

Abstract
This protocol assesses proinflammatory properties of nucleic acid nanoparticles (NANPs) using a validated preclinical model, peripheral blood mononuclear cells (PBMCs), that is highly predictive of cytokine responses. The experimental procedure details the preparation of pyrogen-free NANPs, isolation of PBMCs from freshly collected human blood, and analysis of characteristic biomarkers (type I and III interferons) produced by PBMCs transfected with NANPs. Although representative NANPs with high and low immunostimulatory potential are used as standards throughout the procedure, this protocol can be adapted to any NANPs or therapeutic nucleic acids, irrespective of whether they are carrier based or carrier free; additional cytokine biomarkers can also be included. We test several commercial platforms and controls broadly accessible to the research community to quantify all biomarkers in either single- or multiplex format. The continuous execution of this protocol takes <48 h; when immediate analysis is not feasible, single-use aliquots of the supernatants can be frozen and stored (−20 °C; 12 months).
Introduction
Nucleic acid–based technologies
A variety of rapidly evolving nucleic acid–based technologies benefit from the ability of RNA and DNA to form both canonical and non-canonical base pairings1. Currently, existing libraries of RNA (and DNA) motifs2–5 can be rationally combined to assemble various NANPs (Fig. 1a) that can be further decorated with therapeutic nucleic acids (TNAs)6 and used as next-generation TNA delivery platforms7,8. At its essence, the design strategy combines different motifs, analogous to the assembly of LEGO blocks, and follows empirically rationalized rules to achieve a remarkable degree of structural control in bottom-up assemblies9–13. The one-pot assembly of NANPs designed with this approach requires two-step incubation, enabling the correct folding of individual strands and further activation of magnesium-dependent long-range interactions (Fig. 1b). Another common strategy (Fig. 1c) is based solely on canonical Watson–Crick interactions with ssRNAs and/or ssDNAs computationally programmed to interact only with their partner strands while avoiding any intramolecular pairings14. Similar design principles are widely used in structural DNA nanotechnology15 and DNA origami16 for the construction of a wide variety of functional DNA assemblies.
Fig. 1 |. The main strategies for NANP design.
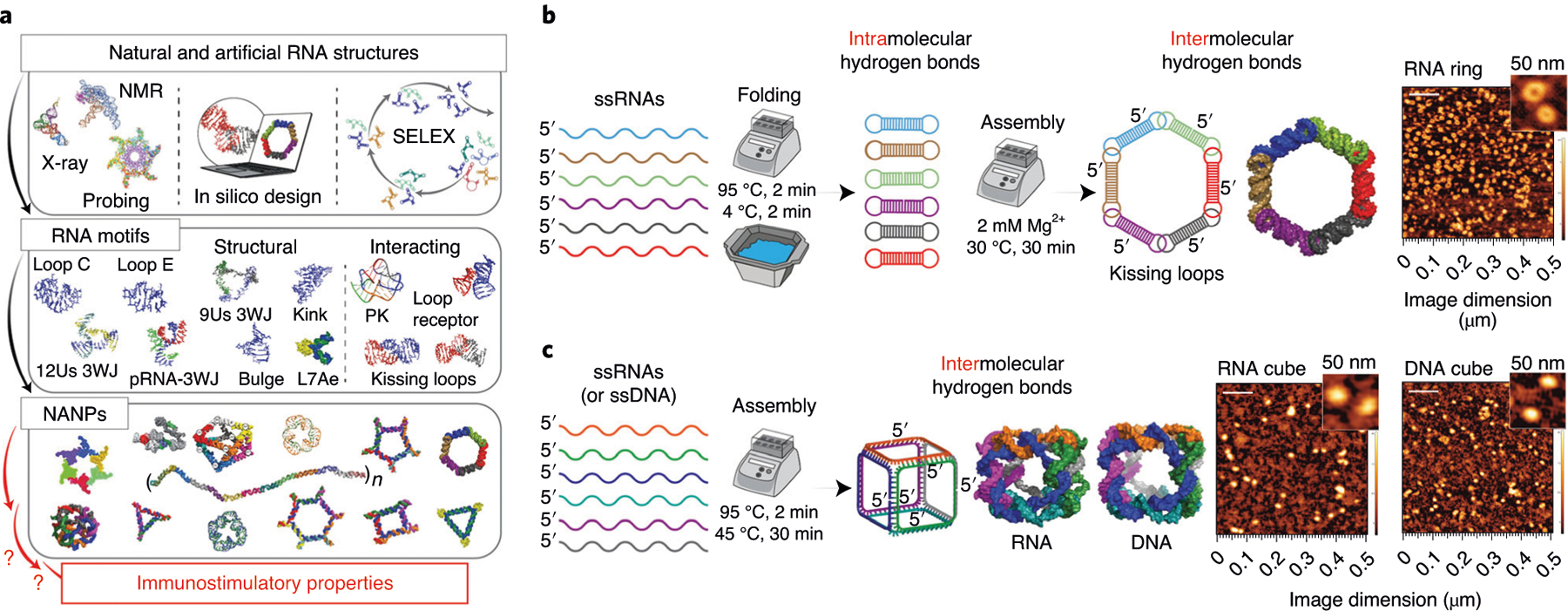
a, Schematics showing the workflow for NANP production. b,c, Two main design strategies: the first relies on intramolecular bonds within each monomer that promote the formation of magnesium-dependent interacting motifs required for intermolecular interactions (b), and the second is based on assembly of NANPs and individual monomers designed to form intermolecular bonds only with their cognate partner strands (c). Scale bars, 500 nm (main); 100 nm (insets). 3WJ, three-way junction; PK, pseudoknot; SELEX, systematic evolution of ligands by exponential enrichment; Us, uracils.
Therapeutic NANPs
The increasing appreciation and use of biocompatible NANPs has led to the establishment of a new field, in which the innate biological functions of nucleic acids are reprogrammed to tackle specific biomedical challenges8. In addition, the burgeoning NANP technology presents many advantages: (i) NANPs can be designed to deliver cocktails of different TNAs to diseased cells17–22, enabling simultaneous targeting of several biological pathways with higher synergistic effects; (ii) the thermal and chemical stabilities of NANPs are tuned with chemical modifications23,24, and NANPs’ immunorecognition is controlled by their structure and composition24–26; (iii) TNAs can be programmed into NANPs to induce responsive behavior23,27; (iv) pyrogen-free NANPs can be produced with high batch-to-batch consistency7, permitting their industrial-scale manufacture (endotoxin, a component of the cell wall of Gram-negative bacteria, is a common pyrogen that contaminates nanoformulations28, precluding their clinical use); (v) introduction of small molecules, aptamers, or antibodies into NANP structures makes them amenable to targeted delivery29; and (vi) carrier-free NANPs are immunoquiescent and safe for systemic administration25.
NANPs’ safety and immune-mediated efficacy considerations
To further advance the translation of NANPs from bench to clinic, the field is in great need of reliable experimental protocols for the assessment of both the safety and the efficacy of these novel nanomaterials. The assessment of immunological responses to NANPs has twofold importance. First, it contributes to the understanding of NANPs’ safety in that the systemic induction of inflammation is associated with the cytokine storm toxicity. Such assessment is especially critical in light of several recent announcements of halted clinical trials and biotech companies’ consequent withdrawal of TNAs formulated with lipid-based carriers because of adverse immune-mediated toxicities of those formulations (e.g., https://pubmed.ncbi.nlm.nih.gov/32238921/). Second, an understanding of immunological responses to NANPs would be helpful in exploring the alternative indications and routes of administration to define conditions in which the local induction of inflammation is beneficial to the host because it improves the efficacy of vaccines and immunotherapies30.
Development of the protocol
Our labs initiated the very first systematic investigation of NANP recognition by immune cells25. In that work, as well as in our other related studies17,23,31–33, we used primary human PBMCs collected from healthy human donors. We specifically developed and optimized the experimental settings for this particular model because PBMCs have been shown to produce the most predictive and reliable results in regard to potential cytokine storm toxicity as compared to common preclinical animal models such as rodents and primates. We base this conclusion on the recent experience of TeGenero Immuno Therapeutics’ product TGN1412, which caused cytokine storm in patients after successfully passing all preclinical safety studies in rats and non-human primates. The cytokine storm toxicity of TGN1412, however, was predicted in vitro in PBMCs derived from healthy donors34. PBMCs have also proven to be predictive of the quality of vaccine adjuvants35,36. Consequently, we identified PBMCs as a sensitive and affordable model that can be used to understand how particular NANPs can trigger the immune system. We confirmed the reliable performance of this model for >60 different NANPs analyzed in PBMCs isolated from >100 healthy human donors17,23,25,26,31,33,37.
Advantages
The key advantage of this protocol is its modularity, which enables various NANPs to be studied together with a broad panel of induced cytokines assessed in clinically relevant settings. Other advantages include the ability to gain mechanistic insights into NANPs’ recognition by human immune cells, standardization of the most critical procedures for generating supernatants, and identification of appropriate controls, along with selection of critical biomarkers, while enabling individual researchers to choose the cytokine detection platform on the basis of the resources available in their labs. Therefore, we anticipate that this protocol can be applied to a broader range of nucleic acid–containing materials38–42.
Limitations
Although this Procedure is predictive of systemic effects of NANPs followed by injection into the blood, it does not assess the ability of NANPs to induce an inflammatory response in other cell types, which are responsible for the local responses to NANPs upon distribution to certain tissues. To analyze the response in other cell types (e.g., fibroblasts and adipocytes), this Procedure can be easily adapted by substituting another cell type of interest to the user for PBMCs. Likewise, to understand cytokine responses in PBMCs of patients with certain diseases, the Procedure can be adapted to PBMCs derived from whole blood of patients. Institutional review board and biosafety requirements would need to be considered for this adjustment, as discussed in the ‘Applications’ section.
Overview of the procedure
Based on extensive experimental work over the course of the past 6 years17,23,25,26,31,33, we introduce the comprehensive Procedure required to accurately assess the critical biomarkers of immunostimulation, as well as cells and molecular pathways responsible for the NANPs’ immunorecognition. We detail the protocols for pyrogen-free assembly and characterization of representative NANPs, isolation and handling of human PBMCs, transfection of NANPs, and assessment of biomarkers of NANPs’ proinflammatory responses, including type I and III interferons (IFN-α, IFN-β, IFN-ω, and IFN-λ). These cytokine responses were confirmed to be consistent for a large cohort (~100) of healthy human donors, even when the described procedures were performed by different technicians who had never worked with NANPs before17,23,25,26,31,33.
In addition, a set of proinflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-8), also known as markers of pyrogenic responses, can be reproducibly measured in PBMCs using this protocol23,25. The regulated induction of proinflammatory cytokines and IFNs has become a powerful tool used in vaccines and immunotherapies43,44. However, excessive and uncontrolled production of cytokines, particularly TNF-α, may cause tissue necrosis at the site of injection45. Therefore, decreasing injection site reactions in patients is considered to be an essential safety goal46–50. The procedure, schematically summarized in Fig. 2, can potentially be used to detect any other cytokine in response to PBMC treatment with NANPs or other TNAs.
Fig. 2 |. Schematic representation of the experimental design required to assess the immunological properties of NANPs and their interactions with PBMCs.
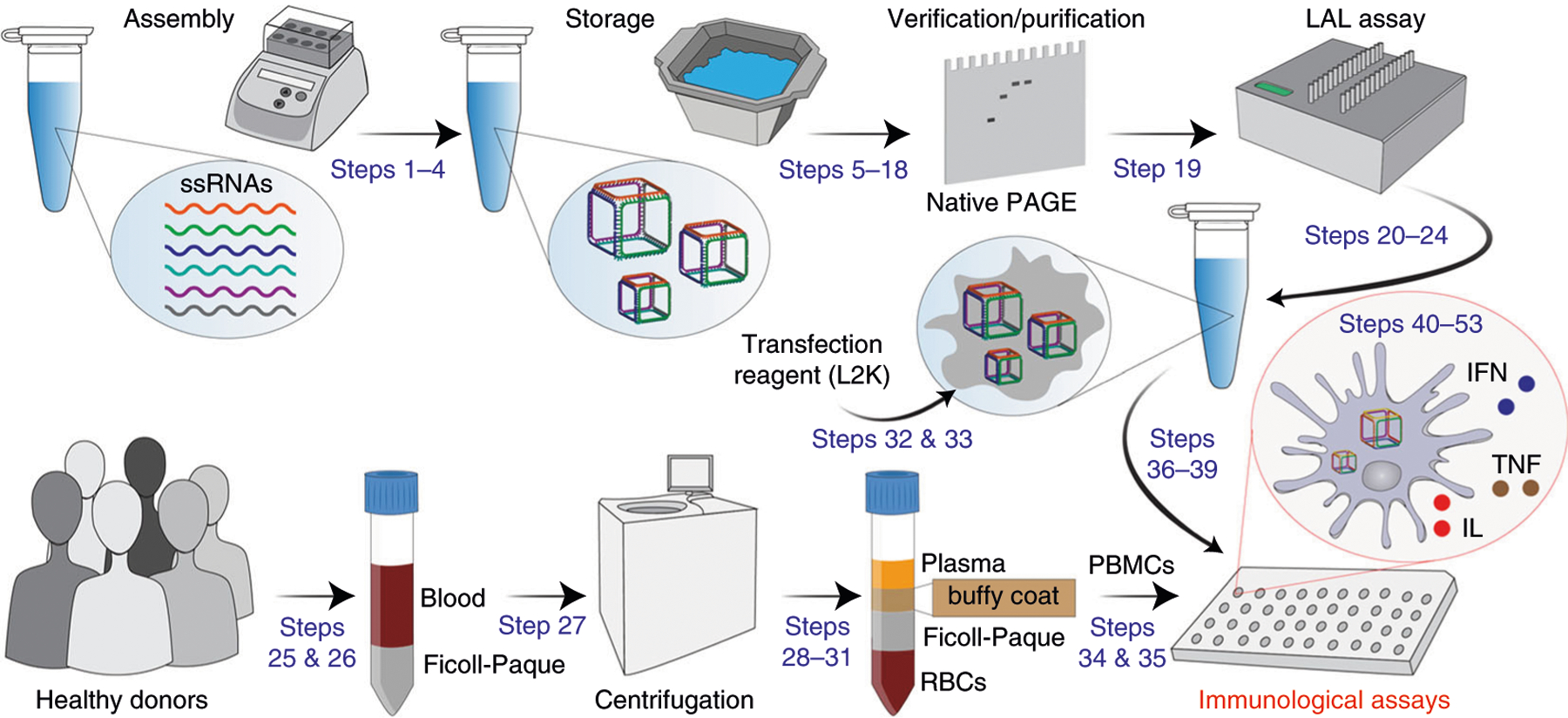
Corresponding Procedure steps are indicated in blue. Steps 54–57, which describe mechanistic studies, are not shown.
Applications
The empirically gained knowledge of relative immunostimulation contributes to establishing the safety profile of NANPs and opens endless possibilities for their use, not only as a drug delivery platform, but also as adjuvants for vaccines and immunotherapies. In addition, despite recent clinical successes (Onpattro51 and Givlaari52), the development of RNAi therapies has encountered several hurdles, of which severe inflammation and cytokine storm (https://pubmed.ncbi.nlm.nih.gov/32238921/) are not the least. The current protocol offers a quick and affordable preclinical tool aimed at addressing the gaps in the understanding of immunological recognition of NANPs and TNAs, thus further accelerating their clinical translation. The described experimental settings can be easily adopted and used by key stakeholders of NANP and TNA technologies—including the global community of basic researchers, regulatory scientists, international standard development organizations (e.g., ISO and ASTM International), biotechnology companies, big pharma, and environmental and occupational health scientists—to provide important information about NANP safety and, in relevant applications such as vaccines efficacy, to physicians, patients, and their families. If the user desires to investigate immunological responses to NANPs in the blood of patients with a particular disease (e.g., cancer), this protocol can be adapted and would require collection of blood from relevant donors under approval by the relevant institutional review board. Although the minimum recommended number of donors in the described protocol is three, one may increase the number of donors to that desirable for the given experiment.
Alternative methods
Human cell lines and engineered reporter cells (e.g., HEK-293 cells overexpressing human Toll-like receptor (TLR)3, TLR7, TLR8, and TLR9) can be used in preliminary studies to reveal common trends in immunostimulation by NANPs24,53,54. However, the cytokines’ pleiotropy55 and the genetic diversity of immune responses in human patients26 cannot be accurately addressed with these models, thus making their clinical relevance incomparable to studies with human PBMCs.
Experimental design
PBMCs
It is important to use freshly isolated PBMCs because cryopreservation procedures may alter biological responses of immune cells. For statistically meaningful results, we recommend using PMBCs isolated from at least three different donors; we also advise using at least three technical replicates per sample per donor. The number of donors can be increased to address specific needs of the user’s laboratory.
Controls
We recommend the use of RNA and DNA cubes (Fig. 1c) as controls for all immunological studies because RNA cubes reliably demonstrate the highest immunostimulatory activity among all tested NANPs25,26, whereas their DNA analogs trigger only minimal activation of cytokine production. We also recommend using RNA rings as controls. Together, these three NANPs represent alternative design strategies, chemical composition (RNA versus DNA), and dimensionality (globular cubes versus planar rings). The immunological profiles of these NANPs are well studied. We summarize general trends in Table 1. We suggest that Lipofectamine 2000 (L2K) be used as a standard carrier because it has been investigated most extensively, and a change in carrier may result in modified inflammatory response to NANPs.
Table 1 |.
Examples of IFN responses to NANPs with various physicochemical properties
| Amount detected (pg/ml) | ||||||
|---|---|---|---|---|---|---|
| Supernatant | Donor 1 | Donor 2 | Donor 3 | |||
| IFN-α | ||||||
| Untreated | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| ODN2216 | 29,287 | 28,034 | 16,963 | 15,605 | 15,179 | 13,193 |
| L2K | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| DNA cube/L2K | 4,889 | 4,602 | 2,375 | 2,262 | 4,198 | 5,121 |
| RNA cube/L2K | 25,530 | 19,965 | 17,564 | 17,204 | 14915 | 15,091 |
| RNA ring/L2K | 6,937 | 5,985 | 4,552 | 5,721 | 9,398 | 7,531 |
| IFN-β | ||||||
| Untreated | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| ODN2216 | 141 | 118 | 82 | 82 | 69 | BLOQ |
| L2K | BLOQ | BLOQ | BLOQ | BLOQ | 162 | BLOQ |
| DNA cube/L2K | BLOQ | BLOQ | BLOQ | BLOQ | 162 | BLOQ |
| RNA cube/L2K | 450 | 343 | 369 | 311 | 362 | 299 |
| RNA ring/L2K | 64 | BLOQ | 77 | BLOQ | 132 | BLOQ |
| IFN-ω | ||||||
| Untreated | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | 12.4 |
| ODN2216 | 1,254 | 1,209 | 738 | 774 | 633 | 607 |
| L2K | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| DNA cube/L2K | 201 | 202 | 118 | 105 | 214 | 231 |
| RNA cube/L2K | 875 | 730 | 623 | 587 | 599 | 686 |
| RNA ring/L2K | 194 | 223 | 132 | 178 | 290 | 291 |
| IFN-λ | ||||||
| Untreated | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| ODN2216 | 729 | 764 | BLOQ | BLOQ | BLOQ | BLOQ |
| L2K | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| DNA cube/L2K | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ | BLOQ |
| RNA cube/L2K | 887 | 698 | 382 | 417 | 600 | 482 |
| RNA ring/L2K | BLOQ | BLOQ | 146 | BLOQ | 147 | 146 |
BLOQ, below the lower limit of quantification. The data are reproduced with permission from ref.25, American Chemical Society. Each value in the table represents an independent sample tested in duplicate on an ELISA plate and is the mean of two responses (%CV <25).
Preparation of nucleic acid components
All DNAs, fluorescently labeled oligos, and RNAs can be purchased from Integrated DNA Technologies (IDT). Alternatively, RNAs can be synthesized via in vitro run-off transcription with T7 RNA polymerase, as detailed elsewhere7. If purchased, we recommend including a purification step (denaturing PAGE) for each strand, as detailed elsewhere7. In addition, chemically synthesized RNAs have 5′-OH groups, whereas in vitro transcribed RNAs contain 5′-triphosphates. This nuance needs to be considered before the immunological studies since different pattern recognition receptors can recognize these moieties within RNA.
NANP design and assembly
The design principles of the six-stranded cubes and rings used as control NANPs are detailed in a previous publication7. All sequences required for these NANPs assemblies are listed in Box 1. Importantly, because contamination with endotoxin (common for nanotechnology-based formulations28) can induce false-positive responses, the assembly of endotoxin-free NANPs must be confirmed7.
Box 1 |. Sequences of control nucleic acid nanoparticles.
The sequences of the NANPs we propose for use as controls have been described earlier25 and are listed below. Each set of NANP sequences contains a fluorescently labeled (with Alexa Fluor 488 (AF488)) strand for NANP visualization. All NANP sequences are custom-ordered from Integrated DNA
RNA cube 5′–3′
rA: GGCAACUUUGAUCCCUCGGUUUAGCGCCGGCCUUUUCUCCCACACUUUCACG
rB: GGGAAAUUUCGUGGUAGGUUUUGUUGCCCGUGUUUCUACGAUUACUUUGGUC
rC: GGACAUUUUCGAGACAGCAUUUUUUCCCGACCUUUGCGGAUUGUAUUUUAGG
rD: GGCGCUUUUGACCUUCUGCUUUAUGUCCCCUAUUUCUUAAUGACUUUUGGCC
rE: GGGAGAUUUAGUCAUUAAGUUUUACAAUCCGCUUUGUAAUCGUAGUUUGUGU
rF: GGGAUCUUUACCUACCACGUUUUGCUGUCUCGUUUGCAGAAGGUCUUUCCGA
rD-AF488: GGCGCUUUUGACCUUCUGCUUUAUGUCCCCUAUUUCUUAAUGACUUUUGGCC-AF488
DNA cube 5′–3′
dA: GGCAACTTTGATCCCTCGGTTTAGCGCCGGCCTTTTCTCCCACACTTTCACG
dB: GGGAAATTTCGTGGTAGGTTTTGTTGCCCGTGTTTCTACGATTACTTTGGTC
dC: GGACATTTTCGAGACAGCATTTTTTCCCGACCTTTGCGGATTGTATTTTAGG
dD: GGCGCTTTTGACCTTCTGCTTTATGTCCCCTATTTCTTAATGACTTTTGGCC
dE: GGGAGATTTAGTCATTAAGTTTTACAATCCGCTTTGTAATCGTAGTTTGTGT
dF: GGGATCTTTACCTACCACGTTTTGCTGTCTCGTTTGCAGAAGGTCTTTCCGA
dD-AF488: GGCGCTTTTGACCTTCTGCTTTATGTCCCCTATTTCTTAATGACTTTTGGCC-AF488
RNA ring 5′–3′
nrA: GGGAACCGUCCACUGGUUCCCGCUACGAGAGCCUGCCUCGUAGC
nrB: GGGAACCGCAGGCUGGUUCCCGCUACGAGAGAACGCCUCGUAGC
nrC: GGGAACCGCGUUCUGGUUCCCGCUACGAGACGUCUCCUCGUAGC
nrD: GGGAACCGAGACGUGGUUCCCGCUACGAGUCGUGGUCUCGUAGC
nrE: GGGAACCACCACGAGGUUCCCGCUACGAGAACCAUCCUCGUAGC
nrF: GGGAACCGAUGGUUGGUUCCCGCUACGAGAGUGGACCUCGUAGC
nrC-AF488: GGGAACCGCGUUCUGGUUCCCGCUACGAGACGUCUCCUCGUAGC-AF488
Cytokine detection
There is no harmonized approach for what cytokine detection platform to use, nor for the choice between single- and multiplex analysis. Protocol users should rely on their scientific judgment and the critical path of the project, focusing on particular types of nanoparticles to determine which types of cytokines and which method to use for analysis of supernatants. As an example, for this protocol, we show data generated using multiplex chemiluminescent assays (Quansys Biosciences). This protocol is used to detect type I and type III interferons (e.g., IFN-α, IFN-β, IFN-ω, and IFN-λ) when one needs to estimate the proinflammatory properties of NANPs because they stimulate these biomarkers according to their physicochemical properties and the responses are consistent between individual donors25. The detection of other proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6, and IL-8) is recommended when one wants to screen NANP formulations for their pyrogenic properties. The analysis with and without a carrier, and comparison with the benchmark carrier L2K are recommended because immunological responses may change depending on the physicochemical properties of the NANPs and the routes of particles’ entry into immune cells. The supernatants prepared in Steps 32–39 could also be used for the detection of any other biomarkers produced by PBMCs. For example, IL-2 and IFN-γ are the markers of T-cell activation and can be tested when the information regarding the ability of NANPs to activate T-lymphocytes is wanted. When the analysis of other cytokines and secondary messengers produced by PBMCs in response to NANPs is desired, Steps 1–31 are performed without modifications; in Steps 32–39, one may need to add an additional positive control (i.e., substance known to induce the biomarker of interest in PBMCs; some examples are provided in Table 2), and the incubation time may also need to be adjusted from 24 h to a shorter (e.g., 6 or 8 h) or longer (e.g., 48 or 72 h) duration, depending on the current knowledge about that particular biomarker.
Table 2 |.
Positive Controls to verify functionality of the test model
| Description/control | E.coli K12 LPS | ODN2216 | PHA-M |
|---|---|---|---|
| Primary purpose | Positive control for Inflammatory cytokines (TNF-α, IL1β, IL-6, IL-8, IL-10, IL-12) | Positive control for Type I interferons (IFN-α, IFN-β, IFN-ω) and type III (IFN-λ) | Positive control for Type II interferon (IFN-γ) |
| Final concentration in assay | 20 ng/mL | 5 μg/mL | 10 μg/mL |
Positive controls shown in this table trigger different receptors and molecular pathways in PBMCs and therefore lead to the expression of different inflammatory markers. These controls provide important information about the functionality of the cells used in the study. More details are in Step 35.
Any traditional single- or multiplex ELISA assay can be used. In the Materials section, we provide details about both commercial ELISA assays and low-cost, self-assembled single-plex ELISA kits that researchers can choose to use on the basis of their resources and the laboratory equipment available. Owing to the differences in ranges and detection limits, one should follow the instructions from the cytokine detection kit to prepare a standard curve and sample dilution, and follow the instructions for the reagent dilution and incubation time. Detailed protocols for self-assembled single-plex ELISA validated according to the International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use have been described by our labs earlier56; these protocols provide tenfold reduction in cost per plate, thereby making these ELISAs more accessible to researchers with limited resources.
Understanding the mechanism of NANP recognition
To determine whether endosomal TLRs are involved in the inflammatory response to NANPs, one can use ODN2088 at a final concentration of 5 μg/ml. This inhibitor is added to the RPMI in Steps 35 and 55. The results generated for NANPs alone and NANPs with ODN2088 are compared. To determine whether the uptake of NANPs requires scavenger receptors, one can use specific inhibitors such as fucoidan. This inhibitor is added to the cells in Step 35. In addition to or instead of fucoidan, one can also use poly-I or dextran sulfate as inhibitors of scavenger receptors. Poly-C and chondroitin sulfate are used as nonspecific controls for poly-I and dextran sulfate, respectively.
Materials
Biological materials
Healthy human donor blood anti-coagulated with lithium (Li)–heparin, obtained from at least 3 healthy donors. Collect whole blood from healthy donor volunteers who have not been on medication and have been clear of infection for at least 2 weeks before blood donation. Use Li-heparin tubes and discard the first 10 cc. For the best results, whole blood should be used within 1 h after collection. Prolonged storage (> 2 h) of whole blood will lead to a decrease in cell function ! CAUTION The blood used in this protocol was obtained under the National Cancer Institute-at-Frederick protocol OH9-CN046; equivalent approval from the user’s home institution is required to work with human blood. Collection of blood requires institutional approval and certified personnel ▲CRITICAL Blood from donors with certain types of diseases (e.g., cancer) can be used with appropriate institutional approval. ▲CRITICAL The use of cryopreserved blood or PBMCs is not recommended because cytokine response in these cells may be altered by the cryopreservation and storage procedures.
Reagents
DNA and RNA strands (Box 1; custom-ordered from IDT (https://www.idtdna.com) or an equivalent supplier)
HyClone HyPure Water (cell-culture grade; GE Life Sciences, cat. no. SH30529.01)
Acrylamide (acrylamide–bisacrylamide (37.5:1; 40% (vol/vol); VWR, cat. no. 218 Q31Q32 97064–542) ! CAUTION Acrylamide–bisacrylamide is toxic if ingested or absorbed through the skin.
Glycerol (Life Technologies, cat. no. 5514UA)
Xylene cyanol (Sigma, cat. no. X-4126) ! CAUTION Avoid contact with and inhalation of xylene cyanol,
Bromophenol blue (Sigma, cat. no. B-8026) ! CAUTION Avoid contact with and inhalation of bromophenol blue.z
Trypan blue
Magnesium chloride (MgCl2; Fisher, cat. no. M35–500)
Tris–borate (TB) buffer (10×; Sigma, cat. no. T1503–5KG; or VWR, cat. no. 97061–978)
Triton X-100 (VWR, cat. no. 97062–208)
Lipopolysaccharide (LPS) from K12 Escherichia coli (ultrapure; InvivoGen, cat. no. tlrl-peklps)
ODN2216 (a CpG DNA oligonucleotide with mixed backbone and the following sequence 5ʹ-gg GGGACGATCGTCGggggG-3ʹ, where lowercase letters show phosphorothioate linkage and capital letters refer to phosphodiester linkage between nucleotides; InvivoGen, cat. no. tlrl-2216)
Phytohemagglutinin (PHA-M; Sigma, cat. no.L8902)
ODN2088 (a CpG oligonucleotide that inhibits activity of endosomal TLRs; Miltenyi Biotec, cat. no. 130-105-815)
Fucoidan (inhibitor of scavenger receptor; Sigma-Aldrich, cat. no. F8190–500MG)
Dextran sulfate (inhibitor of scavenger receptor; Sigma-Aldrich, cat. no. D6001–1G)
Chondroitin sulfate (control for dextran sulfate; Sigma-Aldrich, cat. no. C9819–5G)
Poly-I (scavenger receptor inhibitor; Sigma-Aldrich, cat. no. P4154)
Poly-C, control for poly I (Sigma-Aldrich, cat. no.P4903)
Phosphate-buffered saline (PBS; GE Life Sciences, cat. no. SH30256.01)
RPMI 1640 (Invitrogen, cat. no. 11835–055)
Fetal bovine serum (FBS; HyClone; GE Life Sciences, cat. no. SH30070.03)
Penicillin–streptomycin (Invitrogen, cat. no.15140–148)
l-Glutamine (GE Life Sciences, cat. no. SH30034.01)
Ficoll-Paque Premium (GE Healthcare, cat. no.17-5442-02)
Hank’s Balanced Salt Solution (HBSS; Invitrogen, cat. no. 24020–117)
OptiMEM (Thermo Fisher, cat. no. 31985062)
Lipofectamine 2000 (L2K; Thermo Fisher, cat. no.11668019)
(Optional) Heparin (Sigma, cat. no. H3393)
(Optional) Ethidium bromide for total gel staining (0.5 μg/mL; VWR, cat. no. 97062–736)
(Optional) SYBR Green (Invitrogen, cat. no. S7564)
(Optional) Fluorescently labeled oligonucleotides (IDT, custom synthesis)
Potassium chloride (KCl; Sigma, cat. no. P9333)
(Optional) Cavicide (Metrex, cat.no. 13–1002)
ELISA kits
Custom 4-plex type I and III interferon assays (Quansys Biosciences) are recommended for labs that have chemiluminescence imaging multiplex plate readers.
(Optional) Single- or multiplex ELISA kit that enables measurement of TNF-α, IL-1β, IL-6, IL-8, IL-2, IL-12, IFN-α, IFN-β, IFN-ω and IFN-λ (Q-Plex Human Cytokine–Inflammation kit (e.g., Quansys Biosciences, cat. no. 110433HU). ▲CRITICAL The Procedure detailed in this protocol is for the multiplex IFN kit from Quansys. The volumes, incubation times, incubation temperatures, and type and settings of plate reader may vary when kits from other manufacturers are used.
(Optional) A multiplex cytokine and type II interferon (IFN-γ) panel (MSD, cat. no. K15049D) and custom type I and type III panels (e.g., Quansys Biosciences) are recommended for labs that have fluorescence- and luminescence-based multiplex readers, respectively.
(Optional) ELISA kits (R&D Systems, cat. nos. DY9345–05, DIFNB0, D6050, and DIA00D) and low-cost self-assembled ELISAs (e.g., ITA-22, ITA-23, ITA-24 and ITA-25, https://ncl.cancer.gov/resources/assay-cascade-protocols) are recommended for labs that have regular single-plex readers capable of detecting absorbance at 450 nm.
Equipment
Lithium (Li)–heparin tubes
Heating blocks with heated lids
Vortex mixer
Ice machine
Short-wave UV lamp
Tube rotator
Vertical electrophoresis system for native-PAGE
UV spectrophotometer (Thermo Scientific, model no. NanoDrop 2000)
Imaging system (Bio-Rad, ChemiDoc MP model)
Thin-layer chromatography (TLC) plate
Scalpel
Cutting board
Plastic wrap
Refrigerated microcentrifuge
Pipettes in a range from 0.05 to 10 mL
U-bottom cell culture–grade 96-well plates (VWR, cat. no. 10861–668)
Polypropylene tubes, 50 and 15 mL
Microcentrifuge tubes
Centrifuge
Refrigerator, 2–8 °C
Freezer, −20 °C
Cell culture incubator with 5% CO2 and 95% humidity
Biosafety cabinet approved for biosafety level 2 (BSL-2) handling of biological materials
Inverted microscope
Hemocytometer
Plate reader for cytokine detection (Quansys Biosciences ImagePro multiplex reader or Molecular Devices SpectraMax M5 reader for single-plex ELISA)
Mini-PROTEAN Tetra Vertical Electrophoresis Cells (Bio-Rad)
Prism 8 (GraphPad)
Reagent setup
▲CRITICAL Box 2 provides strategies for avoiding contamination of NANPs with pyrogens.
Box 2 |. How to avoid contamination of NANPs with pyrogens.
Always wear disposable gloves (nitrile preferred) and avoid reusing them.
Never touch your face or unclean surfaces; change gloves as needed during NANP preparation.
Use pyrogen-free reagents and water.
Use autoclaved sterile pipette tips and avoid any cross-contamination with other samples.
Do not use cellulose-based filters, which can be a source of beta-glucan that will interfere with the LAL (Limulus amebocyte lysate) assay and further immunological studies.
Do not breathe, talk, cough, or sneeze around the open tubes during the preparation of the NANPs.
5× NANP assembly buffer
5× NANP assembly buffer is 5× TB buffer, 10 mM MgCl2, and 250 mM KCl. Once made, this buffer can be stored at room temperature (20–22 °C) for 2–3 months.
1× native-PAGE loading buffer
1× native-PAGE loading buffer is 50% (vol/vol) glycerol, 2× NANP assembly buffer, 0.01% (vol/vol) bromophenol blue, and 0.01% (vol/vol) xylene cyanol tracking dyes. Once made, this buffer can be stored at room temperature for 1 year.
1× native-PAGE running buffer
1× native-PAGE running buffer is 1× TB buffer, 2 mM MgCl2. Once made, this buffer can be stored at 4 °C for 1 year.
Complete RPMI 1640 medium
The complete RPMI 1640 medium should contain 10% (vol/vol; heat inactivated) FBS, 2 mM l-glutamine, and 100 U/mL penicillin–streptomycin. Store at 2–8 °C protected from light for no longer than 1 month. Before use, warm the medium to 37 °C in a water bath.
Lipopolysaccharide for positive control in proinflammatory cytokine and chemokine analysis
To make a 1-mg/mL stock of LPS, add 1 mL of sterile water to 1 mg of LPS in the vial and vortex to mix. Make 20-μL aliquots and store at a nominal temperature of −20 °C. Avoid repeated freeze–thaw cycles. On the day of the experiment, thaw one aliquot and use it such that its final concentration in PBMCs culture is 20 ng/mL.
Phytohemagglutinin for positive control in type II interferon analysis
To make a 1-mg/mL stock of PHA-M, add 1 mL of sterile PBS or cell culture medium per 1 mg of PHA-M to the vial and gently rotate to mix. Store daily-use aliquots at a nominal temperature of −20 °C. Avoid repeated freeze–thaw cycles. On the day of the experiment, dilute the stock PHA-M solution in cell culture medium so that its final concentration in the positive control sample is 10 μg/mL.
ODN2216 for positive control in type I and type III interferon analysis
This mixed-backbone oligonucleotide activates TLR9 and is supplied as a lyophilized powder. Reconstitute the powder in pyrogen-free, nuclease-free water to a final concentration of 1 mg/mL. Prepare single-use, 5-μL aliquots and store at −20 °C. On the day of the experiment, thaw an aliquot at room temperature and dilute it in culture medium so that its final concentration in the test sample is 5 μg/mL.
ODN2088 for mechanistic study to reveal potential involvement of endosomal TLRs
This oligonucleotide is a pan-TLR inhibitor; it is supplied as a lyophilized powder. Reconstitute the powder in pyrogen-free, nuclease-free water to a final concentration of 1 mg/mL. Prepare single-use, 5-μL aliquots and store at −20 °C. On the day of the experiment, thaw an aliquot at room temperature and dilute it in culture medium so that its final concentration in the test sample is 5 μg/mL.
Fucoidan, dextran sulfate, poly-I, chondroitin sulfate, and poly-C preparation for mechanistic studies
Each of these materials is supplied as a lyophilized powder. Dissolve the powder in PBS to prepare a stock with a nominal concentration of 1 mg/mL. Prepare single-use 5-μL aliquots and store them at −20 °C. On the day of the experiment, thaw aliquots at room temperature and dilute them in culture medium so that the final concentration of each in the test sample is 50 μg/mL. Fucoidan, poly-I, and dextran sulfate inhibit scavenger receptors, whereas chondroitin sulfate and poly-C do not. Chondroitin sulfate and poly-C are used as controls for dextran sulfate and poly-I, respectively.
Heat-inactivated fetal bovine serum
Thaw a 50-mL aliquot of FBS and equilibrate it to room temperature. Place the tube in a water bath set to 56 °C and incubate with mixing for 35 min. The heat inactivation takes 30 min, and the initial 5 min is used to bring the entire contents of the vial to 56 °C. Chill the serum and use it to prepare complete RPMI 1640 medium.
Procedure
Assembly of NANPs ● Timing 1 h
-
1
Measure the concentrations of solutions of individual RNA (or DNA) strands required for NANP assembly (Box 1; ref.7) with a NanoDrop 2000 UV spectrophotometer; the extinction coefficients of individual strands are calculated using the IDT tools (https://www.idtdna.com/calc/analyzer).
-
2
Specify the final concentration of NANPs and mix individual strands at equimolar concentrations in pyrogen-free water (e.g., Lonza LAL-grade water).
-
3
Place the tubes with samples on a heating block (95 °C) and incubate for 2 min.
▲CRITICAL STEP Do not incubate the samples at 95 °C for longer than 2 min; avoid adding the assembly buffer to the hot samples because it can promote the degradation of RNAs.
▲CRITICAL STEP We recommend the use of heating blocks with heated lids to avoid condensation and changes in the final concentration of NANPs.
-
4
For intramolecular NANP assemblies (e.g., RNA and DNA cubes), snap-cool the samples to 45 °C and incubate for 2 min, add 5× NANP assembly buffer to reach the appropriate concentration, and incubate at 45 °C for an additional 30 min; for intra/intermolecular NANP assemblies (e.g., RNA rings), snap-cool the samples on ice for 2 min, add 5× NANP assembly buffer, and incubate for 30 min at 30 °C.
■PAUSE POINT Once assembled, NANPs can be stored at 4 °C for several weeks. However, continuous execution of the protocol is recommended to avoid any potential degradation of the NANPs
Verification of NANP assemblies by native-PAGE ● Timing ≥2 h
-
5
Pre-cast a mini gel for non-denaturing polyacrylamide gel electrophoresis (native-PAGE) (8% (vol/vol) acrylamide (37.5:1), 1× TB buffer, 2 mM MgCl2). We recommend using Mini-PROTEAN Tetra Vertical Electrophoresis Cells.
-
6
Pre-run the gel in a cold room or refrigerator (4 °C) in 1× native-PAGE running buffer for 3–5 min at 150 V.
-
7
While pre-running the gel, mix NANPs (final concentration of ≥1 μM) with equal volumes of 1× native-PAGE loading buffer, up to a 10-μL of final volume.
-
8
Load samples into individual lanes of a gel (5 μL per lane) and run the system for 30 min at 300 V at 4 °C.
▲CRITICAL STEP Wash the loading wells several times with running buffer before loading.
-
9
Visualize the gel with a ChemiDoc MP System (or similar) using total staining with ethidium bromide (or SYBR Green) or fluorescently labeled oligonucleotides entering the NANPs’ composition. NANPs are expected to migrate as a single band on non-denaturing gels.
▲CRITICAL STEP The presence of any other bands should, in total, constitute no more than 20% of the entire band percentage as determined by following the manufacturer’s instructions for the ChemiDoc analysis software.
? TROUBLESHOOTING
Purification of NANPs by native-PAGE ● Timing ≥12 h
-
10
If—after Step 9—the NANPs need to be further purified, prepare larger volumes and achieve a ≥10 μM concentration by following Steps 1–4 above.
-
11
Pre-cast a gel for non-denaturing polyacrylamide gel electrophoresis (native-PAGE) (8% (vol/vol) acrylamide, 37.5:1). For purification, we recommend using a vertical gel of 16.5 cm × 22 cm with a spacer thickness of 1.5 mm for 10 wells.
-
12
Pre-run the gel in a cold room or refrigerator (4 °C) in 1× native-PAGE running buffer for 10 min at 150 V.
-
13
While pre-running the gel, mix NANPs with equal volumes of 1× native-PAGE loading buffer, up to a 100-μL final volume.
-
14
Load samples into individual lanes of the gel (100 μL per lane) and run it for 90 min at 300 V at 4 °C.
-
15
Disassemble gel and place it into plastic wrap.
-
16
Place the TLC plate underneath the plastic-wrapped gel and visualize the NANP bands with a UV lamp on the short-wavelength setting.
-
17
Use a scalpel to cut out the major NANP band in each lane and place them into separate aliquots of 1× NANP assembly buffer; use as much buffer as needed to completely cover the gel pieces.
-
18
Allow elution overnight; then assess the concentration of the purified NANPs with a NanoDrop 2000 UV spectrophotometer; the extinction coefficients of NANPs are calculated as the sum of extinction coefficients of the individual sequences that comprise them.
-
19
Confirm the absence of endotoxins in the assembled NANPs by LAL assay, using protocols developed by our groups7.
■PAUSE POINT Once purified, the NANPs can be stored at 4 °C for several weeks. However, continuous execution of the protocol is recommended to avoid any potential degradation of the NANPs.
? TROUBLESHOOTING
Assessment of structural integrity of the NANPs upon complexation with delivery reagents
● Timing ~2 h
-
20
Complex the NANPs with a carrier (Fig. 3a,b; refs.24,25,53. For complexation with L2K in a separate tube, combine 10 μL of NANP stock (1 μM concentration) and 2 μL of L2K; mix well by pipetting up and down repeatedly.
-
21
Incubate at room temperature for 30 min.
-
22
Take a 6-μL aliquot of NANP–L2K solution and add 2 μL of 10% (vol/vol) commercial Triton X-100.The remaining 6 μL of NANP–L2K solution will be used as a control in Step 24 and should be incubated at room temperature until then.
? TROUBLESHOOTING
-
23
Incubate the solution at room temperature for an additional 30 min.
-
24
Analyze all samples (NANPs, NANP–L2K, and NANP–L2K + Triton X-100 solutions) on native-PAGE as described in Steps 5–9. NANP–L2K solution is expected to stay in the loading well, whereas Triton X-100–treated NANP–L2K solution will migrate in the gel according to its molecular weight, as would free, untreated NANPs; the structural integrity of the NANPs, therefore, is confirmed by comparing the free NANPs with the NANP–L2K and NANP–L2K + Triton X-100 samples.
■PAUSE POINT Once mixed with L2K, NANPs can be stored at 4 °C for hours. However, continuous execution of the protocol is recommended to avoid any potential aggregation.
Fig. 3 |. Verification that NANPs retain structural integrity upon complexation with Lipofectamine 2000, and of their cellular uptake.
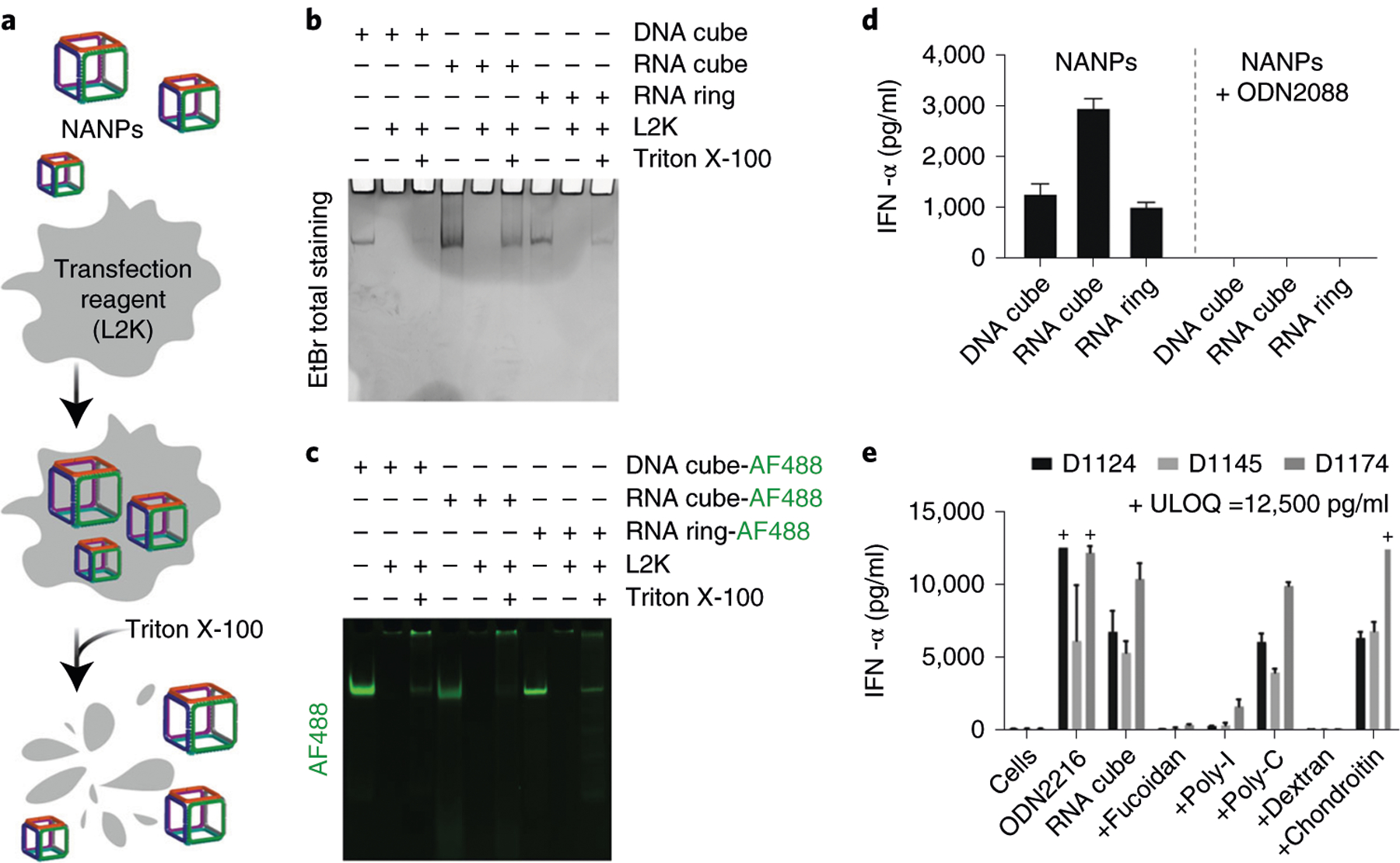
a, Schematic representation of NANPs’ association with L2K, followed by their release upon detergent (Triton X-100) treatment. b, Ethidium bromide (EtBr) total staining native PAGE, indicating formation of NANPs, their complexation with L2K, and successful release upon treatment with Triton X-100. c, NANPs labeled with Alexa Fluor 488 (AF488) form complexes with L2K and retain their structural integrity upon detergent-mediated release. Note that in b and c, NANPs’ complexation with L2K prevents their entering the gel, whereas treatment with Triton X-100 restores NANPs’ electrophoretic mobility. d, Inhibition of NANPs’ inflammatory response due to TLR recognition. ODN2088 is the oligonucleotide that blocks the function of all endosomal TLRs. Each bar shows a mean response of three independent samples (N) and a standard deviation (N = 3) for each of three donors. e, Inhibition of the inflammatory response due to NANP uptake via scavenger receptor. ODN2216 is an oligonucleotide known to stimulate the IFN response and is used as a positive control; fucoidan, poly-I and dextran are known inhibitors of the scavenger receptor, whereas poly-C and chondroitin are non-specific (i.e., non-inhibitory) controls for poly-I and dextran, respectively. Each bar shows a mean response of three independent samples and a standard deviation (N = 3) for each of three donors. Each sample was tested in duplicate on an ELISA plate (%CV <25). The data used in d and e are reproduced with permission from ref.25, American Chemical Society. ODN2208 is a CpG DNA oligonucleotide with mixed backbone; ODN 2216 is a CpG oligonucleotide and TLR9 agonist; fucoidan is a complex polysaccharide; poly-I is polyinosinic acid; poly-C is polycytidylic acid; chondroitin is a glycosaminoglycan; dextran is a complex branched glucan. D, donor number; ULOQ, upper limit of quantification.
Isolation of PBMCs ● Timing ~4 h
▲CRITICAL Human blood may contain blood-borne pathogens. Therefore, Biosafety in Microbiological and Biomedical Laboratories57 recommends following BSL-2 precautions during blood work. Laboratory staff should wear pants, close-toed shoes, disposable gloves, a laboratory coat, and eye protection. In addition, all procedures with human blood in which splashes or aerosols may be created should be conducted inside a biological safety cabinet. Blood-borne pathogen awareness training may be required and is strongly recommended. Any laboratory personnel working with human-derived blood should refer to the relevant national and regional guidelines and regulations for handling of blood-borne pathogens for specific required precautions. Always follow your local and institutional policies for working with human blood and, in case of any concerns or questions, contact your biosafety officer or institutional biosafety committee. Obtaining blood from patients with certain types of diseases also requires institutional review board approval.
-
25
Place freshly drawn blood into 15- or 50-mL conical centrifuge tubes separated by donor. Add an equal volume of room-temperature PBS and mix well.
-
26
Use 3 mL of Ficoll-Paque solution per 4 mL of blood–PBS mixture, e.g., 15 mL of Ficoll-Paque per 20 mL of diluted blood in a 50-mL tube. Slowly layer the blood–PBS mixture over the Ficoll-Paque solution. Alternatively, the Ficoll-Paque solution can be slowly layered underneath the blood–PBS mixture by placing the tip of the pipette containing Ficoll-Paque at the bottom of the blood sample tube.
▲CRITICAL STEP To maintain the Ficoll-Paque–blood interface, hold the tube at a 45° angle.
-
27
Centrifuge for 30 min at 900g at 18–20 °C, without braking.
▲CRITICAL STEP Turning off the brakes on the centrifuge is needed to avoid altering the gradient, which would result in the loss of the buffy coat. Depending on the type of centrifuge, one may also need to set the acceleration speed to the minimum.
-
28
Using a sterile pipette, remove the upper layer containing plasma and platelets and discard it. Using a fresh sterile pipette, transfer the mononuclear cell layers to fresh 15- or 50-mL centrifuge tubes (separated by donor).
-
29
Wash the cells by adding an excess (~3 times the volume of mononuclear layer) of HBSS and centrifuging for 10 min at 400g at 18–20 °C.
▲CRITICAL STEP The removal of excess Ficoll-Paque and platelet-rich plasma collected along with the PBMC fraction is critical to provide optimal cell viability and biological response. Usually 4 mL of blood–PBS mixture results in an ~2 mL fraction containing the cells of interest and requires at least 6 mL of HBSS for the wash step. We use 10 mL of HBSS per each 2 mL of cells.
-
30
Discard the supernatant by pipetting or using vacuum aspiration and repeat the wash step one more time.
-
31
Resuspend the cells in 1 mL of complete RPMI 1640 medium. Dilute the cells 1:5 or 1:10 with trypan blue, count the cells, and determine their viability using trypan blue exclusion58. If viability is ≥90%, continue to next steps.
? TROUBLESHOOTING
Exposing PBMCs to NANPs and collecting supernatants ● Timing ~24 h
-
32
Complex NANPs with a carrier. For complexation with L2K, in a separate tube, combine 20 μL of 1 μM NANP stock and 4 μL of L2K (for a 96-well plate; adjust the volume on the basis of the manufacturer’s recommendations if needed). Mix well by pipetting up and down repeatedly.
-
33
Incubate at room temperature for 5–30 min, then add 376 μL of OptiMEM.
-
34
While waiting for complexation in Step 32, adjust the PBMC concentration to 1.3 × 106 viable cells/mL with complete RPMI 1640 medium.
-
35
Dispense 20 μL of complete RPMI 1640 medium (baseline), negative control (PBS), vehicle control (L2K in OptiMEM at the same concentration as that used for particle complexation), positive controls (PCs) and test samples (NANPs from Step 32) into the corresponding wells of a U-bottom 96-well plate. We advise doubling the number of wells with NANPs in order to prepare cell-free controls (CFCs) in Step 36. We advise setting up 4–6 extra replicates of positive controls. Supernatants collected from these extra PC samples will be used to prepare inhibition enhancement controls (IECs) in ELISA assays (Steps 40–53) to identify potential false-negative results. See Supplementary Fig. 1 for a diagram of the plate layout. Refer to Table 2 for information regarding positive controls and Table 3 for information regarding IECs and CFCs.
▲CRITICAL STEP If one wants to understand whether NANPs may potentiate or inhibit the cellular response to the assay positive control (LPS, PHA-M, or ODN2216), wells should also be set up to co-culture the positive control with NANPs in the presence of cells. In this case, each test well will receive 20 μL of the positive control, 20 μL of NANPs, 20 μL of complete RPMI 1640 medium and 40 μL of PBMCs from Step 34 (to contain 2.6 × 106 viable cells/mL).
-
36
Dispense 80 μL of PBMCs from Step 34 per well into the 96-well plate containing 20 μL per well of NANPs, controls, or medium in wells intended for CFC. Add 80 μL of complete RPMI 1640 medium instead of PBMC suspension to the CFC wells. The final number of cells per well is 100,000, and the total volume per well is 100 μL.
-
37
Repeat Steps 25–36 for cells obtained from each individual donor.
▲CRITICAL STEP There is no limit to the number of donors used in this test. We advise testing each NANP formulation with blood derived from at least three healthy donors.
-
38
Incubate the cells for 20 h in a humidified 37 °C, 5% CO2 incubator.
-
39
Spin the plate in a centrifuge at 400–700g for 10 min at room temperature. Transfer the supernatants to a fresh plate. Alternatively, the supernatants can be collected into Eppendorf tubes and cleared of cells by a brief centrifugation (18,000g, 20–24 °C, 5 min) and then transferred to fresh tubes for storage.
▲CRITICAL STEP CFCs from Step 35 are processed the same way as PBMC samples and serve as a control for false-positive results. To test for potential false-negative results, supernatants from the positive control (Step 39) are spiked with NANPs at a final concentration identical to that of the test sample. Alternatively, supernatants from CFCs are spiked with relevant cytokine standards used in an ELISA or multiplex assay. If NANPs inhibit detection of cytokine, a decrease in the cytokine level will be seen as compared to the level of cytokine in the positive control or quality control.
▲CRITICAL STEP If the user plans on using multiple ELISA assays, the volumes described in the protocol can be scaled up proportionally to generate higher volumes of supernatants. In this case, to avoid repeated freeze–thaw cycles, it is better to prepare multiple, single-use aliquots of each supernatant. The size of such aliquots depends on the volume and sample dilution that are specific to a given ELISA.
■PAUSE POINT After Step 39, one can either proceed with ELISA analysis or store supernatants in aliquots at −20 or −80 °C for up to 1 year.
? TROUBLESHOOTING
Table 3 |.
Controls to test for potential false-negative or false-positive results
| Description/control | IEC | CFC |
|---|---|---|
| Primary purpose | Rule out false-negative results | Rule out false-positive results |
| Content | Positive-control supernatant from Step 39 spiked with NANPs | Complete cell culture medium and NANPs. See Step 35 for the details of preparation |
| CFC supernatant from Step 39 spiked with cytokine standards from ELISA kit |
Inhibition enhancement control (IEC) and cell-free control (CFC) help reveal the ability of NANPs present in the culture supernatants to alter cytokine detection and cause false-positive or false-negative results. More details are in Step 35.
Detection of biomarkers ● Timing ~4–6 h
-
40
Prepare the assay diluent, sample diluent, and wash buffer from the 4-plex interferon kit (Quansys Biosciences). Dilute the stock wash buffer provided with the kit 20-fold with distilled water. The assay and sample diluents are already at the ready-to-use concentrations.
▲CRITICAL STEP Store all buffers at room temperature for use on the same day that they are prepared.
-
41
Prepare calibration standards. First, add 200 μL of the stock calibrator (from the kit) to the first tube, labeled Standard or Calibrator 1. Next, add 120 μL of the assay diluent to the additional six tubes labeled sequentially as Standard or Calibrator 2 through 7. After that, perform serial, threefold dilution of the first standard by transferring 60 μL of Standard 1 to the Standard 2 tube, mixing by repeated up-and-down pipetting, and transferring 60 μL to the next tube, and so on until Standard 7 is reached.
-
42
Thaw frozen culture supernatants from Step 39 at room temperature or use freshly collected ones. To prepare IECs, spike PC supernatants with NANP–L2K solution prepared as described in Steps 20 and 21 and diluted with complete RPMI 1640 medium to achieve 2× the final concentration used to prepare NANP–L2K culture supernatants (e.g., if the final concentration tested in cells was 10 nM, then dilute to 20 nM). Use 30 μL of NANP–L2K for each 30 μL of PC supernatant. Alternatively, use CFC supernatant from Step 39 to prepare calibration standard 3 or 4 (i.e., the one that has a cytokine concentration falling in the middle of calibration curve).
-
43
Dilute the culture supernatants twofold with sample diluent by mixing 60 μL of the culture supernatants with 60 μL of the sample diluent.
-
44
Add 50 μL of the assay diluent to each well on a multiplex ELISA plate.
-
45
Load 50 μL of standards from Step 41 and culture supernatants from Step 43 per well onto the 96-well multiplex plate loaded with assay diluent in Step 44 and incubate it at room temperature on a shaker set to ~500 r.p.m. for 2 h.
-
46
Wash the plate three times with the wash buffer prepared in Step 40. Use 300 μL of the wash buffer per well. Tap the plate on a paper towel to remove excess buffer and immediately proceed to the next step.
-
47
Add 50 μL per well of the detection mix, prepared by the reconstitution of lyophilized stock (from the 4-plex interferon kit, Quansys Biosciences) in 6 mL of distilled water.
-
48
Incubate the plate at room temperature on a shaker set to ~500 r.p.m. for 1 h.
-
49
Repeat Step 46.
-
50
Add 50 μL per well of the ready-to-use streptavidin–HRP (horseradish peroxidase) conjugate provided with the kit.
-
51
Incubate the plate at room temperature on a shaker set to ~500 r.p.m. for 15 min.
-
52
Repeat Step 46 two times and immediately proceed to the next step.
-
53
Add 50 μL per well of the detection reagent prepared by mixing equal volumes of detection reagent A and detection reagent B, and read the plate using a Quansys ImagePro reader equipped with Q-Viewv.3.112 or equivalent software. Process and analyze the data with Prism 8 or equivalent software.
▲CRITICAL STEP Analyze the supernatants from CFC samples prepared in Step 39 to account for potential false-positive responses. Analyze IECs from Step 42. This step will account for potential false-negative results.
▲CRITICAL STEP When analyzing the data, do not forget to account for the twofold dilutions of all study samples and the fourfold dilution for the IEC.
? TROUBLESHOOTING
Mechanistic insight into NANP uptake routes and immune receptor recognition
● Timing ≥24 h to prepare culture supernatants; 4–6 h for ELISA analysis
-
54
Follow the same procedure described in Steps 32–34 to set up culture supernatants.
-
55
When setting up treatments in Step 35, add additional samples containing NANPs and inhibitors as follows: If the involvement of TLRs is of interest, add ODN2088 to the cells before adding NANPs. If the involvement of scavenger receptors is of interest, then apply fucoidan, dextran sulfate, chondroitin sulfate, poly-I, and poly-C to separate wells and then add NANPs.
-
56
Complete the Procedure by following Steps 36–39.
-
57
Follow Steps 40–53 to analyze the culture supernatants prepared in Steps 54–57.
■PAUSE POINT After Step 56 is complete, one can either proceed with ELISA analysis or store aliquots of the supernatants at −20 or −80 °C.
? TROUBLESHOOTING
Troubleshooting
Troubleshooting advice can be found in Table 4.
Table 4 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 9 | NANP bands are not seen on total staining native-PAGE gel | Absence of divalent ions (Mg2+) | Verify that Mg2+ was added to NANP assembly buffer, native-PAGE gel, and native-PAGE running buffer |
| Additional bands besides the NANP band are observed on native-PAGE gel | Individual monomers (ssRNAs and ssDNAs) tend to stick to the test-tube walls | Vortex all stock solutions of individual monomers for 5–10 s before NANP assembly | |
| Monomer concentrations were not measured correctly | Measure the UV absorbance and calculate the monomer concentrations right before NANP assembly | ||
| NANPs stuck in the native-PAGE wells | Loading wells were not washed well enough | Wash the wells thoroughly before loading your samples | |
| 19 | NANPs contain endotoxin | Pyrogen-free reagents and depyrogenated tools were not used | Use pyrogen-free water and sterile disposable consumables; depyrogenate non-disposable reagents and supplies by either baking at 230 °C for ≥30 min or by cleaning them with Cavicide, followed by rinsing with an excess of pyrogen-free water |
| 22 | NANPs are not efficiently released from the carrier | Triton X-100 is a detergent that may not be an efficient releasing agent because of the non-lipid-like nature of the carrier | Use heparin (instead of Triton X-100) to outcompete electrostatic interactions between NANPs and other polymeric or inorganic carriers53,75 |
| 25–31 | No buffy coat | Centrifuge acceleration and or deceleration speed was not adjusted to be minimal | Check centrifuge settings to set the acceleration speed to minimal and turn deceleration speed off |
| Low PBMC yield | Incomplete collection of the buffy coat | Collect buffy coat using Eppendorf or equivalent pipette set for 1-mL volume. Avoid using serological pipettes, even when their volumes are 1–2 mL | |
| Low cell viability | Blood was exposed to extreme temperatures during transportation or was stored for >2 h after the collection and before the experiments | During summer, it is better to transport the blood using cold packs equilibrated to room temperature to avoid overheating the blood. During winter months, it is better to transport the blood on warm packs equilibrated to 37 °C to avoid overcooling the blood | |
| Expired Vacutainers | Check Vacutainer expiration dates | ||
| Diluted blood was held at room temperature for >2 h | Dilute blood with PBS immediately before loading onto Ficoll-Paque | ||
| Purified cells were kept in HBSS for >1 h | After the last wash, replace wash buffer with complete RPMI 1640 culture medium. If the cells cannot be treated immediately after the isolation, place them into an incubator set to 5% CO2 and 37 °C until ready to treat with controls and NANPs | ||
| Altered cell morphology | Same as described above in the ‘Low cell viability’ section | Same as described above in the ‘Low cell viability’ section | |
| 32–39 | High level of cytokines in the negative control | Blood was exposed to extreme heat during transportation | Avoid overheating the blood |
| Cell culture medium or reagents used to prepare the medium were contaminated with endotoxin | Prepare the medium using fresh reagents | ||
| Low signal in the positive control | Expired, degraded, or inappropriately stored controls | Prepare controls using fresh reagents. Avoid repeatedly subjecting stocks to freeze-thaw cycles. After preparation, prepare single-use aliquots of control stocks and store them at −20 °C | |
| Repeated freeze-thaw cycles | To avoid repeated freeze-thaw of supernatants, prepare small aliquots at the time of supernatant collection and store them at −20 °C | ||
| No positive response in NANP samples | Can be expected with small, planar, and fibrous NANPs | When unexpected, use fresh carrier (e.g., L2K) and store freshly prepared NANPs at 4 °C (for up to 1 week) or −20 °C (for longer times). Verify NANP stability by gel-electrophoresis | |
| When unexpected, this can be due to NANP degradation or failure to complex with a carrier. It can also be caused by repeated freeze-thaw cycles | To avoid repeated freeze-thaw of supernatants, prepare small aliquots at the time of supernatant collection and store them at −20 °C | ||
| Unexpectedly high levels of IFNs and other cytokines in NANP supernatants | Contamination of NANPs, L2K, or cell culture medium with endotoxin and other innate immunity-modulating impurities | Test all components for the presence of endotoxin using LAL assay and replace contaminated materials with pyrogen-free ones | |
| 40–53 | High variability between replicates (%CV >25) | Incubation was not performed on the shaker; loose pipette tips, especially when multichannel pipettes are used; the plate was allowed to dry after wash steps | Perform incubations on a shaker; verify that tips are tight on pipettes; avoid long (>5 min) intervals between plate wash and addition of reagents, and keep the plate on a paper towel upside-down until ready to add the reagents to the plate |
| 54–57 | No decrease in cytokine level in the presence of an inhibitor | NANP complexation with L2K did not work, a different carrier or no carrier was used, or the inhibitor quality was compromised during the storage or repeated freeze-thaw cycles | Verify that fresh L2K is used for the complexation. Use fresh inhibitors from commercial reagents as described in Steps 54–57, then prepare single-use aliquots. Discard any unused inhibitor at the end of experiment; in order to avoid repeated freeze-thaw cycles, do not store |
Timing
Steps 1–4, assembly of NANPs: 1 h
Steps 5–9, verification of NANP assemblies by native-PAGE: 2 h
Steps 10–18, gel purification of NANPs: ≥12 h
Step 19, confirmation of the absence of endotoxins in assembled NANPs by LAL assay: ~2h
Steps 20–24, assessment of structural integrity of NANPs upon complexation with delivery reagents: ~2 h
Steps 25–31, PBMC isolation: ~4 h
Steps 32–39, exposing PBMCs to NANPs and collecting supernatants: ~24 h
Steps 40–53, detection of biomarkers: ~4–6 h
Steps 54–57, mechanistic studies into NANP uptake routes and immune receptor recognition: ≥28–30 h
Anticipated results
Anticipated results for NANP assembly and retention of NANP structural integrity upon interaction with transfection reagents
We recommend carrying out native-PAGE experiments (Fig. 3) as a quick and low-cost way to verify the successful assembly of NANPs. The example shown in Fig. 3 demonstrates the correct formation of three representative NANPs. As controls, previously extensively characterized NANPs (e.g., RNA rings or RNA and DNA cubes) can be used. All bands can be quantified using the ChemiDoc software; as an alternative, the publicly available software ImageJ (https://imagej.nih.gov/ij/) can be used.
Anticipated results for endotoxin screening (Step 19)
The results should demonstrate that when the precautions described in Box 2 are followed, the endotoxin in NANP samples is undetectable (i.e., the measured levels are below the assay lower limit of quantification of 0.001 endotoxin units/mL/200 nM).
Anticipated results for cytokine screening (Steps 32–57)
The results shown in Table 1 demonstrate that when NANP quality is achieved by appropriate performance of Steps 1–19, as well as when the complexation with a carrier and Steps 20–24 are followed, the induction of primary biomarkers of NANP immunostimulation (IFN-α, IFN-β, IFN-ω, and IFN-λ) detected in Steps 40–53 is observed (Fig. 3). When a different carrier is used, the spectrum and the magnitude of cytokine response may be different and therefore will provide novel information about the influence of the NANPs’ delivery carrier on their immunological recognition. The magnitude of the biomarker is determined by the physicochemical properties of the NANPs, in that RNA-based NANPs, specifically RNA cubes, are the most potent immunostimulants, whereas DNA cubes and RNA rings are less immunostimulatory. Despite the difference in the magnitude of the interferon biomarker induction by NANPs with identical physicochemical properties, the type I interferons (IFN-α, IFN-β, IFN-ω) and type III interferon (IFN-λ) are consistently observed in all donors, as confirmed in our studies of >100 healthy donors, provided NANP delivery was achieved by a control carrier L2K.
These interferons have beneficial therapeutic properties. For example, they were shown to induce dendritic cell (DC) maturation and support DC function59–61. Type I interferons are also used for therapy of viral infections (e.g., hepatitis C), cancer (e.g., chronic myeloid leukemia), and immune-mediated disorders such as multiple sclerosis62–66. Type III interferons also have anti-cancer activity67. Clinical use of recombinant interferons is associated with fever-like reactions that are due to their systemic distribution and their ability to activate the immune cells in the blood68, and is also complicated by an anti-drug antibody (ADA) response in some patients69. When the ADA is neutralizing, it both affects the drug efficacy and contributes to toxicity70. These toxicities occur despite protein engineering attempts and conjugation with hydrophilic poly(ethylene glycol) (PEG)70. Not only does PEGylation of recombinant proteins not prevent immunogenicity of these products, but it may also create additional hurdles because the blood of healthy individuals contains pre-existing anti-PEG antibodies that may affect both the safety and efficacy of PEGylated drug products71–74. Owing to their ability to induce type I and type III interferons, NANPs could potentially address these problems by stimulating the hosts’ own type of interferon response.
If NANPs are tested without a carrier, and a broad spectrum of pyrogenic cytokines (TNFα, IL-1β, IL-6, IL-8) is detected, it would be highly suggestive of endotoxin contamination that was either undetected or introduced during handling and storage of the NANPs after the initial synthesis and analysis by LAL.
The protocol presented herein enables the screening of multiple NANPs, selection of one with a desirable magnitude of IFN response, understanding of potential contamination issues, and investigating the role of various carriers and their impact on the magnitude and spectrum of NANP-mediated cytokines in comparison to the control carrier L2K.
Anticipated results for mechanistic analysis
If the addition of ODN2088 results in a decrease in the cytokine response to the given NANPs, this response is due to NANP recognition by endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) (Fig. 3d). It is important to emphasize that this inhibitor cannot distinguish between individual endosomal TLRs.
If the addition of fucoidan results in a decrease in the cytokine response to the given NANPs, this response is due to NANP recognition by scavenger receptors. If the addition of either poly-I or dextran sulfate results in a decrease in the cytokine response to the given NANPs, whereas the addition of poly-C or chondroitin sulfate does not influence the result, this response is due to NANP recognition by scavenger receptors (Fig. 3e). When an alternative to the L2K carrier is used to deliver NANPs to the immune cells, the resulting spectrum of cytokines and/or magnitude of the cellular response may be different and will provide novel information about mechanisms of NANP recognition by immune cells. Most important, other inhibitors specific to the pathway of interest to the user of this protocol can also be used. Therefore, this protocol provides a versatile research tool to NANP researchers.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data presented in this article have been published before17,23,25,26,31,33 and are available to users without restrictions other than the copyright.
Supplementary Material
Related links
Key references using this protocol
Hong, E. et al. Nano Lett. 18, 4309–4321 (2018): https://doi.org/10.1021/acs.nanolett.8b01283
Rackley, L. et al. Adv. Funct. Mater. 28, 1805959 (2018): https://doi.org/10.1002/adfm.201805959
Hong, E. et al. Molecules 24, 1094 (2019): https://doi.org/10.3390/molecules24061094
Afonin, K. A. et al. Nat. Protoc. 6, 2022–2034 (2011): https://doi.org/10.1038/nprot.2011.418
Acknowledgements
This study was supported in part by federal funds from the National Cancer Institute, National Institutes of Health, under contracts HHSN261200800001E and 75N91019D00024 (to M.A.D.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Research reported in this publication was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under award no. R01GM120487 (to K.A.A.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank J. Halman and M. Chandler of the University of North Carolina at Charlotte and E. Hong and E. Cedrone of the Nanotechnology Characterization Lab, National Cancer Institute, for excellent technical assistance.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-020-0393-6.
Peer review information Nature Protocols thanks Remi Creusot, Chunhai Fan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Leontis NB, Stombaugh J & Westhof E The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 30, 3497–3531 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bindewald E, Hayes R, Yingling YG, Kasprzak W & Shapiro BA RNAJunction: a database of RNAjunctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 36, D392–D397 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parlea L et al. Ring Catalog: a resource for designing self-assembling RNA nanostructures. Methods 103, 128–137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parlea LG, Sweeney BA, Hosseini-Asanjan M, Zirbel CL & Leontis NB The RNA 3D Motif Atlas: computational methods for extraction, organization and evaluation of RNA motifs. Methods 103, 99–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geary C, Chworos A, Verzemnieks E, Voss NR & Jaeger L Composing RNA nanostructures from a syntax of RNA structural modules. Nano Lett. 17, 7095–7101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weng Y et al. Improved nucleic acid therapy with advanced nanoscale biotechnology. Mol. Ther. Nucleic Acids 19, 581–601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afonin KA et al. Design and self-assembly of siRNA-functionalized RNA nanoparticles for use in automated nanomedicine. Nat. Protoc 6, 2022–2034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jasinski D, Haque F, Binzel DW & Guo P Advancement of the emerging field of RNA nanotechnology. ACS Nano 11, 1142–1164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaeger L, Westhof E & Leontis NB TectoRNA: modular assembly units for the construction of RNA nano-objects. Nucleic Acids Res. 29, 455–463 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaeger L & Leontis NB Tecto-RNA: one-dimensional self-assembly through tertiary interactions. Angew. Chem. Int. Ed. Engl 39, 2521–2524 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Ohno H et al. Synthetic RNA-protein complex shaped like an equilateral triangle. Nat. Nanotechnol 6, 116–120 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Dibrov SM, McLean J, Parsons J & Hermann T Self-assembling RNA square. Proc. Natl Acad. Sci. USA 108, 6405–6408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boerneke MA, Dibrov SM & Hermann T Crystal-structure-guided design of self-assembling RNA nanotriangles. Angew. Chem. Int. Ed. Engl 55, 4097–4100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Afonin KA et al. In vitro assembly of cubic RNA-based scaffolds designed in silico. Nat. Nanotechnol 5, 676–682 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chidchob P & Sleiman HF Recent advances in DNA nanotechnology. Curr. Opin. Chem. Biol 46, 63–70 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Rothemund PWK Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Afonin KA et al. Triggering of RNA interference with RNA-RNA, RNA-DNA, and DNA-RNA nanoparticles. ACS Nano 9, 251–259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Afonin KA et al. Multifunctional RNA nanoparticles. Nano Lett. 14, 5662–5671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol 7, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H et al. RNA as a stable polymer to build controllable and defined nanostructures for material and biomedical applications. Nano Today 10, 631–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu D, Shu Y, Haque F, Abdelmawla S & Guo P Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol 6, 658–667 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khisamutdinov EF et al. Enhancing immunomodulation on innate immunity by shape transition among RNA triangle, square and pentagon nanovehicles. Nucleic Acids Res. 42, 9996–10004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halman JR et al. Functionally-interdependent shape-switching nanoparticles with controllable properties. Nucleic Acids Res. 45, 2210–2220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson MB et al. Programmable nucleic acid based polygons with controlled neuroimmunomodulatory properties for predictive QSAR modeling. Small 13, 1701255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong E et al. Structure and composition define immunorecognition of nucleic acid nanoparticles. Nano Lett. 18, 4309–4321 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong E et al. Toll-like receptor-mediated recognition of nucleic acid nanoparticles (NANPs) in human primary blood cells. Molecules 24, 1094 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bindewald E et al. Multistrand structure prediction of nucleic acid assemblies and design of RNA switches. Nano Lett. 16, 1726–1735 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobrovolskaia MA Pre-clinical immunotoxicity studies of nanotechnology-formulated drugs: challenges, considerations and strategy. J. Control. Release 220, 571–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panigaj M et al. Aptamers as modular components of therapeutic nucleic acid nanotechnology. ACS Nano 13, 12301–12321 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandler M & Afonin KA Smart-responsive nucleic acid nanoparticles (NANPs) with the potential to modulate immune behavior. Nanomaterials (Basel) 9, 611 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sajja S et al. Dynamic behavior of RNA nanoparticles analyzed by AFM on a mica/air interface. Langmuir 34, 15099–15108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ke W et al. RNA-DNA fibers and polygons with controlled immunorecognition activate RNAi, FRET and transcriptional regulation of NF-kB in human cells. Nucleic Acids Res. 47, 1350–1361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rackley L et al. RNA fibers as optimized nanoscaffolds for siRNA coordination and reduced immunological recognition. Adv. Funct. Mater 28, 1805959 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vessillier S et al. Cytokine release assays for the prediction of therapeutic mAb safety in first-in man trials—whole blood cytokine release assays are poorly predictive for TGN1412 cytokine storm. J. Immunol. Methods 424, 43–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregg KA et al. Rationally designed TLR4 ligands for vaccine adjuvant discovery. mBio 8, e00492–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh DY et al. Adjuvant-induced human monocyte secretome profiles reveal adjuvant- and age-specific protein signatures. Mol. Cell. Proteom 15, 1877–1894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandler M, Johnson B, Panigaj M & Afonin KA Innate immune responses triggered by nucleic acids inspire the design of immunomodulatory nucleic acid nanoparticles (NANPs). Curr. Opin. Biotechnol 63, 8–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei M et al. Polyvalent immunostimulatory nanoagents with self-assembled CpG oligonucleotide-conjugated gold nanoparticles. Angew. Chem. Int. Ed. Engl 51, 1202–1206 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Radovic-Moreno AF et al. Immunomodulatory spherical nucleic acids. Proc. Natl Acad. Sci. USA 112, 3892–3897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J et al. Self-assembled multivalent DNA nanostructures for noninvasive intracellular delivery of immunostimulatory CpG oligonucleotides. ACS Nano 5, 8783–8789 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Liu X et al. A DNA nanostructure platform for directed assembly of synthetic vaccines. Nano Lett. 12, 4254–4259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S et al. Rational vaccinology with spherical nucleic acids. Proc. Natl Acad. Sci. USA 116, 10473–10481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong E & Dobrovolskaia MA Addressing barriers to effective cancer immunotherapy with nanotechnology: achievements, challenges, and roadmap to the next generation of nanoimmunotherapeutics. Adv. Drug Deliv. Rev 141, 3–22 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Dobrovolskaia MA Nucleic acid nanoparticles at a crossroads of vaccines and immunotherapies. Molecules 24, 4620 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kondo S & Sauder DN Tumor necrosis factor (TNF) receptor type 1 (p55) is a main mediator for TNF-alpha-induced skin inflammation. Eur. J. Immunol 27, 1713–1718 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Phillips A, Patel C, Pillsbury A, Brotherton J & Macartney K Safety of human papillomavirus vaccines: an updated review. Drug Saf. 41, 329–346 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Miller ER et al. Post-licensure safety surveillance of zoster vaccine live (Zostavax®) in the United States, Vaccine Adverse Event Reporting System (VAERS), 2006–2015. Hum. Vaccin. Immunother 14, 1963–1969 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woo EJ, Moro PL, Cano M & Jankosky C Postmarketing safety surveillance of trivalent recombinant influenza vaccine: reports to the Vaccine Adverse Event Reporting System. Vaccine 35, 5618–5621 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Gause KT et al. Immunological principles guiding the rational design of particles for vaccine delivery. ACS Nano 11, 54–68 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Di Franco S, Turdo A, Todaro M & Stassi G Role of type I and II interferons in colorectal cancer and melanoma. Front. Immunol 8, 878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams D et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med 379, 11–21 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Chan A et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol. Ther. Nucleic Acids 4, e263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halman JR et al. A cationic amphiphilic co-polymer as a carrier of nucleic acid nanoparticles (NANPS) for controlled gene silencing, immunostimulation, and biodistribution. Nanomedicine 23, 102094 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bui MN et al. Versatile RNA tetra-U helix linking motif as a toolkit for nucleic acid nanotechnology. Nanomedicine 13, 1137–1146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ozaki K & Leonard WJ Cytokine and cytokine receptor pleiotropy and redundancy. J. Biol. Chem 277, 29355–29358 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Potter TM, Neun BW, Rodriguez JC, Ilinskaya AN & Dobrovolskaia MA Analysis of pro-inflammatory cytokine and type II interferon induction by nanoparticles. Methods Mol. Biol 1682, 173–187 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Centers for Disease Control and Prevention. Biosafety in Microbiological and Biomedical Laboratories 5th edn (US Department of Health and Human Services, 2009). [Google Scholar]
- 58.Strober W Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol 21, A.3B.1–A.3B.2 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Radvanyi LG, Banerjee A, Weir M & Messner H Low levels of interferon-α induce CD86 (B7.2) expression and accelerates dendritic cell maturation from human peripheral blood mononuclear cells. Scand.J. Immunol 50, 499–509 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Tam MA & Wick MJ MyD88 and interferon-α/β are differentially required for dendritic cell maturation but dispensable for development of protective memory against Listeria. Immunology 128, 429–438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trepiakas R, Pedersen AE, Met O & Svane IM Addition of interferon-alpha to a standard maturation cocktail induces CD38 up-regulation and increases dendritic cell function. Vaccine 27, 2213–2219 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Floros T & Tarhini AA Anticancer cytokines: biology and clinical effects of interferon-α2, interleukin(IL)-2, IL-15, IL-21, and IL-12. Semin. Oncol 42, 539–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheknev SB, Kobyakina NA, Mezentseva MV & Skvortsova VI Long-term study of interferon system state in patients with multiple sclerosis received the individual immune therapy with human recombinant IFN-alpha. Russ. J. Immunol 6, 39–46 (2001). [PubMed] [Google Scholar]
- 64.Bongioanni MR et al. Systemic high-dose recombinant-alpha-2a-interferon therapy modulates lymphokine production in multiple sclerosis. J. Neurol. Sci 143, 91–99 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Kujawski LA & Talpaz M The role of interferon-alpha in the treatment of chronic myeloid leukemia. Cytokine Growth Factor Rev. 18, 459–471 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Rong L & Perelson AS Modeling HIV persistence, the latent reservoir, and viral blips. J. Theor. Biol 260, 308–331 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stiff A & Carson W III Investigations of interferon-lambda for the treatment of cancer. J. Innate Immun 7, 243–250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Filipi ML et al. Nurses’ perspective on approaches to limit flu-like symptoms during interferon therapy for multiple sclerosis. Int. J. MS Care 16, 55–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenberg AS Immunogenicity of biological therapeutics: a hierarchy of concerns. Dev. Biol. (Basel) 112, 15–21 (2003). [PubMed] [Google Scholar]
- 70.Baker MP, Reynolds HM, Lumicisi B & Bryson CJ Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself 1, 314–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang CJ et al. A genome-wide association study identifies a novel susceptibility locus for the immunogenicity of polyethylene glycol. Nat. Commun 8, 522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen BM et al. Measurement of pre-existing IgG and IgM antibodies against polyethylene glycol in healthy individuals. Anal. Chem 88, 10661–10666 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Chen WW, Zhang X & Huang WJ Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep 13, 3391–3396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsieh YC et al. Pre-existing anti-polyethylene glycol antibody reduces the therapeutic efficacy and pharmacokinetics of PEGylated liposomes. Theranostics 8, 3164–3175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cruz-Acuna M, Halman JR, Afonin KA, Dobson J & Rinaldi C Magnetic nanoparticles loaded with functional RNA nanoparticles. Nanoscale 10, 17761–17770 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this article have been published before17,23,25,26,31,33 and are available to users without restrictions other than the copyright.