

Abstract
Infectious diseases caused by viruses such as SARS-CoV-2 and HPV have greatly endangered human health. The nucleic acid detection is essential for the early diagnosis of diseases. Here, we propose a method called PLCR (PfAgo coupled with modified Ligase Chain Reaction for nucleic acid detection) which utilizes PfAgo to only use DNA guides longer than 14-mer to specifically cleave DNA and LCR to precisely distinguish single-base mismatch. PLCR can detect DNA or RNA without PCR at attomolar sensitivities, distinguish single base mutation between the genome of wild type SARS-CoV-2 and its mutant spike D614G, effectively distinguish the novel coronavirus from other coronaviruses and finally achieve multiplexed detection in 70 min. Additionally, LCR products can be directly used as DNA guides without additional input guides to simplify primer design. With desirable sensitivity, specificity and simplicity, the method can be extended for detecting other pathogenic microorganisms.
Keywords: Prokaryotic argonaute, Ligase chain reaction, SARS-CoV-2, HPV, Nucleic acid detection
Graphical abstract
1. Introduction
Many diseases are caused by DNA and RNA viruses. The specific, sensitive, simple, and multiplexed methods to detect the viruses are important. Feng zhang et al. developed a method called SHERLOCK (Specific High-Sensitivity Enzymatic Reporter UnLOCKing) which can detect Zika and Dengue virus down to 2 aM [1]. Subsequently, they improved this method, called it SHERLOCKv2 [2] and combined it with HUDSON (heating unextracted diagnostic samples to obliterate nucleases) for directly detecting samples from bodily fluids [3]. Following, they reported a method called STOPCovid (SHERLOCK Testing in One Pot for detecting SARS CoV-2) which can detect SARS-CoV-2 in 1 h and used lateral flow readout to simplify equipment [4]. Also, Jennifer A. Doudna et al. proposed a method called DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter) which can rapidly and specifically detect HPV [5]. Recently, James P. Broughton et al. just completed the detection of SARS-CoV-2 within 40 min by utilizing CRISPR-Cas12 [6]. Similarly, In 2019, our team proposed a method called PAND (PfAgo-mediated Nucleic acid Detection) which can accomplish attomolar sensitivities and multiplexed detection using PCR coupled with PfAgo (Pyrococcus furiosus Argonaute) [7]. Above all, these methods utilized polymerases to achieve amplification for increasing sensitivity to meet detection requirements. Differently, many researchers chose ligase to achieve amplification, just because ligase chain reaction (LCR) has the best feature of accurately distinguishing single-base mutations in gene sequences, compared with other polymerase-based amplification technologies [8]. Moreover, LCR only needs two temperatures to complete the amplification cycle, which simplifies the equipment requirements. It was first applied to the molecular diagnosis of sickle prize cell anemia by Landegree in 1988 [9]. LCR is used for amplification based on DNA ligase and two pairs of complementary primers. The two pairs of primers in LCR cannot be jointed together for amplification when there is no template or there is a mutation at the junction sites. The ability of DNA ligase to distinguish single-base difference has been used for many genotyping assays [10]. Also, LCR has been applied in many kinds of nucleic acids detection by combining with other technologies. For example, LCR could be chose to detect DNA integrated with polyacrylamide gel electrophoresis [11,12], gold nanoparticles [13,14], single quantum dot analysis [15]. LCR was also united with a printed circuit board (PCB) technology [16] and real-time quantification [17,18] for mRNA detection, electrophoresis and lambda exonuclease-assisted cationic conjugated polymer biosensing for detecting microRNA [[19], [20], [21]], microparticle enzyme immunoassay (MEIA) for detecting HCV RNA [22]. Basically, the sensitivity of detecing RNA was usually relatively lower than that of detecting DNA (Table S1).
PfAgo is a DNA-guided DNA cleavage programmable enzyme that has high levels of activity at temperatures between 80 °C and 100 °C [23,24]. Compared with CRISPR programmable enzymes, Prokaryotic Argonautes (pAgos) has no limit of PAM (protospacer adjacent motif). The DNA guides can be easily designed to cleave complementary ssDNA or dsDNA. Traditionally, PfAgo cannot cleave target DNA using DNA guides shorter than 15-mer [23]. The restriction features of PfAgo and the single-base specificity of LCR are combined to create a novel detection method called PLCR (PfAgo coupled with modified LCR for nucleic acid detection). Compared with PAND or other programmable enzyme-based methods, PLCR can directly use modified LCR products as DNA guides rather than adding additional DNA guides after amplification to target molecular beacon which can simplify experiment design. Especially, PAND which also uses PfAgo for detection requires three additional input guides which is more complicated. Also, qPCR is the gold standard of nucleic acid detection, but it may cause a false positive and false negative [25]. Theoretically, the combination of PfAgo and LCR could be utilized to achieve double assurance of specificity.
Undoubtedly, SARS-CoV-2 is one of the biggest global crises encountered this year. As reported by the World Health Organization, the cumulative number of confirmed cases has reached more than 30 million, and the cumulative death toll has reached more than one million by November 2020. Due to the rapid evolution of the SARS-CoV-2, its various mutants have appeared [26,27]. There is a kind of mutant called spike D614G which reduces S1 shedding and increases infectivity [28,29]. Detection and diagnosis of the SARS-CoV-2 and its mutant are urgent. There is a one-base difference in the genome between wild type SARS-CoV-2 and its mutant spike D614G. The ability of LCR to distinguish single-base difference can be used to discriminate SARS-CoV-2 and its mutant. HPV is an abbreviation of Human Papilloma Virus which can be divided into the low-risk type and high-risk type [30]. In 2018, approximately 311 000 women died from cervical cancer; more than 85% of these deaths occurring in low- and middle-income countries. Cervical cancer could be cured if diagnosed at an early stage [31]. A specific, sensitive, simple, and multiplexed method that can detect both SARA-CoV-2 and HPV is urgently needed. Here, SARS-CoV-2 and HPV will be good samples to confirm the flexibility of PLCR, indicating our method could be applied to detect both DNA and RNA viruses.
2. Material and methods
2.1. Material
The PfAgo gene was synthesized by Gene Create (China, Wuhan) (Table S2). The sequences containing oligonucleotides (Tables S3 and S4), molecular beacons (Table S5), mock templates (Table S6), the sequence about the single-base difference (Table S7), different length of DNA guides (Table S8), and different Tm value of oligonucleotides (Table S9) were synthesized by Sangon (China, Wuhan). The SplintR ligase and Taq DNA ligase were purchased from New England Biolabs (NEB, United States). The Ampligase® Thermostable DNA Ligase was purchased from lucigen (epicentre, United States). The reverse transcriptase was purchased from ABclonal (China, Wuhan). The dNTPs and the Blood Directed Polymerase Chain Reaction (PCR) Kit V2 was purchased from Vazyme (China, Nanjing).
2.2. Expression and purification of PfAgo protein
PfAgo gene was cloned into pET23a vector. The plasmid pET23a-PfAgo was transformed into Escherichia coli BL21 (DE3) pLysS and cultured at 37 °C overnight. Then the transformant was inoculated in LB medium containing 100 μg/ml ampicillin and cultured at 37 °C. The medium was added 1 mM IPTG when the OD600 reached 0.6–0.8. Collected cells were resuspended in Buffer I (20 mM Tris/HCl, pH 8.0, 300 mM NaCl, 2 mM MnCl2) and lysed by ultrasonic sterilizer (Scientz, China, Ningbo). The cell lysate was centrifuged at 15 000×g for 20 min. The supernatant was heated at 80 °C for 30 min and shaken every 5 min. The treated supernatant was purified with Ni-NTA affinity purification. The column was washed three times using wash buffer (20 mM Tris/HCl, pH 8.0, 300 mM NaCl, 2 mM MnCl2, 25 mM imidazole) with 3 times column volume, then target protein was eluted using elution buffer (20 mM Tris/HCl, pH 8.0, 300 mM NaCl, 2 mM MnCl2, 250 mM imidazole) with 3 times column volume (Fig. S1). The sample was concentrated with a Millipore 50 kD membrane at 4 °C and resuspended in storage buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.5 mM MnCl2, 15% [v/v] glycerol). Purified proteins were flash-frozen in liquid nitrogen and stored storage at −80 °C.
2.3. LCR reaction system
For HPV DNA detection, LCR was carried out in a volume of 10 μl, which contained 10 U Ampligase® Thermostable DNA Ligase, 1 × DNA ligase buffer, 1 μM probe A, B, A′, B’, 2 μg salmon sperm DNA and different amount of target HPV DNA. Then 30 thermal cycles (80 °C for 5 s and 40 °C for 1 min) were performed.
For SARS-CoV-2 RNA detection, LCR was carried out in a volume of 10 μl, which contained 10 U Ampligase® Thermostable DNA Ligase, 1 × DNA ligase buffer, 1 μM probe A, B, A′, B’, 20 U reverse transcriptase, 0.5 mM dNTP, 2 μg salmon sperm DNA and different amount of target SARS-CoV-2 RNA. The reaction was performed at 42 °C for 5 min, followed by 30 thermal cycles (80 °C for 5 s and 40 °C for 1 min).
2.4. PfAgo reaction system
Above mentioned LCR reaction product was transferred to PfAgo reaction with 10 μl volume containing 1 × PfAgo reaction buffer (20 mM HEPES pH 7.5, 250 mM NaCl, and 0.5 mM MnCl2), 2 μg PfAgo protein and 1 μM molecular beacon. After reaction at 95 °C for 20 min, the fluorescence intensity was detected.
2.5. Gel electrophoresis and imaging
10 μl of LCR reaction product was mixed with 10 μl 2 × Loading Buffer followed by reaction at 95 °C for 5 min. Then the mixture was resolved by 20% polyacrylamide gel. Electrophoresis was performed at 150 V for 1 h with 0.5 × TBE buffer. Finally, the gel was visualized using a gel image analysis system (BioRad, Gel Doc XR + system, USA).
2.6. Determining the sensitivity and specificity of PLCR
For preparation of mock HPV, we cloned the gene sequence of HPV11E6 to the pLVX-IRSE vector. Then the recombinant plasmid was used to analyze the sensitivity of HPV detection. For mock SARS-CoV-2 RNA, we synthesized a short RNA with the same sequence as part of the N gene. The recombinant plasmid and the short RNA were diluted with non-ribozyme water from 100 pM to 1 aM and non-ribozyme water as the negative control. We used three different fluorophores to distinguish different subtypes of HPV and two different fluorophores to perform multiplexed tests of the N and ORF1ab gene of SARS-CoV-2. The spike D614G mutant was distinguished with wild type novel coronavirus using different fluorophores. The excitation wavelength and emission wavelength of the fluorophores were shown in Table S10.
2.7. Preparation of clinical samples of SARS-CoV-2 and HPV
Clinical oropharyngeal swab samples (Table S11) from patients infected with SARS-CoV-2 were collected and tested by the Centers for Disease Control and Prevention (CDC) Wuhan, China. The viral RNA extraction was performed using a KingFisher Flex nucleic acid extraction system (ThermoFisher, USA). Clinical cervical swab samples (Table S11) from patients infected with different HPV subtypes were collected by the Medical College of Hubei University of Arts and Sciences. The genomic DNA was extracted using Takara MiniBEST Whole Blood Genomic DNA Extraction Kit (Takara Biomedical Technology). The extracted nucleic acids were amplified with the Blood Directed PCR Kit V2 to confirm the subtypes of HPV.
2.8. Multi-channel and clinical samples detection of HPV with PLCR method
PfAgo and molecular beacons labeled with different fluorophores were combined to accomplish multiplexed detection of different HPV subtypes. Three kinds of human papillomavirus subtype HPV11, HPV16, HPV18 were detected in a volume of 15 μl containing 1 μM 11/16/18-A, B, A′, B’, 15 U Ampligase® Thermostable DNA Ligase, 1 × DNA ligase buffer, 2 μg salmon sperm DNA and 1 pM HPV11/HPV16/HPV18 DNA or clinical samples, followed with the above-mentioned LCR reaction. Whereafter PfAgo reaction containing 1 × PfAgo reaction buffer, 2 μg PfAgo protein, and 1 μM HPV11/16/18 molecular beacons was added to the LCR reaction.
2.9. Multi-channel and clinical samples detection of SARS-CoV-2 with PLCR method
To achieve two-channel detection for N/ORF1ab gene or clinical samples of SARS-CoV-2, LCR was carried out in a volume of 15 μl which contained 1 μM N/O-A, B, A′, B’, 15 U Ampligase® Thermostable DNA Ligase, 1 × DNA ligase buffer, 40 U reverse transcriptase and 1 mM dNTPs, 2 μg salmon sperm DNA and 1 pM SARS-CoV-2 N/O RNA or clinical samples, followed with the above-mentioned LCR reaction. Whereafter PfAgo reaction containing 1 × PfAgo reaction buffer, 2 μg PfAgo protein, and 1 μM SARS-CoV-2 N/O molecular beacons was added to the LCR reaction.
2.10. Statistical analysis
Statistical analyses were carried out using GraphPad Prism 7 and Microsoft Excel. The background fluorescence value was measured when the blank control didn't react. The actual measured fluorescence value subtracted from the background fluorescence value was taken as the normalized photoluminescent (PL) Intensity. The two-tailed Student's t-test was used to compare differences between two groups, with a p-value <0.05 taken as the threshold for significance. All data are shown as mean ± standard deviation from three technical replicates.
3. Results and discussion
3.1. PfAgo coupled with modified LCR for nucleic acid detection
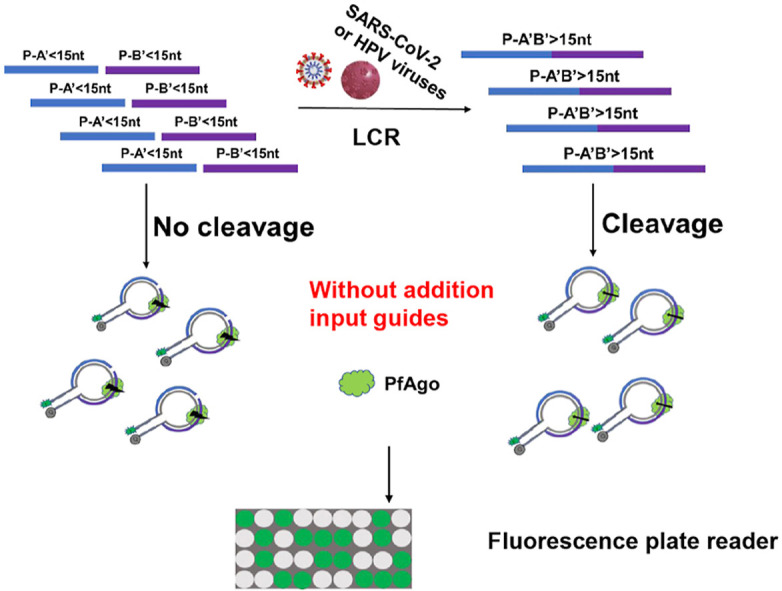
To achieve the early diagnosis of diseases, we proposed a method called PLCR (PfAgo coupled with modified LCR for nucleic acid detection) (Scheme 1 ). Usually, LCR selects two phosphorylated and two non-phosphorylated oligonucleotides longer than 20-mer for amplification which may make separation temperature of double-strand is 95 °C. Owing to that PfAgo can only cleave target DNA guided by phosphorylated DNA guides longer than 14-mer (Fig. 1 A, Table S8) [23]. Once two phosphorylated oligonucleotides shorter than 15-mer are jointed together by LCR, the formed phosphorylated oligonucleotide longer than 14-mer can guide PfAgo to cleave target molecular beacons. We selected one non-phosphorylated oligonucleotide and three phosphorylated oligonucleotides shorter than 15-mer which not only can't guide PfAgo to cleave target DNA unless jointed together by LCR but also can reduce the denaturation temperature to 70–80 °C for saving time of thermal cycling. Firstly, with dsDNA or ssDNA as a template, non-phosphorylated oligonucleotide A and phosphorylated B were linked to produce AB, and phosphorylated A′, B′ were linked to produce phosphorylated A’B’. The Tm value of these probes was close to each other. Then four oligonucleotides were linked in pairs with AB and A’B’ as the template in the next cycle. This process was exponentially amplified. Next, PfAgo protein and molecular beacon were added. PfAgo loading with produced phosphorylated A’B’ can cleave fluorescently labeled molecular beacon. The fluorescence value after the reaction was measured with a fluorescence plate reader. If no template, AB and A’B’ can't be jointed together which would result in PfAgo can't cleave molecular beacon for fluorescence detection. For RNA detection, we added reverse transcriptase and dNTP to the reaction to transform RNA to DNA.
Scheme 1.

Schematic diagram of PLCR of SARS-CoV-2 or HPV. The four oligonucleotides are highlighted in different colors. The oligonucleotide A is non-phosphorylated and oligonucleotide B, A′, B′ are phosphorylated. RT-LCR, reverse transcriptase–LCR.
Fig. 1.

Preliminary verification of PLCR. (A) The cleavage efficiency of PfAgo guided by DNA guides of different lengths. (B) Normalized PLCR photoluminescent (PL) signal from SARS-CoV-2 RNA/HPV11 plasmid positive (1 pM/sample) and negative control (ddH2O) samples. (C) SARS-CoV-2 genome map showing PLCR target sequences. (D) Normalized two-channel PLCR photoluminescent (PL) signal from N and ORF1ab gene of SARS-CoV-2. Data are presented as mean ± SD of three technical replicates.
We firstly selected two ssDNA corresponding with HPV11E6 and SARS-CoV-2 N gene for preliminary experiments (Fig. S2, Table S6). To verify the feasibility of the experiment, we selected HPV11 plasmid and SARS-CoV-2 RNA as the template of PLCR. The results showed that PLCR can detect both DNA and RNA (Fig. 1B). The assay target sites of the Chinese CDC of SARS-CoV-2 are N and ORF1ab gene (Fig. 1C). The two sites were selected for PLCR of SARS-CoV-2 (Table S6). The result demonstrated that PLCR was capable of SARS-CoV-2 detection (Fig. 1D). The PLCR of mock HPV and SARS-CoV-2 samples demonstrated the viability of this platform to detect DNA and RNA viruses.
3.2. The optimization of LCR
The largest disadvantage of LCR is that it can amplify independent of the template [21]. To solve this problem, we optimized it from cycle numbers, the concentration of salmon sperm DNA, Tm value of four oligonucleotides, different ligases, and ligation time of one cycle. For 10 pmol target ssDNA, 2 μg PfAgo can cleave most of the target DNA in 10 min and all target DNA in 20 min (Fig. S3A). The prerequisite was that the amount of DNA guides was 10 nM or more (Fig. S3B). So appropriate cycles were required to get enough DNA guides. We used DNA or RNA as a template to explore the optimal cycle numbers. When the cycle numbers were less than 30, there was almost no template-independent amplification. When the cycle numbers were more than 35, significant template-independent amplification may be observed. Therefore, we selected 30 cycles for the modified LCR. Some research showed that salmon sperm DNA can mitigate template-independent amplification [22]. Thus we explored the influence of concentration of salmon sperm DNA on modified LCR. The specificity was determined to be best at 2 μg salmon sperm DNA (Fig. 2 A and B, Fig. S4A). Next, we analyzed the influence of the average Tm value of four oligonucleotides on modified LCR. This result showed that if the average Tm value of oligonucleotides exceeded 50 °C or was close to 50 °C, the specificity of LCR may be improved (Fig. S4B, Table S9). Following, we explored the influence of ligase including Taq DNA ligase and Ampligase® Thermostable DNA Ligase on modified LCR. The result showed that Ampligase® Thermostable DNA Ligase was better than Taq DNA ligase in terms of template-dependent amplification (Fig. S4C). Finally, we made the denaturation time of each cycle as 5 s, but changed the ligation time in a cycle from 1 min to 5 min. The results showed that when the ligation time was 3 min or more, the fluorescence value had little change. Surprisingly, there was clear distinction between mock positive and negative samples when the ligation time of each cycle was 1 min (Fig. 2C) which would shorten the time required to detection. We tried to use SplintR ligase to detect RNA directly without reverse transcription process. The result showed that the fluorescence value of PLCR for SARS-CoV-2 using SplintR ligase was significantly lower than using reverse transcriptase. Meanwhile, there was little change in the negative control (Fig. 2D). In general, we selected 30 cycles, 2 μg salmon sperm DNA, four oligonucleotides whose Tm value exceeded 50 °C or was close to 50 °C and Ampligase® Thermostable DNA Ligase for modified LCR to avoid amplification independent of the template. The reverse transcriptase rather than SplintR ligase was utilized for modified LCR to enhance detection sensitivity. The 1 min ligation time in each cycle can be selected to detect clinical samples for saving detection time.
Fig. 2.

LCR assay optimization. (A) The influence of salmon sperm DNA concentrations and PfAgo reaction times on PLCR of SARS-CoV-2. (B) The influence of salmon sperm DNA concentrations and PfAgo reactions times on PLCR of HPV11. (C) The normalized photoluminescent (PL) signal after PLCR for different ligation times of one cycle and different reaction times of PfAgo. (D) The normalized photoluminescent (PL) signal after PLCR linking two oligonucleotides with SplintR ligase or performing reverse transcription using RNA as a template. Data are presented as mean ± SD of three technical replicates.
3.3. The sensitivity and specificity of PLCR
For avoiding false-negative results, attomolar sensitivity is required for detecting trace amounts of viruses. To evaluate the sensitivity of PLCR, we diluted HPV11 and SARS-CoV-2 with non-ribozyme water from 100 pM to 1 aM. The results demonstrated that PLCR can detect concentrations as low as 10 aM HPV11 DNA or SARS-CoV-2 RNA in 70 min (Fig. 1, Fig. 3 aM in 100 min (Fig. S5). In 70 min, 10 aM was estimated as the limit of detection (LOD). To shorten detection time, we chose 1 min as the ligation time in each cycle, which may cause fluorescence value increase modestly in different template concentrations, while a significant increase compared to blank control could be observed. As shown in Fig. S4, when the ligation time of each cycle was increased up to 2 min, prominent fluorescence intensity increase can be observed in different template concentrations. The plot of the normalized photoluminescent (PL) intensity versus logarithmical concentration of SARS-CoV-2 N gene (10 aM–10 pM) and HPV11E6 (10 aM–1 pM) showed a strong linear relationship with a correlation coefficient of 0.96 and 0.99 (Fig. 3C and D). There are various types of coronaviruses which are difficult to distinguish. To verify the specificity of PLCR, we selected the homologous sequences on the N gene of SARS-CoV-2, MERS, and SARS by sequence alignment to distinguish (Fig. S6A, Tables S3 and 6). This result proved that PLCR can effectively differentiate SARS, MERS, and SARS-CoV-2 (Fig. 4 A, Figs. S7A and B). As mentioned above, LCR can precisely distinguish single-base difference at the junction sites. We firstly used two single-stranded DNA as a template for preliminary verification of the ability of modified LCR to distinguish single-base difference (Fig. S7C, Table S7). Recent studies have found that the novel coronavirus has rapid evolution and genetic diversity [32]. The spike D614G which is more infectious has a single base difference from the wild-type novel coronavirus in genome sequence [28]. To explore the potential use of PLCR to distinguish single-base mutation, we designed four oligonucleotides and different fluorophore for S gene of SARS-CoV-2 and its mutant separately (Tables S3,5,6). Experimental results proved that PLCR can distinguish mock wild type novel coronavirus and its spike D614G mutant samples (Fig. 4B). These results declared the ability of PLCR of high sensitivity and high specificity.
Fig. 3.

Analysis of the sensitivity of PLCR. (A) The normalized photoluminescent (PL) signal after PLCR for different concentrations of SARS-CoV-2. The LCR was carried out at 80 °C for 5 s, 40 °C for 1 min for 30 thermal cycles, followed with PfAgo digestion for 20 min. (B) The normalized photoluminescent (PL) signal after PLCR for different concentrations of HPV11. The LCR was carried out at 80 °C for 5 s, 40 °C for 1 min for 30 thermal cycles, followed with PfAgo digestion for 20 min. (C) Calibration plot of the normalized photoluminescent (PL) intensity versus logarithmical concentration of SARS-CoV-2 N gene (from 10 aM to 10 pM). (D) Calibration plot of the normalized photoluminescent (PL) intensity versus logarithmical concentration of HPV11E6. (from 10aM to 1pM). Data are presented as mean ± SD of three technical replicates. (P < 0.05; **, P < 0.01; ***, P < 0.001; ****).
Fig. 4.

Analysis of the specificity of PLCR. (A) The ability of PLCR to distinguish SARS-CoV-2 and other coronaviruses. The nucleic acid concentrations of SARS-CoV-2, MERS, and SARS used in the experiment were 1 pM. The LCR was carried out at 80 °C 5 s, 40 °C 1 min for 30 thermal cycles, followed with PfAgo digestion for 20 min. (B) The ability of PLCR to distinguish wild type SARS-CoV-2 and its spike D614G mutant. The nucleic acid concentrations of SARS-CoV-2 and its mutant used in the experiment were 1 pM. The LCR was carried out at 80 °C 5 s, 40 °C 1 min for 30 thermal cycles, followed with PfAgo digestion for 40 min. (C) The results of PLCR with three-channel detection about HPV different subtypes. The nucleic acid concentrations of HPV11, HPV16 and HPV18 used in the experiment were 1 pM. The LCR was carried out at 80 °C 5 s, 40 °C 1 min for 30 thermal cycles, followed with PfAgo digestion for 40 min. Data are presented as mean ± SD of three technical replicates.
3.4. Multiplexed nucleic acid detection using PLCR
With the guarantee of specificity and sensitivity, we sought to determine if PLCR could detect multiplexed genes in a reaction using different fluorophores. To ensure the authenticity of the results, two sites are usually selected for the detection of SARS-CoV-2. We designed four probes and different fluorophore for N and ORF1ab gene of SARS-CoV-2 respectively (Tables S3,5,6). The result showed that PLCR can achieve two-channel detection of N and ORF1ab gene of SARS-CoV-2 in a reaction (Fig. 1D). Infection of HPV of different genotypes can cause different clinical lesions. Through the HPV genotyping, the risk of cervical cancer can be estimated. The HPV11, HPV16, and HPV18 subtypes were chosen to attempt three-channel PLCR detection. Four primers were designed for HPV11E6, HPV16L1 and HPV18L1 separately (Fig. S6B, Tables S4 and 6). Since different linked products after LCR can correspond to different fluorescently labeled molecular beacons (MB), we used FAM, ROX and 6-HEX as fluorophores of HPV11, HPV16, HPV18 MB respectively (Table S5). The results demonstrated that PLCR can distinguish mock HPV subtypes samples (Fig. 4C, Figs. S7B and D).
3.5. Testing of clinical samples
Finally, we evaluated whether PLCR was effective in clinical sample testing. Firstly, we extracted HPV genomic DNA from the cervical swabs and SARS-CoV-2 RNA from the throat swabs (Table S11). The N gene and ORF1ab gene of 16 clinical samples for SARS-CoV-2 were tested with two-channel PLCR in a reaction. The PLCR results obtained were consistent with the RT-qPCR results (Fig. 5 A). The 9 clinical samples for HPV were detected with three-channel PLCR the results of which were consistent with PCR (Fig. 5B). The results highlighted the power of PLCR for the detection of actual samples and multi-channel detection.
Fig. 5.

The clinical sample assay of SARS-CoV-2 or HPV. (A) SARS-CoV-2 clinical samples detection with PLCR and RT-qPCR. (B) HPV clinical samples detection with PLCR and PCR.
4. Conclusion
Here, we propose a method called PLCR which can detect DNA and RNA viruses. This method takes advantage of the characteristic that PfAgo can only cleave target DNA guided by phosphorylated DNA guides longer than 14-mer to design a series of experiments. The method can achieve high sensitivity, high specificity, and multiplexed detection. Compared with CRISPR-based detection, PLCR don't need to use RNA as guides which are unstable and need higher cost. Compared with PAND and other programmable enzyme-based detection methods, PLCR can directly use LCR products as DNA guides without additional input guides to target MB which simplifies the experiment. The method can detect HPV or SARS-CoV-2 as low as 10 aM in 70 min which requires less time than qPCR and has comparable accuracy to qPCR. The N gene and ORF1ab gene of the novel coronavirus can be detected with two-channel PLCR detection in one reaction by designing four oligonucleotides for each gene. The ability to detect single-base differences opens the opportunity for using PLCR to detect the novel coronavirus and its mutant spike D614G which is more infectious [28]. This method can also achieve three-channel detection of different HPV subtypes. The experiments have not been carried out for the detection of more channels, but it is theoretically possible. Of course, this method also has some drawbacks. It cannot accomplish one-tube reaction so that absolute quantification cannot be performed. We will continue to improve it in the future. We have shown that PLCR is a versatile method that can detect DNA and RNA viruses, achieve high sensitivity, high specificity, multi-channel detection, and discrimination of single base mutant. This method also reminds us that more pAgos properties should be explored to expand its application.
Author contribution
Longyu Wang: Designing scheme, Experiment, Data processing, Writing; Ruyi He: Designing scheme, Writing; Bin Lv: Experiment, Writing; Xiao Yu: Experiment, Data processing; Yang Liu: Writing; Jun Yang: Experiment; Wenqiang Li: Writing; Yuan Wang: Experiment; Hang Zhang: Experiment; Guangbo Yan: Experiment; Wuxiang Mao: Review; Linlin Liu: Project administration; Fei Wang: Project administration, Review; Lixin Ma: Designing scheme, Project administration, Review.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was supported by a major Technological Innovation project in Hubei Province (2017ACA174) and the National Natural Science Foundation of China (21708006). Hubei University has patents pending for detection of SARS-CoV-2 and HPV with PLCR on which the authors are inventors.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.talanta.2021.122154.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Gootenberg J.S., et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gootenberg J.S., et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myhrvold C., et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018;360:444–448. doi: 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joung J., et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. medRxiv. 2020 doi: 10.1101/2020.05.04.20091231. [DOI] [Google Scholar]
- 5.Chen J.S., et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:436–439. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broughton J.P., et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020;38:870–874. doi: 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He R., et al. Pyrococcus furiosus Argonaute-mediated nucleic acid detection. Chem. Commun. 2019;55:13219–13222. doi: 10.1039/c9cc07339f. [DOI] [PubMed] [Google Scholar]
- 8.Gibriel A.A., Adel O. Advances in ligase chain reaction and ligation-based amplifications for genotyping assays: detection and applications. Mutat. Res. 2017;773:66–90. doi: 10.1016/j.mrrev.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landegren U., Kaiser R., Sanders J., Hood L. A ligase-mediated gene detection technique. Science. 1988;241:1077–1080. doi: 10.1126/science.3413476. [DOI] [PubMed] [Google Scholar]
- 10.Shin G.W., Chung B., Jung G.Y., Jung G.Y. Multiplexed ligase-based genotyping methods combined with CE. Electrophoresis. 2014;35:1004–1016. doi: 10.1002/elps.201300361. [DOI] [PubMed] [Google Scholar]
- 11.Minamitani S., Nishiguchi S., Kuroki T., Otani S., Monna T. Detection by ligase chain reaction of precore mutant of hepatitis B virus. Hepatology. 1997;25:216–222. doi: 10.1053/jhep.1997.v25.pm0008985293. [DOI] [PubMed] [Google Scholar]
- 12.Barany F. Genetic disease detection and DNA amplification using cloned thermostable ligase. Proc. Natl. Acad. Sci. U. S. A. 1991;88:189–193. doi: 10.1073/pnas.88.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen W., Deng H., Gao Z. Gold nanoparticle-enabled real-time ligation chain reaction for ultrasensitive detection of DNA. J. Am. Chem. Soc. 2012;134:14678–14681. doi: 10.1021/ja306265n. [DOI] [PubMed] [Google Scholar]
- 14.Yin H., et al. Ligation Chain Reaction based gold nanoparticle assembly for ultrasensitive DNA detection. Biosens. Bioelectron. 2014;52:8–12. doi: 10.1016/j.bios.2013.07.064. [DOI] [PubMed] [Google Scholar]
- 15.Song Y., Zhang Y., Wang T.H. Single quantum dot analysis enables multiplexed point mutation detection by gap ligase chain reaction. Small. 2013;9:1096–1105. doi: 10.1002/smll.201202242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez J.L., et al. Multiplexed PCB-based electrochemical detection of cancer biomarkers using MLPA-barcode approach. Biosens. Bioelectron. 2016;82:224–232. doi: 10.1016/j.bios.2016.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Su F., Ji J., Zhang P., Wang F., Li Z. Real-time quantification of fusion transcripts with ligase chain reaction by direct ligation of adjacent DNA probes at fusion junction. Analyst. 2020;145:3977–3982. doi: 10.1039/d0an00163e. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson M., Antson D.O., Barbany G., Landegren U. RNA-templated DNA ligation for transcript analysis. Nucleic Acids Res. 2001;29:578–581. doi: 10.1093/nar/29.2.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang P., et al. Highly sensitive and specific multiplexed microRNA quantification using size-coded ligation chain reaction. Anal. Chem. 2014;86:1076–1082. doi: 10.1021/ac4026384. [DOI] [PubMed] [Google Scholar]
- 20.Yuan Z., Zhou Y., Gao S., Cheng Y., Li Z. Homogeneous and sensitive detection of microRNA with ligase chain reaction and lambda exonuclease-assisted cationic conjugated polymer biosensing. ACS Appl. Mater. Interfaces. 2014;6:6181–6185. doi: 10.1021/am500883q. [DOI] [PubMed] [Google Scholar]
- 21.Yan J., Li Z., Liu C., Cheng Y. Simple and sensitive detection of microRNAs with ligase chain reaction. Chem. Commun. 2010;46:2432–2434. doi: 10.1039/b923521c. [DOI] [PubMed] [Google Scholar]
- 22.Marshall R.L., et al. Detection of HCV RNA by the asymmetric gap ligase chain reaction. PCR Methods Appl. 1994;4:80–84. doi: 10.1101/gr.4.2.80. [DOI] [PubMed] [Google Scholar]
- 23.Swarts D.C., et al. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015;43:5120–5129. doi: 10.1093/nar/gkv415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enghiad B., Zhao H. Programmable DNA-guided artificial restriction enzymes. ACS Synth. Biol. 2017;6:752–757. doi: 10.1021/acssynbio.6b00324. [DOI] [PubMed] [Google Scholar]
- 25.Tahamtan A., Ardebili A. Real-time RT-PCR in COVID-19 detection: issues affecting the results. Expert Rev. Mol. Diagn. 2020;20:453–454. doi: 10.1080/14737159.2020.1757437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M., et al. International expansion of a novel SARS-CoV-2 mutant. J. Virol. 2020;94 doi: 10.1128/JVI.00567-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pachetti M., et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020;18:179. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korber B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827. doi: 10.1016/j.cell.2020.06.043. e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv. 2020 doi: 10.1101/2020.06.12.148726. [DOI] [Google Scholar]
- 30.Goodman A. HPV testing as a screen for cervical cancer. BMJ. 2015;350:h2372. doi: 10.1136/bmj.h2372. [DOI] [PubMed] [Google Scholar]
- 31.World Health Organization. https://www.who.int/en/newsroom/fact-sheets/detail/human-papillomavirus-(HPV)-and cervical cancer.
- 32.Lu J., et al. On the origin and continuing evolution of SARS-CoV-2. National Science Review. 2020;7:1012–1023. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.