

Graphical abstract

Keywords: Corona virus, COVID-19, Mpro, SARS-CoV-2, Vanadium, Vanadate, Consensus, Docking, Autodock metal parameters, PTP1B
Abstract
In silico techniques helped explore the binding capacities of the SARS-CoV-2 main protease (Mpro) for a series of metalloorganic compounds. Along with small size vanadium complexes a vanadium-containing derivative of the peptide-like inhibitor N3 (N-[(5-methylisoxazol-3-yl)carbonyl]alanyl-l-valyl-N1-((1R,2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl] methyl }but-2-enyl)-l-leucinamide) was designed from the crystal structure with PDB entry code 6LU7. On theoretical grounds our consensus docking studies evaluated the binding affinities at the hitherto known binding site of Chymotrypsin-like protease (3CLpro) of SARS-CoV-2 for existing and designed vanadium complexes. This main virus protease (Mpro) has a Cys-His dyad at the catalytic site that is characteristic of metal-dependent or metal-inhibited hydrolases. Mpro was compared to the human protein-tyrosine phosphatase 1B (hPTP1B) with a comparable catalytic dyad. HPTP1B is a key regulator at an early stage in the signalling cascade of the insulin hormone for glucose uptake into cells. The vanadium-ligand binding site of hPTP1B is located in a larger groove on the surface of Mpro. Vanadium constitutes a well-known phosphate analogue. Hence, its study offers possibilities to design promising vanadium-containing binders to SARS-CoV-2. Given the favourable physicochemical properties of vanadium nuclei, such organic vanadium complexes could become drugs not only for pharmacotherapy but also diagnostic tools for early infection detection in patients. This work presents the in silico design of a potential lead vanadium compound. It was tested along with 20 other vanadium-containing complexes from the literature in a virtual screening test by docking to inhibit Mpro of SARS-CoV-2.
1. Introduction
At the end of 2019 the world-wide spread of a new virus strain of the coronavirus family [1] took its origin in Wuhan, China, which quickly developed pandemic dimensions. Vaccination campaigns of the world’s human population are a first remedy, along with efforts to develop oral drug treatments [2]. In spring 2020 the World Health Organization named the epidemic disease caused by this virus “Corona virus disease 2019″, or COVID-19 for short [3]. The International Committee on Taxonomy of Viruses denominated the new virus as “severe acute respiratory syndrome coronavirus-2”, for short: SARS-CoV-2 [4]. Clinically, after infection by inhalation, COVID-19 has been associated with manifestations of the respiratory tract, ranging from harmless symptoms including coughing, headaches, fever to pneumonia and severe impairment of breathing, sometimes leading to fulminant death in certain types of patients, even when treated in intensive care units (ICUs) of hospitals. The limited capacities of ICU has ushered the rise of intensive research & development efforts in the field of Medicinal Chemistry, including all well-established strategies from de novo design to drug repurposing in the hope of finding some new and reliable cures against the pandemic threat very fast [5], [6], [7]. Anti-inflammatory drugs have been reviewed recently as comedication to treat infected persons [8]. In addition to its infective ability by inhalation [9], [10], the SARS-CoV-2 virus has shown that it can easily mutate into new variants with higher transmission rates [11]. The viral genome has been sequenced [12], [13] and in the meantime a growing part of its proteins has been characterized [14], [15], so that three-dimensional molecular structures have become already available as targets for the ongoing drug research [16], [17] and development of vaccines [18], [19], [20].
A significant amount of drug candidates has been targeted towards the main protease (Mpro) of SARS-CoV-2 [21], [22], [23]. This enzyme plays a pivotal role in the propagation of the virus, since it is responsible for a large part of the proteolytic cleavage required to obtain the functional proteins that are essential for the replication and transcription of the virus, because these proteins are synthesized in the form of polyproteins that need to be released to produce mature proteins [21]. SARS-CoV. Mpro is found in a mixture of monomers and dimers in solution, its active state has been associated with its dimeric form [22].
In 2020, liganded and unliganded crystal structures of SARS-CoV Mpro were published [16], [21], [23]. Moreover, the enzyme characterizes itself through a typically flexible protein structure, a fact that also concerns the known conformations at its catalytic or active site [14]. Its primary sequences are highly conserved in segments around the active site with a 96% identity score [15], [24]. Intriguingly, its active site shows unusual residues with respect to other “more conventional” chymotrypsin-type enzymes or to serine and cysteine hydrolases. In particular, it shows a Cys-His dyad [21] and not a canonical Ser/Cys-His-Asp/Glu triad [25]. In this context, it is possible to take advantage of modeling strategies that have been developed for inhibitors of enzymes with Cys-His/Asp dyads, among them the protein family of protein tyrosine phosphatases (PTPs) [26]. With the experience gathered in the field of design and modeled inhibitors derived from vanadium complexes [27], [28], [29], [30], our group aims at contributing with a computational study to evaluate the potential affinity of vanadium complexes to the Mpro target.
Vanadium is a versatile metal that can be present as central atom in organic complexes operating as potential drug candidates against diabetes or even cancer [31], [32], [33], [34], [35]. Vanadium’s biomedical versatility is mainly due to speciation in combination with changing geometries, all of which enable oxygen-containing vanadium ions to successfully mimic phosphate anions in biochemical signaling pathways [30], [31]. It is particularly avid to interact with proteins [36] at both sides of cellular membranes [37], [38]. Numerous studies suggested that vanadium compounds impact the immune system – for example by stimulating and activating T cells – and vanadium compounds have been used as complementary pharmacological agents to oncolytic-virotherapy mediated immunotherapy [39].
Antivirus activity has been reported for vanadium polyoxido-clusters [40], and a vanadium-substituted polyoxoidotungstate possesses antiviral activity against FluV A, RSV, parainfluenza virus (PfluV) type 2, Dengue fever virus, HIV-1 and SARS coronavirus in vitro [41]. Of note, the original papers speak of polyoxo, polyoxotungstate or polyoxovanadates compounds, yet we follow the official inorganic nomenclature settings [40], [41].
In particular, those polyoxidovanadates have been proven to act as broad spectrum agents and non-toxic anti-RNA virus agents in both in vitro bioassays and in vivo studies. Finally, the mechanism of action against HIV was studied by Shigeta and coworkers [41]. Apparently, it affected HIV binding to cell membranes as well as syncytia formations between HIV-infected and uninfected cells.
As a most valuable asset for our study, a great wealth of the extent COVID-19 literature hints at the potential role of vanadium-containing compounds as promising candidates to treat acute respiratory diseases. Besides other metal compounds, vanadium-based substances not only offers pharmaceutical possibilities as therapeutics with a wide spectrum of actions, but also thanks to its nuclear properties, such as distinguishable electromagnetic propensities, it is well-suited as a contrast medium agent for magnetic resonance imaging [42], for the development of radiological diagnosis.
Recently, the antiproliferative effects of vanadium 8-hydroxyquinoline against cancer cell lines were reported to promote cellular detachment and the development of encapsulated nanoparticles of an oxidovanadium (IV) nalidixic acid (NA) complex for drug delivery with stronger antimicrobial activity against E. coli, B. cereus, S. aureus and P. aeruginosa than NA [43], [44]. Both reports open the possibility of exploiting vanadium compounds for the development of novel preventive and therapeutic agents against numerous diseases, including COVID-19.
2. Equipment and methods
PC Hewlett Packard, EliteDesk 800G1 TWR, 1 TB Solid state HD, 3 GB video card, 12 GB RAM; ArgusLab 4.0.1 (build 20.19.15.4531); Gaussian 16 [45]; Discovery Studio Visualizer (v19.1.0.18287) [46]; GaussView version 6.0.16. We also applied in the present study the following modeling programs: Autodock 4 and MGL-Tools [47], Chimera X [48], Swiss PDB Viewer [49] and Vega ZZ [50].
Four crystal structures of our target protein were retrieved at RCSB PDB: (i) the viral main protease (Mpro) in its unliganded or apo form (PDB code: 6M03) [16]; (ii) the structure of this main protease in complex with an inhibitor ligand which was labeled N3 for short by the authors (PDB code: 6LU7) [16]. Its chemical name is N-[(5-methylisoxazol-3-yl)carbonyl]alanyl-l-valyl-N ~ 1~-((1R,2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl]methyl}but-2-enyl)-l-leucinamide; (iii) an dimeric unliganded structure of 3CL Mpro (PDB code: 6Y84) [21]; as well as (vi) a ligand-free structure of 3CL Mpro (PDB code: 6Y2E) [23]. Other PDB entries were also inspected like 6WQF, 6XB0, 6XB1, 6XB2 or 6XBH (Supplemental Material, SM).
As reference model for a liganded protein tyrosine phosphatase 1B complex we chose a crystal structure where the active site is occupied by vanadate (PDB code: 3QKQ) [51]. The protein constitutes a single-point mutant (W179F) and was not used as docking target but merely as reference to identify all those amino acids which typically interact with bound vanadium atoms.
In our chimeric drug design study, we created a vanadium-containing derivative of the peptide-like ligand called “N3” which constitutes a strong binder to viral main protease Mpro (PDB code: 6LU7). The geometries of the vanadium organic chelate were refined under the Quantum chemistry software Gaussian 16, with the APFD method, using the base 6–311 + g(d,2p). The calculation was carried out in the same software with the parameters corresponding to a flexible binder, using the calculation type “docking” under the docking tool AD4. Other methods than docking have also been applied to the very same target, e.g. data base screening and molecular dynamics [52].
2.1. Computational settings for docking by Autodock4
Aiming at a consensus docking study, we carried out preliminary docking simulations under different free software packages (Autodock Vina, Autodock 4, Yeti, PyRx, LeDock and iGMDock in SM) but only one program in particular was able to handle satisfactorily vanadium compounds, namely Autodock4 (AD4) [Molecular Graphic Laboratory, Department of Molecular Biology, Scripps Research Institute, La Jolla, CA] [47]. This software runs under MS-DOS as a stand-alone tool or implemented through a graphical user interface like Vega ZZ or Autodock-Tools (ADT), aka MGL-Tools. Prior to docking of the vanadium-containing molecules into the target, the different program settings were applied for exploration (Table S1 in SM) since vanadium and other heavy metals have not been parametrized in standard Autodock 4 (Table S1 in SM). The missing vanadium descriptors were implemented in the core part of AutodockD 4 (parameter file named AD4_parameters.dat). They were necessary to generate the so-called grid box which is placed at the binding site to evaluate the interaction between the ligand‘s atoms and the protein residues [Grid Parameter File GPF (Table S2 in SM)]. In addition to the parameter settings to run docking simulations, input data were stored into the so-called Docking Parameter File with the file extension DPF (Table S3 in SM). In 2007, a computational study was published to dock a vanadium-containing compound (decavanadate) to ATPase. To this end the vanadium atom was replaced by an iron atom (Fe) in Autodock 4, and here we cite: “ … The non-bond parameters for vanadium were copied from the corresponding iron values in the AutoDock library, a reasonable assumption given the similarity between these two elements.….” [53].
In the present in silico study Autodock4 (AD4) was also used for back docking of the original co-crystalized ligand N3 and the blind docking of its V, S, P, Fe and Ni derivatives.
So-called “back docking” is an essential means to locate and confirm the protein active site by reproducing the crystallographically observed binding mode. The crystal structure of the target protein was retrieved from the RCSB protein data base with PDB code 6LU7. The protein structure was cleaned from crystal water molecules and other co-crystalized moieties probably related to crystallogenesis as part of the standard procedure under ADT to prepare the ligand and protein target. In particular, polar hydrogen atoms were added and Kollman united atom charges were loaded for all amino acids. ADT created the PDBQT input files prior to launching autogrid4 and autodock4 [47].
Although the vanadium-containing derivative of ligand N3 was the initially proposed compound to be studied, four other coordination complexes with the same overall structure (scaffold) were included in the docking study. The only difference between the molecules is the vanadium central atom, which was successively replaced by S, P, Fe or Ni. These four additional test molecules helped evaluate the behaviour of the docking program under nonstandard conditions in a systematic way. In particular, our approach required implementation of missing parameters for V, Fe and Ni. Upon consulting the official help and FAQ online pages of AD4 at the Web site of Scripps Institute, USA, they were manually added to the so-called “AD4_parameters” file [54], [55].
For the PDBQT input models of all studied ligands, the maximal number of rotatable bonds had to be limited and was set to eight. With more predefined rotatable bonds, the stochastic procedure and genetic algorithm of AD4 would not exhaustively explore the conformational search space and spurious results would miss the best solutions of occupying the binding site (a so-called docking pitfall). All rotatable bonds were defined in staggered positions around the central part (first active, second inactive, third active and so forth). This way only those bonds were rotatable which could significantly contribute to structural changes on the scaffold of ligand N3. During docking, all amide bonds were held rigid as well as all terminal groups, i.e. the few end point atoms of the N3 scaffold. For all ligands atomic partial charges were assigned by the Gasteiger approach under VEGA ZZ [50].
The three-dimensional grid box with a box size of 60 × 60 × 60 Ångstroms was ligand-centred on the crystal structure position of ligand N3. The default distance value of 0.375 Å was set between all grid points to generate the evaluation points in space. Again, a too large a search space (grid box size) or inter-point distance for evaluation would give rise to non-systematic results with the risk of missing relevant interaction at an atomic scale between ligand and amino acids (another docking pitfall, see above). The number of evaluations to collect solutions for each docking study was raised to 1000 under the Lamarckian genetic algorithm in order to elucidate the ligand conformations with the highest receptor affinities. ADT was used to analyse the numeric output date and DS-Visualizer from Discovery Studio [49] was used to display the resulting 3D models and to produce the figures [56].
3. Results
Upon comparison, the initial consensus docking idea was put aside. The programs fell soon short of expectation, i.e. without the possibility to describe vanadium compound geometries correctly. The option to work-around under geometrically similar atom types was also not possible due to missing metal parameter implementations (details in SM). The first part of this study was the demonstration of similar catalytic sites for the main protease (Mpro) of SARS-CoV-2 and protein tyrosine phosphatase 1B, PTP1B, both using the dyad Cys-His/Asp. To this end, 2D or 3D alignments of sequences or structures were carried out concerning 6LU7 [16] and 3QKQ [51], respectively (Fig. 1 ). It can be speculated that the ligand could be the monoanionic H2VO4 − vanadate as hydrogens are not recognizable by crystallographic imaging techniques.
Fig. 1.

Display of two crystal structures for viral target Mpro and hPTP1B. Both structures were rotated sideway to show the respective active sites with the catalytic dyad with cysteine and histidine/aspartate. Both amino acids are displayed in space-fill models with four colors: C in grey, O in red, N in blue, S in yellow and H omitted). (Left) The crystal structure of 3CL Mpro from SARS-CoV-2 (PDB code: 6LU7) is the monomeric complex with an oligopeptide derivative called N3. This ligand is displayed at the top as stick model in horizontal orientation. (Right) The vanadium-liganded crystal structure of human PTP-1B (PDB code: 3QKQ). Surprisingly, neither HVO42− nor H2VO4− are present but vanadate VO43− and this fact might hint at crystallographic or crystallogenesis issues (resolution: 2.20 Å, see also label “VO4”). Both proteins are displayed as ribbon models for the protein backbones with three colors: α-helices in red, β-strands in blue and hairpin turns and loops in green.
The structure of the 3CL Mpro protein (Fig. 1) embraces three domains: (i) domain I (residues 8 to 101); (ii) domain II (residues 102 to 184) with the catalytic site in an antiparallel β-barrel structure; and (iii) domain III (residues 201 to 303) of each monomer forms the interface for intermolecular interactions to produce the active dimeric unit. Structurally speaking, it is a region of five α-helices. The catalytic dyad with cysteine (C145) and histidine (H41) is located at the bottom of a superficial rim (elongated cleft or crevice) between domains I and II [16].
Intrigued by the fact that the active site of PTP1B (Fig. 1) also possesses a catalytic dyad – an insight knowledge from our prior antidiabetic vanadium research work [27], [28], [29], [30] – we hypothesized that vanadium compounds which strongly inhibit PTP1B may also act as Mpro inhibitors.
Fig. 1 shows the catalytic dyad, here with Cys215 and Asp181. It is assisted by three adjacent residues of Arg221, Trp179, and Pro180. They stabilize the phosphotyrosine moiety (which belongs to a natural substrate protein) in its position at the so-called WDP (tryptophan, aspartate, proline). Cys215 carries out a nucleophilic attack on the phosphate group of the substrate, while aspartate (Asp181) transfers a proton to the oxygen of substrate tyrosine to complete the reaction and allows the tyrosine moiety to leave – and with it the substrate protein [26].
The human tyrosine-protein phosphatase was selected as reference protein. It belongs to the same hydrolase superfamily as our viral target protein. The cleavage reactions catalyzed by the hydrolases are mediated by a water moiety and correspond to the third class of enzymes in the EC classification system (EC 3, for short). Hence, we studied the vanadium-ligand interacting residues at the active site. Other features are far too distant on an evolutionary scale, so that no topological or structural similarities can be expected, other than the observation how vanadium compounds are recognized on an atomic level by binding site residues. Of note, the Mpro viral target is classified as EC: 3.4.22.69, while the human tyrosine-protein phosphatase (hPTP1B) is a member of the EC 3.1.3.48 subclass of hydrolases. Another source of inspiration constitutes the Influenza A neuraminidase, which also belongs to the third enzyme class (EC 3.2.1.18) [57]. Research groups have predicted that inhibitors of Influenza A neuraminidase – such as oseltamivir, zanamivir and zidovudine – may be effective clues in the treatment of COVID-19: either through complete genomic sequence similarity between the viruses and chemical structure similarity among the drugs [57]; or through molecular screening and docking against the SARS-CoV-2 Mpro enzyme (Fig. 2 ) [58]. Interestingly, there are no pre-clinical trials in COVID19-positive patients with these agents or analogs with new scaffolds [59].
Fig. 2.

Schematic display of the interacting amino acids at the binding site of the viral target protein Mpro in complex with its ligand N3 (PDB code: 6LU7). This crystal structure was selected because it shows the liganded active site. Ligand N3 is a peptide-like elongated inhibitor of Mpro (center). Color code: H bonds in dark green. Van der Waals interactions in light green. Pi -stacked amide bonds in magenta, and alkyl interactions in pink (light magenta). Labels: the residue names are given in three-letter code and the ID numbers were taken from the PDB entry 6LU7.
As expected, the sequence similarity between both proteins lies low the threshold for protein homology of 25% for a domain length with 100 residues (Table S4 in SM) [60], [61]. The domains containing the active sites are well conserved within each EC subclass [62]. But residue conservation above this threshold is vital to keep the catalytic general mechanism functioning, i.e. to catalyze bond cleavage for specific (or limited variation of) substrates. In hPTP1B the active site is embedded into a region of U-turn loops and α-helices with flexibility to open and close the access to the catalytic dyad. Precisely, the general access mechanism for inhibitors embrace molecules with chemical composition for successful recognition. They can open the cavity, unless they do not show suitable conformations or binding affinities to cysteine residues for keeping the cavity open to inhibit the enzyme.
In the second part of this work, we took advantage of the knowledge about inhibition of PTP1B to design a proper inhibitor for Mpro of SARS-CoV-2, based on a chimeric approach and repurposing vanadium-containing inhibitors of hPTP1B. The idea was to take the ligand known as N3 which is displayed in Fig. 2 [16], [63]. It possesses affinity to SARS-CoV-2 Mpro and serves as reference ligand for ligand docking of a series of vanadium-containing ligands for docking to viral target Mpro. A first round of docking simulations tested our hypothesis that vanadium compounds are druggable, i.e. as inhibitors of PTP1B they should be strong binders to Mpro as well. After evaluating the results of the first trial round, the simulation settings were improved for the second screening round with refined docking parameters for vanadium. This way we screened the viral protein Mpro for inhibition by our proposed vanadium compounds.
The first round of AD4 docking had the following molecular components: (i) the designed ethylendiamminacetate chelating-derivative of N3 (for short: N3modif(COO−)2) in an oxidoaquavanadium(V) complex: VO(H2O)N3modif(COO−)2; (ii) the previously designed vanadium inhibitor of PTP1B [29] named TSAG0101; (iii) together with proposed vanadium complexes as potential PTP1B inhibitors; followed by a series of well-known or classical vanadium compounds without hitherto known inhibitor properties (Table 1 ). The numeric results of the docking simulations for all molecules used in the first round of this study constitute crude estimates. In direct consequence, concentration differences of the inhibition constants (Ki) are truly insignificant below a ten to hundred-fold threshold, e.g. 4 micromolar vs. 0.04 concentration (40 nanomolarity). Hence, all target affinity values in Table 1 were rounded off to distinguish meaningful values, here: 0.04 < 0.4 < 4 < 40 ≪ 200 [μM]. The lower the concentration (Ki) the higher the affinity (E) and the more likely the perspective of promising druggability. The Ala-Val-Leu tripeptide scaffold of 01-VL (second and third rows in Table 1) with its monocyclic isoxazole head group is identical to N3 (first row in Table 1). Hence, the structure of 01-VL is closely related to N3. Based on our structure - activity analysis it can be expected that 01-VL will also be a strong binder to target in a similar binding mode as N3. It becomes evident that the nonidentical substructures will occupy different locations in the binding rim on the target surface (Figs. 3 and 4 ).
Table 1.
Listing of chemical structures with activities and bibliographic sources. Second row: 3D drawing of our proposed ligand 01-VL. Third row: additional 2D drawing of 01-VL scaffold without vanadium complexation. Asterisks (*) mark four atoms for vanadium central atom coordination. Symbols: [E; Ki] represent the computed Gibbs Free Energy of Binding (kcal/mol) and the micromolar inhibition constant. (Ref) indicate reference numbers. Abbreviations:ID: identification code; N3: ligand in 6LU7; 01-VL: VO(H2O)-N3modif(COO−)2; 04-VL: trans-BMOV; 11-VL: cis-VO(H2O)(acac)2; 03-VL: cis-BMOV; 05-VL: VO(EDT)2; 02-VL: TSAG0101; 09-VL: VO(EBIP); 14-VL: cis-VO(H2O)(ox)2; 12-VL: trans-VO(acac)2; 10-VL: VO(H2O)2DPC; and 15-VL: trans-VO(ox)2; BMOV: bis-maltolateoxidovanadate; acac: acetylacetonate; EDT: ethane-1,2-dithiol; TSAG0101: pyridine-2-carboxamideaquaoxidovanadate; EBIP: ethanebis-(2Z)-4-iminopent-2-en-2-ol; Ox: oxalate; DPC: pyridine-2,6-dicarboxylate.
| ID [E; Ki] (Ref) | Drawing of all studied vanadium-containing structures |
|---|---|
| N3 [-10; 0.04][16] | 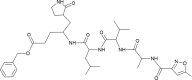 |
| 01-VL [-9; 0.4] (this work) | 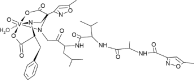 |
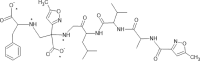 |
|
| 04-VL [-7; 4][31] | 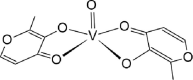 |
| 11-VL [-7; 4][28] | 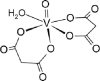 |
| 03-VL [-7; 4][28] | 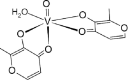 |
| 05-VL [-7; 4][28] | 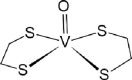 |
| 02-VL [-6; 40][29] | 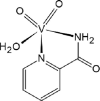 |
| 09-VL [-6; 40][28] | 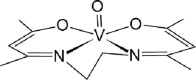 |
| 14-VL [-6; 40][28] | 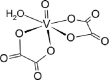 |
| 12-VL [-6; 40][35] | 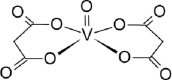 |
| 10-VL [-6; 40][64] | 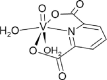 |
| 15-VL [-5; 200][65] | 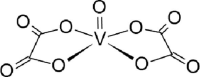 |
Fig. 3.

Display of the final docked poses for N3 and two of its derivatives. The target protein is Mpro (PDB code: 6LU7). Its N3 ligand is shown in its natural position (mustard-green). The other two docked poses were computed for 01-VL with the user-defined vanadium parameters from the literature (dark green) or the AD4 built-in sulphur parameters replacing the atom type V by S (blue). The V (S) atom type is displayed as a dark green (below and behind the blue) ball. The elongated binding site (rim) is in top-down view in diagonal orientation from bottommost left to topmost right filled with the docked ligands. It is contoured by its surface with semi-transparent colours: from red (negative charge) over grey (neutral) to blue (positive charge). The inlay panel shows the partial atomic charge distribution between 0.1 and −0.1 (bottom left). The ribbons represent parts of the protein backbone in red for α-helical segments, green for loops and light blue for β-strands.
Fig. 4.

Display of docked poses at the active site of the main protease target. The mustard-green structure is the crystal pose of ligand N3. The superposed stick models show the four back docked poses of N3 with 2, 4, 6 or 8 rotatable bonds (four bluish colours). The blind docked vanadium-containing derivative (01-VL) is coloured in dark green. By eyesight it can be judged that back docking of N3 was successful, since all docking solutions are in good superposition with the crystallographically observed Mpro complex with ligand N3 (PDB code: 6LU7). Blind docking of 01-VL results in a totally identical orientation of the N3 ligand’s tripeptide backbone with alanine, leucine and valine (Ala2, Leu3, Val4 on chain C) inside the binding rim. Only the head group is tilted along the tripeptide backbone with the protruding toluene sidechain (top rightmost dark green sticks, sitting in front of a light blue β-strand).
The numerical docking results [47] were analysed under MGL-Tools [51] and listed (Table 1). All results from this first round are of significance in the sense that coordination compounds could be competitive inhibitors, as is evident by comparing the docking free energy of N3 versus 01-VL. This hints at the druggability of such vanadium-organic ligands in general. Concerning our hypothesis that vanadium compounds designed to be inhibitors of PTP1B may act as Mpro inhibitors as well – the computed data reflect that the stronger the PTP1B-inhibitor (TSAG0101, BMOV) the higher also the affinity to the active centre of Mpro. We observed differences with respect to the specific ligands for Mpro and some structure–activity relationships that could increase the affinity: (i) larger size, (ii) low symmetry, and (iii) the presence of aromatic rings.
Almost all studied vanadium-containing ligands (VL, for short) have none or only one rotatable bond, with two obvious exceptions: (i) the elongated ligand N3 of target Mpro [16], [63], which has 19 rotatable groups, and (ii) our proposed new ligand 01-VL, which has 16 rotatable bonds (RoBos).
The AD4 runs were limited to 100 cycles for all ligands under scrutiny. Docking analyses under ADT revealed that the docked solutions for all except two ligands could be sorted by RMSD into one or two clusters. And again, the two exceptions concern the aforementioned large compound with the many rotatable bonds. AD4 docking is not amenable to treat elongated flexible scaffolds with more than 10 rotatable bonds or so. We tried to work around with recommended Autodock Vina which is documented to be suited to handle more rotatable bonds, but it fell short of expectation because the ligand was placed out of the active site-centred search box (data not shown). Under AD4 all “VL”-labelled ligands had negative docking free energies which means that they are strong binders to target in the lower micromolar range (Table 1). And all of them were bound to residues by noncovalent interactions at the active site (Fig. 3).
For the second round of AD4 docking assays we refined the docking procedure to take into account the influence of rotatable bonds and the presence of heteroatoms in the results for docking. For the first problem, we undertook blind docking to target with the N3 ligand and its (modified) vanadium derivative whose structure can be formulated as VO(H2O)-N3modif(COO−)2, and designate as 01-VL.
For the particular case of the heteroatoms we had to test the parametrization of vanadium to give reliable docking results. Vanadium is a “d” block element and the electronic fine structure cannot be reflected in AD4 docking procedures and the affinity could be underestimated. To avoid this bias from happening, we substitute vanadium for sulfur.
For the first problem, we carried out a “back docking” procedure (sometimes called re- or self-docking) to observe the dependencies of results from the numbers of rotatable bonds (RB). To this end, different number of RoBos were selected manually and docked again against 6LU7. All RoBos were located at the side chains. Results are given in Table S5 in SM. After calibration, our docking protocol was set with eight RoBos to be the best choice for a trade-off between spurious search results of AD4 due to the sheer amount of RoBos (cf. the strong increment in RMSD clusters for 100 runs) and molecular conformational flexibility to adopt a meaningful conformation. Since the “natural” N3 was already “frozen-in” into a crystal complex with the same target, the search for an active conformation could be fairly reduced to eight (in staggered order).
Fig. 4 shows the final poses of N3 at the active site after the docking with a variety of RB numbers. N3 has a total number of RoBos with conformational relevance of RB = 19. From Table 2 , it became evident that a setting of RB = 8 was the best choice, and it was taken henceforth as default value for our docking study here.
Table 2.
AD4 docking results for ligands N3, 01-VL and ligands of type 01-ML with M = V, S, P, Fe or Ni. Settings: elitism = 3, runs = 1000, precise; 250,000 = minimum lowest, grid dimensions 60x60x60. For all ligands, the number of RB was set equal to 8. Of note, here the Ki values were not rounded off (cf. Table 1). The designed vanadium-containing derivative of N3 (VO(H2O)N3modif(COO−)2) was docked by replacing V parameters by S, P, Ni, Fe, respectively to test their docking performance.
| CODE and (Method of replacing V by = …) | Estimated free ENERGY of binding [kcal/mol] | INHIBITION CONSTANT Ki [molarities] |
|---|---|---|
| N3 | −10 | 25 nanoM |
| 01-ML (M=S) | −9 | 333 nanoM |
| 01-ML (M=P) | −8 | 2 microM |
| 01-ML (M=Ni) | −7 | 6 microM |
| 01-ML (M=Fe) | −7 | 7 microM |
| 01-ML (M=V), 01-VL | −6 | 60 microM |
To test the effect of the “d” block elements, we repeated the docking procedure changing the atom V for S in the structure of 01-VL ligand (Table 1). Results yielded −8.5 kcal/mol using AD4 and −7.5 kcal/mol using Autodock Vina [66]. Both values differ from the value −9.07 kcal/mol in Table 1. The difference clearly reflects to what quantitative degree AD4 is capable of differentiating “electronic” factors (predefined constant partial charge values) through its molecular mechanistic approach to determine atom-to-atom interactions. Therefore, we modified the docking protocol to test with V and its replacement by S as well. The values for the binding free energy and inhibition constant (Ki) are given in Table 2. In the next step, we performed blind docking for six ligands, N3, its vanadium-containing derivative 01-VL along with its variations where the vanadium element was formally replaced by sulfur, phosphor, iron and nickel atoms (S, P, Fe or Ni) for complex formation (Table 2). The following AD4 settings were used: the precision level was limited to 2,500,000 energy evaluations; survival rate (elitism) was limited to 1, the number of final poses was raised to 100 solutions (cf. histograms for both runs are given in Table S6 in SM).
Upon inspection of the docked poses in superposition (Fig. 5 ) it became evident that the best poses of 01-VL lied in good keeping to the overall orientation of reference ligand N3 from the crystal structure of Mpro (Table 2). Precisely, the docked ligand occupied the same binding rim as N3. The orientation of the V=O group is directed towards Cys145 to block its function in the same way as the CH2- group of ligand N3 which is crystallographically observed in the liganded crystal complex of Mpro (PDB code: 6LU7). This cysteine (C145) constitutes the conserved residue of the catalytic dyad for the proton exchange step during substrate cleavage. The back docked poses differ from the crystal pose by a rotation along their longitudinal axes to hide their V=O or S=O groups (Fig. 5). As a direct result of the rotated positions an aromatic ring hydrogen atom of both ligands covers the access of the reactive thiol group (-SH) of catalytic cysteine (C145). Both tilted positions block again the enzyme function in the same way as the aforementioned CH2- group of N3. This means that the vanadium complexation of N3 (VO(H2O)N3modif(COO−)2) constitutes an effective modification for viral enzyme inhibition with a strong affinity. Moreover, this tilted position avoids steric hindrance and reduces the intermolecular repulsion energy (penalty score of AD4). This way it comes as no surprise that its final free energy of binding estimate was found top ranked. Hence, vanadium-containing derivatives of N3 constitute strong binders. Occupying an essential part of the catalytical center they effectively act as blockers or inhibitors. In our best-case scenario only between ten- to hundred-fold higher concentrations are needed to achieve the same receptor effect as the literature reference N3 (PDB code: 6LU7).
Fig. 5.

Display of 3D stick models for two docked poses of modified N3 as well as crystal pose of ligand N3. The vanadium and sulfur atoms are displayed by their van der Waals volumes (top rightmost dark green and blue balls). Color codes: mustard-green N3 (PDB code: 6LU7); blue 01-ML with M = V (VO(H2O)N3modif(COO−)2); dark-green 01-ML with M = S (where an S atom formally replaces the V element in VO(H2O)N3modif(COO−)2). Hydrogen atoms are not displayed.
According to Table 2 it becomes evident that the vanadium parameter implementation does not perform well, actually it was borrowed from AD3, an older version of Autodock [53] whose hydrophilic versus hydrophobic contributions to the final energy calculation had to be rebalanced giving birth of the AD4 release [67]. In AD4, V, Ni and Fe are non-standard elements, so prior to docking we had implemented their parameters to the AD4 parameters file. Their parameters improved the outcome of AD4 somewhat through the choice of these elements in the order S > P > Ni = Fe (Table 2), albeit a true electronic effect of transition elements is not implemented in AD4 (Table 2 and Fig. 6 ). The order strongly hints at the fact that prior to their release of AD4, the program developers carried out intensive calibration work with a larger training set with many more significant liganded complexes to cover enthalpy-driven (H bonds, salt bridges, electrostatics) as well as entropy-driven (linearly scaled solvent effects, hydrophobic) forces etc. It can be seen from Table 2 that the free binding energy and inhibition constant values all lie within the same range, i.e. they were not really affected by the element choice of either P or S. Both belong to the parametrized atom set of AD4. Smaller fluctuations stem from Ni of Fe with user-implemented parameters. In terms of the final positions, again no significant differences were observed for the scaffolds (Fig. 6), although the side chains apparently could adopt different conformations in space (cf. terminal aromatic rings of ligands in Fig. 6).
Fig. 6.

Display of docking results for our proposed N3 derivative. For docking different parameter sets for the coordination atom M in MO(H2O)N3modif(COO−)2 were applied (here: V, S and Fe replacing M). M is shown as balls in the 3D stick models (centre). The best performing result was with M = S (mustard-green). The protein surface is displayed at the active site of the target protein (PDB code: 6LU7) and coloured: from bluish (positive partial charges) over grey (neutral) to reddish (negative partial charges).
Finally, we searched for lead compounds among the vanadium metalorganic structures in the Cambridge Crystallographic Data Centre (CCDC), assisted by the free service of CCDC and FIZ Karlsruhe, Germany, to view and retrieve structures. The search template was based on our vanadium coordination complexes selected of the first round of docking (Table 1). The search for potential inhibitors of Mpro resulted in selected structures on intuitional ground (Table 3 ).
Table 3.
Listing of chemical structure, ID code, literature reference and AD4 docking results of the final docked poses for all molecules used in the second round of this study. Non-default settings of AD4: medium precision with elitism = 1 and 100 runs. The “M” variable in the ID code alludes to the replacement of element V by S during alternative calculations, see last column: VO- or SO-ligands. First column names are either from the literature (Ref) or the Cambridge CSD entries.
| Name (Ref) and internal Ligand Code | Structure drawing | Estimated free Energy of binding [kcal/mol]; Inhibition Constant Kiµ |
|
|---|---|---|---|
| VO-ligand | SO-ligand | ||
| VO(H2O)-N3modif(COO−)2 (this work) 01-ML | 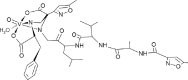 |
−6 60 [µM] |
−9 333 [nM] |
| cis-BMOV [28]03-ML | 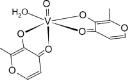 |
−7 4 [µM] |
−8 925 [nM] |
| trans-BMOV [31]04-ML | 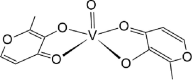 |
−8 2 [µM] |
−9 301 [nM] |
| ABOBOK [68]16-ML | 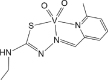 |
−8 3 [µM] |
−8 797 [nM] |
| CAWKUI [69]17-ML | 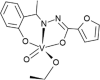 |
−9 478 [µM] |
−8 977 [nM] |
| AROJOG [70]18-ML | 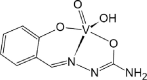 |
−7 10 [µM] |
−8 1 [µM] |
| AREYAY [71]19-ML | 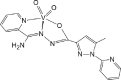 |
−9 619 [nM] |
−9 173 [nM] |
| ACUYOP* [72]20-ML | 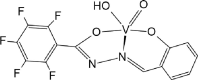 |
−7 8 [µM] |
−8 2 [µM] |
| ACILIH [73]21-ML | 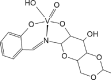 |
−7 9.7 [µM] |
−8 1 [µM] |
| BEGHAX [74]22-ML | 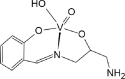 |
−7 19 [µM] |
−9 604 [nM] |
Abbreviations: BMOV, bis-maltolateoxidovanadate; ABOBOK, (N-Ethyl-N′-((6-methylpyridin-2-yl)methylene)-carbamohydrazonothioato)-(dioxido)-vanadium(v); CAWKUI, (Ethanolato)(N-(1-(2 (hydroxy)phenyl)ethylidene)furan-2-carbohydrazonato)oxidovanadium(v); AROJOG, cis-dioxido-(salicylaldehyde semicarbazonato)-vanadium(v); (N-(amino(pyridin-2-yl)methylene)-5-methyl-1-(pyridin-2-yl)-1H-pyrazole-3-carbohydrazonato-N,N′,O)-dioxido-vanadium(v) dihydrate; ACUYOP*, hydroxo-oxido-(2,3,4,5,6-pentafluoro-N-((2-oxidophenyl)methylidene)benzene-1-carbohydrazonato) vanadium(v); ACILIH, dimethylammonium (N-(2-oxybenzylidene)-4,6-O-ethylidene-β-d-glucopyranosylamine)-methanol-dioxido-vanadium(v); BEGHAX, (N-(3-ammonio-2-oxypropyl)salicylideniminato)-dioxido-vanadium(v) dihydrate. * see also SM.
After meeting difficulties for consensus docking due to missing atom types of metals for systematic exploitation and result interpretation of the retrieved molecules under scrutiny, two AD4 docking protocols were established: (i) docking the ligands after adding the vanadium parameters to the AD4 program was named “VO”; whereas (ii) docking the ligands after replacing the V atom by S was named “SO”. Of note, under (ii) recalculating the Gasteiger charges by Vega ZZ was necessary prior to docking. The retrieved structures had only few rotatable bonds ranging between RB = 0 to 3 what reduces the challenge of the genetic algorithm to search active conformations and suites AD4.
At that stage we were able to interpret the VO and SO models of each ligand. The VO model corresponds to the strongest ligand binding inside the cavity and SO could be related to the highest affinities a ligand could achieve in this study. Therefore, taking together all observations we could interpret the docking results of all ligands on the same footing.
In particular, ligand 01-VL kept one of the highest binding free energies (−8.84 kcal/mol, SO-ligand model), and it badly occupied the cavity, although its contribution to steric hindrance was high (−5.76 kcal/mol, VO-ligand model). All compounds in Table 3 show better binding free energies (VO-ligand models) than 01-VL – and it comes as no surprise as a logic consequence of size and bulk (steric hindrance) of the molecules. In the same way, higher values of binding free energy (VO-ligand model) were found for molecule 17-VL, followed by 19-VL. Both compounds share aromatic rings at either side of the chain. In contrast to the longer N3 and 01-VL, the other candiats are smaller but still bind along the key catalytic core segment from His41 to Met49 and prominet Cys145 (Fig. S2 to S5 in SM). As strong binders their function as blocking agents (inhibitors) like N3 and 01-VL. A similar structure–activity relationship holds true also for 20-VL, but the presence of fluorine atoms in the benzene ring is most likely a source of calculation bias in (AD4 or ADV) leading to an insufficient interaction of the fluorine-containing ligand inside the cavity [75], [76].
The SO-ligand column in Table 3 lists data for binding free energy (−8.84 kcal/mol, SO-ligand model) of 01-SO as reference. It lends most valuable insight about how strong the affinity of the molecules for the active site might be. The first molecule to outperform the reference is trans-BMOV (04-SL) with a value of −8.90 kcal/mol. Our computed finding is in excellent keeping with data that report that this compound is one of the strongest inhibitors of hPTP1B. An intriguing detail here is that for inhibition of hPTP1B an increased action stems from the cis-BMOV isomer, a result that is certainly related to steric differences in the cavity’s geometries of those enzymes [28].
Following the table data in the SO-ligand column, the highest value is found for compound compound 19-SL with −9.22 kcal/mol. This molecule is a base with a high content in nitrogen atoms, similar in this respect to 01-SL. Therefore, since 19-VL is also second best hit in binding free energy for our VO-ligand models, this compound could be considered the best inhibitor for targeting Mpro of SARS-CoV-2 in the group of compounds analyzed in this work, and probably a competitive one. Our experience reported here helps develop better inhibitors. For instance, it could be predicted that, for molecule 17-ML, the chimeric replacement of its furan ring to the pyridine-pyrazole chain of 19-ML could result in a desired increase of binding free energy.
4. Discussion
On computational ground it is safe to utter that the question raised in the title about the druggability of vanadium complexes can be answered positively. The strategy of repurposing inhibitors of SARS-CoV-2 proteins, such as Mpro-related hydrolases, could be considered a valid one [77], [78], [79], especially in the light that, to date, no protease inhibitors targeting SARS-CoV 3CL Mpro have been officially approved as drugs for the commercial markets, despite intensive research efforts.
In our case of drug complexes, which are based on transition elements such as vanadium, an increased binding affinity to target can be expected in the light of the known structural flexibility of viral proteins [14].
Structure–activity-related features for vanadium-based inhibitors of Mpro can be drawn from the inspection of the docked poses as follows: (i) molecular size in the range 20–40 heavy atoms (no H); (ii) lower symmetry; (iii) with presence of aromatic rings on both sides; (iv) the VO group clearly exposed at one end (and not in the centre); (v) electronically delocalized systems with a higher basic nitrogen content as the coordination environment for vanadium.
A lead compound for targeting SARS-CoV 3CL Mpro could be 19-ML with the chemical name: (N-(Amino(pyridin-2-yl)methylene)-5-methyl-1-(pyridin-2-yl)-1H-pyrazole-3-carbohydrazonato-N,N′,O)-dioxido-vanadium(V) dihydrate [71].
With respect of the methods used for docking of transition metal compounds, electronic contributions could mark the difference, but this cannot be taken into account by docking here. All docking results where V was replaced by Fe, Ni, P, or S were practically the same. This comes to no surprise because predefined atom types for ligands and amino acids evaluate the atom-to-atom interactions at the ligand/protein interface in space (grid box). While steric effects are reproduced with atom radii or bond distances, angles and torsions between atoms, electronic effects are oversimplified for the sake of speed and model complexity and reduced to electrostatic attraction and repulsion forces based on the concept of total and partial atom charges. In more general terms, the AD4 method is an extension of the molecular mechanic approaches with conformational search and (docked energy) scoring functions. It becomes clear that metal ions, radii and their geometries do not vary much, and their subtle differences in electronic, binding and geometric properties cannot be taken into account in details. Moreover, the precision threshold of AD4 excludes the distinction of Ki values with less than hundred-fold differences.
If time and hardware resources are not limited, the properties of the “d” shield electrons can be studied in details by so-called ONIOM-type docking simulations which applies ab initio calculations in the core part (for only tens of heavy atoms) and semi-empirical molecular mechanics methods around this hot spot of interest to cover the hundreds to thousands of remaining atoms of a huge protein complex or other biopolymer like DNA or RNA. Of note, ONIOM is a mnemotechnical acronym for “Our own N-layered Integrated molecular Orbital and Molecular mechanics”.
Our findings are summarized here: (i) applying vanadium-containing complexes for drug design against COVID-19; (ii) targeting the catalytic site of the main protease of SARS-CoV-2, which resembles protein tyrosine phosphatase 1B. Both structures have catalytic mechanisms based on a dyad (cysteine -[histidine or aspartate]) which is highly conserved among the members of related hydrolase subfamilies; (iii) using metalorganic compounds as inhibitors of SARS-CoV-2, with better stability at the active site; (iv) identifying possible lead compounds; (v) vanadium complexes could be developed as drugs not only for pharmacotherapy but also as diagnostic tools like contrast media agents for early infection detection in patients.
5. Conclusions
Drug repurposing strategy of inhibitors from related hydrolase enzymes could be applied in future studies for viral 3CL Mpro. The approach promises to identify new potential ligands thanks to the exploitation of the hitherto known ligands bound to the target. The design of metalorganic compounds as potential inhibitors of SARS-CoV 3CL Mpro will probably usher a new area of metal-containing drug development against the coronavirus in the future. The proposed vanadium complexes belong to these potential candidates for pharmacotherapy and diagnosis for SARS-CoV-2.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ica.2021.120287.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Listing of the online web sources to implement the missing vanadium central atom parameters into AD4, GPF settings to run the grid calculation under AD4, the input data settings for the binding poses of all 17 ligands of round 1 docking assays, results for of the redocking of N3 ligand in 6LU7 with different RB = 2, 4, 6 or 8 and the AD4 setting for 01-VL ligand which is the proposed vanadium-containing derivative of N3.
References
- 1.C. Chakraborty, AR. Sharma, M. Bhattacharya, G. Sharma, SS. Lee. The 2019 novel coronavirus disease (COVID-19) pandemic: A zoonotic prospective. Asian Pac J Trop Med [serial online] 2020 [cited 2021 Jan 27];13:242-6. Available from: https://www.apjtm.org/text.asp?2020/13/6/242/281613.
- 2.Rothan H.A., Byrareddy S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020;109 doi: 10.1016/j.jaut.2020.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization (WHO), (2020). https://covid19.who.int/ (last visit 19 October 2020).
- 4.International Committee on Taxonomy of Viruses (ICTV), (2020). https://talk.ictvonline.org/ (last visit 19 October 2020).
- 5.Robson B. COVID-19 Coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed achilles’ heel conserved region to minimize probability of escape mutations and drug resistance. Comput. Biol. Med. 2020;121 doi: 10.1016/j.compbiomed.2020.103749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborty C., Sharma A.R., Sharma G., Bhattacharya M., Lee S.S. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur. Rev. Med. Pharmacol. Sci. 2020;24(7):4016–4026. doi: 10.26355/eurrev_202004_20871. [DOI] [PubMed] [Google Scholar]
- 7.P. Saha Rudra, A.R. Sharma, K. Singh Manoj, S. Saikat, B. Swarnav, M. Snehasish, B. Manojit, L. Sang-Soo, C. Chakraborty Chiranjib. Repurposing Drugs, Ongoing Vaccine, and New Therapeutic Development Initiatives Against COVID-19 . Frontiers in Pharmacology, 2020, 11, 1258. URL=https://www.frontiersin.org/article/10.3389/fphar.2020.01258. [DOI] [PMC free article] [PubMed]
- 8.H. Gilzad-Kohan, F. Jamali, Anti-Inflammatory properties of drugs used to control COVID-19 and their effects on the Renin-Angiotensin system and Angiotensin-Converting enzyme-2, J. Pharm. Pharm. Sci. 23 (2020) 259–277. 10.18433/jpps31346. [DOI] [PubMed]
- 9.M. Chinazzi, J.T. Davis, M. Ajelli, C. Gioannini, M. Litvinova, S. Merler, A. Pastore y Piontti, K. Mu, L. Rossi, K. Sun, C. Viboud, X. Xiong, H. Yu, M.E. Halloran, I.M. Longini, A. Vespignani, The effect of travel restrictions on the spread of the 2019 novel coronavirus (COVID-19) outbreak, Science 368 (2020) eaba9757. 10.1126/science.aba9757. [DOI] [PMC free article] [PubMed]
- 10.Leung K., Wu J.T., Liu D., Leung G.M. First-wave COVID-19 transmissibility and severity in China outside Hubei after control measures, and second-wave scenario planning: a modelling impact assessment. Lancet. 2020;395:1382–1393. doi: 10.1016/S0140-6736(20)30746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grubaugh N.D., Hanage W.P., Rasmussen A.L. Making Sense of Mutation: What D614G means for the COVID-19 pandemic remains unclear. Cell. 2020;182:794–795. doi: 10.1016/j.cell.2020.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang, N. Zhu, Y. Bi, X. Ma, F. Zhan, L. Wang, T. Hu, H. Zhou, Z. Hu, W. Zhou, L. Zhao, J. Chen, Y. Meng, J. Wang, Y. Lin, J. Yuan, Z. Xie, J. Ma, W.J. Liu, D. Wang, W. Xu, E.C. Holmes, G.F. Gao, G. Wu, W. Chen, W. Shi, W. Tan, Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding, Lancet. 395 (2020) 565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed]
- 13.Mousavizadeh L., Ghasemi S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2020 doi: 10.1016/j.jmii.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.R. Römer, N. Römer, A.K. Wallis, Flexibility and mobility of SARS-CoV-2-related protein structures, (2020) 2–13. 10.1101/2020.07.12.199364. [DOI] [PMC free article] [PubMed]
- 15.Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 17.C. Chakraborty, AR . Sharma, M. Bhattacharya, G. Sharma, SS . Lee, G. Agoramoorthy. Consider TLR5 for new therapeutic development against COVID-19. J Med Virol. 2020 Nov;92(11):2314-2315. doi: 10.1002/jmv.25997. Epub 2020 May 22. PMID: 32391920; PMCID: PMC7272826. [DOI] [PMC free article] [PubMed]
- 18.Sørensen B., Susrud A., Dalgleish A.G. Biovacc-19: A candidate vaccine for Covid-19 (SARS-CoV-2) developed from analysis of its general method of action for infectivity. QRB Discov. 2020;1 doi: 10.1017/qrd.2020.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.M. Bhattacharya, AR. Sharma, P. Patra, P. Ghosh, G. Sharma, BC. Patra, SS. Lee, C. Chakraborty. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J Med Virol. 2020 Jun;92(6):618-631. doi: 10.1002/jmv.25736. Epub 2020 Mar 5. PMID: 32108359; PMCID: PMC7228377. [DOI] [PMC free article] [PubMed]
- 20.Bhattacharya M., Ranjan Sharma A., Patra P., Ghosh P., Sharma G., Chandra Patra B., Saha R.P., Lee S.-S., Chakraborty C. A SARS-CoV-2 vaccine candidate: In-silico cloning and validation. Inf. Med. Unlocked. 2020;20 doi: 10.1016/j.imu.2020.100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kneller D.W., Phillips G., O’Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu W.-C., Chang H.-C., Chou C.-Y., Tsai P.-J., Lin P.-I., Chang G.-G. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005;280:22741–22748. doi: 10.1074/jbc.M502556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020:eabb3405. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z., Peng C., Shi Y., Zhu Z., Mu K., Wang X., Zhu W. Nelfinavir was predicted to be a potential Inhibitor of 2019-NCov main protease by an integrative approach combining homology modelling. Mol. Docking Binding Free Energy Calculation. 2020 doi: 10.1101/2020.01.27.921627. [DOI] [Google Scholar]
- 25.Gorbalenya A.E., Snijder E.J. Viral cysteine proteinases. Perspect. Drug Discov. Des. 1996;6:64–86. doi: 10.1007/BF02174046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma B., Xie L., Yang F., Wang W., Zhou Q., Xiang M., Zhou S., Lv W., Jia Y., Pokhrel L., Shen J., Xiao Q., Gao L., Deng W. Recent advance on PTP1B inhibitors and their biomedical applications. Eur. J. Med. Chem. 2020;199 doi: 10.1016/j.ejmech.2020.112376. [DOI] [PubMed] [Google Scholar]
- 27.Scior T., Guevara-Garcia A., Bernard P., Do Q.-T., Domeyer D., Laufer S. Are vanadium compounds drugable? Structures and effects of antidiabetic vanadium compounds: a critical review. Mini-Reviews Med. Chem. 2005;5:995–1008. doi: 10.2174/138955705774575264. [DOI] [PubMed] [Google Scholar]
- 28.Scior T., Mack H.-G., García J.A.G., Koch W. Antidiabetic bis-maltolato-oxovanadium(IV): conversion of inactive trans- to bioactive cis-BMOV for possible binding to target PTP-1B. Drug Des. Devel. Ther. 2008;2:221–231. doi: 10.2147/DDDT.S3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scior T., Guevara-García J.A., Melendez F.J., Abdallah H.H., Do Q.-T., Bernard P. Chimeric design, synthesis, and biological assays of a new nonpeptide insulin-mimetic vanadium compound to inhibit protein tyrosine phosphatase 1B. Drug Des. Devel. Ther. 2010;4:231. doi: 10.2147/DDDT.S8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scior T., Guevara-Garcia J., Do Q.-T., Bernard P., Laufer S. Why antidiabetic vanadium complexes are not in the pipeline of “Big Pharma” drug research? A critical review. Curr. Med. Chem. 2016;23:2874–2891. doi: 10.2174/0929867323666160321121138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rehder D. The potentiality of vanadium in medicinal applications. Inorganica Chim. Acta. 2020;504 doi: 10.1016/j.ica.2020.119445. [DOI] [Google Scholar]
- 32.Sinha A., Banerjee K., Banerjee A., Sarkar A., Ahir M., Adhikary A., Chatterjee M., Choudhuri S.K. Induction of apoptosis in human colorectal cancer cell line, HCT-116 by a vanadium-Schiff base complex. Biomed. Pharmacother. 2017;92:509–518. doi: 10.1016/j.biopha.2017.05.108. [DOI] [PubMed] [Google Scholar]
- 33.Petanidis S., Kioseoglou E., Domvri K., Zarogoulidis P., Carthy J.M., Anestakis D., Moustakas A., Salifoglou A. In vitro and ex vivo vanadium antitumor activity in (TGF-β)-induced EMT. synergistic activity with carboplatin and correlation with tumor metastasis in cancer patients. Int. J. Biochem. Cell Biol. 2016;74:121–134. doi: 10.1016/j.biocel.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 34.Martínez-Valencia B., Corona-Motolinia N.D., Sánchez-Lara E., Noriega L., Sánchez-Gaytán B.L., Castro M.E., Meléndez-Bustamante F., González-Vergara E. Cyclo-tetravanadate bridged copper complexes as potential double bullet pro-metallodrugs for cancer treatment. J. Inorg. Biochem. 2020;208 doi: 10.1016/j.jinorgbio.2020.111081. [DOI] [PubMed] [Google Scholar]
- 35.Treviño S., Díaz A., Sánchez-Lara E., Sanchez-Gaytan B.L., Perez-Aguilar J.M., González-Vergara E. Vanadium in biological action: Chemical, pharmacological aspects, and metabolic implications in diabetes mellitus. Biol. Trace Elem. Res. 2019;188:68–98. doi: 10.1007/s12011-018-1540-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costa Pessoa J., Garribba E., Santos M.F.A., Santos-Silva T. Vanadium and proteins: Uptake, transport, structure, activity and function. Coord. Chem. Rev. 2015;301–302:49–86. doi: 10.1016/j.ccr.2015.03.016. [DOI] [Google Scholar]
- 37.Treviño S., Diaz A. Vanadium and insulin: partners in metabolic regulation. J. Inorg. Biochem. 2020;208 doi: 10.1016/j.jinorgbio.2020.111094. [DOI] [PubMed] [Google Scholar]
- 38.Samart N., Althumairy D., Zhang D., Roess D.A., Crans D.C. Initiation of a novel mode of membrane signaling: vanadium facilitated signal transduction. Coord. Chem. Rev. 2020;416 doi: 10.1016/j.ccr.2020.213286. [DOI] [Google Scholar]
- 39.Selman M., Rousso C., Bergeron A., Son H.H., Krishnan R., El-Sayes N.A., Varette O., Chen A., Le Boeuf F., Tzelepis F., Bell J.C., Crans D.C., Diallo J.-S. Multi-modal potentiation of oncolytic virotherapy by vanadium compounds. Mol. Ther. 2018;26:56–69. doi: 10.1016/j.ymthe.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shigeta S., Mori S., Yamase T., Yamamoto N., Yamamoto N. Anti-RNA virus activity of polyoxometalates. Biomed. Pharmacother. 2006;60:211–219. doi: 10.1016/j.biopha.2006.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shigeta S., Mori S., Kodama E., Kodama J., Takahashi K., Yamase T. Broad spectrum anti-RNA virus activities of titanium and vanadium substituted polyoxotungstates. Antiviral Res. 2003;58:265–271. doi: 10.1016/S0166-3542(03)00009-3. [DOI] [PubMed] [Google Scholar]
- 42.Mustafi D., Ward J., Dougherty U., Bissonnette M., Hart J., Vogt S., Karczmar G.S. X-ray fluorescence microscopy demonstrates preferential accumulation of a vanadium-based magnetic resonance imaging contrast agent in murine colonic tumors. Mol. Imaging. 2015;14 doi: 10.2310/7290.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choroba K., Raposo L.R., Palion-Gazda J., Malicka E., Erfurt K., Machura B., Fernandes A.R. In vitro antiproliferative effect of vanadium complexes bearing 8-hydroxyquinoline-based ligands – the substituent effect. Dalt. Trans. 2020;49:6596–6606. doi: 10.1039/D0DT01017K. [DOI] [PubMed] [Google Scholar]
- 44.Bueloni B., Sanna D., Garribba E., Castro G.R., León I.E., Islan G.A. Design of nalidixic acid-vanadium complex loaded into chitosan hybrid nanoparticles as smart strategy to inhibit bacterial growth and quorum sensing. Int. J. Biol. Macromol. 2020;161:1568–1580. doi: 10.1016/j.ijbiomac.2020.07.304. [DOI] [PubMed] [Google Scholar]
- 45.Gaussian 16, Revision C.01, M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, G.A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A.V. Marenich, J. Bloino, B.G. Janesko, R. Gomperts, B. Mennucci, H.P. Hratchian, J.V. Ortiz, A.F. Izmaylov, J.L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V.G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J.A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M.J. Bearpark, J.J. Heyd, E.N. Brothers, K.N. Kudin, V.N. Staroverov, T.A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A.P. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, J.M. Millam, M. Klene, C. Adamo, R. Cammi, J.W. Ochterski, R.L. Martin, K. Morokuma, O. Farkas, J.B. Foresman, and D.J. Fox, Wallingford CT, Gaussian, Inc., (2016).
- 46.Dassault Systèmes BIOVIA . San Diego; Dassault Systèmes: 2018. Discovery Studio, v19. [Google Scholar]
- 47.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E., ChimeraX U.C.S.F. Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-Pdb viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 50.Pedretti A., Mazzolari A., Gervasoni S., Fumagalli L., Vistoli G. The VEGA suite of programs: an versatile platform for cheminformatics and drug design projects. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa774. [DOI] [PubMed] [Google Scholar]
- 51.Brandão T.A.S., Johnson S.J., Hengge A.C. The molecular details of WPD-loop movement differ in the protein-tyrosine phosphatases YopH and PTP1B. Arch. Biochem. Biophys. 2012;525:53–59. doi: 10.1016/j.abb.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathpal S., Joshi T., Sharma P., Joshi T., Pundir H., Pande V., Chandra S. A dynamic simulation study of FDA drug from zinc database against COVID-19 main protease receptor. J. Biomol. Struct. Dyn. 2020:1–17. doi: 10.1080/07391102.2020.1821785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tiago T., Martel P., Gutiérrez-Merino C., Aureliano M. Binding modes of decavanadate to myosin and inhibition of the actomyosin ATPase activity. Biochim. Biophys. Acta - Proteins Proteomics. 2007;1774:474–480. doi: 10.1016/j.bbapap.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 54.AutoDock, http://autodock.scripps.edu/ (last visit 21 October 2020).
- 55.Quiroga R., Villarreal M.A. Vinardo: A scoring function based on Autodock Vina improves scoring, docking, and virtual Screening. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.You Z.-L., Cui Y.-M., Ma Y.-P., Wang C., Zhou X.-S., Li K. Synthesis, characterization and urease inhibitory activity of oxovanadium(V) complexes with similar Schiff bases. Inorg. Chem. Commun. 2011;14:636–640. doi: 10.1016/j.inoche.2011.01.038. [DOI] [Google Scholar]
- 57.L. Zhou, J. Wang, G. Liu, Q. Lu, R. Dong, G. Tian, J. Yang, L. Peng, Probing antiviral drugs against SARS-CoV-2 through virus-drug association prediction based on the KATZ method, Genomics. 112 (2020) 4427–4434. 10.1016/j.ygeno.2020.07.044.J. Gasteiger, M. Marsili, Prediction of proton magnetic resonance shifts: The dependence on hydrogen charges obtained by iterative partial equalization of orbital electronegativity, Org. Magn. Reson. 15 (1981) 353–360. 10.1002/mrc.1270150408. [DOI] [PMC free article] [PubMed]
- 58.Gurung A.B., Ali M.A., Lee J., Farah M.A., Al-Anazi K.M. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci. 2020;255 doi: 10.1016/j.lfs.2020.117831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.L. Márquez-Domínguez, J. Reyes-Leyva, I. Herrera-Camacho, G. Santos-López, T. Scior. Five Novel Non-Sialic Acid-Like Scaffolds Inhibit In Vitro H1N1 and H5N2 Neuraminidase Activity of Influenza a Virus. Molecules. 2020, 16, 25(18), 4248. doi: 10.3390/molecules25184248. PMID: 32947893; PMCID: PMC7571124. [DOI] [PMC free article] [PubMed]
- 60.Clustal Omega, Multiple Sequence Alignment, https://www.ebi.ac.uk/Tools/msa/clustalo/ (last visit 21 October 2020).
- 61.Wahab H.A., Scior T. In: Drug Des. Res. Perspect. Kaplan S.P., editor. Nova Science Publishers; New York: 2007. Structure prediction of proteins with very low homology: a comprehensive introduction and a case study on aminopeptidase; pp. 275–306. [Google Scholar]
- 62.Joosten R.P., te Beek T.A.H., Krieger E., Hekkelman M.L., Hooft R.W.W., Schneider R., Sander C., Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39:D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hatada R., Okuwaki K., Mochizuki Y., Handa Y., Fukuzawa K., Komeiji Y., Okiyama Y., Tanaka S. Fragment molecular orbital based interaction analyses on COVID-19 main protease − inhibitor N3 complex (PDB ID: 6LU7) J. Chem. Inf. Model. 2020;60:3593–3602. doi: 10.1021/acs.jcim.0c00283. [DOI] [PubMed] [Google Scholar]
- 64.Pessoa J.C., Etcheverry S., Gambino D. Vanadium compounds in medicine. Coord. Chem. Rev. 2015;301–302:24–48. doi: 10.1016/j.ccr.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakurai H., Kojima Y., Yoshikawa Y., Kawabe K., Yasui H. Antidiabetic vanadium(IV) and zinc(II) complexes. Coord. Chem. Rev. 2002;226:187–198. doi: 10.1016/S0010-8545(01)00447-7. [DOI] [Google Scholar]
- 66.Trott O., Olson A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009;31 doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O.V Buzko, A.C. Bishop, K.M. Shokat, Modified AutoDock for accurate docking of protein kinase inhibitors, (2002) 113–127. 10.1023/A:1016366013656. [DOI] [PubMed]
- 68.Elsayed S.A., El-Hendawy A.M., Mostafa S.I., Jean-Claude B.J., Todorova M., Butler I.S. Antineoplastic activity of new transition metal complexes of 6-methylpyridine-2-carbaldehyde-N(4)-ethylthiosemicarbazone: X-ray crystal structures of [VO2(mpETSC)] and [Pt(mpETSC)Cl] Bioinorg. Chem. Appl. 2010;2010:1–11. doi: 10.1155/2010/149149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.L. Li, H. Cui, Z. Yang, X. Tao, H. Yang, 4-{2-[4-(Dimethylamino)phenyl]ethenyl}-1-methylpyridinium 3,5-dicarboxybenzenesulfonate methanol monosolvate, Acta Crystallogr. Sect. E Struct. Reports Online. 68 (2012) o281–o281. 10.1107/S1600536811054419. [DOI] [PMC free article] [PubMed]
- 70.Noblía P., Baran E.J., Otero L., Draper P., Cerecetto H., González M., Piro O.E., Castellano E.E., Inohara T., Adachi Y., Sakurai H., Gambino D. New vanadium(V) complexes with salicylaldehyde semicarbazone derivatives: Synthesis, characterization, and in vitro insulin-mimetic activity−crystal structure of [VvO2(salicylaldehyde semicarbazone)] Eur. J. Inorg. Chem. 2004;2004:322–328. doi: 10.1002/ejic.200300421. [DOI] [Google Scholar]
- 71.Mandal T.N., Roy S., Gupta S., Paul B.K., Butcher R.J., Rheingold A.L., Kar S.K. Syntheses, characterization, x-ray crystal structures and emission properties of five oxovanadium(V) complexes with pyridyl/pyrimidyl–pyrazole derived ditopic ligands. Polyhedron. 2011;30:1595–1603. doi: 10.1016/j.poly.2011.03.018. [DOI] [Google Scholar]
- 72.Gazi S., Đokić M., Moeljadi A.M.P., Ganguly R., Hirao H., Sen Soo H. Kinetics and DFT studies of photoredox carbon–carbon bond cleavage reactions by molecular vanadium catalysts under ambient conditions. ACS Catal. 2017;7:4682–4691. doi: 10.1021/acscatal.7b01036. [DOI] [Google Scholar]
- 73.Sah A.K., Rao C.P., Saarenketo P.K., Wegelius E.K., Kolehmainen E., Rissanen K. First crystallographic investigation of complexes of cis-VO2+, cis-MoO22+, and trans-UO22+ species with schiff-base molecules derived from 4,6-O-ethylidene-β-D-glucopyranosylamine. Eur. J. Inorg. Chem. 2001;2001:2773. doi: 10.1002/1099-0682(200111)2001:11<2773::AID-EJIC2773>3.0.CO;2-T. [DOI] [Google Scholar]
- 74.Smith K.I., Borer L.L., Olmstead M.M. Vanadium(IV) and vanadium(V) complexes of salicyladimine ligands. Inorg. Chem. 2003;42:7410–7415. doi: 10.1021/ic034640p. [DOI] [PubMed] [Google Scholar]
- 75.Kolář M.H., Hobza P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016;116:5155–5187. doi: 10.1021/acs.chemrev.5b00560. [DOI] [PubMed] [Google Scholar]
- 76.Costa P.J., Nunes R., Vila-Viçosa D. Halogen bonding in halocarbon-protein complexes and computational tools for rational drug design. Expert Opin. Drug Discov. 2019;14:805–820. doi: 10.1080/17460441.2019.1619692. [DOI] [PubMed] [Google Scholar]
- 77.Tachoua W., Kabrine M., Mushtaq M., Ul-Haq Z. An in-silico evaluation of COVID-19 main protease with clinically approved drugs. J. Mol. Graphics Modell. 2020;101:107758. doi: 10.1016/j.jmgm.2020.107758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sencanski Molecules. 2020;25(17):3830. doi: 10.3390/molecules25173830. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7503980/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar J. Infect. Public Health. 2020 doi: 10.1016/j.jiph.2020.06.016. https://pubmed.ncbi.nlm.nih.gov/32561274/ [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Listing of the online web sources to implement the missing vanadium central atom parameters into AD4, GPF settings to run the grid calculation under AD4, the input data settings for the binding poses of all 17 ligands of round 1 docking assays, results for of the redocking of N3 ligand in 6LU7 with different RB = 2, 4, 6 or 8 and the AD4 setting for 01-VL ligand which is the proposed vanadium-containing derivative of N3.