

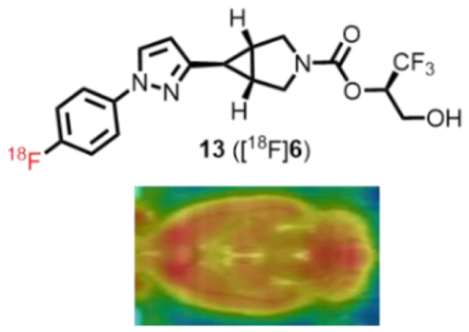
Abstract
Dysfunction of monoacylglycerol lipase (MAGL) is associated with several psychopathological disorders, including drug addiction and neurodegenerative diseases. Herein we design, synthesize and evaluate several irreversible fluorine-containing MAGL inhibitors for positron emission tomography (PET) ligand development. Compound 6 (identified from a therapeutic agent) was advanced for 18F-labeling via a novel spirocyclic iodonium ylide (SCIDY) strategy, which demonstrated high brain permeability and excellent specific binding. This work supports further development of novel 18F-labeled MAGL PET probes.
Keywords: monoacylglycerol lipase (MAGL), endocannabinoid, positron emission tomography (PET), spirocyclic iodonium ylide (SCIDY), PF06795071
Graphical Abstract
INTRODUCTION
The endocannabinoid (eCB) system is a lipid signaling network, mainly consisting of two membrane-bound G-coupled protein receptors, CB1 and CB2, and two endocannabinoids, N-arachidonoylethanolamine (anandamide; AEA) and 2-arachidonoylglycerol (2-AG).1–5 eCB is well prominent in both the central nervous system (CNS) and peripheral organs, and controls diverse physiological processes in mammals, such as pain, inflammation, feeding, cognition and neurodegeneration via finely tuning the endocannabinoid levels.6 While AEA is mainly degraded by fatty acid amide hydrolase (FAAH), 2-AG is primarily (ca. 85%) hydrolyzed by monoacylglycerol lipase (MAGL) to release the proinflammatory eicosanoid precursor arachidonic acid (AA).7, 8
MAGL, equipped with a Ser122-His269-Asp239 catalytic triad, is a membrane-bound serine hydrolase found not only in the endocannabinoid system, but also in the eicosanoid and other lipid signaling pathways.9 Recently, MAGL has attracted increasing attention, due to the growing awareness of its significant functions and the fact that 2-AG is much more abundant than AEA in the CNS.9–12 As an endogenous ligand for CB1 and CB2, 2-AG can exert beneficial impacts on neuroinflammation.13 Inhibition of MAGL enhances 2-AG signaling and lowers AA levels, thus generating anti-inflammatory effects. Also, inhibition of MAGL can protect against cognitive impairment, epileptogenesis and neurodegenerative diseases in the CNS, as well as play a vital role in the control of nociception, energy metabolism, cardiovascular and gastrointestinal function and cancer pathogenesis in peripheral organs.14 To date, a series of MAGL inhibitors bearing irreversible or reversible binding mechanism has been disclosed from academic and industry research community, both of which are equally important in pharmacology and drug discovery.12 While reversible MAGL inhibitors feature with transient blockade and non-covalent modification of the target protein, irreversible MAGL inhibitors enable sustained blockade, which is highly desirable for the pursuit of efficacy in neuroinflammation.15
A positron emission tomography (PET) probe for MAGL is not only an ideal tool to investigate its expression and better understand its biology in vivo, but also highly desirable to accelerate the translation of MAGL-related drugs by providing information about target occupancy and dose selection.16–18 To date, several potent irreversible MAGL PET probes based on the piperidyl carbamate, azetidinyl oxadiazole, or piperazinyl azetidine scaffold, including [11C]SAR127303, [11C]MA-PB-1, [11C]MAGL-0519 and [11C]MAGL-2–11, have been disclosed by our19–21 and other groups22–24, and some of them exhibited high brain uptake and good binding specificity in rodents as well as non-human primates (Figure 1A). Another irreversible 11C-labeled MAGL PET probe was recently reported in a conference abstract by Pfizer research.25 While several irreversible PET ligands have been reported for studying MAGL activity in the living brain, the fact that they are radiolabeled with the short-lived radionuclide carbon-11 (11C; t½ = 20.4 min) limits their use to facilities with an in-house cyclotron and expertise to work with 11C. On the other hand, fluorine-18 (18F; t½ = 109.7 min) can be produced and handled by all facilities, and also can be shipped over fairly large distances to sites which do not possess a cyclotron, and a 18F-labeled MAGL ligand can be used several hours after its synthesis. In addition, due to its slower radioactive decay and concomitant improved counting statistics, radiotracers labeled with fluorine-18 allow for a more accurate determination of the plasma input function than radiotracers labeled with carbon-11. Therefore, we envision that a novel 18F-labeled MAGL irreversible inhibitor will enable widespread use to study brain activity and function affected by variability in MAGL (Figure 1C). During the preparation of this manuscript, Koike et al.26 and our group27 reported reversible MAGL probe [18F]T-401 and [18F]MAGL-4- 11, respectively, as a complementary imaging tool to irreversible binding mechanism (Figure 1B).
Figure 1.
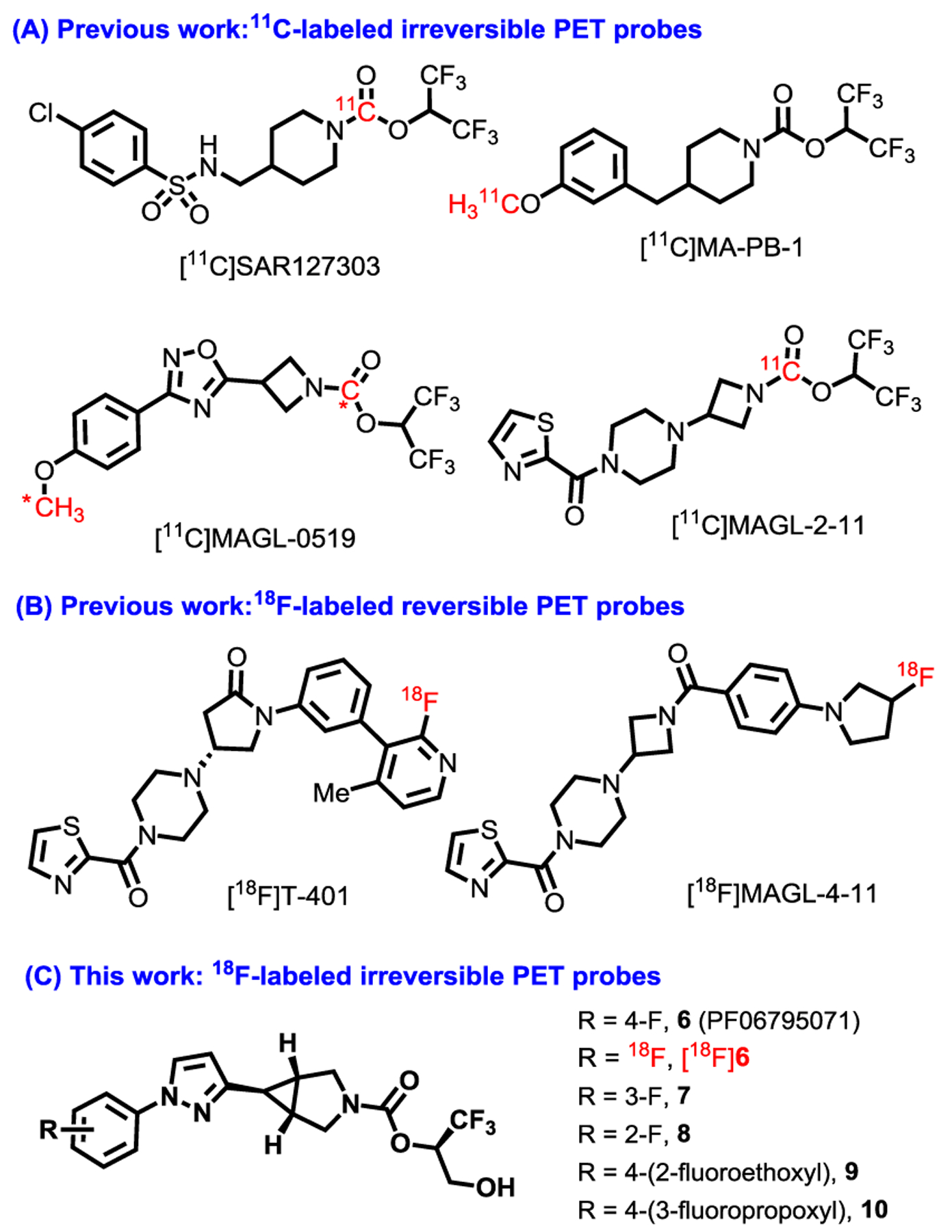
Representative irreversible MAGL PET probes.
As our continuous interest and effort in the development of 18F-labeled MAGL PET ligands, we were inspired by a recently-disclosed MAGL therapeutic agent PF06795071 (6) bearing azabicyclo[3.1.0]hexane scaffold, which demonstrated excellent potency, selectivity and brain permeability.28 In this work, we designed an array of fluorinated MAGL inhibitors (6-10) based on this new structural feature with amenability for radiofluorination. The preliminary pharmacological, physiochemical and molecular docking studies were carried out for these inhibitors, followed by an efficient isotopologue 18F-labeling of 6 based on our novel spirocyclic iodonium ylide (SCIDY) strategy.29–32 18F-Labeled MAGL ligand 13 ([18F]6) demonstrated excellent blood-brain-barrier (BBB) penetration and regional brain distribution, which is in line with the expression pattern of MAGL in the brain. Excellent in vitro and in vivo binding specificity was also validated by autoradiography and PET studies, respectively. This work not only showcases design, synthesis and evaluation of a focused library of fluorinated MAGL inhibitors amenable for labeling, but also demonstrates the successful transition from a therapeutic agent to its isotopologue, i.e., a 18F-labeled irreversible MAGL inhibitor, as a validated PET probe for MAGL.
RESULTS AND DISCUSSION
Chemistry.
We synthesized an array of fluorinated irreversible MAGL inhibitor candidates 6–10 based on 3-azabicyclo[3.1.0]hexane scaffold,28 as shown in Scheme 1, and all these compounds can be potentially labeled with fluorine-18. Briefly, key pyrazole intermediate 1–5 was synthesized in 30–58% yield with different fluorine-substituted patterns (Scheme S1, SI). Deprotection of tert-butyloxycarbonyl (Boc) group in 1–5 followed by coupling reaction with (R)-1,1,1-trifluoro-3-(4-methoxybenzyloxy)propan-2-ol proceeded smoothly to provide desired fluorinated MAGL inhibitor candidates 6–10 in 9–18% yields over 8 steps from commercially available (1R,5S,6r)-3-(tert-butoxycarbonyl)-3-azabicyclo [3.1.0]hexane-6-carboxylic acid.
Scheme 1. Synthesis of fluorinated MAGL inhibitorsa.
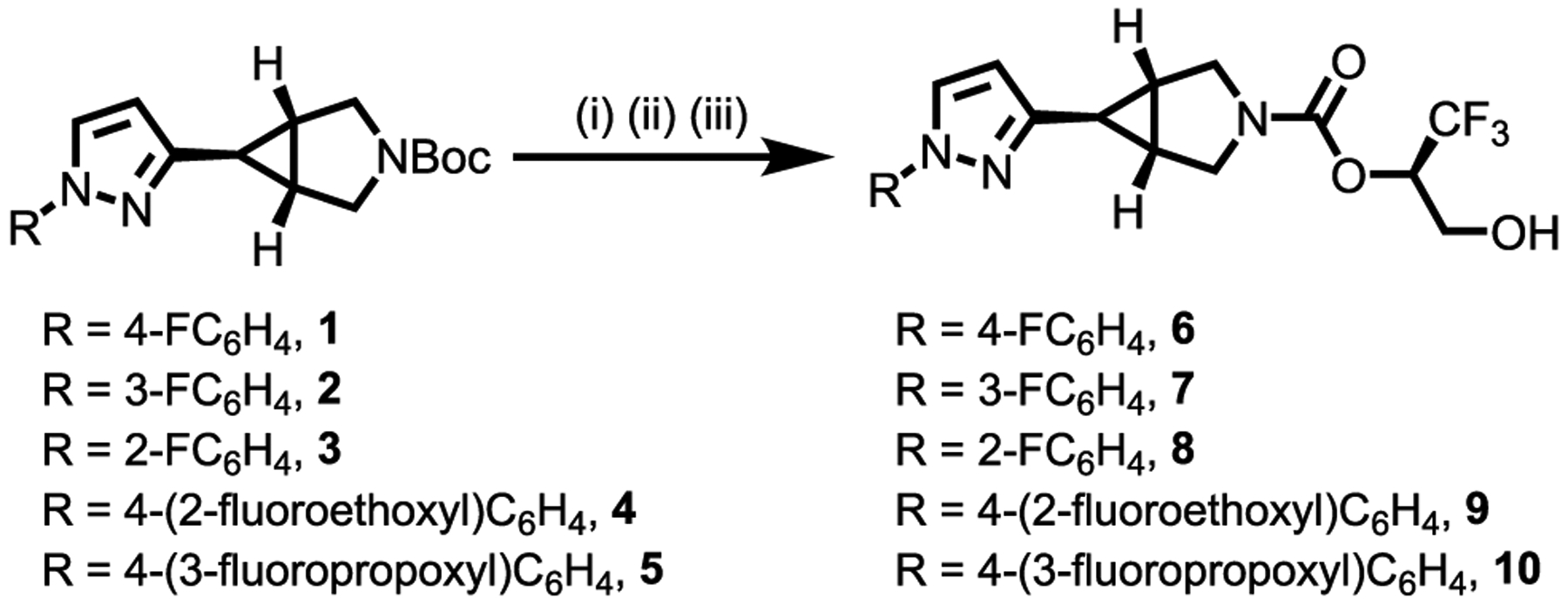
aReagents and conditions: (i) TFA, rt, 1 h; (ii) (R)-1,1,1-trifluoro-3-(4-methoxybenzyloxy)propan-2-ol, 1,1’-[carbonylbis(oxy)]di-pyrrolid-ine-2,5-dione, Et3N, CH2Cl2, 30 °C, 30 h; (iii) TFA, CH2Cl2, rt, 4 h; 34% yield for 6; 35% yield for 7; 22% yield for 8; 18% yield for 9; 31% yield for 10.
Molecular Docking Studies.
A preliminary molecular docking study was conducted to probe the possible interactions of compounds 6–10 within the binding domain of MAGL. While compound 10 only partially entered the binding domain (probably due to its bulky fluoropropoxyl end) of the pre-covalent MAGL structure (PDB ID: 3PE6) (Figure S2, SI)27, 33, all the other four compounds 6–9 were well-docked into the active sites (Figure 2). Therefore, compounds 6–9 hold great promise to reduce the possibility of 2-AG to approach this catalytic site, and thereafter inhibit its hydrolysis. Within the binding pocket, H-bonding interactions were found between compounds 6–8 and the Met123, Ala51 or Try194 residue. Detailed interaction was outlined in the supplementary information (section 2.4 & Figure S2).
Figure 2.
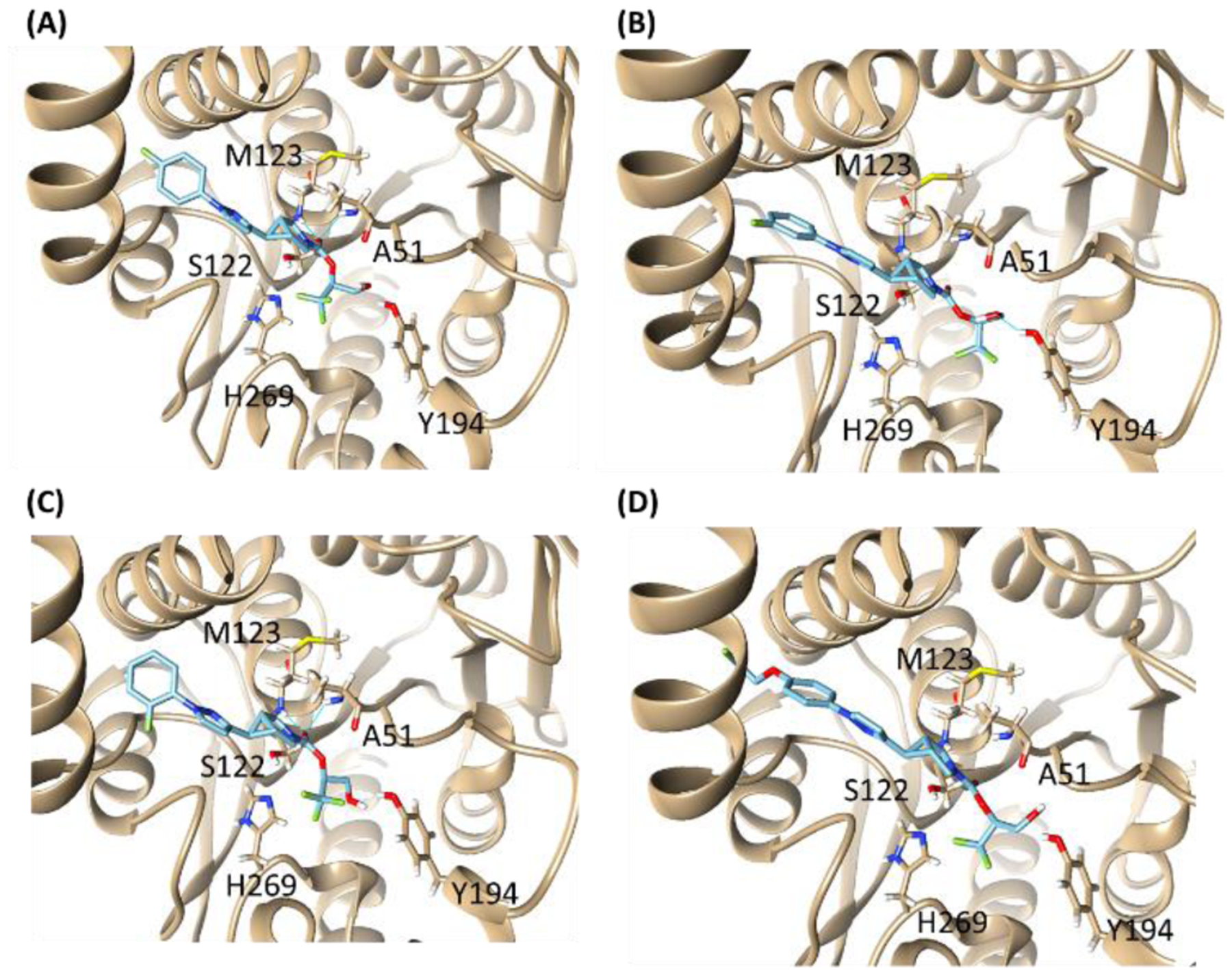
Molecular docking structures of compounds 6 (A), 7 (B), 8 (C) and 9 (D) onto MAGL (PDB ID: 3PE6).
Pharmacology.
Motivated by the promising docking results, we then investigated the in vitro potency and selectivity of compounds 6–10 towards MAGL via activity-based protein profiling (ABPP) assays.34 A preliminary screening commenced at a single concentration of 5 μM, and all these candidates exhibited excellent potency towards inhibition of MAGL activity (Figure 3A). However, remarkable inhibition of FAAH activity was also observed for candidate 7 and 10 (>70% inhibition), whereas candidates 6 & 8–9 implicated promising selectivity towards MAGL over FAAH. Subsequently, the IC50 value towards inhibition of MAGL activity for 6 & 8–9 was investigated, and 6 demonstrated to be the most promising with the IC50 as 2.3 nM (Figure 3B). The selectivity of candidate 6 towards MAGL over other serine hydrolases including FAAH, KIAA1363, ABHD6 and ABHD12, was also evaluated, and no noticeable interactions between 6 and these targets were detected (Figure 3C). In addition, no substantial off-target interaction, including two major cannabinoid receptors in the eCB signaling, namely CB1 (EC50 = 1.5 μM) and CB2 (> 10 μM), was observed for compound 6.28 To confirm the irreversible binding mechanism for 6, we performed time-dependent ABPP studies. As shown in Figure 3D, in contrast to MAGL activity in samples treated with FEPAD,35 a known reversible MAGL inhibitor, samples treated with 6 demonstrated much slower recovery of MAGL activity, implicating candidate 6 as an irreversible inhibitor and a slow hydrolysis accompanied with the enzyme-inhibitor adduct over time.
Figure 3.
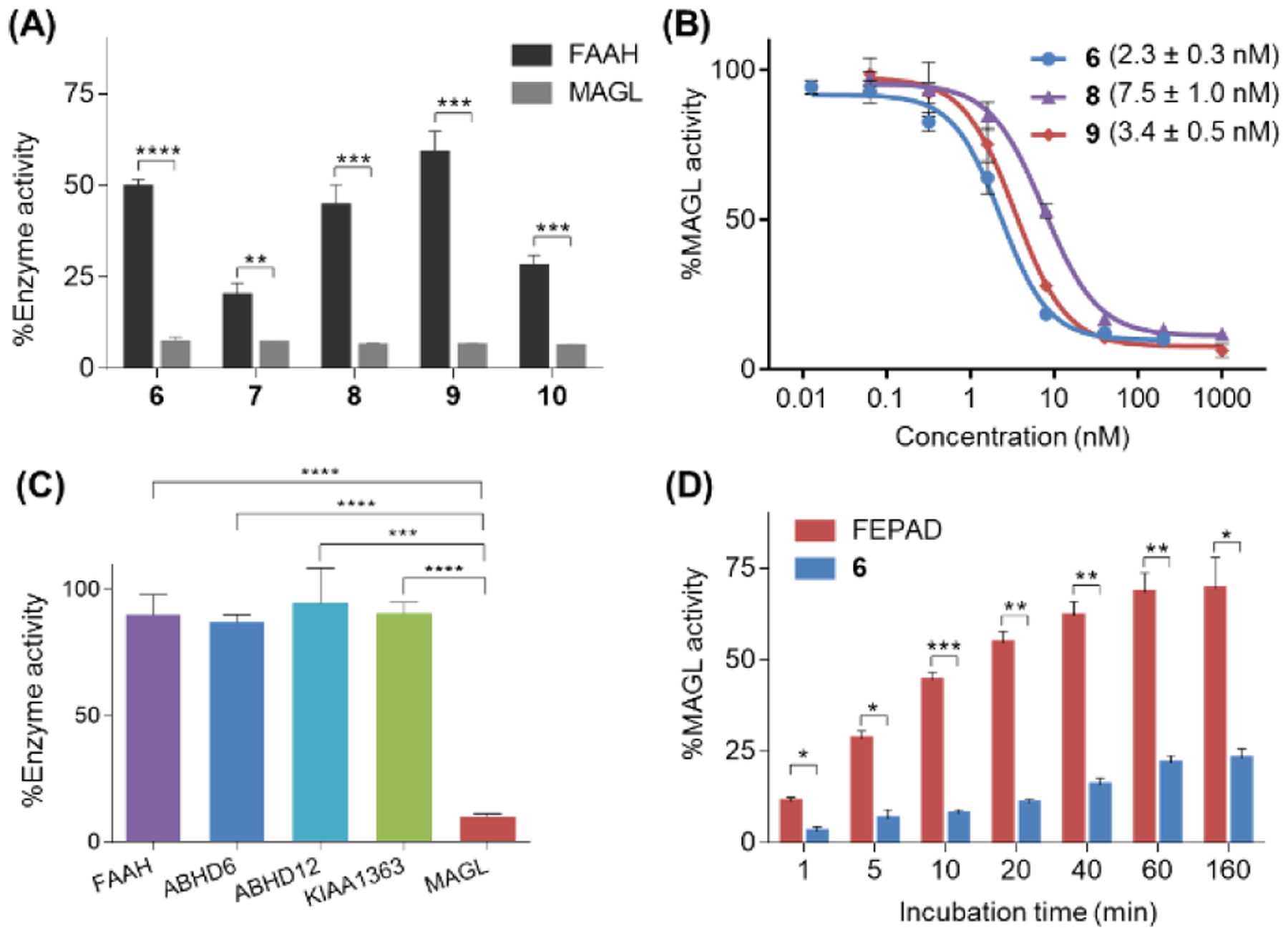
Inhibition properties of MAGL activity by compounds 6–10 determined by ABPP assay. (A) Inhibition of mouse brain MAGL and FAAH activity with compounds 6-10 (5 μM); (B) Concentration-response curves of 6, 8 and 9 for inhibition of mouse brain MAGL; (C) Subtype selectivity of 6 (at 200 nM) among MAGL and other serine hydrolases in the endocannabinoid system including KIAA1363, FAAH, ABHD6 and ABHD12; (D) MAGL activity determined at different time points with 6 (at 200 nM). A reversible inhibitor FEPAD (at 1 μM) was used as a control. No significant time-dependent changes of MAGL activity indicates that the binding of compound 6 is irreversible while the reversible inhibitor FEPAD exhibits recovered MAGL activity over time. All data are mean ± SD, n = 3. *p < 0.05, **p ≤0.01, and ***p ≤0.001.
Lipophilicity is an important parameter for estimating the ability of a candidate compound to cross the blood-brain barrier (BBB).36 By using the ‘shake flask method’, also known as liquid-liquid partition between PBS and n-octanol,37 the LogD value of compound 6 was determined to be 3.32 ± 0.04 (n = 3). This result, together with reasonable topological polar surface area value (tPSA = 65.37) (Table S1, SI), indicated a high likelihood of high brain penetration for compound 6.
Radiochemistry.
The promising results of pharmacology and physiochemical property encouraged us to label 6 with fluorine-18 via a novel spirocyclic iodonium ylide (SCIDY) strategy.29–32 The SCIDY precursor 12 for 18F-labeling was obtained in 35% yield via oxidation of 11 with meta-chloroperoxybenzoic acid (mCPBA) followed by ligand exchange with spirocyclic auxiliary SPIAd (Scheme 2 and Figure S3 in SI). With the SCIDY precursor 12 in hand, we then carried out the radiosynthesis of 13 ([18F]6). As shown in Figure S3A, initial attempt for the preparation of radioligand 13 commenced with optimization of reaction parameters for [18F]fluorination. Upon heating the mixture of 12 (2.0 mg), tetraethylammonium bicarbonate (TEAB, 1.0 mg) and [18F]fluoride in acetonitrile (MeCN) at 100 °C for 10 min, desired [18F]fluorinated intermediate was obtained in 31% radiochemical conversion (RCC) (entry 1). Increasing or reducing the amount of TEAB lowered RCCs (entries 2 and 3). Although similar RCC could be achieved in N,N-dimethylformamide instead of MeCN (31%, entry 4), temperature elevation from 100 °C to 120 °C remarkably improved the RCC (59%, entry 5). Using 1.0 mg of precursor 12 resulted in a slightly decreased RCC (43%, entry 6). Ultimately, radioligand 13 was prepared by nucleophilic substitution of the SCIDY precursor 12 (2.0 mg) with [18F]fluoride in DMF at 120 °C for 10 minutes in the presence of TEAB (1.0 mg), followed by hydrochloric acid-promoted deprotection of the PMB group. This method exhibited several advantages for direct late-stage radiofluorination of non-activated aromatics, and reasonable radiochemical yield (8% RCY, non-decay corrected), high molar activity (33.3–225.7 GBq/μmol) as well as high radiochemical purity (>99%). Besides, radioligand 13 demonstrated excellent in vitro stability, as evidenced by no radiolysis detected up to 4 h in saline containing 5% ethanol (Figure S3B, SI).
Scheme 2. Synthesis of MAGL PET probe 13 ([18F]6)a.

aReagents and conditions: (i) mCPBA, CHCl3, rt, 12 h; (ii) SPIAd, 10% Na2CO3, EtOH, rt, 4 h; 35% yield over 2 steps. (iii) [18F]Et4NF, DMF, 120 °C, 10 min; (iv) 6 N HCl, 100 °C, 4 min, 8% non-decay corrected RCY.
In vitro autoradiography.
To evaluate the in vitro specific binding of 13 towards MAGL, autoradiography was carried out in sagittal rat brain sections (20 μM) and the representative autoradiograms were shown in Figure 4. Briefly, the brain sections were pre-incubated with Tris-HCl buffer (50 mM) containing 0.01% Triton X-100 for 20 min at ambient temperature, followed by incubation with 13. For blocking studies, the brain sections were incubated with KML29 (10 μM) or PF06795071 (10 μM) prior to incubation with 13 (see detailed experimental procedures in Supplementary Information, section 2.6). As shown in Figure 4A, the control experiment exhibited heterogeneous distribution for the bound radioactivity, and showed high uptake in the cerebral cortex, hippocampus, cerebellum and striatum (Figure 4D), which is in line with the enzyme expression level of MAGL in rodents.26 Pretreatment with unlabeled PF06795071 resulted in 26–40% reduction of the bound activity in these brain regions and abolishment of the heterogeneous distribution (Figure 4B). In blocking studies with KML29, a known MAGL inhibitor, higher levels of blockade (35–49% reduction) was observed (Figure 4C).38 These results suggested that ligand 13 had good to excellent in vitro binding specificity towards MAGL.
Figure 4.
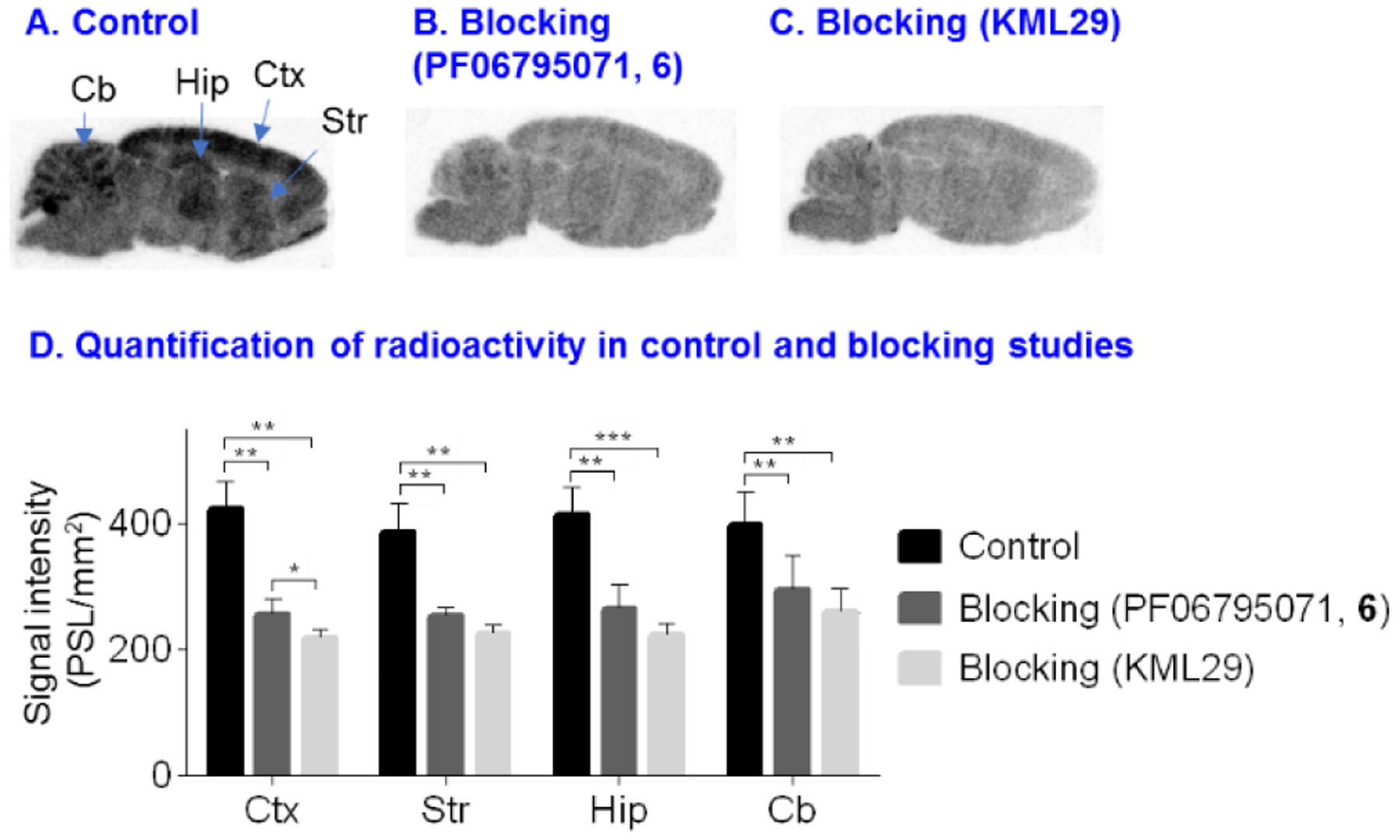
In vitro autoradiography of 13 in rat brain sections. (A) Brain sections were treated with 13; (B) Brain sections were pre-treated with PF06795071 (6, 10 μM) followed by 13; (C) Brain sections were pre-treated with KML29 (10 μM) followed by 13; (D) Quantification of radioactivity under control and blocking conditions. The data are expressed as radioactivity per mm2 (n = 5). Ctx = cerebral cortex; Str = striatum; Hip = hippocampus; Cb = cerebellum. Asterisks indicate statistical significance. *p < 0.05, **p ≤0.01, and ***p ≤0.001.
PET imaging studies of 13 ([18F]6) in rat brain and whole-body biodistribution.
Encouraged by the promising results of in vitro autoradiography, we then performed dynamic PET acquisitions of tracer 13 in Sprague-Dawley (SD) rats for 90 mins. Representative PET images in rat brains (summed images 0–90 min) and the corresponding time-activity curves are depicted in Figure 5 and Figure S4 in the Supporting Information. Radioligand 13 readily passed the BBB and the standard uptake value (SUV) reached its max value of 1.75 in the whole brain at 2.5 min (Figure S4). Heterogeneous distribution was observed with the highest radioactivity in the striatum, followed by hippocampus, cerebral cortex, and cerebellum, and the lowest in pons (Figure 5A). Pretreatment with KML29 (3 mg/kg) led to a significant decrease of bound activity in various brain regions and the whole-brain uptake was reduced by 56% based on the area under the curve (AUC) (Figure 5B and S4). Blocking studies with non-radioactive PF06795071 (3 mg/kg, 6) was also performed, and remarkably decreased uptake of 13 in whole rat brain (61% reduction based on AUC) and abolishment of heterogeneous distribution was observed (Figure 5C and S4). These results indicated excellent in vivo binding specificity for 13. We then carried out whole-body distribution studies in mice to profile the uptake, biodistribution and clearance of tracer 13 and four time points (5, 15, 30 and 60 min) after tracer injection was selected. As depicted in Figure 6 and Table S2, high radioactivity accumulation was observed in several organs including heart, lungs, liver and kidneys (>10% ID/g, injected dose per gram of wet tissue). The radioactivity levels in small intestine and liver both reached a plateau at 30 min followed by slow washout, indicating that 13 was eliminated possibly via the hepatobiliary and urinary pathway. No significant in vivo de-radiofluorination was observed during the imaging study. To evaluate in vivo stability, radiometabolic analysis of 13 in SD rat brain and plasma homogenate was conducted at 5 min and 30 min post injection, respectively. By adopting an established method for irreversible radiotracers,39 we first confirmed that the majority of radioactivity was bound to the rat brain tissue, and average 76% and 73% of the radioactivity was irreversibly bound to the rat brain tissues at 5 min and 30 min post injection (n=3), respectively. For unbound radioactivity, we determined that average 89% and 54% of the radioactivity was parent fraction in the rat brain at 5 min and 30 min post injection (n=3), respectively, and only other polar metabolites were found (see Supplementary information, Figure S5). In parallel, we also measured radiometabolites in plasma and found that average 77% and 40% of the radioactivity was parent fraction in the rat plasma at 5 min and 30 min post injection (n=3), respectively (Figure S5). Although our lead ligand 13 showed reasonable in vivo stability, further study in higher species would be desirable due to possible species difference in metabolism rate and pathway.
Figure 5.
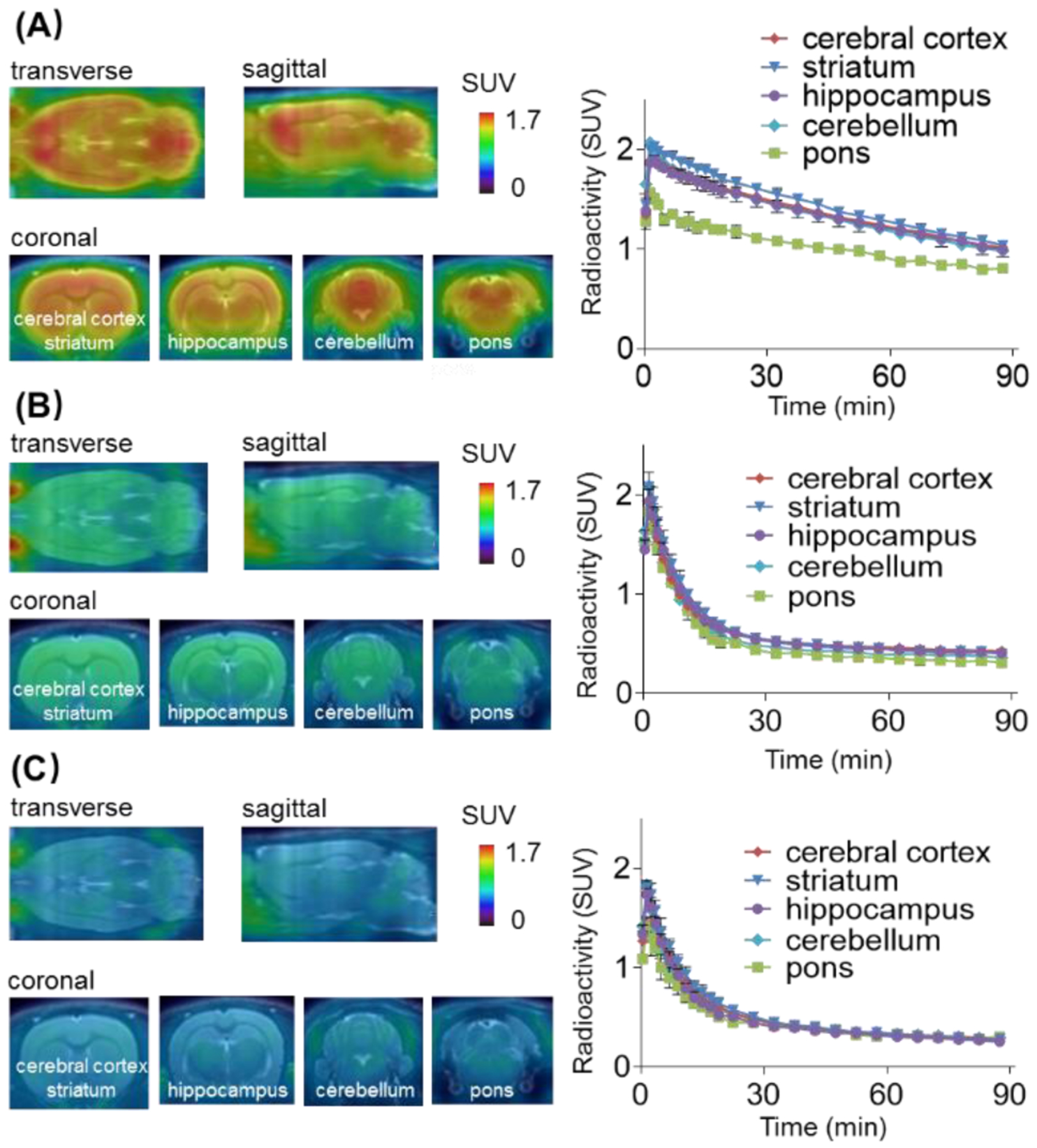
Representative PET images and time-activity curves of 13 in rat brain: (A) Baseline; (B) Blocking studies with KML29 (3 mg/kg); (C) Blocking studies with PF06795071 (3 mg/kg).
Figure 6.
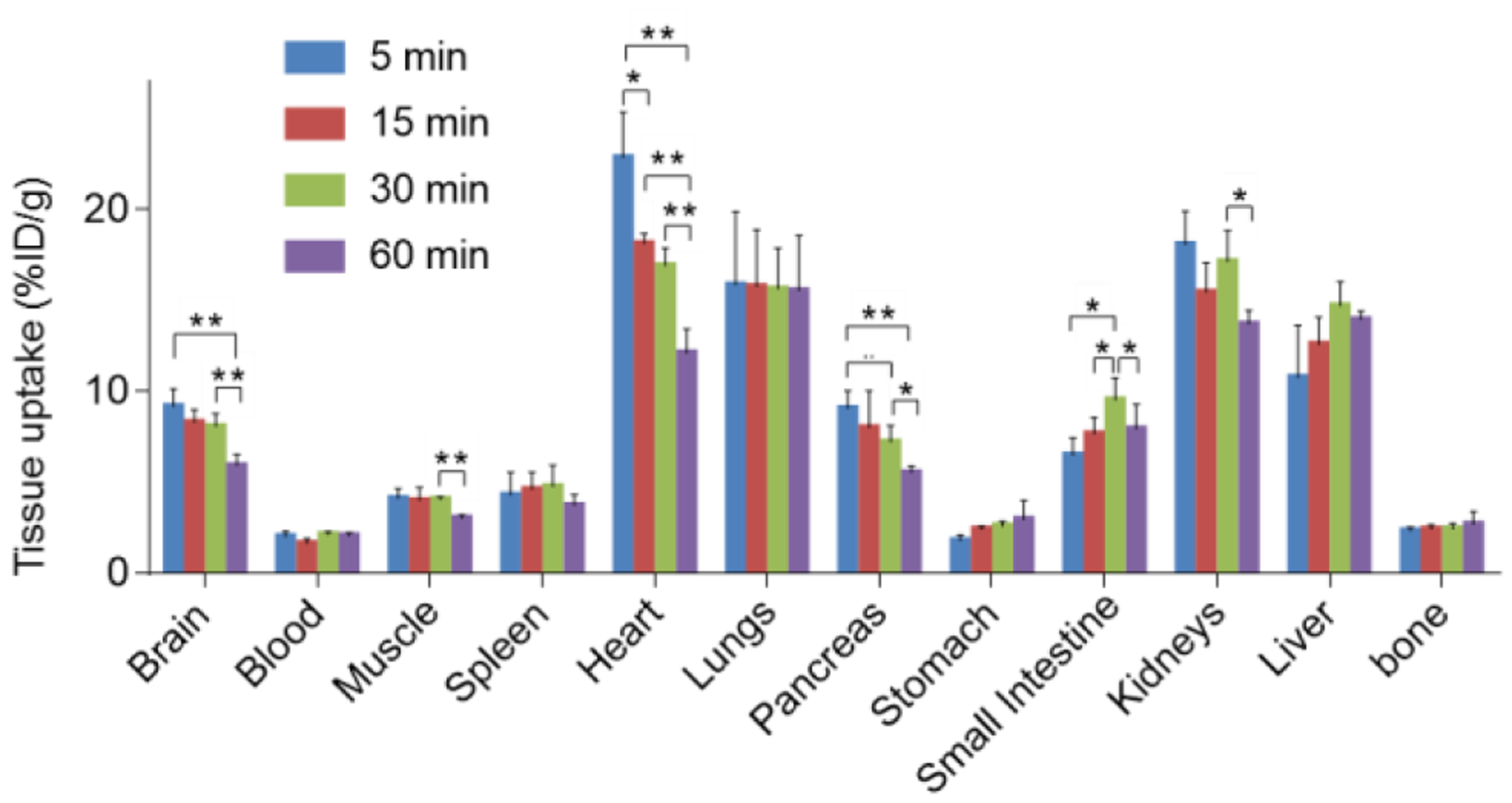
Whole-body ex vivo biodistribution studies of 13. Asterisks indicate statistical significance. * p < 0.05 and ** p ≤ 0.01. %ID/g = injected dose per gram of wet tissue.
CONCLUSIONS
We have established a focused library of fluorinated irreversible MAGL inhibitors based on a novel therapeutic agent bearing azabicyclo[3.1.0]hexane scaffold. Potency, selectivity as well as irreversible binding profiles of these compounds were determined in vitro by the ABPP assay. Molecular docking studies were also carried out to probe possible interactions of these molecules with the binding domain of MAGL. Lipophilicity (LogD) of lead compound 6 was measured by the “shake-flask” method, which indicated reasonable brain permeability. The corresponding 18F-isotopologues 13 ([11C]6; also known as [18F]PF06795071) was synthesized in good radiochemical yield, high molar activity and high radiochemical purity using our spirocyclic iodonium ylide (SCIDY) strategy. The pharmacokinetic profiles of 13 including binding specificity, brain uptake, and clearance were further evaluated by in vitro autoradiography, PET imaging study, as well as ex vivo whole-body distribution studies in rodents. PET probe 13 readily crossed the BBB and exhibited excellent in vitro and in vivo binding specificity and heterogenous regional brain uptake, which was consistent with MAGL distribution. In conclusion, this work identifies a novel 18F-labeled irreversible MAGL PET probe and provides preliminary results that support further translation in higher species, such as nonhuman primates, for brain uptake, specificity and radiometabolism validation.
ASSOCIATED CONTENT
Preparation of precursor 12.
To solution of aryl iodide 11 (0.49 mmol) in chloroform (5 mL), mCPBA (0.64 mmol, 75% max content) was added. The resulting mixture was stirred at room temperature overnight before removal of the solvent in vacuo. The residue was re-desolved in ethanol (5 mL), and (1r,3r,5r,7r)-spiro-[adamantane-2,2′-[1,3]dioxane]-4′,6′-dione (SPIAd, 0.5 mmol) was added followed by 10% aqueous solution of Na2CO3 (w/v, 5 mL, 0.33 M solution). The resulting suspension was stirred at room temperature for 5 h until full conversion of to the iodoinium ylide as monitored by TLC. The reaction mixture was then diluted with water and extracted with CH2Cl2. The organic layers were successively combined, washed with water and brine, dried with anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified on a silica gel chromatography (ethyl acetate as eluent) to give the desired product as a white solid.
Radiosynthesis of [18F]6 (13).
The cyclotron-produced [18F]F− was separated from H218O using the Sep-Pak Accell Plus QMA Plus Light cartridge (Waters; Milford, Ma). The produced [18F]F− was eluted from the cartridge with an aqueous solution of tetraethylammonium bicarbonate (2 mg in 300 μL) and CH3CN (700 μL), and transferred to a reaction vessel in the hot cell. After drying [18F]Et4NF solution at 120 °C for 30 min to remove water and CH3CN, a solution of SCIDY precursor 12 (2.0 mg) in anhydrous DMF (400 μL) was then added. The vessel was heated at 120 °C for 10 min. After cooling to 100 °C, 6 N HCl (300 μL) was added, and the resulting mixture was heated at 100 °C for 4 min. Then the reaction mixture was allowed to cool to room temperature, neutralized with saturated aqueous solution of NaOH, diluted with HPLC mobile phase, and then injected into an HPLC column. HPLC purification was performed on an X-Select® Prep C18 column (10 × 250 mm, 5 μm) using a mobile phase of CH3CN / 0.1 M aqueous solution of ammonium formate (50/50, PH = ~6.5) at a flowrate of 5.0 mL/min. The reaction time of 13 was 15.4 min. The radioactive fraction corresponding to the desired product was collected in a sterile flask, evaporated to dryness in vacuo, and reformulated in a saline solution (3 mL) containing 100 μL of 25% ascorbic acid in sterile water and 100 μL of 20% Tween 80 in ethanol. (Note: We added ascorbic acid to prevent potential radiolysis and Tween 80 to improve aqueous solubility.) The synthesis time was 71 min from end of bombardment. Radiochemical and chemical purity was measured by analytical HPLC (X Bridge Prep C18 column (4.6 × 250 mm, 5 μm) using a mobile phase of CH3CN / 0.1 M aqueous solution of ammonium formate (50/50) at a flow rate of 1.0 mL/min. The identity of 13 was confirmed by the co-injection with unlabeled 6. Radiochemical yield was 8% non-decay-corrected based on [18F]F− with >99% radiochemical purity, and the molar activity was higher than 37 GBq/μmol (1.0 Ci/μmol).
Supplementary Material
ACKNOWLEDGMENT
We thank Professor Thomas J. Brady for helpful discussion. Financial support from the NIH grants (DA038000 and DA043507 to S.L., DA033760 to B.F.C.), the Swiss National Science Foundation (No. P2EZP3_175137 to M.A.S) and CSC scholarship (No. 201606250107 to Z.C.) is gratefully acknowledged.
ABBREVIATIONS USED
- PET
positron emission tomography
- SCIDY
spirocyclic iodonium ylide (SCIDY)
- eCB
endocannabinoid
- AEA
anandamide
- 2-AG
2-arachidonoylglycerol
- CNS
central nervous system
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- AA
arachidonic acid
- SUV
standardized uptake value
- TAC
time−activity curve
- %ID/g
percentage of injected dose per gram of wet tissue
Footnotes
The authors declare no competing financial interest.
Supporting Information
Experimental procedures, compound characterization, computation details, NMR spectra for new compounds (PDF)
Molecular formula strings (CSV)
REFERENCES
- 1.Matsuda LA; Lolait SJ; Brownstein MJ; Young AC; Bonner TI Structure of a cannabinoid receptor and function expression of the cloned cDNA. Nature 1990, 346, 561–564. [DOI] [PubMed] [Google Scholar]
- 2.Cravatt BF; Lichtman AH The endogenous cannabinoid system and its role in nociceptive behavior. J. Neurobio 2004, 61, 149–160. [DOI] [PubMed] [Google Scholar]
- 3.Pertwee R The pharmacology of cannabinoid receptors and their ligands: an overview. Int. J. Obes 2006, 30, S13–S18. [DOI] [PubMed] [Google Scholar]
- 4.Ahn K; McKinney MK; Cravatt BF Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev 2008, 108, 1687–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aaltonen N; Ribas CR; Lehtonen M; Savinainen JR; Laitinen JT Brain regional cannabinoid CB1 receptor signalling and alternative enzymatic pathways for 2-arachidonoylglycerol generation in brain sections of diacylglycerol lipase deficient mice. Eur. J. Pharm. Sci 2014, 51, 87–95. [DOI] [PubMed] [Google Scholar]
- 6.Di Marzo V; Bisogno T; De Petrocellis L Endocannabinoids and related compounds: walking back and forth between plant natural products and animal physiology. Chem. Biol 2007, 14, 741–756. [DOI] [PubMed] [Google Scholar]
- 7.Piomelli D The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci 2003, 4, 873–884. [DOI] [PubMed] [Google Scholar]
- 8.Chevaleyre V; Takahashi KA; Castillo PE Endocannabinoid-mdediated synaptic plasticity in the CNS. Annu. Rev. Neurosci 2006, 29, 37–76. [DOI] [PubMed] [Google Scholar]
- 9.Mechoulam R; Ben-Shabat S; Hanus L; Ligumsky M; Kaminski NE; Schatz AR; Gopher A; Almog S; Martin BR; Compton DR; Pertwee RG; Griffin G; Bayewitch M; Barg J; Vogel Z Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol 1995, 50, 83–90. [DOI] [PubMed] [Google Scholar]
- 10.Sugiura T; Kondo S; Sukagawa A; Nakane S; Shinoda A; Itoh K; Yamashita A; Waku K 2-Arachidonoylgylcerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophy. Res. Commun 1995, 215, 89–97. [DOI] [PubMed] [Google Scholar]
- 11.Stella N; Schweitzer P; Piomelli D A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [DOI] [PubMed] [Google Scholar]
- 12.Gil-Ordóñez A; Martín-Fontecha M; Ortega-Gutiérrez S; López-Rodríguez ML Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol 2018, 157, 18–32. [DOI] [PubMed] [Google Scholar]
- 13.Chiurchiù V; Battistini L; Maccarrone M Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulvihill MM; Nomura DK Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 2013, 92, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh J; Petter RC; Baillie TA; Whitty A The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- 16.Ametamey SM; Honer M; Schubiger PA Molecular imaging with PET. Chem. Rev 2008, 108, 1501–1516. [DOI] [PubMed] [Google Scholar]
- 17.Willmann JK; van Bruggen N; Dinkelborg LM; Gambhir SS Molecular imaging in drug development. Nat. Rev. Drug Discov 2008, 7, 591–607. [DOI] [PubMed] [Google Scholar]
- 18.Miller PW; Long NJ; Vilar R; Gee AD Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew. Chem., Int. Ed 2008, 47, 8998–9033. [DOI] [PubMed] [Google Scholar]
- 19.Wang L; Mori W; Cheng R; Yui J; Rotstein B; Fujinaga M; Hatori A; Vasdev N; Zhang M-R; Liang S A novel class of sulfonamido [11C-carbonyl]-labeled carbamates and ureas as radiotracers for monoacylglycerol lipase. J. Nucl. Med 2016, 57, 4. [Google Scholar]
- 20.Wang L; Mori W; Cheng R; Yui J; Hatori A; Ma L; Zhang Y; Rotstein BH; Fujinaga M; Shimoda Y; Yamasaki T; Xie L; Nagai Y; Minamimoto T; Higuchi M; Vasdev N; Zhang M-R; Liang SH Synthesis and preclinical evaluation of sulfonamido-based [11C-carbonyl]-carbamates and ureas for imaging monoacylglycerol lipase. Theranostics 2016, 6, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng R; Mori W; Ma L; Alhouayek M; Hatori A; Zhang Y; Ogasawara D; Yuan G; Chen Z; Zhang X; Shi H; Yamasaki T; Xie L; Kumata K; Fujinaga M; Nagai Y; Minamimoto T; Svensson M; Wang L; Du Y; Ondrechen MJ; Vasdev N; Cravatt BF; Fowler C; Zhang M-R; Liang SH In vitro and in vivo evaluation of 11C-labeled azetidinecarboxylates for imaging monoacylglycerol lipase by PET imaging studies. J. Med. Chem 2018, 61, 2278–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C; Placzek MS; Van de Bittner GC; Schroeder FA; Hooker JM A novel radiotracer for imaging monoacylglycerol lipase in the brain using positron emission tomography. ACS Chem. Neurosci 2016, 7, 484–489. [DOI] [PubMed] [Google Scholar]
- 23.Ahamed M; Attili B; van Veghel D; Ooms M; Berben P; Celen S; Koole M; Declercq L; Savinainen JR; Laitinen JT; Verbruggen A; Bormans G Synthesis and preclinical evaluation of [11C]MA-PB-1 for in vivo imaging of brain monoacylglycerol lipase (MAGL). Eur. J. Med. Chem 2017, 136, 104–113. [DOI] [PubMed] [Google Scholar]
- 24.Yamasaki T; Mori W; Zhang Y; Hatori A; Fujinaga M; Wakizaka H; Kurihara Y; Wang L; Nengaki N; Ohya T; Liang SH; Zhang M-R First demonstration of in vivo mapping for regional brain monoacylglycerol lipase using PET with [11C]SAR127303. NeuroImage 2018, 176, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butler CR; Takano A; Nag S; Arakawa R; Maresca KP; Piro JR; Samad T; Smith D; Nason D; O’Neil S; McAllister L; Grimwood S; Trapa P; McCarthy T; Villalobos A; Halldin C; Zhang L Identification and development of a highly specific monoacylglycerol lipase (MAGL) PET tracer 11C-PF-06809247. Eur. J. Nucl. Med. Mol. Imaging 2017, 44 (Suppl 2), S366. [Google Scholar]
- 26.Hattori Y; Aoyama K; Maeda J; Arimura N; Takahashi Y; Sasaki M; Fujinaga M; Seki C; Nagai Y; Kawamura K; Yamasaki T; Zhang M-R; Higuchi M; Koike T Design, synthesis, and evaluation of (4R)-1-{3-[2-(18F)fluoro-4-methylpyridin-3-yl]phenyl}-4-[4-(1,3-thiazol-2-ylcarbonyl)piperazin-1-yl]pyrrolidin-2-one ([18F]T-401) as a novel positron-emission tomography imaging agent for monoacylglycerol lipase. J. Med. Chem 2019, 62, 2362–2375. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z; Mori W; Deng X; Cheng R; Ogasawara D; Zhang G; Schafroth MA; Dahl K; Fu H; Hatori A; Shao T; Zhang Y; Yamasaki T; Zhang X; Rong J; Yu Q; Hu K; Fujinaga M; Xie L; Kumata K; Gou Y; Chen J; Gu S; Bao L; Wang L; Collier TL; Vasdev N; Shao Y; Ma J-A; Cravatt BF; Fowler C; Josephson L; Zhang M-R; Liang SH Design, synthesis and evaluation of reversible and irreversible monoacylglycerol lipase positron emission tomography (PET) tracers using a ‘tail switching’ strategy on a piperazinyl azetidine skeleton. J. Med. Chem 2019, 62, 3336–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAllister LA; Butler CR; Mente S; O’Neil SV; Fonseca KR; Piro JR; Cianfrogna JA; Foley TL; Gilbert AM; Harris AR; Helal CJ; Johnson DS; Montgomery JI; Nason DM; Noell S; Pandit J; Rogers BN; Samad TA; Shaffer CL; da Silva RG; Uccello DP; Webb D; Brodney MA Discovery of trifluoromethyl glycol carbamates as potent and selective covalent monoacylglycerol lipase (MAGL) inhibitors for treatment of neuroinflammation. J. Med. Chem 2018, 61, 3008–3026. [DOI] [PubMed] [Google Scholar]
- 29.Rotstein BH; Stephenson NA; Vasdev N; Liang SH Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun 2014, 5, 4365. [DOI] [PubMed] [Google Scholar]
- 30.Wang L; Jacobson O; Avdic D; Rotstein BH; Weiss ID; Collier L; Chen X; Vasdev N; Liang SH Ortho-stabilized 18F-azido click agents and their application in PET imaging with single-stranded DNA aptamers. Angew. Chem., Int. Ed 2015, 54, 12777–12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rotstein BH; Wang L; Liu RY; Patteson J; Kwan EE; Vasdev N; Liang SH Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(iii) ylides. Chem. Sci 2016, 7, 4407–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for positron emission tomography: recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions. Angew. Chem., Int. Ed 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schalk-Hihi C; Schubert C; Alexander R; Bayoumy S; Clemente JC; Deckman I; DesJarlais RL; Dzordzorme KC; Flores CM; Grasberger B; Kranz JK; Lewandowski F; Liu L; Ma H; Maguire D; Macielag MJ; McDonnell ME; Mezzasalma Haarlander T; Miller R; Milligan C; Reynolds C; Kuo LC Crystal structure of a soluble form of human monoglyceride lipase in complex with an inhibitor at 1.35 Å resolution. Protein Sci. 2011, 20, 670–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cravatt BF; Wright AT; Kozarich JW Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z; Cheng R; Yang J; Shao T; Vasdev N; Ran C; Zhang M-R; Liang SH A novel 18F-labeled MAG lipase biomarker for differentiating brown and white adipose tissue in the lipid network. J. Nucl. Med 2018, 59, 262. [Google Scholar]
- 36.Waterhouse RN Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol 2003, 5, 376–389. [DOI] [PubMed] [Google Scholar]
- 37.OECD. Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method OECD Publishing, Paris. [Google Scholar]
- 38.Chang JW; Niphakis MJ; Lum KM; Cognetta AB; Wang C; Matthews ML; Niessen S; Buczynski MW; Parsons LH; Cravatt BF Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol 2012, 19, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson AA; Garcia A; Parkes J; Houle S; Tong J; Vasdev N [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl. Med. Biol 2011, 38, 247–253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.