

EPIC1-EZH2 axis activation emerges as a targetable mechanism for tumor immune evasion and immunotherapy resistance.
Abstract
Through an integrative analysis of the lincRNA expression and tumor immune response in 9,626 tumor samples across 32 cancer types, we developed a lincRNA-based immune response (LIMER) score that can predict the immune cells infiltration and patient prognosis in multiple cancer types. Our analysis also identified tumor-specific lincRNAs, including EPIC1, that potentially regulate tumor immune response in multiple cancer types. Immunocompetent mouse models and in vitro co-culture assays demonstrated that EPIC1 induces tumor immune evasion and resistance to immunotherapy by suppressing tumor cell antigen presentation. Mechanistically, lincRNA EPIC1 interacts with the histone methyltransferase EZH2, leading to the epigenetic silencing of IFNGR1, TAP1/2, ERAP1/2, and MHC-I genes. Genetic and pharmacological inhibition of EZH2 abolish EPIC1’s immune-related oncogenic effect and its suppression of interferon-γ signaling. The EPIC1-EZH2 axis emerges as a potential mechanism for tumor immune evasion that can serve as therapeutic targets for immunotherapy.
INTRODUCTION
The immune system plays a fundamental role in recognizing and eliminating neoplastic cells. However, the neoplastic cells can usually develop multiple strategies to evade immune surveillance and hence promote tumor progression (1). Immune evasion has been considered as one of the hallmarks of cancer and a significant event for tumor development (2). Generally, cancer cells can evade immune surveillance through eliminating effector T cells, recruiting regulatory immune cells, and reducing immune recognition to develop an immunosuppressive tumor microenvironment (TME) (3). Although tremendous progress has been made, the molecular mechanism of tumor immune evasion is incompletely understood.
Recent studies have shown that long noncoding RNA genes (lncRNAs) play essential roles in the development, differentiation, apoptosis, and activation of multiple types of immune cells (4, 5). For instance, previous research has revealed that lncRNAs H19, lncHSC-1, and lncHSC-2 can regulate innate immune response in the maintenance of hematopoietic stem cells and the differentiation of myeloid lineages (6). In addition to innate immune response, lncRNAs have been demonstrated to regulate adaptive immune response. Lnc-DC, a dendritic cell–specific lncRNA, blocks signal transducer and activator of transcription 3 (STAT3) dephosphorylation through directly binding with STAT3 and increases dendritic cell differentiation and activation (7). More studies have reported that lncRNAs MAF-4 (8), NeST (9), Ccr2-5’AS (10), and Rmrp (11) regulate the differentiation of CD4+ T cells into T helper 1 cell (TH1), TH2, TH7, and regulatory T cells (Tregs), which are critical for the initiation of adaptive immune responses (12).
A growing body of genomic analysis indicates that genomic/epigenetic alterations in tumor tissues may play essential roles in tumor immune response and checkpoint blockade efficacy (13–19). However, majority of previous genotype-immunophenotype association studies have neglected the potential impact of lncRNAs on tumor immune microenvironment, which have emerged as important modulators of oncogenesis (12, 20–23). Here, we characterized a comprehensive landscape of 7528 intergenic lncRNAs (linRNAs)–tumor immunity interaction by analyzing the lincRNA expression and immunogenomics profile of the TME across 9626 tumor samples. By delineating lincRNAs’ tissue specificity, we aimed to discover lincRNA-based biomarkers for predicting clinically relevant immune responses in patients with cancer. By further integrating the genome-wide DNA methylation data in the same tumor samples, we hope to identify the potential “driver” lincRNAs that can regulate the tumor immune response in multiple cancer types. By further using in vivo and in vitro cancer models, we aimed to identify and mechanistically validate lincRNAs that are master regulators of tumor immune response.
RESULTS
Integrative analysis of the lincRNA expression and tumor immune response characterized the landscape of lincRNA–cancer immunity interactions
Our analysis focused on 7528 lincRNAs whose expression and DNA methylation levels are more reliable to be inferred from RNA sequencing (RNA-seq) and DNA methylation data, respectively. We first performed correlation analysis between the expression of 7528 lincRNAs and tumor immune signature expression (24) across 9626 tumor samples from The Cancer Genome Atlas (TCGA) database (fig. S1A and table S1). This analysis identified 3491 lincRNAs, whose expression was significantly correlated with immune signatures across multiple cancer types (Materials and Methods). Further t-distributed stochastic neighbor embedding (t-SNE) and clustering analysis (Materials and Methods) identified four lincRNA clusters with distinct association patterns with anticancer immune response: C1 (382 lincRNAs, negatively correlated with immune response in multiple cancer types), C2 (1178 lincRNAs, negatively correlated with immune response in specific cancer types), C3 (737 lincRNAs, positively correlated with immune response in multiple cancer types), and C4 (1194 lincRNAs, positively correlated with immune response in specific cancer types) (Fig. 1A, fig. S1B, and table S2).
Fig. 1. The landscape of immunity-associated lincRNAs in cancer.
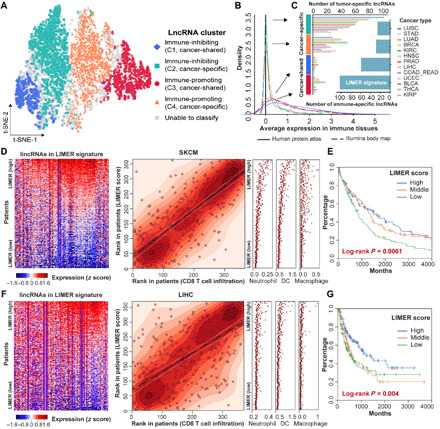
(A) t-SNE embedding of the four identified cancer immunity–associated lincRNA clusters. (B) LincRNA expression distribution in immune-related organs (bottom left). (C) Number of tumor-specific lincRNAs in each cancer type (left) and number of immune-specific lincRNAs in each cluster (right). (D and F) Association between lincRNA-based immune response (LIMER) score and cytotoxic T cell infiltration in melanoma (SKCM) (D) and liver cancer (LIHC) (F). Left: The expression of each LIMER lincRNAs (columns) in tumor patients (rows). Middle: The correlation between LIMER score (y axis) and cytotoxic T cell infiltration (x axis). Right: The infiltration (x axis) of other immune cells. All three panels share the same order of patients, which were sorted by descending the LIMER score. (E and G) Kaplan-Meier plot shows the association between LIMER score and patient progression-free intervals. Patients are equally stratified into three groups based on LIMER scores in the same cohort as (D) and (F), respectively.
One plausible explanation for the observed correlation between lincRNA expression and immune response scores could be that some tumor-infiltrating lymphocyte (TIL)–expressing lincRNAs are also detected by the bulk tumor RNA-seq data. Their overexpression in the tumor tissue represents the increased percentage of the immune component in the TME. Further tissue specificity analysis (Materials and Methods) for each of the 3491 immune-associated lincRNAs revealed that 96.07% of lincRNAs in C1 and 92.99% in C3 clusters were expressed in healthy immune tissues (i.e., spleens and lymph nodes) (Fig. 1, B and C). These observations suggested that C1 and C3 clusters are likely enriched with immune-specific lincRNAs.
Using the expression of 105 immune-specific lincRNAs from the C3 cluster, we developed an lincRNA-based immune response (LIMER) score to estimate tumor immune cell infiltration from bulk tumor RNA-seq data (Materials and Methods). Tumors with higher LIMER scores tend to have a higher immune response, indicated by higher infiltration of CD8+ T cells [median Spearman correlation coefficient rho = 0.55; immune cell infiltrations were estimated by Tumor IMmune Estimation Resource (TIMER) (25, 26)], CD4+ T cells (median rho = 0.63), dendritic cells (median rho = 0.67), neutrophils (median rho = 0.64), macrophages (median rho = 0.42), and B cells (median rho = 0.50) (Fig. 1, D to F, and fig. S2B). Moreover, patients with higher LIMER scores in their tumors have a significant beneficial progression-free survival in multiple cancer types (fig. S2C) including melanoma (Fig. 1E), liver cancer (Fig. 1G), bladder cancer (fig. S3A), breast cancer (fig. S3B), cervical cancer (fig. S3C), and head and neck cancer (fig. S3D).
DNA methylation analysis of tumor-specific lincRNAs revealed EPIC1 as a master suppressor of tumor immune response
Besides the immune-specific lincRNAs, our analysis also identified 263 lincRNAs in the C2 cluster that are exclusively expressed in tumor cells but showed a strong negative correlation with tumor immune response (table S2). These tumor-specific lincRNAs are highly expressed in tumor tissues compared with adjacent normal tissues (Fig. 2A). However, unlike the immune-specific lincRNAs, these 263 lincRNAs are not expressed [i.e., fragments per kilobase of transcript per million mapped reads (FPKM) = 0] in immune tissues (Fig. 1, B and C). In an effort to further identify lincRNA genes that have somatic genomic or epigenetic alterations in tumor genome, we integrated with DNA methylation data and characterized 11 epigenetically activated (EA) lincRNAs (27), of which the expression is driven by epigenetic alterations in tumors cells (Materials and Methods and table S2). Tumors with these EA lincRNA activation appeared to have decreased immune response, indicated by underexpression of genes in immune checkpoint (median rho = 0.40, Spearman’s correlation), chemokine signaling (median rho = 0.37), and cytotoxic T cell activity (median rho = 0.27) (Fig. 2, A, C, D, F, and G, and fig. S2A). Moreover, patients with this EA lincRNA activation in their tumors have significantly worse survival (Fig. 2, E and H).
Fig. 2. DNA methylation analysis on tumor-specific lincRNAs revealed EPIC1 as a suppressor of tumor immune response.
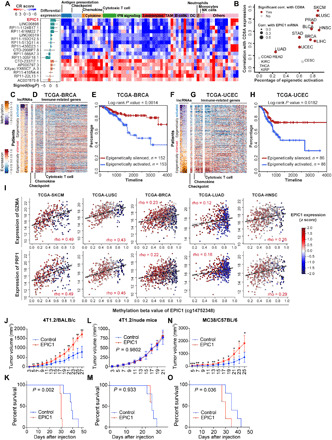
(A) Consensus regulation (CR) score of 11 EA tumor-specific lincRNAs (C2 cluster) and 6 epigenetically silenced (ES) tumor-specific lincRNAs (C4 cluster) that are correlated with tumor immune response (heatmap) and their average differential expression across 23 cancer types (bar plots). (B) Correlation between epigenetic activation fraction of epigenetically induced lincRNA 1 (EPIC1) and its association with CD8A expression. (C, D, F, and G) DNA methylation (z score normalized beta value) of tumor-specific lincRNAs (C and F) and the expression of immune signature genes (D and G) in TCGA-BRCA and TCGA-UCEC patients. BRCA, Breast Cancer; UCEC, Uterine Corpus. (E and H) Survival curves of the patients with top and bottom 20% epigenetic activity in (C) and (F). (I) Correlation between EPIC1 methylation and GZMA and PRF1 expression. (J to O) Tumor volume (J, L, and N) and overall survival (K, M, and O) of BALB/c mice, BALB/c nude mice, and C57BL/6 mice that are inoculated with 4T1.2 cells or MC38 cells stably expressing empty vector (control) or EPIC1 (n = 5 animals per group). EC, Endometrial Carcinoma; GZMA, Granzyme A; PRF1, Perforin 1. Data are means ±SD. *P <0.05; **P <0.01; ***P < 0.001.
Among the 11 EA lincRNAs that are highly correlated with immune response, lincRNA EPIC1 (epigenetically induced lincRNA 1) was the top lincRNA that is differentially expressed between tumor and normal samples (Fig. 2, A and B). To further demonstrate EPIC1’s tissue specificity, we analyzed a published single-cell ATAC-seq (assay for transposase-accessible chromatin sequencing) data (28) from 28,274 TILs and ~5000 cancer cells. This analysis revealed that EPIC1’s promoter is only activated in tumor cells (as indicated by chromatin openness) but not in TIL cells, indicating that EPIC1 is a cancer cell–specific lincRNA that is EA (fig. S3E). Furthermore, tumors with epigenetic activation of EPIC1 have a remarkable decreased expression of CD8+ T cell markers, including perforin 1 and granzyme A in multiple cancer types such as melanoma, lung cancer, and breast cancer (Fig. 2I and fig. S4, B to D). Although we have recently reported that the expression of EPIC1 is positively associated with poor prognosis and drug resistance in ER+ breast cancer (27, 29), the role of EPIC1 in the regulation of antitumor immunity is unknown. To determine how tumor cell–expressed EPIC1 regulates antitumor immune responses, we orthotopically injected empty vector (control) or EPIC1-overexpressed 4T1.2 cells in BALB/c mice. Compared with the control group, overexpression of EPIC1 significantly promoted tumor growth and led to poorer survival (Fig. 2, J and K). However, there is no difference in tumor growth and survival between control and EPIC1-overexpressing 4T1.2 tumors in immune-deficient nude mice (Fig. 2, L and M), suggesting that the protumor effect of EPIC1 in the 4T1.2 model is dependent on the adaptive immune system. To further confirm EPIC1’s immune-dependent protumor effect, we injected the control and EPIC1-overexpressed MC38 (Fig. 2, N and O) and CT26 (fig. S4, E and F) colorectal cancer cells into back flanks of C57BL/6 and BALB/c mice, respectively. Consistent with the 4T1.2 tumor model, EPIC1 overexpression in these tumor cells led to significantly increased tumor growth rates and reduced overall survival.
EPIC1 suppresses cytotoxic T lymphocyte infiltration and activation in TME
Histopathological analysis revealed that EPIC1-overexpressed 4T1.2 tumors were infiltrated by fewer total immune cells and T cells compared with control tumors (Fig. 3, A and B, and fig. S4G). In addition, we observed that percentages of tumor-infiltrating CD8+ T cells and tumor-associated macrophage 1 (M1) cells decreased in both 4T1.2-EPIC1 (Fig. 3, C and D) and MC38-EPIC1 (Fig. 3, E and F) tumors when compared with control tumors. Moreover, EPIC1 overexpression increased tumor-associated macrophage 2 (M2) cells in both 4T1.2 and MC38 models (Fig. 3, D and F). There was no difference in the percentage of CD4+ T cells, γδ T cells, B cells, and FoxP3+ regulatory T cells between EPIC1-overexpressed tumors and control tumors in either mouse models (Fig. 3, C, E, G, and H). Furthermore, EPIC1 overexpression led to a reduction in the percentage of interferon-γ+ (IFN-γ+) CD8+, IFN-γ+ CD4+, and granzyme B+ CD8+ T cells, but it did not change the population of CD8+ LAG3+, CD8+ PD1+, CD4+ LAG3+, or CD4+ PD1+ cells (Fig. 3, I to K and fig. S4, H and I). Together, these results indicate that EPIC1 overexpression in tumor cells can suppress cytotoxic T lymphocyte (CTL) infiltration and activation in the TME.
Fig. 3. EPIC1 decreases cytotoxic T lymphocyte infiltration and activation.
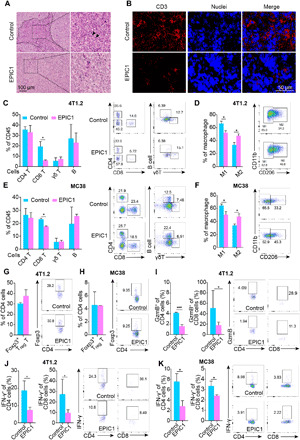
(A) Control and EPIC1-overexpressed 4T1.2 tumors were paraffin embedded and applied with hematoxylin and eosin staining. Black arrows indicate the immune cells. (B) Frozen sections from the empty vector (control) and EPIC1 overexpressed (EPIC1) 4T1.2 tumors were subjected to immunostaining analysis of CD3 (red) along with 4′,6-diamidino-2-phenylindole for nuclei (blue). (C to H) Quantification of indicated TILs from control and EPIC1-overexpressed 4T1.2 or MC38 tumors in BALB/c mice or C57BL/6 mice, respectively (n = 5 for control, n = 5 for EPIC1 overexpression). TILs were analyzed by flow cytometry on the 15th day after transplantation. The right panel of each figure indicates the representative flow cytometry profiles. (I) The production of granzyme B by CD4+ and CD8+ TILs in 4T1.2 tumors was analyzed by flow cytometry, respectively. The right panel shows the representative flow cytometry profiles. (J and K) The production of interferon-γ (IFN-γ) by CD4+ and CD8+ TILs in 4T1.2 and MC38 tumors was analyzed by flow cytometry, respectively. The right panel shows the representative flow cytometry profiles. Data are means ± SD. *P < 0.05 **P < 0.01; ***P < 0.001.
EPIC1 enhances tumor immune evasion via suppressing antigen presentation in tumor cells
Emerging evidence showed that immune cell–expressed lincRNAs play important roles in tumor immune response by regulating immune cell activation and differentiation (4, 12). However, few studies were able to identify tumor cell–expressed lincRNAs that can modulate tumor immune response (30). To determine how EPIC1 expression in tumor cells lead to CTLs suppression, we knocked down EPIC1 with two small interfering RNAs (siRNAs) individually or together in four cancer cell lines and performed RNA-seq analyses (Materials and Methods). EPIC1 knockdown significantly up-regulated major histocompatibility complex (MHC) genes (Fig. 4A). Consistent with our observation in EPIC1 knockdown cell lines, EPIC1’s expression was significantly associated with lower expression of MHC genes in 9 of 13 cancer types (Fig. 4A). Notably, knockdown of EPIC1 did not change the expression of interleukin-2 (IL-2), IL-12, IL-4, and IL-8 in cancer cell lines because they are immune cell–secreted cytokines. However, these cytokines were inversely correlated with EPIC1 expression in bulk tumor tissues (Fig. 4A). These observations suggest that EPIC1 expression in tumor cells suppresses immune cell activation through the tumor-intrinsic mechanism, such as antigen presentation.
Fig. 4. EPIC1 suppresses antigen presentation of tumor cells.
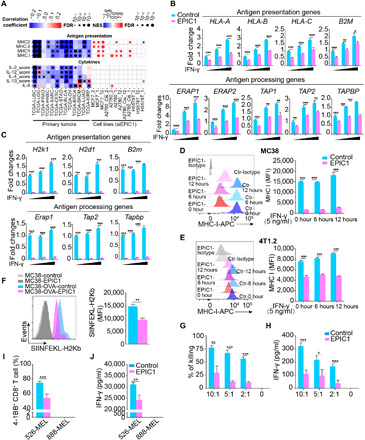
(A) Left: Heatmap shows the association between EPIC1 expression and MHC-I and cytokine signatures in TCGA patients. Right: Heatmap shows the changes of same signatures after EPIC1 knockdown in different cell lines. (B and C) The expression of antigen presentation and antigen processing genes in control and EPIC1-overexpressed MCF-7 cells (B) and MC38 cells (C). Cells were treated with IFN-γ (0, 1, and 5 ng/ml) for 24 hours. (D and E) Cell surface levels of MHC-I in control and EPIC1-overexpressed MC38 (D) and 4T1.2 (E) cells. Cells were treated with IFN-γ (5 ng/ml) for indicated time points. (F) SIINFEKL-H2Kb presentation by the empty vector (control) or EPIC1-overexpressed MC38 cells. The quantification of MFI (mean fluorescence intensity) of SIINFEKL-H2Kb is shown on the right panel. Cells were treated with IFN-γ (5 ng/ml) for 24 hours. (G and H) Killing effect of MC38 (OVA+) cells overexpressed with empty vector (vehicle) or EPIC1 after coculture with OT-1 T cells (G). The production of IFN-γ of OT-1 cells was determined by enzyme-linked immunosorbent assay (H). (I and J) Activation (I) and the production of IFN-γ (J) of gp100 TCR-transduced CD8+ T cells were cocultured with 888-MEL (gp100+) and 526-MEL (gp100+) cells transduced with empty vector (Control) or EPIC1 for 24 hours. Data are means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
Silencing of EPIC1 promoted the expression of antigen presentation genes [human leukocyte antigen A (HLA-A), HLA-B, HLA-C, and B2M] as well as antigen processing genes (ERAP1/2, TAP1/2, and TAPBP) in human breast cancer cell MCF-7 (fig. S5A), lung cancer cell H1299 (fig. S5B), and colorectal cancer cells HCT116 (fig. S5C). Consistently, EPIC1 overexpression resulted in lower expression of antigen presentation and processing genes in these human cancer cell lines (Fig. 4B and fig. S5, D and E) as well as in murine cell lines MC38 and 4T1.2 (Fig. 4C and fig. S5F). The cell surface level of MHC-I was also reduced in EPIC1-overexpressing MC38 and 4T1.2 cells (Fig. 4, D and E).
Previous studies have shown that decreasing the expression of antigen processing and presentation genes in tumor cells significantly contributes to the tumor evasion from cytotoxic T cell recognition (31). To investigate whether EPIC1’s inhibition of tumor immune response is mediated by its suppression of antigen presentation and processing in tumor cells, we ectopically expressed the full length of ovalbumin (OVA) in control or EPIC1-overexpressed MC38 cells. The cell surface of OVA peptide SIINFEKL bound to MHC-I (H2Kb) was then quantified by flow cytometry using a specific monoclonal antibody. This analysis demonstrated that the OVA peptide-loaded MHC-I (SIINFEKL-H2Kb) was significantly decreased in EPIC1-overexpressed MC38 cells (Fig. 4F).
Control and EPIC1-overexpressed MC38 (OVA+) cells were further cocultured with OT-1 CD8+ T cells. As we expected, EPIC1-overexpressed MC38 cells (OVA+) induced lower levels of IFN-γ production and were more resistant to cytotoxicity by OT-1 CD8+ T cells compared with control MC38 cells (Fig. 4, G and H). We further used a human melanoma cell and CD8+ T cell coculture system to determine EPIC1’s effects on tumor antigen presentation and T cell activation. Specifically, the control or EPIC1-overexpressed human melanoma cell line 526-MEL, which expressed HLA-A*02 and tumor antigen gp100, was cocultured with HLA-A*02–restricted gp100 T cell receptor (TCR)–transduced human CD8+ T cells (Materials and Methods). We observed that EPIC1 overexpression in 526-MEL cells suppressed activation and IFN-γ production by gp100-specific CD8+ T cells (Fig. 4, I and J, and fig. S6B). As a negative control, neither control nor EPIC1-overexpressing 888-MEL cells, a human melanoma cell line expressing gp100 but not HLA-A*02, were able to stimulate gp100-specific CD8+ T cells (Fig. 4, I and J, and fig. S6B). Together, these data demonstrated that EPIC1 overexpression in tumor inhibits CTL infiltration and activation through suppressing tumor antigen processing and presentation.
EPIC1 inhibited IFN-γ–Janus kinase–STAT1 signaling pathway through suppressing IFNGR1 expression
To further explore the mechanism of EPIC1’s regulation of tumor cell antigen presentation, we performed pathway analyses in the RNA-seq data from EPIC1-silenced cells and TCGA cancer patients’ data. These analyses revealed that EPIC1 suppresses the IFN-γ signaling in both tumor tissues and cancer cells (Fig. 5, A and B). For example, in TCGA lung cancer tissues, EPIC1 activation has significant inverse correlation with IFNG signaling (r = −0.45, P = 7.14 × 10−20, Lung squamous cell carcinoma (LUSC); r = −0.17, P = 0.00023, Lung Adenocarcinoma (LUAD), Pearson’s correlation). In MCF-7, NCI-H1299, and HCT116 cells, EPIC1 knockdown led to higher expression of the IFN-γ receptor, IFNGR1, and its downstream targets, IRF1 and IRF9 (fig. S7, A to C).
Fig. 5. EPIC1 inhibits IFNGR1 expression and type II interferon signaling.
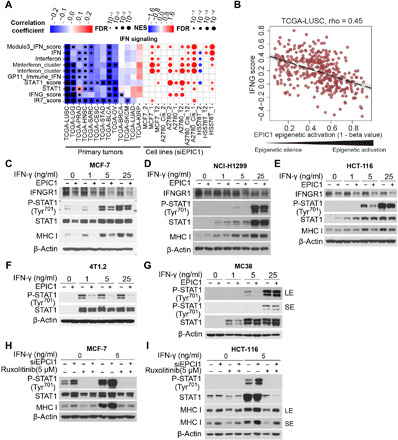
(A) Heatmap (left) shows the association between EPIC1 expression and interferon signatures in TCGA patients. Color in the heatmap indicates the effect size. Dots indicate the logarithmic false discovery rate. Heatmap (right) shows the pathway changed after EPIC1 knockdown in different cell lines. The color of the dots in the heatmap indicates the enrichment score. The size of the dots indicates the false discovery rate. (B) Correlation between EPIC1 epigenetic activation and IFNG response score in immune response signature. (C to E) Immunoblot of IFNGR1, p-STAT, and MHC-I in human cancer cell lines MCF-7 (C), NCI-H1299 (D), and HCT116 (E) cells stably expressing empty vector (Control) or EPIC1 further treated with the indicated concentration of IFN-γ for 24 hours. (F and G) Immunoblot of p-STAT in murine breast cancer cells 4T1.2 and colorectal cancer cells MC38 stably expressing empty vector (control) or EPIC1 further treated with the indicated concentration of IFN-γ for 24 hours. LS, long exposure; SE, short exposure. (H and I) The measurement of p-STAT1 and MHC-I protein expression by immunoblot in MCF-7 (H) and NCI-H1299 (I) cells transduced with EPIC1 siRNA and further treated by JAK1/2 inhibitor ruxolitinib (5 μm) with/without IFN-γ (5 ng/ml). LS, long exposure; SE, short exposure.
It has been well studied that IFN-γ can bind to IFNGR1 and lead to STAT1 phosphorylation (p-STAT1), which further activates Janus kinase (JAK)–STAT1 signaling in tumor immunity (32). The IFN-γ is involved in cancer immune surveillance by inducing MHC-I genes expression for CTL recognition and elimination of tumor cells (32, 33). We found that overexpression of EPIC1, by either a lentiviral system or a CRISPR/Cas9 Synergistic Activation Mediator (SAM) system (see Materials and Methods), suppressed both IFNGR1 protein and mRNA expression (Fig. 5, C to E, and fig. S7, D to I) in human breast, lung, and colorectal cancer cell lines, as well as in murine breast cell line 4T1.2 and colorectal cancer line MC38 (Fig. 5, F and G). As a result of IFNGR1 suppression, EPIC1 overexpression led to the decreased STAT1 phosphorylation (Fig. 5, C and E, and fig. S8, A and B). Consistently, siRNA silencing of EPIC1 up-regulated IFN-γ–JAK–STAT1 signaling and MHC-I expression (fig. S8, C to E). Furthermore, JAK1 inhibitor (i.e., ruxolitinib) treatment partially restored EPIC1 inhibition of MHC-I in MCF-7, HCT116, and NCI-H1299 cells (Fig. 5, H and I, and fig. S8F). These data demonstrated that EPIC1 inhibited the IFN-γ–JAK–STAT1 signaling pathway through suppressing the IFNGR1 expression.
EPIC1’s regulation of type II interferon signaling is mediated by its interaction with EZH2 protein
It is apparent to us that EPIC1’s inhibition of the IFN-γ–JAK–STAT1 signaling pathway and MHC-I in tumor cells contributes to its suppression of tumor immune response as we observed in both in vitro (Fig. 3) and in vivo (Fig. 2) tumor models. We set out to investigate the underlying molecular mechanism of how EPIC1 regulates IFNGR1 and antigen presentation genes’ expression. Inspired by our recent study that using cell line pharmacogenomics database to identify lincRNA’s functional interacting protein (29), we analyzed EPIC1 expression profiles in 582 cell lines and the cell lines’ drug responses to 545 compounds from the Cancer Therapeutics Response Portal (CTRP) database (34–36). This analysis revealed that the expression of EPIC1 was highly correlated with polycomb repressive complex 2 (PRC2) inhibitor (BRD1835) resistance in 32 breast cancer cell lines (rho = −0.41, P = 0.02; see Materials and Methods, Fig. 6A, and table S3). Moreover, the pathway analysis of EPIC1 knockdown RNA-seq data further revealed that the enhancer of zeste homolog 2 (EZH2) targets are enriched in the differentially expressed genes after EPIC1 knockdown (Fig. 6B).
Fig. 6. EPIC1’s regulation of type II interferon signaling is mediated by its interaction with EZH2 protein.
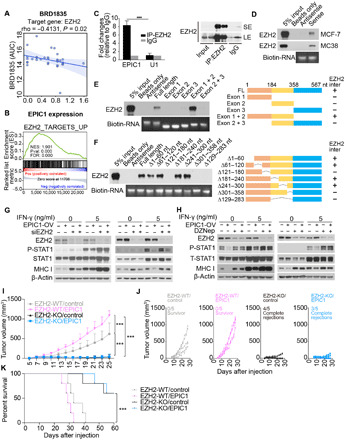
(A) Correlation between EPIC1 expression and PRC2 inhibitor response in breast cancer cell lines. (B) Gene set enrichment analysis of enhancer of zeste homolog 2 (EZH2) targets in siEPIC1-treated MCF-7 cell lines. (C) The enrichment of EPIC1 and U1 by EZH2 RNA immunoprecipitation assay analyzed by real-time quantitative polymerase chain reaction (qPCR). Immunoblot of EZH2 indicates the immunoprecipitation efficiency of EZH2 (right). LS, long exposure; SE, short exposure. (D) Immunoblot of EZH2 protein retrieved by in vitro transcribed EPIC1 from MCF-7 and MC38 cells’ nuclear extracts. (E and F) Immunoblot of EZH2 pulled down by indicated EPIC1 truncations (E) and deletions (F). The right panels are the schematic of indicated EPIC1 deletions and their binding with EZH2 (i.e., EZH2-inter). (G and H) Measurement of p-STAT and MHC-I protein levels by immunoblot in MCF-7 (left) and HCT116 (right) stable cell line overexpressing EPIC1 and rescued with EZH2 siRNA treatment (G) or EZH2 inhibitor DZNep treatment (H). (I and J) Tumor size of EZH2 wild-type (WT) or knocked out (KO) 4T1.2 cells stably expressed by empty vector (control) and EPIC1 (n = 5 animals per group). Tumor size was measured every other day. ***P < 0.001. (K) Survival curve for each group in (I).
EZH2 is the functional enzymatic subunit of PRC2, which plays critical roles in cell differentiation (37) and tumor metastasis (38). Most recent evidence suggests that its activation in tumor cells can also regulate tumor immune response through inhibition immunogenicity (39) and TH1 chemokine expression (40). However, how EZH2 itself is activated in solid tumor remains unclear. To investigate the role of EZH2 in EPIC1’s regulation of immune response, we performed RNA immunoprecipitation (RIP) and revealed that EPIC1 could be enriched by EZH2 RIP (Fig. 6C). In addition, RNA pull-down assay showed that EZH2 protein from both human breast cancer cell line MCF-7 and murine colorectal cancer cell line MC38 can be pulled down by in vitro–transcribed EPIC1 RNA (Fig. 6D). We constructed truncated mutants of EPIC1 to further map the EZH2-binding sequence in EPIC1. This analysis indicated that exon 1 [1 to 184 nucleotides (nt)] and exon 2 (185 to 358 nt) together are enough for EPIC1 to bind with EZH2 protein (Fig. 6E). We further constructed seven deleted mutants within the exon 1 and exon 2 regions of EPIC1 and have revealed that the 121- to 180-nt and 301- to 358-nt regions are necessary for EPIC1’s binding to the EZH2 protein (Fig. 6F). EPIC1 lacking EZH2 binding sequence (i.e., 301- to 358-nt region) failed to suppress STAT1 activation and MHC-I expression (fig. S9, A and B). The silencing of EZH2 reversed the effect of EPIC1 and abolished its inhibition of both IFN-γ–JAK–STAT1 signaling and MHC-I expression (Fig. 6G and fig. S8C), which was further confirmed by treatment with an EZH2 inhibitor DZNep to EPIC1-overexpressed cells (Fig. 6H and fig. S8D). Moreover, knockout of EZH2 in 4T1.2 cells blocked EPIC1’s tumor-promoting effect in vivo (Fig. 6, I to K). Together, these data demonstrated that the inhibition of EPIC1 to IFN-γ–JAK–STAT1 signaling and antigen presentation pathway depends on EZH2.
Activation of the EPIC1-EZH2 axis leads to anti-PD1 resistance through epigenetically silencing IFNGR1 and antigen presentation genes
EZH2 can suppress gene expression by trimethylating lysine 27 on histone H3 (H3K27me3) at the promoter region of its target genes (41–43). Given that EPIC1 can interact with EZH2 and inhibits IFNGR1 on both the mRNA and protein level, we performed chromatin immunoprecipitation (ChIP) assays to examine the occupancy of EZH2- and H3K27me3- at IFNGR1 promoters. This analysis revealed that the overexpression of EPIC1 promoted EZH2 and H3K27me3 occupancy at the promoter of the IFNGR1 gene (Fig. 7A). A recent study showed that EZH2 can directly silence antigen processing and presentation genes through trimethylating H3K27 at the promoters of those genes (39). We also noticed that EPIC1 could down-regulate the basal level of MHC-I without IFN-γ treatment (Fig. 4, B to E). We thus further examined if the EPIC1-EZH2 axis can directly regulate antigen processing and presentation genes’ expression. The ChIP assays demonstrated that EPIC1 enhanced the binding of EZH2 and H3K27me3 on the promoters of antigen presentation genes and directly regulated their expression (Fig. 7, A and B, and fig. S10, A and B). These observations suggest that EPIC1 can, directly and indirectly, inhibit antigen presentation, which is mediated by EZH2 (Fig. 7F).
Fig. 7. Activation of EPIC1-EZH2 axis leads to anti-PD1 resistance through epigenetically silencing IFNGR1 and antigen presentation genes.
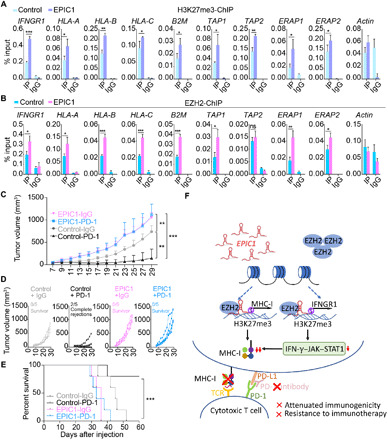
(A and B) ChIP-qPCR analysis of H3K27me3 (A) and EZH2 (B) occupancy on the promoters of indicated genes in MCF-7 cells stably overexpressed with empty vector (control) or EPIC1. (C and D) Tumor size of 4T1.2 cells stably expressed by the empty vector (control) and EPIC1 after anti–PD-1 and isotype control antibody (IgG). Mice were treated with a control IgG antibody (n = 5) or anti–PD-1 antibody (n = 5). Tumor size was measured every other day and plotted individually in (D). *P < 0.05; **P < 0.01; ***P <0.001. (E) Survival curve for each treated group in (C). (F) Proposed model depicting the regulation of antitumor immunity and resistance to checkpoint blockade therapy by lincRNA EPIC1 in tumor cells.
Recent clinical studies revealed that deficiency in antigen presentation and IFN-γ–JAK–STAT1 pathways resulted in the acquired resistance to immune-checkpoint blockade therapies (44–46). Given the observation that EPIC1 overexpression decreased the MHC-I–mediated antigen presentation and JAK-STAT1 signaling, we sought to determine if EPIC1 can induce the resistance to anti–PD-1 therapy. For the control 4T1.2 mouse model, consistent with a previous study (47), anti–PD-1 antibody treatment delayed 4T1.2 tumor progression compared with isotype control antibody (Fig. 7, C and D). In contrast, there is no significant therapeutic difference between anti–PD-1 and isotype control antibodies in EPIC1-overexpressed 4T1.2 tumors (Fig. 7, C and D). Moreover, anti–PD-1 antibody therapy resulted in significant improvement of overall survival for control 4T1.2 tumors, but not for EPIC1-overexpressed tumors (Fig. 7E). These data suggest the critical roles of EPIC1 in promoting resistance of checkpoint blockade therapy in vivo. Together, our finding indicated that EPIC1 enhanced the epigenetic silencing of IFNGR1 and antigen presentation genes via specific binding with EZH2. This EPIC1-mediated inhibition of the IFN-γ–JAK–STAT1 signaling and antigen presentation enables tumor cells to evade the immune surveillance and develop resistance to immune checkpoint blockade therapy (Fig. 7F).
DISCUSSION
In the past decade, enormous genomic efforts have been invested in identifying the clinically actionable biomarkers and regulators for the tumor immune response and immunotherapy response. Multiple computational platforms (26, 48) have been developed to quantitatively estimate tumor immune cell infiltration using bulk tumor RNA-seq data. Despite using different algorithms and statistical frameworks, all of the existing computational platforms rely on immune-specific protein-coding gene markers that are selected on the basis of prior knowledge. In this study, by integrating the noncoding transcriptome with the immune response profile of patients from 32 cancer types, we have characterized four clusters of lincRNAs that show distinct correlation patterns with tumor immune response in multiple cancer types. We have shown that a great number of tumor immune associative lincRNAs are expressed in a highly immune tissue-specific manner, which may be involved in the differentiation and function of innate and adaptive immune cells. On the basis of these immune-specific lincRNAs, we have further developed a LIMER score that can predict the immune cell infiltration and tumor patient prognosis in multiple cancer types. To the best of our knowledge, the LIMER is the first LIMER score that can be applied to the bulk-tumor RNA-seq data and robustly estimate the antitumor immune response in tumor tissue.
On top of the immune-specific lincRNA that can predict tumor immune response, our integrative analysis has also identified tumor cell–specific lincRNAs, which may be involved in the regulation of tumor immune response. Few genotype-immunophenotype association studies can establish causal relationships regarding whether and how a genetic/epigenetic change in tumor cells regulates immune response (16). In this study, by further integrating with DNA methylation data in the same patients with cancer, we have identified candidate lincRNAs that are specifically expressed in tumor cells and are associated with tumor immune response. We envision that some of these tumor cell–specific lincRNAs may play important roles in the tumor-intrinsic mechanism for immune suppression. Tumor-intrinsic mechanisms for immune suppression have been recently demonstrated to be key drivers for CTL suppression (26, 49). Previous studies have shown that loss of MHC, inhibition of IFN-γ, tumor necrosis factor, nuclear factor κB, and Wnt pathways enable tumor cells to evade recognition or to be resistant to the cytotoxic effect from immune cells (50). Most recently, lincRNAs are also emerging to have important roles in tumor-intrinsic mechanism for immune suppression. For instance, lincRNA LINK-A down-regulates antigenicity and intrinsic tumor suppression by enhancing the degradation of antigen peptide-loading complex, which inhibits tumor-associated antigen presentation and T cell recognition (30). In this study, our computational analysis in patients with cancer revealed that epigenetic activation of EPIC1 inversely correlates with the decreased MHC expression and decreased CD8+ T activation and infiltration in multiple cancer types. Our in vitro and in vivo models demonstrated that EPIC1 plays an important role in tumor immune evasion and immunotherapy resistance by simultaneous suppression of the IFN-γ–JAK–STAT1 and antigen presentation pathways in tumor cells. We have further revealed that EPIC1 recruited EZH2 to epigenetically silence the expression of IFN-γ receptor IFNGR1 and antigen presentation genes in tumor cells. The simultaneous inhibition of IFN-γ–JAK–STAT1 signaling and antigen presentation pathway by EPIC1-EZH2 axis emerges as a novel mechanism for tumor immune evasion in solid tumors. In our future study, we will identify the mouse EPIC1 ortholog and create mouse models to study the in vivo functions of EPIC1 in tumor immune evasion.
EZH2 has been well established to play a critical role in cancer initiation and metastasis (51). However, its role in tumor immunity has only been recently revealed. Ennishi and colleagues (19) have recently performed an integrative genomics study of 457 diffuse large B cell lymphoma (DLBCL) cases and found that EZH2 mutated in DLBCL tumors are characterized with decreased MHC-I and MHC-II expression compared with EZH2 wild-type tumors. Moreover, a recent study has also shown that EZH2 can epigenetically silence chemokine CXCL10 expression in tumor cells and results in the suppression of T cell infiltration (40).
Although its roles in tumorigenesis and immune modulation has been established to be important, how EZH2 is activated in solid tumor remains unclear. In follicular lymphoma and DLBCL, gain-of-function mutations in the EZH2 catalytic domain (e.g., SET domain) have been identified in 10 to 25% of patients (52, 53). However, in the solid tumor, the EZH2 gain-of-function mutations are at very low frequeqncy. Alternatively, the activation of EZH2 in solid tumors seems to be achieved by the genomic/epigenetic alterations of its regulators. For example, loss of function of INI1, which is a core component of the SWI/SNF (SWItch/Sucrose Non-Fermentable) complex that acts in opposition to EZH2, leads to an oncogenic dependency on EZH2 in solid tumors (54–57). In this study, we have shown that EZH2 in the solid tumor can be activated by an oncogenic lincRNA, EPIC1, which has been previously demonstrated to be EA in up to 50% of tumor samples across multiple cancer types (27). This reveals a novel epigenetic mechanism for EZH2’s activation in the solid tumor and its subsequent suppression of TME and the facilitation of immune evasion. Notably, we observed that EPIC1 overexpression also decreased the chemokine CXCL10 expression in human cancer cells and murine cancer cells (fig. S6A). In addition, because programmed death ligand 1 (PD-L1) expression is dependent on IFN-γ, resistance to programmed cell death protein 1 (PD-1) treatment could also result from lack of PD-L1 expression on tumor cells. Future studies are necessary to determine whether EPIC1 can inhibit T cell infiltration and PD-1 response through inhibition of tumor CXCL10 secretion (40) and PD-L1 expression. Although we have shown that EPIC1 binds to EZH2 and increases EZH2 localization on IFNGR1 and MHC-1 promoter loci, it is not clear whether EPIC1 can regulate EZH2 chromatin recruitment globally or only a subset of EZH2 targets. Recently, a number of studies (58) have demonstrated that EZH2’s chromatin binding is dependent on its interaction with chromatin-associated RNA. In our future study, we will explore the mechanism how EPIC1 recruits and enhances EZH2’s binding of its target genes.
Collectively, the current study established a comprehensive landscape of the lincRNA–tumor immunity landscape. This should facilitate the ongoing effort in understanding lincRNAs’ role in tumor immune response. Moreover, the functional characterization of the EPIC1-EZH2 axis and their essential role in the epigenetic reprogramming of the TME will pave the way for developing novel cancer immunotherapy by providing promising therapeutic markers and targets.
MATERIALS AND METHODS
Clinical and transcriptome data in patients
We collected the clinical information and the transcriptome data of 11,080 TCGA (59) patients across 32 cancer types from the Genomic Data Commons (GDC) data portal. The analyses in this study were mostly restricted to primary tumor samples. For the cancer types where the primary tumor samples were not available, those that were metastatic were selected, resulting in a total number of 9626 tumor samples. The transcriptome data from GDC were annotated based on human reference genome GRCh38 and were quantified as the gene-level expression in FPKM upper quartile. On the basis of the annotated gene biotypes, 19,668 protein-coding genes and 7528 long intergenic noncoding RNAs were selected for the following analyses. To ensure cross-sample comparability, we normalized the expression level for all the genes by logarithmic transformation and normalization using the average expression of 98 housekeeping genes (60).
Genomic alteration data in patients
For DNA methylation, we used the same probe annotation for lincRNA regions as described in our previous publication (27) and obtained the Illumina 450K Human Methylation microarray beta values from 6616 TCGA patients.
Immune signature, immune infiltration, and nontumor fraction
The 68 immune signatures were obtained from previous studies (24), and an enrichment score was calculated for each signature and each patient using the single-sample gene set enrichment analysis (ssGSEA) (61). Within each cancer type, the relative immune infiltration abundance of T cells, B cells, macrophages, dendritic cells, and natural killer cells was estimated by TIMER (25) for each individual sample.
Identification of lincRNA-immunity regulatory clusters
To study the relationship between lincRNA and immune responses in cancer, we first calculated the Spearman correlation between lincRNA expression and immune signature enrichment scores across 32 cancer types. To identify significant genes in the correlation analysis, we applied the Benjamini-Hochberg method to convert the correlation P values to false discovery rates (FDRs) and set FDR <0.1 as a threshold of significance. Next, we created a consensus regulation (CR) score to measure the extent to which an lincRNA associates with a corresponding immune signature in a pan-cancer manner: For each lincRNA m with a given signature n, the CR score is calculated by
βi and FDRi represent the coefficient and FDR of the Spearman correlation in the cancer type i, respectively. This procedure generated a raw CR matrix and was further reduced to 4292 lincRNAs whose absolute CR scores were higher or equal to 5 in at least 10 signatures, resulting in a final core CR matrix. To identify patterns between the lincRNAs in the core CR matrix, we applied t-SNE to embed these lincRNAs into a low-dimensional space. Specifically, t-SNE converted the similarity in CR profiles between lincRNAs to joint probabilities. It embedded each lincRNA in a way that lincRNAs showing similar CR profile will have a higher probability of being picked as neighbors than those of the dissimilar ones. To increase the stableness of embedding, t-SNE was initialized by the principal components analysis. This procedure finally led to a two-dimensional t-SNE map embedded with 4292 lincRNAs.
Subsequently, a clustering algorithm named density-based spatial clustering of applications with noise (DBSCAN) was implemented to identify lincRNA-immune regulatory clusters from the t-SNE map. The parameters of DBSCAN were set to min_samples = 150 and eps = 0.077, with the aim that each high-density region on the map is assigned to a separate cluster. This procedure revealed four lincRNA-immunity regulatory clusters with distinct CR profiles. Using these four clusters as initial assignment and the core CR matrix as the training set, a K-nearest neighbor classifier was trained with leaf_size = 75 and n_neighbours = 10. Using the trained classifier, the identity of all lincRNAs was predicted and validated based on fivefold cross-validation showing a high accuracy (98.5%) for the classification of the initial four clusters. LincRNAs with a posterior probability lower than 0.99 were marked as “noise” and were excluded from the following analyses.
To characterize the lincRNA-immunity regulatory clusters, Spearman correlation analysis was applied to the lincRNA expression, nontumor fraction, and immune cell infiltration. GSEA was applied to identify representative signatures for each cluster. The final resulting classification and representative signatures are shown in fig. S1B.
LincRNA selection for LIMER signature and prediction of clinically beneficial immune response
To identify lincRNAs that are specifically expressed in immune cells, we obtained the expression profiles of immune-related organs from Human Protein Atlas (62) and Illumina Body Map (63). Five immune-related organs were selected for the downstream analyses: leukocyte, lymph node, bone marrow, spleen, and tonsil. We required the lincRNAs to have an average expression level among the top 10% in immune-related organs from both databases and have a positive correlation with immune response in multiple cancer types, i.e., the lincRNAs should be in the C3 (positively correlated with immune response in multiple cancer types) cluster. These criteria resulted in a list of 105 lincRNAs, which were used as the LIMER signature. Next, we defined the LIMER score of a given patient as the median expression of lincRNAs in the LIMER signature. We computed the LIMER score for each patient from 17 TCGA cancer types with sufficient tumor sample size (n > = 200), available immune infiltration estimation, and survival information. To evaluate whether the LIMER score can predict immune response in patients, we correlated LIMER score with the immune infiltration fraction estimated by the TIMER algorithm (25, 26) using Spearman correlation methods. To test whether the LIMER score can indicate clinically beneficial immune response, we applied the Cox regression, log-rank test, and Kaplan-Meier estimators to LIMER score and used progression-free intervals as survival indicators for each cancer type. To compare the LIMER score between tumor and adjacent/normal samples, we excluded two cancer types [SKCM (Skin Cutaneous Melanoma) and LGG (lower-grade glioma)] from the above 17 cancer types due to the insufficient number of normal samples.
Identification of tumor-intrinsic regulators of the immune response
To identify potential tumor-intrinsic regulators of the immune response, we filtered out the lincRNAs that are expressed in any of the following immune-related organs: leukocyte, lymph node, bone marrow, spleen, and tonsil. Next, we selected lincRNAs that are overexpressed in tumor compared with adjacent normal with the below criteria: The lincRNA should have a P value from Wilcoxon test less than 0.01; if more than 20% of total lincRNAs can pass this criterion, then only top 20% lincRNAs with most differential expression will be selected. Last, to exclude those lincRNAs that are regulated by the immune response, we further overlapped the tumor-specific immune-associated lincRNAs with the EA lincRNAs in patients with cancer as defined in the previous publication (27).
RNA-seq analysis
We used STAR (Spliced Transcript Alignment to a Reference) and RSEM (RNA-Seq by Expectation Maximization) to profile RNA-seq data of A2780, A2780cis, MCF-7, and Hs578T cell lines after EPIC1 knockdown. The gene expression was quantified in log2-transformed FPKM and annotated based on the human reference genome GRCh38. The RNA-seq data can be downloaded from the Gene Expression Omnibus (GEO) (GSE98538). To interpret the function of regulated genes after EPIC1 siRNA treatment, GSEA was performed using the 50 cancer hallmark gene sets and 68 immune signatures on the log2 fold change of protein-coding genes. Significance was defined by FDR <0.1.
Single-cell ATAC-seq analysis
To address the tissue specificity of EPIC1, we have downloaded the single-cell ATAC-seq data from 28,274 TILs and ~5000 cancer cells from a published study (28). We used the established cell type information from the study and mapped the chromosome coordinate of EPIC1’s promoter region to the single-cell chromatin accessibility profile of tumor-infiltrated immune cells and tumor cells.
Correlation analysis of drug response
Drug response data of 545 agents tested across 722 cancer cell lines are downloaded from the CTRP (35, 36). The drug response in each cell line is indicated by area under the curves. Expression data of cell lines are from the Cancer Cell Line Encyclopedia (64) and were downloaded from the Expression Atlas (65). The correlation analysis between EPIC1 expression and drug response was only performed on 582 cell lines (all cancer types) and 32 breast cancer cell lines with both drug response and genomic alteration data available.
Cell lines and reagents
Human breast cancer cell MCF-7, lung cancer cell NCI-H1299, and colorectal cancer cell HCT116 cell were purchased from the American Type Culture Collection. Murine breast cancer cell line 4T1.2 is a gift from S. Li’s laboratory. Murine colorectal cancer cell line MC38 was obtained from B. Lu’s laboratory. Peripheral blood mononuclear cells (PBMCs) transduced with TCR gp100, 526-MEL, and 888-MEL are from Dr. U. Kammula’s laboratory. PBMCs were cultured in complete medium [RPMI-1640, 10% heat-inactivated human AB serum (Gemini Bio-Products, Woodland, CA), 2 mM l-glutamine (Invitrogen, Carlsbad, CA), 50 U/penicillin (Invitrogen), streptomycin (50 μg/ml) (Invitrogen), gentamicin (50 μg/ml) (Invitrogen), 10 mM Hepes (Invitrogen), and Amphotericin B (250 ng/ml) (Invitrogen)]. MCF-7, NCI-H1299 cell, HCT116 cell, 4T1.2 cell, and MC38 cell were cultured in Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with 10% fetal bovine serum (FBS) (Gibco). 526-MEL and 888-MEL were cultured in RPMI-1640 (Hyclone) supplemented with 10% FBS (Gibco).
Human and mouse IFN-γ was purchased from Sigma-Aldrich. To treat the cells for 24 hours, 1, 5, and 25 ng/ml were used. JAK1/2 inhibitor ruxolitinib and EZH2 inhibitor DZNep were purchased from Selleckchem. Ruxolitinib (5 μM) was used to treat the cells for 24 hours before detection. DZNep (5 μM) was applied to treat the cells for 72 hours before detection. We use a CRISPR strategy for the deletion of EZH2. Cells were infected with the lentivirus packaged by Cas9 and single-guide RNA (sgRNA) expression plasmid encoding puromycin resistance (Addgene plasmid, #52961). The guide sequences used were 5′-ACACGCTTCCGCCAACAAAC-3′, 5′-TTCCAATCGTCAGAAAATTT-3′, and 5′-AGAACTTCATCCCCCATATA-3′ for mouse EZH2. The successfully knocked out cells were selected by immunoblot analysis for the lack of EZH2 proteins. CRISPR-Cas9 SAM 3-vector [lenti dCAS-VP64, lentiMPH, and lenti sgRNA (MS2)] system was used to up-regulate endogenous EPIC1 expression.
The control guide sequences used were 5′-CTGAAAAAGGAAGGAGTTGA-3′, and the guide RNA targeting the promoter of EPIC1 was 5′-GATTCTCTCTGCCTGACCCA-3′.
Mice
Six- to 8-week old female C57BL/6 mice, BALB/c mice, and BALB/c nude mice were purchased from the Jackson laboratory. These mice were used for tumor implantation experiments. C57BL/6-Tg (TcraTcrb)1100Mjb/J OT-1 transgenic mice were purchased from the Jackson laboratory and used for OT-1 T cell isolation. For in vivo tumor implantation, 1 × 105 4T1.2, 1 × 106 MC38, or 1 × 106 CT26 cells stably expressing empty vector or humanized EPIC1 resuspended in 50 μl of phosphate-buffered saline (PBS) were injected into the mammary fat pad of BLAB/c mice, C57BL/6 mice flank, or BLAB/c mice flank, respectively. The tumors were measured every other day beginning on day 3 from challenge to time of death. Death was defined when the volume of tumor size reaches 1000 mm3. For the TIL analysis, the tumors were harvested at day 15 after cell injection. For anti–PD-1 therapy, 4T1.2 control and EPIC1-overexpressed mice were grouped into immunoglobulin G (IgG) antibody (BioXcell, MPC-11) treatment and anti–PD-1 antibody (BioXcell, Clone J43) treatment. IgG and anti–PD-1 antibodies were conducted by intraperitoneal injection of 200 μg per mouse from day 10 every 4 days for a total of three injections. All animal studies were performed in accordance with the institutional guidelines, and the experiments followed the protocols approved by the Institutional Animal Care and Use Committee of University of Pittsburgh.
Antibodies
For immunoblotting, these antibodies are used: rabbit anti-STAT1 (CST, #14994), rabbit anti–phospho-STAT1 (CST, #7649), rabbit anti-EZH2 (CST, #5246), mouse anti–MHC-I class I (Santa Cruz Biotechnology, #sc-55582), rabbit anti-IFNGR1 (Millipore, #MABF753), and mouse anti–β-actin (Sigma-Aldrich, #A5441).
For flow cytometry, the following fluorochrome-conjugated antibodies are used: anti-mouse H-2Kb/H-2Db (clone 28-8-6, BioLegend), anti-mouse H-2Kb bound to SIINFEKL (clone 25-D1.16, BioLegend), anti-mouse CD3 (clone 17A2, BioLegend), anti-mouse CD28 (clone 37.51, BioLegend), anti-mouse granzyme B (clone GB11, BioLegend), anti-mouse B220 (clone RA3-6B2, BioLegend), anti-mouse CD49b (clone DX5, BioLegend), anti-mouse Gr-1 (clone RB6-8C5, BioLegend), anti-mouse MHC-II (clone M5/114.15.2, BioLegend), anti-mouse F4/80 (clone BM8, BioLegend), anti-mouse IFN-γ (XMG1.2, eBioscience), anti-mouse CD11b (M1/70, eBioscience), anti-mouse NK1.1 (clone PK136, BD), anti-mouse CD103 (clone M290, BD), anti-mouse CD206 (clone MR5D3, BD), anti-mouse CD24 (clone M1/69, BD), anti-mouse CD4 (clone GK1.5, BD), anti-mouse CD45 (clone 30-F11, BD), anti-mouse CD8a (clone 53-6.7, BD), anti-mouse Foxp3 (clone MF23, BD), anti-mouse γδ TCR (clone GL3, BD), anti-human CD3 (SK7, BioLegend), anti-human CD4 (SK3, BioLegend), anti-human CD8 (SK1, BioLegend), and anti-human 137(4-1BB) (4B4-1, BioLegend).
Vectors, RNA interference, and lentiviral transfection
The full length of EPIC1 was amplified from the plasmid described before (27) and inserted into the PCDH-CMV vector, which was then named PCDH-EPIC1. The deletion mutants and truncated EPIC1 were constructed by using QuickChange II XL Site-Direct Mutagenesis Kit (Agilent Technologies). All of the abovementioned plasmids were verified by DNA sequencing at Genomics Research Core, University of Pittsburgh. The full length of OVA was purchased from the Addgene. These constructs, together with pMD2.G and psPAX2, were transfected into 293T cells to produce lentivirus, the supernatant-containing lentivirus was collected at 48 hours after transfection. Cells were infected by the lentiviruses for 24 hours and selected with puromycin to establish stable expressing cell lines.
The siRNA targeting EPIC1, EZH2, and scramble was transfected with Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. The RNA or proteins were extracted at 48 to 72 hours after transcription. The sequences of these siRNAs are listed as follows: EPIC1, 5′-TCTAGAAGTCCGCCATTGCAAACACG-3′ (forward) and CTCGAGGCACCAGCAATTTTTTTTAT (reverse). The scrambled siRNA sequence was 5′-GUGCGUUGUUAGUACUAAU-3′. The siRNA sequences for EPIC1 knockdown were 5′-CCUUCAGACUGUCUUUGAA-3′ and 5′-GCUUUCUCUCGGAAACGUG-3′. The siRNA sequence for human EZH2 knockdown was 5′-GACUCUGAAUGCAGUUGCU-3’.
In vitro T cell killing assays
CD8+ T cells were isolated from spleens of OT-1 TCR transgenic mice using a CD8a+ T Cell Isolation Kit (Miltenyi Biotec) with a MidiMACS separator. Purified OT-1 T cells were then suspended in RPMI medium (Gibco) containing 10% FBS, anti-mouse CD3 (clone: 2C11, 2 μg/ml), and anti-mouse CD28 (clone: 37.51, 2 μg/ml). After 48 hours of stimulation, activated OT-1 T cells were seeded into a new plate with fresh medium for 2 days of further culture. Afterward, T cells were collected for the coculture assay. MC38-OVA cells (5 × 104) stained with CellTrace Far Red dye (Thermo Fisher Scientific) were exposed to stimulated OT-1 T cells at E:T ratios of 10:1, 5:1, 2:1, and 0:1 at 37°C for 24 hours. IFN-γ in the supernatant was measured by enzyme-linked immunosorbent assay, and the remaining tumor cells were analyzed by flow cytometry.
Gp100 TCR-transduced CD8+ T cells together with gp100+ 526-MEL/888-MEL were used for coculture assay. gp100+ 526-MEL/888-MEL cells (1 × 105) and T cells (1 × 105) were well mixed in T cell and seeded into 96-well plates. After 24 hours of coculture, T cells were collected and stained with propidium iodide (BD) followed by human anti-CD3+ (SK7, BioLegend), anti-CD4+ (SK3, BioLegend), anti-CD8+ (SK1, BioLegend), and anti-CD137 (4B4-1, BioLegend), which were then analyzed by flow cytometry. Deidentified human CD8+ T cells were obtained from UPMC Hillman Cancer Center under an exempt institutional review board protocol.
Analysis of TILs by flow cytometry
Mice were euthanized around 15 days after transplantation when the diameter of the tumor reached 8 to 10 mm. Isolated tumors were cut into small pieces and digested with Liberase TL (0.25 mg/ml) (Roche) and deoxyribonuclease (DNase) (0.33 mg/ml) (Sigma-Aldrich) in 37°C for 30 min. Until now, single-cell suspensions were obtained. For IFN-γ and granzyme B staining, cells were stimulated for 4 hours with phorbol 12-myristate 13-acetate (50 ng/ml) (Sigma-Aldrich) and ionomycin (1 μg/ml) (Sigma-Aldrich) in the presence of brefeldin A (10 μg/ml). After stimulation, cells were stained for indicated antibodies of surface marker, followed by treatment with fixation and permeabilization buffer (eBioscience) according to the manufacturer’s instructions. Cells were further stained with antibodies of intracellular markers. All the samples were applied to Fortessa FACS (BD Biosciences) and analyzed by Flowjo software (TreeStar).
Reverse transcription quantitative polymerase chain reaction
Total RNA from cultured cells were isolated using TRIzol. cDNA was synthesized from 2 μg of RNA of each cell line by Reverse Transcription Kit (Applied Biosystems, #4368813). Quantitative polymerase chain reaction (qPCR) was performed as reported previously (Cancer Cell paper). Primer sequences used for qPCR are listed as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (human), 5′-GGTGAAGGTCGGAGTCAACG-3′ (forward) and 5′- TGGGTGGAATCATATTGGAACA-3′ (reverse); IFNGR-1 (human), 5′-GTCAGAGTTAAAGCCAGGGTTG-3′ (forward) and 5′-CTTCCTGCTCGTCTCCATTTAC-3′ (reverse); HLA-A (human), 5′-AAAAGGAGGGAGTTACACTCAGG-3′ (forward) and 5′-GCTGTGAGGGACACATCAGAG-3′ (reverse); HLA-B (human), 5′-CTACCCTGCGGAGATCA-3′ (forward) and 5′-ACAGCCAGGCCAGCAACA-3′ (reverse); HLA-C (human), 5′-CACACCTCTCCTTTGTGACTTCAA-3′ (forward) and 5′-CCACCTCCTCACATTATGCTAACA-3′ (reverse); B2M (human), 5′-ATGTCTCGCTCCGTGGCCTT-3′ (forward) and 5′-GACTTTCCATTCTCTGCTGG-3′ (reverse); TAP1 (human), 5′-GCTGTTCCTGGTCCTGGTGG-3′ (forward) and 5′-TTTCGAGTGAAGGTATCGGC-3′ (reverse); TAP2 (human), 5′-CAATAGCAGCGGAGAAGGTG-3′ (forward) and 5′-CTCGGCCCCAAAACTGCGAA-3′ (reverse); TAPBP (human), 5′-CTCAGCCTCTCCAGCCTCTT-3′ (forward) and 5′-GAGCATCTTGTCCCAGTCTC-3′ (reverse); ERAP1 (human), 5′-CGGAGACTTTCCACGGATTT-3′ (forward) and 5′-GAAGGCAGGTTCATCAAAGC-3′ (reverse); ERAP2 (human), 5′-GGTGATGGCTTTGAAGGGT-3′ (forward) and 5′-CCTGCTCTCTCTTCGTATC-3′ (reverse); IRF1 (human), 5′-CATGGCTGGGACATCAACAA-3′ (forward) and 5′-TTGTATCGGCCTGTGTGAATG-3′ (reverse); IRF9 (human), 5′-AGGACCAGGATGCTGCCTTC-3′ (forward) and 5′-TAGGGCTCAGCAACATCCA-3′ (reverse); CXCL-10 (human), 5′-GCTGCCTTATCTTTCTGACT-3′ (forward) and 5′-GGACAAAATTGGCTTGCAGG-3′ (reverse); Gapdh (mouse), 5′-CCTCGTCCCGTAGACAAAAT-3′ (forward) and 5′-GAGGTCAATGAAGGGGTCGT-3′ (reverse); H2d1 (mouse), 5′-AAGAACGGGAACGCGACGCT-3′ (forward) and 5′-AACTGCCAGGTCAGGGTGAT-3′ (reverse); H2k1 (mouse), 5′-ATACCTGAAGAACGGGAACG-3′ (forward) and 5′-GTCAGCAGGGTAGAAGCCCA-3′ (reverse); B2m (mouse), 5′-CTGGTGCTTGTCTCACTGAC-3′ (forward) and 5′-GTATGTTCGGCTTCCCATTC-3′ (reverse); Tap1 (mouse), 5′-CCACTCCTGCTTATCTTGGA-3′ (forward) and 5′-GATAAGAAGAACCGTCCGAG-3′ (reverse); Tap2 (mouse), 5′-GCCATCTTTTTCATGTGCCT-3′ (forward) and 5′-TCTCGTATCCGCAGGTTGA-3′ (reverse); Erap1 (mouse), 5′-CCTGTCTGAGAGTTTCCATG-3′ (forward) and 5′-ATCAAAGCAGGGAAAAGCCA-3′ (reverse); Tapbp (mouse), 5′-CTGCGGGAGCCTGTCGTCAT-3′ (forward) and 5′-CAGGGCGGAGGGTGCGTAGG-3′ (reverse); Cxcl-10 (mouse), 5′-ATCCTGCTGGGTCTGAGTGG-3′ (forward) and 5′-TATGGCCCTCATTCTCACTG-3′ (reverse).
The relative mRNA expression was normalized to GAPDH and presented as fold changes.
Western blot
The total protein was extracted from indicated cells with radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with 1× protease inhibitor (Sigma-Aldrich) and phosphatase inhibitors (Thermo Fisher Scientific) for 30 min. The protein concentration was determined by a BCA Protein Assay Kit (Thermo Fisher Scientific). Afterward, the cell lysates were supplemented with 5× SDS loading buffer and then denatured at 98°C for 10 min, further resolved on SDS–polyacrylamide gel electrophoresis, and transferred onto polyvinylidene difluoride membranes (Bio-Rad). The membrane then was incubated with appropriate primary antibodies overnight at 4°C, followed by detection with horseradish peroxidase–conjugated secondary antibodies for another for 1 hour at room temperature. Signal was visualized by enhanced chemiluminescence substrate (Thermo Fisher Scientific) and exposed by films with the AX700LE film processor (Alphatek).
ChIP analysis
MCF-7 and 4T1.2 cells were cross-linked with 37% formaldehyde, and the final concentration of 1.42% was obtained for 15 min at room temperature, which was quenched with 125 mM glycine for further 5 min at room temperature. Afterward, the cells were collected and washed with cold PBS twice and lysed with 1 ml of IP buffer [150 mM NaCl, 50 mM tris-HCl (pH 7.5), 5 mM EDTA, NP-40 (0.5% v/v), Triton X-100 (1.0% v/v)] containing protease inhibitors (Sigma-Aldrich). The lysates were then sonicated to shear the chromatin to yield DNA fragment around 0.5 to 1.0 kb and cleared by centrifuging at 12,000g for 10 min, which were then incubated with 2 μg of indicated antibodies and IgG overnight at 4°C, respectively. Meanwhile, 10% of cleared lysates were taken as input. Protein A/G agarose beads (40 μl) (Thermo Fisher Scientific) were supplied into the lysates for a further 2-hour incubation at 4°C. The beads were washed with 1 ml of cold IP buffer for six times. Last, 100 μl of 10% Chelex 100 slurry was directly added into washed beads. After briefly mixing the samples, they were boiled for 10 min to isolate the DNA. Real-time qPCR detection was performed to evaluate the Ct value of targets in IP, normal IgG, and input DNA.
The primers used for target genes and control are listed as follows: human IFNGR1, 5′-TGACGGAAGTGACGTAAGGC-3′ (forward) and 5′-TACCGACGGTCGCTGGCTCCAA-3′ (reverse); human actin, 5′-AGTGTGGTCCTGCGACTTCTAAG-3′ (forward) and 5′-CCTGGGCTTGAGAGGTAGAGTGT-3′ (reverse); human HLA-A, 5′-ACAGGAGCAGAGGGGTCAGG-3′ (forward) and 5′-CAATCCATACACCGCCTTCG-3′ (reverse); human HLA-B, 5′-ACGAACTGCGTGTCGTCCAC-3′ (forward) and 5′-CTGCTCTTCTCCAGAAGGCA-3′ (reverse); human HLA-C, 5′-ACATTCAGGTGCCTTTGCAG-3′ (forward) and 5′-CCTGTGTGGGACTGAGATGC-3′ (reverse); human B2M, 5′-CACTCACCTGATTTTTGGTTC-3′ (forward) and 5′-ATAGACGCCTCCACTAATCCT-3′ (reverse); human TAP1, 5′-AAAAGGGAGGGAGATGGAGT-3′ (forward) and 5′-GAAAAAGGGGTGCTACTGGG-3′ (reverse); Mouse h2d1, 5′-TTGTATTCCCGGAAGTGACCTT-3′ (forward) and 5′-TCACTGTTTCCTAACCTCCACC-3′ (reverse); mouse h2k1, 5′-ACTTTAAGGAAAAGCCTCTCTCTCC-3′ (forward) and 5′-AAAGCCTCTTCCGGGAATACAA-3′ (reverse); mouse b2m, 5′-AATAAATGAAGGCGGTCCCAGG-3′ (forward) and 5′-TGGTGCCCTACTATCTAGGGTG-3′ (reverse); mouse tap1, 5′-GAGAAGAACACGACAGGCCA-3′ (forward) and 5′-TCAGGCTGTTCTGGAAGCTG-3′ (reverse); mouse tap2, 5′-GGCTCAGGCAAGTTTTCTCAAC-3′ (forward) and 5′-GACCTCCGAGCATGTTTTAAGAAG-3′ (reverse); mouse tapbp, 5′-TCCCAACACCCCTCTGTTTG-3′ (forward) and 5′-CGCCACCTCCCTTAAAACCA-3 (reverse); mouse cxcl9, 5′-ACTCCCCGTTTATGTGAAATGGA-3′ (forward) and 5′-ACCACAAATTGATCGTCCTGGG-3′ (reverse); mouse cxcl10, 5′-AGGAGCACAAGAGGGGAGAG-3′ (forward) and 5′-GGGAAGTCCCCTGTAAACCG-3′ (reverse).
RNA immunoprecipitation
Cells were washed with 1 × PBS twice and then cross-linked with 0.3% formaldehyde for 10 min at room temperature. The cross-link was quenched with 0.125 M glycine for 5 min at room temperature. Cells were washed with cold 1 × PBS two more times, and 1 ml of RIPA buffer [150 mM NaCl, 50 mM tris (pH 7.4), 1.0 mM EDTA, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.5 mM dithiothreitol, protease inhibitor (PMSF), and ribonuclease inhibitor] was applied to lysate the cells. The lysis was then cleared by centrifuging at 13,000 rpm for 10 min. Ten percent of the supernatants were taken as input, and the remaining lysates were collected and supplied with indicated antibodies and IgG for overnight incubation at 4°C. Afterward, 40 μl of protein A/G beads was added into lysates for a further 1 hour of incubation. The beads were washed with cold RIPA buffer for five to six times.
In vitro RNA pull-down assay
RNA pull-down assay was performed as described before with minor modification (27). Briefly, the biotinylated EPIC1 RNA was transcribed with a Biotin RNA Labeling Mix Kit (Roche, #11685597910) and T7 RNA polymerase (Roche, #10881775001). Three micrograms of transcribed RNA was applied for further pull-down assay. Meanwhile, the MCF-7 and MC38 cells were harvested and washed with sterilized and cold PBS twice, and then nuclei were collected by nuclear isolation buffer [1.28 M sucrose, 40 mM tris-HCl (pH 7.5), 20 mM MgCl2, and 4% Triton X-100]. One milligram of cleared nuclear lysates was used to incubate with in vitro transcribed RNA for overnight at 4°C. Afterward, 50 μl of magnetic beads (Thermo Fisher Scientific, catalog no. 65001) was added for another 1 hour of incubation at room temperature. The beads were washed six times and boiled with 1×SDS loading buffer for 10 min at 95°C. The RNA bound proteins were analyzed by Western blot.
Acknowledgments
We thank the Center for Simulation and Modeling (SaM) at the University of Pittsburgh for computing assistance. Funding: This study was supported by the Shear Family Foundation (to D.Y.), the American Cancer Society Research Scholar Award (132632-RSG-18-179-01-RMC to D.Y.), and the National Cancer Institute (1R01CA222274-01 to D.Y.). This project is supported in part by award P30CA047904. Author contributions: D.Y., W.G., and B.L. conceived the project and designed the experiments. D.Y., W.G., Yue W., and B.L. drafted the manuscript. W.G. performed most of the experiments with help from M.Y., Zehua W., Yue W., Zhiyuan W., and S.C., Yifei W. performed most of the bioinformatics analysis with help from S.L. and M.Z. M.Y. assisted with flow cytometry analysis. S.C., G.S.Y., and U.S.K. contributed key reagents and experiments. B.L., S.R., F.C.-B., C.F., S.L., W.X., R.L.F., and U.S.K. contributed to the discussions and editing of the manuscript. D.Y. supervised the work and acquired funding. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The source code in this study for analysis and visualization is available at http://github.com/dangoDANGO/EPIC1. The authors declare that all the other scripts generating the figures and supporting the findings of this study are available from the corresponding author upon reasonable request. RNA-seq of EPIC1 knockdown cell lines can be obtained from GEO (http://www.ncbi.nlm.nih.gov/gds) under accession number GSE98538 in a preprocessed format. The authors declare that all the other data supporting the findings of this study are available in the article and its supplementary data files or from the corresponding author upon reasonable request. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/7/eabb3555/DC1
REFERENCES AND NOTES
- 1.Schreiber R. D., Old L. J., Smyth M. J., Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Mittal D., Gubin M. M., Schreiber R. D., Smyth M. J., New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 27, 16–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atianand M. K., Caffrey D. R., Fitzgerald K. A., Immunobiology of long noncoding RNAs. Annu. Rev. Immunol. 35, 177–198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X., Yan C. C., Zhang X., You Z. H., Long non-coding RNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 18, 558–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hadjicharalambous M. R., Lindsay M. A., Long non-coding RNAs and the innate immune response. Noncoding RNA 5, 34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P., Xue Y., Han Y., Lin L., Wu C., Xu S., Jiang Z., Xu J., Liu Q., Cao X., The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 344, 310–313 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Ranzani V., Rossetti G., Panzeri I., Arrigoni A., Bonnal R. J. P., Curti S., Gruarin P., Provasi E., Sugliano E., Marconi M., de Francesco R., Geginat J., Bodega B., Abrignani S., Pagani M., The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat. Immunol. 16, 318–325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomez J. A., Wapinski O. L., Yang Y. W., Bureau J. F., Gopinath S., Monack D. M., Chang H. Y., Brahic M., Kirkegaard K., The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 152, 743–754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu G., Tang Q., Sharma S., Yu F., Escobar T. M., Muljo S. A., Zhu J., Zhao K., Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 14, 1190–1198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang W., Thomas B., Flynn R. A., Gavzy S. J., Wu L., Kim S. V., Hall J. A., Miraldi E. R., Ng C. P., Rigo F., Meadows S., Montoya N. R., Herrera N. G., Domingos A. I., Rastinejad F., Myers R. M., Fuller-Pace F. V., Bonneau R., Chang H. Y., Acuto O., Littman D. R., DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature 528, 517–522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Chen Y. G., Satpathy A. T., Chang H. Y., Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 18, 962–972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balli D., Rech A. J., Stanger B. Z., Vonderheide R. H., Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin. Cancer Res. 23, 3129–3138 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Wang Q., Hu B., Hu X., Kim H., Squatrito M., Scarpace L., deCarvalho A. C., Lyu S., Li P., Li Y., Barthel F., Cho H. J., Lin Y.-H., Satani N., Martinez-Ledesma E., Zheng S., Chang E., Sauvé C.-E. G., Olar A., Lan Z. D., Finocchiaro G., Phillips J. J., Berger M. S., Gabrusiewicz K. R., Wang G., Eskilsson E., Hu J., Mikkelsen T., De Pinho R. A., Muller F., Heimberger A. B., Sulman E. P., Nam D.-H., Verhaak R. G. W., Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quigley D., Silwal-Pandit L., Dannenfelser R., Langerød A., Vollan H. K. M., Vaske C., Siegel J. U., Troyanskaya O., Chin S.-F., Caldas C., Balmain A., Børresen-Dale A.-L., Kristensen V., Lymphocyte invasion in IC10/basal-like breast tumors is associated with wild-type TP53. Mol. Cancer Res. 13, 493–501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z., Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Ock C. Y., Hwang J.-E., Keam B., Kim S.-B., Shim J.-J., Jang H.-J., Park S., Sohn B. H., Cha M., Ajani J. A., Kopetz S., Lee K.-W., Kim T. M., Heo D. S., Lee J.-S., Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat. Commun. 8, 1050 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spranger S., Bao R., Gajewski T. F., Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Ennishi D., Takata K., Béguelin W., Duns G., Mottok A., Farinha P., Bashashati A., Saberi S., Boyle M., Meissner B., Ben-Neriah S., Woolcock B. W., Telenius A., Lai D., Teater M., Kridel R., Savage K. J., Sehn L. H., Morin R. D., Marra M. A., Shah S. P., Connors J. M., Gascoyne R. D., Scott D. W., Melnick A. M., Steidl C., Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov. 9, 546–563 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Slack F. J., Chinnaiyan A. M., The role of non-coding RNAs in oncology. Cell 179, 1033–1055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyer M. K., Niknafs Y. S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T. R., Prensner J. R., Evans J. R., Zhao S., Poliakov A., Cao X., Dhanasekaran S. M., Wu Y.-M., Robinson D. R., Beer D. G., Feng F. Y., Iyer H. K., Chinnaiyan A. M., The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans J. R., Feng F. Y., Chinnaiyan A. M., The bright side of dark matter: LncRNAs in cancer. J. Clin. Invest. 126, 2775–2782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahu A., Singhal U., Chinnaiyan A. M., Long noncoding RNAs in cancer: From function to translation. Trends Cancer 1, 93–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorsson V., Gibbs D. L., Brown S. D., Wolf D., Bortone D. S., Yang T.-H. O., Porta-Pardo E., Gao G. F., Plaisier C. L., Eddy J. A., Ziv E., Culhane A. C., Paull E. O., Sivakumar I. K. A., Gentles A. J., Malhotra R., Farshidfar F., Colaprico A., Parker J. S., Mose L. E., Vo N. S., Liu J., Liu Y., Rader J., Dhankani V., Reynolds S. M., Bowlby R., Califano A., Cherniack A. D., Anastassiou D., Bedognetti D., Mokrab Y., Newman A. M., Rao A., Chen K., Krasnitz A., Hu H., Malta T. M., Noushmehr H., Pedamallu C. S., Bullman S., Ojesina A. I., Lamb A., Zhou W., Shen H., Choueiri T. K., Weinstein J. N., Guinney J., Saltz J., Holt R. A., Rabkin C. S.; Cancer Genome Atlas Research Network, Lazar A. J., Serody J. S., Demicco E. G., Disis M. L., Vincent B. G., Shmulevich I., The immune landscape of cancer. Immunity 48, 812–830.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T., Fan J., Wang B., Traugh N., Chen Q., Liu J. S., Li B., Liu X. S., TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77, e108–e110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B., Severson E., Pignon J.-C., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J. C., Rodig S., Signoretti S., Liu J. S., Liu X. S., Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z., Yang B., Zhang M., Guo W., Wu Z., Wang Y., Jia L., Li S.; Cancer Genome Atlas Research Network, Xie W., Yang D., lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell 33, 706–720.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satpathy A. T., Granja J. M., Yost K. E., Qi Y., Meschi F., McDermott G. P., Olsen B. N., Mumbach M. R., Pierce S. E., Corces M. R., Shah P., Bell J. C., Jhutty D., Nemec C. M., Wang J., Wang L., Yin Y., Giresi P. G., Chang A. L. S., Zheng G. X. Y., Greenleaf W. J., Chang H. Y., Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Wang Z., Xu J., Li J., Li S., Zhang M., Yang D., Systematic identification of non-coding pharmacogenomic landscape in cancer. Nat. Commun. 9, 3192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Q., Ye Y., Chan L.-C., Li Y., Liang K., Lin A., Egranov S. D., Zhang Y., Xia W., Gong J., Pan Y., Chatterjee S. S., Yao J., Evans K. W., Nguyen T. K., Park P. K., Liu J., Coarfa C., Donepudi S. R., Putluri V., Putluri N., Sreekumar A., Ambati C. R., Hawke D. H., Marks J. R., Gunaratne P. H., Caudle A. S., Sahin A. A., Hortobagyi G. N., Meric-Bernstam F., Chen L., Yu D., Hung M.-C., Curran M. A., Han L., Lin C., Yang L., Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression. Nat. Immunol. 20, 835–851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castro F., Cardoso A. P., Gonçalves R. M., Serre K., Oliveira M. J., Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9, 847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou F., Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 28, 239–260 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Rees M. G., Seashore-Ludlow B., Cheah J. H., Adams D. J., Price E. V., Gill S., Javaid S., Coletti M. E., Jones V. L., Bodycombe N. E., Soule C. K., Alexander B., Li A., Montgomery P., Kotz J. D., Hon C. S.-Y., Munoz B., Liefeld T., Dančík V., Haber D. A., Clish C. B., Bittker J. A., Palmer M., Wagner B. K., Clemons P. A., Shamji A. F., Schreiber S. L., Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seashore-Ludlow B., Rees M. G., Cheah J. H., Cokol M., Price E. V., Coletti M. E., Jones V., Bodycombe N. E., Soule C. K., Gould J., Alexander B., Li A., Montgomery P., Wawer M. J., Kuru N., Kotz J. D., Hon C. S.-Y., Munoz B., Liefeld T., Dančík V., Bittker J. A., Palmer M., Bradner J. E., Shamji A. F., Clemons P. A., Schreiber S. L., Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu A., Bodycombe N. E., Cheah J. H., Price E. V., Liu K., Schaefer G. I., Ebright R. Y., Stewart M. L., Ito D., Wang S., Bracha A. L., Liefeld T., Wawer M., Gilbert J. C., Wilson A. J., Stransky N., Kryukov G. V., Dancik V., Barretina J., Garraway L. A., Hon C. S. Y., Munoz B., Bittker J. A., Stockwell B. R., Khabele D., Stern A. M., Clemons P. A., Shamji A. F., Schreiber S. L., An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Y., Deng P., Yu B., Szymanski J. M., Aghaloo T., Hong C., Wang C. Y., Inhibition of EZH2 Promotes Human Embryonic Stem Cell Differentiation into Mesoderm by Reducing H3K27me3. Stem Cell Reports 9, 752–761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zingg D., Debbache J., Schaefer S. M., Tuncer E., Frommel S. C., Cheng P., Arenas-Ramirez N., Haeusel J., Zhang Y., Bonalli M., McCabe M. T., Creasy C. L., Levesque M. P., Boyman O., Santoro R., Shakhova O., Dummer R., Sommer L., The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 6, 6051 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Burr M. L., Sparbier C. E., Chan K. L., Chan Y.-C., Kersbergen A., Lam E. Y. N., Azidis-Yates E., Vassiliadis D., Bell C. C., Gilan O., Jackson S., Tan L., Wong S. Q., Hollizeck S., Michalak E. M., Siddle H. V., Mc Cabe M. T., Prinjha R. K., Guerra G. R., Solomon B. J., Sandhu S., Dawson S.-J., Beavis P. A., Tothill R. W., Cullinane C., Lehner P. J., Sutherland K. D., Dawson M. A., An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36, 385–401.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng D., Kryczek I., Nagarsheth N., Zhao L., Wei S., Wang W., Sun Y., Zhao E., Vatan L., Szeliga W., Kotarski J., Tarkowski R., Dou Y., Cho K., Hensley-Alford S., Munkarah A., Liu R., Zou W., Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margueron R., Reinberg D., The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim K. H., Roberts C. W. M., Targeting EZH2 in cancer. Nat. Med. 22, 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Croce L., Helin K., Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Zaretsky J. M., Garcia-Diaz A., Shin D. S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D. Y., Abril-Rodriguez G., Sandoval S., Barthly L., Saco J., Homet Moreno B., Mezzadra R., Chmielowski B., Ruchalski K., Shintaku I. P., Sanchez P. J., Puig-Saus C., Cherry G., Seja E., Kong X., Pang J., Berent-Maoz B., Comin-Anduix B., Graeber T. G., Tumeh P. C., Schumacher T. N. M., Lo R. S., Ribas A., Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin D. S., Zaretsky J. M., Escuin-Ordinas H., Garcia-Diaz A., Hu-Lieskovan S., Kalbasi A., Grasso C. S., Hugo W., Sandoval S., Torrejon D. Y., Palaskas N., Rodriguez G. A., Parisi G., Azhdam A., Chmielowski B., Cherry G., Seja E., Berent-Maoz B., Shintaku I. P., le D. T., Pardoll D. M., Diaz L. A. Jr., Tumeh P. C., Graeber T. G., Lo R. S., Comin-Anduix B., Ribas A., Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7, 188–201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao J., Shi L. Z., Zhao H., Chen J., Xiong L., He Q., Chen T., Roszik J., Bernatchez C., Woodman S. E., Chen P.-L., Hwu P., Allison J. P., Futreal A., Wargo J. A., Sharma P., Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu J., Blake S. J., Yong M. C. R., Harjunpää H., Ngiow S. F., Takeda K., Young A., O’Donnell J. S., Allen S., Smyth M. J., Teng M. W. L., Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 6, 1382–1399 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benci J. L., Xu B., Qiu Y., Wu T. J., Dada H., Victor C. T.-S., Cucolo L., Lee D. S. M., Pauken K. E., Huang A. C., Gangadhar T. C., Amaravadi R. K., Schuchter L. M., Feldman M. D., Ishwaran H., Vonderheide R. H., Maity A., Wherry E. J., Minn A. J., Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wellenstein M. D., de Visser K. E., Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 48, 399–416 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Arenas-Ramirez N., Sahin D., Boyman O., Epigenetic mechanisms of tumor resistance to immunotherapy. Cell. Mol. Life Sci. 75, 4163–4176 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morin R. D., Johnson N. A., Severson T. M., Mungall A. J., An J., Goya R., Paul J. E., Boyle M., Woolcock B. W., Kuchenbauer F., Yap D., Humphries R. K., Griffith O. L., Shah S., Zhu H., Kimbara M., Shashkin P., Charlot J. F., Tcherpakov M., Corbett R., Tam A., Varhol R., Smailus D., Moksa M., Zhao Y., Delaney A., Qian H., Birol I., Schein J., Moore R., Holt R., Horsman D. E., Connors J. M., Jones S., Aparicio S., Hirst M., Gascoyne R. D., Marra M. A., Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bodor C., Grossmann V., Popov N., Okosun J., O’Riain C., Tan K., Marzec J., Araf S., Wang J., Lee A. M., Clear A., Montoto S., Matthews J., Iqbal S., Rajnai H., Rosenwald A., Ott G., Campo E., Rimsza L. M., Smeland E. B., Chan W. C., Braziel R. M., Staudt L. M., Wright G., Lister T. A., Elemento O., Hills R., Gribben J. G., Chelala C., Matolcsy A., Kohlmann A., Haferlach T., Gascoyne R. D., Fitzgibbon J., EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 122, 3165–3168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Italiano A., Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol. Ther. 165, 26–31 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Wilson B. G., Wang X., Shen X., McKenna E. S., Lemieux M. E., Cho Y.-J., Koellhoffer E. C., Pomeroy S. L., Orkin S. H., Roberts C. W. M., Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18, 316–328 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mora-Blanco E. L., Mishina Y., Tillman E. J., Cho Y.-J., Thom C. S., Pomeroy S. L., Shao W., Roberts C. W. M., Activation of β-catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene 33, 933–938 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jagani Z., Mora-Blanco E. L., Sansam C. G., McKenna E. S., Wilson B., Chen D., Klekota J., Tamayo P., Nguyen P. T. L., Tolstorukov M., Park P. J., Cho Y.-J., Hsiao K., Buonamici S., Pomeroy S. L., Mesirov J. P., Ruffner H., Bouwmeester T., Luchansky S. J., Murtie J., Kelleher J. F., Warmuth M., Sellers W. R., Roberts C. W. M., Dorsch M., Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 16, 1429–1433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long Y., Hwang T., Gooding A. R., Goodrich K. J., Rinn J. L., Cech T. R., RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat. Genet. 52, 931–938 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R. M., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tirosh I., Izar B., Prakadan S. M., Wadsworth M. H., Treacy D., Trombetta J. J., Rotem A., Rodman C., Lian C., Murphy G., Fallahi-Sichani M., Dutton-Regester K., Lin J. R., Cohen O., Shah P., Lu D., Genshaft A. S., Hughes T. K., Ziegler C. G. K., Kazer S. W., Gaillard A., Kolb K. E., Villani A.-C., Johannessen C. M., Andreev A. Y., van Allen E. M., Bertagnolli M., Sorger P. K., Sullivan R. J., Flaherty K. T., Frederick D. T., Jane-Valbuena J., Yoon C. H., Rozenblatt-Rosen O., Shalek A. K., Regev A., Garraway L. A., Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barbie D. A., Tamayo P., Boehm J. S., Kim S. Y., Moody S. E., Dunn I. F., Schinzel A. C., Sandy P., Meylan E., Scholl C., Fröhling S., Chan E. M., Sos M. L., Michel K., Mermel C., Silver S. J., Weir B. A., Reiling J. H., Sheng Q., Gupta P. B., Wadlow R. C., le H., Hoersch S., Wittner B. S., Ramaswamy S., Livingston D. M., Sabatini D. M., Meyerson M., Thomas R. K., Lander E. S., Mesirov J. P., Root D. E., Gilliland D. G., Jacks T., Hahn W. C., Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uhlen M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I. M., Edlund K., Lundberg E., Navani S., Szigyarto C. A.-K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P.-H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Farrell C. M., O’Leary N. A., Harte R. A., Loveland J. E., Wilming L. G., Wallin C., Diekhans M., Barrell D., Searle S. M. J., Aken B., Hiatt S. M., Frankish A., Suner M.-M., Rajput B., Steward C. A., Brown G. R., Bennett R., Murphy M., Wu W., Kay M. P., Hart J., Rajan J., Weber J., Snow C., Riddick L. D., Hunt T., Webb D., Thomas M., Tamez P., Rangwala S. H., Garvey K. M. M., Pujar S., Shkeda A., Mudge J. M., Gonzalez J. M., Gilbert J. G. R., Trevanion S. J., Baertsch R., Harrow J. L., Hubbard T., Ostell J. M., Haussler D., Pruitt K. D., Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res. 42, D865–D872 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A., The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petryszak R., Keays M., Tang Y. A., Fonseca N. A., Barrera E., Burdett T., Füllgrabe A., Fuentes A. M. P., Jupp S., Koskinen S., Mannion O., Huerta L., Megy K., Snow C., Williams E., Barzine M., Hastings E., Weisser H., Wright J., Jaiswal P., Huber W., Choudhary J., Parkinson H. E., Brazma A., Expression Atlas update—An integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44, D746–D752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/7/eabb3555/DC1