 and NBD-2 contain distinct Walker A and B motifs for ATP binding and hydrolysis, respectively. We found that ClpL predominantly exists as a trimer in solution; however, upon ATP binding, it rapidly forms a hexameric structure. To study structure-function activity, we constructed several substitution and deletion mutants. We found that mutations in Walker A and B motifs interfered with the ATP hydrolysis and oligomerization. Similarly, deletions of N-, M-, and C-domains abolished both the ATPase activity and oligomerization. Since we previously found that ClpL acts as a chaperone, we analyzed the chaperone activity. Surprisingly, we found that the NBD-2 mutants did not display any chaperone activity indicating the essentiality of ATP binding and hydrolysis by NBD-2 for chaperone. However, NBD-1 mutants showed chaperone activities, but the activities were variable depending on the nature of the mutations. Our results indicate that unlike other HSP100 family chaperones, ClpL is a novel chaperone that does not require any additional secondary chaperones for its activity.
Keywords: Streptococcus mutans, chaperone, AAA+ ATpase, HSP100, ClpL, ClpB
INTRODUCTION
Molecular chaperones play a major role in various cellular functions and are essential for cell survival. Chaperone function is needed for the folding of newly synthesized proteins, refolding and reactivation of unfolded and misfolded proteins. Successful assembly and disassembly of macromolecular protein complexes also require chaperone function. Furthermore, chaperones are also required for targeting abnormal or inactive proteins for degradation1, 2. In bacteria, the two major chaperone systems are the GroEL and DnaK3–5. In addition, another chaperone called ClpB (caseinolytic peptidase B), which is homologous to yeast heat-shock protein 104 (HSP104), is also present in bacteria6. The GroEL chaperone system functions by providing a protected environment in which protein folding of individual protein molecules occurs. On the other hand, the DnaK chaperone system, which includes DnaK, DnaJ and GrpE, functions by binding and protecting exposed regions on unfolded or partially folded proteins7, 8. DnaK also interacts with trigger factor during protein translation and with ClpB in reactivating proteins which have become aggregated after heat shock8, 9. While chaperones are generally not required for survival under normal growth conditions, during heat or other environmental stresses, chaperones are necessary for survival.
Both ClpB and yeast HSP104 belong to the HSP100 family of AAA+ proteins6. The family is defined by the presence of a basic core of ~ 200–250 amino acids that comprises an α-helical domain and a Walker-type (AAA+) nucleotide-binding domain10–12. Structural studies have shown that ClpB forms a hexameric ring complex with each monomer having three main domains: N-terminal domain (NTD) for substrate binding and two tandem nucleotide-binding domains, NBD1 and NBD2, responsible for ATP-driven peptide translocation6, 13–15. ClpB also has a coiled-coil middle domain (MD) inserted in the NBDs that distinguishes it from other AAA+ ATPases. For disaggregation activity, ClpB requires synergistic interaction with DnaK that initially binds to the exposed region of aggregated proteins and then transfers the aggregated polypeptide to the ClpB complex. Using the energy derived from ATP hydrolysis, ClpB disentangles the aggregate by threading single polypeptide chains through the central pore of its hexameric ring6, 16, 17. Once released from the aggregate, the unfolded polypeptides either refold spontaneously or fold with the help of additional cellular chaperones6.
While GroEL, DnaK and ClpB are the major chaperone systems in Gram-negative bacteria, in many low GC Gram-positive bacteria, in addition to ClpB, a homolog of ClpB called ClpL is present as well. ClpL, which also belongs to the HSP100 family, shares all the major domains with the ClpB protein. At the physiological level, ClpL is involved in stress tolerance response such as heat and acid stresses18, 19. ClpL is also shown to repress adherence of pathogens to the host cell and to induce TNF-a secretion, which modulates the virulence outcome20. Furthermore, ClpL plays a role in resistance to various antibiotics that specifically target cell-wall biosynthesis21–23. The detailed molecular mechanisms by which ClpL is involved with the observed phenotypes remained to be explored.
Over 200 ClpL homologs have been identified in Gram positive bacteria. While most of these bacteria harbor a single chromosomally encoded clpL gene, some encode multiple homologs. For example, Lactobacillus rhamnosus, an oral organism, encodes two ClpL homologs and both genes are induced during heat stress and osmotic stress24. The second clpL gene, clpL2, appears to be part of a mobile genetic element that L. rhamnosus acquired recently24. Additional clpL genes are also often found on plasmids as well. In Lactococcus lactis, a second clpL gene, is located on a cryptic plasmid but its function was not evaluated25. Some clinical isolates of Listeria monocytogenes, an opportunistic pathogen, also contain a plasmid-borne clpL gene that is required for heat, osmotic and acid stress responses26, 27.
Streptococcus mutans is a low GC Gram positive oral bacterium associated with dental caries formation in humans. It also harbors a chromosomally encoded ClpL homolog that is induced during various stresses28. A S. mutans strain lacking a functional ClpL displays acid-shock response29 and increased sensitivity to some antibiotics28; however, its exact molecular function is currently unknown. S. mutans ClpL is highly homologous to its counterpart from Streptococcus pneumoniae and Streptococcus thermophilus30, 31. ClpL is required for virulence expression and antibiotic resistance in S. pneumoniae30, 32 and in acid tolerance in S. thermophilus31. Thus, it appears that the cellular function of ClpL is highly variable among streptococci. We recently uncovered that in S. mutans ClpL acts as a chaperone to properly fold CtsR, a stress response repressor, and prevents it from forming protein aggregates33. We showed that ClpL was able to successfully refold urea-denatured CtsR but not aggregated proteins and proposed that ClpL recognizes primarily soluble but denatured substrates and prevents formation of large protein aggregates28, 33. Recently, it was shown that ClpL of S. pneumoniae also functions as a chaperone and can refold heterologous proteins in vitro34. In this study, we wanted to further explore the biochemical functions of the S. mutans ClpL protein to gain insights into its molecular roles.
MATERIALS AND METHODS
Mutagenesis and protein purification:
The clpL gene of S. mutans was first cloned into pGEM-T Easy vector (Promega). Point mutations were generated using site-directed mutagenesis method with overlapping primers35. The deletion derivatives of ClpL were constructed using standard PCR technique with appropriate primers. All the mutations were confirmed by DNA sequencing. For purification of ClpL derivatives, the mutant clpL constructs were sub-cloned from the pGEM-T vector into an anhydrotetracycline inducible expression vector pASK43+ (IBA) for His-tagged protein purification. The proteins were purified using Ni-NTA affinity chromatography following manufacturer’s protocol (Novagen). After elution from Ni-NTA column, samples were dialyzed in buffer A (40 mM Tris pH 7.5, 10% Glycerol, 150 mM KCl) and stored at −20°C for future use.
Gel filtration analysis:
Purified wild type ClpL protein (~500 μg) was loaded onto Superdex S-200 10/300 GL (GE healthcare) column equilibrated in 40 mM Tris-Cl (pH7.5), 150 mM KCl, 10% Glycerol, 20 mM MgCl2. To determine the oligomerization status in the presence of ATP the column was equilibrated with the same buffer supplemented with 1 mM ATP. ClpL protein was incubated with 1 mM ATP for 20 min at room temperature to form higher oligomeric structures. The complexes were loaded onto ATP equilibrated Superdex S-200 column and run with a flow rate of 0.5 ml/min at 4°C. Elution volume was determined by measuring the protein amount in the different elution fractions by Bradford method. Molecular weight standards (Sigma) used for the determination of oligomerization are: Thyroglobulin (670 kD), Apoferritin (440 kD), β-Amylase (220 kD), and BSA (66 kD). Blue dextran (2000 kD) was used to determine the void volume.
Limited proteolytic digestion of ClpL:
Wild type or mutant ClpL proteins (1μM) were incubated with trypsin or chymotrypsin at 100:1 (w/w) ratio for desired time points at 25°C in buffer A supplemented with 20 mM MgCl2 in the presence or absence of 5 mM ATP or ADP. The reaction was terminated by the addition of SDS sample buffer followed by incubation at 100°C for 3 min. The samples were analyzed on a 10% Tris-Tricine SDS-PAGE.
Native gel electrophoresis of ClpL:
To identify the oligomeric status of ClpL and its mutant derivative, we performed native polyacrylamide gel electrophoresis as described previously36. We prepared a 10% polyacrylamide gel in 0.5 M Tris-Cl buffer (pH 8.8) and the electrophoresis was carried out using Tris-glycine running buffer at 100 V for 4h. MgCl2 was supplemented in the running buffer as well as in polyacrylamide gel at a final concentration of 20 mM. Similarly, to determine the oligomerization in the presence of ATP, 1mM ATP was added to running buffer and polyacrylamide gel, in addition to MgCl2.
ATP hydrolysis assay:
ATP hydrolysis was performed using standard protocol (REF36). Briefly 1μM ClpL proteins were incubated with desired concentrations of ATP for 10 min. The released phosphate was measured using Phosphate Colorimetric Assay kit (Sigma). The buffer contains 40 mM Tris, 50 mM KCl, 10% glycerol, and 20 mM MgCl2. Using standard phosphate solution, the released phosphate amount was measured. The ATPase activity rates of different ClpL variants were determined by measuring the amount of released phosphate, protein concentration and time of incubation with ATP. Hill coefficients were determined by fitting the equation below with the rates of ATP hydrolysis in different ATP concentrations using GraphPad software.
| (Equation 1) |
The parameters are: v is the velocity of ATP hydrolysis, Vmax is the maximal velocity, [S] represents the molar concentration of ATP, Km values are [S] where v is 50% of Vmax. And nH is the Hill coefficient.
MANT-ATP binding:
Wild type or mutant ClpL proteins (8μM) were titrated with MANT-ATP (0–16 μM) (SIGMA) in buffer A supplemented with 20 mM MgCl2. After addition of MANT-ATP, the mixture was incubated for 5 minutes at room temperature. The fluorescence value was recorded on a Synergy-H1 microtiter plate reader (BIOTEK). The excitation and emission wavelengths were set at 356 nm and 450 nm, respectively. Fluorescence value for MANT-ATP alone was subtracted from the corresponding MANT-ATP and ClpL protein mixture. The resulting fluorescence values were plotted with corresponding MANT-ATP concentrations. The curve was fitted with Hill equation to calculate Kd value using GraphPad software.
| (Equation 2) |
Where, y is fluorescence intensity represent MANT-ATP and protein complex, x is the MANT-ATP concentration, nH denotes Hill coefficient, Kd is dissociation constant. y = y0, when x = 0.
Chaperone activity:
The refolding activity of ClpL was measured using luciferase as a substrate as described previously34. Luciferase (4 μM) was denatured by incubating 6 M urea in 40 mM Tris-Cl (pH 7.5), 50 mM KCl, 1mM dithiothreitol (DTT), and 20 mM MgCl2 for 1h at 30°C. Reactivation (refolding) reactions were carried out in 50 nM in the presence of 40 mM Tris-Cl (pH 7.5), 100 mM KCl, 10% glycerol, 10mM creatine phosphate, 200 μg/ml creatine kinase, 0.015% BSA, 1 mM DTT, 4 mM ATP and 20 mM MgCl2 for 3h at 30°C. The final luciferase concentration in refolding reaction was 50 nM. Luciferase activity was measured using luciferin (Thermo Scientific) as a substrate by measuring the luminance using Synergy-H1. To determine aggregation prevention activity of ClpL and its mutant derivatives, luciferase protein (0.5 μM) was heated in 40 mM Tris-Cl (pH7.5), 100 mM KCl, 10% Glycerol, 8 mM ATP, and 20 mM MgCl2 with or without ClpL proteins (3 μM) at 48°C for 20 min and the luciferase activity was measured as described above.
RESULTS:
Primary sequence analysis suggests that ClpL shares high similarity with the E. coli ClpB protein and contains five domains: N-domain, nucleotide binding domains −1 (NBD-1) and −2 (NBD-2), M-domain, and C-domain (Fig 1A). However, we noticed that the size of the M domain of ClpL is relatively small as compared to ClpB (Fig 1A). Since the structural information for the ClpL protein is not available, we used ITASSER37 to build a homology model of ClpL (Fig 1B). Modelling indicates that in the monomer the NBD domains are aligned similarly to the ClpB monomer15. To gain insight into the functions of these two domains, we first determined the role of various divalent cations on ATP hydrolysis activity. As shown in Fig.1C, ClpL showed ATP hydrolyzing activity in the presence of Mg2+ and Mn2+, but not in the presence of Zn2+ and Ca2+ (Fig. 1C and Fig S1). We also observed similar ATPase activity in the presence of Mn2+ and Mg2+. The ATPase activity of ClpB is highly dependent on salt concentrations, where higher concentrations drastically reduced the activity38. To compare the effect of salt concentrations, we measured the ATPase activity of ClpL in the presence of increasing concentrations of KCl. Surprisingly, we observed that the ATPase activity of ClpL is unaffected at higher KCl concentrations (Fig. 1D and Fig S1).
Figure 1: Features of S. mutans ClpL protein.

(A) Schematic representation of ClpL domains. Mutation sites in the Walker A and B motifs and the conserved arginine residues are shown. (B) The structure of ClpL from S. mutans was modelled using ITASSER software. ClpL contains five domains: N domain (blue), NBD-1 (pink), M domain (yellow), NBD-2 (cyan), and C-domain (grey). (C) ATPase activity of ClpL (1μM) in the presence of various divalent cations. (D) ATP hydrolase activity of ClpL with increasing concentrations of KCl. Concentrations of ClpL and ATP were 1μM and 1mM, respectively.
To understand the role of individual domains of ClpL on the structure and function, we constructed several point mutations and deletion mutations using E. coli ClpB as a template (Fig 1A and Fig S2). Like other AAA proteins (ATPases associated with diverse cellular activities), both NBDs of ClpL contain several motifs and residues that are responsible for ATP binding and hydrolysis. The highly conserved lysine (K128 of NBD1 and K459 of NBD2) residue of the walker A motif (Fig 1A) is assumed to be responsible for ATP binding. In contrast, the conserved glutamine residues of the Walker B motif (E194 of NBD-1 and E527 of NBD-2) are predicted to be responsible for ATP hydrolysis (Fig 1A). There are two conserved arginine residues, termed as sensor 2, which lie at the C-terminal end of each NBD domain. These residues (R250 and R601 correspond to R332 and R756 of ClpB, respectively) (Fig 1A) are assumed to be responsible for γ-phosphate interaction38. Furthermore, to understand the role of the N-, M- and C- domains on the structure and function of ClpL, we also constructed various deletion constructs spanning these domains. Based on our model we determined that the N-, M- and C- domains correspond to the 1–117, 323–389 and 624–701 residues, respectively (Fig 1A) and we made appropriate deletion constructs and purified the proteins as described in the text.
To study the oligomerization pattern of ClpL we performed gel filtration chromatography and native PAGE analysis. As shown in Fig. 2A, the elution profile of ClpL suggests that it exists as a mixture of two different oligomers when nucleotides were absent. The bigger peak corresponds to the molecular weight of ~220 KD which indicates that ClpL predominantly exists as a trimer in solution (the size of a monomer is 79-kD). We also observed a smaller peak that eluted early and corresponds to an ~530 kD complex, indicating the hexameric form of ClpL. Thus, the elution profiles indicate that in the absence of ATP, ClpL predominantly exists as a trimer but a small fraction also exists as hexamer. However, when ATP is present, we found only one major peak corresponding to ~530 kD, indicating that ClpL predominantly exists as a hexamer when nucleotides are present.
Figure 2: Oligomerization of ClpL.

(A) Elution profile of ClpL protein in the absence (solid line) or presence of 1 mM ATP (dotted line). The elution positions of the molecular weight standard are shown on the top. (B) Oligomerization of ClpL wild type and the mutant derivatives were analyzed by NATIVE-PAGE. ClpL and mutant proteins (~1.5μg) were loaded to 10% NATIVE-PAGE in the presence or absence of 1 mM ATP. Tris-glycine buffer supplemented with 20 mM MgCl2 was used as running buffer.
We next assessed whether binding and hydrolysis of ATP to both the domains are required for the observed hexamerization of ClpL. For this, we performed native PAGE analysis with the wild type and the mutant variants of ClpL. We found that in the absence of ATP, all the point mutant derivatives showed similar migration, suggesting similar oligomeric forms, which is most probably a trimeric form. In contrast, in the presence of ATP, we observed two major and some minor differences in migration patterns. When ATP was present, E194A, E527A, R601A and E194A/E527A ClpL variants showed slower migration indicating higher oligomeric form, which is similar to wild type ClpL. Since our gel filtration chromatography data suggested that wild type ClpL forms a hexamer when ATP is present, this slower migration band should correspond to the hexameric form of the proteins (Fig. 2B). Therefore, taken together, we concluded that E194A, E527A, R601A and E194A/E527A ClpL derivatives exist as a trimer when ATP is absent, but form a hexamer in the presence of ATP, similar to the wild type ClpL. It is noteworthy that for these above-mentioned mutants, ATP is able to bind to both the NBDs (see Fig. 5). For the rest of the mutants, such as K128A, K459A, K128A/K459A, K128A/E527A and E194A/K459A, we were unable to detect any slow migrating species that correspond to the hexamer when ATP is present. Additionally, for some mutants such as K459A, we observed a trimer and another slower species that could be a tetrameric form. For two other mutants, R250A and K128A/E527A, we observed a single species that migrated slower than the trimeric species; we speculate that this is also a tetrameric form (Fig. 2B). Since for all these mutants, at least one NBD-domain is unable to bind ATP, we concluded that ATP binding to both domains is essential for nucleotide induced hexamer formation. One exception of this assumption is the mutant R250A. Since R250 and R601 are responsible for interaction with the γ-phosphate of ATP, ATP is able to bind to both the NBD domains (see Fig. 5) and yet R250A is unable to form hexamer in the presence of ATP. As for the deletion mutant variants (ΔN, ΔM, ΔC), all show a similar migration pattern in the presence and absence of ATP (Fig 2 B). This indicates that all three domains (N, M and C) are essential for hexamer formation. Moreover, among the three deletion mutants only the ΔC mutant was unable to bind ATP, but the other two mutants, ΔN and ΔM, were able to bind ATP (see below).
Figure 5: Binding of MANT-ATP with various ClpL mutants.

Titration of ClpL derivatives with MANT-ATP in the buffer as described in the text. The plots showed MANT-ATP binding to wild type and NBD-1 mutants (A), NBD-2 mutants (B), NBD-1& NBD-2 mutants (C), and deletion mutants (D) of ClpL. The plot was fitted with Hill equation and dissociation constant (Kd) and Vmax were obtained from the curve. Data are the means ± SD of at least three repeats.
We then used limited proteolysis to probe whether structural changes occur during ATP binding and hydrolysis by ClpL. ClpL contains 79 trypsin sites which are distributed throughout the entire protein sequence (Fig 3A). We found that ClpL is highly sensitive to trypsin digestion and produces a unique digestion pattern when exposed to trypsin. In the absence of ATP, limited proteolysis generated two stable fragments (Fig 3B, fragments i and ii). The molecular weights of the larger (i) and the smaller (ii) fragments are ~ 45 kD and ~25 kD, respectively, suggesting that the protein contains two major compact regions. When ClpL was incubated with trypsin for a longer time, three additional fragments were generated (fragments iii, iv and v). Addition of ATP or ADP dramatically increased the stability of ClpL to trypsin digestion. In the presence of ATP and after longer incubation with trypsin, we observed only fragment i (Fig. 3B middle). Furthermore, we found that when ADP is present, the banding pattern is very similar to the ATP-bound banding pattern, suggesting that both ATP- and ADP-bound ClpL form different structures than the ATP-unbound form. We also found similar banding pattern changes in the presence and absence of ATP when chymotrypsin was used (data not shown).
Figure 3: Partial proteolysis of ClpL protein.

(A). Domain organizations of ClpL with putative trypsin cleavage sites as shown with vertical lines. (B) Partial proteolysis of ClpL protein by trypsin to probe nucleotide dependent conformational changes. ClpL (1μM) was treated with trypsin at room temperature for 0 to 60 min in the presence or absence of 1 mM ATP or ADP as indicated. Samples were electrophoresed in 10% SDS-PAGE.
To understand the structural rearrangements of the ClpL mutants we performed partial proteolysis with trypsin in the presence and absence of ATP (Fig. 4). We found that only four mutants K459A, K128A/K459A, E194A/K459A and ΔC showed no protection against trypsin digestion in the presence of ATP (Fig 4A and B). Of note, the K459 residue of ClpL is responsible for ATP binding to the NBD-2 domain. Overall, the results indicate that ATP binding to NBD-2 protects ClpL against trypsin cleavage and impairment of ATP binding to NBD-2 abolishes this protection.
Figure 4: ATP binding to NBD-2 but not NBD-1 regulates the conformational change of ClpL.

A. Wild type ClpL and various single mutants. B. ClpL double mutants and various deletion derivatives. ClpL mutants were subjected to partial trypsin digestion in the presence or absence of 1 mM ATP for 20 min and electrophoresed in 10% SDS-PAGE. Samples are: lane 1, without trypsin and ATP; lane 2, with trypsin but no ATP; lane 3, with trypsin and ATP.
We next measured the nucleotide binding affinity of the wild type ClpL and its mutant variants by titrating the proteins with fluorescent nucleotide analog (MANT-ATP). The ClpL is unable to hydrolyze MANT-ATP under experimental conditions (data not shown), and when protein binds to MANT-ATP, the fluorescence intensity increases39. We used the K128A/K459A mutant, where both the NBD domains are mutated, as the negative control (Fig 5C). The ClpL variants where nucleotide binding to NBD-2 is unaffected (such as mutants K128A, E194A, R250A, E527A, R601A, K128A/E527A, E194A/E527A) showed similar binding affinity towards MANT-ATP as the wild type protein (Figure 5A–C Table-1).
Table 1:
Biochemical activities of the CIpL mutants
| Domains | Variants | ATPase activity (nM phosphate/μM min) | MANT- ATP binding | Co-operativity | Relative chaperone activity | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1mM ATP | 2mM ATP | nH | Kd | Vmax | nH | Kd | Refolding ability (% WT) | Aggregation prevention activity (% WT) | ||
| WT | 0.82±0.01 | 0.60±0.04 | 2.02±0.19 | 5.3±0.27 | 0.96±0.01 | 3.02±0.32 | 96.45±3.33 | 100 | 100 | |
| NBD-1 | K128A | 0.19±0.01 | 0.33±0.01 | 2.54±0.34 | 4.14±0.27 | 0.56±0.8 | 0.67±0.27 | 2229±1230 | 13.78±7.07 | 7.02±2.23 |
| E194A | 0.89±0.05 | 0.60±0.01 | 2.80±0.67 | 4.28±0.44 | 0.93±0.02 | 2.7±0.22 | 152±6.86 | 101.3±10.6 | 24.67±3.30 | |
| R250A | 0.54±0.005 | 0.49±0.007 | 2.44±0.29 | 4.72±0.27 | 0.71±0.03 | 1.62±0.11 | 338.6±24.9 | 47.15±9.33 | 15.46±4.28 | |
| NBD-2 | K459A | 0.025±0.028 | 0.19±0.01 | 0.67±0.10 | 25.56±4.6 | ND* | ND* | ND* | 2.87±1.94 | 4.93±0.53 |
| E527A | 0.82±0.03 | 0.67±0.007 | 2.27±0.46 | 3.02±0.47 | 0.94±0.01 | 3.27±0.32 | 129.4±5.65 | 2.14±0.16 | 4.46±0.53 | |
| R601A | 0.07±0.02 | 0.18±0.002 | 2.81±0.41 | 5.59±0.34 | ND* | ND* | ND* | 4.24±2.87 | 4.28±0.4 | |
| NBD-1 & NBD-2 | K128A/K459A | 0±0.02 | 0.01±0.006 | ND* | ND* | ND* | ND* | ND* | 0.0±0.14 | 0.08±0.04 |
| K128A/E527A | 0±0.02 | −0.02±0.004 | 2.21±0.31 | 4.22±0.32 | ND* | ND* | ND* | −0.01±0.16 | 0.8±0.04 | |
| E194A/K459A | −0.02±0.005 | −0.02±0.002 | 0.6±0.19 | 186.1±175 | ND* | ND* | ND* | 0.19±0.39 | 0.5±0.13 | |
| E194A/E527A | 00.3±0.01 | −0.04±0.01 | 2.25±0.47 | 3.43±0.4 | ND* | ND* | ND* | 0.22±0.41 | 0.34±0.08 | |
| N | ΔN | −0.03±0.02 | −0.01±0.001 | 1.73±0.16 | 7.7±0.34 | ND* | ND* | ND* | 0±0.07 | 0.1±0.05 |
| M | ΔM | −0.03±0.023 | −0.03±0.02 | 0.82±0.14 | 21.78±3.69 | ND* | ND* | ND* | −0.04±0.22 | −0.23±0.01 |
| C | ΔC | −0.02±0.02 | 0.003±0.03 | ND* | ND* | ND* | ND* | ND* | −0.30±0.53 | 0.07±0.05 |
However, the affinity reduced drastically when NBD-2 was unable to bind ATP, such as in mutants K459A, E194A/K459A and K128A/K459A (Fig. 5B–C). We found very high Kd values for mutants K459A and E194A/K459, indicating very low affinity to nucleotide under the experimental nucleotide concentration (0–16μM) (Table 1). Some ClpL mutants, such as K128A, R250A, K128A/E527A, showed significant affinity towards ATP. It is important to mention that some mutants, such as K459A and E194A/K459A, form similar oligomerization states in the presence of ATP (Fig. 2). The data indicate that the lower affinity towards ATP of those ClpL mutants is not due to the oligomerization status of the proteins. Thus, nucleotide binding affinity and oligomer formation in the presence of ATP are independent. The data also suggested that NBD-2 has a higher affinity towards ATP compared to NBD-1. At present, we do not know whether both the NBDs (NBD-1 and NBD-2) or only the NBD-2 of ClpL is in the ATP bound state under the experimental conditions. We were unable to determine whether there is an increase in nucleotide binding affinity of NBD-1 upon nucleotide binding to NBD-2. Furthermore, our results also suggested that the conserved arginine of NBD-2 (R601) is not involved in nucleotide binding. We also found that deletion of the M domain severely affected the affinities of ClpL towards the nucleotide (Fig 5D). However, deletion of the C domain completely abolished the nucleotide binding (Fig 5D). This result also supports the proteolytic data where we found no conformational changes for the ΔC mutant in the presence or absence of ATP (Fig. 2B).
To measure the ATPase activity of ClpL and its derivative, we used two different ATP concentrations. We found that wild type ClpL hydrolyzes ATP with a rate of 0.82 nM phosphate/μM/min and 0.60 nM phosphate/μM/min in the presence of 1 mM and 2 mM ATP, respectively (Fig 6 and Table 1). We do not know the exact reason behind the lower ATPase activity rate in the presence of 2 mM ATP compared to 1mM ATP. As expected, the double mutant variants harboring mutations in the catalytic residues of both NBD-1 and NBD-2 did not exhibit any ATPase activity. On the other hand, single mutations in the Walker A or B or R motifs resulted in the complete loss of ATPase activity in the corresponding domain. Mutants of NBD-1, such as K128A, E194A andR250A, all have ATPase deficient NBD1; therefore, these mutants represent ATPase activity of NBD-2 only. We found different ATPase activities depending on the mutations (Fig 6 and Table 1). For example, the K128A mutant, which has ATP binding deficiency in NBD-1, showed significantly lower ATPase activity (~25% of WT) compared to WT. However, ATP hydrolysis deficient Walker B mutants (E194A and R250A) showed higher activity compared to the K128A mutant. While the ATPase activity of the E194A mutant is comparable to the wild type, the R250A mutant exhibited ~75% ATPase activity as compared to the wild type. The data indicate that ATP binding to NBD-1 increases the ATPase activity of NBD-2. By comparing ATPase activity between E194A (where γ-P can interact with NBD-1) and R250A (where γ-phosphate cannot interact with NBD-1), we concluded that γ-phosphate interaction with NBD-1 has a significant effect on conformational changes of NBD-1 that is responsible for higher ATPase activity of NBD-2. Similarly, NBD-2 mutants, such as K459A, E527A, and R601A where we measured the ATPase activity of NBD-1, also exhibited different ATPase activities. ATP binding deficient NBD-2 mutants showed very low ATPase activity at 1mM ATP (Fig 6A). The R601A mutant, in which ATP can bind to NBD-2 but γ-P of ATP cannot interact with NBD-2, showed similar lower activity than in the NBD-1 mutants. Mutant E527A showed similar ATPase activity to the wild type. Like NBD-1, γ- phosphate interaction with NBD-2 seems very important for ATPase activity of NBD-1, because R601A (where γ-P cannot interact with NBD-2) showed significantly lower ATPase activity compared to E527A (where γ- phosphate can interact with NBD-2); although both the mutants exist as a hexamer in the presence of ATP (Fig 2B). Furthermore, removal of the N-domain (ΔN), M-domain (ΔM), and C-domain (ΔC) completely abolished the ATPase activity of the ClpL protein (Fig 6 Table 1). Although a ΔN mutant had similar binding affinity for MANT-ATP as compared to WT, it did not exhibit any measurable ATPase activity, indicating that the N-domain is essential for ATPase activity. Likewise, deletion of the M-domain, which decreases the ATP binding activity, also completely abolished the ATPase activity of ClpL. The ΔC mutant neither exhibited ATP binding activity nor any ATP hydrolysis activity.
Figure 6: ATPase activity of ClpL mutants.
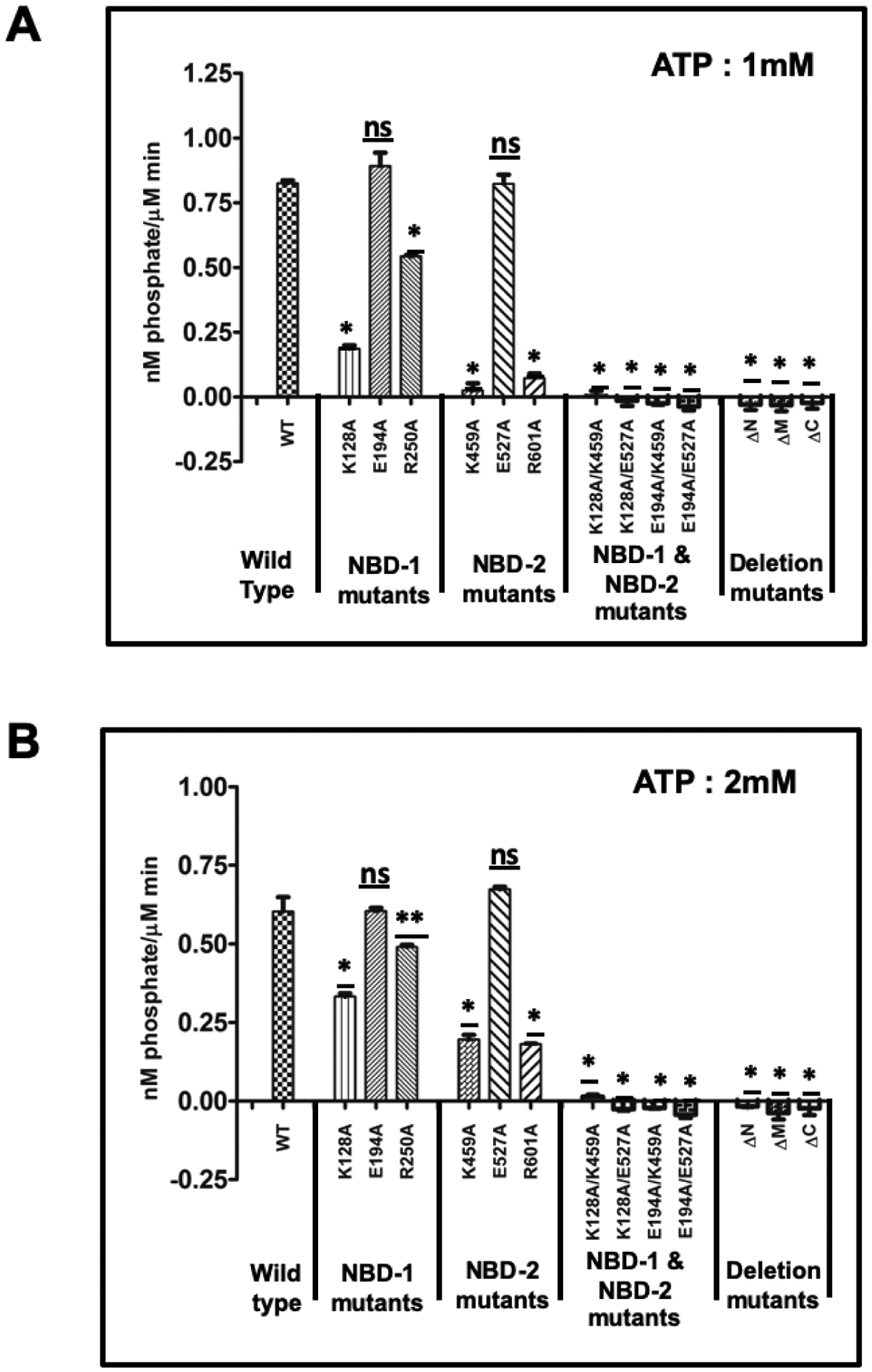
ClpL variants (1μM) were incubated with 1 mM ATP (A) or 2 mM ATP (B) for 10 min. The released phosphate was measured using phosphate colorimetric kit. The rate of hydrolysis was calculated from the nanomoles phosphate released by 1μM ClpL variants during 10 min incubation with ATP. Asterisks denote statistical significance between the activity of wild type and mutant proteins by unpaired, two-tailed Student’s t test (*P < 0.01; **P < 0.05); n.s. no significant difference.
Next, we measured the kinetics of ATP hydrolysis activity of ClpL. For this, we measured the rate of ATP hydrolysis at different ATP concentrations40. As shown in Fig. 7, the Hill coefficient of the wild type ClpL was calculated to be 3.02 ± 0.32 (Table 1). The estimated Hill coefficient for the NBD-1 mutants, such as K128A, E194A and R250A, were found to be 0.67 ± 0.27, 2.7 ± 0.22 and 1.62 ± 0.11, respectively. These results indicate that ATPase activity of NBD-2 is somewhat positively cooperative when ATP is able to bind to NBD-1, except for K128 mutant. However, this positive cooperativity is lost when ATP is unable to bind to NBD-1 (Fig. 6A). Mutating the arginine residue of NBD-1 (R250A) also resulted in decreased positive cooperativity (Fig 5A and Table 1). We also verified the effect of the mutations in the NBD-2 on the ATPase activity of NBD-1. However, we were unable to calculate the Hill coefficient of the K459A and R601A mutants due to very low ATPase activity suggesting that positive cooperativity is lost in these mutants. In contrast, in the E194A mutant, where ATP can bind to NBD-1, we saw a higher Hill coefficient (2.77 ± 0.22) comparable to the wild type value. Taken together, these results suggest that the ATP-bound state of one NBD domain enhances the ATPase activity (cooperativity) of the other domain. The data also suggest that the nucleotide binding deficiency of one NBD domain affects the ATP hydrolysis activity of the other domain.
Figure 7: Cooperativity of ATP hydrolysis.

ATP hydrolysis activity of ClpL (1μM) and its mutant derivatives were measured in the presence of increasing concentrations of ATP. The rate of ATPase activity was plotted against ATP concentrations. The solid line was obtained by fitting the result with equation 1 using GraphPad program. Hill coefficient (nH) and other kinetic parameters were calculated from the equation. N.D indicates not determined.
We previously found that ClpL is required for the folding of the CtsR protein in S. mutants33 and others have also shown that ClpL is able to fold heterologous protein30, 34. Therefore, to understand the mechanism of protein folding by ClpL, we measured the chaperone activity of the ClpL and its mutants by measuring reactivation of urea denatured luciferase protein and also quantified luciferase deactivation at a high temperature in the presence and absence of ClpL variants. To compare the refolding ability of different ClpL mutants, the refolding ability of wild type ClpL protein was set at 100% (Fig 8A). We found that all the NBD-2 mutants did not display any chaperone activity, indicating the essentiality of ATP binding and hydrolysis by NBD-2 for chaperone (Fig 8A). On the other hand, NBD-1 mutants showed chaperone activities, but the activities were variable depending on the mutations. For example, mutant E194A displayed wild type level refolding activity whereas the other mutants exhibited lower chaperone activity. We found that the chaperone activity of NBD-1 mutants is proportional to ATP hydrolysis activity. The order of ATPase and chaperone activity of the wild type and the NBD-1 mutants are: wild type/E194A >R250A >K128A (Fig 6, Fig 8 & Table1). We also observed that all deletion mutants and the double mutants did not display any chaperone activity (Fig 8B).
Figure 8: Chaperone activity of ClpL.

(A) Reactivation of urea denatured luciferase by ClpL mutants. Urea denatured luciferase (50 nM) was allowed to refold in the presence and absence of 3 μM ClpL variants for 4h at room temperature. Luciferase activity was measured using luciferin as a substrate. (B) ClpL prevents heat-induced inactivation of luciferase. 0.5μM luciferase was incubated with or without 3 μM ClpL variants for 20 min at 48°C followed by measuring the luciferase activity. The activity of wild type ClpL protein was set as 100%. Asterisks denote statistical significance between the activity of wild type and mutant proteins by unpaired, two-tailed Student’s t test (*P < 0.01); n.s. no significant difference.
In agreement with previous results, we also found that NBD-1 mutants are able to prevent heat induced deactivation of luciferase to some extent. Surprisingly, the E194A mutant showed less activity (24.67% compared to WT) in preventing heat induced deactivation of luciferase as compared to the wild type (Fig 8B and Table1). The other two NBD-1 mutants (R250A and K128A) were also able to prevent heat induced inactivation of luciferase, but the activities were even lower than the E194A mutant (Fig 8B Table 1). All other mutants displayed very little or no activity against preventing aggregation. Taken together these observations suggest that ATP hydrolysis is strictly required for chaperone activity and impairment of ATPase activity at NBD-2 completely abolishes the chaperone activity of the protein.
DISCUSSION
While ClpB chaperone is present in both gram-negative and gram-positive bacteria, ClpL is uniquely present in gram-positive bacteria. Despite its importance in cellular physiology, at the biochemical level, ClpL is poorly characterized. Here we attempted to understand how this chaperone functions in vivo by studying various biochemical activities of purified S. mutans ClpL protein. Our study has revealed some notable differences between the well-studied ClpB chaperone and the ClpL protein from S. pneumoniae.
We found that the S. mutans ClpL protein predominantly exists as a trimer in solution in the absence of ATP. This observation is somewhat surprising since ClpL of S. pneumoniae exists as a monomer when nucleotides are absent34. However, in the presence of ATP, ClpL from both organisms forms a hexameric ring structure which acts as a chaperone. It is interesting to note that ClpB from various organisms also presents as different oligomeric states in solution when nucleotides are absent41, 42. We found that divalent cations have drastic effects on the ATPase activity. While both Mg2+ and Mn2+ ions were equally effective for the ATP hydrolysis activity, the presence of Zn2+ and Ca2+ cations did not stimulate ATP hydrolysis activity. In contrast, ClpL of S. pneumoniae showed higher activity in the presence of Mn2+ compared to Mg2+ 34. Since the structure for ClpL is currently unavailable, the exact reason for this difference in ion dependency is unknown. In one of the ClpB crystal structures, the Mg2+ ion was found to be coordinated with the residues near the N-terminal region and not at the Walker A or B motifs43. Thus, in addition to NBD domains, other regions of the ClpL might be necessary for metal ion dependence.
Oligomerization of ClpB in the presence of ATP is dependent on both the NBD domains, and ATP binding to these domains is essential for hexamer formation38. Our mutational studies were designed such that at least one of the NBD domains was unable to bind ATP. Our results indicate that ATP binding to both the domains is essential for nucleotide induced hexamer formation, similar to the ClpB chaperone (Fig. 2). However, we also observed some surprising results. The two key arginine residues in the NBD domains (R250 and R601) are responsible for the interaction with the γ-phosphate of ATP38. We found that ClpL R250A is unable to form hexamer in the presence of ATP, but the ClpL R601A mutant could form a hexamer when ATP was present (Fig. 2). From the partial proteolysis assay, we concluded that ATP is able to bind NBD-2, but we do not know whether ATP is able to bind to NBD-1 in the ClpL R250A mutant (Fig. 3). It is possible that the R250 residue has another role in addition to γ-phosphate interaction. Alternatively, γ-phosphate interaction of the NBD-1 is indeed essential for ClpL hexamer formation. Our limited proteolysis assays also shed light on conformational changes of ClpL. Most notably, we found that ClpL mutants with defects in nucleotide binding of NBD-2 (K459A, K128A/K459A and E194A/K459A) failed to show protection against proteolytic digestion. This observation suggests that a major conformational change occurs upon ATP binding to NBD-2 that confers protection against proteolysis. This is in contrast to the ClpB chaperone where ATP binding to NBD-1 is essential for protection against proteolysis38.
All the deletion mutants showed a similar oligomerization status in the presence or absence of ATP, suggesting that all three domains (N, M, and C) are essential for hexamer formation. We also found that the ClpL ΔC mutant was unable to bind ATP, but two other deletion mutants could not form a hexamer in spite of their binding ability with ATP. Our current results cannot differentiate whether deletion of the N or M domains affect the ATP binding to both the NBD domains or only affect binding to one domain. We can only conclude all the domains are essential for ATP dependent oligomerization of the ClpL chaperone. Of note, it is also possible that the deletion mutants are unable to fold properly, therefore they could be biologically inactive. While it is a possibility, we found that at least ΔN mutant was biologically active while ΔC mutant was inactive33.
ATP binding is necessary for the oligomerization of the ClpL chaperone since the 94A/E274A mutant does not have ATPase activity but exists as a hexamer in the presence of ATP. The deviations of the ATPase activities of different ClpL mutants (K128A vs E194A and K459A vs E527A) indicate active communication between the two NBD domains. The ATP bound conformation of one NBD increased the ATPase activity of the other NBD. Surprisingly, in ClpL mutants such as E194A or E527A, where only one NBD is active, a wild type level of ATPase activity was shown. We also found that mutating the conserved arginine residues in the NBD domains (R250A or R601A) showed lower ATPase activity compared to the Walker B mutants (E194A or E527A). This observation suggests that the γ-phosphate interaction significantly affects the conformation of the NBD domains. We also found that nucleotide binding deficient mutants of NBD-2 (K459A and R601A) showed very low ATPase activity compared to the nucleotide binding deficient mutants in NBD-1 (K128A or R250A). The result suggested that nucleotide binding with NBD-2 is more important for ATPase activity compared to nucleotide binding at NBD-1.
Our results indicate that the individual NBD domain shows a pronounced difference in affinity for the nucleotides. We found that NBD-2 has higher affinity than NBD-1. Currently, we do not have any experimental evidence that suggests whether only NBD-2 or both the domains are occupied by MANT-ATP in the wild type ClpL. Interestingly, when ATP was present in the solution, both the NBD-1 mutants (K128A and E194A) showed equal affinity for the nucleotides; although ClpL K128A is present as a trimer whereas E194A exists as a hexamer in solution. These results also indicate that the affinity for nucleotides of NBD-2 does not depend on the oligomerization status of the protein.
We observed lower cooperativity for ATPase activity in the ATP binding-deficient Walker A mutants K128A and K459A; the positive cooperativity was completely abolished in these mutants. On the other hand, the ATP hydrolysis-deficient Walker B mutants (E194A and E527A) showed positive cooperativity as well as ATPase activity at the wild type level (Fig. 7). Based on our ATP binding studies, we speculate that ATP binding to NBD-2 (E527A) enhances the affinity of NBD-1 for the nucleotide. Alternatively, it is also possible that oligomerization might play a role since the E527A mutant can form a hexamer in presence of ATP whereas the K459A mutant is unable to form a hexamer. However, as we mentioned earlier, ATP binding to ClpL is independent of the oligomerization status of the protein. Nevertheless, our data suggest that there is an allosteric communication between the two NBD domains and ATP binding to the high affinity binding site NBD-2 generates an allosteric signal which increases the affinity of NBD-1 for the nucleotide. Our results also confirmed that nucleotide bound conformation of NBD-1 increases the ATPase activity of NBD-2 and vice-versa.
We previously found that in vivo ClpL is needed for proper folding of a heat-shock regulator, CtsR33. Others have also shown that ClpL indeed displays chaperone activity34. Unlike ClpB, the chaperone activity of ClpL does not require any co-chaperone or accessory factors33, 34. However, ATPase activity is a prerequisite for the chaperone activity of ClpL. Surprisingly, we found that hexamer formation is not essential for the observed chaperone activity. For example. the ClpL R250A mutant displays a nearly 50% level of wild type chaperone activity (Fig. 8). This is in contrast to the ClpB chaperone, where hexamer formation is required38. While NBD-1 mutants showed chaperone activity, some nearly wild type level (mutant E194A; Fig. 8), all the NBD-2 mutants did not exhibit any chaperone activity. We speculate that ATP hydrolysis by NBD-2 is absolutely essential for chaperone activity.
While our results indicate that molecular functions of ClpL is different from the other well-studied chaperones such as ClpB, we also noticed that ClpL of S. mutans behaves differently from ClpL of S. pneumoniae28. For example, ClpL of S. mutans exists as a trimer in solution whereas ClpL of S. pneumoniae exists as a monomer34. ATPase activity of ClpL of S. pneumoniae is Mn2+ dependent and both the NBD domains are required for the ATPase activity, oligomerization and chaperone functions, unlike ClpL of S. mutans34. To further understand the differences in activities, we compared the ClpL sequences from S. mutans and S. pneumoniae (Fig. S3). We found that the sequences are highly homologous (90% identity); however, the NBD domains are not identical although they both contain the identical Walker A or B motifs (data not shown). We found differences in other domains as well. Sequence comparison with other streptococci indicated that both N-terminal and C-terminal regions are highly variable compared to the central region. At present, we are not certain whether the dissimilarity in the sequences are responsible for the observed differences.
ClpL is a unique chaperone since it does not require any additional factors for its chaperone activity. So far, CtsR is the only substrate that is directly recognized by ClpL in vivo. Moreover, we previously found that several other proteins are also differentially expressed in a clpL-deficient S. mutans strain, suggesting that ClpL, although not essential, may play an important role in the bacterial physiology. Our study indicates that while ClpL might be necessary for maintaining cellular homeostasis, at the molecular level how this chaperone functions might be different in different organisms. Further biochemical and structural studies involving additional ClpL proteins from different species are necessary to fully understand this chaperone.
Supplementary Material
ACKNOWLEDGEMENT:
This work was supported in part by National Institute of Dental and Craniofacial Research grant DE026995 and by National Institute of General Medical Sciences grant GM128241 awarded to I.B.
Footnotes
Supporting Information
List of oligonucleotides used in the study (Table S1)
ATPase activity of ClpL protein and its derivatives (Figure S1)
Alignment of E. coli ClpB with ClpB and ClpL of S. mutans (Figure S2)
Multiple sequence alignment of ClpL homologs from streptococci (Figure S3)
Conflict of interest:
The authors declare that they have no conflicts of interest with the contents of this article.
ACCESSION CODES
UniProt Accession IDs for S. mutans ClpP protein is Q8DUH3_STRMU.
REFERENCES:
- 1.Gottesman S; Wickner S; Maurizi MR, Protein quality control: triage by chaperones and proteases. Genes Dev 1997, 11 (7), 815–23. [DOI] [PubMed] [Google Scholar]
- 2.Wickner S; Maurizi MR; Gottesman S, Posttranslational quality control: folding, refolding, and degrading proteins. Science 1999, 286 (5446), 1888–93. [DOI] [PubMed] [Google Scholar]
- 3.Hayer-Hartl M; Bracher A; Hartl FU, The GroEL-GroES Chaperonin Machine: A Nano-Cage for Protein Folding. Trends Biochem Sci 2016, 41 (1), 62–76. [DOI] [PubMed] [Google Scholar]
- 4.LaRossa RA; Van Dyk TK, Physiological roles of the DnaK and GroE stress proteins: catalysts of protein folding or macromolecular sponges? Molecular microbiology 1991, 5 (3), 529–34. [DOI] [PubMed] [Google Scholar]
- 5.Lund PA, Microbial molecular chaperones. Adv Microb Physiol 2001, 44, 93–140. [DOI] [PubMed] [Google Scholar]
- 6.Doyle SM; Wickner S, Hsp104 and ClpB: protein disaggregating machines. Trends Biochem Sci 2009, 34 (1), 40–48. [DOI] [PubMed] [Google Scholar]
- 7.Harrison C, GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperones 2003, 8 (3), 218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genest O; Wickner S; Doyle SM, Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. The Journal of biological chemistry 2019, 294 (6), 2109–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhandari V; Houry WA, Substrate Interaction Networks of the Escherichia coli Chaperones: Trigger Factor, DnaK and GroEL. Adv Exp Med Biol 2015, 883, 271–94. [DOI] [PubMed] [Google Scholar]
- 10.Erzberger JP; Berger JM, Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 2006, 35, 93–114. [DOI] [PubMed] [Google Scholar]
- 11.Ogura T; Whiteheart SW; Wilkinson AJ, Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol 2004, 146 (1–2), 106–12. [DOI] [PubMed] [Google Scholar]
- 12.Hanson PI; Whiteheart SW, AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 2005, 6 (7), 519–29. [DOI] [PubMed] [Google Scholar]
- 13.Hodson S; Marshall JJ; Burston SG, Mapping the road to recovery: the ClpB/Hsp104 molecular chaperone. J Struct Biol 2012, 179 (2), 161–71. [DOI] [PubMed] [Google Scholar]
- 14.Gates SN; Yokom AL; Lin J; Jackrel ME; Rizo AN; Kendsersky NM; Buell CE; Sweeny EA; Mack KL; Chuang E; Torrente MP; Su M; Shorter J; Southworth DR, Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 357 (6348), 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S; Sowa ME; Watanabe YH; Sigler PB; Chiu W; Yoshida M; Tsai FT, The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 2003, 115 (2), 229–40. [DOI] [PubMed] [Google Scholar]
- 16.Mogk A; Kummer E; Bukau B, Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci 2015, 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barends TR; Werbeck ND; Reinstein J, Disaggregases in 4 dimensions. Curr Opin Struct Biol 2010, 20 (1), 46–53. [DOI] [PubMed] [Google Scholar]
- 18.Suokko A; Poutanen M; Savijoki K; Kalkkinen N; Varmanen P, ClpL is essential for induction of thermotolerance and is potentially part of the HrcA regulon in Lactobacillus gasseri. Proteomics 2008, 8 (5), 1029–41. [DOI] [PubMed] [Google Scholar]
- 19.Wall T; Bath K; Britton RA; Jonsson H; Versalovic J; Roos S, The early response to acid shock in Lactobacillus reuteri involves the ClpL chaperone and a putative cell wall-altering esterase. Applied and environmental microbiology 2007, 73 (12), 3924–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu le N; Jeong HY; Kwon HY; Ogunniyi AD; Paton JC; Pyo SN; Rhee DK, Modulation of adherence, invasion, and tumor necrosis factor alpha secretion during the early stages of infection by Streptococcus pneumoniae ClpL. Infection and immunity 2007, 75 (6), 2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao C; Wang J; Liu Y; Kwok LY; Zhang H; Zhang W, Adaptation of Lactobacillus plantarum to Ampicillin Involves Mechanisms That Maintain Protein Homeostasis. mSystems 2020, 5 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran TD; Kwon HY; Kim EH; Kim KW; Briles DE; Pyo S; Rhee DK, Decrease in penicillin susceptibility due to heat shock protein ClpL in Streptococcus pneumoniae. Antimicrobial agents and chemotherapy 2011, 55 (6), 2714–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran TD; Kwon HY; Kim EH; Kim KW; Briles DE; Pyo S; Rhee DK, Heat-shock protein ClpL/HSP100 increases penicillin tolerance in Streptococcus pneumoniae. Adv Otorhinolaryngol 72, 126–8. [DOI] [PubMed] [Google Scholar]
- 24.Suokko A; Savijoki K; Malinen E; Palva A; Varmanen P, Characterization of a mobile clpL gene from Lactobacillus rhamnosus. Applied and environmental microbiology 2005, 71 (4), 2061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang DC; Huang XF; Novel G; Novel M, Two genes present on a transposon-like structure in Lactococcus lactis are involved in a Clp-family proteolytic activity. Molecular microbiology 1993, 7 (6), 957–65. [DOI] [PubMed] [Google Scholar]
- 26.Pontinen A; Aalto-Araneda M; Lindstrom M; Korkeala H, Heat Resistance Mediated by pLM58 Plasmid-Borne ClpL in Listeria monocytogenes. mSphere 2017, 2 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hingston P; Brenner T; Truelstrup Hansen L; Wang S, Comparative Analysis of Listeria monocytogenes Plasmids and Expression Levels of Plasmid-Encoded Genes during Growth under Salt and Acid Stress Conditions. Toxins (Basel) 2019, 11 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khara PB, I, ClpL ATPase: a novel chaperone in bacterial stress responses. Wiley and Sons. Inc: 2016. [Google Scholar]
- 29.Kajfasz JK; Martinez AR; Rivera-Ramos I; Abranches J; Koo H; Quivey RG; Lemos JA, Role of Clp Proteins in Expression of Virulence Properties of Streptococcus mutans. Journal of bacteriology 2009, 191 (7), 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwon HY; Kim SW; Choi MH; Ogunniyi AD; Paton JC; Park SH; Pyo SN; Rhee DK, Effect of heat shock and mutations in ClpL and ClpP on virulence gene expression in Streptococcus pneumoniae. Infection and immunity 2003, 71 (7), 3757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varcamonti M; Arsenijevic S; Martirani L; Fusco D; Naclerio G; De Felice M, Expression of the heat shock gene clpL of Streptococcus thermophilus is induced by both heat and cold shock. Microb Cell Fact 2006, 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le NT; Jeong HY; Kwon HY; Ogunniyi AD; Paton JC; Pyo SN; Rhee DK, Modulation of Adherence, Invasion, and Tumor Necrosis Factor Alpha Secretion during the Early Stages of Infection by Streptococcus pneumoniae ClpL (vol 75, pg 2996, 2007). Infection and immunity 2015, 83 (3), 1224–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tao L; Biswas I, ClpL is required for folding of CtsR in Streptococcus mutans. Journal of bacteriology 2013, 195 (3), 576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SS; Kwon HY; Tran TD; Choi MH; Jung SH; Lee S; Briles DE; Rhee DK, ClpL is a chaperone without auxiliary factors. Febs J 2015, 282 (8), 1352–67. [DOI] [PubMed] [Google Scholar]
- 35.Kunkel TA, Rapid and efficient site-specific mutagenesis without phenotypic selection. Proceedings of the National Academy of Sciences of the United States of America 1985, 82 (2), 488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ihara M; Matsuura N; Yamashita A, High-resolution Native-PAGE for membrane proteins capable of fluorescence detection and hydrodynamic state evaluation. Anal Biochem 2011, 412 (2), 217–23. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, I-TASSER server for protein 3D structure prediction. BMC bioinformatics 2008, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mogk A; Schlieker C; Strub C; Rist W; Weibezahn J; Bukau B, Roles of individual domains and conserved motifs of the AAA plus chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. Journal of Biological Chemistry 2003, 278 (20), 17615–17624. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Higuero JA; Acebron SP; Taneva SG; Del Castillo U; Moro F; Muga A, Allosteric communication between the nucleotide binding domains of caseinolytic peptidase B. The Journal of biological chemistry 2011, 286 (29), 25547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishikori S; Esaki M; Yamanaka K; Sugimoto S; Ogura T, Positive cooperativity of the p97 AAA ATPase is critical for essential functions. The Journal of biological chemistry 2011, 286 (18), 15815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akoev V; Gogol EP; Barnett ME; Zolkiewski M, Nucleotide-induced switch in oligomerization of the AAA+ ATPase ClpB. Protein Sci 2004, 13 (3), 567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zolkiewski M; Kessel M; Ginsburg A; Maurizi MR, Nucleotide-dependent oligomerization of ClpB from Escherichia coli. Protein Sci 1999, 8 (9), 1899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeymer C; Barends TR; Werbeck ND; Schlichting I; Reinstein J, Elements in nucleotide sensing and hydrolysis of the AAA+ disaggregation machine ClpB: a structure-based mechanistic dissection of a molecular motor. Acta Crystallogr D Biol Crystallogr 2014, 70 (Pt 2), 582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.