

Abstract
Purpose:
To determine if mosaic tuberous sclerosis complex (TSC) can be stratified into subtypes that correspond with prognosis and extent of disease.
Methods:
Next-generation sequencing of skin tumor and other samples was used to identify patients with mosaic pathogenic variants in TSC1 or TSC2. Extent of disease, onset age, and family history of TSC were determined through retrospective analysis of patient records.
Results:
The median number of disease findings and age at penetrance differed between mosaic patients with asymmetrically distributed facial angiofibromas (4 findings, 24y, n=7), mosaic patients with bilaterally symmetric facial angiofibromas (8 findings, 10y, n=12), and germline TSC patients (10 findings, 4y, n=29). Cutaneous and internal organ involvement positively correlated in mosaic (R=0.62, p=0.005), but not germline (R=−0.24, p=0.24) TSC. Variant allele fraction (VAF) in the blood (range: 0-19%) positively correlated with the number of major features (R=0.55, p=0.028). Five had a TSC2 variant identified in the skin that was below detection in the blood. One of 12 children from a mosaic parent had TSC.
Conclusion:
The phenotype of mosaic TSC ranged from mild to indistinguishable from germline disease. Patients with mosaicism and asymmetric facial angiofibromas exhibited fewer findings, later onset, and lower VAF in the blood.
Keywords: Tuberous sclerosis complex, Mosaic, Segmental, Angiofibroma, Genodermatosis
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant neurocutaneous syndrome characterized by hamartomas in multiple organ systems1. TSC results from a pathogenic variant of TSC1 or more commonly, TSC2. One-third of patients inherit the disease from a parent with a germline variant and the remaining two-thirds of cases result from a sporadic (de novo) variant 2. Many of these sporadic cases represent mosaicism due to a post-zygotic mutation resulting in an individual comprised of wild-type and heterozygous cells. We have previously used next-generation sequencing (NGS) to identify mosaic TSC1/TSC2 variants in patients with TSC who had no mutation identified (NMI) by conventional genetic testing 3. About 15% of patients with a clinical TSC diagnosis, of which half are mosaic, have NMI by conventional sequencing methods 3; yet little is known about diagnosis or prognosis in this significant proportion of TSC patients.
The spectrum of disease documented to date in those with genetically proven mosaic TSC overlaps extensively with that of individuals with germline TSC. As a group, patients with mosaic TSC tend to exhibit a lower overall severity 3, but distinctive clinical features are incompletely defined. This is in contrast to neurofibromatosis type 1 (NF1), another autosomal dominant neurocutaneous syndrome. Mosaicism in NF1 has historically centered on a phenotypically distinct form called segmental NF1, characterized by localized or asymmetric distribution of lesions, particularly in the skin 4,5. Those with segmental NF1 tend to have a lower disease severity 4 and lower risk of disease transmission 6 than those with germline NF1.
Patients with TSC have several types of skin lesions that could serve as markers of segmental disease, including facial angiofibromas (AFs), fibrous cephalic plaques (FCP), hypomelanotic macules (HM), shagreen patches (SP), and ungual fibromas (UF). Among these, facial AFs have the greatest potential to be markers of mosaicism, since they occur in most patients and are typically multiple and bilateral. This symmetrical pattern is not seen in rare individuals in whom there is unexpected sparing of one side of the face7. Unilateral AFs have been hypothesized to represent a segmental mosaic form of TSC, and this has been genetically confirmed in one patient 8. Some of the patients with unilateral AFs reported to date exhibit low disease severity 7, but this has not been characterized in any detail.
In our earlier study testing cultured skin tumor cells to elucidate pathogenetic mechanisms underlying the formation of TSC skin tumors, we unexpectedly identified eight individuals with TSC who had bilaterally symmetric AFs but were nonetheless genetically mosaic 9. This prompted the current study to look specifically for patients with unilateral or asymmetrically distributed AFs as a potential marker for mosaic TSC. Skin lesions were again used as the target tissue to identify mosaicism, this time using mostly whole tissue rather than cultured tumor cells as the DNA source. The genetic findings of an additional 12 patients are reported herein, together with extensive phenotyping of these and our previous patients, including those with germline TSC. These studies broaden our understanding of the range of phenotypes displayed in mosaic TSC and improve our clinical and genetic diagnostic capabilities.
Materials and Methods
Patients with TSC were recruited to participate in studies at the National Institutes of Health Clinical Center in Bethesda, Maryland between 2005 and 2018. Written informed consent was obtained according to IRB-approved protocols 00-H-0051, 95-H-0186, 96-H-0100, and/or 82-H-0032 (ClinicalTrials.gov identifiers: NCT00001975, NCT00001465, NCT00001532, and NCT00001183, respectively). Patients seen under these protocols consented to skin biopsies of cutaneous tumors for research purposes. NGS was performed on DNA isolated from fibroblast cultures, whole tumor tissue, or components of whole tumor split by dispase (Supplemental Methods). Unaffected skin, blood, buccal cells, saliva and/or urine were also collected. Our cohort of 112 patients with TSC were screened for asymmetric or unilateral AFs, and those suspected of having mosaic TSC on this basis had NGS performed on skin tumor and control tissue samples. Three patients exhibited absence of tubers and subependymal nodules (SENs) as a potential marker for mosaicism10. Mosaicism was defined as a variant allele fraction (VAF) of less than 40%11. Results from newly identified patients with mosaic or germline TSC were combined with our previously reported cases for phenotype-genotype analysis.
A retrospective review of clinical records from all patients with mosaic or germline TSC was performed. This included a review of history, patient photography and radiographic imaging. The presence of major and minor features for the clinical diagnosis of TSC12 was determined, including eight mucocutaneous findings (AF, UF, FCP, SP, HM, confetti, dental pitting (DP), and oral fibromas (OF)), and six internal findings (tuber, SEN, subependymal giant cell astrocytoma (SEGA), lymphangioleiomyomatosis (LAM), angiomyolipoma (AML), and retinal hamartoma (RH)). The radiologist was blinded to patient’s genotype. The term features is used in analyses where the presence of AF or FCP is combined, as it comprises one major feature for diagnosis12. The term findings is used herein when AF and FCP are counted separately. Mucocutaneous findings that were present but insufficient in number to meet diagnostic criteria (AF≥3, HM≥3, UF≥2, DP ≥3, and OF≥2) were also recorded. AF distribution was determined to be symmetric or asymmetric (estimated as more than ¾ of lesions on one side of the nose and cheeks) upon clinical examination, and quantified retrospectively using patient photography. Patients with genetically proven mosaicism and similar numbers of AFs on each side of the cheeks and nose were categorized as having symmetrical-AF (Sym-AF) mosaicism, and those with unilateral or asymmetrically distributed AFs were classified as having asymmetrical-AF (Asym-AF) mosaicism. Patient-reported ages of onset and diagnoses were recorded. The age at first TSC finding was defined as the age when the first TSC-associated lesion presented, and the age at TSC penetrance was the age at which features were sufficient to diagnose TSC. In patients with LAM, baseline pulmonary function testing (PFT) results; specifically the percent predicted forced expiratory volume in one second (%FEV1) and diffusing capacity of the lungs for carbon monoxide (%DLCO), were recorded. Pulmonary cyst burden was quantified using a computer aided diagnostic system to measure percentage of the total lung volume occupied with cysts (cyst score) 13.
Continuous characteristics of phenotypes were compared using two-sided t-tests following a validation of normality and equal variances. Ages of onset and diagnosis variables, and the sum of findings present were assessed using two-sided Mann-Whitney test. Bivariate correlation was assessed using Pearson’s correlation coefficient. Due to the nature of these data lending to small observed cell counts, nominal characteristics of the phenotypes were assessed using Fisher’s exact test. The Chi-squared test was supplemented where appropriate. Means were compared across genotypes using one-way analysis of variance. All analyses were conducted in IBM SPSS Statistics software and R statistical software. Type I error was controlled at 5%.
Results
Patient Characteristics
A total of 52 patients were included in this study from our cohort of 112 patients. Fifty were female and two male, reflecting enrichment of the cohort with patients diagnosed with LAM, a TSC-associated lung disease that occurs primarily in women. A flow diagram of all patients included in this study, including those previously published9,14,15 is provided in Figure S1. The average age at initial evaluation was 38.0 ± 11.7 years, and did not differ between patients with germline and mosaic TSC (p=0.39).
Pathogenic Variant Identification using NGS
Skin biopsies were obtained from 12 new patients and three patients previously reported as NMI. NGS analysis identified 11 with mosaicism, 1 with germline disease, and 3 with NMI (Table I). Twenty-four skin tumor samples from these 11 new mosaic patients were evaluated by NGS (8 cultured fibroblast, 15 whole tumor, 1 dermis). Mosaic variants were detected in 21/24 (88%) samples, and the VAF ranged from 0.7-23.7%. Second-hit variants were identified in 8/24 (33%) samples and 7/11 (64%) patients. They were identified as second-hit variants due to presence in the skin tumor and absence in control tissues, and two of these had a UV signature mutation, as we have previously observed 9. Once the variant was identified in the affected skin, we looked for the same variant in additional tissues utilizing amplicon NGS. These patients and our 8 previously reported patients with mosaicism 9 carried mosaic variants in TSC2, and the identical variant was confirmed in control and/or different skin tumor samples. In one case (P49), the variant was identified only in the skin tumor.
Table 1.
Variants in TSC2 in Patients with Tuberous Sclerosis Complex
| ID# | Tumor Sampleb |
TSC2 Variantd | Variant Classificatione |
Skin Tumor | Blood | Otherl | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NGS | GREP Analysis | Amplicon Validation | |||||||||||||||
| VAF | WT Reads |
Variant Reads |
VAF | WT Reads |
Variant Reads |
VAFk | WT Reads |
Variant Reads |
VAFk | Sample Type |
WT Reads | Variant Reads |
VAFk | ||||
| P14 | AF-Cx2 | c.2602delG p.(Val868Cysfs*26) | Del [p] | 3.4% | 434 | 18 | 4.0% | 43791 | 1224 | 2.7% | 671614 | 71544 | 9.6% | Control Skin | 64225 | 340 | 0.5% |
| Saliva | 188166 | 28111 | 13% | ||||||||||||||
| c.1599+2T>G, p.? | S [p] | 3.0% | 461 | 15 | 3.2% | 77252 | 2258 | 2.8% | 2246273 | 2409 | 0%* | Control Skin | 94605 | 39 | 0%* | ||
| Saliva | 808185 | 692 | 0%* | ||||||||||||||
| AF-W | c.2602delG p.(Val868Cysfs*26) | Del [p] | 24.3% | 374 | 124 | 24.9% | 14582 | 4537 | 23.7% | ||||||||
| c.1599+2T>G, p.? | S [p] | 0% | 461 | 0 | 0% | 54843 | 39 | 0%* | |||||||||
| P20 | AF-W | c.2784_2785delinsTT p.(Glu929*) | N [p] | 2.9% | 697 | 18 | 2.5% | 22704 | 776 | 3.3% | 1065396 | 4272 | 0.4% | Buccal | 731572 | 15385 | 2.1% |
| c.5160+2delT, p.? | S [p] | 2.2% | 655 | 17 | 2.5% | 10183 | 190 | 1.8% | 926123 | 0 | 0% | Buccal | 751367 | 0 | 0% | ||
| P25a | AF-Cx1 | c.2627_2633delCCAACCC p.(Thr876fs*16) | Del [p] | 48% | 240 | 203 | 46% | NA# | NA# | 50% | 69671 | 869 | 1.2% | Buccal | 55930 | 235 | 0.4% |
| c.3611G>A, p.(Gly1204Glu) | M [p]f | 26% | 111 | 356 | 24% | 376706 | 144705 | 28% | 40973 | 28 | 0%* | ||||||
| AF-W1 | c.2627_2633delCCAACCC p.(Thr876fs*16) | Del [p] | 3.3% | 1050 | 53 | 4.8% | 8023 | 142 | 1.7% | ||||||||
| c.1512_1513delinsTT, p.(Arg505*) | N [p] | 3.2% | 978 | 36 | 3.6% | 36683 | 1073 | 2.8% | 92267 | 0 | 0% | ||||||
| Nipple AF-W2 | c.2627_2633delCCAACCC p.(Thr876fs*16) | Del [p] | 4.3% | 1092 | 82 | 7.0% | 15921 | 266 | 1.6% | ||||||||
| c.1512_1513delinsTT, p.(Arg505*) | N [p] | 0% | 1146 | 0 | 0% | 43510 | 0 | 0% | |||||||||
| P26 | AF-Cx1 | c.3230delC p.(Thr1087fs*4) | Del [p] | 97% | 194 | 233833 | 97% | 71219 | 71643 | 50% | Control Skin | 103722 | 103476 | 50% | |||
| P29 | AF-Cx1 | c.5024C>T, p.(Pro1675Leu) | M [p]g | 6.9% | 361 | 19 | 5.0% | 275704 | 26341 | 8.7% | 777154 | 886 | 0%* | ||||
| c.4786_4787delinsAA, p.(Gly1596Asn) | M [lp]h | 2.4% | 378 | 8 | 2.1% | 44881 | 1301 | 2.8% | 682726 | 603 | 0%* | Urine | 103059 | 0 | 0% | ||
| AF-Cx2 | c.5024C>T, p.(Pro1675Leu) | M [p]g | 2.5% | 1337 | 31 | 2.3% | 17390 | 469 | 2.6% | 79057 | 117 | 0%* | |||||
| P30 | SP-Wc | c.1513C>T (p.Arg505*) | N [p] | 1.1% | 955 | 12 | 1.2% | 110111 | 1123 | 1.0% | 65675 | 1019 | 1.5% | Control Skin | 1.3% | ||
| Urine | 140205 | 8185 | 5.8% | ||||||||||||||
| SP-Dc | c.1513C>T (p.Arg505*) | N [p] | 1.4% | 1520 | 22 | 1.4% | 136622 | 1912 | 1.4% | ||||||||
| P31 | AF-W1 | c.5034C>G (p.Tyr1678*) | N [p] | 1.3% | 1112 | 13 | 1.2% | 7578 | 103 | 1.3% | 169886 | 20 | 0%* | Control Skin | 132554 | 196 | 0.2% |
| Buccal | 102955 | 2752 | 2.6% | ||||||||||||||
| AF-W2 | c.5034C>G (p.Tyr1678*) | N [p] | 1.8% | 1098 | 19 | 1.7% | 37878 | 720 | 1.9% | ||||||||
| P32 | SP-W1 | c.5238_5255del p.(His1746_Arg1751del) | I [p]i | 9.4% | 578 | 98 | 14.5% | 130512 | 5974 | 4.4% | 99714 | 4126 | 4.0% | Buccal | 134447 | 1052 | 0.8% |
| Urine | 44781 | 570 | 1.3% | ||||||||||||||
| UF-W2 | c.5238_5255del p.(His1746_Arg1751del) | I [p]i | 2.4% | 989 | 30 | 2.9% | 91651 | 1777 | 1.9% | ||||||||
| P33 | AF-W1 | c.975-15G>A p.(Ala326_Gln373del, Met327Hisfs*5) | S [p]j | 3.0% | 1116 | 27 | 2.4% | 24952 | 923 | 3.6% | |||||||
| c.3412C>T, p.(Arg1138*) | N [p] | 0% | 898 | 0 | 0% | 123757 | 213 | 0%* | 1334832 | 1734 | 0%* | ||||||
| AF-W2 | c.3412C>T, p.(Arg1138*) | N [p] | 1.7% | 1120 | 16 | 1.4% | 193785 | 3813 | 1.9% | Saliva | 1282079 | 1174 | 0%* | ||||
| Urine | 20395 | 20 | 0%* | ||||||||||||||
| c.975-15G>A p.(Ala326_Gln373del, Met327Hisfs*5) | S [p]j | 5.4% | 1109 | 51 | 4.4% | 16420 | 1134 | 6.5% | 65787 | 3499 | 5.1 | Saliva | 111250 | 2836 | 2.5% | ||
| P39 | AF-W1 | c.1108C>T, p.(Gln370*) | N [p] | 10.2% | 1048 | 108 | 9.3% | 185047 | 23174 | 11.1% | 1479095 | 1032 | 0%* | Saliva | 1592188 | 1101 | 0%* |
| Buccal | 1013198 | 596 | 0%* | ||||||||||||||
| c.1714C>T, p.(Gln572*) | N [p] | 0% | 1341 | 0 | 0% | 170835 | 176 | 0%* | 3448295 | 2677 | 0%* | Saliva | 2461808 | 2229 | 0%* | ||
| Buccal | 2817203 | 2993 | 0%* | ||||||||||||||
| AF-W2 | c.1108C>T, p.(Gln370*) | N [p] | 3.5% | 984 | 31 | 3.1% | 90243 | 3772 | 4.0% | ||||||||
| c.1714C>T, p.(Gln572*) | N [p] | 3.1% | 1135 | 31 | 2.7% | 29240 | 1082 | 3.6% | |||||||||
| P40 | AF-W | c.4375C>T, p.(Arg1459*) | N [p] | 5.7% | 1232 | 78 | 6.0% | 288622 | 16626 | 5.5% | 3037476 | 15857 | 0.5% | Control Skin | 3849197 | 19660 | 0.5% |
| Saliva | 3073759 | 22291 | 0.7% | ||||||||||||||
| c.2251C>T, p.(Arg751*) | N [p] | 4.1% | 1278 | 48 | 3.6% | 39195 | 1895 | 4.6% | 363675 | 571 | 0%* | Control Skin | 731920 | 876 | 0%* | ||
| Saliva | 405575 | 502 | 0%* | ||||||||||||||
| AF-Cx4 | c.4375C>T, p.(Arg1459*) | N [p] | 1.3% | 741 | 5 | 0.7% | 360514 | 7393 | 2.0% | ||||||||
| c.2251C>T, p.(Arg751*) | N [p] | 0% | 847 | 0 | 0% | 23073 | 38 | 0%* | |||||||||
| AF-Cx2 | c.4375C>T, p.(Arg1459*) | N [p] | 0% | 160849 | 336 | 0%* | |||||||||||
| c.2251C>T, p.(Arg751*) | N [p] | 0% | 63437 | 91 | 0%* | ||||||||||||
| AF-Cx5 | c.4375C>T, p.(Arg1459*) | N [p] | 0% | 333180 | 963 | 0%* | |||||||||||
| c.2251C>T, p.(Arg751*) | N [p] | 0% | 30966 | 43 | 0%* | ||||||||||||
| P49 | AF-Cx1 | c.2251C>T, p.(Arg751*) | N [p] | 21% | 230 | 54 | 19% | 68007 | 19219 | 22% | 102623 | 200 | 0%* | Control Skin | 102940 | 89 | 0%* |
| AF-Cx2 | c.2251C>T, p.(Arg751*) | N [p] | 0% | 170029 | 226 | 0%* | |||||||||||
Abbreviations: AF, Angiofibroma; SP, Shagreen Patch; UF, Ungual Fibroma; NMI, no mutation identified; NGS, Next-Generation Sequencing; VAF, Variant Allele Fraction; WT, Wild Type; NA#, confirmation done by Sanger sequencing.
Patients with NMI were excluded from this table: P16 (AF-W), P41 (AF-Cx1), and P70 (AF-Cx1). Two of three patients previously reported as NMI had a pathogenic variant detected in a newly analyzed tissue (P14, P20). P41 remained NMI.
Patient with germline tuberous sclerosis complex.
Tissue was process as whole tissue (W), cultured fibroblast (Cx), or split with Dispase to isolate the dermis (D).
Histologically, the lesion exhibited thickened and abnormally arranged collagen fibers, and overlying epidermal changes reminiscent of epidermal nevus.
The reference transcripts used are: Genomic refseq ID NG_005895.1; Transcript refseq ID NM_000548.5.
ACMG interpretation of variant classification was assessed by standard criteria as in Genetics in Medicine (2015) 17:405; with reference to the TSC gene mutation database LOVD, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2. Variant classification was abbreviated as follows: Del, deletion; S, splice; N, nonsense; M, missense; I, inframe deletion; [p], pathogenic; [lp], likely pathogenic.
c.3611G>A, p.(Gly1204Glu): Missense mutation shown to be functionally inactivating, TSC gene mutation database LOVD, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2
c.5024C>T, p.P1675L: Highly recurrent missense mutation, known to occur de novo, and shown to be functionally inactivating, TSC gene mutation database LOVD, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2
c.4786_4787GG>AA, p.(Gly1596Asn): Novel missense mutation, likely pathogenic, based on occurrence of two de novo variants, p.(Gly1596Asp) and p.(Gly1596Val), reported at this position in the TSC gene mutation database LOVD, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2, one of which has been shown to be functionally inactivating, p.(Gly1596Val).
c.5238_5255del p.(His1746_Arg1751del): Highly recurrent in-frame del mutation, known to occur de novo, and shown to be inactivating, TSC gene mutation database LOVD, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2
c.975-15G>A (p.Ala326_Gln373del, Met327Hisfs*5): Highly recurrent splice mutation, known to occur de novo, and shown to lead to aberrant splicing, TSC gene mutation database LOVD, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2
0%* means that the detected VAF was similar to that seen in unrelated control DNA samples, and hence is equivalent to 0%.
Control skin refers to normal appearing skin. Control skin was processed as cultured fibroblasts except in patients P30 and P31.
Angiofibroma Distribution in Asym-AF and Sym-AF Mosaicism
By definition, AF distribution was bilateral and symmetric in patients with Sym-AF, and unilateral or asymmetric in those with Asym-AF (Figure 1A, 1B). The side of AF predominance contained 52 ± 1% of the AFs in Sym-AF (n=12) and 87 ± 9% in Asym-AF (n=7), (p<0.001) (Table S1). The total number of AFs on the nose and cheeks ranged from 6 to greater than 600 in Sym-AF and 10 to 91 in Asym-AF. One patient (P26) selected for study based on asymmetric AF distribution had germline disease. This patient had 127 AFs on the nose and hundreds on the cheeks, which was much greater than the number observed in those with Asym-AF mosaicism. Since prior data indicates that AFs develop through a two-hit mechanism with biallelic inactivating mutations in TSC2 (or TSC1), it is possible this germline patient had a generalized second-hit mutation affecting skin fibroblasts on one side of the face (type 2 segmental mosaicism16), causing this asymmetric presentation.
Figure 1. The Clinical Picture of Mosaicism in Tuberous Sclerosis Complex.

(A) Mosaicism with an asymmetric distribution of angiofibromas (AFs) on the nose and cheeks. (AR, AL) Right and left lateral views of the nose highlight the left sided predominance of AFs. (B) Mosaicism with numerous AFs distributed symmetrically on the nose and cheeks, indistinguishable from a patient with germline TSC. (BR, BL) Right and left lateral views of the nose reveal more numerous and symmetric distribution of AFs.
The Clinical Phenotype of Germline Disease, Asym-AF Mosaicism, and Sym-AF Mosaicism
The overall range of clinical phenotypes observed in each individual with germline and mosaic TSC is summarized in Figure 2 and Figure S2. Twenty-nine had germline disease, including those identified through NGS (TSC1:1, TSC2: 16), routine genetic testing (TSC1: 3), or parental transmission without genetic testing (9). Of the 17 patients identified with germline TSC by NGS, there were five with and twelve without a family history of TSC in a parent or sibling. Nineteen (12 Sym-AF, 7 Asym-AF) had mosaicism. Many with Sym-AF were indistinguishable from patients with germline disease, whereas those with Asym-AF were among the most mildly affected (Figure 2, Figure S2).
Figure 2. Phenotypic Spectrum of TSC Patients with Mosaic Subtypes or Germline Disease.

Patients were first sorted by the variant allele fraction in the blood from highest to lowest, and then from the highest to lowest sum of total findings (major & minor mucocutaneous and major internal), color-coded from dark red to yellow. Those with germline disease tended to have a greater extent of disease than those with mosaic TSC. The phenotype of mosaic TSC ranged from very mild to indistinguishable from germline TSC. Bolded text indicates those with Asymmetrical-Angiofibroma mosaicism. Patients with Asymmetrical-Angiofibroma mosaicism have fewer findings (enriched toward the bottom), whereas those with Symmetrical-Angiofibroma mosaicism tended to have more findings (enriched towards the top). Symbols and abbreviations used: U, Unknown; MCF, Mucocutaneous Findings; IF, Internal Findings; NA, Not Applicable – patients who lack UF but are under age 30 years.
Extent of disease was assessed by counting the number of mucocutaneous, internal, and total findings. Patients with Asym-AF had fewer mucocutaneous (Figure 3A), internal (Figure 3B), and total findings (Figure 3C) than those with germline disease or Sym-AF (Table S2). Those with Sym-AF had significantly fewer mucocutaneous and total findings than in those with germline TSC, but more than those with Asym-AF. There were 11/12 (92%) with Sym-AF and only 1/7 (14%) with Asym-AF with at least three of eight mucocutaneous findings (p=0.002). All but one patient with germline disease had at least three mucocutaneous findings. The number of major internal (tuber, SEN, SEGA, LAM, AML) and cutaneous findings (AF, FCP, HM, UF, SP) correlated significantly in mosaic (R=0.62, n=19, p=0.005) but not germline TSC (R=−0.24, n=26, p=0.24), (Figure 3E, 3F).
Figure 3. The Number TSC Findings and Neurological Tumor Status Differs Between Patients with Asymmetrical-Angiofibroma Mosaicism, Symmetrical-Angiofibroma Mosaicism, or Germline Disease.
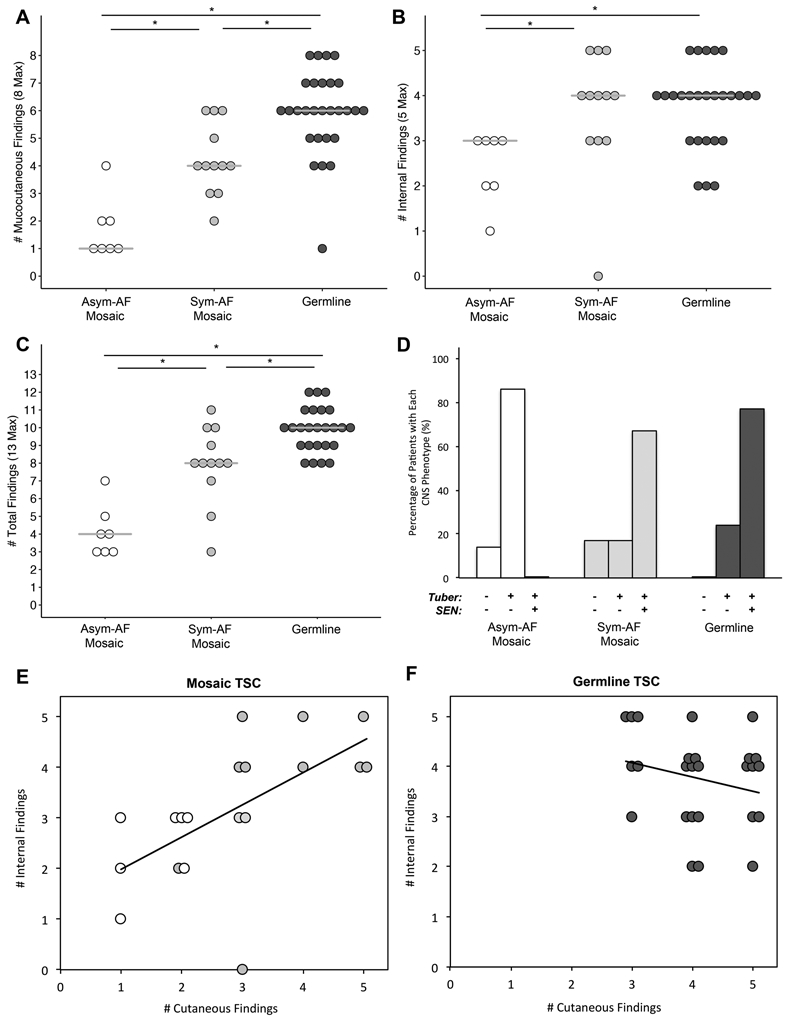
(A) The number of mucocutaneous findings increased sequentially from Asymmetrical-Angiofibroma (Asym-AF) mosaicism, to Symmetrical-Angiofibroma (Sym-AF) mosaicism, to germline TSC. (B) The number of internal findings was significantly lower in patients with Asym-AF mosaicism than Sym-AF mosaicism and germline TSC. (C) The number of total findings increased sequentially from Asym-AF mosaicism, to Sym-AF mosaicism, to germline TSC. (D) The most common CNS phenotypes were tubers without SENs in Asym-AF mosaicism, and tubers with SENs in Sym-AF mosaicism and germline TSC. (E) The number of major internal and cutaneous findings correlated significantly in mosaic TSC (R=0.62, n=19, p=0.005). Those with Asym-AF mosaicism (white) clustered in the lower left quadrant whereas those with Sym-AF mosaicism (light grey) clustered in the upper right quadrant as observed in germline TSC. (F) In patients with germline TSC, the number of internal and cutaneous findings did not correlate (R=−0.24, n=26, p=0.24). These patients clustered in the upper right quadrant of the figure.
Many mucocutaneous findings were less frequent in mosaic TSC (Table S3). UF, DP, and OF were less common in both Sym-AF and Asym-AF than in germline disease. Additionally, patients with Asym-AF less frequently had HM, FCP, and SP than those with germline TSC and were less likely to have HM, SP, and UF than those with Sym-AF.
Tubers were common in all groups, but SENs were less frequent in Asym-AF than in Sym-AF and germline disease (Table S3). The most common CNS phenotype was tubers without SENs in Asym-AF (6/7, 86%), (Figure 3D). This phenotype was more frequent in patients with Asym-AF than in those with germline TSC (6/26, 23%) or Sym-AF (2/12, 17%), (p=0.005, p=0.006). The most common CNS phenotype was tubers with SENs in Sym-AF (8/12, 67%) and germline TSC (20/26, 77%) while this phenotype was not seen in any subject with Asym-AF (p<0.001, p=0.013). There were 2/12 (17%) with Sym-AF, and 1/7 (14%) with Asym-AF without tubers or SENs. None of the patients with germline disease had this phenotype, and all patients with SENs also had tubers.
The prevalence of LAM and AMLs did not differ between groups (Table S3). A past history of surgical interventions (embolization, renal transplant, or nephrectomy) also did not differ between germline and mosaic TSC. However, those with Asym-AF were more likely to have undergone renal embolization than those with germline disease (p=0.012). Cyst burden and pulmonary function, assessed by %FEV1 and %DLCO, did not differ between groups (Table S4).
Patients with Asym-AF were older than those with germline disease at age of first TSC finding onset and TSC penetrance (p=0.042, p=0.043 respectively), (Table S5). The onset of UF and AF tended to be later in patients with mosaic TSC compared to those with germline TSC. UF onset was before the age of 15 years in 11/26 (42%) with germline and 2/13 (15%) with mosaic TSC (p=0.15). Likewise, AF onset was before the age of 5 years in 10/27 (37%) with germline and 3/16 (19%) with mosaic TSC (p=0.31). There was no difference in the age at TSC diagnosis, LAM symptom onset, LAM diagnosis, or AML diagnosis between the three groups.
Fourteen germline patients had a total of 16/26 (62%) children with TSC. In contrast, one of 12 (8%) children from a parent with mosaic TSC developed germline TSC (p=0.002). Five with Sym-AF had a total of seven children, one of whom was diagnosed with TSC. Three with Asym-AF had a total of five unaffected children.
Correlation of VAF with the Extent of Organ Involvement
Blood samples from 16/19 patients with mosaicism were available for NGS, and the median VAF was 1.35% (range: 0 - 19%). The VAF correlated positively with the number of major TSC features present (R=0.55, p=0.028) (Figure 4A). The average VAF in the blood differed between Sym-AF (6.3 ± 6.1%) and Asym-AF (1.0 ± 1.8%), (p=0.043). The VAF in the blood was also greater in those with mosaicism and SENs (9.1 ± 6.8%) than in those without (1.7 ± 2.2%), (p=0.005).
Figure 4. The Variant Allele Fraction (VAF) in the Blood Correlates with the Number of Major Features in Patients with Mosaic TSC.
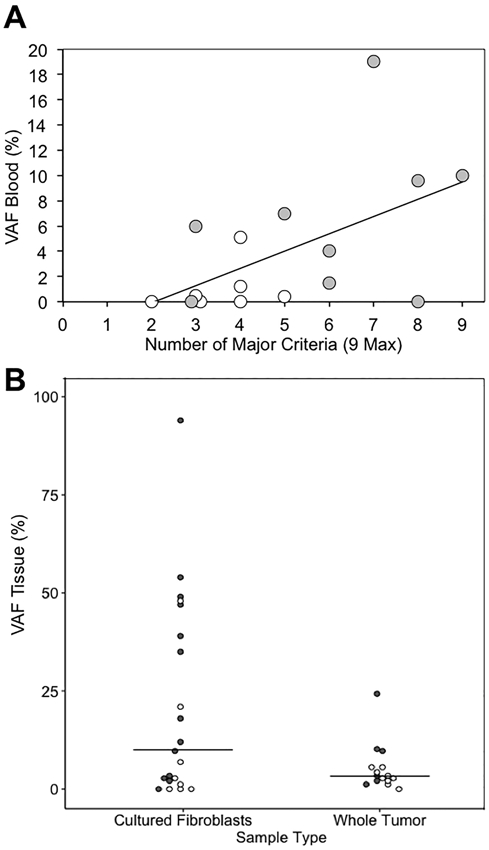
Patients with Asym-AF and Sym-AF mosaicism are represented by white and grey circles respectively. (A) The VAF detected in the blood of patients with mosaicism correlated significantly with the number of major TSC features present (R=0.55, n=16, p=0.028). (B) The range of the VAF detected in cultured fibroblast and whole skin tumor DNA samples. Samples included 29 angiofibromas, 3 ungual fibromas, 3 shagreen patches, and 1 oral fibroma. Four cultured fibroblast samples from three patients, and one whole tissue sample had NMI. These samples include our newly analyzed and previously published samples.
Variant Detection is Enhanced in Lesion Fibroblast Cultures and in the Dermis
Mosaic variants were identified in 31/36 (86%) samples (15 whole tumor, 21 cultured fibroblast) derived from 19 patients with mosaicism. Cultured fibroblast samples had a median VAF of 10% (range 0-94%), whereas the median VAF in whole tumor samples was 3.3% (range 0-24%), (Figure 4B). Eight AFs from a patient with Sym-AF (P05) were analyzed to determine if mutated cells were more prevalent within the epidermis or dermis (Table S6). The VAF of TSC2 from four whole tumor samples was compared to that of four tumors split at the dermal-epidermal junction using dispase. The mean VAF was higher in the dermis (10.8%) than whole tumor (5.7%), (p=0.013).
The majority of blood and whole tissue skin tumor samples evaluated from patients with mosaicism fell below the usual detection limit of Sanger sequencing (20%), (Figure S3). None of the 16 patients that underwent NGS of the blood had a VAF greater than 20%, whereas NGS was able to detect variants above a frequency of 1% in 9/16 (56%). The VAF in the blood was <1% in 2/9 (22%) with Sym-AF and 5/7 (71%) with Asym-AF. Only 1/10 (10%) patient with whole tumor samples exhibited a VAF greater than 20%, compared to 7/14 (50%) of patients with skin tumors processed as cultured fibroblasts.
Discussion
In this cohort of germline and mosaic TSC patients, we present extensive information on the occurrence, genetic consequences, and clinical importance of Asym-AF and Sym-AF mosaicism in TSC. Although earlier case reports had noted unilateral AFs in TSC, the clinical presentations and significance had not been reported in detail previously. Those with Asym-AF manifested the fewest median clinical findings and oldest median age of TSC penetrance; while those with Sym-AF were intermediate by both of these measures relative to germline disease. This is remarkable considering the high variability in disease expression among those with germline disease. One source of variability in germline disease is that individuals with variants in TSC1 tend to be less severe than those with variants in TSC2 1,17-19. Our germline patients included four with TSC1 variants whereas all those with mosaicism had variants in TSC2, making the observation of fewer manifestations in the mosaic group even more surprising. A milder phenotype and/or later onset have also been observed in mosaic NF120, NF221,22, and Turner syndrome23.
In those with mosaic TSC, the number of cutaneous findings correlated with the number of internal findings, as previously reported in a genetic condition that is always mosaic, Proteus syndrome24. External features may provide clues about the extent of internal organ involvement in mosaic TSC, but it should be cautioned that individuals with germline disease may exhibit minimal or no skin manifestations, particularly in early childhood25. It is the unilateral or asymmetric pattern of AFs that is a marker for mosaicism, rather than the absence of cutaneous findings. Those with Asym-AF mosaicism had a minimum ratio of 3:1 for one side of the nose and cheeks versus the other, and typically had fewer AFs. There may a spectrum from the mildest disease in those with a single patch of strictly unilateral AFs as the sole cutaneous manifestation to those with more extensive internal disease in someone with asymmetric AFs combined with other skin findings. It may be possible to use other TSC skin manifestations as markers for mosaic disease, but confidence that these reflect mosaicism is low for lesions that are few in number, particularly if they are sometimes unilateral in germline disease.
The VAF detected in the blood in mosaic TSC correlated positively with the number of major features present. A higher fraction of affected cells, presumably from an earlier post-zygotic mutation, would be expected to result in a greater number and variety of disease manifestations 26,27. Correspondingly, individuals with Asym-AF mosaicism, likely arising from a later post-zygotic mutation, had a lower VAF in the blood and a milder phenotype than those with Sym-AF. This observation suggests that the proportion of affected cells is the major factor influencing extent of disease and age of onset. Since tumor development in TSC fits the Knudson two-hit mechanism, it is not surprising that a lower prevalence of mutation means that there are fewer cells susceptible to tumor development through a second hit mutation. However, even those with Asym-AF in our cohort, which is biased through ascertainment towards those with LAM and AMLs, were not spared from potentially life-threatening TSC manifestations. Hence, mosaic TSC patients should have a complete evaluation and periodic surveillance28.
A clinical feature enriched in those with mosaicism was the occurrence of cortical tubers without SENs, seen in almost all (6/7) of our patients with Asym-AF. Mosaicism cannot be ruled out in patients with both tubers and SENs since most of our patients with Sym-AF mosaicism had this phenotype. Mosaic TSC patients who lacked SENs had a lower VAF in the blood than those with SENs. Likewise, the VAF in the blood or buccal samples of patients with a mosaic PIK3CA variant was lower in those without than with brain overgrowth/malformation (p<0.001)29. Furthermore, two of our patients without cutaneous signs of mosaicism had no tubers or SENs. The lack of tubers and SENs could represent a subtype of mosaic TSC limited to the neural crest lineage 10. Thus, the neurological phenotype is another useful clue for mosaic disease, and may be the only sign of mosaicism in patients with a generalized pattern of cutaneous manifestations.
One patient of five (20%) with Sym-AF mosaicism in our cohort had an affected child, out of seven children total. Further, none of five children born to those with Asym-AF mosaicism had TSC. Although a very conservative approach is to estimate the risk of transmission to offspring at up to 50% for an individual with mosaicism, it is clearly much lower than that in this aggregate though limited series (1 of 12 (8%) children). The risk of transmission is lower in sporadic NF2 patients, and decreases with increasing age at presentation 30,31. The risk is also lower if the NF2 variant is only detectable in the tumor and not the blood 32. Enhanced methods using NGS for detection of mosaic disease using a patient’s blood or cutaneous tumor may improve genetic counseling and allow for pre-implantation screening or presymptomatic detection in offspring 33,34.
Although our sample size is relatively small, this study is the first to describe differences in overall extent of organ involvement between Asym-AF and Sym-AF mosaic TSC. Our patients with mosaicism were identified through genetic testing of affected skin, however other tissues such as AMLs may also be used to identify the mosaic variant 35. Our population consisted mostly of adult women with LAM, and therefore may not reflect findings in the general TSC population. Although LAM may not be as prevalent in mosaic TSC as within our cohort, this study shows that patients with Asym-AF may develop LAM. Future studies are warranted to determine if the VAF in children with mosaic TSC predicts the extent of organ involvement in adult life, and to further investigate the severity of disease in mosaic TSC.
We note that a parallel study on mosaicism in TSC, also reported in this issue, came to similar observations on the prevalence and clinical significance of mosaicism but with some distinct differences. In that report, individuals with mosaic TSC had a high prevalence of facial AFs and kidney angiomyolipoma, but a low incidence of most other TSC clinical findings, and LAM was rare. These two sets of mosaic TSC patients have many similarities as well as some differences which we suspect are due to the methods by which they were ascertained.
In conclusion, the spectrum of mosaic TSC ranges from a mild phenotype with localized cutaneous findings to a more generalized phenotype that is indistinguishable from patients with inherited germline variants. Clinical clues suggestive of low-level mosaicism in an adult with TSC include asymmetric distribution of AFs, disease penetrance in adulthood, and lack of tubers and SENs. Patients with these features are likely to have no pathogenic variant identified when non-NGS approaches are used for genetic testing of the blood, and thus require NGS of their blood or affected skin to detect the disease-causing variant. Patterns of mosaic disease observed in TSC provide clues about prognosis and risk of disease transmission to offspring that may hold true in many other mosaic genetic conditions.
Supplementary Material
Acknowledgements:
Research reported in this publication was supported in part by the Intramural Research Program, National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI); the NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases, under Award Number R01AR062080; NIH, NHLBI under Award Number 1U01HL131022; the Doris Duke Charitable Foundation Clinical Research Mentorship grants #2014088 and #2018042; the Tuberous Sclerosis Alliance, Engles Fund for Research in TSC and LAM. Additionally, this work was made possible through the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, and other private donors. For a complete list, visit the foundation website at http://www.fnih.org. Written consent was obtained to publish the photography of the patients included in Figure 1.
Footnotes
Disclaimer:
The opinions and assertions expressed herein are those of the author(s) and do not necessarily reflect the official policy or position of the Uniformed Services University, the Department of Defense or the National Institutes of Health.
Conflict of Interest Statement
The authors of this manuscript have no conflicts of interest to report.
References
- 1.Peron A, Au KS, Northrup H. Genetics, genomics, and genotype-phenotype correlations of TSC: Insights for clinical practice. Am J Med Genet C Semin Med Genet. 2018;178(3):281–290. [DOI] [PubMed] [Google Scholar]
- 2.Sampson JR, Scahill SJ, Stephenson JB, Mann L, Connor JM. Genetic aspects of tuberous sclerosis in the west of Scotland. J Med Genet. 1989;26(1):28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyburczy ME, Dies KA, Glass J, et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet. 2015;11(11):e1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56(11):1433–1443. [DOI] [PubMed] [Google Scholar]
- 5.Messiaen L, Vogt J, Bengesser K, et al. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Hum Mutat. 2011;32(2):213–219. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Romero MT, Parkin P, Lara-Corrales I. Mosaic Neurofibromatosis Type 1: A Systematic Review. Pediatr Dermatol. 2016;33(1):9–17. [DOI] [PubMed] [Google Scholar]
- 7.Hall MR, Kovach BT, Miller JL. Unilateral facial angiofibromas without other evidence of tuberous sclerosis: case report and review of the literature. Cutis. 2007;80(4):284–288. [PubMed] [Google Scholar]
- 8.Bessis D, Malinge MC, Girard C. Isolated and unilateral facial angiofibromas revealing a type 1 segmental postzygotic mosaicism of tuberous sclerosis complex with c.4949_4982del TSC2 mutation. Br J Dermatol. 2018;178(1):e53–e54. [DOI] [PubMed] [Google Scholar]
- 9.Tyburczy ME, Wang JA, Li S, et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet. 2014;23(8):2023–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boronat S, Shaaya EA, Doherty CM, Caruso P, Thiele EA. Tuberous sclerosis complex without tubers and subependymal nodules: a phenotype-genotype study. Clin Genet. 2014;86(2):149–154. [DOI] [PubMed] [Google Scholar]
- 11.Acuna-Hidalgo R, Bo T, Kwint MP, et al. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am J Hum Genet. 2015;97(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao J, Taveira-DaSilva AM, Colby TV, Moss J. CT grading of lung disease in lymphangioleiomyomatosis. AJR Am J Roentgenol. 2012;199(4):787–793. [DOI] [PubMed] [Google Scholar]
- 14.Nathan N, Tyburczy ME, Hamieh L, et al. Nipple Angiofibromas with Loss of TSC2 Are Associated with Tuberous Sclerosis Complex. J Invest Dermatol. 2016;136(2):535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J, Tyburczy ME, Moss J, Darling TN, Widlund HR, Kwiatkowski DJ. Tuberous sclerosis complex inactivation disrupts melanogenesis via mTORC1 activation. J Clin Invest. 2017;127(1):349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Happle R The categories of cutaneous mosaicism: A proposed classification. Am J Med Genet A. 2016;170A(2):452–459. [DOI] [PubMed] [Google Scholar]
- 17.Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13(6):731–741. [DOI] [PubMed] [Google Scholar]
- 19.Au KS, Williams AT, Roach ES, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9(2):88–100. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Shachar S, Dubov T, Toledano-Alhadef H, et al. Predicting neurofibromatosis type 1 risk among children with isolated cafe-au-lait macules. J Am Acad Dermatol. 2017;76(6):1077–1083 e1073. [DOI] [PubMed] [Google Scholar]
- 21.Spyra M, Otto B, Schon G, Kehrer-Sawatzki H, Mautner VF. Determination of the mutant allele frequency in patients with neurofibromatosis type 2 and somatic mosaicism by means of deep sequencing. Genes Chromosomes Cancer. 2015;54(8):482–488. [DOI] [PubMed] [Google Scholar]
- 22.Halliday D, Emmanouil B, Pretorius P, et al. Genetic Severity Score predicts clinical phenotype in NF2. J Med Genet. 2017;54(10):657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuke MA, Ruth KS, Wood AR, et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen D, Turner JT, Olsen C, Biesecker LG, Darling TN. Cutaneous manifestations of proteus syndrome: correlations with general clinical severity. Arch Dermatol. 2004;140(8):947–953. [DOI] [PubMed] [Google Scholar]
- 25.Jozwiak S, Schwartz RA, Janniger CK, Michalowicz R, Chmielik J. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol. 1998;37(12):911–917. [DOI] [PubMed] [Google Scholar]
- 26.Normand EA, Crandall SR, Thorn CA, et al. Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron. 2013;78(5):895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol Clin. 2017;35(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuentz P, St-Onge J, Duffourd Y, et al. Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genet Med. 2017;19(9):989–997. [DOI] [PubMed] [Google Scholar]
- 30.Evans DG, Ramsden RT, Shenton A, et al. Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet. 2007;44(7):424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans DG, Wallace A. An update on age related mosaic and offspring risk in neurofibromatosis 2 (NF2). J Med Genet. 2009;46(11):792. [DOI] [PubMed] [Google Scholar]
- 32.Moyhuddin A, Baser ME, Watson C, et al. Somatic mosaicism in neurofibromatosis 2: prevalence and risk of disease transmission to offspring. J Med Genet. 2003;40(6):459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kluwe L, Friedrich RE, Tatagiba M, Mautner VF. Presymptomatic diagnosis for children of sporadic neurofibromatosis 2 patients: a method based on tumor analysis. Genet Med. 2002;4(1):27–30. [DOI] [PubMed] [Google Scholar]
- 34.Consoli C, Moss C, Green S, Balderson D, Cooper DN, Upadhyaya M. Gonosomal mosaicism for a nonsense mutation (R1947X) in the NF1 gene in segmental neurofibromatosis type 1. J Invest Dermatol. 2005;125(3):463–466. [DOI] [PubMed] [Google Scholar]
- 35.Han MK, Tyburczy ME, Darling TN, et al. Apparent Sporadic Lymphangioleiomyomatosis in a Man as a Result of Extreme Mosaicism for a TSC2 Mutation. Ann Am Thorac Soc. 2017;14(7):1227–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.