

Abstract
Class switch recombination (CSR) generates isotype-switched antibodies with distinct effector functions essential for mediating effective humoral immunity. CSR is catalyzed by activation-induced deaminase (AID) that initiates DNA lesions in the evolutionarily conserved switch (S) regions at the immunoglobulin heavy chain (Igh) locus. AID-initiated DNA lesions are subsequently converted into DNA double stranded breaks (DSBs) in the S regions of Igh locus, repaired by non-homologous end-joining to effect CSR in mammalian B lymphocytes. While molecular mechanisms of CSR are well characterized, it remains less well understood how upstream signaling pathways regulate AID expression and CSR. B lymphocytes express multiple receptors including the B cell antigen receptor (BCR) and co-receptors (e.g., CD40). These receptors may share common signaling pathways or may use distinct signaling elements to regulate CSR. Here, we discuss how signals emanating from different receptors positively or negatively regulate AID expression and CSR.
Keywords: class switch recombination, activation-induced deaminase, signal transduction, B cell antigen receptor, CD40, toll-like receptor, PI3K, PTEN
1. Overview of antibody-mediated humoral immunity and class switch recombination
1.1. Overview of antibody-mediated immune responses
Antibody-mediated humoral immunity plays a crucial role in protecting us against pathogens and toxins. In fact, protective effects of more than 90% of current vaccines are mediated by high-affinity isotype-switched antibodies (Plotkin, 2010). Mature naïve B cells initially secrete IgM or express IgM as their surface antigen receptor, known as the B cell antigen receptor (BCR). The BCR recognizes antigens and transduces signals to initiate antigen-specific antibody-mediated immune responses. The secreted IgM antibodies can directly neutralize pathogens and toxins by agglutinating bacteria or viruses at the onset of infections, thereby providing instant protection against invading pathogens. However, IgM’s affinity to antigen is very low and its function is limited in the bloodstream since it cannot efficiently cross vascular barriers to clear pathogens systemically (Matter and Ochsenbein, 2008; Pone et al., 2012a). Thus, upon pathogen infection or immunization, antigen and cognate ligands activate the BCR and various co-receptors including CD40, toll-like receptors (TLRs), B cell activating factor receptor (BAFF-R) and transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) as well as cytokine receptors to elicit signals that induce a secondary diversification process at the antibody gene loci.
In mammalian B cells, this secondary diversification process includes somatic hypermutation (SHM) and class switch recombination (CSR) (Figure 1) that are essential for generating antigen-specific high-affinity isotype-switched antibodies (Kato et al., 2012). SHM enables B cells to acquire point mutations into the variable (V) region exons of antibody genes, and allows the selection of B cell clones producing higher affinity antibodies (Di Noia and Neuberger, 2007). However, due to the lack of robust in vitro systems to study the signaling regulation of SHM, little is known about how different signaling pathways coordinate to regulate SHM. Thus, this review focuses on the signaling control of CSR. During CSR, the constant (C) region of antibody heavy chain is switched; thus, CSR enables B cells to produce isotype-switched IgG, IgA or IgE antibody, thereby diversifying antibody effector functions. For example, IgG can activate natural killer cells and phagocytes to more effectively clear pathogen-infected cells; moreover, IgG can protect fetus against infection by passing through placenta (Casadevall and Pirofski, 2004; Simister, 2003).
Figure 1. CSR model for IgG1 production at the mouse Igh locus.
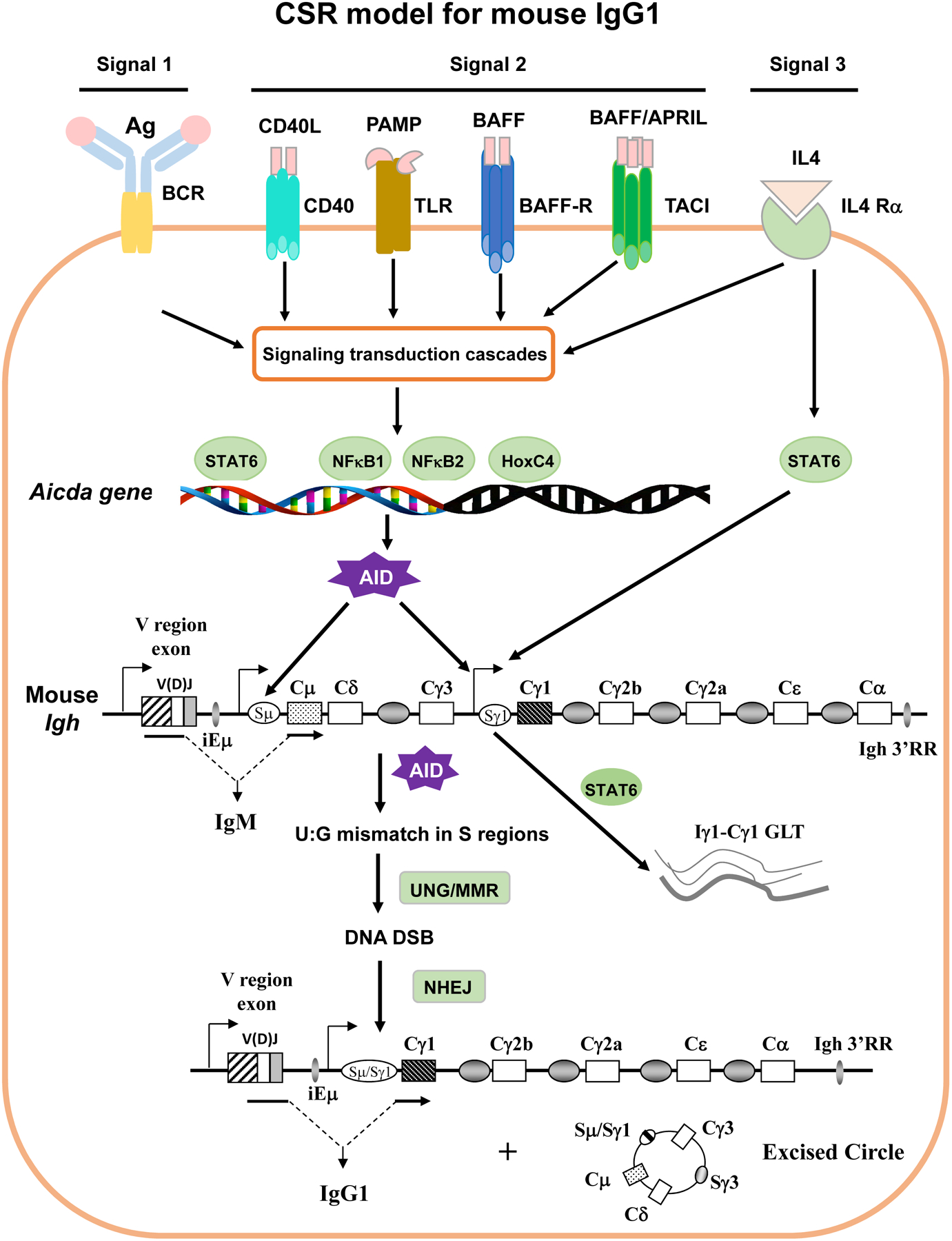
Antigen-specific antibody responses are mediated by stimulating multiple receptors expressed on B cells with their cognate ligands. The signals emanating from these receptors can be categorized into three major types. Signal 1 is the initiating signal generated by BCR upon recognizing antigen. Signal 2 is generated by co-receptors (CD40, TLRs, etc.) upon recognizing their individual ligands. Signal 3 is generated by cytokine receptors (e.g., IL-4R) upon binding to specific cytokines. Signals originating from different receptors are transduced through signaling cascade and eventually lead to activation of different transcription factors (e.g., NF-κB, HoxC4 etc). The activated transcription factors induce the expression of AID and the Igh GLT (e.g., Cγ1 GLT) that allows AID to access Sγ1 region. The genomic configuration of the rearranged mouse Igh locus is shown. AID introduces point mutations into variable (V) region exon during SHM (not depicted). During CSR, AID initiates U:G mismatches in the donor Sμ and the downstream acceptor Sγ1 regions. AID-initiated U:G mismatches are processed and converted into DNA double strand breaks (DSBs) by UNG and mismatch repair (MMR) pathways. Broken S regions are rejoined via non-homologous end-joining (NHEJ), while intervening DNA is excised as a circle. Transcription is required for both SHM/CSR with promoters delineated for both V and S regions (arrows). Upon CSR, originally expressed Cμ exons are replaced by Cγ1 exons so that naïve IgM+ B cells switch to antigen experienced IgG1+ B cells (See details in text).
While isotype-switched antibodies more effectively protect the host against infection, they can also aggravate autoimmune diseases, such as IgG2a in systemic lupus erythematosus (SLE) (Boes et al., 2000; Ehlers et al., 2006; Ehrenstein et al., 1995; Jiang et al., 2007; Jiang et al., 2011; Korganow et al., 1999; Ohnishi et al., 1994; Peng, Szabo and Glimcher, 2002; Radic et al., 1995; Tsao et al., 1992; Vaughan, 1993), or cause allergic reactions, such as IgE in asthma (Platts-Mills, 2001). Defects in CSR/SHM also result in immunodeficiency, such as hyper-IgM syndrome, manifested by increased susceptibility to infections (Allen et al., 1993; DiSanto et al., 1993; Dobbs et al., 2007; Imai et al., 2003; Jain et al., 2001; Kasahara et al., 2003). Furthermore, to initiate CSR, B cells need to express a unique enzyme, activation-induced cytidine deaminase (AID) (Muramatsu et al., 2000). AID is a genome mutator and, if dysregulated, can cause genome-wide DNA double-stranded breaks (DSBs) that lead to chromosomal translocations and B cell lymphomagenesis (Alt et al., 2013; Chen and Wang, 2014; Daniel and Nussenzweig, 2013; Nussenzweig and Nussenzweig, 2010; Wang, 2013). Thus, to mount effective humoral immunity, signals emanating from the BCR, co-receptors and cytokine receptors need to be well-controlled to promote protection yet avoid detrimental effects. To achieve this goal, the signals from activating and inhibitory pathways need to be well-balanced. Hence, this review focuses on signal mechanisms regulating CSR.
1.2. Overview of molecular mechanisms of CSR
Antibodies are composed of heavy (H) and light (L) chains, encoded by immunoglobulin H (Igh) and L (Igl) chain genes, respectively, which can be divided into the N-terminal variable (V) region and C-terminal constant (C) region. The V region exon is assembled from variable (V), diversity (D), and joining (J) segments during V(D)J recombination that contains the antigen binding domain; thus, the V region of an antibody molecule determines the specificity of humoral immune responses. The C region of the mouse Igh locus consists of eight sets of CH exons, Cμ, Cδ, Cγ3, Cγ1, Cγ2b, Cγ2a, Cε, and Cα (Figure 1), encoding the C region of each corresponding isotype of antibody. Each set of CH exons is preceded with a switch (S) region except Cδ, termed Sμ, Sγ3, Sγ1, Sγ2b, Sγ2a, Sε, and Sα, respectively. An intronic promoter is located at the 5′ of each S region, whose activation is differentially induced by distinct transcription factors activated by signals originating from different co-receptors or cytokine receptors. The intronic promoter-initiated transcripts do not encode any protein, thus, are termed sterile germline transcripts (GLT) (Chaudhuri et al., 2007; Stavnezer and Schrader, 2014).
During pathogen infection or immunization, the BCR recognizes antigen and elicits the first signal to activate B cells. Meanwhile, B cells are also activated by the second signal delivered by CD40 interacting with CD40 ligand (CD40L) expressed on activated CD4 T cells, or by TLR interacting with pathogen associated molecular patterns (PAMP). The third signal is provided by cytokine receptors (e.g., IL-4R). These three signals coordinate to trigger AID expression and turn on the germline transcription of a specific set of CH exons (Figure 1). GLT facilitates the generation of single-stranded (ss) DNA in the S region, thereby allowing AID access. AID converts cytosine (C) to uracil (U) (Neuberger et al., 2003), generating U:G mismatch lesions in DNA. Subsequently, AID-initiated U:G mismatches are recognized by base excision repair (BER) and mismatch repair (MMR) and converted to DSBs at S regions (Chaudhuri and Alt, 2004). The broken upstream donor Sμ and downstream acceptor S regions are rejoined by non-homologous end-joining (NHEJ), while the intervening DNA sequence is excised as a circle (Figure 1) (Boboila, Alt and Schwer, 2012). Consequently, the antigen-binding V region is juxtaposed to a different downstream C region (e.g., Cγ1). This specific DNA recombination process at the Igh locus is termed CSR. CSR does not affect antigen specificity of antibody molecules since V region exons are not altered during CSR, but it generates different isotypes of antibodies that interact with different effector molecules (Wang, 2013).
The targeting mechanisms of AID to specific loci (Ig or non-Ig) remain incompletely understood, and may be influenced by many factors, e.g., unique features of DNA sequences, choice of downstream repair mechanism, transcription, cis regulatory elements, or AID co-factors (e.g. RPA, Spt5 etc.) (Basu et al., 2005; Basu et al., 2011; Chaudhuri, Khuong and Alt, 2004; Chen et al., 2016; Chen et al., 2014; Chen et al., 2012; Cheng et al., 2009; Nowak et al., 2011; Pavri et al., 2010; Wang, 2013; Xu et al., 2010; Yamane et al., 2011). The functions or proposed operating mechanisms of these co-factors have been reviewed extensively (Pavri and Nussenzweig, 2011; Stavnezer, 2011). The DNA repair elements are constitutively expressed and recruited to the Igh locus by AID-initiated DNA lesions. In contrast, AID expression and Igh GLT are induced only upon B cell activation by BCR, CD40, TLR or cytokine receptors (Xu et al., 2012; Zan and Casali, 2013). Thus, signals emanating from various receptors to induce CSR are convergent at regulating AID expression and Igh GLT. In addition, CSR-initiating signals may also regulate AID activity by phosphorylation, ubiquitination and subcellular localization (Montamat-Sicotte et al., 2015; Orthwein and Di Noia, 2012; Orthwein et al., 2010; Vaidyanathan et al., 2014).
2. Different receptors that signal to regulate CSR
Pathogen infection or antigen immunization can induce CSR via activating multiple receptors on B cells, including BCR, CD40, TLRs, BAFF-R and TACI as well as cytokine receptors for interleukin 4 (IL-4), interferon-γ (IFN-γ) and transforming growth factor-β(TGF-β), etc. CSR can be induced by in vivo immunization with T cell-dependent (TD) or -independent (TI) antigens. While such in vivo antigen immunization is of biological relevance, it has limitations for studying individual receptor contribution to CSR. First, in vivo immunization may activate multiple receptors on B cells such as BCR, CD40, and TLRs simultaneously. Second, these receptors may share common signaling pathways or may use distinct signaling elements to regulate CSR. For example, BAFF-R can activate BCR’s adaptor Igα and its downstream tyrosine kinase Syk (Schweighoffer et al., 2013), while TACI directly associates with TLR’s adaptor MyD88 to activate NF-κB transcription factor to induce CSR (He et al., 2010). Third, the limited numbers of B cells activated in vivo preclude in-depth biochemical analysis. Due to these limitations, it is difficult to pinpoint how signals originating from different receptors regulate CSR in vivo. Thus, in vitro induction of CSR using cultured naïve mature B cells with different stimuli is essential for dissecting how a single receptor individually or multiple receptors cooperatively signal to induce CSR. The beauty of an in vitro system is that one can dissect the specific role of each receptor and separate the effects of an individual signaling pathway.
CSR can be induced in vitro by engaging CD40 or TLRs in the presence of cytokines (e.g., IL-4) (Stavnezer and Schrader, 2014; Xu et al., 2012). Different cytokines direct switching to particular isotypes by inducing GLT of a specific CH gene (e.g., Iγ1-Cγ1) (Stavnezer and Schrader, 2014). For instance, TD or TI antigen-induced IgG1 CSR can be mimicked by anti-CD40/IL-4 or anti-Ig/TLR ligands/IL-4 stimulation, respectively (Matthews et al., 2014; Pone et al., 2012a; Pone et al., 2012b; Stavnezer and Schrader, 2014; Xu et al., 2012). Prior studies also employed the in vitro culture system to study how BCR regulates CSR. Using anti-Ig to mimic antigen-induced BCR activation, it was shown that neither anti-IgM/IL-4 (Heltemes-Harris et al., 2008) nor anti-δ/IL-4 (Pone et al., 2012b) induced CSR in WT B cells. On the other hand, engaging BCR does synergize TLRs or TACI to induce more robust CSR (Pone et al., 2012b). However, a high dose of anti-IgM inhibits lipopolysaccharide (LPS)/IL-4-induced CSR (Heltemes-Harris et al., 2008; Jabara et al., 2008) and delays the expression of AID (Jabara et al., 2008). It is unknown whether BCR cross-linking with anti-IgM accurately reflects antigen stimulation, namely, it remains to be addressed how different doses of antigenic stimulation modulates CD40 or LPS/IL-4-induced CSR. Taken together, these findings suggest that the BCR, however essential in vivo for humoral immunity, is not able to induce CSR.
While the BCR plays a central role in activating B cells and determining the specificity of isotype-switched antibodies, how BCR signaling regulates CSR is poorly understood. Why cannot stimulating BCR induce CSR in the presence of cytokines? Is it because BCR-elicited signal is not qualitatively or quantitatively sufficient to induce CSR? Alternatively, are there any regulatory mechanisms that prohibit the BCR from inducing CSR? These are fundamental questions for B cell biology and humoral immunity; however, it has received little attention so far due to the lack of proper models. To address these questions, we need to develop novel in vitro systems that allow engaging BCR to induce CSR in the presence of cytokines as engaging CD40 or other co-receptors does.
Lastly, it was reported that adenosine receptors can be activated by adenosine produced by CD39 and CD73 in a B cell autonomous manner to induce CSR (Schena et al., 2013). Estrogen receptor can directly bind the AID promoter to induce AID expression (Pauklin et al., 2009), or directly bind the promoter of transcription factor HoxC4 to turn on HoxC4 expression (Mai et al., 2010), which in turn promotes AID expression and CSR.
2.1. The role of BCR signaling components in CSR
The BCR of mature naïve B cells is composed of the antigen recognition part, IgM/IgD, and signal transduction part, Igα (CD79A) and Igβ (CD79B) heterodimer. The intracellular domains of Igα/Igβ contain immune receptor tyrosine-based activation motifs (ITAM) responsible for transducing signals. The BCR can be activated during infection or immunization by interacting with antigen, or by engaging BCR in vitro with anti-IgM, anti-IgD or specific antigens, which leads to the clustering of BCR complex in membrane lipid rafts to form the BCR signalosome. Formation of the BCR signalosome subsequently recruits and activates protein tyrosine kinases, Src and Syk (spleen tyrosine kinase) (Rickert, 2013). Src kinase is a non-receptor membrane-associated tyrosine kinase. Src family kinases contain six conserved domains, and the N-terminal myristoylated domain attaches Src to the cellular membrane (Schamel and Reth, 2000). Src family kinases include Src, Yes, Fyn, Fgr, Lck, Hck, Blk, and Lyn. Lck is predominantly expressed in T cells, while Lyn is mainly expressed in B cells. Syk family kinases are cytosolic protein tyrosine kinases and consist of two members, Syk and ZAP70. Syk is mainly expressed in B cells and ZAP70 in T cells. Lyn and Syk phosphorylate the ITAM of Igα/Igβ, which in turn further activates Syk. The function of Lyn is more complicated in BCR signaling because Lyn also phosphorylates immune receptor tyrosine-based inhibitory motifs (ITIM) of inhibitory receptors such as CD22, CD72 and FcγRIIb (CD32B). The inhibitory receptors, via their ITIM, recruit signaling suppressors, SH2 containing protein tyrosine phosphatase 1 (SHP1) to inactivate Src and Syk, and SH2 containing inositol 5′-polyphosphatase 1 (SHIP1) to inactivate phosphatidylinositol 3,4,5-triphosphate (PIP3) generated by phosphatidylinositol 3-kinases (PI3Ks). Signal transduction downstream of Syk can be classified into three major signaling pathways that could be activated simultaneously, whose ultimate outcome is to activate different transcription factors.
One of the BCR signaling pathways is composed of: BTK→BLNK→PLC-γ2→PKCβ→CBM complex→TRAF6/TRAF2→TAK1→NF-κB1 (Figure 2). Syk phosphorylates bruton tyrosine kinase (BTK) and B cell linker (BLNK). Btk mutation or deletion causes severe human immunodeficiency X-linked agammaglobulinemia (XLA) (Rawlings et al., 1993; Tsukada et al., 1993). XLA patients are unable to produce isotype-switched antibody (Kinnon et al., 1993), and this defect in XLA patients is attributed to a severe block in B cell development at the pre-B transition stage (Desiderio, 1997). However, it remains unknown whether BTK regulates CSR per se in mature B cells.
Figure 2. BCR signaling pathways.
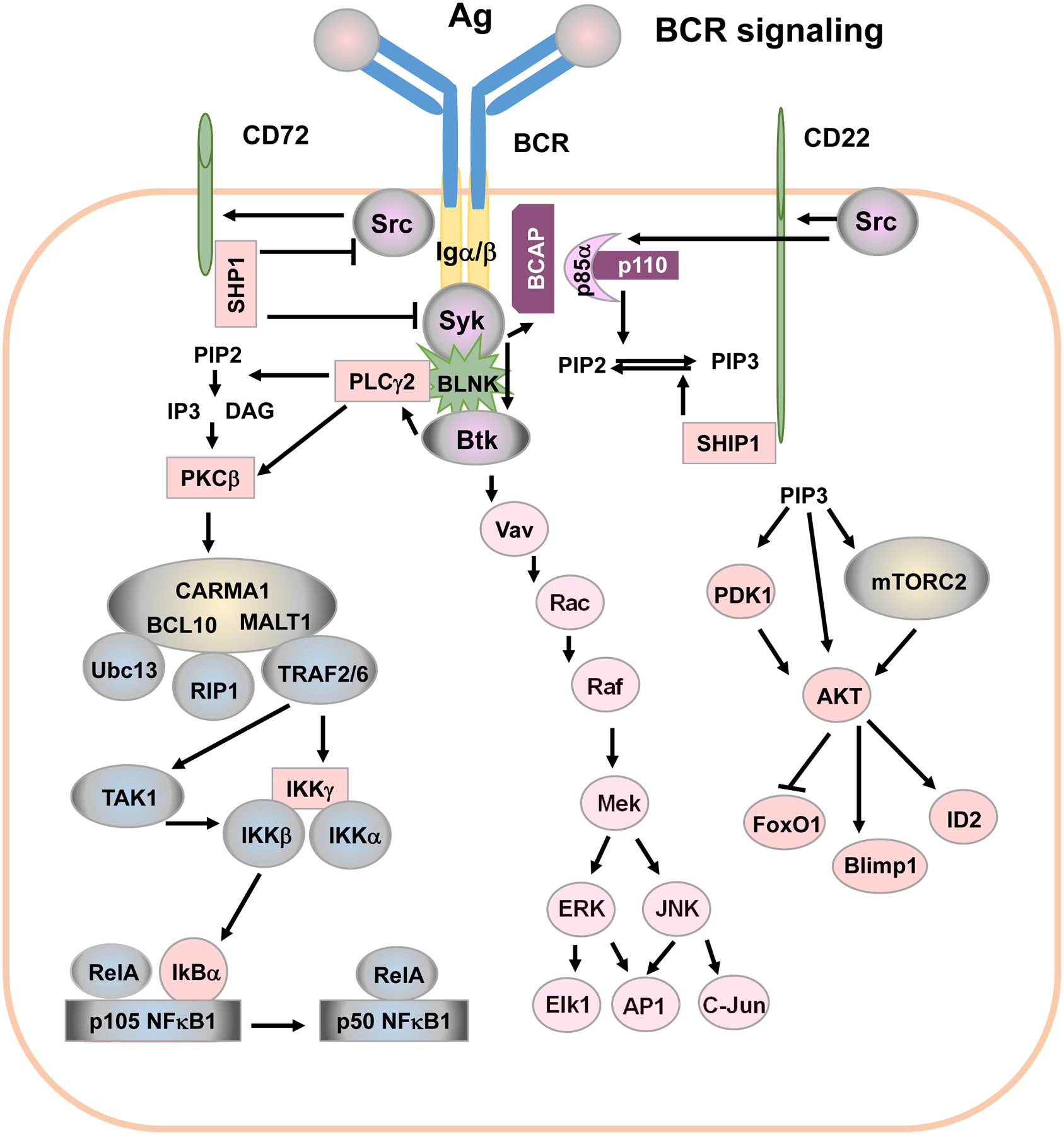
BCR is activated by antigens. BCR’s adaptor Igα and Igβ contain ITAM. Engaging BCR by antigen recruits Src and Syk to phosphorylate Igα β ITAM. Phosphorylated ITAM recruits BCAP to activate the PI3K/AKT signaling pathway. Src also phosphorylates ITIM of CD22 and CD72, which recruits signaling suppressors SHP1 and SHIP1 to enforce a negative feedback regulation of BCR signaling. Signal transduction downstream of Syk can be classified into three major pathways: (1): BTK→BLNK→PLC-γ2→PKCβ→CBM complex→TRAF6/TRAF2→TAK1→NF-κB1; (2): Vav→Rac→Raf→JNK→c-Jun/AP-1; (3) BCAP→PI3K→AKT→Foxo1/Blimp-1/ID2 (See details in text).
BTK phosphorylates BLNK to fully activate BLNK (Figure 2). BLNK then functions as a scaffold to orchestrate multiple signaling elements; for example, BLNK recruits Syk and BTK to phosphorylate PLC-γ2. PLC-γ2 then converts 1-phosphatidyl-1D-myo-inositol 4,5-bisphosphate to 1D-myo-inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 and DAG are second messenger molecules important for transmitting signals of growth factor receptors and immune system receptors. Genetic mutations of PLC-γ2 result in antibody deficiency in human patients (Ombrello et al., 2012). PLCγ2-deficient mouse B cells do not flux calcium, and PLCγ2-deficient mice have significantly reduced serum levels of IgM, IgG2a, and IgG3, likely due to a decreased number of peripheral B cells (Hashimoto et al., 2000; Wang et al., 2000). Again, it remains unknown whether PLCγ2 regulates CSR process intrinsically in mature B cells.
IP3 upregulates intracellular calcium, which, together with DAG, activates protein kinase C β (PKC-β). PKC-β plays an important role in BCR-mediated NF-κB1 activation (also called canonical NF-κB) by phosphorylating caspase recruitment domain 11 (CARD11) (also known as CARMA1) (Shinohara et al., 2005; Sommer et al., 2005). The phosphorylation of CARMA1 facilitates the assembly of the CBM complex (CARMA1, B cell leukemia 10 (BCL10) and mucosa-associated lymphoid tissue 1 (MALT1)) (Figure 2). Interaction of BCL10 and MALT1 recruits receptor interacting protein 1 (RIP1). RIP1 interacts with ubiquitin-conjugating enzyme 13 (Ubc13) as well as tumor necrosis factor receptor associated factor 2 (TRAF2) and/or TRAF6 to mediate K63 polyubiquitination of inhibitor of kappa B (I0κB) kinase-γ (IKKγ, also called NEMO) to activate IKKγ. Activated IKKγ initiates IKK complex assembly. The CBM complex can also bring TGF-β activated kinase 1 (TAK1) to IKKβ, and TAK1 subsequently phosphorylates IKKβ. Activated IKKβ then phosphorylates IκBα and triggers ubiquitination-mediated IκBα degradation, which promotes the conversion of p105 to p50 (NF-κB1) and releases p50/RelA or p50/c-Rel. The heterodimer of p50/RelA or p50/c-Rel translocates to the nucleus and activates transcription of many genes. NF-κB1 is implicated in inducing AID transcription (Gourzi, Leonova and Papavasiliou, 2007; Park et al., 2009) and BCR engagement activates NF-κB1 (Patterson et al., 2006; Schulze-Luehrmann and Ghosh, 2006); however, BCR engagement cannot induce AID expression (Kuraoka et al., 2017; Pone et al., 2012b) (see more discussion below). This is likely because AID expression needs additional transcription factors (see below). Prior studies reported that dextran-conjugated anti-IgD induced NF-κB2 activation (non-canonical or alternative NF-κB) (Pone et al., 2012b). However, the detailed signaling pathways remain to be elucidated by which engaging BCR induces NF-κB2 expression and activation.
One of the BCR signaling pathways consists of Vav→Rac→Raf→JNK→c-Jun/AP-1 (Figure 2). Through BLNK and/or PIP3 generated by PI3K, BCR engagement recruits Vav, a guanine nucleotide exchange factor, to the plasma membrane, where Vav is phosphorylated and activated by Syk. Through Rac/Rho small G proteins, the Vav pathway leads to the activation of c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinases (ERK). Activated JNK phosphorylates and activates transcription factors, c-Jun and AP-1. JNK activation was shown to be critical for AID expression and IgE CSR induced by CD40 (Jabara and Geha, 2005). Although BCR signaling can also induce JNK activation, it is unclear why BCR cannot induce CSR.
One of the signaling pathways includes: BCAP→PI3K→AKT→Foxo1/Blimp-1/ID2 (Figure 2). PI3K is composed of an regulatory subunit, predominantly, p85α, and an catalytic subunit p110 (Koyasu, 2003) (see details below). Syk phosphorylates B cell adaptor protein (BCAP), which recruits p85, a regulatory subunit of PI3Ks, and activates PI3K catalytic subunit p110. Then, the catalytic subunit p110 phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate PIP3. PIP3 activates many signaling elements that contain pleckstrin homology (PH) domain via recruiting them to the plasma membrane, including BTK, protein kinase B (AKT) and 3-phosphoinositide-dependent protein kinase-1 (PDK1), etc. AKT is activated by phosphorylation at threonine 308 by PDK1 and serine 473 by the mammalian target of rapamycin complex 2 (mTORC2). Subsequently, activated AKT targets numerous proteins downstream that regulate cell proliferation, differentiation, survival and apoptosis (Manning and Cantley, 2007; Zhang et al., 2011).
The PI3K/AKT pathway regulates the activity of two important transcription factors, fork head box O1 (FOXO1) and B lymphocyte-induced maturation protein-1 (BLIMP-1), as well as inhibitor of DNA binding 2 (ID2). AKT phosphorylates FOXO1 and inactivates FOXO1 by promoting its export from the nucleus (Zhang et al., 2011). FOXO1 is a transcription factor that positively regulates AID expression since the deletion of foxo1 in B cells reduces the level of AID expression (Dengler et al., 2008). PI3K/AKT signaling activates BLIMP-1 (Setz et al., 2018). BLIMP-1 is a transcription factor that regulates B cell differentiation to antibody-producing plasma cells, triggers apoptosis of activated B cells and inhibits AID transcription (Minnich et al., 2016; Shaffer et al., 2002). The PI3K/AKT pathway upregulates ID2 expression (Belletti et al., 2001). ID2 was reported to prevent several transcription factors from binding the AID promoter, thereby, inhibiting AID expression (Gonda et al., 2003). Taken together, although BCR induced-PI3K/AKT activation is required for B cell activation, the PI3K/AKT axis negatively regulates AID expression and CSR, and inhibits antibody responses (see more discussion below).
In summary, the BCR is essential for B cell survival (Kraus et al., 2004; Srinivasan et al., 2009), activation and specific antigen recognition. However, signaling by the BCR alone cannot induce CSR in vitro (Heltemes-Harris et al., 2008; Pone et al., 2012b), and the current paradigm is that CSR activation requires co-stimulation of BCR with ligands for CD40 or TLRs, etc (Matthews et al., 2014; Pone et al., 2012a; Pone et al., 2012b; Stavnezer and Schrader, 2014; Xu et al., 2012). It remains to be addressed why the BCR does not induce CSR without co-stimulation.
2.2. CD40 and its downstream signaling molecules
CD40 is a member of the tumor necrosis factor (TNF) receptor (TNFR) family and is expressed on various cell types including B cells (Bishop et al., 2007). CD40 is a co-receptor of B cells and plays a critical role in B cell-mediated humoral immunity. CD40 ligand (CD40L) or CD154 belongs to TNF family and is expressed on activated CD4 T cells. When B cells present antigens to CD4 T cells, CD40L interacts with CD40 to elicit signal transduction. CD40 signaling promotes germinal center (GC) formation and induces SHM for antibody affinity maturation and CSR for isotype-switching in activated B cells. Thus, defects in CD40/CD40L abrogate TD antigen-induced CSR and IgG or IgA production in mice (Kawabe et al., 1994; Xu et al., 1994), and cause Hyper-IgM syndrome (HIGM) in humans (Allen et al., 1993; Nonoyama et al., 1993). HIGM is a subtype of primary immunodeficiency disease (PID) characterized by normal or elevated serum IgM and low or absent serum IgG, IgA and IgE (Etzioni and Ochs, 2004). HIGM is caused by genetic mutations in molecules that regulate CSR (Allen et al., 1993; Etzioni and Ochs, 2004; Nonoyama et al., 1993; Qamar and Fuleihan, 2014). Although CD40 mutations are rare, CD40L mutations are the most common cause of HIGM (Etzioni and Ochs, 2004; Qamar and Fuleihan, 2014). Together, CD40 and CD40L defects account for more than 60% of HIGM cases (Etzioni and Ochs, 2004).
The role of TRAFs in CD40-induced CSR
TD antigen-mediated CD40-induced CSR can be mimicked in culture by stimulating naïve B cells with CD40L or agonist anti-CD40 antibody, together with cytokines (e.g., IL-4). Upon activation, monomers of CD40 cluster to form trimeric CD40s in membrane lipid rafts that recruit downstream signaling molecules, among which are TRAFs, including TRAF1, TRAF2, TRAF3, TRAF5 and TRAF6 (Figure 3). TRAF1-null B cells have no CSR defect (Tsitsikov et al., 2001). TRAF5 knockout (KO) mice can produce IgG in response to TD antigen immunization but TRAF5 KO B cells produce less IgG upon CD40 engagement in vitro (Nakano et al., 1999). Apart from genetic deletion of TRAFs, wild-type (WT) or mutant CD40 lacking the ability to bind TRAF6 were employed to restore CD40 expression in B cells of CD40−/− mice (Jabara et al., 2002). TRAF6 binding ability of CD40 was not required for antibody isotype-switching in response to in vivo TD antigen immunization or in vitro CD40 engagement (Jabara et al., 2002), suggesting a dispensable role of TRAF6 in CSR. However, B cell-specific deletion of TRAF6 impaired IgG isotype-switching in response to TD antigen immunization and TRAF6 KO B cells responded poorly to in vitro stimulation of CD40 and TLRs (Lomaga et al., 1999). Taken together, these studies suggest that TRAF6 binding ability of CD40 does not accurately represent TRAF6’s function, at least, in transducing signals to induce AID expression and CSR. One possibility is that, when CD40 loses its binding ability to TRAF6, TRAF2 and/or TRAF3 can recruit TRAF6 to CD40 signalosome to activate downstream signals to induce AID expression and CSR.
Figure 3. CD40 signaling pathways.
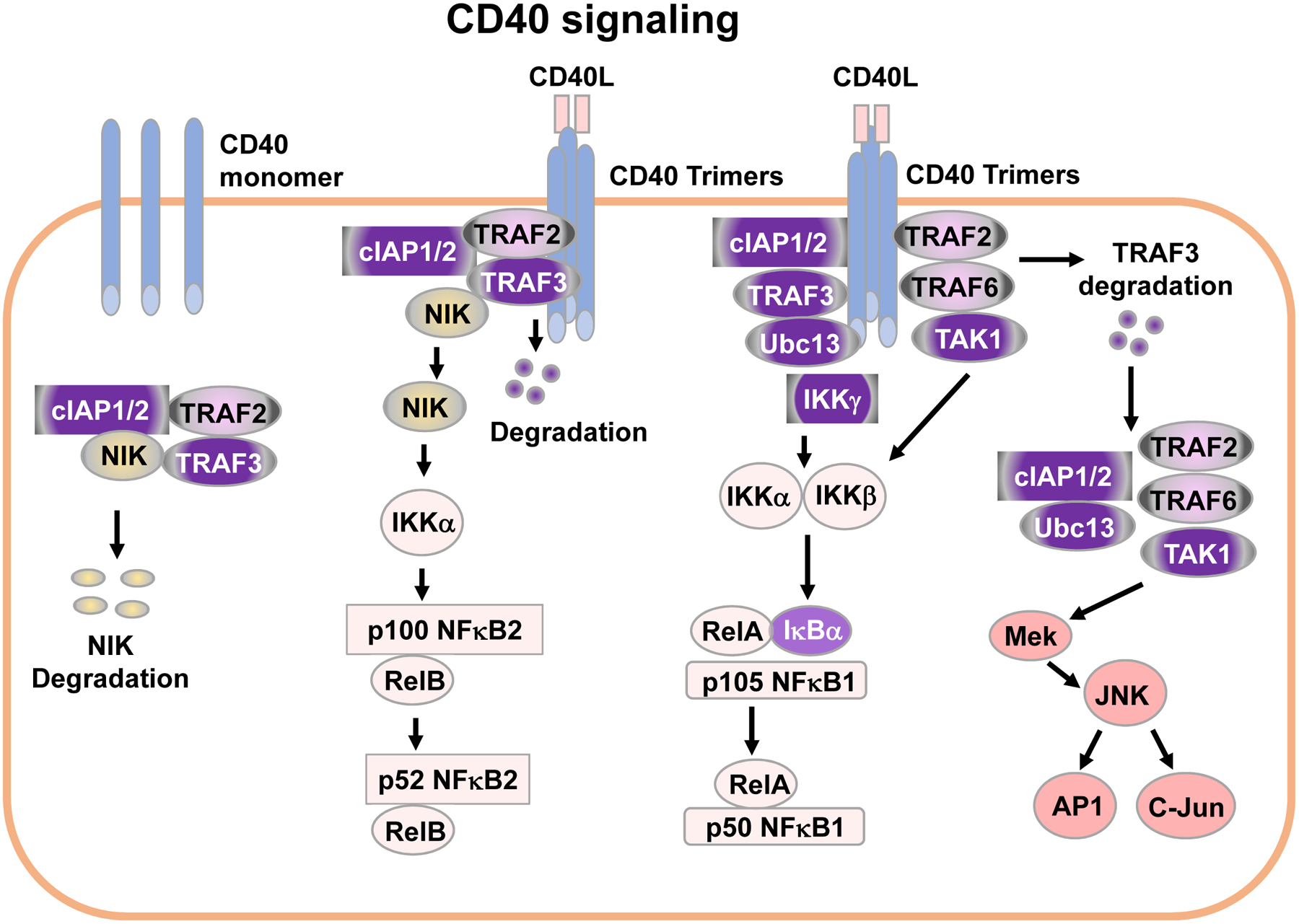
In resting B cells, NIK is constitutively degraded. Upon engagement by CD40L, monomers of CD40 cluster to form CD40 trimers in membrane lipid rafts, which recruit downstream signaling molecules including TRAF1, TRAF2, TRAF3, TRAF5 and TRAF6. TRAFs transduce signals leading to activation of NF-κB1 and NF-κB2 as well as MAPK pathways (See details in text).
CD40 mutants that lacked the ability to bind TRAF2 or TRAF3 or both were also employed to restore CD40 expression in B cells of CD40−/− mice (Jabara et al., 2009). This study reported that both B cell-specific TRAF2 (B-TRAF2) and TRAF3 are required for CD40-induced antibody isotype-switching and the two proteins independently signal to induce CSR upon CD40 engagement (Jabara et al., 2009). Contrary to these findings, B cell-specific TRAF3 (B-TRAF3) KO mice showed no defects in isotype-switching in response to TD antigen immunization (Xie et al., 2007); moreover, engaging CD40 in vitro induced significantly elevated secretion of IgG1 in TRAF3-KO B cells compared to WT B cells (Xie et al., 2011). These studies suggested that B-TRAF3 is dispensable for CD40-induced isotype-switching. Thus, the role of B-TRAF3 in TD humoral immune responses and CD40-induced CSR remained controversial. It is necessary to ascertain TRAF3’s role in the CD40 signaling pathway because therapeutic approaches are emerging in PID, inflammation, and cancer by targeting CD40 signaling (Beatty, Li and Long, 2017; Meng, Yang and Suen, 2018). Despite of a number of previous studies investigating the role of TRAF2 in immune responses, CD40 signaling, B cell development and survival as well as lymphoid organ homeostasis (Gardam et al., 2008; Grech et al., 2004; Hostager et al., 2003; Jabara et al., 2009; Nguyen et al., 1999), it remained unknown whether and how TRAF2 deletion in B cells affects CD40-induced CSR and TD humoral immune responses. In this regard, our recent studies clarify the controversial role of B-TRAF3 and confirm its dispensability in CD40-induced CSR (Woolaver et al., 2018). Furthermore, we found that TRAF2 is essential for TD humoral immunity and CD40-induced CSR (Woolaver et al., 2018). Mechanistically, CD40-induced AID expression was markedly impaired by B-TRAF2 deficiency, whereas B-TRAF3 deficiency did not affect CD40-induced AID expression (Woolaver et al., 2018). Thus, our studies provide significant biological bases for optimizing therapeutic strategies to treat B cell-associated immune disorders by targeting CD40 signaling.
TRAF-mediated downstream CD40 signaling
Engaging CD40 activates TAK1 by recruiting TRAF2/TRAF6 (Figure 3) (Arcipowski and Bishop, 2012; Sato et al., 2005; Song et al., 2014; Wang et al., 2012). When TRAF2 and TRAF6 are recruited to membrane receptor complex, they function together to enhance the K63 ubiquitin ligase activity of cellular inhibitor of apoptosis 1/2 (cIAP1/2). cIAP1/2 then function together with Ubc13 to add K63 polyubiquitin to IKK-γ to activate IKK-γ. Then, activated IKKγ recruits TAK1 to phosphorylate IKKβ and promote IKKα/β/γ complex assembly, which phosphorylates IκBα and triggers ubiquitination-mediated IκBα degradation. Removal of IκBα promotes the cleavage of NF-κB1 precursor p105 into active p50, and facilitates the nuclear translocation of the RelA/p50 heterodimer to initiate target gene transcription.
In resting B cells, TRAF3 associates with NF-κB inducing kinase (NIK), while TRAF2 associates with cIAP1/2 (Figure 3). Within this cytoplasmic complex, TRAF2 and TRAF3 heteromeric interaction allows cIAP1/2 to induce NIK polyubiquitination and target NIK for degradation (Figure 3). Thus, in resting B cells, the steady level of NIK is very low. Upon CD40 clustering, the TRAF2/TRAF3/cIAP1/2/NIK complex is recruited to the CD40 signalosome in membrane rafts where TRAF2/TRAF3 interact with CD40 that directs cIAP1/2 to degrade TRAF3 (Hostager et al., 2003; Liao et al., 2004) (Figure 3). TRAF3 degradation releases the TRAF2/TRAF3/cIAP1/2/NIK signaling complex from membrane-associated CD40 to activate the downstream MAPK pathway (e.g., JNK), and releases NIK to allow cytoplasmic NIK accumulation. NIK in turn phosphorylates IKKα. Activated IKKα phosphorylates NF-κB2 p100 and triggers p100 proteolytic cleavage into active NF-κB2 p52. The heterodimer of RelB/p52 then translocates into the nucleus and initiates target gene transcription. NF-κB2 is constitutively activated in B-TRAF2 or B-TRAF3 KO B cells (Gardam et al., 2008; Woolaver et al., 2018; Xie et al., 2007). Therefore, both TRAF2 and TRAF3 are negative regulators of CD40-induced NF-κB2 activation. NIK level is also negatively regulated by TANK binding kinase 1 (TBK1) in a TRAF3-independent manner (Jin et al., 2012). TBK1 deletion in B cells causes hyper-activation of NF-κB2, which leads to abnormally increased IgA production and IgA nephropathy (Jin et al., 2012).
Both NF-κB1 and NF-κB2 are implicated in inducing AID expression (Zan and Casali, 2013). Specifically, the NF-κB2 (p52) and the NF-κB1 (p50) subunits can be recruited to the AID promoter region (Gourzi, Leonova and Papavasiliou, 2007; Park et al., 2009); and the RelA subunit can be recruited to an upstream enhancer element (Tran et al., 2010). In addition, p50−/− primary mouse B cells exhibited defects in CSR and AID expression (Gourzi, Leonova and Papavasiliou, 2007). Consistently, deletion of either NF-κB1 or NF-κB2 markedly impairs TD humoral immune responses (Caamano et al., 1998; Sha et al., 1995). However, it remained incompletely understood how TRAF2 and TRAF3 regulate CD40-induced AID expression. Our recent studies showed that B-TRAF2 deficiency impairs AID expression and CSR induced by engaging CD40 but not by engaging TLRs (Woolaver et al., 2018). Mechanistically, B-TRAF2 deficiency causes defective activation of the NF-κB1 complex in a CD40-autonomous manner, whereas TRAF3 is dispensable for CD40-induced NF-κB1 activation (Woolaver et al., 2018). Moreover, restoring CD40-induced NF-κB1 activation in TRAF2-deficient B cells rescues AID expression and CSR (Woolaver et al., 2018). Taken together, both TRAF2 and TRAF6 contribute to CD40-induced NF-κB1 activation, while our study suggests that each of them plays a non-redundant role in this process. Our study also implies that simultaneous activation of NF-κB1 and NF-κB2 may be essential for inducing AID expression and CSR.
CD40-induced AKT activation
Engaging CD40 also activates the AKT pathway (Deregibus et al., 2003); however, the mechanism directing signal transduction of CD40-induced AKT activation is poorly understood. Prior studies showed that the neural precursor cell expressed developmentally downregulated protein 4 (NEDD4), an E3 ubiquitin ligase, is a component of the CD40 signaling complex (Fang et al., 2014). NEDD4 constitutively interacts with CD40 and mediates K63-linked ubiquitination of TRAF3 (Fang et al., 2014). Furthermore, NEDD4-mediated TRAF3 ubiquitination is critical for CD40-induced AKT activation (Fang et al., 2014). NEDD4−/− splenic B cells expressed a higher level of AID transcripts and exhibited a higher level of IgG CSR, and NEDD4−/− mice produced a higher level of total IgG and antigen-specific IgG (Fang et al., 2014). These results are consistent with a negative role of AKT activation in AID expression and CSR. However, B-TRAF3 KO B cells had no defects in CD40-induced AKT activation (unpublished data), suggesting that embryonic fibroblast cells (Fang et al., 2014) and primary B cells might employ differential mechanisms to regulate CD40-induced AKT activation. In line with this notion, other studies report that engaging CD40 in B cells barely induces K63-linked ubiquitination of TRAF3 (Matsuzawa et al., 2008). Nevertheless, it is important to notice that CD40 stimulation not only activates NF-κB pathways to induce AID transcription but also activates the AKT pathway that inhibits AID expression and CSR.
2.3. The role of TLRs in regulating CSR
Structure and functions of TLRs
TLR family is a major subgroup of conserved pattern recognition receptors (PRRs). Human cells express 10 (TLR1–TLR10) and mouse cells express 12 (TLR1–TLR9, TLR11–TLR13) members of TLR family (Kawai and Akira, 2010). TLRs are expressed in innate and adaptive immune cells as well as non-immune cells (Kawai and Akira, 2010). Based on their cellular compartmentalization, TLRs can be categorized into two groups, cell surface TLRs and intracellular TLRs. The former group includes TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, and the latter group includes TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13 (Kawai and Akira, 2010).
Structurally, all TLRs contain an ectodomain, transmembrane and cytoplasmic domain (Botos, Segal and Davies, 2011). The ectodomain recognizes microbial membrane components such as lipids, lipoproteins, or proteins. TLRs recognize their respective PAMPs as a homo- or heterodimer (Botos, Segal and Davies, 2011). Different TLRs recognize different types of PAMPs (Kawai and Akira, 2010). For example, TLR4 recognizes bacterial LPS. TLR3 recognizes viral double-stranded RNA (dsRNA) and self-RNAs derived from damaged host cells (Bernard et al., 2012; Takemura et al., 2014; Zhang et al., 2007). TLR9 recognizes bacterial and viral DNA enriched in unmethylated CpG-DNA motifs (Dalpke et al., 2006; Takeshita et al., 2001). The transmembrane domain anchors TLRs either to plasma membrane or endosome or lysosome membrane. TLRs and IL-1Rs have a conserved region in their cytoplasmic tails, termed Toll/IL-1R (TIR) domain (Kawasaki and Kawai, 2014). This TIR domain of TLRs associates with TIR domain containing adaptors to transduce TLR signaling. TLR adaptors include myeloid differentiation primary-response protein 88 (MyD88), TIR-domain-containing adaptor protein (TIRAP) (also known as MyD88-adaptor-like protein), TIR-domain-containing adaptor protein inducing IFN-β (TRIF) (also known as TIR-domain-containing molecule 1 (TICAM1)), and TRIF-related adaptor molecule (TRAM) (also known as TICAM2) (Kawasaki and Kawai, 2014).
TLRs act as a critical link between innate and adaptive immunity, because TLR stimulation by PAMPs not only activates innate immunity but also contributes to adaptive immunity (Pone et al., 2012a). Mutations in genes encoding TLRs impair antibody responses (Andersen-Nissen et al., 2007; Bochud et al., 2007; Echchannaoui et al., 2002; Hawn et al., 2003; Kesh et al., 2005; Kimman et al., 2008; Thuong et al., 2007). In B cells, TLRs interact with PAMPs (e.g., LPS or CpG-DNA) to induce antibody responses against TI antigens (Pone et al., 2012a). TLR activation also plays a critical role during the early stage of TD antigen-induced antibody responses, when antigen-specific T cell help is not available to B cells yet. Moreover, TLRs in B cells can synergize with BCR signaling to induce more robust antibody responses by promoting AID expression and CSR (Pone et al., 2012b). The TLR9 ligand CpG DNA substantially boosted IgG2b and IgG2c antibody responses to protein antigens administered in the virus-like particles (Hou et al., 2011). However, engaging TLR alone in B cells does not induce CSR efficiently, except for TLR4 (Pone et al., 2012a) (see more discussion below). While it is controversial whether human B cells express TLR4 (Bekeredjian-Ding and Jego, 2009; Ganley-Leal et al., 2010; Hornung et al., 2002), TLR4 in mouse B cells plays an important role in inducing AID expression and CSR (Pone et al., 2012a).
TLR signaling pathways
All TLRs transduce signals via MyD88 except TLR3 that transduces signals via TRIF (Kawasaki and Kawai, 2014). TIRAP is a bridging adaptor that recruits cytosolic MyD88 to interact with the activated TIR domain of TLR4 (Figure 4). TLR4 can also transduce signals via TRIF that needs another bridging adaptor, TRAM (Figure 4). Other TLRs signal directly and only through MyD88 or TIRAP/MyD88. For MyD88-dependent signaling (Figure 4), once cytosolic MyD88 is recruited to membrane-associated TLRs, MyD88 recruits IL-1R-associated kinase 4 (IRAK4). IRAK4 interacts with IRAK1 and induces IRAK1 auto-phosphorylation. Phosphorylated IRAK1 recruits TRAF6 that eventually activates TAK1. TAK1 then activates two different pathways: the IKK complex-NF-κB pathway and MAPK pathway. TAK1 interacts with IKK-γ and phosphorylates IKK-β. The activated IKKα/β/γ complex then phosphorylates IκBα, leading to IκBα degradation. IκBα degradation allows the p50(NF-κB1)/RelA heterodimer to translocate into the nucleus and induce transcription of target genes, including AID.
Figure 4. TLR4 signaling pathways.
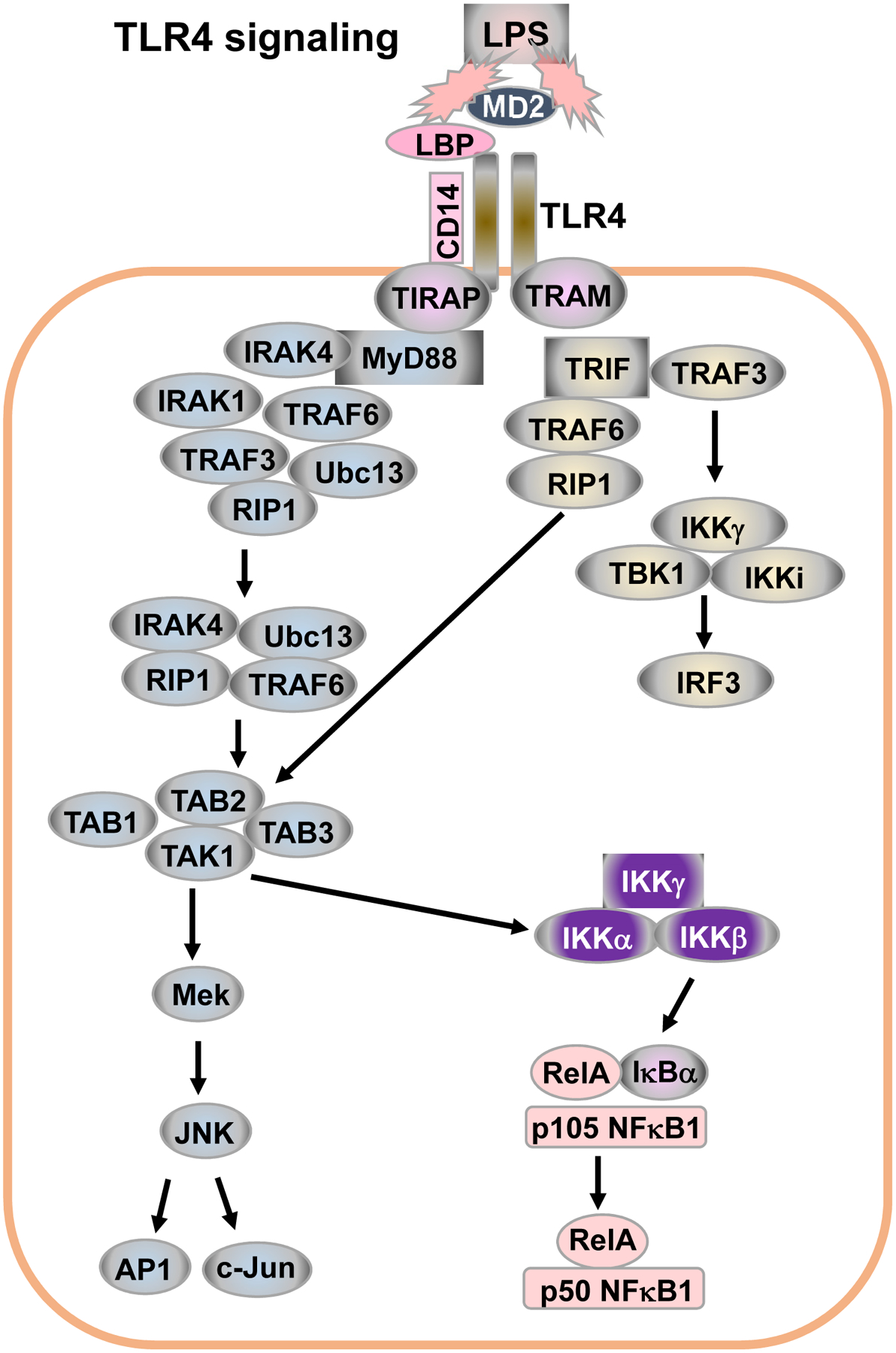
LPS binding protein (LBP) and MD2 facilitate the binding of LPS to CD14 and TLR4. TLR4 transduces signals via two pathways, namely, MyD88 dependent- and TRIF-dependent pathways, to activate NF-κB, MAPK and IRF3. (See details in text).
For TRIF-dependent signaling (Figure 4), TRIF interacts with TRAF6 and TRAF3. TRAF6 activates the TAK1 complex, leading to activation of NF-κB and MAPKs. TRAF3 recruits the IKK-related kinases TBK1, IKKi and IKK-γ for the phosphorylation of IFN-regulatory factor 3 (IRF3). Overall, TRAF6 activates MyD88-dependent and TRIF-dependent signaling pathways, whereas TRAF3’s role in TLR signaling is more complicated (Kawasaki and Kawai, 2014). TRAF3 promotes the activation of the TRIF-dependent pathway. TRAF3 was also shown to be recruited to the MyD88 complex in TLR4 signaling, and subsequently degraded, which led to TAK1 activation (Tseng et al., 2010). Thus, TRAF3 functions as a suppressor of the MyD88-dependent signaling pathway.
Patients deficient in MyD88 or IRAK4 exhibited impaired antibody responses to capsular bacteria, and presented with recurrent pyogenic infections (Picard, Casanova and Puel, 2011; Picard et al., 2010). In addition, deficiency in MyD88 or IRAK4 leads to a decreased IgG response to polysaccharidic antigens (Picard et al., 2010), supporting a role of the dual BCR/TLR engagement by PAMPs in IgG responses. B cell-specific MyD88 KO mice exhibited defective antibody responses to inactivated influenza virus (Hou et al., 2011). Consistent with a role of TRAF3 in suppressing MyD88-dependent signaling, B-TRAF3 deficiency leads to elevated IgG production in response to TI antigen immunization in vivo (Woolaver et al., 2018; Xie et al., 2011; Xie et al., 2007), and B-TRAF3 KO B cells exhibit a higher level of IgG1 CSR in response to LPS/IL-4 stimulation in vitro (Woolaver et al., 2018).
TLRs can also activate the PI3K pathway by recruiting PI3K regulatory subunit p85 (Akira and Takeda, 2004). TLR cytoplasmic domains contain YXXM motifs that can directly interact with p85 (Akira and Takeda, 2004). Due to the association of p85 with TLR, PI3K catalytic subunit p110 can be recruited to the plasma membrane where activated PI3Ks synthesize PIP3. PIP3 then activates AKT. MyD88 also contains an YXXM motif that can recruit p85 (Laird et al., 2009), thereby leading to PI3K/AKT activation. Taken together, TLR stimulation activates not only the NF-κB pathway but also the PI3K/AKT pathway that can negatively regulate AID expression and CSR.
Engaging TLR alone on mature naïve B cells induces AID expression and CSR to different classes of isotype-switched antibodies (IgG, IgA or IgE) in the presence of corresponding cytokines; however, the level of AID expression and CSR remains relatively low (Pone et al., 2012a). Co-engaging BCR and TLR causes synergy to induce a much higher level of AID expression and CSR, and the synergy of co-engaging BCR and TLR can more effectively activate NF-κB1 and NF-κB2 in a p85-dependent manner (Pone et al., 2012b). In the presence of cytokines (e.g., IL-4 or TGF-β), LPS stimulates mature naïve mouse B cells to induce a maximal level of AID expression and CSR comparable to that induced by engaging CD40 or co-engaging TLRs and BCR (Pone et al., 2012b; Woolaver et al., 2018). This robustness of LPS-induced CSR may be attributed to the ability of LPS to activate the BCR, TLR4 and co-receptor CD14 simultaneously to induce stronger downstream signals (Lu, Yeh and Ohashi, 2008; Pone et al., 2012a). Interestingly, LPS without additional cytokines induces IgG3 CSR, whereas, LPS plus IL-4 induces IgG1 and IgE CSR, directed by IL-4.
TLRs can synergize with the BCR to induce the production of IgG and IgA against specific antigens, which is especially important for combating early viral and bacterial infections, given that antigen-specific T cell help is not available to B cells at this point (Pone et al., 2012a). However, when autoantigen is present, TLRs may exaggerate autoimmune manifestations by synergizing with an autoreactive BCR to induce isotype-switched autoantibody, such as IgG2a seen in SLE of humans and mouse models (Avalos, Busconi and Marshak-Rothstein, 2010; Suurmond and Diamond, 2015).
2.4. TACI and BAFF-R
BAFF and a proliferation-inducing ligand (APRIL) are TNF family members. Both BAFF and APRIL interact with three specific receptors, TACI, B cell maturation antigen (BCMA), and BAFF-R, which are TNFR family members (Zhang et al., 2015). In the presence of cytokines, APRIL and BAFF can stimulate TACI to induce AID expression and CSR, while only BAFF can stimulate BAFF-R to do so (Castigli et al., 2005). Neither APRIL nor BAFF can stimulate BCMA to induce AID expression and CSR in the presence of cytokines (Castigli et al., 2005). TACI activates NF-κB and synergizes with BCR, CD40 or TLR signaling to induce AID expression and CSR (Pone et al., 2012b). Reciprocally, BCR and TLRs potentiate TACI and BAFF-R signaling by promoting the expression of receptors and their ligands (Abu-Rish, Amrani and Browning, 2013; Smith and Cancro, 2003; Uslu et al., 2014).
TACI and BAFF-R both recruit TRAFs for downstream signal transduction; BAFF-R only interacts with TRAF3 while TACI can interact with TRAF2, TRAF5 and TRAF6 (Zhang et al., 2015). Of note, TRAF2 is also required for BAFF-R to induce NF-κB2 activation, and TRAF2 mediates this effect by recruiting TRAF3 to cIAP1/2 for TRAF3 K48-linked ubiquitination and degradation. TRAF3 degradation allows cytoplasmic NIK accumulation that eventually leads to NF-κB2 activation. TACI and BAFF-R function similarly in inducing AID expression and CSR (Castigli et al., 2005). However, TACI and BAFF-R exhibit different effects on B cell survival, proliferation, expansion, and differentiation as well as GC formation, because TACI and BAFF-R compete for BAFF (Ou, Xu and Lam, 2012; Zhang et al., 2015). BAFF-R signaling promotes B cell survival, proliferation and expansion as well as GC formation. In contrast, TACI inhibits B cell proliferation and expansion, and promotes apoptosis (Sakurai et al., 2007; Seshasayee et al., 2003; Zhang et al., 2015). TACI upregulates the expression of cIAP1/2 in GC B cells, thereby inhibiting the BAFF-R-mediated NF-κB2 activation and GC formation (Seshasayee et al., 2003). TACI signaling induces the expression of transcription factor BLIMP-1 that arrests B cell activation, promotes B cell terminal differentiation into antibody-producing plasma cells and inhibits AID transcription (Minnich et al., 2016; Shaffer et al., 2002).
An intriguing observation is that TACI intracellular domain has a conserved motif that can directly interact with MyD88 to transduce signals to induce AID expression and CSR (He et al., 2010). Upon engagement by BAFF or APRIL, TACI can associate with MyD88 that recruits IRAK1 and IRAK4, then this assembled receptor complex recruits TRAF6 that eventually activates TAK1 by K63-linked poly-ubiquitination. As discussed above in TLR signaling, TAK1 promotes IKK complex assembly and activation, leading to activation and nuclear translocation of the p50(NF-κB1)/RelA heterodimer. TACI can also activate p52(NF-κB2)/RelB via interacting with TRAF2 and TRAF3. Both NF-κB1 and NF-κB2 are implicated in inducing AID transcription (Zan and Casali, 2013). On the other hand, MyD88 can also activate the PI3K/AKT pathway (Laird et al., 2009) that inhibits AID expression and CSR. MyD88 can directly interact with PI3K regulatory subunit p85 (Laird et al., 2009), which activates the PI3K signaling pathway. Thus, PI3K/AKT may serve as a negative feedback regulator of TACI-induced AID expression and CSR.
2.5. Other receptors
CD73 is a plasma membrane protein and functions as an ecto-5′-nucleotidase (Deaglio and Robson, 2011). CD73 deficiency is associated with common variable immune deficiency (CVID) (Schena et al., 2013), a subtype of PID. Notably, CVID patients with impaired isotype-switched antibody responses exhibited defects in CD73 expression in B cells selectively (Schena et al., 2013). CD39 is a member of the ectonucleoside triphosphate diphosphohydrolase (E-NTPDase) family that includes four plasma membrane-bound members: CD39 (NTPDase1), NTPDase2, NTPDase3, and NTPDase8 (Robson, Sevigny and Zimmermann, 2006). These E-NTPDase enzymes can hydrolyze, with varying affinities, nucleoside triphosphates and diphosphates to corresponding monophosphate derivatives (e.g., ATP and ADP to AMP). Co-engagement of BCR and TLR results in the release of ATP stored in Ca2+-sensitive secretory vesicles, then CD73 and CD39 function together to hydrolyze ATP to adenosine, which induces CSR in a B cell autonomous manner (Schena et al., 2013). Adenosine initiates its biological effects via binding four adenosine receptors, A1, A2A, A2B and A3 subtypes, which are G protein-coupled receptors (Sheth et al., 2014). Activation of the A2A and A2B adenosine receptors increases cyclic AMP (cAMP) production, resulting in activation of the protein kinase A (PKA) and phosphorylation of the cAMP response element binding protein (CREB) (Sheth et al., 2014). CD73-deficient human B cells have impaired CSR induced by engaging BCR and TLRs (Schena et al., 2013). PKA-mediated phosphorylation at serine 38 of AID is essential for an optimal level of CSR (Basu et al., 2005; Cheng et al., 2009; McBride et al., 2008). It would be of interest to elucidate whether adenosine receptor signaling regulates AID’s activity by activating PKA during B cell stimulation for CSR induction in vitro and antibody responses in vivo. Studies as such may lead to mechanistic insight to explain why CD73 deficiency is associated with CVID.
Clinical and epidemiological studies have implicated that females tend to develop stronger and more rapid immune responses when encountering antigens (Butterworth, McClellan and Allansmith, 1967; Eidinger and Garrett, 1972). This gender bias is also observed in immune disorders such as allergic disease (e.g., asthma), and autoimmune disease (e.g., SLE) (Favalli et al., 2018; Gold et al., 2018; Li and McMurray, 2007; Nalbandian and Kovats, 2005; Peeva and Zouali, 2005). Estrogen, a female-specific hormone, enhances antibody and autoantibody responses, possibly through upregulating AID expression. It was initially reported that estrogen receptor directly bound and activated the AID promoter to induce AID expression and CSR (Pauklin et al., 2009). Later, it was found that estrogen receptor directly bound the promoter of transcription factor HoxC4 and activated HoxC4 expression (Mai et al., 2010), which in turn induced AID expression. The later study also found that binding to the AID promoter is not necessary for estrogen receptor signaling to synergize AID expression and CSR induced by CD40 and TLR (Mai et al., 2010). Administrating estrogen in vivo also significantly enhances CSR and SHM for antigen-specific antibody responses (Mai et al., 2010).
3. AID and its regulation
3.1. Structure and function of AID
AID is encoded by Aicda gene located on chromosome 6 or 12 in mice or humans, respectively. The AID protein has 198 amino acids, consisting of N-terminus, cytidine deaminase catalytic domain, linker sequence and C-terminus (Chaudhuri and Alt, 2004; Ta et al., 2003). The N terminus has a nuclear localization signal motif and the C-terminus has a nuclear export signal motif. Genetic deletion of Aicda in mouse completely abrogated CSR and SHM (Muramatsu et al., 2000). Consistently, human patients with Aicda mutations lack IgG and IgA antibodies but have a high level of IgM, classified as HIGM2, and present in childhood with recurrent infection (Revy et al., 2000). Further mutagenesis studies show that the C-terminus of AID is responsible for CSR, and N-terminus responsible for SHM (Shinkura et al., 2004).
When AID was initially discovered, it was proposed to function as an RNA editing enzyme (Muramatsu et al., 2000). Although it remains formally possible that AID might target cellular or viral RNAs to mediate deamination (Liang et al., 2013), convincing genetic and biochemical studies have shown that AID functions as a DNA deaminase during SHM/CSR to convert C to U (Neuberger et al., 2003), thereby generating U:G mismatch lesions in DNA. Moreover, AID only acts on ssDNA and cannot access double-stranded (ds) DNA (Bransteitter et al., 2003; Chaudhuri, Khuong and Alt, 2004; Chaudhuri et al., 2003; Dickerson et al., 2003; Pham et al., 2003; Ramiro et al., 2003; Sohail et al., 2003). During SHM, ssDNA is probably generated during transcription in the form of transcription bubbles (Chaudhuri, Khuong and Alt, 2004). During CSR, ssDNA might be generated via a special structure termed “R-loop” (Tian and Alt, 2000; Yu et al., 2003). R-loops are nucleic acid structures in which a RNA strand forms a RNA/DNA hybrid molecule by displacing one strand of DNA in a duplex DNA molecule for a limited length. R-loop structures are formed at sequences that generate a G-rich transcript such as prokaryotic origins of replication (Masukata and Tomizawa, 1990) or mitochondrial origins of replication (Lee and Clayton, 1996). Mammalian S regions are unusually G-rich on the non-template strand (Chaudhuri et al., 2007; Stavnezer, Guikema and Schrader, 2008), thus producing G-rich RNA transcripts that can stably associate with the template strand of the DNA molecule to form R-loops. In the R-loop structure of S regions, the non-template DNA strand is displaced and exists as ssDNA (Chaudhuri and Alt, 2004; Tian and Alt, 2000; Yu et al., 2003). It has been proposed that evolutionarily conserved mammalian S regions are prone to form ssDNA and thus serve as the main targets of AID (Chaudhuri et al., 2007).
AID-initiated U:G mismatches can be resolved via several competing pathways (Di Noia and Neuberger, 2007; Martomo, Yang and Gearhart, 2004; Neuberger and Rada, 2007; Rada, Di Noia and Neuberger, 2004; Rada et al., 1998; Rada et al., 2002; Shen et al., 2006): 1) The general DNA replication machinery can interpret the U as if it were a thymine (T). One of the daughter cells will acquire a C→T transition mutation; 2) Uracil glycosylase (UNG), a component of the BER pathway, can remove the U, creating an abasic site. Then, error-prone polymerases such as Rev1 can incorporate any nucleotide in place of the removed U, causing transitions or transversions at C:G base pairs; 3) MSH2/MSH6 (mutS homolog 2/6), components of the MMR pathway, can recognize the U:G mismatch. The U-bearing strand is excised and, at loci that undergo SHM, error-prone polymerases are recruited to fill the gap, causing transition or transversion mutations at A:T base pairs. Therefore, the mutations in the V region are not sheer outcome of AID deamination, but rather rely on how UNG and MMR recognize and process the AID-initiated mismatches; 4) After MMR or UNG recognition, error-free repair could also correct the U:G mismatched DNA lesions so that no mutations are generated in this scenario. If both MSH2 and UNG are deleted, AID-initiated U:G mismatches cannot be recognized by either pathway, thus, are converted to C→T or G→A mutations during DNA replication. Indeed, in MSH2−/−UNG−/− mice, almost all the mutations are either C→T or G→A transitions that represent the footprint of AID deamination (Rada, Di Noia and Neuberger, 2004; Xue, Rada and Neuberger, 2006). How AID-initiated U:G mismatches are converted into DSBs during CSR is reviewed elsewhere (Chen and Wang, 2014). While AID is essential for SHM/CSR to produce high affinity isotype-switched antibodies, AID has also been implicated in generating chromosomal translocations of both Ig and non-Ig loci in leukemia and lymphoma (Alt et al., 2013; Daniel and Nussenzweig, 2013; Nussenzweig and Nussenzweig, 2010; Swaminathan et al., 2015; Tsai et al., 2008; Wang, 2013).
3.2. The role of target DNA sequences in regulating AID targeting specificity
Although AID is capable of deaminating any transcribed substrate in vitro and could potentially access the genome widely to cause genomic instability in B cells, its physiological targets during SHM and CSR are almost exclusively restricted to V or S regions of Ig loci. Major unresolved questions are how AID-induced DNA alterations are specifically targeted to Ig loci, and what regulatory mechanisms refrain AID from causing genome-wide damages in B lymphocytes.
During SHM, it remains to be addressed how AID specifically targets the V region exons of Igh and Igl loci. Although AID indeed targets a group of non-Ig genes during SHM, the mutation frequency of these genes is several order of magnitudes lower than that of V regions (Di Noia and Neuberger, 2007; Gordon et al., 2003; Liu et al., 2008; Longerich et al., 2006; Odegard and Schatz, 2006; Pasqualucci et al., 1998; Peng et al., 1999; Shen et al., 1998; Storb et al., 2001). The specificity and efficiency of AID targeting to the V region may be regulated at multiple levels (Delker, Fugmann and Papavasiliou, 2009; Stavnezer, 2011), including but not limited to regulation by specific sequence motifs, cis regulatory elements, histone modification pattern, and AID-cofactors. Correlative studies have long suggested that certain hotspot motifs such as RGYW or AGCT may influence mutation frequency (Di Noia and Neuberger, 2007; Rogozin and Kolchanov, 1992; Rogozin et al., 2001). However, it remains unclear whether and how target DNA sequences regulate mutation frequency or influence the outcome of AID-initiated DNA lesions. In a series of recent studies, we attempted to address these fundamental questions by establishing several knock-in models via a gene-targeting approach to introduce S regions into different genomic loci to test how different target DNA sequences regulate the outcomes of AID-initiated U:G mismatch DNA lesions.
Bcl6Sγ1 knock-in model
In the first knock-in model, we introduced a core Sγ1 region into the first intron of Bcl6 (Chen et al., 2012). Consistent with previous analysis of a similar region (Muto et al., 2006), we found that the mouse Bcl6 first intron region mutated at a frequency of approximately 2×10−4 (Chen et al., 2012). We discovered that the mutation frequency of the inserted Sγ1 region was 10 fold higher than that of the adjacent Bcl6 intron sequence (Chen et al., 2012). Hence, our results demonstrate that S region sequence per se, independent of Igh cis regulatory elements, enhances AID targeting efficiency (Chen et al., 2012). Mechanistically, we showed that the mutational phenotypes of the inserted S region were probably due to the enhanced recruitment of RNA polymerase II (RNAPII) and AID (Chen et al., 2012). More interestingly, we found that the higher level of RNAPII accumulation was detected at both the 5′ and 3′ ends of the inserted Sγ1 region (Chen et al., 2012), implicating a higher density of RNAPII across the entire knock-in S region. These results are highly consistent with the previous findings of the endogenous S region in the Igh locus (Wang et al., 2009b). We propose that RNAPII pausing at S regions, together with AID cofactors such as Spt5 or RPA, likely facilitates the repositioning of repressive nucleosomes to establish a permissive chromatin architecture that allows AID to access target DNA sequences (Chen et al., 2012). Taken together, we propose that nucleotide sequences, as the targets of AID, function actively to determine their own mutability, possibly by forming higher-order structures, recruiting sequence-specific co-factors, modifying chromatin context, and/or regulating the transcriptional process. In addition, the nucleotide sequence preference may serve as an additional layer of AID regulation by restricting its mutagenic activity to specific sequences such as evolutionarily conserved S regions. This regulatory mechanism may ensure that AID deamination frequency remains relatively low at most loci in the genome, below the threshold of repair capacity, so that these AID-initiated lesions can be efficiently repaired, thereby protecting the integrity of the B cell genome. Such a notion is also consistent with the observation that although AID is recruited to 5910 target genes in in vitro activated B cells (Yamane et al., 2011), most of these loci would not display mutations in the presence of a normal repair mechanism.
c-mycSγ2b knock-in model
In the second knock-in model, we introduced an Sγ2b region, a bona fide AID target, into the first intron of c-myc (c-mycSγ2b knock-in) (Chen et al., 2014). c-myc is a frequent translocation partner of Igh and Igl loci in human mature B cell lymphomas (e.g., Burkitt’s lymphomas) (Janz, 2006). Most of the Ig-c-myc translocations are thought to derive from GC B cells (Kuppers, 2005). However, extensive sequencing studies of the c-myc locus in human memory B cells showed little SHM activity (Shen et al., 2000; Shen et al., 1998; Storb et al., 2001), suggesting that c-myc is not an efficient AID target in B cells. If so, then how can c-myc become a frequent target of Ig translocations and does AID actually access the c-myc locus? In this regard, prior studies showed that the mutation frequency of the c-myc locus is much higher in MSH2−/−UNG−/− or UNG−/− Peyer’s patch (PP) GC B cells than in WT controls (Liu et al., 2008). These data suggest that AID-initiated lesions at certain non-Ig loci, such as c-myc, are normally processed by error-free repair so that no mutations are generated, only in the absence of MSH2 and UNG or UNG alone, these AID-initiated lesions are converted to mutations at the c-myc locus in GC B cells (Liu et al., 2008). Thus, it was proposed that a differential DNA repair mechanism may protect non-Ig loci from an excessive amount of mutations (Liu et al., 2008). In particular, AID-initiated lesions are repaired in an error-free manner in most of the tested non-Ig loci, including c-myc, whereas a few of them appear to undergo error-prone repair, including Bcl6 and Cd83 (Liu et al., 2008). The error-free repair mechanism operating at most non-Ig loci probably protects the genome of GC B cells.
Using our c-mycSγ2b knock-in mouse model, we detected a high level of c-myc locus DSBs or translocations in the absence of ATM in the in vitro cytokine-activated B cells (Chen et al., 2014). The increased c-myc locus DSBs are dependent on both AID and the inserted Sγ2b region in our c-mycSγ2b/ATM−/− model (Chen et al., 2014). ATM (Ataxia-telangiectasia mutated) is a member of PI3K-related kinases, and a DNA damage response factor essential for repairing DSBs (Marechal and Zou, 2013). We found that ATM is required for optimal GC formation since the percentage of GC B cells is much lower in ATM−/− mice than control mice (Chen et al., 2014). Intriguingly, we observed no significant level of c-myc genomic instability in the GC B cells of c-mycSγ2b/ATM−/− mice (Chen et al., 2014). Thus, our findings reveal a sharp contrast between the populations of GC and in vitro cytokine-activated B cells in their ability to acquire genomic instability at the c-myc locus. These results suggest that conclusions drawn from studies using one population of B cells may not be generalizable to others. Since previous studies in the field often rely on in vitro activation of naïve B cells as the model system (Chiarle et al., 2011; Klein et al., 2011; Wang et al., 2009a; Yamane et al., 2011), we may need to take into consideration the difference between the two experimental models when investigating CSR and translocation mechanisms.
It remains unknown how the differential DNA repair of AID-initiated U:G lesions is regulated at non-Ig loci. It has been well documented that B cells activated with different stimuli (e.g. TD or TI antigens or cytokines) undergo distinct differentiation pathways and display unique signatures of gene expression (Shaffer et al., 2001). Hence, we propose that the AID-initiated DNA lesions at non-Ig loci are differentially processed in distinct B cell populations (e.g., GC vs. in vitro cytokine-activated B cells) (Chen et al., 2014). Of interest, the AID-initiated DNA lesions at the Igh locus can be processed in an error-prone manner that leads to mutations or DSBs at the Igh locus (Alt et al., 2013; Wang, 2013). This error-prone processing is independent of B cell populations because both GC and cytokine-activated B cells harbor frequent mutations in the S regions of the Igh locus (Nagaoka et al., 2002; Rada, Di Noia and Neuberger, 2004; Xue, Rada and Neuberger, 2006). Consistently, we found that both GC and cytokine-activated B cells exhibited Igh locus DSBs in the absence of ATM (Chen et al., 2014). We suggest that locus-specific regulatory elements might regulate not only AID targeting efficiency but also the repair manner of AID-initiated lesions (Chen et al., 2014). It is likely that the DNA repair mechanisms at the c-myc locus become dysregulated in GC B cells, and instead of error-free repair, the AID-initiated lesions are repaired in an error-prone manner, thereby leading to DSBs or translocations at the c-myc locus. It is highly relevant to elucidate the mechanisms that regulate population-specific processing of AID-initiated lesions at the non-Ig loci, since many of these loci such as c-myc are frequently targeted by translocations in human B cell lymphomas (Kuppers and Dalla-Favera, 2001).
V region knock-in models
In the third knock-in model, we inserted a portion of core Sμ region into the V region of the endogenous Igh locus (Chen et al., 2016). While we show that AID’s mutagenic activity depends on its target sequence at a non-Ig locus (Chen et al., 2012), the role of target DNA sequence in regulating AID activity has not been addressed in the most physiologically relevant locus, the endogenous Igh locus. Moreover, whether AID-initiated lesions lead to point mutations or DSBs is confounded by a complex interplay between AID-deamination and the processing of AID-initiated lesions by UNG or MMR pathway. To specifically dissect out the role of target DNA sequences in regulating AID deamination and subsequent repair pathway choice, we employed two knock-in models in which a portion of core Sμ region (cSμ) or a rearranged VDJ exon (VB1–8) was placed into the endogenous V region locus via gene-targeting, termed V-cSμ or VB1–8 knock-in, respectively (Chen et al., 2016). Both of these two sequences were inserted into the exactly same genomic location and driven by the same VH186.2 promoter (Chen et al., 2016). Thus, our experimental system allows a direct comparison between the mutability of cSμ and VDJ exon sequences and their ability to regulate AID-deamination and subsequent repair process. We report several important findings: 1) S region sequence is an intrinsically more efficient AID deamination target than V region sequence; 2) the AID-initiated lesions at the Igh locus are processed by both error-free and error-prone repair; 3) S region harbors more UNG-dependent deletions, an indicator of DSB formation. These S region deletional events are significantly enhanced by MSH2 deficiency; 4) V region mutational hotspots are largely determined by AID deamination; and 5) recurrent and conserved S region motifs potentially function as spacers between AID-deamination hotspots (Chen et al., 2016). Overall, we conclude that target DNA sequences directly modulate AID-deamination frequency and promote differential accessibility of repair factors (UNG vs MMR) to AID-initiated lesions, thereby leading to distinct outcomes of AID deamination.
We observed that the mutation frequency of the cSμ sequence is significantly higher than that of the VB1–8 exon sequence in the absence of both UNG and MSH2 (Chen et al., 2016). In MSH2−/−UNG−/− mice, almost all the mutations are either C→T or G→A transitions that represent the footprint of AID deamination. Thus, our data demonstrate that the cSμ sequence is indeed a more efficient AID deamination target. One potential caveat of our model is that the difference in SHM of the two sequences might be influenced by antigen selection within GCs. However, under our short-term immunization conditions, SHM patterns of VB1–8 productive allele are not biased by antigen selection (Weiss, Zoebelein and Rajewsky, 1992). Furthermore, VB1–8 exon sequence exhibited a similar mutation frequency and pattern including hotspot distribution regardless of whether it serves as a productive or passenger allele (Yeap et al., 2015). The productive and passenger VB1–8 alleles share the identical transcription control elements and essentially identical sequence except that translation termination codons are introduced in the passenger VB1–8 allele (Yeap et al., 2015). Therefore, these data demonstrated that the SHM pattern of VB1–8 productive allele shows no influence of antigen selection (Yeap et al., 2015). Taken together, we conclude that the SHM difference between cSμ and VDJ exon sequence is driven by sequence-intrinsic mechanisms (Chen et al., 2016; Yeap et al., 2015). Of note, such sequence intrinsic mechanisms of somatic mutation also contribute to affinity maturation of broadly neutralizing antibodies against HIV-1 (Hwang et al., 2017).
AID deamination leads to U:G mismatches that are subsequently recognized by MMR or UNG pathways. After MMR or UNG recognition, in theory, both error-free and error-prone repair can be recruited to the lesions. It has been suggested that error-prone repair might be preferentially recruited to Ig loci, whereas, error-free repair functions predominantly at non-Ig loci (Liu et al., 2008; Liu and Schatz, 2009). However, based on our data, we propose that error-free repair is also involved in the processing of AID-initiated lesions at Ig loci (Chen et al., 2016). We found that the mutation frequencies of both V and S regions were significantly higher in MSH2−/−UNG−/− mice than in WT mice (Chen et al., 2016). Since the mutation frequency in the absence of MSH2 and UNG reflects the frequency of AID deamination, we reason that the lower mutation frequency in UNG/MSH2 proficient mice is due to the error-free repair which can correct the U:G mismatches and generate no mutations (Chen et al., 2016). These data led us to conclude that AID-initiated lesions at Igh locus can be processed by error-free repair, similar to the non-Ig loci, and that the mutation level at Igh locus probably exceeds the capacity of error-free repair, thereby resulting in the recruitment of error-prone repair, which in turn causes mutations (Chen et al., 2016). Our studies reveal an evolutionarily conserved role for target DNA sequences in regulating antibody gene diversity, AID targeting specificity and differential recognition of AID-initiated lesions by different repair pathways (Chen et al., 2016; Chen et al., 2014; Chen et al., 2012; Yeap et al., 2015).
3.3. Signaling pathways that regulate AID expression
Non-Hodgkin’s lymphomas (NHL) are a heterogeneous group of malignancies affecting lymphocytes. Collectively, NHL are the fifth most common cancers in the US, and more than 90% of NHL are of B cell origin (Scott and Gascoyne, 2014). There are roughly equal number of T and B cells in spleen and more T cells in lymph nodes. Why are B cells so prone to lymphomagenesis? This is probably due to B cell-specific DNA mutagenesis processes, SHM and CSR (Chen and Wang, 2014; Wang, 2013). SHM and CSR are required to produce high affinity isotype-switched antibodies that are essential for immunity against pathogens. However, B cells pay a high price for utilizing AID to generate point mutations or DSBs during SHM/CSR (Chen and Wang, 2014; Wang, 2013). AID is a genome mutator and, if dysregulated, can cause genome-wide DSBs that lead to chromosomal translocations and lymphomas (Alt et al., 2013; Chen and Wang, 2014; Chiarle et al., 2011; Gazumyan et al., 2012; Robbiani et al., 2009; Robbiani et al., 2015; Wang, 2013). Hence, AID expression level is tightly controlled and a high level of AID expression only occurs in activated B cells during infection or immunization.
AID expression is regulated at the transcriptional level (Matthews et al., 2014; Vaidyanathan et al., 2014; Zan and Casali, 2013). There are four cis-regulatory regions that induce AID expression specifically in activated B cells and inhibit AID expression in other types of cells (Nagaoka et al., 2010). These 4 regions are termed region 4, region 1, region 2 and region 3, arranged in this genomic order at the Aicda locus (Figure 5). The transcription of the Aicda gene is controlled by multiple transcription activators and repressors that bind the sites located in the four cis-regulatory regions in and around the Aicda gene locus (Figure 5). AID expression is also regulated at the post-transcriptional and post-translational level, extensively reviewed elsewhere (Matthews et al., 2014; Vaidyanathan et al., 2014; Vuong and Chaudhuri, 2012).
Figure 5. Transcriptional Regulation of AID.
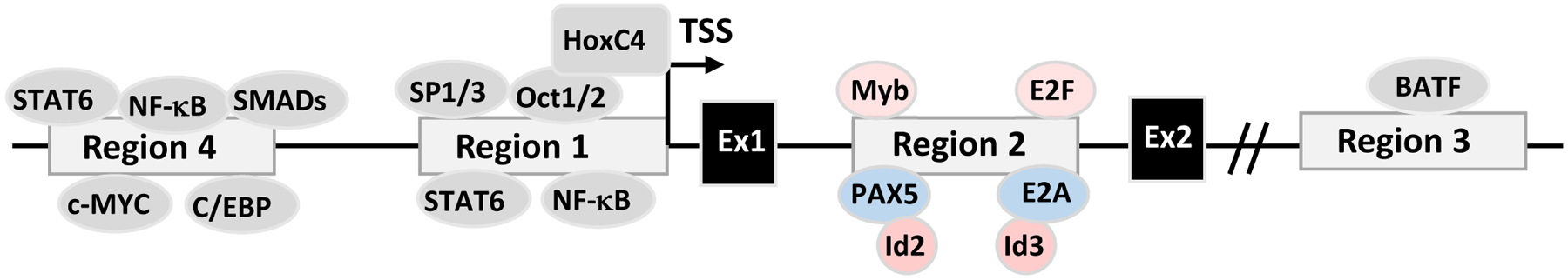
The Aicda gene locus contains 4 conserved regulatory regions (Region 1–4). The exon 1 and 2 of AID and transcription factors that potentially bind to these regions are shown.
Region 1 consists of the Aicda promoter and the sequences immediately upstream of the promoter. Region 1 also contains enhancer elements that bind HoxC4, Oct1/2 and SP1/3 (Lee-Theilen and Chaudhuri, 2010), and elements responding to estrogen and progesterone that, respectively, induce or suppress AID expression (Mai et al., 2010; Pauklin and Petersen-Mahrt, 2009; Pauklin et al., 2009). It was initially reported that estrogen-bound estrogen receptor induced AID transcription directly by binding and activating the AID promoter (Pauklin et al., 2009); however, later studies showed that estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to enhance HoxC4-mediated AID induction (Mai et al., 2010). In contrast, progesterone-bound progesterone receptor suppressed the transcription of the Aicda gene by binding region 1 sequence upstream of the AID promoter (Pauklin and Petersen-Mahrt, 2009). Hormonal factor-mediated AID regulation might have implications in hormone-driven oncogenesis and autoimmunity; however, their involvement in the normal CSR process remains to be determined (Incorvaia et al., 2013; Maul and Gearhart, 2009; Pauklin and Petersen-Mahrt, 2009; Pauklin et al., 2009). Lastly, region 1 contains the binding sites for NF-κB (p50) and STAT6 transcription factors (Park et al., 2009; Yadav et al., 2006); thus, region 1 could respond to signals triggered by IL-4 together with CD40 (Dedeoglu et al., 2004), TLR (Pone et al., 2012b), BAFF-R or TACI ligation (Castigli et al., 2005; Pone et al., 2012b). Some of these stimulating conditions can also induce HoxC4 expression in activated B cells that enhances the activity of AID promoter (Park et al., 2013; Park et al., 2009; Zan and Casali, 2013).
Region 2 contains the regulatory elements in the first intron of the Aicda gene. This intronic regulatory region contains the binding sites for PAX5, E2A, c-Myb and E2F (Gonda et al., 2003; Huong le et al., 2013; Sayegh et al., 2003; Tran et al., 2010). PAX5 and E2A proteins are B cell-specific that promote AID expression, while c-Myb and E2F, whose expression is not restricted to B cells, repress AID expression (Gonda et al., 2003; Sayegh et al., 2003; Tran et al., 2010). Deletion of the silencer elements that bind repressors c-Myb and E2F drastically enhances AID expression, without inducing AID transcription in non-B cells, supporting the notion that AID expression is tightly controlled in a B cell-specific manner (Huong le et al., 2013). Apart from the binding of c-Myb and E2F to region 2, suppressing AID transcription can be achieved via inhibitor of DNA binding (Id) proteins (Gonda et al., 2003; Sayegh et al., 2003). Id2 and Id3 might form heterodimers with PAX5 and E2A proteins, respectively, and impair the binding of PAX5 and E2A to region 2 of the Aicda locus, thereby inhibiting AID transcription (Gonda et al., 2003; Sayegh et al., 2003). Of note, Id2 can be upregulated by PI3K/AKT signaling (Belletti et al., 2001); it remains to be addressed whether the PI3K/AKT pathway inhibits AID expression by modulating Id2 and/or Id3 expression. The PI3K/AKT pathway can also upregulate BLIMP-1 (Setz et al., 2018) that inhibits AID expression and CSR, possibly by repressing PAX5 expression or directly repressing AID transcription (Lin et al., 2002; Minnich et al., 2016; Omori et al., 2006; Shaffer et al., 2002).
Region 3 is located about 7kb or 24kb downstream of the exon 5 of the Aicda gene in mouse or human genomes, respectively. Region 3 contains the binding sites for AP1 family transcription factor BATF, and may act as an enhancer element for AID expression since Batf deletion leads to abrogation of AID expression and CSR (Betz et al., 2010; Crouch et al., 2007; Ise et al., 2011). Region 4 is located about 8kb or 16 kb upstream of the transcription start site of the Aicda gene in mouse or human genomes, respectively (Crouch et al., 2007; Nagaoka et al., 2010; Yadav et al., 2006). Region 4 contains enhancers that have the binding sites for transcription factors, STAT6, SMAD3/4, C/EBP and NF-κBs that are activated during B cell stimulation (Huong le et al., 2013; Tran et al., 2010). In addition, c-Myc has been implicated in binding region 4 to induce AID expression (Dominguez-Sola et al., 2012; Fernandez et al., 2013). Region 4 is mainly responsible for responding to stimulatory signals through cytokine receptors or costimulatory molecules.
AID expression was initially reported in GC B cells (Muramatsu et al., 1999). Studies using AID reporter mice confirmed the initial findings and showed that AID expression occurred predominantly in activated mature B cells (Crouch et al., 2007). Later studies found that AID could be expressed in B cells outside GCs, such as immature B cells in bone marrow, driven by BCR and TLR-mediated signals (Han et al., 2007; Kuraoka et al., 2017). Prior studies also showed that AID was expressed in human immature B cells that expressed recombination-activating gene 2 (RAG2); most of these AID+ immature B cells were deleted by apoptosis (Cantaert et al., 2015). These data suggest that AID expression in immature B cells might contribute to central B cell tolerance potentially through its RAG-coupled genotoxic activity (Cantaert et al., 2015). However, AID expression in mature B cells did not lead to apoptosis since B cell-specific AID transgenic mice had normal number and percentage of B cells in secondary lymphoid organs (Muto et al., 2006). Although AID transcripts can be detected in immature B cells, mature naïve B cell have little AID expression and do not upregulate AID without stimulation (Crouch et al., 2007). Since humoral immune responses are largely mediated by mature B cells stimulated by TD or TI antigens, we focus on discussing the signal pathways that can induce naïve mature B cells to express AID.
As discussed above, in vivo antigen immunization may activate multiple receptors on B cells simultaneously, thus, it is difficult to define which receptor is able to signal to induce AID expression. Ex vivo stimulation of B cells in culture provides an essential alternative to overcome this difficulty. In culture system, LPS is the only stimulus that can activate CSR on its own, probably due to its ability to stimulate BCR, CD14 and TLR4 altogether (Lu, Yeh and Ohashi, 2008; Pone et al., 2012a). None of other stimuli (e.g., CD40 or TLR ligands) can activate CSR efficiently (Pone et al., 2012a), likely because none of them can fully support B cell proliferation in vitro and a sufficient level of cellular proliferation is required for AID expression. To resolve this issue, cytokines or costimulatory ligands, such as IL-4 or BAFF, were included in B cell culture to induce AID expression and CSR. In the presence IL-4 cytokine, CD40, TLRs, TACI, BAFF-R all can induce the expression of AID protein, consistently, B cells stimulated under these conditions can undergo CSR (Castigli et al., 2005; Dedeoglu et al., 2004; Pone et al., 2012a). However, stimulating BCR by anti-IgD in the presence of IL-4 does not induce AID expression and CSR (Pone et al., 2012a). Recently, it was reported that anti-IgM in the presence of BAFF did not induce AID transcription in naïve mature B cells (Kuraoka et al., 2017). Another study showed that stimulating BCR by anti-IgM in the presence of IL-4 induced AID expression and Igh GLT; however, these stimulated B cells failed to undergo CSR (Heltemes-Harris et al., 2008). These prior studies consistently showed that engaging BCR did not induce CSR; however, their results were inconsistent about whether engaging BCR could induce AID expression. We stimulated splenic B cells purified from C57BL/6, BALB/cJ and S129 mice with anti-IgM in the presence of IL-4 and found that anti-IgM/IL-4 stimulation did not induce AID expression in WT naïve mature B cells regardless of their genetic background (unpublished data). Based on previous reports and our unpublished observation, we conclude that engaging BCR in the presence of IL-4 does not induce AID expression. Since the BCR is essential for recognizing antigen and for determining the specificity of isotype-switched antibody, it is counterintuitive that it cannot promote CSR on its own. Is it because there are regulatory mechanisms in play that prevent the BCR from inducing AID? Or simply because downstream signaling pathways activated by BCR engagement alone are not qualitatively or quantitatively sufficient? These are fundamental questions for B cell biology and humoral immunity that have not been explored before and remain to be addressed.
4. Signaling balance for CSR
We have discussed multiple co-receptors of B cells, including CD40, TLRs, TACI and BAFF-R, all of which can transduce positive signals to induce AID expression and CSR. Intriguingly, all of these co-receptors can also activate the PI3K/AKT pathway that inhibits AID expression and CSR. We propose that the balance between activating and inhibitory signaling may serve as an evolutionarily conserved mechanism to ensure that the immune system would not overreact to antigen stimulation and humoral immune responses are carried to a proper level without causing collateral damage, such as AID-induced genomic instability. In addition, the BCR does not induce AID expression and CSR but it does activate the PI3K/AKT pathway; thus, antigen stimulation might elicit stronger inhibitory signals that prevent AID expression. To assure that humoral immune responses initiate productively, PI3K’s negative effects must be counteracted by factors that allow AID expression and CSR.
PI3Ks exert their effects by generating PI(3,4,5)P3 via phosphorylating the 3-position of the inositol ring of PI(4,5)P2 (Figure 6). On the contrary, phosphatase and tensin homolog (PTEN) can dephosphorylate PI(3,4,5)P3 at the 3-position of the inositol ring and converts PI(3,4,5)P3 back to PI(4,5)P2, thus, PTEN is able to antagonize PI3K’s negative effects on AID expression and CSR (Figure 6). In support of this notion, prior studies have demonstrated that deletion of PTEN in B cells reduced AID expression and resulted in decreased levels of isotype-switched antibodies in vivo and defective CSR in vitro (Chen et al., 2015; Omori et al., 2006; Suzuki et al., 2003; Wang et al., 2018). In contrast, hyper-activation of PI3Ks impairs AID expression, CSR and isotype-switched antibody production (Chen et al., 2015).
Figure 6. The signaling balance between PI3Ks and PTEN controls AID expression and CSR.
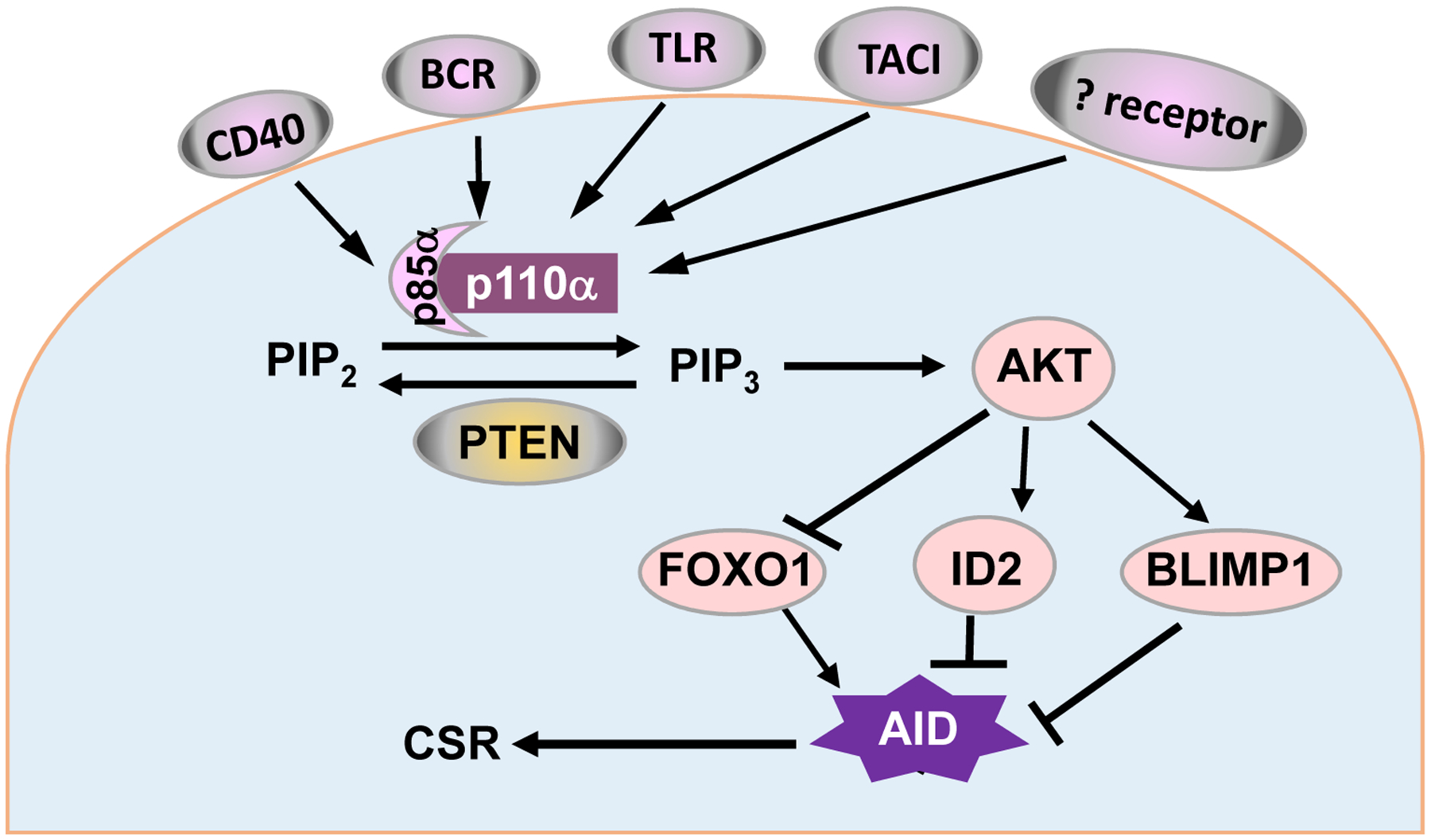
Signaling balance for CSR. PI3Ks catalyze the phosphorylation of PI(4,5)P2 and converts it into PIP3, whereas PTEN counteracts PI3Ks by converting PIP3 into PIP2. Increased PIP3 activates AKT which suppresses AID expression likely by regulating transcription factors FOXO1 and BLIMP-1. Reduced AID expression leads to a decreased level of CSR.
4.1. PI3Ks
According to structural characteristics and substrate specificity, the PI3K family can be classified into class IA, class IB, class II and class III (Deane and Fruman, 2004; Koyasu, 2003). Hematopoietic cells predominantly express class IA PI3Ks, which are heterodimers consisting of a regulatory subunit and a catalytic subunit (Deane and Fruman, 2004; Koyasu, 2003). The catalytic subunits of class IA have three isoforms: p110α, p110β and p110δ (Deane and Fruman, 2004; Koyasu, 2003), encoded by pik3ca, pik3cb and pik3cd gene, respectively. We identify a cooperative role of p110α and p110δ in suppressing CSR, furthermore, they do not compensate for each other on these effects since inhibiting either one with corresponding isoform-specific inhibitors enhances CSR (Chen et al., 2015). At rest state, the class IA PI3Ks exist in an inhibitory conformation. PI3K p110 subunit has a C2 domain and a catalytic domain (Vanhaesebroeck et al., 2001; Walker et al., 1999). The C2 domain of p110 binds phospholipid and targets PI3Ks to the membrane where PI3Ks can be activated. The activity of p110 catalytic subunit is inhibited by the regulatory subunit, which has 5 isoforms, p85α, p55α, p50α, p85β and p55γ, with p85α as the most abundant one (Deane and Fruman, 2004). p85α contains a N-terminal SH2 and a C-terminal SH2 domain (nSH2 and cSH2, respectively) that are connected by an interstitial SH2 domain (iSH2) (Pauls et al., 2012). All of iSH2, nSH2 and cSH2 domains of p85 can associate with p110 subunit and promote an inhibitory conformation (Pauls et al., 2012). Of note, the precise mechanism of p85-mediated regulation may differ among p110 isoforms (Burke et al., 2011). To activate the catalytic activity of p110, nSH2 and cSH2 of p85 subunit bind phosphopeptide motifs such as YXXM (Pauls et al., 2012). YXXM motifs can be found in plasma membrane proteins such as membrane-bound receptors (e.g., TLRs) or adaptors (e.g., MyD88). The binding of SH2 domains to such motifs not only recruits the complex to the membrane but also dissociates these SH2 domains from p110, thereby releasing the inhibitory effect of p85 and activating p110 (Carpenter et al., 1993; Yu et al., 1998b).
The N-terminal domain of p85α functions as a linker domain to interact with other proteins, such as PTEN (Cheung et al., 2011; Cheung et al., 2015). The interaction of free p85α with PTEN stabilizes PTEN and enhances its phosphatase activity (Cheung et al., 2015), which may in turn promote AID expression and CSR in B cells. It was reported that p85α subunit is required for the BCR to synergize with TLRs to induce AID expression and CSR (Pone et al., 2012b). However, it is unknown whether p85α mediates the synergistic effects between BCR and TLR via regulating PTEN’s stability or activity. Taken together, PI3K activation and its net effects are regulated and counteracted by at least two distinct mechanisms: conformational inhibition mediated by PI3K regulatory subunit, and PTEN’s ability to catalyze the reverse reaction to convert PIP3 back to PIP2.
4.2. PTEN
PTEN is a phosphatase with dual activity for both lipid and protein (Lee, Chen and Pandolfi, 2018). PTEN is encoded by a single Pten gene that can be translated into a canonical or long form of PTEN (PTEN-Long) protein through differential usage of start codons (Hopkins et al., 2013; Hopkins et al., 2014). PTEN-Long can be secreted and enter into neighboring cells, while the canonical PTEN can be packaged by vesicular membrane and transported from one cell to another (Hopkins et al., 2014). However, these exogenous agents of PTEN probably do not play a crucial role in antibody responses. Supporting this notion, B cell-specific PTEN deficient mice exhibit a high level of IgM and reduced levels of isotype-switched antibodies (Sander et al., 2015; Suzuki et al., 2003; Wang et al., 2018), suggesting that, during antibody responses, B cells may not acquire exogenous PTEN from other cells to mediate efficient CSR.
The N-terminus of PTEN binds phosphatidylinositol and the catalytic phosphatase domain dephosphorylates PIP3 (Lee, Chen and Pandolfi, 2018). The C-terminus of PTEN regulates PTEN’s activity and mediates protein-protein interaction. Phosphorylation of the C-terminus inactivates PTEN and prevents it from binding membrane lipids so that PTEN cannot access its targets. At the same time, phosphorylation of the C-terminus can also prevent PTEN from interacting with other proteins such as ubiquitin ligases, thus preserving PTEN from ubiquitination-mediated degradation (Lee, Chen and Pandolfi, 2018). Taken together, C-terminal phosphorylation of PTEN maintains a cellular pool of PTEN in an inactive form. Dephosphorylation of the C-terminus activates PTEN’s activity but also promotes ubiquitination-mediated proteasomal degradation of PTEN (Lee, Chen and Pandolfi, 2018). PTEN dephosphorylation facilitates its homo-dimerization to fully activate its PIP3 lipid phosphatase activity (Papa et al., 2014) and allows PTEN to interact with p85α or other proteins (e.g., ubiquitin ligases). Interacting with ubiquitin ligases promotes PTEN degradation, while interacting with p85α stabilizes PTEN and enhances PTEN’s phosphatase activity (Cheung et al., 2011; Cheung et al., 2015). Overall, the activity and stability of PTEN protein are exquisitely regulated in an opposing direction to ensure that PTEN is not over- or under-activated. Lastly, it remains to be elucidated whether p85α mediates the synergy of the BCR with TLRs in inducing AID expression and CSR via interacting with PTEN.
4.3. The opposing effects of PI3Ks and PTEN on CSR regulation
The role of PI3Ks in CSR appears to be complicated, probably due to the fact that B cells express multiple isoforms of PI3Ks (Pauls et al., 2012). Up to date, only the roles of p110δ and p110α in CSR have been reported. Germline p110δ KO B cells have no defects in CSR to IgG1 assessed by an in vitro CSR culture assay (Janas et al., 2008). B cell-specific deletion of p110δ mediated by CD19Cre has no significant effects on TD antigen-induced antibody responses or GC formation, except that it strongly promotes antigen-specific IgE production, implying specific dysregulation of IgE CSR (Rolf et al., 2010). Overall, genetic deletion of p110δ does not significantly affect IgG1 CSR but strongly promotes IgE CSR. In contrast, pharmaceutical inhibition of p110δ in WT B cells robustly increases the percentage of IgG1+ and IgE+ B cells (Zhang et al., 2008). The discrepancy between these studies regarding IgG1 CSR is probably attributed to the compensatory effects of other PI3K isoforms in the p110δ-deleted B cells. To avoid the complex effects of deleting one subunit that may affect expression of other subunits, a knock-in allele was generated that carried an inactive point mutation of p110δ (D910A) (Okkenhaug et al., 2002). p110δD910A (inactive) mutant mice had reduced in vivo Ab responses to TD and TI antigens (Okkenhaug et al., 2002), likely due to poor B and T cell development in these mice. Indeed, p110δD910A mutant B cells displayed an increased level of CSR to IgG1 and IgE in an in vitro CSR assay (Zhang et al., 2008), suggesting that p110δ normally suppresses CSR. It remains unknown whether other PI3K isoforms of catalytic subunit (p110α, β or γ) affect CSR. Prior studies showed that p110α compensates for p110δ in B cell development (Ramadani et al., 2010). Thus, we reason that other PI3K isoforms may also play a critical role in CSR that may explain the inconsistent phenotypes caused by p110δ deletion versus point mutation.
Consistent with our hypothesis, we discovered that p110α hyper-activation strongly inhibits CSR, in contrast, its genetic deletion or pharmacological inhibition promotes CSR (Chen et al., 2015). Thus, our recent studies identify an important role of p110α in CSR regulation, which has not been recognized previously. Furthermore, we show that p110α-or p110δ-specific inhibitor promotes CSR individually, and combined treatment of both inhibitors promotes CSR additively, especially for IgE CSR (Chen et al., 2015). Our findings are consistent with the drastically increased serum IgE level in p110α/p110δ double KO mice (Ramadani et al., 2010). Thus, we conclude that p110α and p110δ act coordinately to suppress CSR (Chen et al., 2015). Our results are also in line with previous reports showing that p110δ pharmacological blockade or p110δD910A point mutation resulted in a higher level of CSR (Zhang et al., 2008). Notably, our kinetic studies also show that the deletion of p110δ gene in CH12F3 cells indeed promotes CSR at early time points; however, such promoting effects diminish at later time points, resulting in a comparable CSR level between WT and p110δ deficient cells (Chen et al., 2015). These results are consistent with the possibility that p110α might exert its compensatory effects in p110δ deficient B cells, especially at later time points, to suppress CSR. Another possibility is that, at later time points, cellular proliferation is arrested and CSR level is saturated. In addition, our data suggest the importance of kinetic analysis of CSR efficiency. Taken together, our study may explain the lack of CSR phenotypes in p110δ deletion mutant and clarify the discrepancy between genetic and pharmacological blockade data. We conclude that p110α and p110δ act cooperatively to antagonize PTEN’s activity to control the efficiency and kinetics of CSR (Chen et al., 2015). In line with our data, recent studies show that hyper-activation of p110α specifically in GC B cells also leads to significantly reduced antibody responses and CSR (Sander et al., 2015). In conclusion, different isoforms of PI3Ks negatively regulate CSR.
Prior studies showed that CD19Cre-mediated Pten deficiency in B cells results in a reduced level of CSR in vivo and in vitro (Omori et al., 2006; Suzuki et al., 2003). However, since CD19Cre mediates efficient deletion at pre-B cell developmental stage (Rickert, Roes and Rajewsky, 1997), it remains formally possible that CD19Cre-mediated deletion of Pten may affect B cell development that subsequently impairs CSR. Moreover, CD19Cre-mediated Pten deletion results in the expansion of B1 and marginal zone B cells (Anzelon, Wu and Rickert, 2003; Suzuki et al., 2003), which may also affect the ability of B cells to undergo CSR. Addressing the unresolved question of PTEN’s effect on CSR requires a better controlled model system. In a recent study, we took an advantage of a newly developed genetic model in which the Pten gene was deleted in mature B cells acutely, with a narrow time window, namely, a few days apart from genetic deletion to CSR assay, thereby unequivocally excluding the indirect effects of Pten deletion (Chen et al., 2015). Consistent with prior studies using CD19Cre (Omori et al., 2006; Suzuki et al., 2003), our studies show that acute deletion of Pten drastically reduces IgG1 CSR induced by engaging CD40 in a mature B cell autonomous manner (Chen et al., 2015). Prior studies have not evaluated the effects of Pten deletion on IgE CSR, which requires a much more complicated procedure for detection. Based on a previous study (Erazo et al., 2007), we developed a reliable method to readily monitor the percentage of IgE switched B cells (Chen et al., 2015). Our data clearly demonstrate that PTEN is required for a normal level of IgE CSR induced by anti-CD40/IL-4 (Chen et al., 2015). In addition, we employed a newly developed transgenic model in which PTEN protein is subtly over-expressed in a mature B cell autonomous manner (Chen et al., 2015). Our results show that even such a modest upregulation of PTEN protein can enhance the level of CSR significantly (Chen et al., 2015). Therefore, we conclude that PTEN directly regulates CSR signaling per se, instead of influencing CSR via modulating B cell development or differentiation of B cell subsets (Chen et al., 2015).
Consistent with our findings, Cγ1Cre-mediated deletion of PTEN or hyper activation of p110α reduces the percentage of IgG1+ or IgA+ B cells in PP GC B cells or antigen-immunized splenocytes (Sander et al., 2015). Another independent study also showed that Cγ1Cre-mediated deletion of PTEN resulted in the reduced IgG1 B cells in GCs (Wang et al., 2018). Mechanistically, gain-of-function of PI3K or loss-of-function of PTEN enhances AKT activation (Chen et al., 2015; Janas et al., 2008; Omori et al., 2006; Suzuki et al., 2003). Reversely, loss-of-function of PI3K (Chen et al., 2015; Janas et al., 2008) or gain-of-function of PTEN (Chen et al., 2015) reduces AKT activity. AKT activation likely inhibits CSR via regulating the expression or activity of transcription factors BLIMP-1 or FOXO1 (Dengler et al., 2008; Omori et al., 2006). Blimp-1−/− B cells show elevated CSR in response to LPS or LPS/IL-4 stimulation, demonstrating a negative role of BLIMP-1 in regulating CSR (Omori et al., 2006). In contrast, CD21Cre-mediated deletion of foxo1 in B cells reduced IgG CSR in response to LPS/IL-4 stimulation (Dengler et al., 2008). Consistently, Cγ1Cre-mediated deletion of foxo1 specifically in GC B cells also drastically reduced CSR (Dominguez-Sola et al., 2015; Sander et al., 2015). Taken together, we conclude that a signaling balance between PTEN and PI3K isoforms is essential to maintain a normal level of CSR and antibody response.
Mutations in the components of PI3K pathway (e.g., p110δ and p85α) predispose to human immunodeficiency (Angulo et al., 2013; Crank et al., 2014; Deau et al., 2014; Lucas et al., 2014a; Lucas et al., 2014b). A recent review extensively summarized the progress in human patients with autosomal dominant gain-of-function (GOF) mutations in Pik3cd and Pik3r1 genes, encoding p110δ and p85α, or loss-of-function mutations in PTEN (Jhamnani et al., 2018). These patients develop combined immunodeficiencies and present with CSR/HIGM syndromes (Jhamnani et al., 2018). To further test how PI3K hyperactivation affects B cell development and function, CRISPR/Cas9 gene editing was employed to introduce a heterozygous GOF mutation (E1020K) in Pik3cd in the germline of C57BL/6 mice (Avery et al., 2018). Consistent with prior studies, Pik3cdGOF mouse B cells exhibited significantly reduced CSR to multiple IgG isotypes, in response to various stimuli including anti-CD40/IL-4 in vitro (Avery et al., 2018). These results demonstrate that patient-derived PI3K mutations affect the CSR process in a B cell intrinsic manner. Taken together, patient data further support the notion that the signaling balance between PTEN and PI3K pathway plays a crucial role in regulating CSR and antibody deficiency.
4.4. PI3Ks negatively and PTEN positively regulates AID expression
CD19Cre-mediated deletion of PTEN significantly reduced AID expression in B cells activated in vitro (Suzuki et al., 2003). We also found that acute deletion of PTEN in mature B cells also impaired AID expression in cytokine-activated B cells (Chen et al., 2015). Consistently, inducible expression of a PTEN transgene elevated AID expression in B cells activated by anti-CD40/IL-4 (Chen et al., 2015). In line with our study, Cγ1Cre-mediated deletion of Pten also markedly reduced AID expression in LPS/IL-4 stimulated B cells (Wang et al., 2018). In addition, this study sorted out IgM-BCR expressing GC B cells to exclude the potential contamination of isotype-switched GC B cells, and RT-PCR results showed that IgM-BCR expressing GC B cells from Pten-deficient mice exhibited markedly reduced AID transcripts (Wang et al., 2018). However, another independent study showed that Cγ1Cre-mediated Pten deficiency did not impair AID expression in total GC B cells (Sander et al., 2015). The discrepancy between these studies may be attributed to the specific populations examined for AID expression. Wang et al. sorted IgM-BCR expressing GC B cells, performed RT-PCR to detect AID, PTEN and Cγ1 transcripts, and found that IgM-BCR+ GC B cells had markedly reduced PTEN and AID transcripts in Ptenfl/flCγ1Cre/+ mice (Wang et al., 2018). Of note, a comparable level of Cγ1 transcripts was detected in IgM-BCR+ GC B cells isolated from control and Ptenfl/flCγ1Cre/+ mice, which explained the occurrence of Cγ1Cre-mediated PTEN deletion in IgM-BCR expressing GC B cells (Wang et al., 2018). Sander et al. employed total GC B cells without further sorting IgM-expressing cells and found that GC B cells from Ptenfl/flCγ1Cre/+ mice showed no defects in AID transcription (Sander et al., 2015). It is likely that total GC B cells consist of isotype-switched B cells (e.g., IgG2a+ or IgG3+) that may express a high level of AID, thereby confounding the result of AID transcription analysis. Based on all the prior studies, we conclude that PTEN is a positive regulator of AID expression.
Contrary to PTEN’s role in promoting AID expression, the PI3K/AKT pathway inhibits AID expression. Our studies showed that AID protein expression was drastically inhibited by p110α hyperactivation in anti-CD40/IL-4 activated mouse B cells (Chen et al., 2015). We found that pharmacological inhibition of AKT with GSK690693 markedly increased AID protein expression in activated mouse B cells (Chen et al., 2015). Our results are consistent with previous reports showing that p110δ pharmacological blockade or p110δD910A point mutation resulted in a higher level of AID transcription in in vitro activated B cells (Zhang et al., 2008). Furthermore, Pik3cdGOF mouse B cells had a reduced level of AID transcripts in response to anti-CD40/IL-4 stimulation (Avery et al., 2018). On the other hand, p110δ inhibitors enhance genomic instability in normal and neoplastic B cells via an AID-dependent mechanism (Compagno et al., 2017). p110δ inhibitors increased AID expression and translocation frequency to Igh locus and AID off-target sites in human chronic lymphocytic leukemia and mantle cell lymphoma cell lines (Compagno et al., 2017). Patients treated with idelalisib, a p110δ inhibitor, had increased SHM in AID off-target genes (Compagno et al., 2017). Overall, these studies collectively demonstrate that the PI3K/AKT pathway inhibits AID expression.
Defective AID expression in PTEN deficient B cells can be partially rescued by genetic deletion or pharmacological inhibition of p110δ (Janas et al., 2008; Omori et al., 2006), indicating that other isoforms of PI3Ks may also function to suppress AID expression. Indeed, we showed that p110α can also inhibit AID expression and CSR (Chen et al., 2015). Interestingly, ectopic expression of AID in PTEN deficient B cells only partially rescued CSR (Omori et al., 2006), suggesting that PTEN or PI3Ks may also regulate the activity of AID or affect other aspects of CSR process. In this regard, PTEN has been shown to translocate into nucleus and nuclear PTEN is involved in DNA repair (Bassi et al., 2013; Shen et al., 2007). While DNA repair is essential for completing CSR (Boboila, Alt and Schwer, 2012), there is no direct evidence showing that PTEN participates in the DNA repair process during CSR. PTEN can be phosphorylated in the nucleus by ATM, and ATM-mediated PTEN phosphorylation promotes the turnover of nuclear PTEN (Bassi et al., 2013). However, it remains unknown whether nuclear PTEN plays a role in regulating AID expression or activity.
In summary, multiple receptors expressed by B cells coordinately trigger activating signals to promote CSR and allow B cells to produce IgG, IgE and IgA for effective humoral immunity. Engaging these receptors also elicits inhibitory signals for negative feedback regulation of AID expression and CSR. The activating and inhibitory signaling are exquisitely balanced to ensure a proper level of humoral immune response that protects against invading pathogens, yet does not cause collateral damages, such as AID-induced genomic instability in B lymphocytes.
Acknowledgments
We apologize to those whose work was not cited due to length restrictions. We thank Rachel A. Woolaver for proofreading of the manuscript. This work was supported by University of Colorado School of Medicine and Cancer Center startup funds, R21-AI133110 and R01-DE027329 to J.H.W., and a fund from the Cancer League of Colorado and American Cancer Society (IRG 57-001-53) to Z.C.
ABBREVIATIONS
- AID
Activation-induced deaminase
- CSR
Class switch recombination
- SHM
Somatic hypermutation
- IL-4
interleukin-4
- BCR
B cell antigen receptor
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensin homolog
- TNFR
Tumor necrosis factor receptor
- TLR
Toll-like receptor
- TRAF2
TNFR associated factor 2
- TRAF3
TNFR associated factor 3
Footnotes
Compliance with Ethics Guidelines
Zhangguo Chen and Jing H. Wang declare no conflict of interest.
Reference
- Abu-Rish EY, Amrani Y, and Browning MJ (2013). Toll-like receptor 9 activation induces expression of membrane-bound B-cell activating factor (BAFF) on human B cells and leads to increased proliferation in response to both soluble and membrane-bound BAFF. Rheumatology (Oxford) 52, 1190. [DOI] [PubMed] [Google Scholar]
- Akira S, and Takeda K (2004). Toll-like receptor signalling. Nat Rev Immunol 4, 499. [DOI] [PubMed] [Google Scholar]
- Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, Bedell MA, Edelhoff S, Disteche CM, Simoneaux DK, and et al. (1993). CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science 259, 990. [DOI] [PubMed] [Google Scholar]
- Alt FW, Zhang Y, Meng FL, Guo C, and Schwer B (2013). Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen-Nissen E, Hawn TR, Smith KD, Nachman A, Lampano AE, Uematsu S, Akira S, and Aderem A (2007). Cutting edge: Tlr5−/− mice are more susceptible to Escherichia coli urinary tract infection. J Immunol 178, 4717. [DOI] [PubMed] [Google Scholar]
- Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, Baxendale H, Coulter T, Curtis J, Wu C, Blake-Palmer K, Perisic O, Smyth D, Maes M, Fiddler C, Juss J, Cilliers D, Markelj G, Chandra A, Farmer G, Kielkowska A, Clark J, Kracker S, Debre M, Picard C, Pellier I, Jabado N, Morris JA, Barcenas-Morales G, Fischer A, Stephens L, Hawkins P, Barrett JC, Abinun M, Clatworthy M, Durandy A, Doffinger R, Chilvers ER, Cant AJ, Kumararatne D, Okkenhaug K, Williams RL, Condliffe A, and Nejentsev S (2013). Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 342, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzelon AN, Wu H, and Rickert RC (2003). Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol 4, 287. [DOI] [PubMed] [Google Scholar]
- Arcipowski KM, and Bishop GA (2012). Roles of the kinase TAK1 in TRAF6-dependent signaling by CD40 and its oncogenic viral mimic, LMP1. PLoS One 7, e42478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos AM, Busconi L, and Marshak-Rothstein A (2010). Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity 43, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery DT, Kane A, Nguyen T, Lau A, Nguyen A, Lenthall H, Payne K, Shi W, Brigden H, French E, Bier J, Hermes JR, Zahra D, Sewell WA, Butt D, Elliott M, Boztug K, Meyts I, Choo S, Hsu P, Wong M, Berglund LJ, Gray P, O’Sullivan M, Cole T, Holland SM, Ma CS, Burkhart C, Corcoran LM, Phan TG, Brink R, Uzel G, Deenick EK, and Tangye SG (2018). Germline-activating mutations in PIK3CD compromise B cell development and function. J Exp Med 215, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, and Stambolic V (2013). Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, and Alt FW (2005). The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature 438, 508. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, and Alt FW (2011). The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 144, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Li Y, and Long KB (2017). Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 17, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekeredjian-Ding I, and Jego G (2009). Toll-like receptors--sentries in the B-cell response. Immunology 128, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belletti B, Prisco M, Morrione A, Valentinis B, Navarro M, and Baserga R (2001). Regulation of Id2 gene expression by the insulin-like growth factor I receptor requires signaling by phosphatidylinositol 3-kinase. J Biol Chem 276, 13867. [DOI] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Borkowski AW, Martinez L, Greidinger EL, Yu BD, and Gallo RL (2012). Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med 18, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz BC, Jordan-Williams KL, Wang C, Kang SG, Liao J, Logan MR, Kim CH, and Taparowsky EJ (2010). Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med 207, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GA, Moore CR, Xie P, Stunz LL, and Kraus ZJ (2007). TRAF proteins in CD40 signaling. Adv Exp Med Biol 597, 131. [DOI] [PubMed] [Google Scholar]
- Boboila C, Alt FW, and Schwer B (2012). Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116, 1. [DOI] [PubMed] [Google Scholar]
- Bochud PY, Hersberger M, Taffe P, Bochud M, Stein CM, Rodrigues SD, Calandra T, Francioli P, Telenti A, Speck RF, Aderem A, and Swiss HIVCS (2007). Polymorphisms in Toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS 21, 441. [DOI] [PubMed] [Google Scholar]
- Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, and Chen J (2000). Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci U S A 97, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I, Segal DM, and Davies DR (2011). The structural biology of Toll-like receptors. Structure 19, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, and Goodman MF (2003). Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A 100, 4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth M, McClellan B, and Allansmith M (1967). Influence of sex in immunoglobulin levels. Nature 214, 1224. [DOI] [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, and Bravo R (1998). Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med 187, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantaert T, Schickel JN, Bannock JM, Ng YS, Massad C, Oe T, Wu R, Lavoie A, Walter JE, Notarangelo LD, Al-Herz W, Kilic SS, Ochs HD, Nonoyama S, Durandy A, and Meffre E (2015). Activation-Induced Cytidine Deaminase Expression in Human B Cell Precursors Is Essential for Central B Cell Tolerance. Immunity 43, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A, and Pirofski LA (2004). New concepts in antibody-mediated immunity. Infect Immun 72, 6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, Bram RJ, Jabara H, and Geha RS (2005). TACI and BAFF-R mediate isotype switching in B cells. J Exp Med 201, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, and Alt FW (2004). Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol 4, 541. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, Manis J, and Alt FW (2007). Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 94, 157. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, and Alt FW (2004). Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature 430, 992. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, and Alt FW (2003). Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 422, 726. [DOI] [PubMed] [Google Scholar]
- Chen Z, Eder MD, Elos MT, Viboolsittiseri SS, Chen X, and Wang JH (2016). Interplay between Target Sequences and Repair Pathways Determines Distinct Outcomes of AID-Initiated Lesions. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Getahun A, Chen X, Dollin Y, Cambier JC, and Wang JH (2015). Imbalanced PTEN and PI3K Signaling Impairs Class Switch Recombination. J Immunol 195, 5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ranganath S, Viboolsittiseri SS, Eder MD, Chen X, Elos MT, Yuan S, Hansen E, and Wang JH (2014). AID-initiated DNA lesions are differentially processed in distinct B cell populations. J Immunol 193, 5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Viboolsittiseri SS, O’Connor BP, and Wang JH (2012). Target DNA sequence directly regulates the frequency of activation-induced deaminase-dependent mutations. J Immunol 189, 3970. [DOI] [PubMed] [Google Scholar]
- Chen Z, and Wang JH (2014). Generation and repair of AID-initiated DNA lesions in B lymphocytes. Front Med 8, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, Phan RT, Datta A, Manis J, Alt FW, and Chaudhuri J (2009). Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proc Natl Acad Sci U S A 106, 2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, and Mills GB (2011). High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 1, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung LW, Walkiewicz KW, Besong TM, Guo H, Hawke DH, Arold ST, and Mills GB (2015). Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. Elife 4, e06866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, Neuberg D, Monti S, Giallourakis CC, Gostissa M, and Alt FW (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagno M, Wang Q, Pighi C, Cheong TC, Meng FL, Poggio T, Yeap LS, Karaca E, Blasco RB, Langellotto F, Ambrogio C, Voena C, Wiestner A, Kasar SN, Brown JR, Sun J, Wu CJ, Gostissa M, Alt FW, and Chiarle R (2017). Phosphatidylinositol 3-kinase delta blockade increases genomic instability in B cells. Nature 542, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, Agharahimi A, Meuwissen H, Stoddard J, Niemela J, Kuehn H, and Rosenzweig SD (2014). Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol 34, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, and Casellas R (2007). Regulation of AID expression in the immune response. J Exp Med 204, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpke A, Frank J, Peter M, and Heeg K (2006). Activation of toll-like receptor 9 by DNA from different bacterial species. Infect Immun 74, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, and Nussenzweig A (2013). The AID-induced DNA damage response in chromatin. Mol Cell 50, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, and Robson SC (2011). Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol 61, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane JA, and Fruman DA (2004). Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol 22, 563. [DOI] [PubMed] [Google Scholar]
- Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, Cavazzana M, Picard C, Durandy A, Fischer A, and Kracker S (2014). A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 124, 3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, and Geha RS (2004). Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol 16, 395. [DOI] [PubMed] [Google Scholar]
- Delker RK, Fugmann SD, and Papavasiliou FN (2009). A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat Immunol 10, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, and Rickert RC (2008). Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol 9, 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregibus MC, Buttiglieri S, Russo S, Bussolati B, and Camussi G (2003). CD40-dependent activation of phosphatidylinositol 3-kinase/Akt pathway mediates endothelial cell survival and in vitro angiogenesis. J Biol Chem 278, 18008. [DOI] [PubMed] [Google Scholar]
- Desiderio S (1997). Role of Btk in B cell development and signaling. Curr Opin Immunol 9, 534. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, and Neuberger MS (2007). Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76, 1. [DOI] [PubMed] [Google Scholar]
- Dickerson SK, Market E, Besmer E, and Papavasiliou FN (2003). AID mediates hypermutation by deaminating single stranded DNA. J Exp Med 197, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, and de Saint Basile G (1993). CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature 361, 541. [DOI] [PubMed] [Google Scholar]
- Dobbs AK, Yang T, Farmer D, Kager L, Parolini O, and Conley ME (2007). Cutting edge: a hypomorphic mutation in Igbeta (CD79b) in a patient with immunodeficiency and a leaky defect in B cell development. J Immunol 179, 2055. [DOI] [PubMed] [Google Scholar]
- Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, and Dalla-Favera R (2015). The FOXO1 Transcription Factor Instructs the Germinal Center Dark Zone Program. Immunity 43, 1064. [DOI] [PubMed] [Google Scholar]
- Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, and Dalla-Favera R (2012). The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 13, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, and Landmann R (2002). Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis 186, 798. [DOI] [PubMed] [Google Scholar]
- Ehlers M, Fukuyama H, McGaha TL, Aderem A, and Ravetch JV (2006). TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 203, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, and Isenberg DA (1995). Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int 48, 705. [DOI] [PubMed] [Google Scholar]
- Eidinger D, and Garrett TJ (1972). Studies of the regulatory effects of the sex hormones on antibody formation and stem cell differentiation. J Exp Med 136, 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo A, Kutchukhidze N, Leung M, Christ AP, Urban JF Jr., Curotto de Lafaille MA, and Lafaille JJ (2007). Unique maturation program of the IgE response in vivo. Immunity 26, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzioni A, and Ochs HD (2004). The hyper IgM syndrome--an evolving story. Pediatr Res 56, 519. [DOI] [PubMed] [Google Scholar]
- Fang DF, He K, Wang N, Sang ZH, Qiu X, Xu G, Jian Z, Liang B, Li T, Li HY, Li AL, Zhou T, Gong WL, Yang B, Karin M, Zhang XM, and Li WH (2014). NEDD4 ubiquitinates TRAF3 to promote CD40-mediated AKT activation. Nat Commun 5, 4513. [DOI] [PubMed] [Google Scholar]
- Favalli EG, Biggioggero M, Crotti C, Becciolini A, Raimondo MG, and Meroni PL (2018). Sex and Management of Rheumatoid Arthritis. Clin Rev Allergy Immunol. [DOI] [PubMed] [Google Scholar]
- Fernandez D, Ortiz M, Rodriguez L, Garcia A, Martinez D, and Moreno de Alboran I (2013). The proto-oncogene c-myc regulates antibody secretion and Ig class switch recombination. J Immunol 190, 6135. [DOI] [PubMed] [Google Scholar]
- Ganley-Leal LM, Liang Y, Jagannathan-Bogdan M, Farraye FA, and Nikolajczyk BS (2010). Differential regulation of TLR4 expression in human B cells and monocytes. Mol Immunol 48, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardam S, Sierro F, Basten A, Mackay F, and Brink R (2008). TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28, 391. [DOI] [PubMed] [Google Scholar]
- Gazumyan A, Bothmer A, Klein IA, Nussenzweig MC, and McBride KM (2012). Activation-induced cytidine deaminase in antibody diversification and chromosome translocation. Adv Cancer Res 113, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SM, Willing A, Leypoldt F, Paul F, and Friese MA (2018). Sex differences in autoimmune disorders of the central nervous system. Semin Immunopathol. [DOI] [PubMed] [Google Scholar]
- Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, Yokota Y, and Shimizu A (2003). The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med 198, 1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MS, Kanegai CM, Doerr JR, and Wall R (2003). Somatic hypermutation of the B cell receptor genes B29 (Igbeta, CD79b) and mb1 (Igalpha, CD79a). Proc Natl Acad Sci U S A 100, 4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourzi P, Leonova T, and Papavasiliou FN (2007). Viral induction of AID is independent of the interferon and the Toll-like receptor signaling pathways but requires NF-kappaB. J Exp Med 204, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grech AP, Amesbury M, Chan T, Gardam S, Basten A, and Brink R (2004). TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity 21, 629. [DOI] [PubMed] [Google Scholar]
- Han JH, Akira S, Calame K, Beutler B, Selsing E, and Imanishi-Kari T (2007). Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity 27, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto A, Takeda K, Inaba M, Sekimata M, Kaisho T, Ikehara S, Homma Y, Akira S, and Kurosaki T (2000). Cutting edge: essential role of phospholipase C-gamma 2 in B cell development and function. J Immunol 165, 1738. [DOI] [PubMed] [Google Scholar]
- Hawn TR, Verbon A, Lettinga KD, Zhao LP, Li SS, Laws RJ, Skerrett SJ, Beutler B, Schroeder L, Nachman A, Ozinsky A, Smith KD, and Aderem A (2003). A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires’ disease. J Exp Med 198, 1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A, Puel A, Reichenbach J, Marodi L, Doffinger R, Vasconcelos J, Issekutz A, Krause J, Davies G, Li X, Grimbacher B, Plebani A, Meffre E, Picard C, Cunningham-Rundles C, Casanova JL, and Cerutti A (2010). The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol 11, 836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltemes-Harris LM, Gearhart PJ, Ghosh P, and Longo DL (2008). Activation-induced deaminase-mediated class switch recombination is blocked by anti-IgM signaling in a phosphatidylinositol 3-kinase-dependent fashion. Mol Immunol 45, 1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, Klein J, Pegno S, Sulis ML, Goldstein H, Amendolara B, Lei L, Maurer M, Bruce J, Canoll P, Hibshoosh H, and Parsons R (2013). A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BD, Hodakoski C, Barrows D, Mense SM, and Parsons RE (2014). PTEN function: the long and the short of it. Trends Biochem Sci 39, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, and Hartmann G (2002). Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168, 4531. [DOI] [PubMed] [Google Scholar]
- Hostager BS, Haxhinasto SA, Rowland SL, and Bishop GA (2003). Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem 278, 45382. [DOI] [PubMed] [Google Scholar]
- Hou B, Saudan P, Ott G, Wheeler ML, Ji M, Kuzmich L, Lee LM, Coffman RL, Bachmann MF, and DeFranco AL (2011). Selective utilization of Toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity 34, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huong le T, Kobayashi M, Nakata M, Shioi G, Miyachi H, Honjo T, and Nagaoka H (2013). In vivo analysis of Aicda gene regulation: a critical balance between upstream enhancers and intronic silencers governs appropriate expression. PLoS One 8, e61433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JK, Wang C, Du Z, Meyers RM, Kepler TB, Neuberg D, Kwong PD, Mascola JR, Joyce MG, Bonsignori M, Haynes BF, Yeap LS, and Alt FW (2017). Sequence intrinsic somatic mutation mechanisms contribute to affinity maturation of VRC01-class HIV-1 broadly neutralizing antibodies. Proc Natl Acad Sci U S A 114, 8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, and Durandy A (2003). Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol 4, 1023. [DOI] [PubMed] [Google Scholar]
- Incorvaia E, Sicouri L, Petersen-Mahrt SK, and Schmitz KM (2013). Hormones and AID: balancing immunity and autoimmunity. Autoimmunity 46, 128. [DOI] [PubMed] [Google Scholar]
- Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, Alt FW, Tang J, Oltz EM, Murphy TL, and Murphy KM (2011). The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol 12, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabara H, Laouini D, Tsitsikov E, Mizoguchi E, Bhan A, Castigli E, Dedeoglu F, Pivniouk V, Brodeur S, and Geha R (2002). The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity 17, 265. [DOI] [PubMed] [Google Scholar]
- Jabara HH, Chaudhuri J, Dutt S, Dedeoglu F, Weng Y, Murphy MM, Franco S, Alt FW, Manis J, and Geha RS (2008). B-cell receptor cross-linking delays activation-induced cytidine deaminase induction and inhibits class-switch recombination to IgE. J Allergy Clin Immunol 121, 191. [DOI] [PubMed] [Google Scholar]
- Jabara HH, and Geha RS (2005). Jun N-terminal kinase is essential for CD40-mediated IgE class switching in B cells. J Allergy Clin Immunol 115, 856. [DOI] [PubMed] [Google Scholar]
- Jabara HH, Weng Y, Sannikova T, and Geha RS (2009). TRAF2 and TRAF3 independently mediate Ig class switching driven by CD40. Int Immunol 21, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Ma CA, Liu S, Brown M, Cohen J, and Strober W (2001). Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2, 223. [DOI] [PubMed] [Google Scholar]
- Janas ML, Hodson D, Stamataki Z, Hill S, Welch K, Gambardella L, Trotman LC, Pandolfi PP, Vigorito E, and Turner M (2008). The effect of deleting p110delta on the phenotype and function of PTEN-deficient B cells. J Immunol 180, 739. [DOI] [PubMed] [Google Scholar]
- Janz S (2006). Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst) 5, 1213. [DOI] [PubMed] [Google Scholar]
- Jhamnani RD, Nunes-Santos CJ, Bergerson J, and Rosenzweig SD (2018). Class-Switch Recombination (CSR)/Hyper-IgM (HIGM) Syndromes and Phosphoinositide 3-Kinase (PI3K) Defects. Front Immunol 9, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, and Diaz M (2007). Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol 178, 7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Zhao ML, Scearce RM, and Diaz M (2011). Activation-induced deaminase-deficient MRL/lpr mice secrete high levels of protective antibodies against lupus nephritis. Arthritis Rheum 63, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, Brittain GC, Chang M, Cheng X, and Sun SC (2012). The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol 13, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Kaneko H, Fukao T, Terada T, Asano T, Kasahara K, and Kondo N (2003). Hyper-IgM syndrome with putative dominant negative mutation in activation-induced cytidine deaminase. J Allergy Clin Immunol 112, 755. [DOI] [PubMed] [Google Scholar]
- Kato L, Stanlie A, Begum NA, Kobayashi M, Aida M, and Honjo T (2012). An evolutionary view of the mechanism for immune and genome diversity. J Immunol 188, 3559. [DOI] [PubMed] [Google Scholar]
- Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, and Kikutani H (1994). The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1, 167. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11, 373. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, and Kawai T (2014). Toll-like receptor signaling pathways. Front Immunol 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesh S, Mensah NY, Peterlongo P, Jaffe D, Hsu K, M VDB, O’Reilly R, Pamer E, Satagopan J, and Papanicolaou GA (2005). TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann N Y Acad Sci 1062, 95. [DOI] [PubMed] [Google Scholar]
- Kimman TG, Banus S, Reijmerink N, Reimerink J, Stelma FF, Koppelman GH, Thijs C, Postma DS, and Kerkhof M (2008). Association of interacting genes in the toll-like receptor signaling pathway and the antibody response to pertussis vaccination. PLoS One 3, e3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnon C, Hinshelwood S, Levinsky RJ, and Lovering RC (1993). X-linked agammaglobulinemia--gene cloning and future prospects. Immunol Today 14, 554. [DOI] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, Casellas R, and Nussenzweig MC (2011). Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, and Mathis D (1999). From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 10, 451. [DOI] [PubMed] [Google Scholar]
- Koyasu S (2003). The role of PI3K in immune cells. Nat Immunol 4, 313. [DOI] [PubMed] [Google Scholar]
- Kraus M, Alimzhanov MB, Rajewsky N, and Rajewsky K (2004). Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117, 787. [DOI] [PubMed] [Google Scholar]
- Kuppers R (2005). Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer 5, 251. [DOI] [PubMed] [Google Scholar]
- Kuppers R, and Dalla-Favera R (2001). Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 20, 5580. [DOI] [PubMed] [Google Scholar]
- Kuraoka M, Snowden PB, Nojima T, Verkoczy L, Haynes BF, Kitamura D, and Kelsoe G (2017). BCR and Endosomal TLR Signals Synergize to Increase AID Expression and Establish Central B Cell Tolerance. Cell Rep 18, 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, and Vogel SN (2009). TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol 85, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Theilen M, and Chaudhuri J (2010). Walking the AID tightrope. Nat Immunol 11, 107. [DOI] [PubMed] [Google Scholar]
- Lee DY, and Clayton DA (1996). Properties of a primer RNA-DNA hybrid at the mouse mitochondrial DNA leading-strand origin of replication. J Biol Chem 271, 24262. [DOI] [PubMed] [Google Scholar]
- Lee YR, Chen M, and Pandolfi PP (2018). The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol 19, 547. [DOI] [PubMed] [Google Scholar]
- Li J, and McMurray RW (2007). Effects of estrogen receptor subtype-selective agonists on autoimmune disease in lupus-prone NZB/NZW F1 mouse model. Clin Immunol 123, 219. [DOI] [PubMed] [Google Scholar]
- Liang G, Kitamura K, Wang Z, Liu G, Chowdhury S, Fu W, Koura M, Wakae K, Honjo T, and Muramatsu M (2013). RNA editing of hepatitis B virus transcripts by activation-induced cytidine deaminase. Proc Natl Acad Sci U S A 110, 2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G, Zhang M, Harhaj EW, and Sun SC (2004). Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 279, 26243. [DOI] [PubMed] [Google Scholar]
- Lin KI, Angelin-Duclos C, Kuo TC, and Calame K (2002). Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol 22, 4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, and Schatz DG (2008). Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841. [DOI] [PubMed] [Google Scholar]
- Liu M, and Schatz DG (2009). Balancing AID and DNA repair during somatic hypermutation. Trends Immunol 30, 173. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, and Mak TW (1999). TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, Basu U, Alt F, and Storb U (2006). AID in somatic hypermutation and class switch recombination. Curr Opin Immunol 18, 164. [DOI] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, and Ohashi PS (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145. [DOI] [PubMed] [Google Scholar]
- Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, Avery DT, Moens L, Cannons JL, Biancalana M, Stoddard J, Ouyang W, Frucht DM, Rao VK, Atkinson TP, Agharahimi A, Hussey AA, Folio LR, Olivier KN, Fleisher TA, Pittaluga S, Holland SM, Cohen JI, Oliveira JB, Tangye SG, Schwartzberg PL, Lenardo MJ, and Uzel G (2014a). Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol 15, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, Butrick M, Matthews H, Price S, Biancalana M, Wang X, Richards M, Pozos T, Barlan I, Ozen A, Rao VK, Su HC, and Lenardo MJ (2014b). Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 211, 2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai T, Zan H, Zhang J, Hawkins JS, Xu Z, and Casali P (2010). Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. J Biol Chem 285, 37797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, and Cantley LC (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martomo SA, Yang WW, and Gearhart PJ (2004). A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J Exp Med 200, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masukata H, and Tomizawa J (1990). A mechanism of formation of a persistent hybrid between elongating RNA and template DNA. Cell 62, 331. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Tseng PH, Vallabhapurapu S, Luo JL, Zhang W, Wang H, Vignali DA, Gallagher E, and Karin M (2008). Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 321, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter MS, and Ochsenbein AF (2008). Natural antibodies target virus-antibody complexes to organized lymphoid tissue. Autoimmun Rev 7, 480. [DOI] [PubMed] [Google Scholar]
- Matthews AJ, Zheng S, DiMenna LJ, and Chaudhuri J (2014). Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol 122, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul RW, and Gearhart PJ (2009). Women, autoimmunity, and cancer: a dangerous liaison between estrogen and activation-induced deaminase? J Exp Med 206, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, and Nussenzweig MC (2008). Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med 205, 2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Yang B, and Suen WC (2018). Prospects for modulating the CD40/CD40L pathway in the therapy of the hyper-IgM syndrome. Innate Immun 24, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, and Busslinger M (2016). Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 17, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montamat-Sicotte D, Litzler LC, Abreu C, Safavi S, Zahn A, Orthwein A, Muschen M, Oppezzo P, Munoz DP, and Di Noia JM (2015). HSP90 inhibitors decrease AID levels and activity in mice and in human cells. Eur J Immunol 45, 2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, and Honjo T (1999). Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 274, 18470. [DOI] [PubMed] [Google Scholar]
- Muto T, Okazaki IM, Yamada S, Tanaka Y, Kinoshita K, Muramatsu M, Nagaoka H, and Honjo T (2006). Negative regulation of activation-induced cytidine deaminase in B cells. Proc Natl Acad Sci U S A 103, 2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, and Honjo T (2002). Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J Exp Med 195, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka H, Tran TH, Kobayashi M, Aida M, and Honjo T (2010). Preventing AID, a physiological mutator, from deleterious activation: regulation of the genomic instability that is associated with antibody diversity. Int Immunol 22, 227. [DOI] [PubMed] [Google Scholar]
- Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, Munechika E, Sakai T, Shirasawa T, Akiba H, Kobata T, Santee SM, Ware CF, Rennert PD, Taniguchi M, Yagita H, and Okumura K (1999). Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Natl Acad Sci U S A 96, 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian G, and Kovats S (2005). Understanding sex biases in immunity: effects of estrogen on the differentiation and function of antigen-presenting cells. Immunol Res 31, 91. [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Harris RS, Di Noia J, and Petersen-Mahrt SK (2003). Immunity through DNA deamination. Trends Biochem Sci 28, 305. [DOI] [PubMed] [Google Scholar]
- Neuberger MS, and Rada C (2007). Somatic hypermutation: activation-induced deaminase for C/G followed by polymerase eta for A/T. J Exp Med 204, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Duncan GS, Mirtsos C, Ng M, Speiser DE, Shahinian A, Marino MW, Mak TW, Ohashi PS, and Yeh WC (1999). TRAF2 deficiency results in hyperactivity of certain TNFR1 signals and impairment of CD40-mediated responses. Immunity 11, 379. [DOI] [PubMed] [Google Scholar]
- Nonoyama S, Hollenbaugh D, Aruffo A, Ledbetter JA, and Ochs HD (1993). B cell activation via CD40 is required for specific antibody production by antigen-stimulated human B cells. J Exp Med 178, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak U, Matthews AJ, Zheng S, and Chaudhuri J (2011). The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol 12, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, and Nussenzweig MC (2010). Origin of chromosomal translocations in lymphoid cancer. Cell 141, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard VH, and Schatz DG (2006). Targeting of somatic hypermutation. Nat Rev Immunol 6, 573. [DOI] [PubMed] [Google Scholar]
- Ohnishi K, Ebling FM, Mitchell B, Singh RR, Hahn BH, and Tsao BP (1994). Comparison of pathogenic and non-pathogenic murine antibodies to DNA: antigen binding and structural characteristics. Int Immunol 6, 817. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, and Vanhaesebroeck B (2002). Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297, 1031. [DOI] [PubMed] [Google Scholar]
- Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, Subramanian N, Bunney TD, Baxendale RW, Martins MS, Romberg N, Komarow H, Aksentijevich I, Kim HS, Ho J, Cruse G, Jung MY, Gilfillan AM, Metcalfe DD, Nelson C, O’Brien M, Wisch L, Stone K, Douek DC, Gandhi C, Wanderer AA, Lee H, Nelson SF, Shianna KV, Cirulli ET, Goldstein DB, Long EO, Moir S, Meffre E, Holland SM, Kastner DL, Katan M, Hoffman HM, and Milner JD (2012). Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med 366, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, and Rickert RC (2006). Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 25, 545. [DOI] [PubMed] [Google Scholar]
- Orthwein A, and Di Noia JM (2012). Activation induced deaminase: how much and where? Semin Immunol 24, 246. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Patenaude AM, Affar el B, Lamarre A, Young JC, and Di Noia JM (2010). Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med 207, 2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X, Xu S, and Lam KP (2012). Deficiency in TNFRSF13B (TACI) expands T-follicular helper and germinal center B cells via increased ICOS-ligand expression but impairs plasma cell survival. Proc Natl Acad Sci U S A 109, 15401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH, Knoblauch N, Guarnerio J, Ito K, Turka LA, Beck AH, Pinton P, Bronson RT, Wei W, and Pandolfi PP (2014). Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Kim PH, Lee KS, Lee SH, Seo GY, Yoo YC, Lee J, and Casali P (2013). APRIL stimulates NF-kappaB-mediated HoxC4 induction for AID expression in mouse B cells. Cytokine 61, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, Xu Z, Mai T, and Casali P (2009). HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol 10, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, Chaganti RS, Klein U, Kuppers R, Rajewsky K, and Dalla-Favera R (1998). BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci U S A 95, 11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson HC, Kraus M, Kim YM, Ploegh H, and Rajewsky K (2006). The B cell receptor promotes B cell activation and proliferation through a non-ITAM tyrosine in the Igalpha cytoplasmic domain. Immunity 25, 55. [DOI] [PubMed] [Google Scholar]
- Pauklin S, and Petersen-Mahrt SK (2009). Progesterone inhibits activation-induced deaminase by binding to the promoter. J Immunol 183, 1238. [DOI] [PubMed] [Google Scholar]
- Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, and Petersen-Mahrt SK (2009). Estrogen directly activates AID transcription and function. J Exp Med 206, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls SD, Lafarge ST, Landego I, Zhang T, and Marshall AJ (2012). The phosphoinositide 3-kinase signaling pathway in normal and malignant B cells: activation mechanisms, regulation and impact on cellular functions. Front Immunol 3, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, and Nussenzweig MC (2010). Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, and Nussenzweig MC (2011). AID targeting in antibody diversity. Adv Immunol 110, 1. [DOI] [PubMed] [Google Scholar]
- Peeva E, and Zouali M (2005). Spotlight on the role of hormonal factors in the emergence of autoreactive B-lymphocytes. Immunol Lett 101, 123. [DOI] [PubMed] [Google Scholar]
- Peng HZ, Du MQ, Koulis A, Aiello A, Dogan A, Pan LX, and Isaacson PG (1999). Nonimmunoglobulin gene hypermutation in germinal center B cells. Blood 93, 2167. [PubMed] [Google Scholar]
- Peng SL, Szabo SJ, and Glimcher LH (2002). T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A 99, 5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P, Bransteitter R, Petruska J, and Goodman MF (2003). Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 424, 103. [DOI] [PubMed] [Google Scholar]
- Picard C, Casanova JL, and Puel A (2011). Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev 24, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, McDonald D, Geha RS, Takada H, Krause JC, Creech CB, Ku CL, Ehl S, Marodi L, Al-Muhsen S, Al-Hajjar S, Al-Ghonaium A, Day-Good NK, Holland SM, Gallin JI, Chapel H, Speert DP, Rodriguez-Gallego C, Colino E, Garty BZ, Roifman C, Hara T, Yoshikawa H, Nonoyama S, Domachowske J, Issekutz AC, Tang M, Smart J, Zitnik SE, Hoarau C, Kumararatne DS, Thrasher AJ, Davies EG, Bethune C, Sirvent N, de Ricaud D, Camcioglu Y, Vasconcelos J, Guedes M, Vitor AB, Rodrigo C, Almazan F, Mendez M, Arostegui JI, Alsina L, Fortuny C, Reichenbach J, Verbsky JW, Bossuyt X, Doffinger R, Abel L, Puel A, and Casanova JL (2010). Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platts-Mills TA (2001). The role of immunoglobulin E in allergy and asthma. Am J Respir Crit Care Med 164, S1. [DOI] [PubMed] [Google Scholar]
- Plotkin SA (2010). Correlates of protection induced by vaccination. Clin Vaccine Immunol 17, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pone EJ, Xu Z, White CA, Zan H, and Casali P (2012a). B cell TLRs and induction of immunoglobulin class-switch DNA recombination. Front Biosci (Landmark Ed) 17, 2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z, and Casali P (2012b). BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nat Commun 3, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar N, and Fuleihan RL (2014). The hyper IgM syndromes. Clin Rev Allergy Immunol 46, 120. [DOI] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, and Neuberger MS (2004). Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell 16, 163. [DOI] [PubMed] [Google Scholar]
- Rada C, Ehrenstein MR, Neuberger MS, and Milstein C (1998). Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity 9, 135. [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, and Neuberger MS (2002). Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12, 1748. [DOI] [PubMed] [Google Scholar]
- Radic MZ, Ibrahim SM, Rauch J, Camper SA, and Weigert M (1995). Constitutive secretion of transgene-encoded IgG2b autoantibodies leads to symptoms of autoimmune disease. J Immunol 155, 3213. [PubMed] [Google Scholar]
- Ramadani F, Bolland DJ, Garcon F, Emery JL, Vanhaesebroeck B, Corcoran AE, and Okkenhaug K (2010). The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci Signal 3, ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Stavropoulos P, Jankovic M, and Nussenzweig MC (2003). Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol 4, 452. [DOI] [PubMed] [Google Scholar]
- Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, and et al. (1993). Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science 261, 358. [DOI] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, and Durandy A (2000). Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 102, 565. [DOI] [PubMed] [Google Scholar]
- Rickert RC (2013). New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol 13, 578. [DOI] [PubMed] [Google Scholar]
- Rickert RC, Roes J, and Rajewsky K (1997). B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res 25, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, Ried T, Nussenzweig A, and Nussenzweig MC (2009). AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell 36, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, Jankovic M, Silva IT, Rommel PC, Bosque D, Eisenreich T, Nussenzweig A, and Nussenzweig MC (2015). Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 162, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson SC, Sevigny J, and Zimmermann H (2006). The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal 2, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, and Kolchanov NA (1992). Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim Biophys Acta 1171, 11. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, and Kunkel TA (2001). Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol 2, 530. [DOI] [PubMed] [Google Scholar]
- Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, Santinelli S, Saunders T, Hebeis B, Killeen N, Okkenhaug K, and Turner M (2010). Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol 185, 4042. [DOI] [PubMed] [Google Scholar]
- Sakurai D, Kanno Y, Hase H, Kojima H, Okumura K, and Kobata T (2007). TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol 37, 110. [DOI] [PubMed] [Google Scholar]
- Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, Li S, Imami K, Selbach M, Di Virgilio M, Bullinger L, and Rajewsky K (2015). PI3 Kinase and FOXO1 Transcription Factor Activity Differentially Control B Cells in the Germinal Center Light and Dark Zones. Immunity 43, 1075. [DOI] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, and Akira S (2005). Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6, 1087. [DOI] [PubMed] [Google Scholar]
- Sayegh CE, Quong MW, Agata Y, and Murre C (2003). E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol 4, 586. [DOI] [PubMed] [Google Scholar]
- Schamel WW, and Reth M (2000). Monomeric and oligomeric complexes of the B cell antigen receptor. Immunity 13, 5. [DOI] [PubMed] [Google Scholar]
- Schena F, Volpi S, Faliti CE, Penco F, Santi S, Proietti M, Schenk U, Damonte G, Salis A, Bellotti M, Fais F, Tenca C, Gattorno M, Eibel H, Rizzi M, Warnatz K, Idzko M, Ayata CK, Rakhmanov M, Galli T, Martini A, Canossa M, Grassi F, and Traggiai E (2013). Dependence of immunoglobulin class switch recombination in B cells on vesicular release of ATP and CD73 ectonucleotidase activity. Cell Rep 3, 1824. [DOI] [PubMed] [Google Scholar]
- Schulze-Luehrmann J, and Ghosh S (2006). Antigen-receptor signaling to nuclear factor kappa B. Immunity 25, 701. [DOI] [PubMed] [Google Scholar]
- Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, and Tybulewicz VL (2013). The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 38, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DW, and Gascoyne RD (2014). The tumour microenvironment in B cell lymphomas. Nat Rev Cancer 14, 517. [DOI] [PubMed] [Google Scholar]
- Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, and Grewal IS (2003). Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity 18, 279. [DOI] [PubMed] [Google Scholar]
- Setz CS, Hug E, Khadour A, Abdelrasoul H, Bilal M, Hobeika E, and Jumaa H (2018). PI3K-Mediated Blimp-1 Activation Controls B Cell Selection and Homeostasis. Cell Rep 24, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, and Baltimore D (1995). Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 80, 321. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, and Staudt LM (2002). Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17, 51. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Rosenwald A, Hurt EM, Giltnane JM, Lam LT, Pickeral OK, and Staudt LM (2001). Signatures of the immune response. Immunity 15, 375. [DOI] [PubMed] [Google Scholar]
- Shen HM, Michael N, Kim N, and Storb U (2000). The TATA binding protein, c-Myc and survivin genes are not somatically hypermutated, while Ig and BCL6 genes are hypermutated in human memory B cells. Int Immunol 12, 1085. [DOI] [PubMed] [Google Scholar]
- Shen HM, Peters A, Baron B, Zhu X, and Storb U (1998). Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 280, 1750. [DOI] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, and Storb U (2006). Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J Immunol 177, 5386. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, and Yin Y (2007). Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157. [DOI] [PubMed] [Google Scholar]
- Sheth S, Brito R, Mukherjea D, Rybak LP, and Ramkumar V (2014). Adenosine receptors: expression, function and regulation. Int J Mol Sci 15, 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkura R, Ito S, Begum NA, Nagaoka H, Muramatsu M, Kinoshita K, Sakakibara Y, Hijikata H, and Honjo T (2004). Separate domains of AID are required for somatic hypermutation and class-switch recombination. Nat Immunol 5, 707. [DOI] [PubMed] [Google Scholar]
- Shinohara H, Yasuda T, Aiba Y, Sanjo H, Hamadate M, Watarai H, Sakurai H, and Kurosaki T (2005). PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J Exp Med 202, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simister NE (2003). Placental transport of immunoglobulin G. Vaccine 21, 3365. [DOI] [PubMed] [Google Scholar]
- Smith SH, and Cancro MP (2003). Cutting edge: B cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J Immunol 170, 5820. [DOI] [PubMed] [Google Scholar]
- Sohail A, Klapacz J, Samaranayake M, Ullah A, and Bhagwat AS (2003). Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res 31, 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, and Rawlings DJ (2005). Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23, 561. [DOI] [PubMed] [Google Scholar]
- Song Z, Zhu X, Jin R, Wang C, Yan J, Zheng Q, Nanda A, Granger DN, and Li G (2014). Roles of the kinase TAK1 in CD40-mediated effects on vascular oxidative stress and neointima formation after vascular injury. PLoS One 9, e101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, and Rajewsky K (2009). PI3 kinase signals BCR-dependent mature B cell survival. Cell 139, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J (2011). Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol 32, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, and Schrader CE (2008). Mechanism and regulation of class switch recombination. Annu Rev Immunol 26, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, and Schrader CE (2014). IgH chain class switch recombination: mechanism and regulation. J Immunol 193, 5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storb U, Shen HM, Michael N, and Kim N (2001). Somatic hypermutation of immunoglobulin and non-immunoglobulin genes. Philos Trans R Soc Lond B Biol Sci 356, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurmond J, and Diamond B (2015). Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125, 2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, and Nakano T (2003). Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med 197, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, Schwarz K, Kogan SC, Casellas R, Schatz DG, Lieber MR, Greaves MF, and Muschen M (2015). Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol 16, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, Kinoshita K, Muramatsu M, and Honjo T (2003). AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol 4, 843. [DOI] [PubMed] [Google Scholar]
- Takemura N, Kawasaki T, Kunisawa J, Sato S, Lamichhane A, Kobiyama K, Aoshi T, Ito J, Mizuguchi K, Karuppuchamy T, Matsunaga K, Miyatake S, Mori N, Tsujimura T, Satoh T, Kumagai Y, Kawai T, Standley DM, Ishii KJ, Kiyono H, Akira S, and Uematsu S (2014). Blockade of TLR3 protects mice from lethal radiation-induced gastrointestinal syndrome. Nat Commun 5, 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, and Klinman DM (2001). Cutting edge: Role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol 167, 3555. [DOI] [PubMed] [Google Scholar]
- Thuong NT, Hawn TR, Thwaites GE, Chau TT, Lan NT, Quy HT, Hieu NT, Aderem A, Hien TT, Farrar JJ, and Dunstan SJ (2007). A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun 8, 422. [DOI] [PubMed] [Google Scholar]
- Tian M, and Alt FW (2000). Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J Biol Chem 275, 24163. [DOI] [PubMed] [Google Scholar]
- Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, Honjo T, and Nagaoka H (2010). B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol 11, 148. [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, and Lieber MR (2008). Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell 135, 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao BP, Ohnishi K, Cheroutre H, Mitchell B, Teitell M, Mixter P, Kronenberg M, and Hahn BH (1992). Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J Immunol 149, 350. [PubMed] [Google Scholar]
- Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, and Karin M (2010). Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol 11, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, and Geha RS (2001). TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity 15, 647. [DOI] [PubMed] [Google Scholar]
- Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, and et al. (1993). Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 72, 279. [DOI] [PubMed] [Google Scholar]
- Uslu K, Coleman AS, Allman WR, Katsenelson N, Bram RJ, Alugupalli KR, and Akkoyunlu M (2014). Impaired B cell receptor signaling is responsible for reduced TACI expression and function in X-linked immunodeficient mice. J Immunol 192, 3582. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan B, Yen WF, Pucella JN, and Chaudhuri J (2014). AIDing Chromatin and Transcription-Coupled Orchestration of Immunoglobulin Class-Switch Recombination. Front Immunol 5, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, and Waterfield MD (2001). Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70, 535. [DOI] [PubMed] [Google Scholar]
- Vaughan JH (1993). 1992 Joseph J. Bunim Lecture. Pathogenetic concepts and origins of rheumatoid factor in rheumatoid arthritis. Arthritis Rheum 36, 1. [DOI] [PubMed] [Google Scholar]
- Vuong BQ, and Chaudhuri J (2012). Combinatorial mechanisms regulating AID-dependent DNA deamination: interacting proteins and post-translational modifications. Semin Immunol 24, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, and Williams RL (1999). Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402, 313. [DOI] [PubMed] [Google Scholar]
- Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, Hoffmeyer A, Jackson CW, Cleveland JL, Murray PJ, and Ihle JN (2000). Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity 13, 25. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Hou B, Yang M, Dong Z, Qi H, and Liu W (2018). PTEN-Regulated AID Transcription in Germinal Center B Cells Is Essential for the Class-Switch Recombination and IgG Antibody Responses. Front Immunol 9, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH (2013). The role of activation-induced deaminase in antibody diversification and genomic instability. Immunol Res 55, 287. [DOI] [PubMed] [Google Scholar]
- Wang JH, Gostissa M, Yan CT, Goff P, Hickernell T, Hansen E, Difilippantonio S, Wesemann DR, Zarrin AA, Rajewsky K, Nussenzweig A, and Alt FW (2009a). Mechanisms promoting translocations in editing and switching peripheral B cells. Nature 460, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wuerffel R, Feldman S, Khamlichi AA, and Kenter AL (2009b). S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med 206, 1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang G, Vogel P, Neale G, Reizis B, and Chi H (2012). Transforming growth factor beta-activated kinase 1 (TAK1)-dependent checkpoint in the survival of dendritic cells promotes immune homeostasis and function. Proc Natl Acad Sci U S A 109, E343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss U, Zoebelein R, and Rajewsky K (1992). Accumulation of somatic mutants in the B cell compartment after primary immunization with a T cell-dependent antigen. Eur J Immunol 22, 511. [DOI] [PubMed] [Google Scholar]
- Woolaver RA, Wang X, Dollin Y, Xie P, Wang JH, and Chen Z (2018). TRAF2 Deficiency in B Cells Impairs CD40-Induced Isotype Switching That Can Be Rescued by Restoring NF-kappaB1 Activation. J Immunol 201, 3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Poovassery J, Stunz LL, Smith SM, Schultz ML, Carlin LE, and Bishop GA (2011). Enhanced Toll-like receptor (TLR) responses of TNFR-associated factor 3 (TRAF3)-deficient B lymphocytes. J Leukoc Biol 90, 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Stunz LL, Larison KD, Yang B, and Bishop GA (2007). Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 27, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, and Flavell RA (1994). Mice deficient for the CD40 ligand. Immunity 1, 423. [DOI] [PubMed] [Google Scholar]
- Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, Steinacker P, Li Z, Yates J 3rd, Herron B, Otto M, Zan H, Fu H, and Casali P (2010). 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nat Struct Mol Biol 17, 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zan H, Pone EJ, Mai T, and Casali P (2012). Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 12, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue K, Rada C, and Neuberger MS (2006). The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med 203, 2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav A, Olaru A, Saltis M, Setren A, Cerny J, and Livak F (2006). Identification of a ubiquitously active promoter of the murine activation-induced cytidine deaminase (AICDA) gene. Mol Immunol 43, 529. [DOI] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, and Casellas R (2011). Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol 12, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeap LS, Hwang JK, Du Z, Meyers RM, Meng FL, Jakubauskaite A, Liu M, Mani V, Neuberg D, Kepler TB, Wang JH, and Alt FW (2015). Sequence-Intrinsic Mechanisms that Target AID Mutational Outcomes on Antibody Genes. Cell 163, 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, and Lieber MR (2003). R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4, 442. [DOI] [PubMed] [Google Scholar]
- Zan H, and Casali P (2013). Regulation of Aicda expression and AID activity. Autoimmunity 46, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, and Casanova JL (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522. [DOI] [PubMed] [Google Scholar]
- Zhang TT, Okkenhaug K, Nashed BF, Puri KD, Knight ZA, Shokat KM, Vanhaesebroeck B, and Marshall AJ (2008). Genetic or pharmaceutical blockade of p110delta phosphoinositide 3-kinase enhances IgE production. J Allergy Clin Immunol 122, 811. [DOI] [PubMed] [Google Scholar]
- Zhang X, Tang N, Hadden TJ, and Rishi AK (2011). Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813, 1978. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li J, Zhang YM, Zhang XM, and Tao J (2015). Effect of TACI signaling on humoral immunity and autoimmune diseases. J Immunol Res 2015, 247426. [DOI] [PMC free article] [PubMed] [Google Scholar]