

Abstract
We have developed an innovative methodology to overcome the lack of techniques for real-time assessment of degradable biomedical polymers at physiological conditions. The methodology was established by combining polymer characterization techniques with electrochemical sensors. The in vitro hydrolytic degradation of a series of aliphatic polyesters was evaluated by following the molar mass decrease and the mass loss at different incubation times while tracing pH and l-lactate released into the incubation media with customized miniaturized electrochemical sensors. The combination of different analytical approaches provided new insights into the mechanistic and kinetics aspects of the degradation of these biomedical materials. Although molar mass had to reach threshold values for soluble oligomers to be formed and specimens’ resorption to occur, the pH variation and l-lactate concentration were direct evidence of the resorption of the polymers and indicative of the extent of chain scission. Linear models were found for pH and released l-lactate as a function of mass loss for the l-lactide-based copolymers. The methodology should enable the sequential screening of degradable polymers at physiological conditions and has potential to be used for preclinical material’s evaluation aiming at reducing animal tests.
1. Introduction
The design and synthesis of degradable polymeric materials that find usability as temporary devices in biomedical applications, such as drug delivery carriers and scaffolds for tissue engineering, rely on the ability to program the degradation rate and profile, as well as the resorption profile to the application needs.1,2 Degradation is the process of chemical cleavage of macromolecules to form lower molar mass products, which, for degradable polymers such as aliphatic polyesters and in abiotic conditions, occurs through hydrolysis of the ester bonds. The resorption of aliphatic polyesters in hydrolytic conditions arises instead from the loss of mass owing to oligomers and low-molecular-weight products leaving the polymeric matrix and dissolving in the surrounding environment.3 Degradation is a function encoded in the chemical structure of polymers,4 being largely affected by the bulk properties, shape, thickness, and porosity of the material, together with environmental factors.5,6 This implies that for the real and absolute understanding of the mechanism of degradation and resorption of a polymer in a specific physiological environment, and therefore, to get information about the clinical outcome of a material, the degradation rate and the service lifetime need to be in vivo evaluated. With the analytical tools available to date, this evaluation is commonly predicted by means of a series of in vitro assays that provide information about polymer degradation under simplified conditions compared to the real physiological environment.
Typical in vivo experiments involve a large number of samples collected from different subjects (animals) that are usually analyzed over an appropriate time frame (from weeks to years) to get enough data about the material degradation and resorption process while operating in the in vivo context.7 Despite several studies being reported in the literature for specific polymers,8−11 limitations arise about the data that can be obtained because of ethical issues and the inability to follow the material over time once implanted. Such a kind of experiment requires that the animals involved in the test have to be sacrificed at different time scales over the total scaffold implantation time, with the collected samples being analyzed after their removal from the subject by standard (and expensive) methods centralized in laboratories. Altogether, the effectiveness of current approaches is really inconvenient in terms of the number of sacrificed subjects, time to provide valuable observations, resources, and compliance with ethical issues although they are necessary to answer the requirements for regulatory clearance. Moreover, the current methods prevent sequential and real-time monitoring of exactly the same sample during the implantation period in one subject, which may lead to biased interpretations due to results’ variability.
Because the scientific community is aware of the need for new tools for the in vivo and real-time assessment of polymeric materials’ degradation, some efforts have been recently accomplished toward the development of approaches based on chemical and physical sensors. Artzi et al. reported the in vivo degradation of materials both hydrolytically (poly(ethylene glycol) [PEG]/dextran hydrogel) and enzymatically (collagen) by fluorescence imagining of mice at different time scales (ranging from hours to 80 days).12 For this purpose, the materials were modified with a fluorescent tag and subcutaneously implanted, and then, the loss of fluorescence intensity was followed over time. A correlation between in vitro and in vivo assays was demonstrated, which in principle allows the in vivo resorption of new materials to be predicted. This approach was successfully extended to hyaluronan hydrogels for tissue engineering applications,13 thermosensitive PEGylated polyester hydrogel,14 and poly(lactide-co-glycolide) [PLGA] degradation for regenerative medicine applications.15 Also, fluorescence resonance energy transfer was implemented to follow the assembly and disassembly of micellar, thermoresponsive hydrogels consisting of triblock copolymers; the use of multiple tags allowed tracing the fate of the materials both in vitro and in vivo and at nano- and molecular levels.16 Despite the undeniable utility of these approaches, the chemical modification with fluorescent tags may affect the physicochemical properties of the polymer and the biological safety. Non-invasive approaches based on magnetic resonance imaging have recently been described as methodologies to trace hydrogel degradation,17,18 while photoacoustic tomography enabled the monitoring of the resorption of PLGA scaffolds doped with contrast agents.19
Yu et al. reported on a magnetoelastic-based sensor to wirelessly monitor the in vitro degradation of polylactic acid artificial bones.20 A piece of the artificial bone is formed by covering a strip containing the sensor by means of a three-dimensional (3D) printing technique. Then, the degradation was followed in a different medium, mainly alkaline solution at pH 12 and buffer at physiological pH (7.4), by measuring changes in the output power of the sensor. Although further in vivo application is claimed, this has not been reported yet. Schoning and co-workers developed capacitive field-effect sensors prepared as polymer-modified electrolyte–insulator–semiconductors for in vitro monitoring degradation of poly(d,l-lactide) [PDLLA].21,22 However, the approach is rather complex since (i) the material under study is coated in an electrochemical cell specifically designed for impedance measurements, and (ii) it is necessary to assume the equivalent electrical circuit to interpret the data. While the authors demonstrated the influence of pH and a lipase enzyme on the degradation profile, this approach seems difficult to be translated to in vivo applications. Salpavaara et al. presented inductively coupled passive resonance sensors embedded in the polymer shell for tracing PLGA copolymers presenting different degradation profiles.23 While resonance features were qualitatively compared to conventional polymer characterization methods, no mathematical definition was provided. It was simply claimed that the sensors provide easy-to-access information at the laboratory scale for general screenings.
Our rationale was that to succeed in the development of a real-time in vivo methodology for material degradation monitoring, it is essential to select the appropriate sensor readout providing a tangible and quantitative correlation with the physicochemical events occurring during the degradation and without requiring material labeling or functionalization. Accordingly, the technique should present appropriate selectivity for one analyte linked to the degradation process. Advantageously, electrochemical sensors have largely proved suitability for different types of decentralized measurements, including the clinical analysis of sweat, interstitial fluid, and blood with on-body wearable devices.24−29 Regarding the traced analyte(s), in principle, the best option is the monitoring of a compound that is released from the implanted material over time while degrading and which concentration could be directly correlated to the extent of degradation. For example, aliphatic polyesters, which are among the most used degradable polymers for biomedical applications,30,31 usually degrade by hydrolysis of the ester bonds, eventually forming low-molecular-weight hydroxyl acids as products. Poly(l-lactide) [PLLA] and relative copolymers degrade indeed, forming l-lactic acid (or l-lactate) as a final product of the hydrolytic degradation. l-Lactate represents, therefore, a suitable analyte to be tracked for the monitoring of their degradation process. Besides being a product of the degradation, it has also been demonstrated that the l-lactate released from implanted polymeric scaffolds has a role in the specific tissue regeneration mechanism, i.e., acting as a fuel for neurons32 and regulating chondrocyte proliferation in cartilage regeneration;33 its tracking might provide a further understanding of such biological processes. In addition, because of the formation of carboxylic groups, the pH of the surrounding environment decreases during degradation of polyesters.
Hence, we rationalized that the assessment of the in vivo degradation of degradable polyesters should be possible by means of l-lactate and pH sensing. Accordingly, our aim was to utilize pH and l-lactate electrochemical sensors in a series of in vitro experiments to analyze and monitor the formation of the final degradation products of polyesters with the goal to comprehend the physical and chemical changes that occur to the scaffolds over the degradation time in the service environment. The purpose was to combine traditional polymer characterization techniques with electrochemical sensors tracing the release of degradation products. Thus, we sought to obtain insights into the process of hydrolytic degradation of aliphatic polyesters at a macromolecular level and correlate this with new data obtained by the sensors in controlled in vitro environments. This step is a prerequisite to translate the sensing methodology to the tracing of in vivo performance of degradable devices and, in a near future, bridge indeed the gap between the in vitro and in vivo experiments. Models for pH variation and released l-lactate concentration as a function of traditional parameters (such as mass loss) for the analyzed l-lactide-based copolymers should enable the screening of degradable polymers at physiological conditions during preclinical material’s evaluation.
2. Materials and Methods
2.1. Materials
Poly(l-lactide) [PLLA], resomer L 207 S, poly(d,l-lactide) [PDLLA], resomer R 207 S, poly(l-lactide-co-glycolide) [PLGA], resomer LG 824 S, poly(l-lactide-co-trimethylene carbonate) [PLTMC], resomer LT 706 S, and poly(l-lactide-co-ε-caproalctone) [PCLA], resomer LC 703 S, were purchased from Evonik. Poly(d,l-lactide-co-glycolide) [PDLLGA], PURASORB PDLG 5010, was purchased from Corbion. Poly(ε-caprolactone), PCL, average Mn 80 kg mol–1, was purchased from Sigma-Aldrich. All of the polymers were used as received. Poly(ε-caprolactone-co-p-dioxanone) [PCLDX] was synthesized as previously reported.34 Phosphate-buffered saline (PBS) tablets were purchased from Sigma-Aldrich, and before use, fresh buffer solution (pH = 7.4) was prepared by dissolving each tablet in 200 mL of Milli-Q water.
2.2. Solvent Cast Films
Films for all of the polymers with a thickness of ∼200 μm were prepared by casting a polymer solution in CHCl3 at a concentration of 100 g L–1 in a Petri dish. Samples from the films, with a mass of about 50 mg, were then cut out and used for in vitro degradation tests.
2.3. In Vitro Degradation
Degradation tests were performed in hydrolytic conditions at 60 °C over 15–20 days depending on the degradation rate of the polymer. During this period, four data points were collected; for each data point, triplicate samples were analyzed. Small films with a mass of about 50 mg were dried in a vacuum to constant weight before tests. Afterward, samples were immersed separately in vials containing each 30 mL of sodium phosphate buffer (PBS), pH 7.4, and kept at 60 °C. The buffer solution was not changed over the degradation time. Films were withdrawn from the incubation medium at scheduled periods, washed carefully with distilled water, dried to constant weight to calculate the mass loss (%), and then analyzed by size exclusion chromatography (SEC). The incubation medium was used to measure the pH and l-lactate concentration.
2.4. Polymer Characterization Methods
Polymer composition was determined by 1H NMR spectroscopy. Spectra of polymer samples were obtained in CDCl3 at room temperature using a Bruker Avance 400 spectrometer (1H: 400.13; 13C: 100.62 MHz) and recorded using Bruker TopSpin v2.1 software. 1H NMR spectra were referenced to the residual solvent proton at δ 7.26 ppm. Data processing was performed using MestReNova v9.0.0 software.
Thermal properties of the as-received polymers and the as-synthesized PCLDX were evaluated by differential scanning calorimetry (DSC) using aluminum pans and a Mettler Toledo DSC 1 calibrated with indium. Measurements were performed under nitrogen flow with a heating rate of 10 °C min–1 from −30 to +220 °C. The DSC data are reported for the first heating cycle, the glass-transition temperature is taken as the midpoint ISO, and the melting temperatures are taken as the maximum value of the endothermic peaks.
Number-average molar mass (Mn), mass-average molar mass (Mw), and dispersity (Đ) were measured by size exclusion chromatography (SEC). The measurements were performed at 35 °C on a Malvern GPCMAX system equipped with a refractive index (RI) detector, Viscotek VE 3580, and three columns, one guard column (PLgel 5 μm Guard, 7.5 × 50 mm2) and two linear columns (PLgel 5 μm Mixed-D, 300 × 7.5 mm2), using CHCl3 as the eluent (0.5 mL min–1). Narrow polystyrene standards with molar mass in the range from 1.2 to 940 kg mol–1 were used for calibration, and the flow rate fluctuations were corrected using toluene as an internal standard. Reported data are the average values of at least three measures.
The mass loss of the films over degradation time was calculated using the equation (eq 1)
| 1 |
where m0 is the initial mass of the sample and mt is the mass at the specific time data point. The reported data are the mean value of three samples.
2.5. Electrochemical Measurements for pH and l-Lactate Quantification
All of the pH measurements were accomplished by a glass micro-pH electrode 6.0234.110 (Metrohm Autolab) coupled to pH-meter station 2.914.0220 (Metrohm Autolab). Notably, the electrode was calibrated using the manufacturer’s protocol. l-Lactate measurements were attained by means of a home-made l-lactate biosensor containing a working electrode (WE), a reference electrode (RE), and a counter electrode (CE). Screen-printed electrodes were fabricated on a polyester substrate and based on a conductive rectangular path (15 × 2 mm2) connected to a circular portion (diameter of 2.5 mm). The rectangular path was additionally covered by a rectangular portion of regular adhesive tape (acquired in 3M). A semiautomatic screen-printed machine (SPR-45 Automated SMT Stencil Printer, DDM Novastar, Inc.) was used. The WE and CE electrodes were made of carbon ink (CI-2051, Engineered Conductive Materials, Inc.), being cured at 80 °C for 5 min, while the RE was fabricated with Ag/AgCl ink (CI-1036, Engineered Conductive Materials, Inc.), being cured at 100 °C for 10 min. The RE and CE were used as printed (i.e., Ag/AgCl WE and carbon-based CE), whereas the circular part of the WE was further modified as reported elsewhere and just adapting the deposited volumes to the electrode dimensions.35,36 A volume of 3 μL of 0.1 M potassium ferricyanide in 0.01 M HCl/0.1 M KCl solution and then the same volume of 0.1 M FeCl3 in 0.01 M HCl/0.1 M KCl solution were added, both drops were thoroughly mixed by pipetting and allowed to react for 15–20 min to form the Prussian Blue redox mediator. The excess of volume (not attached to the electrode) was removed, and the surface was rinsed with 0.01 M HCl by drop-casting. Then, the WE was annealed for 1 h at 100 °C in an oven. A volume of 1 μL of 30 mg mL–1 solution of the enzyme lactate oxidase (LOx, EC# 1.13.12.4, purchased in Sorachim) in 10 mM phosphate buffer (PBS) at pH 7.4 was drop-casted on the electrode surface and allowed to dry at room temperature in the fume hood for 20 min. Subsequently, 1 μL of a solution of 1% chitosan (CHI) in 0.1 M acetic acid was drop-casted on top of the enzyme layer and allowed to dry at room temperature in the fume hood for 20 min. The WE electrode was stored in the fridge at 4 °C, with a lifetime of at least 1 month after its preparation. Figure 1a illustrates the l-lactate biosensor prepared as herein described.
Figure 1.
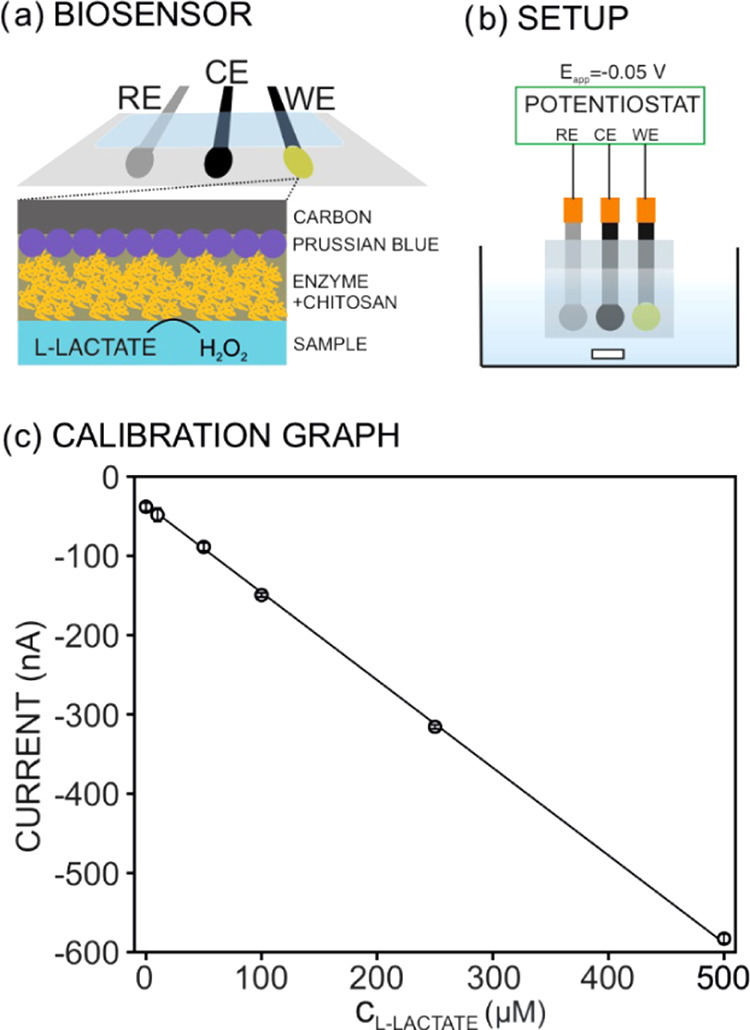
(a) Design of the l-lactate biosensor and the response mechanism: l-lactate in the solution interacts with the enzyme, which is physically immobilized in the electrode surface by means of a chitosan (CHI) layer. The formed hydrogen peroxide (H2O2) is an oxidizing agent of the Prussian Blue film being reduced at −0.05 V. WE, working electrode; RE, reference electrode; CE, counter electrode. (b) Sketch of the electrochemical cell for chronoamperometric measurements. The solution is under constant stirring. (c) Calibration graph observed for increasing l-lactate concentrations in saline phosphate buffer. The biosensor exhibited a slope of 1.01 μM nA–1 and an intercept of 45.1 nA with a limit of detection of 4.5 μM.
For the calibration of the l-lactate biosensor, the WE, RE, and CE were placed in a small beaker with an appropriate cap that allows placing of the electrodes and creating the electrical connections to the potentiostat (Autolab 302N, purchased in Metrohm Nordic). The experimental setup is presented in Figure 1b. For the calibration graph of l-lactate, increasing concentrations were added to a phosphate saline buffer background (PBS; 10 mM PB and 140 mM NaCl, pH 7.4), while the electrode runs under the chronoamperometry mode with an applied potential of −0.05 V. After each l-lactate addition, the steady-state potential was represented against the concentration. The linear fitting of the data corresponds to the calibration graph used to calculate the l-lactate concentration in in vitro experiments during polymeric scaffold degradation. One example of the observed calibration graph is present in Figure 1c. Depending on the degradation degree in the samples, dilution up to 1:50 was employed to maintain the electrochemical measurements within the linear range of response (up to 500 μM). Notably, the linear range of response of the lactate biosensor can be wider by means of slight changes in the tailoring of the sensing element, as has already been demonstrated in the literature.37,38 Thus, in future research toward in vivo measurements, the needed analytical performances will be easily tuned according to real lactate concentrations.
3. Results and Discussion
Herein, we have selected eight different polyesters (Table 1), which undergo degradation by hydrolytic cleavage of ester bonds, and monitored their hydrolysis to demonstrate the feasibility of pH and l-lactate electrochemical sensors as effective analytic devices to follow their degradation profile in real time and at physiological conditions. In particular, we evaluated PLLA and five of its copolymers with d-lactide [d-LA], glycolide [GA], ε-caprolactone [CL], and trimethylene carbonate [TMC] with various compositions but similar Mn, as well as a poly(ε-caprolactone) [PCL] and its copolymer with p-dioxanone [DX], which was recently developed by us.39 These polymers have different compositions and long-range order of the macromolecules, which determine diverse physical and thermal properties,40,41 and therefore, different degradation profiles are expected.5,42
Table 1. Structure, Composition, and Thermal Properties of the Evaluated Polymerse.

Determined by 1H NMR (CDCl3, 400 MHz).
Determined by SEC (CHCl3, 0.5 mL min–1) versus polystyrene standards.
Determined by DSC.
The degree of crystallinity, Xc, was calculated from DSC considering an enthalpy of fusion for an infinitely large PLLA crystal of 93 J g–1 53 and for a PCL crystal of 136.1 J g–1.54
Not detectable under the experimental DSC condition used.
The initial chemical composition, molar mass, dispersity, morphology, and physical state for each polymer were characterized because these are the key factors that determine the degradation and resorption rate of the materials.5 Besides that, the chemical composition is known to affect hydrolysis kinetics of the ester bonds as well as material hydrophilicity and solubility in water, determining both the diffusion degrees of water through the bulk of the specimen. Additionally, water diffusion strongly depends on the physical state of the polymer, which determines the free volume and the mobility of the chains at a macromolecular level, under the experimental conditions in which the hydrolytic degradation occurs. Thus, given a certain semicrystalline polymer, it has been generally observed that its amorphous phase is more prone to water uptake and, as a result, a faster degradation than the crystalline phase is expected.1 Moreover, the water uptake increases from the glassy to the rubbery state, being the degradation rate faster if the material is above the glass-transition temperature (Tg) at hydrolytic conditions. In this regard, under the accelerated hydrolytic conditions used in our experiments (T = 60 °C), PLLA is semicrystalline because the experimental T is below both the Tg and melting temperature (Tm), i.e., the polymer is brittle and in the glassy state. Indeed, it has been recently reported that the time necessary for PLA degradation to reach a Mn of 10 kg mol–1 is reduced by more than a half when the degradation temperature is increased from 40 to 85 °C, therefore crossing the Tg of the material.43 Nevertheless, models based on the time–temperature superimposition can be used to predict the lifetime of materials over a broad range of temperatures, even involving changes of the physical state due to temperature, such as glass transition.1,43
For PDLLA, the experimental T is close to its Tg, while PLGA, PLTMC, and PCLA are semicrystalline with a comparable degree of crystallinity (Xc) and in the rubbery state (experimental T above Tg). Finally, PCL and PCLDX are close to the molten state. The reader is referred to Table 1 for the values of Tg and Tm for each particular polymer.
3.1. Assessment of Hydrolytic Degradation of Biomedical Polymers by Means of Traditional Methods
The experiments of hydrolytic degradation were performed using solvent cast films of each polymer (listed in Table 1) in saline phosphate buffer (PBS) at 60 °C over 15–20 days. Models to predict the degradation time at different temperatures have been reported in the literature.44 During the entire time frame, the average molar mass decrease and the mass loss (%) of the polymers were monitored at different time points, coinciding with the pH and l-lactate detection using electrochemical sensors in the incubation media. The degradation process of the aliphatic polyesters is, in the current case, the result of (i) the abiotic hydrolysis of the ester bonds, which occurs throughout the bulk of the polymer in a random way, eventually leading to the formation of low-molecular-weight oligomers that are soluble in water and able to diffuse out of the matrix, therefore causing (ii) the resorption of the sample.1 Accordingly, to obtain the entire picture of the degradation process, we foresaw a combination of traditional polymer characterization methods to correlate molar mass decrease, dispersity, and mass loss with the new outcomes from the monitoring of the released low-molecular-weight oligomers by means of electrochemical sensors. All this information is expected to enable one to discern closer kinetic hydrolysis models, even during in vivo evaluation of the biomedical materials’ degradation by means of pH and l-lactate measurements. Furthermore, the approach could be applied to trace any analyte formed during material degradation as long as a selective sensing methodology is available.
Figure 2 presents the number-average molar mass (Mn) and dispersity (Đ) over the degradation time for each polymer at five subsequent time points T0–T4: 0, 5, 10, 15, and 20 days for PLLA, PDLLA, PLTMC, PCLA, and PCL; 0, 5, 7, 10, and 15 days for PLGA and PCLDX, and 0, 2, 5, 7, and 10 days for PDLLGA (the reader is referred to Table S1 for the values of Mn and Đ). As observed, PLLA and the relative copolymers with d-LA, GA, and CL, namely, PDLLA, PLGA, PDLLGA, and PCLA, showed an abrupt decrease of the molar mass in the very early stage of degradation (Figure 2a), which is typical of a heterogeneous bulk degradation process caused by random cleavage of the ester bonds along the macromolecules. Indeed, monomodal distributions of the molar mass were observed over the degradation time for all of the polymers, although the dispersity values (Đ) increased from 1.5 up to 5 at the later stage of degradation (Figure 2b). Low molar mass oligomers with Mn below 1 kg mol–1 were formed after 15 days for PDLLA and PLGA and after 5 days for PDLLGA and PLLA. The Mn of PCLA decreased down to 3 and 1.5 kg mol–1 after 15 and 20 days, respectively. For all of these samples, i.e., PLLA, PDLLA, PLGA, PDLLGA, and PCLA, the final Mn decrease was indeed more than 98%, as observed in Figure 3a. Overall, the copolymers of l- and d,l-LA with GA showed the highest decrease of molar mass as a consequence of the stronger interaction with water and the lower energy barrier for the hydrolysis reaction exhibited by the glycolic units compared to lactic units.45 Furthermore, in the case of PDLLGA, the hydrolysis rate was accelerated compared to PLGA, first, because the higher content in GA (50 against 18 mol %) favors both the hydrolysis of the esters and the water uptake capacity of the PDLLGA and, second, the amorphous state of the PDLLGA leads to an even higher water uptake capacity.46
Figure 2.
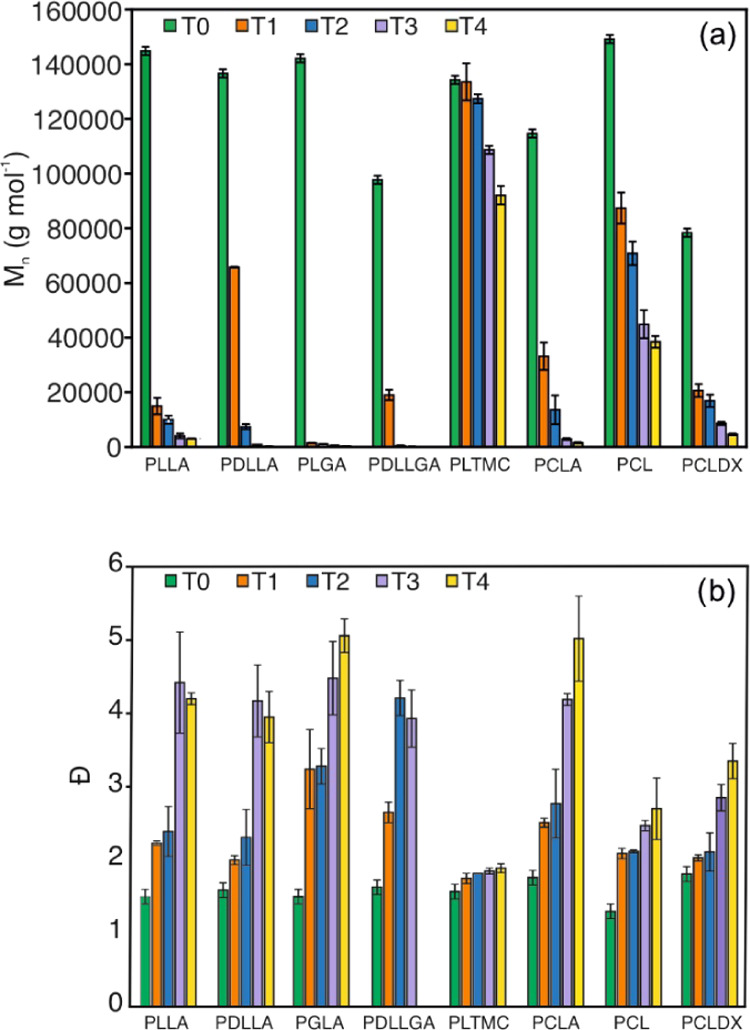
(a) Number-average molar mass, Mn, and (b) dispersity, Đ, values of the polymers over degradation time. T0–T4 are, respectively, 0, 5, 10, 15, and 20 days for PLLA, PDLLA, PLTMC, PCLA, and PCL; 0, 5, 7, 10, and 15 days for PLGA and PCLDX; and 0, 2, 5, 7, and 10 days for PDLLGA.
Figure 3.
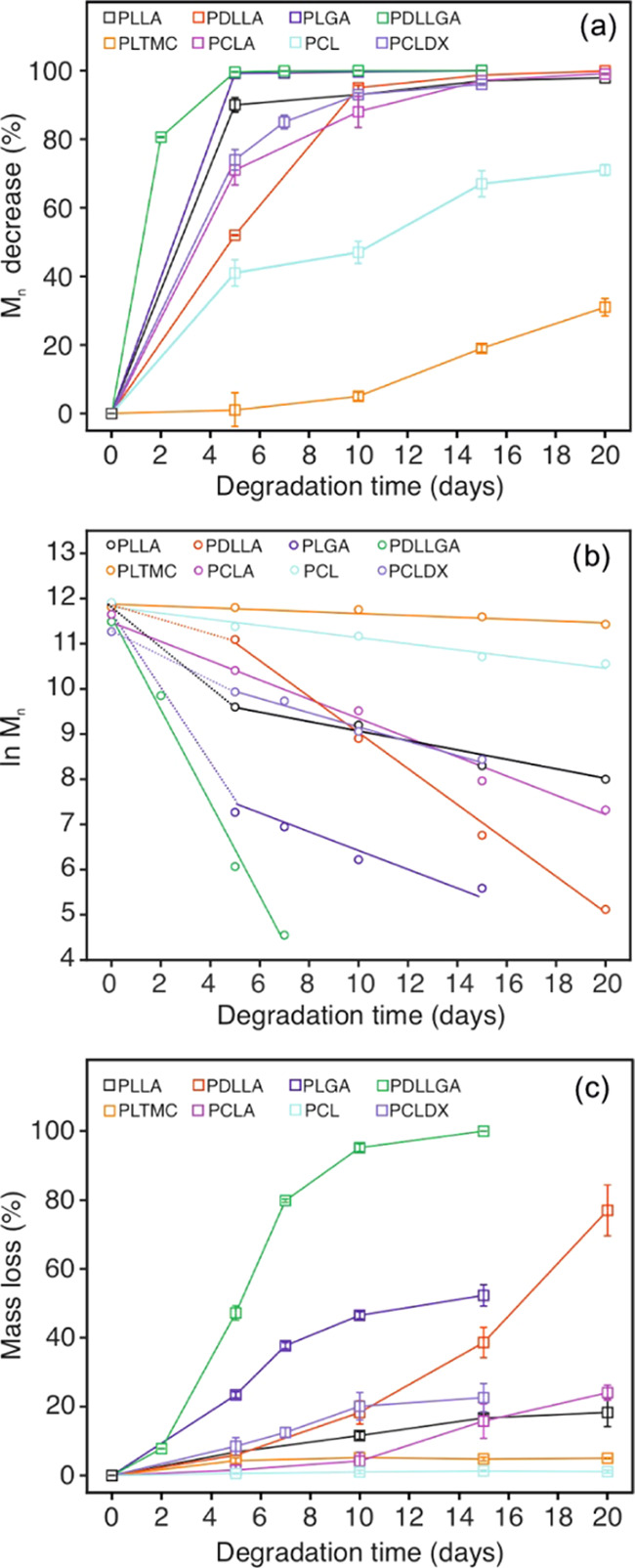
(a) Number-average molar mass decrease, Mn (%), and (b) mass loss (%) of the polymers over degradation time. (c) Kinetic model for the hydrolysis of the polymers over degradation time.
Because of the higher hydrophobicity and more packed long-range order of the macromolecules, PCL showed a slower molar mass decrease than the LA-based copolymers and the PCLDX, for which the inclusion of DX units enhances the hydrolysis rate with respect to PCL.39 The Mn decrease values presented by the PCL and PCLDX samples were 71% after 20 days and 96% after 15 days, respectively, (Figure 3a). This points out a closer behavior of PCLDX to PLLA and the relative copolymers with d,l-LA, GA, and CL (presenting a 98% Mn decrease as discussed above). PCL additionally displayed faster degradation than PLTMC, which was indeed the polymer that degraded the slowest: the Mn was around 90 kg mol–1 after 20 days with an overall decrease of merely 31% (see Figures 2a and 3a) despite the accelerated degradation conditions utilized for the experiments. The slower and more homogeneous decrease of molar mass is a consequence of the presence of TMC units and, therefore, of the carbonate bonds along the polymer chains. Notably, carbonate bonds are more hydrophobic and less susceptible to hydrolytic degradation than the ester bonds, also favoring long-range order of the chains due to the symmetry of the structure. Both aspects potentially prevent water uptake. The high content of TMC (40 mol %; see Table 1) in the copolymer explains the slow and homogenous profile of the molar mass decrease after an induction period of 10 days (Figures 2a and 3a). Indeed, if the amount of TMC in a copolymer of l-LA and TMC is ca. 16–21 mol %, it will slow down the heterogeneous bulk degradation process of PLLA, leading to a more homogenous decrease of the molar mass and longer retention of the physical properties of the material.42,47
Aliphatic polyesters usually present a bulk degradation behavior, and as a consequence of the statistical cleavage of the ester bonds along the macromolecules, the numbers of both carboxyl and hydroxyl chain-ends increase over time, while the number of reactive ester units decreases. Such a mechanism implies that the hydrolysis rate decreases over the experimental time, and therefore, an exponential decrease of the molar mass is observed. Thus, a linear correlation between ln(Mn) and the degradation time has been proposed as a model for the kinetics of hydrolysis of bulk degrading polyesters.48 In this regard, the plot of ln(Mn) versus time for the hydrolytic degradation of each polymer is presented in Figure 3b. In addition, the apparent rate constants of hydrolysis were extrapolated as the slope for the fitting of the portions in each data set showing a clear linearity (see Table S2).
Among the analyzed polymers, the PLTMC, PCL, PCLA, and PDLLGA displayed the best correlation considering the linear model, thus confirming the hypothesized bulk degradation behavior. An evident change in the slope of the curve after 5 days of degradation was observed for the rest of the polymers (i.e., PLLA, PDLLA, PLGA, and PCLDX). This delay in degradation is likely ascribed to the decrease in pH together with a higher concentration of degradation products in the incubation media, as shown below. Indeed, it has been recently reported that for the pH medium below 6, the hydrolysis of LA/GA copolymers is slowed down as a consequence of a decrease of concentration of the hydroxide ions, which are more effective catalysts than hydronium ions in the hydrolysis process.49 It is noticeable that, in the current experimental conditions, the hydrolysis rate is accelerated by the higher temperature compared to regular physiological conditions and, additionally, by the accumulation of degradation products in the incubation media, being able to autocatalyze the reaction.50 The incubation media was not changed over the degradation time with the purpose of monitoring pH changes and l-lactate release over time with the electrochemical sensors (vide infra).
When the hydrolytic degradation rate constants (days–1) were extrapolated as the slope of the curves for all of the assayed polymers, the observed values varied between 0.02 and 1.03 days–1 (see Table S2), with the degradation rate following the order PLTMC < PCL < PLLA < PCLDX < PCLA < PDLLA < PLGA < PDLLGA. The relative hydrolysis rate conjunctly depends on the susceptibility of the ester bonds of each monomeric unit toward hydrolysis, the water uptake capacity of the polymer, the hydrophilicity/hydrophobicity ratio in the experimental reaction conditions (all of these three factors being the constitutional units in the order glycolic > lactic > caproyl > trimethylene carbonate), and on the physical phase of the material at the experimental temperature, which also affects the water uptake capacity of the material. The order found for the assayed materials correlates rather well with these factors.
Because of the bulk degradation process, a faster decrease of molar mass yields a higher mass loss for the samples over the degradation time, as observed in Figure 3c. As a consequence of the lower Mn decrease, PLTMC and PCL were the polymers that showed the slowest resorption rate, with the measured mass loss being almost negligible after 20 days. Then, PLLA and PCLA presented a mass loss around 20% after 20 days, comparable to the observation for PCLDX after 15 days. Furthermore, polymers presented a higher decrease of Mn, also showing a higher resorption, with the PDLLGA being the only material with complete mass loss after 15 days. Pictures of the degraded samples at each time point are presented in Figure 4. The aspect of each polymer gives a visual proof of the measured mass loss, whose values over the degradation time are reported for each of the polymers in Table S3.
Figure 4.

Photographs of film samples at given time points over degradation time.
The relationship between molar mass decrease and mass loss can be in principle generalized independent of the polymer’s composition and expressed as a function of the extent of molar mass decrease to specific threshold values. Thus, mass loss was below 10% when the Mn of the polymer was higher than ca. 20 kg mol–1, while samples presented a loss of up to 40% of their mass when the Mn decreased to ca. 1 kg mol–1. In addition, as observed for PDLLGA, a decrease of Mn to ca. 100 g mol–1 corresponded to a mass loss of 80% or above, meaning a complete degradation in terms of molar mass and a total resorption after 15 days of degradation, as described above. Seemingly, it is necessary to reach a Mn decrease of ca. 80% to observe a mass loss of at least 10%, confirming once more the bulk degradation behavior. Furthermore, mass loss is observed when the random cleavage of ester bonds occurs to the extent that the molar mass of the oligomers formed as hydrolysis products reaches a certain value, which is different and typical for each polymer. This molar mass value is such that the oligomers are soluble in water and can diffuse out of the polymer matrix.
Specifically, it has been reported that oligomers of LA were soluble in water when the number of LA units was less than 13 and, therefore, when the Mn is lower than 936 g mol–1.51,52 A similar number has also been reported for copolymers of LA and GA, for which oligomers containing 12 α-hydroxy acids units were water soluble. On the other hand, PDLLGA having Mn of 1.7 kg mol–1 has per se the tendency to form water-soluble oligomers,49 therefore supporting the higher resorption rate observed for PDLLGA. Then, the negligible mass loss observed for PCL and PLTMC is evidently a consequence of the lower molar mass decrease. For such materials, a more substantial decrease would be indeed necessary before water-soluble oligomers are even formed due to their more hydrophobic nature.
Overall, the evaluated polymers degraded by simple hydrolysis of the ester bonds following a bulk degradation behavior. Random chain scission occurs until lower molar mass chains were formed, which below a certain value allows for diffusion out of the matrix, hence causing the resorption of the polymeric sample. The smallest “fragments” that should be formed, as final products of the hydrolytic degradation, are the hydroxy acids constituting the macromolecules, as depicted in Scheme 1a.
Scheme 1. (a) Mechanism of Hydrolytic Degradation by Random Chain Scission; (b) Structure and Relative pKa Value of the Hydroxy Acids Formed as Final Hydrolysis Products.

3.2. Monitoring of pH Changes and l-Lactate Release during Degradation of Biomedical Polymers by Means of Electrochemical Sensors
Electrochemical detection of pH and l-lactate was accomplished in each degradation medium after the accelerated process was stopped for each polymer at each established time point. We sought to find the correlation between pH and l-lactate concentration and the information extracted by means of traditional techniques about hydrolytic degradation and resorption. We aimed at providing models for such correlations to achieve a methodology for the real-time monitoring of polymer degradation. The pH of the degradation medium was measured for all polymer samples at each time point according to the procedure described in Section 2. In addition, the concentration of l-lactate was detected by means of the developed biosensor in the case of the polymers containing l-LA as a monomeric unit. Accordingly, Figure 5a,b presents pH and l-lactate concentration over the degradation time (the reader is referred to Tables S4 and S5 for the numeric collection of raw pH values and l-lactate concentrations). As observed, negligible variations in the pH of the incubation media were displayed over time for PLTMC and PCL (i.e., a pH really similar to the phosphate buffer initially used as the medium was always measured: 7.4). Then, a slight decrease of less than 0.3 pH units was detected for PLLA and PCLDX over the entire degradation experiment, while the decrease for PCLA from the 10th day was slightly higher, with a final pH of 6.6. In contrast, PDLLA showed an abrupt decrease of the pH after 10 days of degradation, whereas for PLGA and PDLLGA, the pH started to drop after 5 and 2 days of degradation time, reaching final pH values of 3.8 and 3.3, respectively.
Figure 5.

(a) pH and (b) l-lactate concentration in the incubation media as measured by electrochemical sensors over degradation time. (c) pH variation as a function of the l-lactide concentration. (d) l-Lactate release (%) normalized to each polymer composition as a function of the degradation time.
In view of these results, the decrease of the pH can be correlated to both the type and concentration of hydroxy acids released from the polymer sample over the degradation process. Notably, the concentration of the −COOH group formed in the medium, which is in principle responsible for the pH decrease, is a direct consequence of the number of chain scissions that have occurred for each polymer. Furthermore, the number of chain scissions determines the concentration of oligomers that diffuse from the material to the incubation media. Importantly, an in-depth analysis of the data presented in Figure 5a revealed that the trends found in pH variation also depend on the pKa values of the relative hydroxyl acids formed (see Scheme 1b): lower final pH values were detected for polymers comprising GA and LA as constitutional units, for which the pKa values are indeed the lowest ones. Then, on the basis of the pH value detected after 15 days of degradation time for the PLGA, it may be hypothesized that the degradation products exist as protonated species rather than as carboxylates. The same occurs for the PDLLGA after 7 days of degradation. Evidently, the higher pKa value of longer alkyl-chain acids is the reason for the minor pH decrease detected for the PCLDX, despite a Mn decrease of 96%.
The concentration of l-lactate was found to increase over time in agreement with the extent of the hydrolytic degradation of the polymer (Figure 5b and Table S5). Thus, the first indications of the presence of l-lactate were detected on the 5th day for PLGA and PDLLGA, with these two polymers always presenting higher concentrations of l-lactate in the medium. Following a different trend, PDLLA, PCLA, and PLLA showed an abrupt increase in l-lactate concentration after an induction period of 10 days. During these initial 10 days, the l-lactate concentration in the medium was close to the limit of detection of the biosensor (4.5 μM). As observed in Figure 5b, after the mentioned induction period, the concentration of l-lactate increased rather linearly with time for PLLA and PCLA, while slight variations from linearity were observed for PDLLA, PLGA, and PDLLGA. This correlation indeed indicates that the hydrolysis process involved the more susceptible ester moieties of the d-lactic and glycolic units. Finally, negligible l-lactate was detected for PLTMC over the entire degradation time, which means that the l-lactate formed is lower than the limit of detection of the electrochemical sensor (>4.5 μM) or even null in the degradation medium, which is in agreement with the lower extent of degradation of the polymer.
Comparing Figure 5a for the pH changes and Figure 5b for the concentration of l-lactate released to the medium, the same trends but in the opposite direction are evident: i.e., decreasing pH is correlated with l-lactate appearance in the medium. In this sense, the plot of the pH versusl-lactate concentration is shown in Figure 5c. Interestingly, the pH seems to follow a linear function with the l-lactate concentration for the polymers that are able to form only l-lactic and d-lactic acid as the most acidic species in the incubation media (i.e., for PLLA, PCLA, and PDLLA). However, when other acidic species are also formed, i.e., glycolic acid, a deviation from linearity is observed (see the trend for PLGA and PDLLGA in Figure 5c).
Besides the degradation rate, the amount of l-lactate released in the incubation medium of course depends on the initial composition of the polymer, specifically on the amount of l-lactide, which was different for the selected polymers (see Table 1). Thus, the percentage of l-lactate released into the medium normalized to the maximum concentration expected for a total degradation in each polymer was also calculated (see the Supporting Information for equations) and is reported in Figure 5d for each polymer over the degradation time. These data confirmed almost a total degradation of PDLLGA in its constitutional α-hydroxy acid (80% of the total l-lactate release), while the presence of longer oligomers in the incubation media was more likely for the other polymers since the amount of measured l-lactate is lower than the theoretical one calculated from the initial composition of each polymer.
Subsequently, we analyzed the correlations between the pH and the percentage of l-lactate release with the mass loss and the Mn of the polymers over degradation time (Figure 6a,b). As observed in Figure 6a, the mass loss tends to present a threshold value at around 10% of the total mass of the polymer sample before the pH of the degradation media substantially drops, following afterward a linear decrease with increasing mass loss for the most eroding polymers. Indeed, a series of linear models of the pH as a function of mass loss could be extrapolated after the induction period for the PLLA and its copolymers (Figure 6c, equations of the linear fitting are reported in Table S6). Furthermore, since the mass loss is a function of the extent of molar mass decrease in the polymer, pH variations were observed when Mn values were below 10 kg mol–1 and abruptly dropped when Mn decreased to more than about 1 kg mol–1 (Figure 6b). Indeed, this is in agreement with the threshold value of molar mass already reported for water-soluble oligomers comprising lactic and glycolic acids units.51 Consequently, the monitored pH variation provides information about the resorption of the polymer and could be considered as indicative of the extent of the chain scission that leads to the formation of soluble oligomers. Notably, linear models of pH variation as a function of mass loss could be obtained for slower degrading polymers when experiments are performed for longer times and until soluble oligomers are formed. Also, other copolymer compositions could be studied.
Figure 6.

(a) pH and l-lactate release (%) as a function of the mass loss (%). (b) pH and l-lactate release (%) as a function of Mn (log scale). (c) Linear models of pH decrease as a function of mass loss (%) after the induction period for PLLA, PDLLA, PLGA, PDLLGA, and PCLA. (d) Linear models of l-lactate release (%) as a function of mass loss (%) after the induction period for PDLLA, PLGA, and PDLLGA.
Analogous threshold values of mass loss and Mn were identified when inspecting the % of l-lactate release (Figure 6b). The polymers that showed a faster molar mass decrease presented a relationship between mass loss and Mn with the increase of l-lactate concentration after an induction: a series of linear models with increasing mass loss are indeed extrapolated for PDLLA, PLGA, and PDLLGA polymers (Figure 6d, equations of the linear fitting are reported in Table S7). Advantageously, the sensing of l-lactate enables the monitoring of material degradation in terms of detection of the final degradation products as a function of the initial composition, relative hydrolysis, and resorption rate. The outcomes are indicative of the changes that occur at the macromolecular level upon degradation, and they are applicable to all of the copolymers having l-LA as the monomeric unit once the resorption process has started.
Having established pertinent correlations between pH changes and l-lactate release with traditional observations related to hydrolytic degradation and resorption of a representative set of polymers, conveniently, we found linear models for the pH variation and the l-lactate released in/to the polymer surrounding that could be extrapolated as a unique function of the mass loss (Figure 6c,d; Tables S6 and S7). This is indeed independent of the degradation time and the conditions utilized to trigger the chain cleavage. Our further hypothesis is that, when such kinds of models are obtained by in vitro experiments for a particular polymer, the real-time in vivo resorption of the polymer can be determined by measuring the pH and the l-lactate concentration with electrochemical sensors. As a result, the methodology presented herein possesses a strong potential toward the universal in vivo and real-time monitoring of the degradation process of any kind of polyester material, as long as selective sensing methodologies to precisely detect the released compounds during its degradation are available.
4. Conclusions
It is herein demonstrated the potential for the electrochemical detection of pH and l-lactate to establish a route toward real-time assessment of biomedical polymer degradation. In vitro data obtained by traditional characterization techniques and those detecting pH variations and l-lactate released using electrochemical sensors in the surrounding environment of the polymers have been correlated. Specifically, two series of linear models have been found for the variation of pH and l-lactate as a sole function of the mass loss over degradation time in a set of l-lactide-based copolymers. These models signify that the developed methodology has the potential to be further translated in the preclinical evaluation of biomedical materials. Overall, the combination of traditional and sensing approaches has provided unique insights into the in vitro hydrolysis of bulk degrading polymers: a relationship between the rate of the degradation process at the macromolecular level, the formation of short oligomers and hydroxyl acids released as final degradation products, and the material resorption was concluded. Indeed, the relative rates of the hydrolytic degradation process were found to be dependent on the copolymer composition, pKa, of the formed acidic species, hydrophobicity, and physical state of the polymer material. Besides the tracing of the resorption profile, the results also demonstrate that the proposed methodology allows for monitoring of the molar mass decrease of the polymers although threshold values of Mn have to be reached before correlations with pH variations and/or l-lactate are detected. We envisage that the developed methodology can be applied to analyze the degradation process of all possible copolymers of l-lactide with other monomers utilized as biomedical polymers. Thus, it is expected to support research into existing and new implantable (and degradable) materials by facilitating a sequential and comprehensive monitoring of their degradation in the service environment while reducing currently required resources, especially in terms of animal tests and cost.
Acknowledgments
The authors acknowledge financial support from the Swedish Foundation for Strategic Research (RMA15-0010), the Swedish Research Council (Project Grant VR-2017-4887), Stiftelsen Olle Engkvist Byggmästare (204-0214), and Novo Nordisk Fonden (19OC0056171).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c01621.
Molar mass (Mn) and dispersity (Đ) values of the evaluated polymers over the degradation time (Table S1); apparent constants of hydrolysis rate extrapolated for the evaluated polymers (Table S2); mass loss (%) values of the evaluated polymers over the degradation time (Table S3); pH values of the incubation media for each of the evaluated polymers over the degradation time (Table S4); l-lactate concentration in the incubation media for each of the evaluated polymers containing l-lactide as the monomeric unit over the degradation time (Table S5); linear fitting of pH as a function of the mass loss (Table S6); linear fitting of the l-lactate released (%) as a function of the mass loss (Table S7); equations used to calculate l-lactate release % in the incubation media (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Laycock B.; Nikolić M.; Colwell J. M.; Gauthier E.; Halley P.; Bottle S.; George G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. 10.1016/j.progpolymsci.2017.02.004. [DOI] [Google Scholar]
- Hofmann D.; Entrialgo-Castaño M.; Kratz K.; Lendlein A. Knowledge-Based Approach towards Hydrolytic Degradation of Polymer-Based Biomaterials. Adv. Mater. 2009, 21, 3237–3245. 10.1002/adma.200802213. [DOI] [PubMed] [Google Scholar]
- Vert M.; Doi Y.; Hellwich K.-H.; Hess M.; Hodge P.; Kubisa P.; Rinaudo M.; Schué F. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012). Pure Appl. Chem. 2012, 84, 377. 10.1351/PAC-REC-10-12-04. [DOI] [Google Scholar]
- Dharmaratne N. U.; Jouaneh T. M. M.; Kiesewetter M. K.; Mathers R. T. Quantitative Measurements of Polymer Hydrophobicity Based on Functional Group Identity and Oligomer Length. Macromolecules 2018, 51, 8461–8468. 10.1021/acs.macromol.8b01747. [DOI] [Google Scholar]
- Woodard L. N.; Grunlan M. A. Hydrolytic Degradation and Erosion of Polyester Biomaterials. ACS Macro Lett. 2018, 7, 976–982. 10.1021/acsmacrolett.8b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan R. P.; Dove A. P. Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates. Biomater. Sci. 2017, 5, 9–21. 10.1039/C6BM00584E. [DOI] [PubMed] [Google Scholar]
- Dånmark S.; Finne-Wistrand A.; Schander K.; Hakkarainen M.; Arvidson K.; Mustafa K.; Albertsson A. C. In vitro and in vivo degradation profile of aliphatic polyesters subjected to electron beam sterilization. Acta Biomater. 2011, 7, 2035–2046. 10.1016/j.actbio.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Jeong S. I.; Kim B.-S.; Kang S. W.; Kwon J. H.; Lee Y. M.; Kim S. H.; Kim Y. H. In vivo biocompatibilty and degradation behavior of elastic poly(l-lactide-co-ε-caprolactone) scaffolds. Biomaterials 2004, 25, 5939–5946. 10.1016/j.biomaterials.2004.01.057. [DOI] [PubMed] [Google Scholar]
- Pamula E.; Menaszek E. In vitro and in vivo degradation of poly(l-lactide-co-glycolide) films and scaffolds. J. Mater. Sci.: Mater. Med. 2008, 19, 2063–2070. 10.1007/s10856-007-3292-2. [DOI] [PubMed] [Google Scholar]
- Seyednejad H.; Gawlitta D.; Kuiper R. V.; de Bruin A.; van Nostrum C. F.; Vermonden T.; Dhert W. J. A.; Hennink W. E. In vivo biocompatibility and biodegradation of 3D-printed porous scaffolds based on a hydroxyl-functionalized poly(ε-caprolactone). Biomaterials 2012, 33, 4309–4318. 10.1016/j.biomaterials.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Suliman S.; Sun Y.; Pedersen T. O.; Xue Y.; Nickel J.; Waag T.; Finne-Wistrand A.; Steinmüller-Nethl D.; Krueger A.; Costea D. E.; Mustafa K. In Vivo Host Response and Degradation of Copolymer Scaffolds Functionalized with Nanodiamonds and Bone Morphogenetic Protein 2. Adv. Healthcare Mater. 2016, 5, 730–742. 10.1002/adhm.201500723. [DOI] [PubMed] [Google Scholar]
- Artzi N.; Oliva N.; Puron C.; Shitreet S.; Artzi S.; bon Ramos A.; Groothuis A.; Sahagian G.; Edelman E. R. In vivo and in vitro tracking of erosion in biodegradable materials using non-invasive fluorescence imaging. Nat. Mater. 2011, 10, 890. 10.1038/nmat3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Rossi F.; Papa S.; Violatto M. B.; Bigini P.; Sorbona M.; Redaelli F.; Veglianese P.; Hilborn J.; Ossipov D. A. Non-invasive in vitro and in vivo monitoring of degradation of fluorescently labeled hyaluronan hydrogels for tissue engineering applications. Acta Biomater. 2016, 30, 188–198. 10.1016/j.actbio.2015.11.053. [DOI] [PubMed] [Google Scholar]
- Wang W.; Liu J.; Li C.; Zhang J.; Liu J.; Dong A.; Kong D. Real-time and non-invasive fluorescence tracking of in vivo degradation of the thermosensitive PEGlyated polyester hydrogel. J. Mater. Chem. B 2014, 2, 4185–4192. 10.1039/c4tb00275j. [DOI] [PubMed] [Google Scholar]
- Bardsley K.; Wimpenny I.; Yang Y.; El Haj A. J. Fluorescent, online monitoring of PLGA degradation for regenerative medicine applications. RSC Adv. 2016, 6, 44364–44370. 10.1039/C6RA04690H. [DOI] [Google Scholar]
- Huang P.; Song H.; Zhang Y.; Liu J.; Cheng Z.; Liang X.-J.; Wang W.; Kong D.; Liu J. FRET-enabled monitoring of the thermosensitive nanoscale assembly of polymeric micelles into macroscale hydrogel and sequential cognate micelles release. Biomaterials 2017, 145, 81–91. 10.1016/j.biomaterials.2017.07.012. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Chu C.; Kuddannaya S.; Yuan Y.; Walczak P.; Singh A.; Song X.; Bulte J. W. M. In Vivo Imaging of Composite Hydrogel Scaffold Degradation Using CEST MRI and Two-Color NIR Imaging. Adv. Funct. Mater. 2019, 29, 1903753 10.1002/adfm.201903753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Feng Z.; Song H.; Zhang J.; Dong A.; Kong D.; Wang W.; Huang P. 19F magnetic resonance imaging enabled real-time, non-invasive and precise localization and quantification of the degradation rate of hydrogel scaffolds in vivo. Biomater. Sci. 2020, 8, 3301–3309. 10.1039/D0BM00278J. [DOI] [PubMed] [Google Scholar]
- Zhang Y. S.; Cai X.; Yao J.; Xing W.; Wang L. V.; Xia Y. Non-Invasive and In Situ Characterization of the Degradation of Biomaterial Scaffolds by Volumetric Photoacoustic Microscopy. Angew. Chem., Int. Ed. 2014, 53, 184–188. 10.1002/anie.201306282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.; Ren L.; Tan Y.; Wang J. Wireless Magnetoelasticity-Based Sensor for Monitoring the Degradation Behavior of Polylactic Acid Artificial Bone In Vitro. Appl. Sci. 2019, 9, 739 10.3390/app9040739. [DOI] [Google Scholar]
- Schusser S.; Krischer M.; Bäcker M.; Poghossian A.; Wagner P.; Schöning M. J. Monitoring of the Enzymatically Catalyzed Degradation of Biodegradable Polymers by Means of Capacitive Field-Effect Sensors. Anal. Chem. 2015, 87, 6607–6613. 10.1021/acs.analchem.5b00617. [DOI] [PubMed] [Google Scholar]
- Schusser S.; Poghossian A.; Bäcker M.; Krischer M.; Leinhos M.; Wagner P.; Schöning M. J. An application of field-effect sensors for in-situ monitoring of degradation of biopolymers. Sens. Actuators, B 2015, 207, 954–959. 10.1016/j.snb.2014.10.058. [DOI] [Google Scholar]
- Salpavaara T.; Hänninen A.; Antniemi A.; Lekkala J.; Kellomäki M. Non-destructive and wireless monitoring of biodegradable polymers. Sens. Actuators, B 2017, 251, 1018–1025. 10.1016/j.snb.2017.05.116. [DOI] [Google Scholar]
- Parrilla M.; Ortiz-Gómez I.; Cánovas R.; Salinas-Castillo A.; Cuartero M.; Crespo G. A. Wearable Potentiometric Ion Patch for On-Body Electrolyte Monitoring in Sweat: Toward a Validation Strategy to Ensure Physiological Relevance. Anal. Chem. 2019, 91, 8644–8651. 10.1021/acs.analchem.9b02126. [DOI] [PubMed] [Google Scholar]
- Parrilla M.; Cuartero M.; Padrell Sánchez S.; Rajabi M.; Roxhed N.; Niklaus F.; Crespo G. A. Wearable All-Solid-State Potentiometric Microneedle Patch for Intradermal Potassium Detection. Anal. Chem. 2019, 91, 1578–1586. 10.1021/acs.analchem.8b04877. [DOI] [PubMed] [Google Scholar]
- Cuartero M.; Parrilla M.; Crespo G. A. Wearable Potentiometric Sensors for Medical Applications. Sensors 2019, 19, 363 10.3390/s19020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira P. C.; Ataíde V. N.; Silva Chagas C. L.; Angnes L.; Tomazelli Coltro W. K.; Longo Cesar Paixão T. R.; Reis de Araujo W. Wearable electrochemical sensors for forensic and clinical applications. TrAC, Trends Anal. Chem. 2019, 119, 115622 10.1016/j.trac.2019.115622. [DOI] [Google Scholar]
- Wiorek A.; Parrilla M.; Cuartero M.; Crespo G. A. Epidermal Patch with Glucose Biosensor: pH and Temperature Correction toward More Accurate Sweat Analysis during Sport Practice. Anal. Chem. 2020, 92, 10153–10161. 10.1021/acs.analchem.0c02211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Guzmán J. J.; Pérez-Ràfols C.; Cuartero M.; Crespo G. A. Microneedle based electrochemical (Bio)Sensing: Towards decentralized and continuous health status monitoring. TrAC, Trends Anal. Chem. 2021, 135, 116148 10.1016/j.trac.2020.116148. [DOI] [Google Scholar]
- Zhang F.; King M. W. Biodegradable Polymers as the Pivotal Player in the Design of Tissue Engineering Scaffolds. Adv. Healthcare Mater. 2020, 9, 1901358 10.1002/adhm.201901358. [DOI] [PubMed] [Google Scholar]
- Pappalardo D.; Mathisen T.; Finne-Wistrand A. Biocompatibility of Resorbable Polymers: A Historical Perspective and Framework for the Future. Biomacromolecules 2019, 20, 1465–1477. 10.1021/acs.biomac.9b00159. [DOI] [PubMed] [Google Scholar]
- Álvarez Z.; Castaño O.; Castells A. A.; Mateos-Timoneda M. A.; Planell J. A.; Engel E.; Alcántara S. Neurogenesis and vascularization of the damaged brain using a lactate-releasing biomimetic scaffold. Biomaterials 2014, 35, 4769–4781. 10.1016/j.biomaterials.2014.02.051. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wu Y.; Pan Z.; Sun H.; Wang J.; Yu D.; Zhu S.; Dai J.; Chen Y.; Tian N.; Heng B. C.; Coen N. D.; Xu H.; Ouyang H. The effects of lactate and acid on articular chondrocytes function: Implications for polymeric cartilage scaffold design. Acta Biomater. 2016, 42, 329–340. 10.1016/j.actbio.2016.06.029. [DOI] [PubMed] [Google Scholar]
- Fuoco T.; Ahlinder A.; Jain S.; Mustafa K.; Finne-Wistrand A. Poly(ε-caprolactone-co-p-dioxanone): a Degradable and Printable Copolymer for Pliable 3D Scaffolds Fabrication toward Adipose Tissue Regeneration. Biomacromolecules 2020, 21, 188–198. 10.1021/acs.biomac.9b01126. [DOI] [PubMed] [Google Scholar]
- Karyakin A. A. Advances of Prussian blue and its analogues in (bio)sensors. Curr. Opin. Electrochem. 2017, 5, 92–98. 10.1016/j.coelec.2017.07.006. [DOI] [Google Scholar]
- Payne M. E.; Zamarayeva A.; Pister V. I.; Yamamoto N. A. D.; Arias A. C. Printed, Flexible Lactate Sensors: Design Considerations Before Performing On-Body Measurements. Sci. Rep. 2019, 9, 13720 10.1038/s41598-019-49689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathee K.; Dhull V.; Dhull R.; Singh S. Biosensors based on electrochemical lactate detection: A comprehensive review. Biochem. Biophys. Rep. 2016, 5, 35–54. 10.1016/j.bbrep.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaryanov N. V.; Nikitina V. N.; Karpova E. V.; Karyakina E. E.; Karyakin A. A. Nonenzymatic Sensor for Lactate Detection in Human Sweat. Anal. Chem. 2017, 89, 11198–11202. 10.1021/acs.analchem.7b03662. [DOI] [PubMed] [Google Scholar]
- Fuoco T.; Finne-Wistrand A. Enhancing the Properties of Poly(ε-caprolactone) by Simple and Effective Random Copolymerization of ε-Caprolactone with p-Dioxanone. Biomacromolecules 2019, 20, 3171–3180. 10.1021/acs.biomac.9b00745. [DOI] [PubMed] [Google Scholar]
- Ahlinder A.; Fuoco T.; Finne-Wistrand A. Medical grade polylactide, copolyesters and polydioxanone: Rheological properties and melt stability. Polym. Test. 2018, 72, 214–222. 10.1016/j.polymertesting.2018.10.007. [DOI] [Google Scholar]
- Fuoco T.; Mathisen T.; Finne-Wistrand A. Poly(l-lactide) and Poly(l-lactide-co-trimethylene carbonate) Melt-Spun Fibers: Structure–Processing–Properties Relationship. Biomacromolecules 2019, 20, 1346–1361. 10.1021/acs.biomac.8b01739. [DOI] [PubMed] [Google Scholar]
- Fuoco T.; Mathisen T.; Finne-Wistrand A. Minimizing the time gap between service lifetime and complete resorption of degradable melt-spun multifilament fibers. Polym. Degrad. Stab. 2019, 163, 43–51. 10.1016/j.polymdegradstab.2019.02.026. [DOI] [Google Scholar]
- Limsukon W.; Auras R.; Selke S. Hydrolytic degradation and lifetime prediction of poly(lactic acid) modified with a multifunctional epoxy-based chain extender. Polym. Test. 2019, 80, 106108 10.1016/j.polymertesting.2019.106108. [DOI] [Google Scholar]
- Lyu; Schley J.; Loy B.; Lind D.; Hobot C.; Sparer R.; Untereker D. Kinetics and Time–Temperature Equivalence of Polymer Degradation. Biomacromolecules 2007, 8, 2301–2310. 10.1021/bm070313n. [DOI] [PubMed] [Google Scholar]
- Entrialgo-Castaño M.; Lendlein A.; Hofmann D. Molecular Modeling Investigations of Dry and Two Water-Swollen States of Biodegradable Polymers. Adv. Eng. Mater. 2006, 8, 434–439. 10.1002/adem.200600021. [DOI] [Google Scholar]
- Vey E.; Rodger C.; Meehan L.; Booth J.; Claybourn M.; Miller A. F.; Saiani A. The impact of chemical composition on the degradation kinetics of poly(lactic-co-glycolic) acid copolymers cast films in phosphate buffer solution. Polym. Degrad. Stab. 2012, 97, 358–365. 10.1016/j.polymdegradstab.2011.12.010. [DOI] [Google Scholar]
- Cork J.; Whittaker A. K.; Cooper-White J. J.; Grøndahl L. Tensile properties and in vitro degradation of P(TMC-co-LLA) elastomers. J. Mater. Chem. B 2015, 3, 4406–4416. 10.1039/C5TB00202H. [DOI] [PubMed] [Google Scholar]
- Pitt C. G.; Zhong-wei G. Modification of the rates of chain cleavage of poly(ϵ-caprolactone) and related polyesters in the solid state. J. Controlled Release 1987, 4, 283–292. 10.1016/0168-3659(87)90020-4. [DOI] [Google Scholar]
- Machatschek R.; Lendlein A. Fundamental insights in PLGA degradation from thin film studies. J. Controlled Release 2020, 319, 276–284. 10.1016/j.jconrel.2019.12.044. [DOI] [PubMed] [Google Scholar]
- Antheunis H.; van der Meer J.-C.; de Geus M.; Heise A.; Koning C. E. Autocatalytic Equation Describing the Change in Molecular Weight during Hydrolytic Degradation of Aliphatic Polyesters. Biomacromolecules 2010, 11, 1118–1124. 10.1021/bm100125b. [DOI] [PubMed] [Google Scholar]
- Höglund A.; Hakkarainen M.; Edlund U.; Albertsson A.-C. Surface Modification Changes the Degradation Process and Degradation Product Pattern of Polylactide. Langmuir 2010, 26, 378–383. 10.1021/la902166j. [DOI] [PubMed] [Google Scholar]
- Borovikov P. I.; Sviridov A. P.; Antonov E. N.; Dunaev A. G.; Krotova L. I.; Fatkhudinov T. K.; Popov V. K. Model of aliphatic polyesters hydrolysis comprising water and oligomers diffusion. Polym. Degrad. Stab. 2019, 159, 70–78. 10.1016/j.polymdegradstab.2018.11.017. [DOI] [Google Scholar]
- Kalb B.; Pennings A. J. General crystallization behaviour of poly(l-lactic acid). Polymer 1980, 21, 607–612. 10.1016/0032-3861(80)90315-8. [DOI] [Google Scholar]
- Khambatta F. B.; Warner F.; Russell T.; Stein R. S. Small-angle x-ray and light scattering studies of the morphology of blends of poly(ϵ-caprolactone) with poly(vinyl chloride). J. Polym. Sci., Polym. Phys. Ed. 1976, 14, 1391–1424. 10.1002/pol.1976.180140805. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.