

Abstract
Gene tree discordance in large genomic data sets can be caused by evolutionary processes such as incomplete lineage sorting and hybridization, as well as model violation, and errors in data processing, orthology inference, and gene tree estimation. Species tree methods that identify and accommodate all sources of conflict are not available, but a combination of multiple approaches can help tease apart alternative sources of conflict. Here, using a phylotranscriptomic analysis in combination with reference genomes, we test a hypothesis of ancient hybridization events within the plant family Amaranthaceae s.l. that was previously supported by morphological, ecological, and Sanger-based molecular data. The data set included seven genomes and 88 transcriptomes, 17 generated for this study. We examined gene-tree discordance using coalescent-based species trees and network inference, gene tree discordance analyses, site pattern tests of introgression, topology tests, synteny analyses, and simulations. We found that a combination of processes might have generated the high levels of gene tree discordance in the backbone of Amaranthaceae s.l. Furthermore, we found evidence that three consecutive short internal branches produce anomalous trees contributing to the discordance. Overall, our results suggest that Amaranthaceae s.l. might be a product of an ancient and rapid lineage diversification, and remains, and probably will remain, unresolved. This work highlights the potential problems of identifiability associated with the sources of gene tree discordance including, in particular, phylogenetic network methods. Our results also demonstrate the importance of thoroughly testing for multiple sources of conflict in phylogenomic analyses, especially in the context of ancient, rapid radiations. We provide several recommendations for exploring conflicting signals in such situations. [Amaranthaceae; gene tree discordance; hybridization; incomplete lineage sorting; phylogenomics; species network; species tree; transcriptomics.]
The exploration of gene tree discordance has become common in the phylogenetic era (Salichos et al. 2014; Smith et al. 2015; Huang et al. 2016; Pease et al. 2018) and is essential for understanding the underlying processes that shape the Tree of Life. Discordance among gene trees can be the product of multiple sources. These include errors and noise in data assembly and filtering, hidden paralogy, incomplete lineage sorting (ILS), gene duplication/loss (Pamilo and Nei 1988; Doyle 1992; Maddison 1997; Galtier and Daubin 2008), random noise from uninformative genes, as well as misspecified model parameters of molecular evolution such as substitutional saturation, codon usage bias, or compositional heterogeneity (Foster 2004; Cooper 2014; Cox et al. 2014; Liu et al. 2014). Among these potential sources of gene tree discordance, ILS is the most studied in the systematics literature (Edwards 2009), and several phylogenetic inference methods have been developed to accommodate ILS as the source of discordance (reviewed in Edwards et al. 2016; Mirarab et al. 2016; Xu and Yang 2016). More recently, methods that account for additional processes such as hybridization or introgression have gained attention. These include methods that estimate phylogenetic networks while accounting for ILS and hybridization simultaneously (e.g., Solís-Lemus and Ané 2016; Wen et al. 2018), and methods that detect introgression based on site patterns or phylogenetic invariants (e.g., Green et al. 2010; Durand et al. 2011; Kubatko and Chifman 2019). Frequently, multiple processes can contribute to gene tree heterogeneity (Holder et al. 2001; Buckley et al. 2006; Meyer et al. 2017; Knowles et al. 2018). However, at present, no method can estimate species trees from phylogenomic data while modeling multiple sources of conflict and heterogeneity in molecular substitution simultaneously. To overcome these limitations, the use of multiple phylogenetic tools and data partitioning schemes in phylogenomic data sets is essential to disentangle sources of gene tree heterogeneity and resolve recalcitrant relationships at deep and shallow nodes of the Tree of Life (e.g., Alda et al. 2019; Widhelm et al. 2019; Prasanna et al. 2020; Roycroft et al. 2020).
In this study, we evaluate multiple sources of gene tree conflict to test controversial hypotheses of ancient hybridization among subfamilies in the plant family Amaranthaceae s.l. Amaranthaceae s.l. includes the previously segregated family Chenopodiaceae (Hernández-Ledesma et al. 2015; The Angiosperm Phylogeny Group 2016). With ca. 2050 to 2500 species in 181 genera and a worldwide distribution (Hernández-Ledesma et al. 2015), Amaranthaceae s.l. is iconic for the repeated evolution of complex traits representing adaptations to extreme environments such as C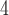 photosynthesis in hot and often dry environments (e.g., Kadereit et al. 2012; Bena et al. 2017), various modes of extreme salt tolerance (e.g., Flowers and Colmer 2015; Piirainen et al. 2017) that in several species are coupled with heavy metal tolerance (Moray et al. 2016), and very fast seed germination and production of multiple diaspore types on one individual (Kadereit et al. 2017). Several important crops are members of Amaranthaceae s.l., such as the pseudocereals quinoa and amaranth, sugar beet, spinach, glassworts, and saltworts. Many species of the family are also important fodder plants in arid regions and several are currently being investigated for their soil remediating and desalinating effects (e.g., Li et al. 2019). Due to their economic importance, reference genomes are available for Beta vulgaris (sugar beet, subfamily Betoideae; Dohm et al. 2014), Chenopodium quinoa (quinoa, Chenopodioideae; Jarvis et al. 2017), Spinacia oleracea (spinach; Chenopodioideae; Xu et al. 2017), and Amaranthus hypochondriacus (amaranth; Amaranthoideae; Lightfoot et al. 2017), representing three of the 13 currently recognized subfamilies of Amaranthaceae s.l. (sensu Kadereit et al. 2003; Kadereit et al. 2017).
photosynthesis in hot and often dry environments (e.g., Kadereit et al. 2012; Bena et al. 2017), various modes of extreme salt tolerance (e.g., Flowers and Colmer 2015; Piirainen et al. 2017) that in several species are coupled with heavy metal tolerance (Moray et al. 2016), and very fast seed germination and production of multiple diaspore types on one individual (Kadereit et al. 2017). Several important crops are members of Amaranthaceae s.l., such as the pseudocereals quinoa and amaranth, sugar beet, spinach, glassworts, and saltworts. Many species of the family are also important fodder plants in arid regions and several are currently being investigated for their soil remediating and desalinating effects (e.g., Li et al. 2019). Due to their economic importance, reference genomes are available for Beta vulgaris (sugar beet, subfamily Betoideae; Dohm et al. 2014), Chenopodium quinoa (quinoa, Chenopodioideae; Jarvis et al. 2017), Spinacia oleracea (spinach; Chenopodioideae; Xu et al. 2017), and Amaranthus hypochondriacus (amaranth; Amaranthoideae; Lightfoot et al. 2017), representing three of the 13 currently recognized subfamilies of Amaranthaceae s.l. (sensu Kadereit et al. 2003; Kadereit et al. 2017).
Within the core Caryophyllales the previously segregated families Amaranthaceae s.s. and Chenopodiaceae have always been regarded as closely related, and their separate family status has long been the subject of phylogenetic and taxonomic debate (Kadereit et al. 2003; Masson and Kadereit 2013; Hernández-Ledesma et al. 2015; Walker et al. 2018; Fig. 1). Their close affinity is supported by a number of shared morphological, anatomical, and phytochemical synapomorphies and has been substantiated by molecular phylogenetic studies (discussed in Kadereit et al. 2003). Amaranthaceae s.s. has a predominantly tropical and subtropical distribution with the highest diversity found in the Neotropics, eastern and southern Africa and Australia (Müller and Borsch 2005), while Chenopodiaceae predominantly occurs in temperate regions and semi-arid or arid environments of subtropical regions (Kadereit et al. 2003). The key problem has always been the species-poor and heterogeneous subfamilies Polycnemoideae and Betoideae, neither of which fit easily within Chenopodiaceae or Amaranthaceae s.s. (cf. Table 5 in Kadereit et al. 2003). Polycnemoideae is similar in ecology and distribution to Chenopodiaceae but shares important floral traits such as petaloid tepals, filament tubes and 2-locular anthers with Amaranthaceae s.s. Morphologically, Betoideae fits into either Chenopodiaceae or Amaranthaceae s.s. but has a unique fruit type---a capsule that opens with a circumscissile lid (Kadereit et al. 2006). Both Betoideae and Polycnemoideae possess only a few species each and each has a strongly disjunct distribution pattern across three continents. Furthermore, the genera of both subfamilies display a number of morphologically dissociating features. Both intercontinental disjunctions of species-poor genera and unique or intermediate morphological traits led to the hypothesis that Betoideae and Polycnemoideae might have originated from hybridization events among early-branching lineages in Amaranthaceae s.l. (Hohmann et al. 2006; Masson and Kadereit 2013). To test this hypothesis, a phylotranscriptomic approach is particularly compelling as it not only provides thousands of low-copy nuclear genes for dissecting sources of phylogenetic discordance but also enables future studies associating gene tree topology with gene function and habitat adaptation.
Figure 1.

Phylogenetic hypotheses of Amaranthaceae s.l. from previous studies. a) Kadereit et al. (2003) using the plastid (cpDNA) rbcL coding region. b) Müller and Borsch (2005); using the cpDNA matK coding region and partial trnL intron. c) Hohmann et al. (2006) using the cpDNA ndhF coding region d) Kadereit et al. (2017) using the cpDNA atpB-rbcL spacer, matK with trnL intron, ndhF-rpL32 spacer, and rps16 intron. e) Walker et al. (2018) using 455 nuclear genes from transcriptome data. Major clades of Amaranthaceae s.l. named following the results of this study. Image credits: Amaranthus hypochondriacus by Picture Partners, Beta vulgaris by Olha Huchek, Chenopodium quinoa by Diana Mower, Nitrophila mohavensis by James M. André, and Salicornia europaea by Homeydesign.
Previous molecular phylogenetic analyses struggled to resolve the relationships among Betoideae, Polycnemoideae and the rest of the Amaranthaceae s.l. (Fig. 1). The first phylogenomic study of Amaranthaceae s.l. using nuclear loci (Walker et al. 2018; Fig. 1e) revealed that gene tree discordance mainly occurred at deep nodes of the phylogeny involving Betoideae. Polycnemoideae was sister to Chenopodiaceae, albeit supported by only 17% of gene trees, which contradicted previous analyses based on plastid data (Fig. 1a--d). However, only a single species of Betoideae (the cultivated beet and its wild relative) was sampled in Walker et al. (2018). Furthermore, Walker et al. (2018) found conflicting topologies between concatenated and coalescent-based analyses, but sources of conflicting signals among gene trees remained unexplored.
In this study, we used a large genomic data set to examine sources of gene tree discordance in Amaranthaceae s.l. Specifically, we tested whether Polycnemoideae and Betoideae result from independent hybridizations between Amaranthaceae s.s. and Chenopodioideae by distinguishing the signal of hybridization from gene tree discordance produced by ILS, uninformative gene trees, hidden paralogy, misspecifications of model of molecular evolution, and hard polytomy.
Materials and Methods
An overview of all data set and phylogenetic analyses can be found in Supplementary Figure S1 available on Dryad at http://dx.doi.org/10.5061/dryad.ns1rn8pq4.
Taxon Sampling, Transcriptome Sequencing
We sampled 92 ingroup species (88 transcriptomes and four genomes) representing 53 genera (out of ca. 181) of all 13 currently recognized subfamilies and 16 out of 17 tribes of Amaranthaceae s.l. [sensu (Kadereit et al. 2003; Kadereit et al. 2017)]. In addition, 13 outgroups across the Caryophyllales were included (10 transcriptomes and 3 genomes; Supplementary Table S1 available on Dryad). We generated 17 new transcriptomes for this study on an Illumina HiSeq2500 platform (Supplementary Table S2 available on Dryad). Library preparation was carried out using either poly-A enrichment or ribosomal RNA depletion. See Supplementary Methods available on Dryad for details on tissue collection, RNA isolation, library preparation, and quality control.
Transcriptome Data Processing, Assembly, Homology and Orthology Inference
Read processing, assembly, translation, and homology and orthology inference followed the ``phylogenomic data set construction'' pipeline (Yang and Smith 2014) with multiple updates. We briefly describe our procedure below, with details in the Supplementary Methods and updated scripts in https://bitbucket.org/yanglab/phylogenomic_dataset_construction/
We processed raw reads for all 88 transcriptome data sets (except Bienertia sinuspersici) used in this study (Supplementary Table S1 available on Dryad). Reads were corrected for errors, trimmed for sequencing adapters and low-quality bases, and filtered for organellar reads. De novo assembly of processed nuclear reads was carried out with Trinity v 2.5.1 (Grabherr et al. 2011) with default settings, but without in silico normalization. Low-quality and chimeric transcripts were removed. Filtered transcripts were clustered into putative genes with Corset v 1.07 (Davidson and Oshlack 2014) and only the longest transcript of each putative gene was retained (Chen et al. 2019). Lastly, transcripts were translated, and identical coding sequences (CDS) were removed. Homology inference was carried out on CDS using reciprocal BLASTN, followed by orthology inference using the ``monophyletic outgroup'' approach (Yang and Smith 2014), keeping only ortholog groups with at least 25 ingroup taxa.
Assessment of Recombination
Coalescent species tree methods assume that there is free recombination between loci and no recombination within loci. To determine the presence of recombination in our data set, we used the pairwise homoplasy index test 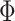 for recombination, as implemented in PhiPack (Bruen et al. 2006). We tested recombination on the final set of ortholog alignments (with a minimum of 25 taxa) with the default sliding window size of 100 bp. Alignments that showed a strong signal of recombination with
for recombination, as implemented in PhiPack (Bruen et al. 2006). We tested recombination on the final set of ortholog alignments (with a minimum of 25 taxa) with the default sliding window size of 100 bp. Alignments that showed a strong signal of recombination with 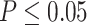 were removed from all subsequent phylogenetic analyses
were removed from all subsequent phylogenetic analyses
Nuclear Phylogenetic Analysis
We used both concatenation and coalescent-based methods to reconstruct the phylogeny of Amaranthaceae s.l. Sequences from final orthologs were aligned using MAFFT v 7.307 (Katoh and Standley 2013) with settings ``---genafpair --maxiterate 1000''. Columns with more than 70% missing data were trimmed with Phyx (Brown et al. 2017), and alignments with at least 1000 characters and 99 out of 105 taxa were retained. We first estimated a maximum likelihood (ML) tree of the concatenated matrix with RAxML v 8.2.11 (Stamatakis 2014) using a partition-by-gene scheme with GTRCAT model for each partition and clade support assessed with 200 rapid bootstrap (BS) replicates. To estimate a coalescent-based species tree, first, we inferred individual ML gene trees using RAxML with a GTRCAT model and 200 BS replicates to assess clade support. Gene trees were then used to infer a species tree with ASTRAL-III v5.6.3 (Zhang et al. 2018) using local posterior probabilities (LPP; Sayyari and Mirarab 2016) to assess clade support.
Detecting and Visualizing Nuclear Gene Tree Discordance
To explore discordance among gene trees, we first calculated the internode certainty all (ICA) value to quantify the degree of conflict on each node of a target tree (i.e., species tree) given individual gene trees (Salichos et al. 2014). In addition, we calculated the number of conflicting and concordant bipartitions on each node of the species trees. Both the ICA scores and conflicting/concordant bipartitions were calculated with Phyparts (Smith et al. 2015), mapping against the inferred ASTRAL species trees, using individual gene trees with BS support of at least 50% for the corresponding node. Additionally, in order to distinguish strong conflict from weakly supported branches, we carried out Quartet Sampling (QS; Pease et al. 2018) with 100 replicates. QS subsamples quartets from the input tree and alignment and assesses the confidence, consistency, and informativeness of each internal branch by the relative frequency of the three possible quartet topologies (Pease et al. 2018). Both ICA and QS scores provide an alternative branch support that reflects underlying gene tree conflict and that is not affected by anomalous high levels of bootstrap support common in phylogenomic data (Kumar et al. 2012).
To further visualize conflict, we built a cloudogram using DensiTree v2.2.6 (Bouckaert and Heled 2014). As DensiTree cannot accommodate missing taxa among gene trees, we reduced the final ortholog alignments to include 41 species (38 ingroup and 3 outgroups) in order to include as many orthologs as possible while representing all main clades of Amaranthaceae s.l. (see Results). Individual gene trees were inferred as previously described. Trees were time-calibrated with TreePL v1.0 (Smith and O'Meara 2012) by fixing the crown age of Amaranthaceae s.l. to 66--72.1 based on a pollen record of Polyporina cribraria from the late Cretaceous (Maastrichtian; Srivastava 1969), and the root for the reduced 41-species data set (most common recent ancestor of Achatocarpaceae and Aizoaceae) was set to 95 Ma based on the time-calibrated plastid phylogeny of Caryophyllales from Yao et al. (2019).
Plastid Assembly and Phylogenetic Analysis
Although DNase treatment was carried out to remove genomic DNA, due to their high copy number, plastid sequences are often carried over in RNA-seq libraries. In addition, as young leaf tissue was used for RNA-seq, the presence of RNA from plastid genes is expected to be represented. To investigate phylogenetic signal from plastid sequences, de novo assemblies were carried out with the Fast-Plast v.1.2.6 pipeline (https://github.com/mrmckain/Fast-Plast) using the filtered organelle reads. Contigs produced by Spades v 3.9.0 (Bankevich et al. 2012) were mapped to the closest available reference plastomes (Supplementary Table S3 available on Dryad), one copy of the Inverted Repeat was removed, and the remaining contigs manually edited in Geneious v.11.1.5 (Kearse et al. 2012) to produce the final oriented contigs.
Contigs were aligned with MAFFT with the setting ``--auto''. Two samples (Dysphania schraderiana and Spinacia turkestanica) were removed due to low sequence occupancy. Using the annotations of the reference genomes (Supplementary Table S3 available on Dryad), the coding regions of 78 genes were extracted and each gene alignment was visually inspected in Geneious to check for potential misassemblies. From each gene alignment, taxa with short sequences (i.e., 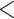 50% of the aligned length) were removed and the remaining sequences realigned with MAFFT. The genes rpl32 and ycf2 were excluded from downstream analyses due to low taxon occupancy (Supplementary Table S4 available on Dryad). For each individual gene, we performed extended model selection (Kalyaanamoorthy et al. 2017) followed by ML gene tree inference and 1000 ultrafast bootstrap replicates for branch support (Hoang and Chernomor 2018) in IQ-TREE v.1.6.1 (Nguyen et al. 2015). For the concatenated matrix, we searched for the best partition scheme (Lanfear et al. 2012) followed by ML gene tree inference and 1000 ultrafast bootstrap replicates for branch support in IQ-Tree. Additionally, we evaluated branch support with QS using 1000 replicates and gene tree discordance with PhyParts. Lastly, to identify the origin of the plastid reads (i.e., genomic or RNA), we predicted RNA editing from CDS alignments using PREP (Mower 2009) with the alignment mode (PREP-aln), and a cutoff value of 0.8.
50% of the aligned length) were removed and the remaining sequences realigned with MAFFT. The genes rpl32 and ycf2 were excluded from downstream analyses due to low taxon occupancy (Supplementary Table S4 available on Dryad). For each individual gene, we performed extended model selection (Kalyaanamoorthy et al. 2017) followed by ML gene tree inference and 1000 ultrafast bootstrap replicates for branch support (Hoang and Chernomor 2018) in IQ-TREE v.1.6.1 (Nguyen et al. 2015). For the concatenated matrix, we searched for the best partition scheme (Lanfear et al. 2012) followed by ML gene tree inference and 1000 ultrafast bootstrap replicates for branch support in IQ-Tree. Additionally, we evaluated branch support with QS using 1000 replicates and gene tree discordance with PhyParts. Lastly, to identify the origin of the plastid reads (i.e., genomic or RNA), we predicted RNA editing from CDS alignments using PREP (Mower 2009) with the alignment mode (PREP-aln), and a cutoff value of 0.8.
Species Network Analysis Using a Reduced 11-Taxon Data Set
We inferred species networks that model ILS and gene flow using a maximum pseudo-likelihood approach (Yu and Nakhleh 2015). Species network searches were carried out with PhyloNet v.3.6.9 (Than et al. 2008) with the command ``InferNetwork_MPL'' and using the individual gene trees as input. Due to computational restrictions, and given our main focus to identify potential reticulating events among major clades of Amaranthaceae s.l., we reduced our taxon sampling to one outgroup and 10 ingroup taxa to include two representative species from each of the five well-supported major lineages in Amaranthaceae s.l. (see Results). We filtered the final 105-taxon ortholog set to include genes that have all 11 taxa [referred herein as 11-taxon(net) data set; Supplementary Fig.S1 available on Dryad]. After alignment and trimming we kept genes with a minimum of 1000 aligned base pairs and individual ML gene trees were inferred using RAxML with a GTRGAMMA model and 200 bootstrap replicates. We carried out five network searches by allowing one to five reticulation events and 10 runs for each search. To estimate the optimum number of reticulations, we optimized the branch lengths and inheritance probabilities and computed the likelihood of the best scored network from each of the five maximum reticulation events searches. Network likelihoods were estimated given the individual gene trees using the command ``CalGTProb'' in PhyloNet (Yu et al. 2012). Then, we performed model selection using the bias-corrected Akaike information criterion (AICc; Sugiura 1978), and the Bayesian information criterion (BIC; Schwarz 1978). The number of parameters was set to the number of branch lengths being estimated plus the number of hybridization probabilities being estimated. The number of gene trees used to estimate the likelihood was used to correct for finite sample size. To compare network models to bifurcating trees, we also estimated bifurcating concatenated ML and coalescent-based species trees and a plastid tree as previously described with the reduced 11-species taxon sampling.
Hypothesis Testing and Detecting Introgression Using Four-Taxon Data Sets
Given the signal of multiple clades potentially involved in hybridization events detected by PhyloNet (see Results), we next conducted quartet analyses to explore a single event at a time. First, we further reduced the 11-taxon(net) data set to six taxa that included one outgroup genome (Mesembryanthemum crystallinum) and one ingroup from each of the five major ingroup clades: Amaranthus hypochondriacus (genome), Beta vulgaris (genome), Chenopodium quinoa (genome), Caroxylon vermiculatum (transcriptome), and Polycnemum majus (transcriptome) to represent Amaranthaceae s.s., Betoideae, ``Chenopods I'', ``Chenopods II'', and Polycnemoideae, respectively. We carried out a total of ten quartet analyses using all 10 four-taxon combinations that included three out of five ingroup species and one outgroup. We filtered the final set of 105-taxon orthologs for genes with all four taxa for each combination and inferred individual gene trees as described before. For each quartet, we carried out the following analyses. We first estimated a species tree with ASTRAL and explored gene tree conflict with PhyParts. We then explored individual gene tree resolution by calculating the Tree Certainty (TC) score (Salichos et al. 2014) in RAxML using the majority rule consensus tree across the 200 bootstrap replicates. Next, we explored potential correlation between TC score and alignment length, GC content and alignment gap proportion using a linear regression model in R v.3.6.1 (R Core Team 2019). Lastly, we tested for the fit of gene trees to the three possible rooted quartet topologies for each gene using the approximately unbiased (AU) tests (Shimodaira 2002). We carried out 10 constraint searches for each of three topologies in RAxML with the GTRGAMMA model, then calculated site-wise log-likelihood scores for the three constraint topologies in RAxML using GTRGAMMA and carried out the AU test using Consel v.1.20 (Shimodaira and Hasegawa 2001). In order to detect possible introgression among species of each quartet, first we estimated a species network with PhyloNet using a full ML approach (Yu et al. 2014) with 100 runs per search while optimizing the likelihood of the branch lengths and inheritance probabilities for every proposed species network. Furthermore, we also carried out the ABBA/BABA test to detect introgression (Green et al. 2010; Durand et al. 2011) in each of four-taxon species trees. We calculated the 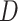 -statistic and associated
-statistic and associated 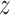 score for the null hypothesis of no introgression (
score for the null hypothesis of no introgression (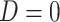 ) following each quartet ASTRAL species tree for taxon order assignment using 100 jackknife replicates and a block size of 10,000 bp with evobiR v1.2 (Blackmon and Adams) in R.
) following each quartet ASTRAL species tree for taxon order assignment using 100 jackknife replicates and a block size of 10,000 bp with evobiR v1.2 (Blackmon and Adams) in R.
Additionally, to detect any non-random genomic block of particular quartet topology (Fontaine et al. 2015), we mapped the physical location of genes supporting each alternative quartet topology onto the Beta vulgaris reference genome using a synteny approach (see Supplementary Information available on Dryad for details).
Assessment of Substitutional Saturation, Codon Usage Bias, Compositional Heterogeneity, and Model of Sequence Evolution Misspecification
Analyses were carried out in a 11-taxon data set [referred herein as 11-taxon(tree); Supplementary Fig.S1 available on Dryad] that included the same taxa used for species network analyses but was processed differently to account for codon structure (see Supplementary Methods available on Dryad for details). Saturation was evaluated by plotting the uncorrected genetic distances of the concatenated alignment against the inferred distances (see Supplementary Methods available on Dryad for details). To determine the effect of saturation in the phylogenetic inferences we estimated individual ML gene trees using an unpartitioned alignment, a partition by first and second codon positions, and the third codon positions, and by removing all third codon positions. All tree searches were carried out in RAxML with a GTRGAMMA model and 200 bootstrap replicates. We then estimated a coalescent-based species tree and explored gene tree discordance with PhyParts.
Codon usage bias was evaluated using a correspondence analysis of the Relative Synonymous Codon Usage (RSCU; see Supplementary Methods available on Dryad for details). To determine the effect of codon usage bias in the phylogenetic inferences, we estimated individual gene trees using codon-degenerated alignments (see Supplementary Methods available on Dryad for details). Gene tree inference and discordance analyses were carried out on the same three data schemes as previously described.
Among-lineage compositional heterogeneity was evaluated on individual genes using a compositional homogeneity test (Supplementary Methods available on Dryad for details). To assess if compositional heterogeneity had an effect in species tree inference and gene tree discordance, gene trees that showed the signal of compositional heterogeneity were removed from saturation and codon usage analyses and the species tree and discordance analyses were rerun.
To explore the effect of sequence evolution model misspecification, we reanalyzed the data sets from the saturation and codon usage analyses using inferred gene trees that accounted for model selection. Additionally, we also explored saturation and model misspecification in phylogenetic trees from amino acid alignments (see Supplementary Methods available on Dryad for details).
Polytomy Test
To test if the gene tree discordance among the main clades of Amaranthaceae s.l. could be explained by polytomies instead of bifurcating nodes, we carried out the quartet-based polytomy test by Sayyari and Mirarab (2018) as implemented in ASTRAL. We performed the polytomy test using the gene trees inferred from the saturation and codon usage analyses [11-taxon(tree) data set]. Because this test can be sensitive to gene tree error (Syyari and Mirarab 2018), we performed a second test using gene trees where branches with less than 75% of bootstrap support were collapsed.
Coalescent Simulations
To investigate if gene tree discordance can be explained by ILS alone, we carried out coalescent simulations similar to Cloutier et al. (2019). An ultrametric species tree with branch lengths in mutational units (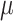 T) was estimated by constraining an ML tree search of the 11-taxon(net) concatenated alignment to the ASTRAL species tree topology with a GTR
T) was estimated by constraining an ML tree search of the 11-taxon(net) concatenated alignment to the ASTRAL species tree topology with a GTR 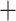 GAMMA model while enforcing a strict molecular clock in PAUP v4.0a (build 165; Swofford 2002). The mutational branch lengths from the constrained tree and branch lengths in coalescent units (
GAMMA model while enforcing a strict molecular clock in PAUP v4.0a (build 165; Swofford 2002). The mutational branch lengths from the constrained tree and branch lengths in coalescent units (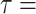 T/4N
T/4N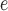 ) from the ASTRAL species trees were used to estimate the population size parameter theta (
) from the ASTRAL species trees were used to estimate the population size parameter theta (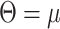 T/
T/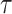 ; Degnan and Rosenberg 2009) for internal branches. Terminal branches were set with a population size parameter theta of one. We used the R package Phybase v. 1.4 (Liu and Yu 2010) that uses the formula from Rannala and Yang (2003) to simulate 10,000 gene trees using the constraint tree and the estimated theta values. Then we calculated the distribution of Robinson and Foulds (1981) tree-to-tree distances between the species tree and each gene tree using the R package Phangorn v2.5.3 (Schliep 2011), and compared this with the distribution of tree-to-tree distances between the species tree and the simulated gene tree. We ran simulations using the species tree and associated gene tree distribution from the original no partition 11-taxon(net).
; Degnan and Rosenberg 2009) for internal branches. Terminal branches were set with a population size parameter theta of one. We used the R package Phybase v. 1.4 (Liu and Yu 2010) that uses the formula from Rannala and Yang (2003) to simulate 10,000 gene trees using the constraint tree and the estimated theta values. Then we calculated the distribution of Robinson and Foulds (1981) tree-to-tree distances between the species tree and each gene tree using the R package Phangorn v2.5.3 (Schliep 2011), and compared this with the distribution of tree-to-tree distances between the species tree and the simulated gene tree. We ran simulations using the species tree and associated gene tree distribution from the original no partition 11-taxon(net).
Test of the Anomaly Zone
The anomaly zone occurs where a set of short internal branches in the species tree produces gene trees that differ from the species tree more frequently than those that are concordant [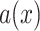 ; as defined in equation 4 of Degnan and Rosenberg (2006)]. To explore if gene tree discordance observed in Amaranthaceae s.l. is a product of the anomaly zone, we estimated the boundaries of the anomaly zone [
; as defined in equation 4 of Degnan and Rosenberg (2006)]. To explore if gene tree discordance observed in Amaranthaceae s.l. is a product of the anomaly zone, we estimated the boundaries of the anomaly zone [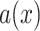 ; as defined in equation 4 of Degnan and Rosenberg (2006)] for the internal nodes of the species tree. Here,
; as defined in equation 4 of Degnan and Rosenberg (2006)] for the internal nodes of the species tree. Here, 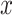 is the branch length in coalescent units in the species tree that has a descendant internal branch. If the length of the descendant internal branch (
is the branch length in coalescent units in the species tree that has a descendant internal branch. If the length of the descendant internal branch (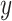 ) is smaller than
) is smaller than 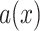 , then the internode pair is in the anomaly zone and is likely to produce anomalous gene trees (AGTs). We carried out the calculation of
, then the internode pair is in the anomaly zone and is likely to produce anomalous gene trees (AGTs). We carried out the calculation of 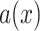 following Linkem et al. (2016) in the same 11-taxon(tree) ASTRAL species tree used for coalescent simulations. Additionally, to establish the frequency of gene trees that were concordant with the estimated species trees, we quantified the frequency of all 105 possible rooted gene trees with Amaranthaceae s.l. being monophyletic.
following Linkem et al. (2016) in the same 11-taxon(tree) ASTRAL species tree used for coalescent simulations. Additionally, to establish the frequency of gene trees that were concordant with the estimated species trees, we quantified the frequency of all 105 possible rooted gene trees with Amaranthaceae s.l. being monophyletic.
Results
Transcriptome Sequencing, Assembly, Translation, and Quality Control
Raw reads for the 17 newly generated transcriptomes are available from the NCBI Sequence Read Archive (BioProject: PRJNA640363; Supplementary Table S2 available on Dryad). The number of raw read pairs ranged from 17 to 27 million. For the 16 samples processed using RiboZero, organelle reads accounted for 15--52% of read pairs (Supplementary Table S2 available on Dryad). For Tidestromia oblongifolia that poly-A enrichment was carried out in library prep with  5% of raw reads were from organelle (Supplementary Table S2 available on Dryad). The final number of orthologs was 13,024 with a mean of 9813 orthologs per species (Supplementary Table S1 available on Dryad). Of those, 82 orthologs had a strong signal of recombination (
5% of raw reads were from organelle (Supplementary Table S2 available on Dryad). The final number of orthologs was 13,024 with a mean of 9813 orthologs per species (Supplementary Table S1 available on Dryad). Of those, 82 orthologs had a strong signal of recombination (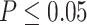 ) and were removed from downstream analyses.
) and were removed from downstream analyses.
Analysis of the Nuclear Data Set of Amaranthaceae s.l.
The final set of nuclear orthologous genes included 936 genes with at least 99 out of 105 taxa and 1000 bp in aligned length after removal of low occupancy columns (the 105-taxon data set). The concatenated matrix consisted of 1,712,054 columns with a gene and character occupancy of 96% and 82%, respectively. The species tree from ASTRAL and the concatenated ML tree from RAxML recovered the exact same topology with most clades having maximal support [i.e., bootstrap percentage (BS) 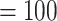 , LPP
, LPP 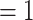 ; Fig. 2; Supplementary Figs S2 and S3 available on Dryad]. Both analyses recovered Chenopodiaceae as monophyletic with the relationships among major clades concordant with the cpDNA analysis from Kadereit et al. (2017, Fig. 1d). Betoideae was placed as sister of Chenopodiaceae, while Polycnemoideae was strongly supported as sister (BS
; Fig. 2; Supplementary Figs S2 and S3 available on Dryad]. Both analyses recovered Chenopodiaceae as monophyletic with the relationships among major clades concordant with the cpDNA analysis from Kadereit et al. (2017, Fig. 1d). Betoideae was placed as sister of Chenopodiaceae, while Polycnemoideae was strongly supported as sister (BS 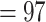 , LPP
, LPP 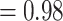 ) to the clade composed of Chenopodiaceae and Betoideae. Amaranthaceae s.s. had an overall topology concordant to Kadereit et al. (2017), with the exception of Iresine, which was recovered among the Aervoids (Fig. 2; Supplementary Figs S2 and S3 available on Dryad).
) to the clade composed of Chenopodiaceae and Betoideae. Amaranthaceae s.s. had an overall topology concordant to Kadereit et al. (2017), with the exception of Iresine, which was recovered among the Aervoids (Fig. 2; Supplementary Figs S2 and S3 available on Dryad).
Figure 2.

Maximum likelihood phylogeny of Amaranthaceae s.l. inferred from RAxML analysis of the concatenated 936-nuclear gene supermatrix, which had the same topology as recovered from ASTRAL. All nodes have maximal support (bootstrap 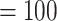 /ASTRAL local posterior probability
/ASTRAL local posterior probability 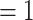 ) unless noted. Pie charts present the proportion of gene trees that support that clade (concordant), support the main alternative bifurcation, support the remaining alternatives, and the proportion (conflict or support) that have
) unless noted. Pie charts present the proportion of gene trees that support that clade (concordant), support the main alternative bifurcation, support the remaining alternatives, and the proportion (conflict or support) that have 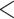 50% bootstrap support (uninformative). Only pie charts for major clades are shown (see Supplementary Fig.S2 available on Dryad for all node pie charts). Branch lengths are in number of substitutions per site. The inset (top left) shows the DensiTree cloudogram inferred from 1242 nuclear genes for the reduced 41-taxon data set.
50% bootstrap support (uninformative). Only pie charts for major clades are shown (see Supplementary Fig.S2 available on Dryad for all node pie charts). Branch lengths are in number of substitutions per site. The inset (top left) shows the DensiTree cloudogram inferred from 1242 nuclear genes for the reduced 41-taxon data set.
The conflict analyses confirmed the monophyly of Amaranthaceae s.l. with 922 out of 930 informative gene trees being concordant (ICA 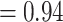 ) and having full QS support (1/--/1; i.e., all sampled quartets supported that branch). Similarly, the monophyly of Amaranthaceae s.s. was highly supported by 755 of 809 informative gene trees (ICA
) and having full QS support (1/--/1; i.e., all sampled quartets supported that branch). Similarly, the monophyly of Amaranthaceae s.s. was highly supported by 755 of 809 informative gene trees (ICA 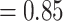 ) and the QS scores (0.92/0/1). However, the backbone of the family was characterized by high levels of gene tree discordance (Fig. 2; Supplementary Figs S2 and S3 available on Dryad). The monophyly of Chenopodiaceae was supported only by 231 out of 632 informative gene trees (ICA
) and the QS scores (0.92/0/1). However, the backbone of the family was characterized by high levels of gene tree discordance (Fig. 2; Supplementary Figs S2 and S3 available on Dryad). The monophyly of Chenopodiaceae was supported only by 231 out of 632 informative gene trees (ICA 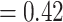 ) and the QS score (0.25/0.19/0.99) suggested weak quartet support with a skewed frequency for an alternative placement of two well-defined clades within Chenopodiaceae, herein referred to as ``Chenopods I'' and ``Chenopods II'' (Fig. 2; Supplementary Figs S2 and S3 available on Dryad). ``Chenopods I'' and ``Chenopods II'' were each supported by the majority of gene trees, 870 (ICA
) and the QS score (0.25/0.19/0.99) suggested weak quartet support with a skewed frequency for an alternative placement of two well-defined clades within Chenopodiaceae, herein referred to as ``Chenopods I'' and ``Chenopods II'' (Fig. 2; Supplementary Figs S2 and S3 available on Dryad). ``Chenopods I'' and ``Chenopods II'' were each supported by the majority of gene trees, 870 (ICA 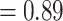 ) and 916 (ICA
) and 916 (ICA 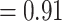 ), respectively and full QS support. Similarly, high levels of conflict among informative gene trees were detected in the placement of Betoideae (126 out of 579 informative genes being concordant, ICA
), respectively and full QS support. Similarly, high levels of conflict among informative gene trees were detected in the placement of Betoideae (126 out of 579 informative genes being concordant, ICA 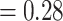 ; QS score 0.31/0.57/1) and Polycnemoideae (116/511; ICA
; QS score 0.31/0.57/1) and Polycnemoideae (116/511; ICA 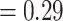 ;0.3/0.81/0.99). The DensiTree cloudogram also showed significant conflict along the backbone of Amaranthaceae s.l. (Fig. 2).
;0.3/0.81/0.99). The DensiTree cloudogram also showed significant conflict along the backbone of Amaranthaceae s.l. (Fig. 2).
Together, analysis of nuclear genes recovered five well-supported clades in Amaranthaceae s.l.: Amaranthaceae s.s., Betoideae, ``Chenopods I'', ``Chenopods II'', and Polycnemoideae. However, relationships among these five clades showed a high level of conflict among genes [ICA scores and gene counts (pie charts)] and among subsampled quartets (QS scores), despite having high support from both BS and LPP scores.
Plastid Phylogenetic Analysis of Amaranthaceae s.l.
RNA editing prediction analysis revealed editing sites only on CDS sequences of reference plastomes (Supplementary Table S3 available on Dryad), suggesting that cpDNA reads in RNA-seq libraries come from RNA rather than DNA leftover from incomplete DNase digestion during sample processing (See Discussion for details in plastid assembly from RNA-seq data).
The final alignment from 76 genes included 103 taxa and 55,517 bp in aligned length. The ML tree recovered the same five main clades within Amaranthaceae s.l. with maximal support (BS 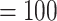 ; Supplementary Figs S4--S6 available on Dryad). Within each main clade, relationships were fully congruent with Kadereit et al. (2017) and mostly congruent with our nuclear analyses. However, the relationship among the five main clades differed from the nuclear tree. Here, the sister relationships between Betoideae and ``Chenopods I'', and between Amaranthaceae s.s. and Polycnemoideae were both supported by BS
; Supplementary Figs S4--S6 available on Dryad). Within each main clade, relationships were fully congruent with Kadereit et al. (2017) and mostly congruent with our nuclear analyses. However, the relationship among the five main clades differed from the nuclear tree. Here, the sister relationships between Betoideae and ``Chenopods I'', and between Amaranthaceae s.s. and Polycnemoideae were both supported by BS 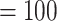 . The sister relationship between these two larger clades was moderately supported (BS
. The sister relationship between these two larger clades was moderately supported (BS 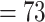 ), leaving ``Chenopods II'' as sister to the rest of Amaranthaceae s.l.
), leaving ``Chenopods II'' as sister to the rest of Amaranthaceae s.l.
Conflict analysis confirmed the monophyly of Amaranthaceae s.l. with 51 out of 69 informative gene trees supporting this clade (ICA 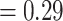 ) and full QS support (1/--/1). On the other hand, and similar to the nuclear phylogeny, significant gene tree discordance was detected among plastid genes regarding placement of the five major clades (Supplementary Figs S4--S6 available on Dryad). The sister relationship of Betoideae and ``Chenopods I'' was supported by only 20 gene trees (ICA
) and full QS support (1/--/1). On the other hand, and similar to the nuclear phylogeny, significant gene tree discordance was detected among plastid genes regarding placement of the five major clades (Supplementary Figs S4--S6 available on Dryad). The sister relationship of Betoideae and ``Chenopods I'' was supported by only 20 gene trees (ICA 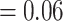 ), but it had a strong support from QS (0.84/0.88/0/94). The relationship between Amaranthaceae s.s. and Polycnemoideae was supported by only 15 gene trees (ICA
), but it had a strong support from QS (0.84/0.88/0/94). The relationship between Amaranthaceae s.s. and Polycnemoideae was supported by only 15 gene trees (ICA 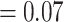 ), while QS showed weak support (0.41/0.21.0.78) with signals of a supported secondary evolutionary history. The clade uniting Betoideae, ``Chenopods I'', Amaranthaceae s.s., and Polycnemoideae was supported by only four gene trees, with counter-support from both QS (
), while QS showed weak support (0.41/0.21.0.78) with signals of a supported secondary evolutionary history. The clade uniting Betoideae, ``Chenopods I'', Amaranthaceae s.s., and Polycnemoideae was supported by only four gene trees, with counter-support from both QS ( 0.29/0.42/0.75) and ICA (
0.29/0.42/0.75) and ICA ( 0.03), suggesting that most gene trees and sampled quartets supported alternative topologies.
0.03), suggesting that most gene trees and sampled quartets supported alternative topologies.
Species Network Analysis of Amaranthaceae s.l.
The reduced 11-taxon(net) data set included 4138 orthologous gene alignments with no missing taxon and a minimum of 1000 bp (aligned length after removal of low occupancy columns). The 11-taxon(net) ASTRAL species tree was congruent with the 105-taxon tree, while both the nuclear and plastid ML trees from concatenated supermatrices had different topologies than their corresponding 105-taxon trees (Fig. 3). Model selection indicated that any species network was a better model than the best bifurcating nuclear or plastid trees (ASTRAL; AICc 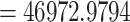 ; Supplementary Table S5 available on Dryad). PhyloNet identified up to five hybridization events among the clades of Amaranthaceae s.l. (Fig. 3), with the best model having five hybridization events involving all five clades (AICc
; Supplementary Table S5 available on Dryad). PhyloNet identified up to five hybridization events among the clades of Amaranthaceae s.l. (Fig. 3), with the best model having five hybridization events involving all five clades (AICc 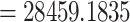 ; Supplementary Table S5 available on Dryad). The best species network did not support the hypothesis of the hybrid origin of Betoideae or Polycnemoideae. Moreover, the best species network showed a complex reticulate pattern that involved mainly ``Chenopods I'' and ``Chenopods II'' (Fig. 3), but none of these reticulations events were supported by
; Supplementary Table S5 available on Dryad). The best species network did not support the hypothesis of the hybrid origin of Betoideae or Polycnemoideae. Moreover, the best species network showed a complex reticulate pattern that involved mainly ``Chenopods I'' and ``Chenopods II'' (Fig. 3), but none of these reticulations events were supported by 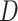 -Statistic or species network results from the four-taxon analyses (see below).
-Statistic or species network results from the four-taxon analyses (see below).
Figure 3.

Species trees and species networks of the reduced 11-taxon(net) data set of Amaranthaceae s.l. Nuclear concatenated phylogeny inferred from 4138-nuclear gene supermatrix with RAxML. ASTRAL species tree inferred using 4138 nuclear genes. cpDNA concatenated tree inferred from 76-plastid gene supermatrix with IQ-tree. Species network inferred from PhyloNet pseudolikelihood analyses with 1 to 5 maximum number of reticulations. Dashed (red) and solid (blue) curved branches indicate the minor and major edges, respectively, of hybrid nodes. Numbers next to curved branches indicate inheritance probabilities for each hybrid node.
Four-Taxon Analyses
To test for hybridization events one at a time, we further reduced the 11-taxon(net) data set to 10 four-taxon combinations that each included one outgroup and one representative each from three out of the five major ingroup clades. Between 7756 and 8793 genes were used for each quartet analysis (Supplementary Table S6 available on Dryad) and each quartet topology can be found in Figure 4. Only five out of the ten bifurcating quartet species trees (H0 and more frequent gene tree) were compatible with the nuclear species tree inferred from the complete 105-taxon data set. The remaining quartets corresponded to the second most frequent gene tree topology in the 105-taxon nuclear tree, except for the quartet of Betoideae, ``Chenopods II'' and Polycnemoideae (PBC2, which correspond to the least frequent gene tree).
Figure 4.

Gene counts from approximate unbiased (AU) topology test of the 10 quartets from the five main clades of Amaranthaceae s.l. AU tests were carried out between the three possible topologies of each quartet. H0 represents the ASTRAL species tree of each quartet. ``Equivocal'' indicates gene trees that fail to reject all three alternative topologies for a quartet with 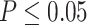 . Gene counts for each of the three alternative topologies represent gene trees supporting unequivocally one topology by rejecting the other two alternatives with
. Gene counts for each of the three alternative topologies represent gene trees supporting unequivocally one topology by rejecting the other two alternatives with 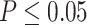 . Insets represent gene counts only for unequivocal topology support. Double arrowed lines in each H0 quartet represent the direction of introgression from the ABBA/BABA test. Each quartet is named following the species tree topology, where the first two species are sister to each other. A
. Insets represent gene counts only for unequivocal topology support. Double arrowed lines in each H0 quartet represent the direction of introgression from the ABBA/BABA test. Each quartet is named following the species tree topology, where the first two species are sister to each other. A  Amaranthaceae s.s. (represented by Amaranthus hypochondriacus), B
Amaranthaceae s.s. (represented by Amaranthus hypochondriacus), B  Betoideae (Beta vulgaris), C1
Betoideae (Beta vulgaris), C1  Chenopods I (Chenopodium quinoa), C2
Chenopods I (Chenopodium quinoa), C2  Chenopods II (Caroxylon vermiculatum), P
Chenopods II (Caroxylon vermiculatum), P  Polycnemoideae (Polycnemum majus). All quartets are rooted with Mesembryanthemum crystallinum.
Polycnemoideae (Polycnemum majus). All quartets are rooted with Mesembryanthemum crystallinum.
In each of the 10 quartets, the ASTRAL species tree topology (H0) was the most frequent among individual gene trees (raw counts) but only accounted for 35--41% of gene trees, with the other two alternative topologies having balanced to slightly skewed frequencies (Supplementary Fig.S7a and Table S7 available on Dryad). Gene counts based on the raw likelihood scores from the constraint analyses showed similar patterns (Supplementary Fig.S7b and Table S7 available on Dryad). When filtered by significant likelihood support (i.e., 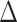 AICc
AICc 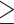 ~2), the number of trees supporting each of the three possible topologies dropped between 34% and 45%, but the species tree remained the most frequent topology for all quartets (Supplementary Fig.S7b and Table S7 available on Dryad). The AU topology tests failed to reject (
~2), the number of trees supporting each of the three possible topologies dropped between 34% and 45%, but the species tree remained the most frequent topology for all quartets (Supplementary Fig.S7b and Table S7 available on Dryad). The AU topology tests failed to reject (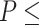 ~0.05) approximately 85% of the gene trees for any of the three possible quartet topologies and rejected all but a single topology in only 3--4.5% of cases. Among the unequivocally selected gene trees, the frequencies among the three alternative topologies were similar to ones based on raw likelihood scores (Supplementary Fig.S7 and Table S7 available on Dryad). Therefore, topology tests showed that most genes were uninformative for resolving the relationships among the major groups of Amaranthaceae s.l.
~0.05) approximately 85% of the gene trees for any of the three possible quartet topologies and rejected all but a single topology in only 3--4.5% of cases. Among the unequivocally selected gene trees, the frequencies among the three alternative topologies were similar to ones based on raw likelihood scores (Supplementary Fig.S7 and Table S7 available on Dryad). Therefore, topology tests showed that most genes were uninformative for resolving the relationships among the major groups of Amaranthaceae s.l.
Across all 10 quartets, we found that most genes had very low TC scores (for any single node the maximum TC value is 1; Supplementary Fig.S8 available on Dryad), showing that individual gene trees also had high levels of conflict among bootstrap replicates, which also indicated uninformative genes and was concordant with the AU topology test results. We were unable to detect any significant correlation between TC scores and alignment length, GC content or alignment gap fraction (Supplementary Table S8 available on Dryad), suggesting that filtering genes by any of these criteria was unlikely to increase the information content of the data set.
Species network analyses followed by model selection using each of the four-taxon data sets showed that in seven out of the ten total quartets, the network with one hybridization event was a better model than any bifurcating tree topology. However, each of the best three networks from PhyloNet had very close likelihood scores and no significant 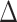 AICc among them (Supplementary Table S6 and Fig S9 available on Dryad). For the remaining three quartets, the species trees (H0) was the best model.
AICc among them (Supplementary Table S6 and Fig S9 available on Dryad). For the remaining three quartets, the species trees (H0) was the best model.
The ABBA/BABA test results showed a significant signal of introgression within each of the ten quartets (Supplementary Table S9 available on Dryad; Fig 4). The possible introgression was detected between six out of the ten possible pairs of taxa. Potential introgression between Betoideae and Amaranthaceae s.s., ``Chenopods I'' or ``Chenopods II'', and between ``Chenopods I'' and Polycnemoideae was not detected.
To further evaluate whether alternative quartets were randomly distributed across the genome, we mapped topologies from the quartet of Betoideae, ``Chenopods II'', and Amaranthaceae s.s. (BC1A) onto the reference genome of Beta vulgaris. We used the BC1A quartet as an example as all four species in this quartet have reference genomes. Synteny analysis between the diploid ingroup reference genome Beta vulgaris and the diploid outgroup reference genome Mesembryanthemum crystallinum recovered 22,179 collinear genes in 516 syntenic blocks. With the collinear ortholog pair information, we found that of the 8258 orthologs of the BC1A quartet, 6941 contained syntenic orthologous genes within 383 syntenic blocks. The distribution of the BC1A quartet topologies along the chromosomes of Beta vulgaris did not reveal any spatial clustering of any particular topology along the chromosomes (Supplementary Fig.S10 available on Dryad).
Gene ontology enrichment analyses (not shown) using alternative topologies of the BC1A quartet did not recover any significant term associated with C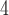 photosynthesis, drought recovery, or salt stress response.
photosynthesis, drought recovery, or salt stress response.
Assessment of Substitutional Saturation, Codon Usage Bias, Compositional Heterogeneity, Sequence Evolution Model Misspecification, and Polytomy Test
We assembled a second 11-taxon(tree) data set that included 5936 genes and a minimum of 300 bp (aligned length after removal of low occupancy columns) and no missing taxon. The saturation plots of uncorrected and predicted genetic distances showed that the first and second codon positions were unsaturated (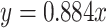 ), whereas the slope of the third codon positions (
), whereas the slope of the third codon positions (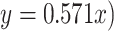 showed a signal of saturation (Supplementary Fig.S11 available on Dryad). The correspondence analyses of RSCU show that some codons are more frequently used in different species, but overall the codon usage was randomly dispersed among all species and not clustered by clade (Supplementary Fig.S12 available on Dryad). This suggests that the phylogenetic signal is unlikely to be driven by differences in codon usage bias among clades. Furthermore, only 549 (
showed a signal of saturation (Supplementary Fig.S11 available on Dryad). The correspondence analyses of RSCU show that some codons are more frequently used in different species, but overall the codon usage was randomly dispersed among all species and not clustered by clade (Supplementary Fig.S12 available on Dryad). This suggests that the phylogenetic signal is unlikely to be driven by differences in codon usage bias among clades. Furthermore, only 549 (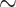 9%) genes showed a signal of compositional heterogeneity (
9%) genes showed a signal of compositional heterogeneity (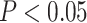 ). The topology and support (LPP
). The topology and support (LPP 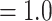 ) for all branches was the same for the ASTRAL species trees obtained from the different data schemes while accounting for saturation, codon usage, compositional heterogeneity, and model of sequence evolution, and was also congruent with the ASTRAL species tree and concatenated ML from the 105-taxon analyses (Supplementary Fig.S13 available on Dryad). In general, the proportion of gene trees supporting each bipartition remained the same in every analysis and showed high levels of conflict among the five major clades of Amaranthaceae s.l. (Supplementary Fig.S13 available on Dryad).
) for all branches was the same for the ASTRAL species trees obtained from the different data schemes while accounting for saturation, codon usage, compositional heterogeneity, and model of sequence evolution, and was also congruent with the ASTRAL species tree and concatenated ML from the 105-taxon analyses (Supplementary Fig.S13 available on Dryad). In general, the proportion of gene trees supporting each bipartition remained the same in every analysis and showed high levels of conflict among the five major clades of Amaranthaceae s.l. (Supplementary Fig.S13 available on Dryad).
The ASTRAL polytomy test resulted in the same bifurcating species tree for the 11-taxon(tree) data set and rejected the null hypothesis that any branch is a polytomy (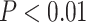 in all cases). These results were identical when using gene trees with collapsed branches.
in all cases). These results were identical when using gene trees with collapsed branches.
Coalescent Simulations and Tests of the Anomaly Zone
The distribution of tree-to-tree distances of the empirical and simulated gene trees to the species tree from the 11-taxon(tree) data set largely overlapped (Fig. 5a), suggesting that ILS alone is able to explain most of the observed gene tree heterogeneity (Maureira-Butler et al. 2008). The anomaly zone limit calculations using species trees from the 11-taxon(tree) data set detected two pairs of internodes among the five major groups in Amaranthaceae s.l. that fell into the anomaly zone (the red pair and the green pair, Fig. 5b; Supplementary Table S10 available on Dryad). Furthermore, gene tree counts showed that the species tree was not the most common gene tree topology, as defined for the anomaly zone (Degnan and Rosenberg 2006; Fig. 5c). The species tree was the fourth most common gene tree topology (119 out of 4425 gene trees), while the three most common gene tree topologies occurred 170, 136, and 127 times (Fig. 5c).
Figure 5.

Coalescent simulations and tests of the anomaly zone from the 11-taxon(tree) data set estimated from individual gene trees. a) Distribution of tree-to-tree distances between empirical gene trees and the ASTRAL species tree, compared to those from the coalescent simulation. b) ASTRAL species tree showing branch length in coalescent units. Bracket indicate the internodes that fall in the anomaly zone (see Supplementary Table S10 available on Dryad for anomaly zone limit values). c) Gene tree counts (top) of the four most common topologies (bottom). Gene trees that do not support the monophyly of any of the five major clades were ignored for the counting.
Discussion
The exploration of gene tree discordance has become a fundamental step to understand recalcitrant relationships across the Tree of Life. Recently, new tools have been developed to identify and visualize gene tree discordance (e.g., Salichos et al. 2014; Smith et al. 2015; Huang et al. 2016; Pease et al. 2018). However, downstream methods that evaluate processes generating observed patterns of gene tree discordance are still in their infancy. In this study, by combining transcriptomes and genomes, we were able to create a rich and dense data set to start to tease apart alternative hypotheses concerning the sources of conflict along the backbone phylogeny of Amaranthaceae s.l. We found that gene tree heterogeneity observed in Amaranthaceae s.l. can be explained by a combination of processes, including ILS, ancient hybridization, and uninformative genes, that might have acted simultaneously and/or cumulatively.
Gene Tree Discordance Detected Among Plastid Genes
Although both our concatenation-based plastid and nuclear phylogenies supported the same five major clades of Amaranthaceae s.l., the relationships among these clades are incongruent (Fig.2 and Supplementary Fig.S4 available on Dryad). Cytonuclear discordance is well-known in plants and it has been traditionally attributed to reticulate evolution (Rieseberg and Soltis 1991; Sang et al. 1995; Soltis and Kuzoff 1995). Such discordance continues to be treated as evidence in support of hybridization in more recent phylogenomic studies that assume the plastome to be a single, linked locus (e.g., Folk et al. 2017; Vargas et al. 2017; Morales-Briones et al. 2018b; Lee-Yaw et al. 2019). However, recent work showed that the plastome might not necessarily act as a single locus and high levels of tree conflict have been detected (Gonçalves et al. 2019; Walker et al. 2019).
In Amaranthaceae s.l., previous studies based on plastid protein-coding genes or introns (Fig. 1; Kadereit et al. 2003; Müller and Borsch 2005; Hohmann et al. 2006; Kadereit et al. 2017) resulted in different relationships among the five main clades and none in agreement with our 76-gene plastid phylogeny. Our conflict and QS analyses of the plastid data set (Supplementary Figs S5 and S6 available on Dryad) revealed strong signals of gene tree discordance among the five major clades of Amaranthaceae s.l., likely due to heteroplasmy, although the exact sources of conflict are yet to be clarified (Gonçalves et al. 2019). Unlike the results found by Walker et al. (2019), our individual plastid gene trees had highly supported nodes (i.e., BS 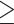 ~70, Supplementary Fig S5 available on Dryad), suggesting that low phylogenetic information content is not the main source of conflict in our plastid data set.
~70, Supplementary Fig S5 available on Dryad), suggesting that low phylogenetic information content is not the main source of conflict in our plastid data set.
Our results support previous studies showing RNA-seq data can be a reliable source for plastome assembly (Smith 2013; Osuna-Mascaró et al. 2018; Gitzendanner et al. 2018). RNA-seq libraries can contain some genomic DNA due to incomplete digestion during RNA purification (Smith 2013). Given the AT-rich nature of plastomes, plastid DNA may survive the poly-A selection during mRNA enrichment (Schliesky et al. 2012). However, RNA editing prediction results showed that our Amaranthaceae s.l. cpDNA assemblies came from RNA rather than DNA contamination regardless of library preparation by poly-A enrichment (71 transcriptomes) or RiboZero (16 transcriptomes). Similarly, Osuna-Mascaró et al. (2018) also found highly similar plastome assemblies (i.e., general genome structure, and gene number and composition) from RNA-seq and genomic libraries, supporting the idea that plastomes are fully transcribed in photosynthetic eukaryotes (Shi et al. 2016). Furthermore, the backbone topology of our plastid tree built mainly from RNA-seq data (97 out of 105 samples) was consistent with a recent complete plastome phylogeny of Caryophyllales mainly from genomic DNA (Yao et al. 2019), showing the utility of recovering plastid gene sequences from RNA-seq data. Nonetheless, RNA editing might be problematic when combining samples from RNA-seq and genomic DNA, especially when resolving phylogenetic relationships among closely related species.
Identifiability in Methods for Detecting Reticulation Events
All methods that we used to detect ancient hybridization inferred the presence of reticulation events. However, our results suggest that these methods all struggle with ancient, rapid radiations. Advances have been made in recent years in developing methods to infer species networks in the presence of ILS (reviewed in Elworth et al. 2019). These methods have been increasingly used in phylogenetic studies (e.g., Wen et al. 2016; Copetti et al. 2017; Morales-Brinones et al. 2018a; Crowl et al. 2020). To date, however, species network inference is still computationally intensive and limited to a small number of species and a few hybridization events (Hejase and Liu 2016; but see Hejase et al. 2018 and Zhu et al. 2019). Furthermore, studies evaluating the performance of different phylogenetic network inference approaches are scarce and restricted to simple hybridization scenarios. Kamneva and Rosenberg (2017) showed that likelihood methods like Yu et al. (2014) are often robust to ILS and gene tree error when symmetric hybridization (equal genetic contribution of both parents) events are considered. While this approach usually does not overestimate hybridization events, it fails to detect skewed hybridization (unequal genetic contribution of both parents) events in the presence of significant ILS. Methods developed to scale to larger numbers of species and hybridizations like the ones using pseudo-likelihood approximations (i.e., Yu and Nakhleh 2015; Solís-Lemus and Ané 2016) are yet to be evaluated independently, but in the case of the Yu and Nakhleh (2015) method based on rooted triples, it cannot distinguish the correct network when other networks can produce the same set of triples (Yu and Nakhleh 2015). On the other hand, the method of Solís-Lemus and Ané (2016), based on unrooted quartets, is better at avoiding indistinguishable networks, but it is limited to only level-1 network scenarios.
Applying the above methods to our data set recovered multiple reticulation events. Analysis of our 11-taxon(net) data set using a pseudo-likelihood approach detected up to five hybridization events involving all five major clades of Amaranthaceae s.l. (Fig. 3). Model selection, after calculating the full likelihood of the obtained networks, also chose the 5-reticulation species as the best model. Likewise, we found that any species network had a better ML score than a bifurcating tree (Supplementary Table S5 available on Dryad). However, further analyses demonstrated that full likelihood network searches with up to one hybridization event are indistinguishable from each other (Supplementary Table S6 available on Dryad), resembling a random gene tree distribution. This pattern can probably be explained by the high levels of gene tree discordance and lack of phylogenetic signal in the inferred quartet gene trees (Fig. 4), suggesting that the 11-taxon(net) network searches can potentially overestimate reticulation events due to high levels of gene tree error or ILS.
Using the 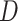 -Statistic (Green et al. 2010; Durand et al. 2011) we also detected signals of introgression in seven possible locations among the five main groups of Amaranthaceae s.l. (Supplementary Table S9 available on Dryad). The inferred introgression events agreed with at least one of the reticulation scenarios from the phylogenetic network analysis. However, the
-Statistic (Green et al. 2010; Durand et al. 2011) we also detected signals of introgression in seven possible locations among the five main groups of Amaranthaceae s.l. (Supplementary Table S9 available on Dryad). The inferred introgression events agreed with at least one of the reticulation scenarios from the phylogenetic network analysis. However, the 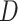 -Statistic did not detect any introgression that involves Betoideae, which was detected in the phylogenetic network analysis with either four or five reticulations events. The
-Statistic did not detect any introgression that involves Betoideae, which was detected in the phylogenetic network analysis with either four or five reticulations events. The 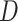 -Statistic has been shown to be robust to a wide range of divergence times, but it is sensitive to relative population size (Zheng and Janke 2018), which agrees with the notion that large effective population sizes and short branches increase the chances of ILS (Pamilo and Nei 1988) and in turn can dilute the signal for the
-Statistic has been shown to be robust to a wide range of divergence times, but it is sensitive to relative population size (Zheng and Janke 2018), which agrees with the notion that large effective population sizes and short branches increase the chances of ILS (Pamilo and Nei 1988) and in turn can dilute the signal for the 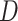 -Statistic (Zheng and Janke 2018). Recently, Elworth et al. (2018) found that multiple or ``hidden'' reticulations can cause the signal of the
-Statistic (Zheng and Janke 2018). Recently, Elworth et al. (2018) found that multiple or ``hidden'' reticulations can cause the signal of the 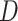 -statistic to be lost or distorted. Furthermore, when multiple reticulations are present, the traditional approach of dividing data sets into quartets can be problematic as it largely underestimates
-statistic to be lost or distorted. Furthermore, when multiple reticulations are present, the traditional approach of dividing data sets into quartets can be problematic as it largely underestimates 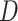 values (Elworth et al. 2018). Given short internal branches in the backbone of Amaranthaceae s.l. and the phylogenetic network results showing multiple hybridizations, it is plausible that our
values (Elworth et al. 2018). Given short internal branches in the backbone of Amaranthaceae s.l. and the phylogenetic network results showing multiple hybridizations, it is plausible that our 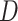 -statistic may be affected by these issues.
-statistic may be affected by these issues.
Our analysis highlights problems with identifiability in relying on 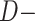 statistic or phylogenetic network analysis alone to detect reticulation events, especially in cases of ancient and rapid diversification. Both analyses resulted in highly complex and inconsistent reticulate scenarios that cannot be distinguished from ILS or gene tree error. Hence, despite the use of genome-scale data and exhaustive hypothesis testing, support is lacking for the hybrid origin of Polycnemoideae or Betoideae, or any particular hybridization event among major groups in Amaranthaceae s.l. In addition to potential hybridization events, rapid speciation, short branches, and large ancestral population size all impacting our ability to resolve relationships among major clades in Amaranthaceae s.l. Simulating combinations of these scenarios is beyond the scope of this manuscript.
statistic or phylogenetic network analysis alone to detect reticulation events, especially in cases of ancient and rapid diversification. Both analyses resulted in highly complex and inconsistent reticulate scenarios that cannot be distinguished from ILS or gene tree error. Hence, despite the use of genome-scale data and exhaustive hypothesis testing, support is lacking for the hybrid origin of Polycnemoideae or Betoideae, or any particular hybridization event among major groups in Amaranthaceae s.l. In addition to potential hybridization events, rapid speciation, short branches, and large ancestral population size all impacting our ability to resolve relationships among major clades in Amaranthaceae s.l. Simulating combinations of these scenarios is beyond the scope of this manuscript.
ILS and the Anomaly Zone
ILS is ubiquitous in multi-locus phylogenetic data sets. In its most severe cases ILS produces the ``anomaly zone'', defined as a set of short internal branches in the species tree that produce AGTs that are more likely than the gene tree that matches the species tree (Degnan and Rosenberg 2006). Rosenberg (2013) expanded the definition of the anomaly zone to require that a species tree contain two consecutive internal branches in an ancestor--descendant relationship in order to produce AGTs. To date, only a few empirical examples of the anomaly zone have been reported (Linkem et al. 2016; Cloutier et al. 2019). Our results show that the species tree of Amaranthaceae s.l. has three consecutive short internal branches that lay within the limits of the anomaly zone (i.e., 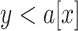 ; Fig. 5; Supplementary Table S10 available on Dryad) and that the species tree is not the most frequent gene tree (Fig. 4). While both lines of evidence support the presence of AGTs, it is important to point out that our quartet analysis showed that most quartet gene trees were equivocal (94--96%; Fig. 4), and therefore, were uninformative. Huang and Knowles (2009) pointed out that the gene tree discordance produced from the anomaly zone can be produced by uninformative gene trees and that for species trees with short branches the most probable gene tree topology is a polytomy rather than an AGT. Our ASTRAL polytomy test, however, rejected a polytomy along the backbone of Amaranthaceae s.l. in any of the gene tree sets used. While we did not test for polytomies in individual gene trees, our ASTRAL polytomy test using gene trees with branches of
; Fig. 5; Supplementary Table S10 available on Dryad) and that the species tree is not the most frequent gene tree (Fig. 4). While both lines of evidence support the presence of AGTs, it is important to point out that our quartet analysis showed that most quartet gene trees were equivocal (94--96%; Fig. 4), and therefore, were uninformative. Huang and Knowles (2009) pointed out that the gene tree discordance produced from the anomaly zone can be produced by uninformative gene trees and that for species trees with short branches the most probable gene tree topology is a polytomy rather than an AGT. Our ASTRAL polytomy test, however, rejected a polytomy along the backbone of Amaranthaceae s.l. in any of the gene tree sets used. While we did not test for polytomies in individual gene trees, our ASTRAL polytomy test using gene trees with branches of 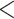 75% bootstrap support collapsed also rejected the presence of a polytomy. Therefore, the distribution of gene tree frequency in combination with short internal branches in the species tree supports the presence of an anomaly zone in Amaranthaceae s.l.
75% bootstrap support collapsed also rejected the presence of a polytomy. Therefore, the distribution of gene tree frequency in combination with short internal branches in the species tree supports the presence of an anomaly zone in Amaranthaceae s.l.
Taxonomic Implications
Despite the strong signal of gene tree discordance, both nuclear and plastid data sets strongly supported five major clades within Amaranthaceae s.l.: Amaranthaceae s.s., ``Chenopods I'', ``Chenopods II'', Betoideae, and Polycnemoideae (Fig. 2 and Supplementary Fig.S4 available on Dryad). These five clades are congruent with morphology and previous taxonomic treatments of the group. However, the relationships among these five lineages remain elusive with our data. Taken together, our tests of sources of incongruence for these early-diverging nodes indicate that no single source such as a particular ancient hybridization event can confidently account for the strong signal of gene tree discordance, suggesting that the discordance results primarily from ancient and rapid lineage diversification. Thus, the backbone of Amaranthaceae s.l. remains, and likely will remain, unresolved even with genome-scale data. The stem age of Amaranthaceae s.l. dates back to the early Tertiary (Paleocene; Kadereit et al. 2012; Di Vincenzo et al. 2018; Yao et al. 2019), but due to nuclear and plastid gene tree along the backbone, the geographic origin of Amaranthaceae s.l. remains ambiguous.
Therefore, for the sake of taxonomic stability, we suggest retaining Amaranthaceae s.l. sensu APG IV (The Angiosperm Phylogeny Group 2016), which includes the previously recognized Chenopodiaceae. Amaranthaceae s.l. is characterized by a long list of anatomical, morphological, and phytochemical characters such as minute sessile flowers with five tepals, a single whorl of epitepalous stamens, and one basal ovule (Kadereit et al. 2003). Here, we recognize five subfamilies within Amaranthaceae s.l. represented by the five well-supported major clades recovered in this study (Fig. 2): Amaranthoideae (Amaranthaceae s.s.), Betoideae, Chenopodioideae (``Chenopods I''), Polycnemoideae, and Salicornioideae (``Chenopods II'').
Conclusions
Our analyses highlight the need to test for multiple sources of conflict in phylogenomic analyses, especially when trying to resolve phylogenetic relationships with extensive phylogenetic conflict. Furthermore, one needs to be aware of the strengths and limitations of different phylogenetic methods and be cautious about relying on any single analysis, for example in the usage of phylogenetics species networks over coalescent-based species trees (Blair and Ané 2020). We make the following recommendation on five essential steps towards exploring heterogeneous phylogenetic signals in phylogenomic data sets in general. 1) Study design: consider whether the taxon sampling and marker choice enable testing alternative sources of conflicting phylogenetic signal. For example, will there be sufficient phylogenetic signal and sufficient taxon coverage in individual gene trees for methods such as phylogenetic network analyses? 2) Data processing: care should be taken in data cleaning, partitioning (e.g., nuclear vs. plastid), and using orthology inference methods that explicitly address paralogy issues (e.g., tree-based orthology inference and synteny information). 3) Species tree inference: select species tree methods that accommodate the data set size and data type [e.g., ASTRAL for gene tree-based inferences or SVDquartet (Chifman and Kubatko 2014) for SNP-based inferences], followed by visualization of phylogenetic conflict using tools such as the pie charts (e.g., PhyParts) and quartet-based tools [e.g., QS; Quadripartition Internode Certainty (Zhou et al. 2020); Concordance Factors (Minh et al. 2020)]. 4) Assessing hybridization: if phylogenetic conflict cannot be explained by processes like ILS, phylogenetic species network analyses (e.g., PhyloNet) reduced taxon sampling can be applied to test hybridization hypotheses given results in step 3; 5) Hypothesis testing: additional tests can be performed given the results of recommendation 3 and 4 depending on the scenario. These could include testing for model misspecification, anomaly zone, uninformative gene tree, and if hybridization is hypothesized, testing putative reticulation events one at a time, as illustrated in this study.
Despite using genome-scale data and exhaustive hypothesis testing, the backbone phylogeny of Amaranthaceae s.l. remains unresolved, and we were unable to distinguish ancient hybridization events from ILS or uninformative gene trees. Similar situations might not be atypical across the Tree of Life. As we leverage more genomic data and explore gene tree discordance in more detail, these steps will be informative in other clades, especially in those that are products of ancient and rapid lineage diversification (e.g., Widhelm et al. 2019; Koenen et al. 2020). Ultimately, such endeavors will be instrumental in gaining a full understanding of the complexity of the Tree of Life.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.ns1rn8pq4.
Acknowledgments
The authors thank H. Freitag, J.M. Bena, and the Millennium Seed Bank for providing seeds; U. Martiné for assisting with RNA extraction; N. Wang and Y.-Y. Huang for sample sequencing. A. Crum, R. Ree, B. Carstens, and three anonymous reviewers for providing helpful comments; the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing access to computational resources.
Funding
This work was supported by the University of Minnesota, the University of Michigan, the US National Science Foundation (DEB 1354048), and the Department of Energy, Office of Science, Genomic Science Program (Contract Number DE-SC0008834).
References
- Alda F., Tagliacollo V.A., Bernt M.J., Waltz B.T., Ludt W.B., Faircloth B.C., Alfaro M.E., Albert J.S., Chakrabarty P.. 2019. Resolving deep nodes in an ancient radiation of neotropical fishes in the presence of conflicting signals from incomplete lineage sorting. Syst. Biol. 68:573–593. [DOI] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., Pyshkin A.V., Sirotkin A.V., Vyahhi N., Tesler G., Alekseyev M.A., Pevzner P.A.. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Bena M.J., Acosta J.M., Aagesen L..
2017.
Macroclimatic niche limits and the evolution of C
 photosynthesis in Gomphrenoideae (Amaranthaceae). Bot. J. Linn. Soc. 184:283–297. [Google Scholar]
photosynthesis in Gomphrenoideae (Amaranthaceae). Bot. J. Linn. Soc. 184:283–297. [Google Scholar] - Blackmon H., Adams R.A.. 2015. EvobiR: Tools for comparative analyses and teaching evolutionary biology. Zenodo. 10.5281/zenodo.30938. [DOI] [Google Scholar]
- Blair C., Ané C.. 2020. Phylogenetic trees and networks can serve as powerful and complementary approaches for analysis of genomic data. Syst. Biol. 69:593–601. [DOI] [PubMed] [Google Scholar]
- Bouckaert R., Heled J.. 2014. DensiTree 2: seeing trees through the forest. BioRxiv. 10.1101/012401. [DOI] [Google Scholar]
- Brown J.W., Walker J.F., Smith S.A.. 2017. Phyx - phylogenetic tools for unix. Bioinformatics. 33:1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen T.C., Philippe H., Bryant D.. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics. 172:2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley T.R., Cordeiro M., Marshall D.C., Simon C.. 2006. Differentiating between hypotheses of lineage sorting and introgression in New Zealand Alpine Cicadas (Maoricicada Dugdale). Syst. Biol. 55:411–425. [DOI] [PubMed] [Google Scholar]
- Chen L.-Y., Morales-Briones D.F., Passow C.N., Yang Y.. 2019. Performance of gene expression analyses using de novo assembled transcripts in polyploid species. Bioinformatics. 35:4314–4320. [DOI] [PubMed] [Google Scholar]
- Chifman J., Kubatko L.. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics. 30:3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier A., Sackton T.B., Grayson P., Clamp M., Baker A.J., Edwards S.V.. 2019. Whole-genome analyses resolve the phylogeny of flightless birds (Palaeognathae) in the presence of an empirical anomaly zone. Syst. Biol. 68:937–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper E.D. 2014. Overly simplistic substitution models obscure green plant phylogeny. Trends Plant Sci. 19:576–582. [DOI] [PubMed] [Google Scholar]
- Copetti D., Búrquez A., Bustamante E., Charboneau J.L.M., Childs K.L., Eguiarte L.E., Lee S., Liu T.L., McMahon M.M., Whiteman N.K., Wing R.A., Wojciechowski M.F., Sanderson M.J.. 2017. Extensive gene tree discordance and hemiplasy shaped the genomes of North American columnar cacti. Proc. Natl. Acad. Sci. USA. 114:12003–12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C.J., Li B., Foster P.G., Embley T.M., Civáò P.. 2014. Conflicting phylogenies for early land plants are caused by composition biases among synonymous substitutions. Syst. Biol. 63:272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowl A.A., Manos P.S., McVay J.D., Lemmon A.R., Lemmon E.M., Hipp A.L.. 2020. Uncovering the genomic signature of ancient introgression between white oak lineages (Quercus). New Phytol. 226:1158–1170. [DOI] [PubMed] [Google Scholar]
- Davidson N.M., Oshlack A.. 2014. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 15:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2006. Discordance of species trees with their most likely gene trees. PLoS Genet. 2:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Di Vincenzo V., Gruenstaeudl M., Nauheimer L., Wondafrash M., Kamau P., Demissew S., Borsch T.. 2018. Evolutionary diversification of the African achyranthoid clade (Amaranthaceae) in the context of sterile flower evolution and epizoochory. Ann. Bot. 122:69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm J.C., Minoche A.E., Holtgräwe D., Capella-Gutiérrez S., Zakrzewski F., Tafer H., Rupp O., Sörensen T.R., Stracke R., Reinhardt R., Goesmann A., Kraft T., Schulz B., Stadler P.F., Schmidt T., Gabaldón T., Lehrach H., Weisshaar B., Himmelbauer H.. 2014. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature. 505:546–549. [DOI] [PubMed] [Google Scholar]
- Doyle J.J. 1992. Gene trees and species trees: molecular systematics as one-character taxonomy. Syst. Bot. 17:144. [Google Scholar]
- Durand E.Y., Patterson N., Reich D., Slatkin M.. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.V. 2009. Is a new and general theory of molecular systematics emerging? Evolution. 63:1–19. [DOI] [PubMed] [Google Scholar]
- Edwards S.V., Xi Z., Janke A., Faircloth B.C., McCormack J.E., Glenn T.C., Zhong B., Wu S., Lemmon E.M., Lemmon A.R., Leaché A.D., Liu L., Davis C.C.. 2016. Implementing and testing the multispecies coalescent model: a valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 94:447–462. [DOI] [PubMed] [Google Scholar]
- Elworth R.A.L., Allen C., Benedict T., Dulworth P., Nakhleh L.K.. 2018. DGEN: A test statistic for detection of general introgression scenarios. WABI. [Google Scholar]
- Elworth R.A.L., Ogilvie H.A., Zhu J., Nakhleh L.. 2019. Advances in computational methods for phylogenetic networks in the presence of hybridization. In: Warnow T., editor. Bioinformatics and Phylogenetics. Computational Biology, vol 29 Cham, Switzerland: Springer; p. 317–360. [Google Scholar]
- Erfan Sayyari, Siavash Mirarab. 2018. Testing for polytomies in phylogenetic species trees using quartet frequencies. Genes. 9:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers T.J., Colmer T.D.. 2015. Plant salt tolerance: adaptations in halophytes. Ann. Bot. 115:327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folk R.A., Mandel J.R., Freudenstein J.V.. 2017. Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 66:320–337. [DOI] [PubMed] [Google Scholar]
- Fontaine M.C., Pease J.B., Steele A., Waterhouse R.M., Neafsey D.E., Sharakhov I.V., Jiang X., Hall A.B., Catteruccia F., Kakani E., Mitchell S.N., Wu Y.-C., Smith H.A., Love R.R., Lawniczak M.K., Slotman M.A., Emrich S.J., Hahn M.W., Besansky N.J.. 2015. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science. 347:1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P.G. 2004. Modeling compositional heterogeneity. Syst. Biol. 53:485–495. [DOI] [PubMed] [Google Scholar]
- Galtier N., Daubin V.. 2008. Dealing with incongruence in phylogenomic analyses. Philos. Trans. R. Soc. B Biol. Sci. 363:4023–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitzendanner M.A., Soltis P.S., Yi T.-S., Li D.-Z., Soltis D.E.. 2018. Plastome phylogenetics: 30 years of inferences into plant evolution. In: Chaw S.-M., Jansen R., editors. Advances in Botanical Research. Plastid Genome Evolution, vol. 85 Cambridge, MA, USA: Academic Press; p. 293–313. [Google Scholar]
- Gonçalves D.J.P., Simpson B.B., Ortiz E.M., Shimizu G.H., Jansen R.K.. 2019. Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol. Phylogenet. Evol. 138:219–232. [DOI] [PubMed] [Google Scholar]
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., di Palma F., Birren B.W., Nusbaum C., Lindblad-Toh K., Friedman N., Regev A.. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.Y., Hansen N.F., Durand E.Y., Malaspinas A.S., Jensen J.D., Marques-Bonet T., Alkan C., Prufer K., Meyer M., Burbano H.A., Good J.M., Schultz R., Aximu-Petri A., Butthof A., Hober B., Hoffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E.S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Doronichev V.B., Golovanova L.V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R.W., Johnson P.L.F., Eichler E.E., Falush D., Birney E., Mullikin J.C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Paabo S.. 2010. A draft sequence of the neandertal genome. Science. 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejase H.A., Liu K.J.. 2016. A scalability study of phylogenetic network inference methods using empirical datasets and simulations involving a single reticulation. BMC Bioinformatics. 17:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejase H.A., VandePol N., Bonito G.M., Liu K.J.. 2018. FastNet: fast and accurate statistical inference of phylogenetic networks using large-scale genomic sequence data. In: Blanchette M., Ouangraoua A., editors. Comparative Genomics. RECOMB-CG 2018. Lecture Notes in Computer Science, vol 11183 Cham, Switzerland: Springer; p. 242--259. [Google Scholar]
- Hernández-Ledesma P., Berendsohn W.G., Borsch T., Mering S.V., Akhani H., Arias S., Castañeda-Noa I., Eggli U., Eriksson R., Flores-Olvera H., Fuentes-Bazán S., Kadereit G., Klak C., Korotkova N., Nyffeler R., Ocampo G., Ochoterena H., Oxelman B., Rabeler R.K., Sanchez A., Schlumpberger B.O., Uotila P.. 2015. A taxonomic backbone for the global synthesis of species diversity in the angiosperm order Caryophyllales. Willdenowia. 45:281. [Google Scholar]
- Hoang D.T., Chernomor O.. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S., Kadereit J.W., Kadereit G.. 2006. Understanding Mediterranean-Californian disjunctions: molecular evidence from Chenopodiaceae-Betoideae. Taxon. 55:67–78. [Google Scholar]
- Holder M.T., Anderson J.A., Holloway A.K.. 2001. Difficulties in detecting hybridization. Syst. Biol. 50:978–982. [DOI] [PubMed] [Google Scholar]
- Huang H., Knowles L.L.. 2009. What is the danger of the anomaly zone for empirical phylogenetics? Syst. Biol. 58:527–536. [DOI] [PubMed] [Google Scholar]
- Huang W., Zhou G., Marchand M., Ash J.R., Morris D., Van Dooren P., Brown J.M., Gallivan K.A., Wilgenbusch J.C.. 2016. TreeScaper: visualizing and extracting phylogenetic signal from sets of trees. Mol. Biol. Evol. 33:3314–3316. [DOI] [PubMed] [Google Scholar]
- Jarvis D.E., Ho Y.S., Lightfoot D.J., Schmöckel S.M., Li B., Borm T.J.A., Ohyanagi H., Mineta K., Michell C.T., Saber N., Kharbatia N.M., Rupper R.R., Sharp A.R., Dally N., Boughton B.A., Woo Y.H., Gao G., Schijlen E.G.W.M., Guo X., Momin A.A., Negrão S., Al-Babili S., Gehring C., Roessner U., Jung C., Murphy K., Arold S.T., Gojobori T., Linden C.G.V.D., van Loo E.N., Jellen E.N., Maughan P.J., Tester M.. 2017. The genome of Chenopodium quinoa. Nature. 542: 307–312. [DOI] [PubMed] [Google Scholar]
-
Kadereit G., Ackerly D., Pirie M.D..
2012.
A broader model for C
 photosynthesis evolution in plants inferred from the goosefoot family (Chenopodiaceae s.s.). Proc. R. Soc. B Biol. Sci. 279: 3304--3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
photosynthesis evolution in plants inferred from the goosefoot family (Chenopodiaceae s.s.). Proc. R. Soc. B Biol. Sci. 279: 3304--3311. [DOI] [PMC free article] [PubMed] [Google Scholar] -
Kadereit G., Borsch T., Weising K., Freitag H..
2003.
Phylogeny of Amaranthaceae and Chenopodiaceae and the evolution of C
 photosynthesis. Int. J. Plant Sci. 164:959–986. [Google Scholar]
photosynthesis. Int. J. Plant Sci. 164:959–986. [Google Scholar] - Kadereit G., Hohmann S., Kadereit J.W.. 2006. A synopsis of Chenopodiaceae subfam. Betoideae and notes on the taxonomy of Beta. Willdenowia 36:9–19. [Google Scholar]
- Kadereit G., Newton R.J., Vandelook F.. 2017. Evolutionary ecology of fast seed germination---a case study in Amaranthaceae/Chenopodiaceae. Perspect. Plant Ecol. Evol. Syst. 29:1–11. [Google Scholar]
- Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamneva O.K., Rosenberg N.A.. 2017. Simulation-based evaluation of hybridization network reconstruction methods in the presence of incomplete lineage sorting. Evol. Bioinforma. 13:117693431769193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A.. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles L.L., Huang H., Sukumaran J., Smith S.A.. 2018. A matter of phylogenetic scale: distinguishing incomplete lineage sorting from lateral gene transfer as the cause of gene tree discord in recent versus deep diversification histories. Am. J. Bot. 105:376–384. [DOI] [PubMed] [Google Scholar]
- Koenen, E., Ojeda, D., Steeves, R., Migliore, J., Bakker, F., Wieringa, J., Kidner, C., Hardy, O., Pennington, R., Bruneau, A., Hughes, C.. 2020. Large-scale genomic sequence data resolve the deepest divergences in the legume phylogeny and support a near-simultaneous evolutionary origin of all six subfamilies. New Phytol. 225: 1355-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatko L.S., Chifman J.. 2019. An invariants-based method for efficient identification of hybrid species from large-scale genomic data. BMC Evol. Biol. 19:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., Filipski, A., Battistuzzi, F., Kosakovsky Pond, S., Tamura, K., 2012. Statistics and truth in phylogenomics. Mol. Biol. Evol. 29:457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R., Calcott B., Ho S.Y.W., Guindon S.. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29:1695–1701. [DOI] [PubMed] [Google Scholar]
- Lee-Yaw J.A., Grassa C.J., Joly S., Andrew R.L., Rieseberg L.H.. 2019. An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytol. 221:515–526. [DOI] [PubMed] [Google Scholar]
- Li B., Wang J., Yao L., Meng Y., Ma X., Si E., Ren P., Yang K., Shang X., Wang H.. Halophyte Halogeton glomeratus, a promising candidate for phytoremediation of heavy metal-contaminated saline soils. Plant Soil. 442:323--331. [Google Scholar]
- Lightfoot D.J., Jarvis D.E., Ramaraj T., Lee R., Jellen E.N., Maughan P.J.. 2017. Single-molecule sequencing and Hi-C-based proximity-guided assembly of amaranth (Amaranthus hypochondriacus) chromosomes provide insights into genome evolution. BMC Biol. 15:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkem C.W., Minin V.N., Leaché A.D.. 2016. Detecting the anomaly zone in species trees and evidence for a misleading signal in higher-level skink phylogeny (Squamata: Scincidae). Syst. Biol. 65:465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Yu L.. 2010. Phybase: an R package for species tree analysis. Bioinformatics. 26:962–963. [DOI] [PubMed] [Google Scholar]
- Liu Y., Cox C.J., Wang W., Goffinet B.. 2014. Mitochondrial phylogenomics of early land plants: mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst. Biol. 63:862–878. [DOI] [PubMed] [Google Scholar]
- Maddison W.P. 1997. Gene trees in species trees. Syst. Biol. 46:532–536. [Google Scholar]
- Masson R., Kadereit G.. 2013. Phylogeny of Polycnemoideae (Amaranthaceae): implications for biogeography, character evolution and taxonomy. Taxon. 62:100–111. [Google Scholar]
- Maureira-Butler I.J., Pfeil B.E., Muangprom A., Osborn T.C., Doyle J.J.. 2008. The reticulate history of Medicago (Fabaceae). Syst. Biol. 57:466–482. [DOI] [PubMed] [Google Scholar]
- Mclean B.S., Bell K.C., Allen J.M., Helgen K.M., Cook J.A.. 2019. Impacts of inference method and data set filtering on phylogenomic resolution in a rapid radiation of ground squirrels (Xerinae: Marmotini). Syst. Biol. 68:298–316. [DOI] [PubMed] [Google Scholar]
- Meyer B.S., Matschiner M., Salzburger W.. 2017. Disentangling incomplete lineage sorting and introgression to re?ne species-tree estimates for Lake Tanganyika Cichlid Fishes. Syst. Biol. 66: 531–550. [DOI] [PubMed] [Google Scholar]
- Minh B.Q., Hahn M., Lanfear R.. 2020. New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. msaa106. 10.1093/molbev/msaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Bayzid M.S., Warnow T.. 2016. Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Syst. Biol. 65:366–380. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Liston A., Tank D.C.. 2018a. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). New Phytol. 218:1668–1684. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Romoleroux K., Koláø F., Tank D.C.. 2018b. Phylogeny and evolution of the neotropical radiation of Lachemilla (Rosaceae): uncovering a history of reticulate evolution and implications for infrageneric classification. Syst. Bot. 43:17–34. [Google Scholar]
- Moray C., Goolsby E.W., Bromham L.. 2016. The phylogenetic association between salt tolerance and heavy metal hyperaccumulation in angiosperms. Evol. Biol. 43:119–130. [Google Scholar]
- Mower J.P. 2009. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 37:W253–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller K., Borsch T.. 2005. Phylogenetics of Amaranthaceae based on matK/trnK sequence data: evidence from parsimony, likelihood, and Bayesian analyses. Ann. Mo. Bot. Gard. 92:66–102. [Google Scholar]
- Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuna-Mascaró C., Rubio de Casas R., Perfectti F.. 2018. Comparative assessment shows the reliability of chloroplast genome assembly using RNA-seq. Sci. Rep. 8:17404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo P., Nei M.. 1988. Relationships between gene trees and species trees. Mol. Biol. Evol. 5:568–583. [DOI] [PubMed] [Google Scholar]
- Pease J.B., Brown J.W., Walker J.F., Hinchliff C.E., Smith S.A.. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105:385–403. [DOI] [PubMed] [Google Scholar]
- Piirainen M., Liebisch O., Kadereit G.. 2017. Phylogeny, biogeography, systematics and taxonomy of Salicornioideae (Amaranthaceae/Chenopodiaceae)---a cosmopolitan, highly specialized hygrohalophyte lineage dating back to the Oligocene. Taxon. 66:109–132. [Google Scholar]
- Prasanna A.N., Gerber D., Kijpornyongpan T., Aime M.C., Doyle V.P., Nagy L.G.. 2020. Model choice, missing data, and taxon sampling impact phylogenomic inference of deep basidiomycota relationships. Syst. Biol. 69:17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2019. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Rannala B., Yang Z.. 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 166:1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg L.H., Soltis D.E.. 1991. Phylogenetic consequences of cytoplasmic gene flow in plants. Evol. Trends Plants. 5:65–84. [Google Scholar]
- Robinson D.F., Foulds L.R.. 1981. Comparison of phylogenetic trees. Math. Biosci. 53:131–147. [Google Scholar]
- Rosenberg N.A. 2013. Discordance of species trees with their most likely gene trees: a unifying principle. Mol. Biol. Evol. 30: 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roycroft E.J., Moussalli A., Rowe K.C.. 2020. Phylogenomics uncovers confidence and conflict in the rapid radiation of Australo-Papuan rodents. Syst. Biol. 69:431–444. [DOI] [PubMed] [Google Scholar]
- Salichos L., Stamatakis A., Rokas A.. 2014. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 31:1261–1271. [DOI] [PubMed] [Google Scholar]
- Sang T., Crawford D.J., Stuessy T.F.. 1995. Documentation of reticulate evolution in peonies (Paeonia) using internal transcribed spacer sequences of nuclear ribosomal DNA: implications for biogeography and concerted evolution. Proc. Natl. Acad. Sci. USA. 92:6813–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E., Mirarab S.. 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep K.P. 2011. phangorn: phylogenetic analysis in R. Bioinformatics. 27:592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliesky S., Gowik U., Weber A.P.M., Bräutigam A.. 2012. RNA-Seq assembly---are we there yet? Front. Plant Sci. 3:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. 1978. Estimating the dimension of a model. Ann. Stat. 6:461–464. [Google Scholar]
- Sharp P.M., Li W.-H.. 1986. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24:28–38. [DOI] [PubMed] [Google Scholar]
- Shi C., Wang S., Xia E.-H., Jiang J.-J., Zeng F.-C., Gao L.-Z.. 2016. Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci. Rep. 6:30135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H. 2002. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51:492–508. [DOI] [PubMed] [Google Scholar]
- Shimodaira H., Hasegawa M.. 2001. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 17:1246–1247. [DOI] [PubMed] [Google Scholar]
- Smith D.R. 2013. RNA-Seq data: a goldmine for organelle research. Brief. Funct. Genomics. 12:454–456. [DOI] [PubMed] [Google Scholar]
- Smith S.A., Moore M.J., Brown J.W., Yang Y.. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.A., O'Meara B.C.. 2012. treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics. 28:2689–2690. [DOI] [PubMed] [Google Scholar]
- Solís-Lemus C., Ané C.. 2016a. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis D.E., Kuzoff R.K.. 1995. Discordance between nuclear and chloroplast phylogenies in the Heuchera group (Saxifragaceae). Evolution. 49:727–742. [DOI] [PubMed] [Google Scholar]
- Srivastava S.K. 1969. Assorted angiosperm pollen from the Edmonton Formation (Maestrichtian), Alberta, Canada. Can. J. Bot. 47:975–989. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8---a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura N. 1978. Further analysts of the data by akaike' s information criterion and the finite corrections. Commun. Stat. Theory Methods. 7:13–26. [Google Scholar]
- Swofford D. 2002. PAUP*. Phylogenetic analysis using parsimony (*and other methods) version 4. Sunderland (MA): Sinauer Associates. [Google Scholar]
- Than C., Ruths D., Nakhleh L.. 2008. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics. 9:322–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Angiosperm Phylogeny Group, Chase M.W., Christenhusz M.J.M., Fay M.F., Byng J.W., Judd W.S., Soltis D.E., Mabberley D.J., Sennikov A.N., Soltis P.S., Stevens P.F.. 2016. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181:1–20. [Google Scholar]
- Vargas O.M., Ortiz E.M., Simpson B.B.. 2017. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 214:1736–1750. [DOI] [PubMed] [Google Scholar]
- Walker J.F., Walker-Hale N., Vargas O.M., Larson D.A., Stull G.W.. 2019. Characterizing gene tree conflict in plastome-inferred phylogenies. PeerJ. 7:e7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.F., Yang Y., Feng T., Timoneda A., Mikenas J., Hutchison V., Edwards C., Wang N., Ahluwalia S., Olivieri J., Walker-Hale N., Majure L.C., Puente R., Kadereit G., Lauterbach M., Eggli U., Flores-Olvera H., Ochoterena H., Brockington S.F., Moore M.J., Smith S.A.. 2018. From cacti to carnivores: improved phylotranscriptomic sampling and hierarchical homology inference provide further insight into the evolution of Caryophyllales. Am. J. Bot. 105: 446–462. [DOI] [PubMed] [Google Scholar]
- Wen D., Yu Y., Hahn M.W., Nakhleh L.. 2016. Reticulate evolutionary history and extensive introgression in mosquito species revealed by phylogenetic network analysis. Mol. Ecol. 25:2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Yu Y., Zhu J., Nakhleh L.. 2018. Inferring phylogenetic networks using PhyloNet. Syst. Biol. 67:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widhelm T.J., Grewe F., Huang J.-P., Mercado-Díaz J.A., Goffinet B., Lücking R., Moncada B., Mason-Gamer R., Lumbsch H.T.. 2019. Multiple historical processes obscure phylogenetic relationships in a taxonomically difficult group (Lobariaceae, Ascomycota). Sci. Rep. 9:8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B., Yang Z.. 2016. Challenges in species tree estimation under the multispecies coalescent model. Genetics. 204:1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Jiao C., Sun H., Cai X., Wang X., Ge C., Zheng Y., Liu W., Sun X., Xu Y., Deng J., Zhang Z., Huang S., Dai S., Mou B., Wang Q., Fei Z., Wang Q.. 2017. Draft genome of spinach and transcriptome diversity of 120 Spinacia accessions. Nat. Commun. 8:15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Moore M.J., Brockington S.F., Timoneda A., Feng T., Marx H.E., Walker J.F., Smith S.A.. 2017. An efficient field and laboratory workflow for plant phylotranscriptomic projects. Appl. Plant Sci. 5:1600128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Smith S.A.. 2014. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 31:3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G., Jin J.-J., Li H.-T., Yang J.-B., Mandala V.S., Croley M., Mostow R., Douglas N.A., Chase M.W., Christenhusz M.J.M., Soltis D.E., Soltis P.S., Smith S.A., Brockington S.F., Moore M.J., Yi T.-S., Li D.-Z.. 2019. Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogenet. Evol. 134:74–86. [DOI] [PubMed] [Google Scholar]
- Yu Y., Degnan J.H., Nakhleh L.. 2012. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 8:e1002660–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Dong J., Liu K.J., Nakhleh L.. 2014. Maximum likelihood inference of reticulate evolutionary histories. Proc. Natl. Acad. Sci. USA. 111:16448–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Nakhleh L.. 2015. A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics. 16:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S.. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 19:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T., Schranz M.E.. 2019. Network-based microsynteny analysis identifies major differences and genomic outliers in mammalian and angiosperm genomes. Proc. Natl. Acad. Sci. USA. 116: 2165–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Janke A.. 2018. Gene flow analysis method, the D-statistic, is robust in a wide parameter space. BMC Bioinformatics. 19:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X., Lutteropp, S., Czech, L., Stamatakis, A., Looz, M. V., Rokas, A.. 2020. Quartet-based computations of internode certainty provide robust measures of phylogenetic incongruence. Syst. Biol. 69:308–324. [DOI] [PubMed] [Google Scholar]
- Zhu J., Liu X., Ogilvie H.A., Nakhleh L.K.. 2019. A divide-and-conquer method for scalable phylogenetic network inference from multilocus data. Bioinformatics. 35:i370–i378. [DOI] [PMC free article] [PubMed] [Google Scholar]