

Abstract
Background:
Tranexamic acid (TXA) is used to reduce bleeding. TXA inhibits plasmin(ogen) binding to fibrin and reduces fibrinolysis. TXA antifibrinolytic activity is typically measured by clot lysis assays; however, effects on plasmin generation (PG) are unclear due to a lack of tools to measure PG in plasma.
Aims:
Develop an assay to measure PG kinetics in human plasma. Determine effects of TXA on PG and compare with fibrinolysis measured by rotational thromboelastometry (ROTEM).
Methods:
We characterized effects of plasminogen, tissue plasminogen activator, fibrinogen, and α2-antiplasmin on PG in vitro. We also studied effects of TXA on PG in plasma from 30 pregnant women administered intravenous TXA (5, 10, or 15 mg/kg) during cesarean delivery. PG was measured by calibrated fluorescence. PG parameters were compared with TXA measured by mass spectrometry and ROTEM of whole blood.
Results:
The PG assay is specific for plasmin and sensitive to tissue plasminogen activator, fibrin(ogen), and α2-antiplasmin. Addition of TXA to plasma in vitro dose dependently prolonged the clot lysis time and delayed and reduced PG. For all doses of TXA administered intravenously, the PG assay detected delayed time-to-peak (≤3 hours) and reduced the velocity, peak, and endogenous plasmin potential (≤24 hours) in plasma samples obtained after infusion. The PG time-to-peak, velocity, and peak correlated significantly with TXA concentration and showed less variability than the ROTEM lysis index at 30 minutes or maximum lysis.
Conclusions:
The PG assay detects pharmacologically relevant concentrations of TXA administered in vitro and in vivo, and demonstrates TXA-mediated inhibition of PG in women undergoing cesarean delivery.
Keywords: fibrin, fibrinolysis, plasmin, pregnancy, tranexamic acid
1 |. INTRODUCTION
Hemorrhage associated with congenital bleeding disorders, trauma, surgical complications, or obstetrical events can be life-threatening. Approximately 40% of trauma-associated deaths are due to bleeding.1 Hemorrhage is responsible for more than 25% of maternal deaths worldwide, and cesarean delivery is particularly associated with increased blood loss.2,3 The incidence of both postpartum hemorrhage (PPH) and severe outcomes from hemorrhage is increasing.2,3
Tranexamic acid (trans 4-[aminomethyl]cyclohexanecarboxylic acid [TXA], Cyklokapron) is a low molecular weight (157 Da) lysine analogue and potent antifibrinolytic (half maximal inhibitory concentration [IC50] ~ 2–5 μg/mL [10–25 μmol/L]4,5). TXA inhibits binding of both zymogen plasminogen and its active form plasmin to fibrin. Because fibrin is a cofactor for tissue plasminogen activator (tPA)-mediated conversion of plasminogen to plasmin,6 TXA reduces both tPA-mediated plasmin generation (PG) and plasmin cleavage of fibrin. Consequently, TXA can prevent clot dissolution (fibrinolysis). Plasma concentrations of ~10 μg/mL are usually targeted to reduce bleeding and prevent morbidity and mortality.7 When administered within 3 hours of injury, TXA reduces bleeding and all-cause mortality in patients with traumatic extracranial bleeding8,9 or acute traumatic brain injury.10 TXA also reduces mortality and the probability of receiving a blood transfusion following surgery.11,12 TXA, alone or in conjunction with additional hemostatic therapies, enhances clot stability in whole blood from people with hemophilia.13 TXA also reduces heavy menstrual bleeding,14 perioperative blood loss following elective vaginal or cesarean delivery,15–17 and death from postpartum hemorrhage.18,19 However, TXA does not show survival benefit in patients with acute gastrointestinal bleeding20 or in some studies of trauma.21 Moreover, many of these situations are also associated with increased risk of thrombotic complications, and TXA has been associated with increased risk of venous thromboembolism.9,21,22 The role, if any, of plasmin and agents that reduce fibrinolytic activity in these situations remains unclear.23,24 In addition, seizures and visual disturbances have been described in patients receiving high doses of TXA.25 Persistent questions regarding the mechanism of action of TXA underscore the need for a reliable assay that accurately detects low concentrations of TXA, and call for increased understanding of the pharmacokinetic and pharmacodynamic relationship between TXA and PG and plasmin activity. This need is especially important for vulnerable populations, including pregnant women at delivery.
Common methods to detect effects of TXA assess clot formation and lysis in whole blood via rotational thromboelastometry (ROTEM) or thromboelastography (TEG), or in plasma via turbidity.7 These assays provide composite information on overall fibrin formation and fibrinolysis, but do not differentiate between TXA’s ability to reduce plasmin cleavage of fibrin from its ability to block tPA-mediated generation of plasmin. Differentiating these effects is important. In addition to fibrin, plasmin has many non-fibrin substrates in blood (eg, complement C5; factors IX(a), V, and VIII) and on the surface of cells, including leukocytes, platelets, and endothelial cells.26–33 Once generated, plasmin cleavage of these substrates may be differently sensitive to the effects of TXA.32,34–36 Thus, TXA concentrations that effectively block fibrin dissolution may still permit the generation of plasmin and the downstream consequences of plasmin activity. Therefore, it is essential to define effective concentrations of TXA not only by its ability to reduce plasmin’s proteolytic activity towards fibrin, but by its ability to inhibit PG.
We recently developed a new method to quantify PG kinetics during fibrinolysis in mouse plasma.37 Herein, we adapted this PG assay to provide information on tPA-mediated PG in human plasma. We then used this assay to measure the impact of intravenous TXA administration on PG in a cohort of healthy women undergoing cesarean delivery, and compared the ability of the PG assay to detect pharmacodynamic effects of TXA with that detected by ROTEM.
2 |. METHODS
2.1 |. Materials
Recombinant tissue factor (Innovin) was from Dade Behring. Phospholipid vesicles (20 mol% phosphatidylserine, 60 mol% phosphatidylcholine, 20 mol% phosphatidylethanolamine) and α2-macroglobulin-plasmin complex (α2M-Pm) were from Synapse Research Institute (Maastricht, the Netherlands). Fluorogenic substrate for plasmin (Boc-Glu-Lys-Lys-AMC) was from Bachem. Fibrinogen-deficient plasma, plasminogen-deficient plasma, and α2-antiplasmin-deficient plasma were from Affinity Biologicals Inc. Recombinant tPA (rtPA) was Actilyse (Boehringer Ingelheim, PG assays) or Alteplase (Genentech, ROTEM assays). TXA was from Sigma Aldrich.
2.2 |. Preparation of normal pooled plasma from healthy, nonpregnant controls
Approval for the use of healthy, nonpregnant human subjects was obtained from the University of North Carolina Institutional Review Board and the Medical Ethics Committee of Maastricht University Medical Center. All participants provided signed informed consent in accordance with the Declaration of Helsinki. Donors had no known bleeding disorder, liver or kidney disease, cancer, or history of surgery or thrombotic event within the past 3 months, and were not on antiplatelet or anticoagulant therapy. Whole blood from 24 volunteers (12 males and 12 females, mean age 40 years [range 24–62]) was obtained by venipuncture and collected into tubes containing 0.105 mol/L citrate (Becton Dickinson, 10% v/v, final). Individual plasmas were centrifuged (2840g, 10 minutes, twice) and pooled. The pool was then ultra-centrifuged (100 000g, 70 minutes). Aliquots were stored at −80°C.
2.3 |. Fibrin formation and lysis
A solution of tissue factor, phospholipids, and rtPA (10 μL, total) was added to a 96-well plate. Plasma (40 μL of 1:2 dilution [20 μL plasma plus 40 μL 20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid pH 7.4, 150 mmol/L NaCl [HBS] containing TXA) was added to the 96-well plate, and incubated at 37°C for 10 minutes. Reactions were then initiated by addition of 10 μL of CaCl2. Final concentrations were: tissue factor (0.5 pmol/L), phospholipids (4 μmol/L), rtPA (0.31 μg/mL), TXA (0–50 μg/mL, as indicated), and CaCl2 (16.6 mmol/L). Clot formation and lysis were monitored at 405 nm (SpectraMax 384Plus plate reader, Molecular Devices) for 1 hour at 37°C. The clot lysis time was defined as the time from 50% of the turbidity increase to 50% of the turbidity decrease.
2.4 |. PG assay
Plasmin generation was measured in plasma using a calibrated, automated method based on cleavage of a plasmin-specific fluorogenic substrate (37,38 and Figure 1A). Briefly, two measurements were collected for each sample: one in which endogenous PG was triggered (reaction wells) and one in which α2M-Pm was added (calibrator wells). To trigger PG, 10 μL of solution containing tissue factor, phospholipids, and rtPA were added to reaction wells. The α2M-Pm calibrator (10 μL) was added to calibrator wells. Plasma was then diluted (40 μL of 1:2 dilution [20 μL plasma: 40 μL 20 mmol/L HBS containing TXA for in vitro experiments as indicated]) and added to each well, and plates were warmed for 10 minutes at 37°C. CaCl2 and fluorogenic substrate (in 10 μL) were dispensed into each well and plates were mixed by shaking for 10 seconds. Final concentrations were: tissue factor (0.5 pmol/L), phospholipids (4 μmol/L), rtPA (0.31 μg/mL unless otherwise indicated), TXA (0–50 μg/mL, as indicated), CaCl2 (16.6 mmol/L), and fluorogenic substrate (0.5 mmol/L). Reactions were monitored every 20 seconds with a fluorometer (Fluoroskan Ascent, Thrombinoscope) equipped with a dispenser and 390/460 filter set (excitation/emission). Data were analyzed as described.39 Briefly, because plasma color can influence fluorescence intensity, each plasma was compared with its own calibrator measurement to correct for the inner filter effect and then transformed into a PG curve. By comparing the extent to which the fluorescence increase in wells containing the α2M-Pm complex deviates from a straight line, the calculation identifies and corrects for fluorescent substrate consumption during the reaction. Fluorescence intensity that accrues at the end of each reaction (which is not inhibitable by adding α2-antiplasmin) provides a relative measure of the amount of plasmin trapped in α2M-Pm complexes, which is then subtracted from the fluorescence curves. This transformation is analogous to calibrated automated thrombography39 and yields quantitative parameters: lag time (time the plasmin concentration reached 6 nmol/L), time to peak (TtPeak), velocity (peak/[TtPeak-lag time]), peak, and endogenous plasmin potential (EPP) (Figure 1B).
FIGURE 1.
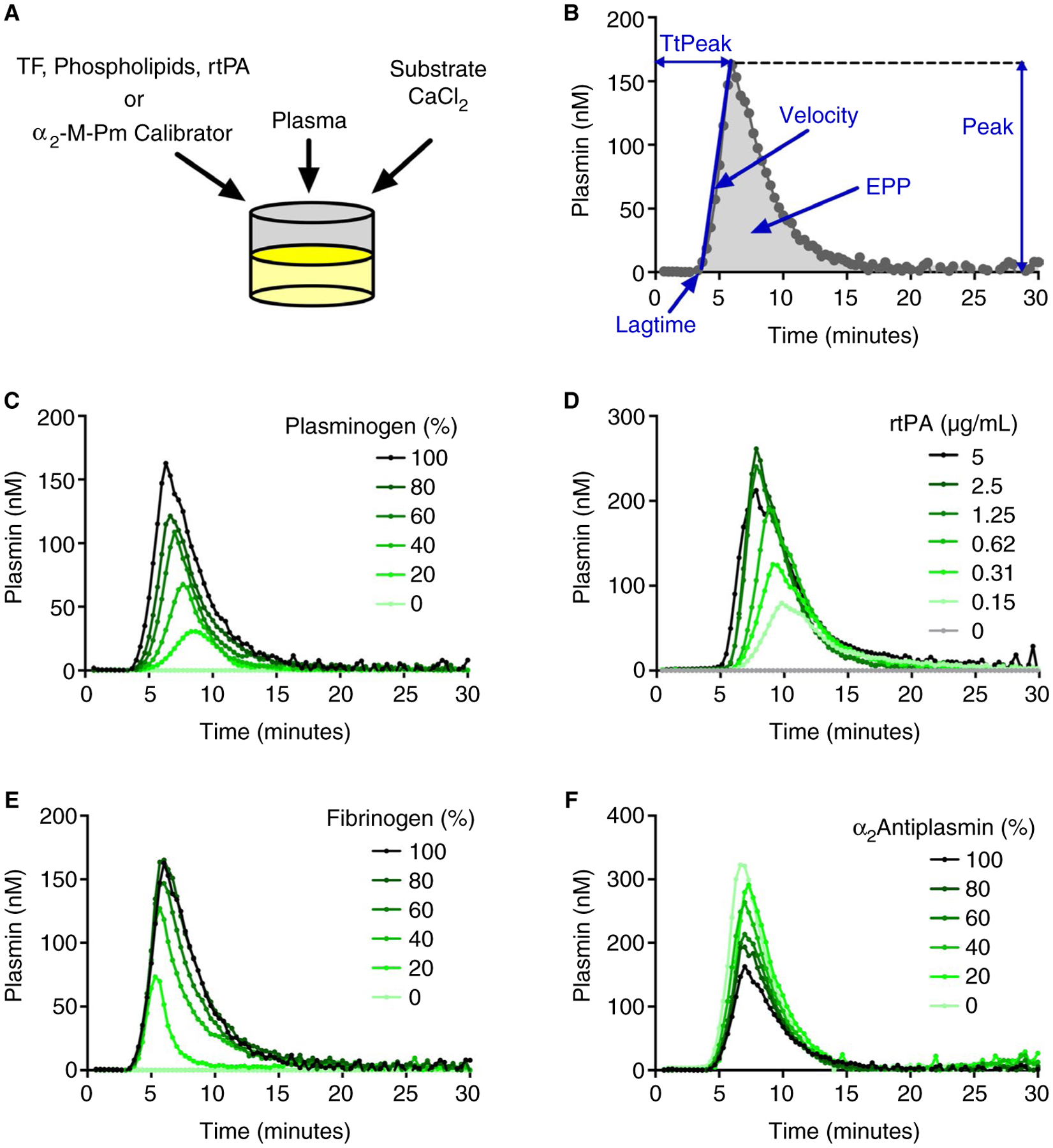
The PG assay is specific for plasmin and sensitive to rtPA, fibrin(ogen), and α2-antiplasmin in human plasma. A, Plasma was mixed with tissue factor (TF), phospholipids, and recombinant tissue plasminogen activator (rtPA) (reaction wells), or α2-macroglobulin/plasmin complex (calibrator wells). Reactions were initiated by automatically dispensing fluorogenic substrate and CaCl2 to each well. B, Fluorescence was monitored over time, and a PG curve was derived mathematically, yielding PG parameters: lag time, time to peak (TtPeak), velocity, peak, and endogenous plasmin potential (EPP). C, PG in normal pooled plasma mixed with plasminogen-deficient plasma. D, PGin normal pooled plasma; reactions were triggered with the indicated concentrations of rtPA. E, PG in normal pooled plasma mixed with fibrinogen-deficient plasma. F, PG in normal pooled plasma mixed with α2-antiplasmin-deficient plasma. Panels C-F show representative curves from three independent experiments.
2.5 |. Preparation of blood and plasma from pregnant women receiving TXA
Plasma samples from 30 pregnant women undergoing scheduled cesarean delivery were obtained from a previous prospective, open-label, dose-finding study approved by the Institutional Review Board of George Washington University (IRB#041737).40 Briefly, women were assigned to one of three cohorts receiving 5, 10, or 15 mg/kg TXA, respectively, via intravenous administration at the time of cord clamp. Mean biometric data, preoperative laboratory assays, and comorbidities were similar between groups (Table S1 and40).
Blood was obtained via venous access before delivery and at time points up to 24 hours after TXA administration. Blood was collected into sodium citrate (Becton Dickinson Vacutainer tubes, Na Citrate 0.109 mol/L, 3.2%) for ROTEM and PG assays. Blood was also collected into potassium ethylenediaminetetraacetic acid (Becton Dickinson Vacutainer tubes, K2 EDTA 7.2 mg) for determination of TXA levels by mass spectrometry. Platelet-poor plasma was prepared by centrifugation (3000g, 15 minutes), flash frozen on dry ice, and stored at −70°C.
2.6 |. Mass spectrometry
Plasma concentrations of TXA were determined by ultrahigh-performance liquid chromatography-tandem mass spectrometry. 4-Aminocyclohexanecarboxylic acid was used as the internal standard41,42 with modifications. Specifically, plasma samples were prepared for analysis using protein precipitation with acetonitrile. Sample extracts were analyzed using normal phase chromatography with a Waters BEH HILIC column (2.1 × 100 mm, 1.7 μm, Waters Corp.) followed by detection with a Waters Xevo TQ-XS mass spectrometer. The mobile phase was A (10 mmol/L ammonium formate in water/isopropanol/formic acid 50/50/0.1):B (10 mmol/L ammonium format in acetonitrile/water/formic acid 90/10/0.1) = 40:60 with 0.25 mL/min flow rate. The mass spectrometer used an electrospray ionization source and a positive ion multiple reaction monitoring mode. For quantification of TXA, mass-to-charge ratios were set to 158.2 > 95.2 for TXA and 144.2 > 109.1 for the internal standard, respectively. Measurements for samples containing over 25 μg/mL TXA were obtained after diluting the samples into control plasma. The lower limit for TXA quantification was 0.04 μg/mL. Intra-assay (within-day) precision (% coefficient of variation [% CV]) and accuracy (% bias) were 1.3% to 8.7% and 0.9% to 7.6%, respectively. Inter-assay (between-day) % CV and % bias were 0.7% to 6.7% and 1.2% to 15.2%, respectively.
2.7 |. ROTEM
Fibrinolysis was measured in whole blood using a modification of a previously described method.40,43,44 Briefly, whole blood was recalcified per manufacturer instructions for EXTEM (Instrument Laboratories) and rtPA (0.3 μg/mL, final) was added ex vivo before starting the ROTEM analysis. Clot formation and lysis were followed by changes in amplitude in a ROTEM delta using software, version 2.7.1. The following parameters were analyzed: clotting time, clot formation time (CFT), alpha angle (alp), amplitude at 10 minutes, amplitude at 20 minutes, maximum clot firmness, lysis index at 30 minutes (LI30), and maximum lysis (ML, percent decrease from maximum clot firmness at 60 minutes).
2.8 |. Statistical methods
Descriptive statistics (means, medians, standard deviations, and range) were calculated using GraphPad Prism, version 8.4.2. PG parameters vs TXA concentration (in vitro experiments) or time after intravenous administration were analyzed by one-way ANOVA with Dunnett’s post hoc testing using Prism 8.4.2. Buffer-treated samples (no TXA) or predelivery samples, respectively, were used as the index condition, and P < .05 was considered statistically significant. The plasma TXA concentration-PG effect relationship following intravenous administration was characterized using a population modeling approach similar to previous reports45,46 carried out by Pumas, version 1.0.5 (www.pumas.ai).47 A likelihood ratio test method was applied to test the significance of the fitted models; P < .05 was used to indicate statistical significance with 95% confidence.
3 |. RESULTS
3.1 |. The PG assay is specific for plasmin and sensitive to tPA, fibrin(ogen), and α2-antiplasmin
Previously we developed an in vitro assay to measure PG kinetic parameters in mouse plasma.37 To adapt this assay to human plasma, we first assessed the sensitivity of the assay to plasminogen, rtPA, α2-antiplasmin, and fibrin(ogen) concentrations. Consistent with that seen in mouse plasma,37 PG was not observed in plasminogen-deficient plasma, demonstrating specificity of this assay to plasmin (Figure 1C). When normal pooled plasma was mixed with plasminogen-deficient plasma, PG was dose dependent with respect to the plasminogen concentration (Figure 1C). PG was also not observed in human plasma in the absence of exogenous rtPA (Figure 1D), indicating neither basal levels of circulating tPA or urokinase plasminogen activator (~1–5 ng/mL each),48,49 nor any contaminating factor XIIa,50,51 were sufficient to trigger measurable PG. Addition of rtPA increased PG in a rtPA concentration-dependent manner (Figure 1D). To assess the sensitivity of PG to fibrin cofactor activity, we mixed normal pooled plasma with fibrinogen-deficient plasma. As expected,6,52 PG was not observed in the absence of fibrinogen, and increasing concentrations of fibrin(ogen) enhanced PG in a dose-dependent manner (Figure 1E). Mixing normal pooled plasma with α2-antiplasmin-deficient plasma increased PG in a dose-dependent manner (Figure 1F). Collectively, these data demonstrate sensitivity of the PG assay to established endogenous mediators of PG in human plasma.
3.2 |. TXA inhibits PG in vitro
To assess sensitivity of the PG assay to TXA, we first added TXA (0–50 μg/mL, final) to human plasma in vitro. These TXA concentrations are achieved in plasma after intravenous administration,7 and turbidity assays confirmed that these concentrations produced a dose-dependent prolongation in the clot lysis time (Figure 2A–B). In PG assays, TXA had a slight, biphasic effect on the lag time, but the lag time was only significantly different at ~15 μg/mL TXA (Figure 2C–D). TXA produced a significant dose-dependent delay in the TtPeak and a decrease in the velocity, peak, and EPP (Figure 2C, E–H). In particular, 10 μg/mL TXA delayed the TtPeak by 86 ± 1.5%, reduced the velocity by 84 ± 1.6%, and decreased the peak by 54 ± 6.7%. Similar inhibitory effects were detected in reactions performed in the presence of lower rtPA concentrations (data not shown). In addition to showing sensitivity of the PG assay to pharmacologically relevant concentrations of TXA, these data specifically show TXA reduces tPA-mediated generation of plasmin in plasma.
FIGURE 2.
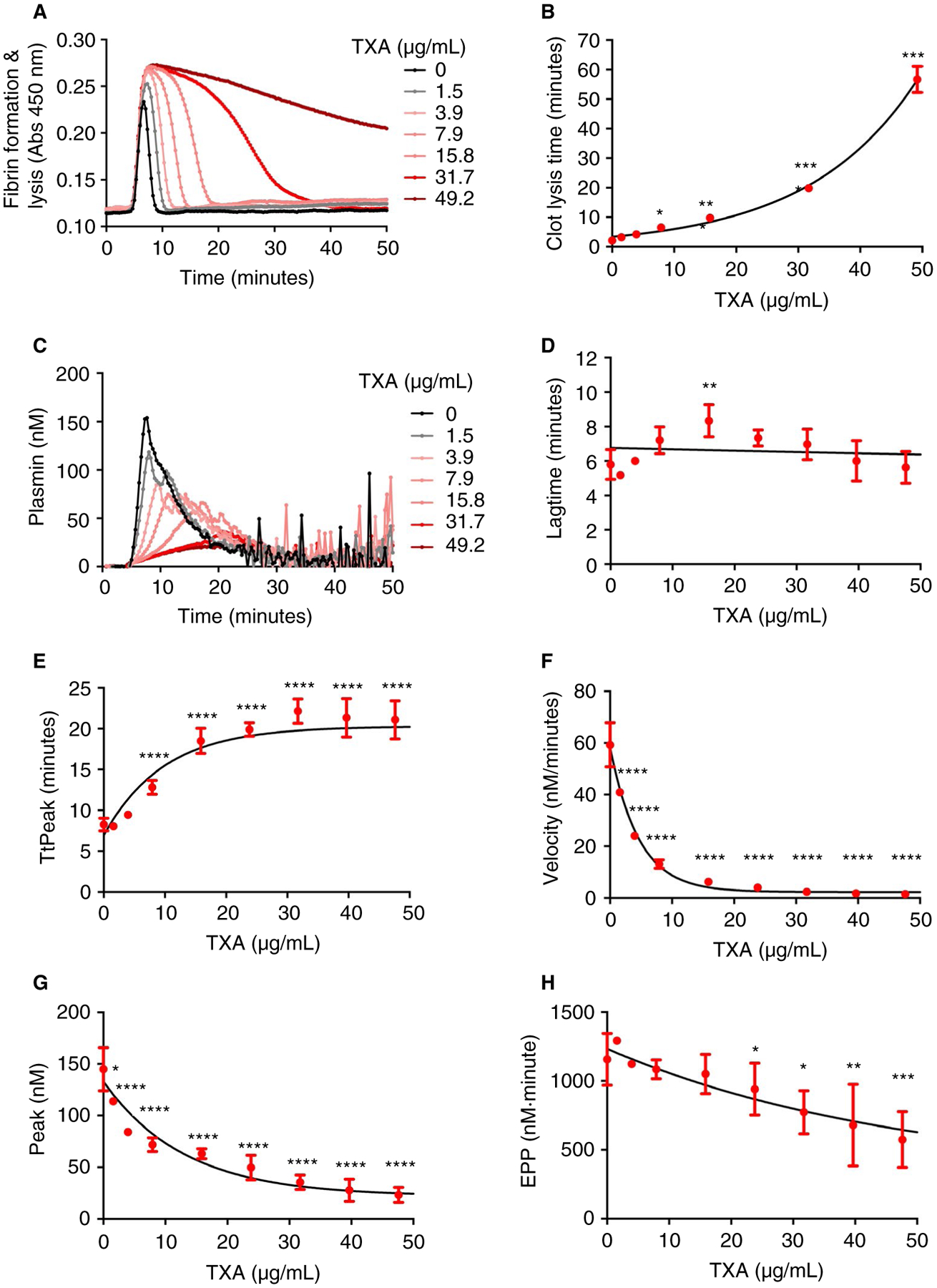
TXA inhibits PG in vitro. Normal pooled plasma was diluted 1:2 and clotted in the presence of tissue factor, phospholipids, rtPA, TXA, and CaCl2. A, Clot formation and fibrinolysis measured by turbidity, representative curves. B, Clot lysis time from turbidity assays. C, PG, representative curves. PG parameters: (D) lag time, (E) TtPeak, (F) velocity, (G) peak, and (H) EPP. Dots show means and standard deviations from four to five independent experiments. *P < .05, **P < .01, ***P < .001, and ****P < .0001
3.3 |. Ex vivo detection of TXA after intravenous administration
To determine the sensitivity of the PG assay to TXA administered intravenously, we measured PG in plasmas obtained from women undergoing cesarean delivery who received TXA (5, 10, or 15 mg/kg) at the time of umbilical cord clamp. Baseline PG parameters before TXA administration were generally similar to that seen in normal pooled plasma (Figures 2 and 3), with the exception of a reduced PG velocity (26.8 ± 4.3 vs 59.2 ± 8.6 nmol/L/min, respectively) and peak (114.7 ± 17.6 vs 144.89 ± 20.8 nmol/L) in plasma from pregnant women. This difference may reflect, in part, increased PAI-1 antigen and activity associated with pregnancy.53 We first analyzed plasma PG as a function of time after TXA administration. As expected, all three TXA dosing levels significantly reduced PG (Figure 3). Similar to that observed following TXA addition to plasma in vitro, compared with pretreatment samples, plasmas from women who received intravenous TXA showed little change in the lag time, but a significantly delayed TtPeak and reduced velocity, peak, and EPP (Figure 3). Effects of TXA were most profound immediately after TXA administration, after which PG increased with time until parameters approached preadministration values. Comparison of the three TXA dosing regimens revealed similar extent of PG inhibition (Figure 3).
FIGURE 3.
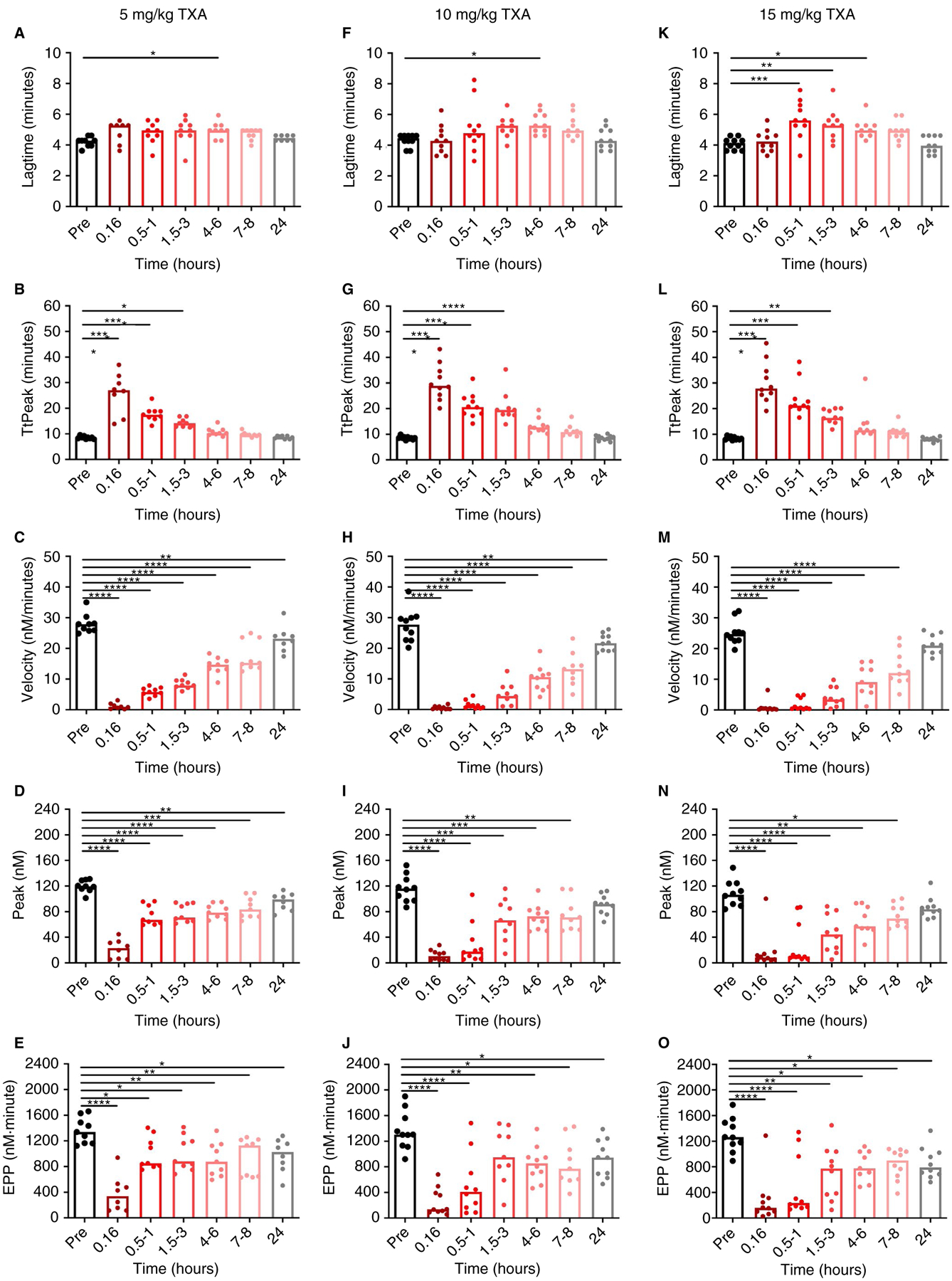
TXA inhibits PG ex vivo. PG was measured in plasmas from women who received (A-E) 5, (F-J) 10, or (K-O) 15 mg/kg TXA. Bars indicate medians, each dot represents a separate subject. *P < .05, **P < .01, ***P < .001, and ****P < .0001
Because ROTEM is considered by many to be a “gold standard” assay for assessing fibrinolysis in a complex milieu, we then compared the time-response of plasma PG parameters with those of ROTEM measured in paired whole blood samples clotted in the presence of rtPA. The ROTEM data are reported elsewhere40 and summarized here. Briefly, the rtPA-initiated ROTEM curve is characterized by an initial increase in amplitude indicating clot formation followed by a decrease in amplitude indicating clot lysis.40,44 Multiple established parameters quantifying this curve, including amplitude at 10 minutes, amplitude at 20 minutes, maximum clot firmness, LI30, and ML detected effects of TXA. Of these parameters, LI30 and ML showed the strongest correlation with time after infusion, with strong antifibrinolytic activity detected immediately after TXA administration followed by a time-dependent decline in activity up to 8 or 3 hours postinfusion, respectively (Figure S1 and data not shown). Suppression of PG velocity, peak, and EPP persisted longer than that detected by the ROTEM LI30, suggesting significantly reduced velocity, peak, and EPP up to 24 hours after administration.
3.4 |. Pharmacodynamic relationship between TXA and PG
Finally, to characterize the relationship between TXA concentration and PG, we used a population modeling approach to correlate plasma concentrations of TXA measured by mass spectrometry with each PG parameter. As expected,54 TXA exhibited a 1.7-hour half-life in plasma following intravenous administration (data not shown). The TXA concentrations significantly describe the changes in ROTEM LI30 and ML as well as PG parameters (P < .001), indicated a strong concentration-effect relationship. Experimental observations vs modeled predictions for each PG parameter are shown in Figure 4. The PG lag time correlated poorly with the plasma TXA concentration (data not shown). However, the TtPeak correlated positively with plasma TXA in an additive Emax model (P < .001, Figure 4A), and the velocity, peak, and EPP correlated negatively with plasma TXA in a fractional Emax model (each P < .001, Figure 4B–D). Consistent with the in vitro spiking experiments (Figure 2), the TtPeak, velocity, peak, and EPP were delayed or reduced, respectively, at TXA concentrations above 10 μg/mL. Of the PG parameters, the velocity demonstrated the strongest dose-relationship at low TXA concentrations (≤10 μg/mL). ROTEM LI30 and ML also correlated strongly with plasma TXA concentration (both P < .001, Figure 4E–F). In general, compared with ROTEM, modeled values for PG parameters adhered better to the measured data (Figure 4A–F).
FIGURE 4.
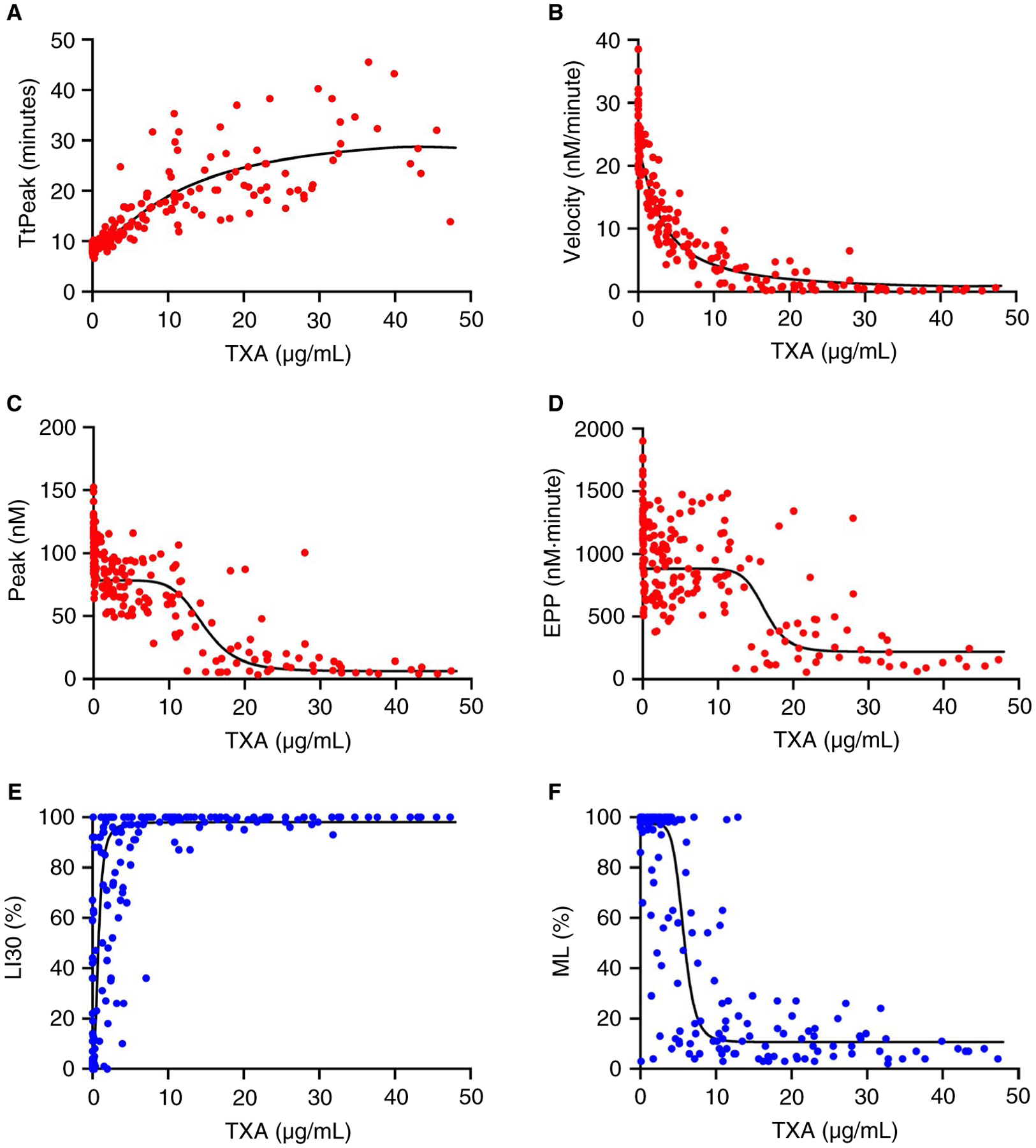
PG parameters correlate with plasma TXA concentration. TXA concentrations measured by mass spectrometry were correlated with (A) PG TtPeak, (B) PG velocity, (C) PG peak, (D) PG EPP, (E) ROTEM LI30, and (F) ROTEM ML. The TXA concentration-effect relationship for each parameter was characterized using a population modeling approach. Each dot represents a separate sample.
Concentrations of TXA that half-maximally reduced PG in plasma and altered ROTEM parameters in whole blood ranged from 3.1 to 16.2 and 0.8 to 6.0 μg/mL, respectively (Table 1). These values are consistent with the concentration of TXA needed to half-maximally inhibit tPA-mediated activation of plasminogen on fibrin in a purified system.5 Compared to LI30 or ML, the PG parameters showed less within-subject variability (measurement error, Table 1). For example, at the therapeutic concentration of 10 μg/mL,7 the predicted ML and velocity responses were 11.8 ± 7.3% and 4.3 ± 2.0 nmol/L/min, respectively, such that the imprecision for the predicted ML was ~ 60% compared with 46% for the PG velocity. Moreover, compared with ROTEM parameters, between-subject variability (% CV) was smaller for PG parameters (Table 1). Collectively, these findings show the PG assay is sensitive to antifibrinolytic activity of TXA in vitro and ex vivo, and reliably detects suppressed PG in plasma from women undergoing cesarean delivery at pharmacologically relevant concentrations of TXA.
TABLE 1.
Measurement error and half-maximal effective concentrations (EC50 or IC50) for PG and ROTEM parameters
| Parameter | EC50 or IC50 (μg/mL) | Predicted Response at 10 μg/mL | Measurement Error at 10 μg/mL | %cv |
|---|---|---|---|---|
| PG | ||||
| Lag time | None | 4.9 min | 0.8 min | - |
| TtPeak | 11.8 | 18.9 min | 3.0 min | 39.5 |
| Velocity | 3.1 | 4.3 nmol/L/min | 2.0 nmol/L/min | 40.9 |
| Peak | 14.6 | 73.0 nmol/L | 13.3 nmol/L | 41.4 |
| EPP | 16.2 | 879.6 nmol/L/min | 124 nmol/L/min | 36.5 |
| ROTEM | ||||
| LI30 | 0.8 | 97.9% | 7.3% | 145.0 |
| ML | 6.0 | 11.8% | 7.3% | 65.3 |
Note: Plasma TXA concentrations measured by mass spectrometry were correlated with PG and ROTEM parameters using a population approach with additive (EC50 for TtPeak and LI30) or fractional (IC50 for velocity, peak, EPP, and ML) Emax models. Measurement error indicates experimental precision of the parameter (within-subject variability). %CV (coefficient of variation) indicates between-subject variability.
Abbreviations: EC50, half-maximal effection concentration; EPP, endogenous plasmin potential; LI30, lysis index at 30 minutes; ML, maximum lysis; PG, plasmin generation; ROTEM, rotational thromboelastometry; TtPeak, time to peak.
4 |. DISCUSSION
Tranexamic acid has been shown to reduce or prevent bleeding in multiple clinical settings. We previously described a calibrated assay to measure the kinetics of PG in mouse plasma37 and have now extended, characterized, and applied this assay to understand effects of TXA on PG in human plasma. The PG assay is sensitive to plasminogen, rtPA, fibrin(ogen), and α2-antiplasmin concentration, and shows a dose-dependent reduction in PG in the presence of pharmacologically relevant concentrations of TXA administered in vitro and ex vivo. Development of this assay and its application to a cohort of women receiving TXA during cesarean delivery yields insight into the pharmacodynamic effects of TXA in this population that has heightened risk of bleeding and thrombosis. Our findings expand the technological arsenal for assessing fibrinolytic mechanisms in vitro and in vivo.
The PG assay is an extension of an assay we previously developed and characterized using plasma from mice with genetically engineered deficiencies in procoagulant and fibrinolytic proteins.37 This assay has similarities to previous methods that used fluorogenic substrates to evaluate PG kinetics in plasma.55–57 Each of these methods used similar concentrations of tissue factor, phospholipids, and rtPA, operate at similar time scales, and test coagulation and fibrinolytic mechanisms. These assays are sensitive to multiple coagulation factors, including fibrinogen and plasminogen. Our assay differs from the previous PG assays via several key features, including its use of a water-soluble substrate, calibration with preformed α2M-Pm complex, and use of a mathematical model to correct for substrate consumption and the inner filter effect.39 These differences enable us to assess the kinetics of plasmin production during fibrinolysis and express PG parameters in molar concentrations, yielding enhanced understanding of plasmin evolution during a fibrinolytic reaction.
Tranexamic acid is a lysine analog capable of blocking both tPA-mediated generation of plasmin and plasmin-mediated cleavage of fibrin. In an experimental system with purified components, ~10 μg/mL (60–70 μmol/L) TXA provides near maximal inhibition of plasminogen conversion to plasmin.5,58 However, effects of TXA in whole blood and plasma have been more difficult to define. In these settings, fibrinolysis is typically measured using so-called “global assays” (eg, ROTEM/TEG, turbidity) that report a composite of reactions, including thrombin generation, fibrin formation, and plasmin generation, that culminate in fibrinolysis. Because most analyses of TXA use these global assays, existing studies have not easily differentiated inhibitory effects of TXA on PG from plasmin cleavage of fibrin. Given the lack of effect of TXA on plasmin-antiplasmin complexes, Godier et al59 suggested TXA primarily inhibits plasmin activity, but not tPA-mediated PG.59 However, because the circulating concentration of plasminogen is higher than α2-antiplasmin (2.4 vs 1.1 μmol/L, respectively), the use of high rtPA (~4 nmol/L, ~2.8 μg/mL) in their reactions may have generated high plasmin concentrations that consumed available α2-antiplasmin.60 Consequently, the use of plasmin-antiplasmin complexes as a biomarker of PG may not have revealed the full dynamics of PG. Our ability to detect plasmin activity in plasma shows that TXA inhibits PG. These findings support the premise that TXA’s antifibrinolytic effects stem from its ability to reduce tPA-mediated conversion of plasminogen to plasmin. Clarifying these effects is important because it indicates that, by reducing PG, TXA can inhibit multiple downstream effects of plasmin, including proteolytic cleavage of nonfibrin substrates that may be differently affected by TXA.
Sensitivity of the PG assay to both endogenous and pharmacologic mediators of fibrinolysis suggests several potential applications, including assessment of other methods of TXA administration (eg, oral delivery) and fibrinolytic abnormalities in individuals with congenital or acquired bleeding disorders. In settings with abnormal lysis, whereas existing technologies like the clot lysis time assay or thromboelastography cannot differentiate between altered fibrin stability vs altered ability to produce plasmin, the PG assay provides this information that is essential for understanding the operant mechanisms. For example, use in plasma from patients experiencing trauma-induced coagulopathy may be especially instructive for characterizing mechanisms implicated in hyperfibrinolysis, hypofibrinolysis, and so-called “fibrinolysis shutdown” as defined by ROTEM or TEG.61–63 Characterization of specific fibrinolytic abnormalities in these settings may optimize the risk-benefit ratio when using antifibrinolytics in selected populations.
Our study has limitations. First, the study design did not include a cohort of women that did not receive TXA at cesarean delivery, so some of the observed reduction in PG attributed to TXA in the ex vivo study may reflect biological changes in PG that accompany pregnancy and cesarean delivery. However, normal tPA-modified ROTEM profiles were previously observed in pregnant women,44 and functional effects of TXA observed ex vivo (Figures 3–4) were consistent with measured concentrations of TXA, as well as TXA effects observed in the in vitro experiments (Figure 2). Second, median estimated blood loss for cohorts receiving 5, 10, or 15 mg TXA was similar (750, 750, and 700 mL, respectively), and we were unable to correlate blood loss with PG in this study. Third, the mathematical derivation of plasmin concentration has caveats: formation of the α2-M-Pm complex is not necessarily linear, substrate cleavage by plasmin differs slightly from that by plasmin trapped by α2-M, and the plasmin substrate is consumed relatively quickly so the decay phase of the reaction curves reflects both plasmin inactivation and substrate consumption, making the EPP parameter less robust. Fourth, although our study evaluated effects of TXA on rtPA-mediated PG, we did not assess the role of TXA on urokinase-mediated PG, which is paradoxically stimulated by TXA.5 Application of the PG assay to understand effects of TXA on urokinase may reveal clues to negative outcomes associated with delayed administration of TXA,8 when urokinase may have a larger role. Finally, we compared the effect of TXA on PG measured in plasma with ROTEM parameters measured in whole blood. Because cells carry plasminogen activators and inhibitors (eg, urokinase, plasminogen activator inhibitor-1) that may affect PG and effect of TXA, caution must be taken when comparing these results.
In summary, we have described a PG assay that is sensitive to pharmacologically relevant concentrations of TXA administered in vitro and in vivo. Our assay, which can detect reduced PG at low TXA concentrations (≤10 μg/mL), may be useful for understanding effects of TXA beyond that which can be appreciated from traditional fibrinolysis assays. Continued development of the PG assay as a potential point-of-care method to assess PG and TXA may reveal pharmacodynamic properties helpful for individualizing therapy.
Supplementary Material
Essentials.
Tranexamic acid (TXA) is an antifibrinolytic drug used to reduce bleeding.
Assaying plasmin generation (PG) in plasma detects clinically relevant TXA levels in vitro and ex vivo.
3.1–16.2 μg/mL TXA half-maximally inhibits PG in plasma from women undergoing cesarean delivery.
PG velocity shows the strongest dose-relationship at low TXA concentrations (≤10 μg/mL).
Funding information
This study was supported by funding from the National Institutes of Health (K23HL141640 and KL2TR001877/UL1TR001876 to H.K.A., R61/R33HL141791 and U01HL143403 to A.S.W.).
Footnotes
CONFLICT OF INTEREST
Dr. Miszta and Dr. de Laat are employed by Synapse Research Institute, a not-for-profit member of the STAGO Diagnostic group that produces calibrated automated thrombography for thrombin generation measurements in plasma. Synapse Research Institute holds the patent on calibrated plasmin generation. The ROTEM was provided by the manufacturer, which did not provide any input on the study design or data interpretation. None of the other authors have relevant potential conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Curry N, Hopewell S, Doree C, Hyde C, Brohi K, Stanworth S. The acute management of trauma hemorrhage: a systematic review of randomized controlled trials. Crit Care. 2011;15(2):R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knight M, Callaghan WM, Berg C, et al. Trends in postpartum hemorrhage in high resource countries: a review and recommendations from the International Postpartum Hemorrhage Collaborative Group. BMC Pregnancy Childbirth. 2009;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Say L, Chou D, Gemmill A, et al. Global causes of maternal death: a WHO systematic analysis. Lancet Glob Health. 2014;2(6):e323–e333. [DOI] [PubMed] [Google Scholar]
- 4.Sperzel M, Huetter J. Evaluation of aprotinin and tranexamic acid in different in vitro and in vivo models of fibrinolysis, coagulation and thrombus formation. J Thromb Haemost. 2007;5(10):2113–2118. [DOI] [PubMed] [Google Scholar]
- 5.Silva MM, Thelwell C, Williams SC, Longstaff C. Regulation of fibrinolysis by C-terminal lysines operates through plasminogen and plasmin but not tissue-type plasminogen activator. J Thromb Haemost. 2012;10(11):2354–2360. [DOI] [PubMed] [Google Scholar]
- 6.Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982;257(6):2912–2919. [PubMed] [Google Scholar]
- 7.Picetti R, Shakur-Still H, Medcalf RL, Standing JF, Roberts I. What concentration of tranexamic acid is needed to inhibit fibrinolysis? A systematic review of pharmacodynamics studies. Blood Coagul Fibrinolysis. 2019;30(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CRASH-2 Trial Collaborators, Roberts I, Shakur H, et al. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet. 2011;377(9771):1096–1101, 1101 e1091–1092. [DOI] [PubMed] [Google Scholar]
- 9.Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military application of tranexamic acid in trauma emergency resuscitation (MATTERs) study. Arch Surg. 2012;147(2):113–119. [DOI] [PubMed] [Google Scholar]
- 10.CRASH-3 trial Collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet. 2019;394(10210):1713–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ker K, Edwards P, Perel P, Shakur H, Roberts I. Effect of tranexamic acid on surgical bleeding: systematic review and cumulative meta-analysis. BMJ. 2012;344:e3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi J, Ji H, Ren F, et al. Protective effects of tranexamic acid on clopidogrel before coronary artery bypass grafting: a multicenter randomized trial. JAMA Surg. 2013;148(6):538–547. [DOI] [PubMed] [Google Scholar]
- 13.Hvas AM, Sorensen HT, Norengaard L, Christiansen K, Ingerslev J, Sorensen B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J Thromb Haemost. 2007;5(12):2408–2414. [DOI] [PubMed] [Google Scholar]
- 14.O’Brien SH. Evaluation and management of heavy menstrual bleeding in adolescents: the role of the hematologist. Blood. 2018;132(20):2134–2142. [DOI] [PubMed] [Google Scholar]
- 15.Sentilhes L, Winer N, Azria E, et al. Tranexamic acid for the prevention of blood loss after vaginal delivery. N Engl J Med. 2018;379(8):731–742. [DOI] [PubMed] [Google Scholar]
- 16.Gai MY, Wu LF, Su QF, Tatsumoto K. Clinical observation of blood loss reduced by tranexamic acid during and after caesarian section: a multi-center, randomized trial. Eur J Obstet Gynecol Reprod Biol. 2004;112(2):154–157. [DOI] [PubMed] [Google Scholar]
- 17.Sekhavat L, Tabatabaii A, Dalili M, Farajkhoda T, Tafti AD. Efficacy of tranexamic acid in reducing blood loss after cesarean section. J Matern Fetal Neonatal Med. 2009;22(1):72–75. [DOI] [PubMed] [Google Scholar]
- 18.WOMAN Trial Collaborators. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post-partum haemorrhage (WOMAN): an international, randomised, double-blind, placebo-controlled trial. Lancet. 2017;389(10084):2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shakur-Still H, Roberts I, Fawole B, et al. Effect of tranexamic acid on coagulation and fibrinolysis in women with postpartum haemorrhage (WOMAN-ETAC): a single-centre, randomised, double-blind, placebo-controlled trial. Wellcome Open Res. 2018;3:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.HALT-IT Trial Collaborators. Effects of a high-dose 24-h infusion of tranexamic acid on death and thromboembolic events in patients with acute gastrointestinal bleeding (HALT-IT): an international randomised, double-blind, placebo-controlled trial. Lancet. 2020;395(10241):1927–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers SP, Kutcher ME, Rosengart MR, et al. Tranexamic acid administration is associated with an increased risk of posttraumatic venous thromboembolism. J Trauma Acute Care Surg. 2019;86(1):20–27. [DOI] [PubMed] [Google Scholar]
- 22.Johnston LR, Rodriguez CJ, Elster EA, Bradley MJ. Evaluation of military use of tranexamic acid and associated thromboembolic events. JAMA Surg. 2018;153(2):169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chornenki NLJ, Um KJ, Mendoza PA, et al. Risk of venous and arterial thrombosis in non-surgical patients receiving systemic tranexamic acid: a systematic review and meta-analysis. Thromb Res. 2019;179:81–86. [DOI] [PubMed] [Google Scholar]
- 24.Myers SP, Neal MD. Venous thromboembolism after tranexamic acid administration: legitimate risk or statistical confounder? ANZ J Surg. 2020;90(4):425–426. [DOI] [PubMed] [Google Scholar]
- 25.Murkin JM, Falter F, Granton J, Young B, Burt C, Chu M. High-dose tranexamic acid is associated with nonischemic clinical seizures in cardiac surgical patients. Anesth Analg. 2010;110(2):350–353. [DOI] [PubMed] [Google Scholar]
- 26.Foley JH, Walton BL, Aleman MM, et al. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine. 2016;5:175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasche B, Ouimet H, Francis S, Loscalzo J. Structural changes in platelet glycoprotein IIb/IIIa by plasmin: determinants and functional consequences. Blood. 1994;83(2):404–414. [PubMed] [Google Scholar]
- 28.Medcalf RL. Fibrinolysis, inflammation, and regulation of the plasminogen activating system. J Thromb Haemost. 2007;5(Suppl 1):132–142. [DOI] [PubMed] [Google Scholar]
- 29.Syrovets T, Simmet T. Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci. 2004;61(7–8):873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol. 2012;92(3):509–519. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt AE, Vadivel K, Whitelegge J, Bajaj SP. Plasmin-mediated proteolysis of human factor IXa in the presence of calcium/phospholipid: conversion of procoagulant factor IXa to a fibrinolytic enhancer. J Thromb Haemost. 2020;18(5):1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett CD, Moore HB, Kong YW, et al. Tranexamic acid mediates proinflammatory and anti-inflammatory signaling via complement C5a regulation in a plasminogen activator-dependent manner. J Trauma Acute Care Surg. 2019;86(1):101–107. [DOI] [PubMed] [Google Scholar]
- 33.Deryugina EI, Quigley JP. Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol. 2012;2012:564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nielsen VG, Ford PM. The ratio of concentrations of aminocaproic acid and tranexamic acid that prevent plasmin activation of platelets does not provide equivalent inhibition of plasmatic fibrinolysis. J Thromb Thrombolysis. 2018;46(3):365–370. [DOI] [PubMed] [Google Scholar]
- 35.Godier A, Roberts I, Hunt BJ. Tranexamic acid: less bleeding and less thrombosis? Crit Care. 2012;16(3):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore EE, Moore HB, Gonzalez E, Sauaia A, Banerjee A, Silliman CC. Rationale for the selective administration of tranexamic acid to inhibit fibrinolysis in the severely injured patient. Transfusion. 2016;56(Suppl 2):S110–S114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miszta A, Kopec AK, Pant A, et al. A high-fat diet delays plasmin generation in a thrombomodulin-dependent manner in mice. Blood. 2020;135(19):1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giesen P, Hemker HC, Al Dieri R, Beguin S, Wagenvoord R. Diagnostic test for determining the concentration of transient proteolytic activity in composite biological media. The Netherlands; 2003. [Google Scholar]
- 39.Hemker HC, Kremers R. Data management in thrombin generation. Thromb Res. 2013;131(1):3–11. [DOI] [PubMed] [Google Scholar]
- 40.Ahmadzia HK, Luban NLC, Li S, et al. Optimal use of tranexamic acid for hemorrhage prevention in pregnant women; Submitted. [DOI] [PMC free article] [PubMed]
- 41.Grassin Delyle S, Abe E, Batisse A, et al. A validated assay for the quantitative analysis of tranexamic acid in human serum by liquid chromatography coupled with electrospray ionization mass spectrometry. Clin Chim Acta. 2010;411(5–6):438–443. [DOI] [PubMed] [Google Scholar]
- 42.Ausen K, Pleym H, Liu J, et al. Serum concentrations and pharmacokinetics of tranexamic acid after two means of topical administration in massive weight loss skin-reducing surgery. Plast Reconstr Surg. 2019;143(6):1169e–1178e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dirkmann D, Hanke AA, Gorlinger K, Peters J. Perioperative use of modified thrombelastography in factor XI deficiency: a helpful method to assess drug effects. Acta Anaesthesiol Scand. 2007;51(5):640–643. [DOI] [PubMed] [Google Scholar]
- 44.Kuiper GJ, Kleinegris MC, van Oerle R, et al. Validation of a modified thromboelastometry approach to detect changes in fibrinolytic activity. Thromb J. 2016;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ordonez AA, Wang H, Magombedze G, et al. Dynamic imaging in patients with tuberculosis reveals heterogeneous drug exposures in pulmonary lesions. Nat Med. 2020;26(4):529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madabushi R, Cox DS, Hossain M, et al. Pharmacokinetic and pharmacodynamic basis for effective argatroban dosing in pediatrics. J Clin Pharmacol. 2011;51(1):19–28. [DOI] [PubMed] [Google Scholar]
- 47.Rachauckas C, Noack A, Dixit V, et al. High performance pharmaceutical modeling and simulation. In preparation.
- 48.Brown NJ, Gainer JV, Stein CM, Vaughan DE. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension. 1999;33(6):1431–1435. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H, Wu X, Lu X, Chen G, Ye X, Huang J. Evaluation of plasma urokinase-type plasminogen activator and urokinase-type plasminogen-activator receptor in patients with acute and chronic hepatitis B. Thromb Res. 2009;123(3):537–542. [DOI] [PubMed] [Google Scholar]
- 50.Goldsmith GH Jr, Saito H, Ratnoff OS. The activation of plasminogen by Hageman factor (factor XII) and Hageman factor fragments. J Clin Invest. 1978;62(1):54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miles LA, Greengard JS, Griffin JH. A comparison of the abilities of plasma kallikrein, beta-factor XIIa, factor XIa and urokinase to activate plasminogen. Thromb Res. 1983;29(4):407–417. [DOI] [PubMed] [Google Scholar]
- 52.Medved L, Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb Haemost. 2003;89(3):409–419. [PubMed] [Google Scholar]
- 53.Kruithof EK, Tran-Thang C, Gudinchet A, et al. Fibrinolysis in pregnancy: a study of plasminogen activator inhibitors. Blood. 1987;69(2):460–466. [PubMed] [Google Scholar]
- 54.Andersson L, Nilsoon IM, Colleen S, Granstrand B, Melander B. Role of urokinase and tissue activator in sustaining bleeding and the management thereof with EACA and AMCA. Ann N Y Acad Sci. 1968;146(2):642–658. [DOI] [PubMed] [Google Scholar]
- 55.van Geffen M, Loof A, Lap P, et al. A novel hemostasis assay for the simultaneous measurement of coagulation and fibrinolysis. Hematology. 2011;16(6):327–336. [DOI] [PubMed] [Google Scholar]
- 56.Simpson ML, Goldenberg NA, Jacobson LJ, Bombardier CG, Hathaway WE, Manco-Johnson MJ. Simultaneous thrombin and plasmin generation capacities in normal and abnormal states of coagulation and fibrinolysis in children and adults. Thromb Res. 2011;127(4):317–323. [DOI] [PubMed] [Google Scholar]
- 57.Matsumoto T, Nogami K, Shima M. Simultaneous measurement of thrombin and plasmin generation to assess the interplay between coagulation and fibrinolysis. Thromb Haemost. 2013;110(4):761–768. [DOI] [PubMed] [Google Scholar]
- 58.Longstaff C Studies on the mechanisms of action of aprotinin and tranexamic acid as plasmin inhibitors and antifibrinolytic agents. Blood Coagul Fibrinolysis. 1994;5(4):537–542. [PubMed] [Google Scholar]
- 59.Godier A, Parmar K, Manandhar K, Hunt BJ. An in vitro study of the effects of t-PA and tranexamic acid on whole blood coagulation and fibrinolysis. J Clin Pathol. 2017;70(2):154–161. [DOI] [PubMed] [Google Scholar]
- 60.Collen D, Verstraete M. alpha 2-Antiplasmin consumption and fibrinogen breakdown during thrombolytic therapy. Thromb Res. 1979;14(4–5):631–639. [DOI] [PubMed] [Google Scholar]
- 61.Da Luz LT, Nascimento B, Shankarakutty AK, Rizoli S, Adhikari NK. Effect of thromboelastography (TEG(R)) and rotational thromboelastometry (ROTEM(R)) on diagnosis of coagulopathy, transfusion guidance and mortality in trauma: descriptive systematic review. Crit Care. 2014;18(5):518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gomez-Builes JC, Acuna SA, Nascimento B, Madotto F, Rizoli SB. Harmful or physiologic: diagnosing fibrinolysis shutdown in a trauma cohort with rotational thromboelastometry. Anesth Analg. 2018;127(4):840–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzalez E, Moore EE, Moore HB. Management of trauma-Induced coagulopathy with thrombelastography. Crit Care Clin. 2017;33(1):119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.