

Abstract
Glaucoma remains only partially understood, particularly at the level of intraocular pressure (IOP) regulation. Trabecular meshwork (TM) and Schlemm’s canal inner wall endothelium (SCE) are key to IOP regulation and their characteristics and behavior are the focus of much investigation, thus, are becoming more apparent with time. We and others have studied the TM and SCE’s extracellular matrix (ECM) extensively and unraveled much about its functions and role in regulating aqueous outflow. Ongoing ECM turnover is required to maintain IOP regulation and several TM ECM manipulations modulate outflow facility.
We have established clearly that the outflow pathway senses sustained pressure deviations and responds by adjusting the outflow resistance correctively to keep IOP within an appropriately narrow range which will not normally damage the optic nerve. The glaucomatous# outflow pathway has in many cases lost this IOP homeostatic response, apparently due, at least in part to loss of TM cells. Depletion of TM cells eliminates the IOP homeostatic response, while restoration of TM cells restores it. Aqueous outflow is not homogeneous, but rather segmental with regions of high, intermediate and low flow. In general, glaucomatous eyes have more low flow regions than normal eyes. There are distinctive molecular differences between high and low flow regions, and during the response to an IOP homeostatic pressure challenge, additional changes in segmental molecular composition occur. In conjunction with these changes, the biomechanical properties of the juxtacanalicular (JCT) segmental regions are different, with low flow regions being stiffer than high flow regions. The JCT ECM of glaucomatous eyes is around 20 times than in normal eyes.
The aqueous humor outflow resistance has been studied extensively, but neither the exact molecular components that comprise the resistance nor their exact location have been established. Our hypothetical model, based on considerable available data, posits that the continuous SCE basal lamina, which lies between 125 and 500 nm beneath the SCE basal surface, is the primary source of normal resistance. On the surface of JCT cells, small and highly controlled focal degradation of its components by Podosome- or invadopodia-like structures, PILS occurs in response to pressure-induced mechanical stretching. Sub-micron sized basement membrane discontinuities develop in the SCE basement membrane and these discontinuities allow passage of aqueous humor to and through SCE giant vacuoles and pores. JCT cells then relocate versican with its highly charged glycosaminoglycan side chains into the discontinuities and by manipulation of their orientation and concentration, the JCT and perhaps the SCE cells regulate the amount of fluid passage. Testing this outflow resistance hypothesis is ongoing in our lab and has the potential to advance our understanding of IOP regulation and of glaucoma.
1. Introduction
1.1. Glaucoma
Glaucoma is a relatively common blinding disease defined by a characteristic pattern of optic nerve damage and associated vision loss (Quigley, 2011; Van Buskirk and Cioffi, 1992; Weinreb et al., 2016). Primary open-angle glaucoma (POAG) is the most common type and is the focus of this manuscript. In the United States, glaucoma affects approximately 3 million people (Friedman et al., 2004) and the financial and personal burden is very large. The estimated direct annual cost in the US is $2.5 billion (Covin et al., 2014). Although there are drugs modulating aqueous humor inflow, uveoscleral outflow and the conventional pathway outflow, none are cures and most have significant side effects resulting in serious compliance issues that are a major concern (Muir and Lee, 2011). These drugs normally require application several times per day and must be administered regularly for the remainder of the patient’s life. Various laser and surgical treatments are also in common usage, but none are optimum, most are invasive, and some require retreatment after time intervals. (Lusthaus and Goldberg, 2019). Consequently, better treatments or cures are needed.
1.2. Outflow and Intraocular Pressure
The major risk factor and only currently treatable parameter for glaucoma is elevated intraocular pressure (IOP) (Stamer and Acott, 2012). Although normal-tension glaucoma is a significant portion of POAG, all of our studies and discussion herein will be focused on the portion of POAG associated with elevated IOP. IOP, which reflects a balance of aqueous humor inflow and outflow rates, is primarily controlled by the resistance to aqueous outflow through the conventional or trabecular meshwork (TM) route. Aqueous humor inflow is relatively pressure insensitive until relatively high pressures are achieved (Brubaker, 1970). Outflow is through both the conventional or trabecular pathway and the alternative or uveoscleral pathway. Since most of the outflow is through the conventional pathway, we will not discuss the uveoscleral pathway herein. In addition, although an ocular pulse of around 3 mm Hg (Kaufmann et al., 2006) and diurnal IOP and episcleral venous pressure patterns are well established, our studies are limited to perfused anterior segment organ culture, hence these parameters are not present. The specific molecular or cellular nature and regulation of this flow resistance has been an active focus of our lab and others’ for many years and is gradually becoming clearer.
1.3. Glaucoma Genetics
Most of glaucoma is genetic and initial locus mapping and gene identification based on large family Mendelian genetics suggested that direct genetic-based therapies would soon be available (Jain et al., 2017; Sarfarazi et al., 1998; Stoilova et al., 1996; Stone et al., 1997; Wirtz et al., 1997b; Wirtz et al., 1999). Since then, primarily using genome wide association studies (GWAS), up to 157 glaucoma associated genes or risk loci have been identified, mostly complex and involving multiple gene-gene interactions (Khawaja et al., 2018; Verma et al., 2016; Youngblood et al., 2019). No gene or gene pair appears to account for over a few percent of total glaucoma and no common convergent pathway(s) have been identified at this point. Hence, gene defect-based treatments would only apply to a small number of individuals (Jain et al., 2017). This dramatically complicates leveraging this vast genetic knowledge to treat or cure the disease. The daunting nature of this task has encouraged more direct and generalized mechanistic approaches, as we present here.
2. Trabecular meshwork (TM) cells – normal and glaucoma
2.1. TM cell origin and development
TM cells are derived primarily from neural crest cells of the periocular mesenchyme with minor contributions from the cranial paraxial mesoderm during eye development (Sowden, 2017). While the iris and cornea are well defined by the 5th month of gestation in humans, the angle undergoes additional maturation to clearly separate the TM from the ciliary body and iris root (Anderson, 1981; Cvekl and Tamm, 2004). At birth, the TM is soft (Raghunathan et al., 2017) and the corneoscleral meshwork beams are defined, but there is little space between the beams and their collagenous core is not apparent. Thus, the TM appears to be a continuous tissue mass (Anderson, 1981). Subsequent post-natal maturation gives rise to the classic fenestrated structure of the TM. Schlemm’s canal inner wall endothelial (SCE) cells, which are an important part of the outflow pathway, have both lymphatic and blood vascular properties and origin (Kizhatil et al., 2014; Truong et al., 2014).
2.2. TM regions and cell types
Different anatomical regions of the TM tissue may display different TM cell phenotypes and possibly different functions, although these functions may not be mutually exclusive (Stamer and Clark, 2017). Cells in the TM insert region, underlying the corneal Schwalbe’s line, are the putative TM stem cells. They divide and migrate to repopulate areas that are damaged by laser trabeculoplasty (LTP)(Acott et al., 1989). Endothelial-like TM cells on corneoscleral beams are flattened and are attached to an underlying continuous basement membrane forming a continuous monolayer (Hogan et al., 1971). The outer uveal and corneoscleral TM beam cells are actively phagocytic and function to scavenge debris present in the aqueous humor so that it does not obstruct the outflow channels deeper in the tissue. While these corneoscleral cells secrete signals and cytokines that can affect outflow resistance, it is unlikely that TM beam cells directly participate in remodeling the outflow resistance in response to sensing sustained elevated pressure. An additional argument against this idea is that juxtacanalicular (JCT), but not TM beam cells express αB-crystallin (Fuchshofer et al., 2006), which is stimulated in TM cells by mechanical stretch/distortion and is not expressed without it (Vittal et al., 2005). This suggests that the TM beam cells do not undergo frequent mechanical stretching, but that JCT cells do.
In contrast to corneoscleral cells, TM cells in the juxtacanalicular region appear stellate and are surrounded by abundant extracellular matrix (ECM) (Acott and Kelley, 2008). TM cells do not form monolayers but have long cellular processes that touch each other as well as extend to Schlemm’s canal inner wall cells (Grierson and Lee, 1974; Grierson et al., 1978; Lai et al., 2017; Lai et al., 2019). Because of their location in the region of the tissue thought to be responsible for regulating outflow resistance (Acott and Kelley, 2008; Grant, 1956, 1958; Johnson, 2006; Maepea and Bill, 1992; Schuman et al., 1999), it is likely that JCT cells, rather than corneoscleral meshwork cells, coordinate the detection of elevated IOP and subsequent remodeling of the outflow resistance. Detection of pressure increases is likely via integrins, which link the ECM to the intracellular cytoskeleton, or other cell-ECM binding proteins.
While these anatomical, phenotypic and functional differences suggest that TM cells are not homogeneous, few studies have investigated differential gene expression between these cells. One study isolated JCT and TM beam cells and compared expression, finding only αB-crystallin expression to be different (Fuchshofer et al., 2006).. Several groups are currently utilizing single cell RNA sequencing to identify cell populations within the region that exhibit different characteristic expression profiles. This is an area of active pursuit but some early data are available. Two recent studies that used single cell RNA sequencing to identify individual populations of outflow pathway cells in adult TM tissue are available in pre-print form: (1) van Zyl et al., https://www.biorxiv.org/content/10.1101/2020.02.04.933911v1; 2) Patel et al., https://www.biorxiv.org/content/10.1101/2020.02.10.942649v1). In these studies, 19 or 12 distinct cell types, respectively, were found by overlapping expression patterns from TM tissue. Specific TM cell types identified were: 1) 2 TM beam cells, 1 JCT cell and 1 SCE cell; or 2) 2 TM cell types and 1 SCE type, both with additional cell types and macrophages. This type of approach should converge eventually, providing evidence and expression patterns for several distinct TM, JCT and SCE cell types with unique expression patterns and perhaps, unique functions.
While more studies are required to confirm these preliminary reports, which have not yet been subjected to peer review, it appears likely that TM beam cells, JCT cells, insert cells and SCE cells represent distinct subpopulations with different primary functions. From our perspective, it will be particularly important to investigate the impact of segmental outflow regions and sustained pressure challenges on these gene expression results.
2.3. TM cell interactions and cross-talk
TM cells in different regions of the tissue are thought to coordinate in order to achieve outflow pathway function. To accomplish this, communication mechanisms must be present and may differ depending on their location. For instance, monolayers of TM beam cells are connected to each other and appear to communicate via gap junctions or possible juxtacrine signaling where cell-surface signals on one cell are bound by receptors on a neighboring cell (Zimmerman et al., 1993). These mechanisms both rely on cells being in close proximity. For signaling between TM cells residing on adjacent TM beams, or in other anatomical regions of the tissue, additional mechanisms may be employed. Classical diffusion-based methods such as autocrine or paracrine signaling are often utilized. Here, soluble signals are secreted from the cell, which then diffuse to find their binding partner. These methods are likely used for growth factor and cytokines signaling as well as exosomes. However, signals are diluted in aqueous humor, which is flowing through the tissue, which may reduce their potential efficacy, and these methods can be somewhat random and lack precision. A direct method of cellular communication was recently described in TM cells called tunneling nanotubes (TNTs) (Keller et al., 2017). These long, cellular processes can transport a diverse array of cargoes over relatively long distances (> 200 μm) (Davis and Sowinski, 2008) Conceptually, this mechanism is an attractive means by which to transport signals from TM beam cells to cells in the JCT, SCE or TM insert region or vice versa.
2.4. Glaucoma and TM cells
While there is some drop out of TM cells observed in glaucoma (section 4.5 later) (Alvarado et al., 1984), those that remain have distinctive differences from normal TM cells. Glaucoma TM (GTM) cells show several differences from normal TM (NTM) cells. For instance, GTM cells are approximately 15% larger in area and have 23% larger volume than NTM cells (Sun et al., 2019b). GTM cells also deposit a stiffer matrix than NTM cells (section 6.3. later) in vitro, suggesting intrinsic differences (Raghunathan et al., 2018). Another well described change is that of the actin cytoskeleton. In cell culture, GTM cells have predominant actin stress fibers, which are significantly thicker than those of NTM cells (Sun et al., 2019a). The actin of GTM cells is also rearranged into cross-linked actin networks (CLANs) (Clark et al., 1994) (Bermudez et al., 2017). These geodesic dome like structures have been characterized in TM cell cultures and in TM tissue, and they are more prevalent in glaucomatous cells and tissue (Clark et al., 1995; Hoare et al., 2009). CLANs are also induced by dexamethasone, which causes steroid-induced glaucoma, and TGFβ2, a growth factor whose levels are increased in the aqueous humor of glaucoma patients (Clark et al., 2005; Filla et al., 2011; O’Reilly et al., 2011). Live cell imaging studies have shown the dexamethasone induction of CLANs in pig TM cells and the less dynamic actin cytoskeleton of GTM cells compared to NTM cells (Fujimoto et al., 2016; Sun et al., 2019a). In addition to these changes in the actin cytoskeleton, GTM cells have reduced phagocytic capacity relative to NTM cells (Sun et al., 2019a; Zhang et al., 2007). This observation suggests that inefficient scavenging may allow debris to flow into the JCT, clogging outflow channels and contributing to elevated IOP in glaucoma patients. A variety of other differences have been observed as well, and this topic and TM cells in general has been reviewed in depth recently (Stamer and Clark, 2017).
3. ECM, ECM turnover and outflow
3.1. Glycosaminoglycans (GAGs) and proteoglycans
For many years, hydrodynamic analyses of transmission electron microscopic (TEM) images have suggested that there is far too much open space throughout the TM and cribriform or juxtacanalicular region to account for the amount of flow resistance that is experimentally observed (Acott and Kelley, 2008; Ethier, 2002; Ethier et al., 1986; Johnson, 2006; Johnson and Erickson, 2000; Johnson et al., 1990). The highly charged glycosaminoglycan (GAG) side chains, which are not generally visible in electron micrographs, are thought to explain this issue (Acott and Kelley, 2008; Francois, 1975). Analysis of TM tissue and cells show that there are numerous GAGs and proteoglycans expressed in this tissue (Acott and Kelley, 2008; Acott et al., 1988; Acott et al., 1985; Wirtz et al., 1997a) Studies specifically degrading outflow pathway GAGs increase outflow facility in animals (Bárány, 1953; Knepper et al., 1984; Van Buskirk and Brett, 1978) but not consistently in primates (Hubbard et al., 1997; Sawaguchi et al., 1992).
3.2. Glycosaminoglycan involvement in outflow resistance - direct GAG manipulations
This inability of GAGases to consistently increase outflow in humans suggests that the GAGs in humans are organized differently and thus less available to enzymes or perhaps due to interactions with binding proteins, are obstructed. An alternative approach would be to inhibit GAG biosynthesis or attachment (Keller et al., 2008). An inhibitor of GAG attachment to proteoglycans, β-xyloside, increased outflow facility, as did a chondroitin sulfate (CS) sulfation inhibitor, sodium chlorate. These two studies indicate clearly that GAGs and proteoglycans are key components of the outflow resistance. To extend this, the involvement of CS GAG chains in outflow facility was assessed. We silenced N-acetylgalactosaminyltransferase-1 (ChGn), the enzyme that catalyzes the first unique step in CS GAG chain biosynthesis (Keller et al., 2011). This increased outflow facility, supporting the notion that TM GAGs, particularly CS GAGs, are important to the outflow resistance (Acott and Kelley, 2008).
3.3. TM GAGs and proteoglycans
GAGs are chains of disaccharide repeats often extending to hundreds of sequential repeats. The subunit saccharides are generally highly space-filling and charged providing very high densities of negative charges, which explains their unique physical properties. With the exception of hyaluronan, GAGs are attached to proteoglycan core proteins with variable numbers of GAG chains of varying length attached to each core protein (Acott and Kelley, 2008). The TM and TM cells express a number of typical GAGs and proteoglycans (Acott and Kelley, 2008; Acott et al., 1988; Acott et al., 1985; Wirtz et al., 1997a). These GAGs and proteoglycans are differentially distributed in various regions of the outflow pathway and are thought to serve a variety of specific functions.
3.4. Versican
Versican is a large proteoglycan with many CS GAG sidechains that is expressed in the TM and many other tissues (Wight, 2002, 2017; Wight et al., 1991; Wight and Mecham, 1987; Wirtz et al., 1997a). A priori, versican is the obvious choice to be the proteoglycan comprising the regulatory role in the outflow resistance (Acott and Kelley, 2008; Gard et al., 1993). It has 5 alternative splice variants (Figure 1) and as many as 23 very long and highly sulfated CS GAG side chains or as few as 0. The 5th isoforms, V4, with no alpha GAG and only a portion of the beta GAG exon has recently been identified (Kischel et al., 2010). Versican has many binding site for a variety of proteins and for the long hyaluronan GAG chain as depicted (Figure 1; taken from (Acott and Kelley, 2008), modified from (Kischel et al., 2010) and based on a figure from Wight (Wight, 2002)). Versican distributes segmentally relative to outflow (Keller et al., 2011), but the significance of this is not yet completely clear. When versican was silenced in porcine TM tissue, outflow facility increased but in humans this caused a decrease in facility (Keller et al., 2011), suggesting complexity in its involvement in the outflow resistance (discussed in section 3.5.2, 5.4.1, 6.2.1 and 6.4 later). Versican isoform expression by TM tissue appears to be related to outflow, since TNFα, IL-1α, TGFβ and mechanical stretch, all of which impact facility, change total amounts and isoform distributions (Acott and Kelley, 2008; Keller et al., 2007; Vittal et al., 2005; Zhao and Russell, 2005). Additional versican data are presented in section 3.4., 3.5.2, 5.4.1, 6.2.1 and 6.4 later.
Figure 1.
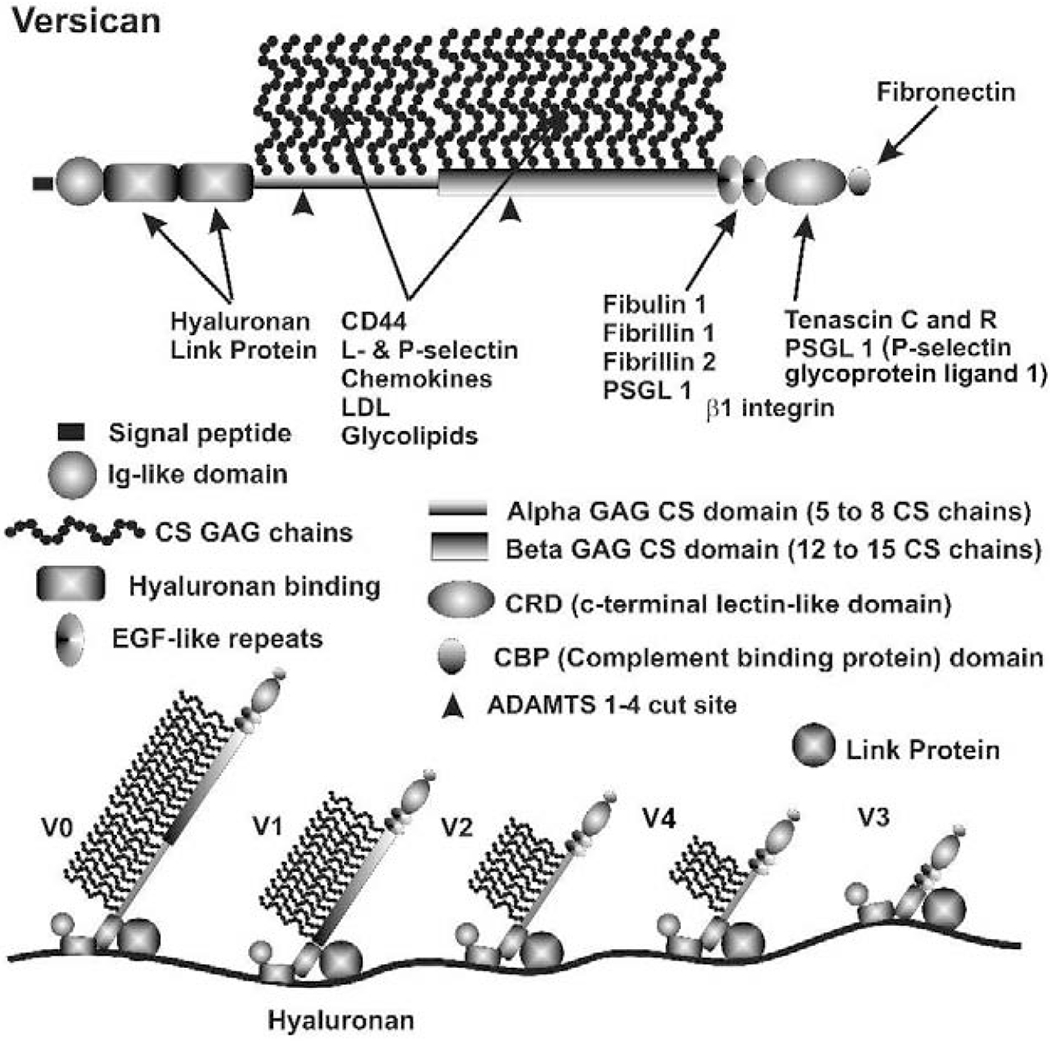
Versican properties, structure, binding sites and isoforms (Acott and Kelley, 2008) and based on a figure from (Wight, 2002). V4 isoform was also added.
3.5. MMPs, ECM turnover, PILS and outflow
3.5.1. MMPs and ongoing ECM remodeling
The drainage of AH from the TM tissue and the rest of the anterior chamber is controlled at least in part by regulation of ECM remodeling. This is accomplished by matrix metalloproteinases (MMPs). In many tissues of the body, MMPs are generally quiescent until activated by signaling, biomechanically or by another homeostatic process. Strikingly, in the TM, some MMPs have high endogenous expression and activity (Keller et al., 2009a; Keller and Acott, 2013). Since the outflow channels in the JCT region are surrounded by and composed of abundant ECM, debris from aqueous humor is easily trapped on the negatively charged GAG chains. Thus, constant remodeling of the ECM in the outflow channels by MMPs would cleave ECM fragments and their associated debris, allowing them to be cleared. Noteworthy here, is our observation that ongoing ECM turnover is required to maintain the IOP (Bradley et al., 1998). Adding or inducing MMPs increased outflow facility and specifically inhibiting endogenous MMPs decreased outflow facility (Bradley et al., 1998). Of course, ECM turnover is complex and after the process is initiated by MMP cleavage, the degraded components are internalized and further degraded. Replacement components are then synthesized, secreted, and incorporated into the ECM. When the outflow resistance is to be adjusted, either there is a difference in the composition or the organization of the replacement ECM (Acott and Kelley, 2008).
3.5.2. MMPs and pressure
In addition to this maintenance remodeling, MMPs are also activated in response to pressure (section 4.2 later). Studies by our laboratory showed that MMP2 and MMP14 activities were up-regulated in response to a pressure challenge in perfusion culture (Bradley et al., 2001). Endogenous levels of the tissue inhibitor of metalloproteinase 2 (TIMP2) were also increased. This is important, as TIMP2 not only participates in the activation of the pro-MMP2 by MMP14, but also, paradoxically, inhibits the active both of these MMPs (Kinoshita et al., 1998). TIMP2 promotes activation of proMMP2 by MMP14 immobilized on agarose beads (Kinoshita et al., 1998). Mechanistically, TIMP2 binds to an MMP14 dimer and to the tail of proMMP2, presenting the propeptide of MMP2 to the active site of one of the MMP14s in the dimer. This allows the MMP14 to cleave the MMP2 propeptide. We have shown the signal transduction pathways involved in the pressure/stretch activation as discussed in section 4.3 and 4.4 later.
3.5.3. MMPs and laser trabeculoplasty
Subsequent studies by us and other groups found that laser trabeculoplasty triggered increases in MMP3 and others in this family mediated by TNFα and IL-1β (Bradley et al., 2000). This involved signal transduction requiring, protein kinase Cμ (Alexander and Acott, 2001), Erk 1/2 map kinases (Alexander and Acott, 2003), JNK map kinase (Hosseini et al., 2006), and p38 map kinase (Kelley et al., 2007a). In addition, IL-1 and TNF work synergistically in this process (Kelley et al., 2007b).
3.5.4. PILS and focal MMP control
While studying MMP distribution within the outflow pathway, we noted a strong colocalization of MMP2 and MMP14 within novel structures in TM cells (Aga et al., 2008). They were also particularly noticeable on TM cells within the JCT region. We further characterized these novel regions, which we named podosome- or invadopodia-like structures (PILS). These unique outflow pathway structures are the site of focal ECM degradation, uptake and secretion. They contain many typical podosome proteins, such as cortactin, and share characteristics of typical podosomes or invadopodia in other tissues (Linder and Kopp, 2005). These structures serve as focal and highly controlled sites of ECM turnover and cell attachment. Rather than just releasing MMPs into the extracellular space without control, these sites provide very discrete and controllable focal ECM turnover. They appear to function to keep ECM degradation regulated and to avoid uncontrolled disruption of the outflow resistance (Aga et al., 2008). This is critical, as uncontrolled ECM degradation in the active outflow route would soon result in the loss of outflow facility control and disruption of the structural integrity of the outflow resistance.
3.5.5. MMPs and glaucoma
Some studies have suggested that MMPs are dysregulated in glaucoma. For instance, reports suggest that MMP and TIMP levels are increased in the aqueous humor of POAG patients (Maatta et al., 2005; Schlotzer-Schrehardt et al., 2003). Several putative glaucoma-related outflow modulators such as TGFβ, Dex, TGFα, IL-1, etc., are associated with changes in MMP or TIMP activity or levels and presumably this impacts some aspects of their mode of action. Most of these types of studies have not been conducted comparing high flow (HF) or low flow (LF) segmental regions, comparing the many different MMPs and their inhibitors together, or carefully analyzing the temporal patterns of expression and activation, so more than an association has not been established. Additional studies of PILS and of specific MMPs and TIMPS as they are involved in IOP modulation, particularly its loss in glaucoma, is of high importance. MMP1 or MMP3 overexpression have been tested as methods of reducing IOP (Borras, 2003; De Groef et al., 2013; O’Callaghan et al., 2017; Spiga and Borras, 2010).
3.6. ADAMTSs and outflow
There are 26 ADAMTSs, a disintegrin and metalloproteinase with thrombospondin repeats (Mead and Apte, 2018). Several ADAMTS’s appear to have roles in glaucoma and/or outflow regulation. In genetic studies of a population of beagles with glaucoma, a Gly661Arg variant in ADAMST10 was found to be causative (Kuchtey et al., 2013; Kuchtey et al., 2011). ADAMTSL4 (ADAMTS like 4) is increased by chronic dexamethasone treatment, is known to associate with microfibril assembly, and may thus contribute to aberrant ECM organization (Raghunathan et al., 2015). ADAMTS4 is an active component of the TM and ADAMTS4 (but not ADAMTS1 or ADAMTS5) was increased in the JCT region with pressure elevation or in TM cells by mechanical stretch. Adding ADAMTS4 increased outflow facility and it localized with PILS (Keller et al., 2009b). Figure 2 shows ADAMTS4 immunostaining in the TM. Another intriguing component that may relate to this outflow process are caveolins. They colocalize with ADAMTS and MMPs and manipulation of Cav 1 or 2 impacts outflow facility, although somewhat differently in humans than in mice (Aga et al., 2014; Gu et al., 2017)
Figure 2.
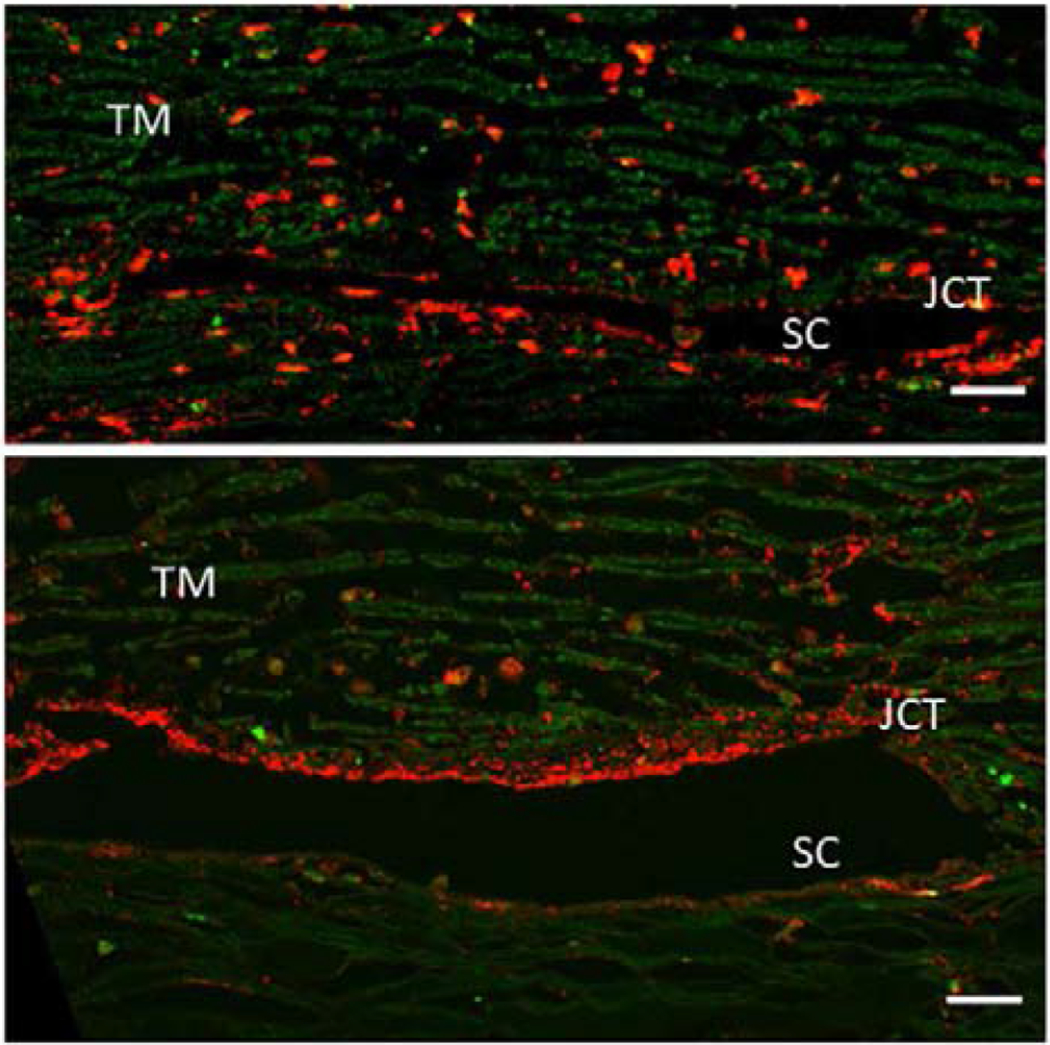
A pair of human eyes was perfused at 1X (top panel) or 2X pressure (bottom panel) for 48 hours. Tissues were fixed, radial paraffin sections were cut and immunostained with an ADAMTS4 polyclonal antibody (red). ADAMTS4 is reduced in the corneoscleral TM but becomes highly upregulated in the juxtacanalicular (JCT) region of the TM tissue in 2X pressure. The immunostaining is also more punctate in 2X tissue compared to 1x tissue. Green = autofluorescence. SC= Schlemm’s canal. Scale bar = 20 μm. (Keller et al., 2009b)
4. IOP homeostasis
4.1. IOP homeostasis in humans
Although glaucoma is a relatively common blinding disease, most people do not ever develop glaucoma (Acott et al., 2014; Acott et al., 2016). One obvious implication of this observation is that there must exist some robust mechanism to maintain IOP within acceptable bounds to avoid triggering optic nerve damage. Of course, normal-tension glaucoma is significant, but evidently a functional IOP homeostatic mechanism is not sufficient to protect these patients. Since we have no data on these individuals, we will not further discuss this population of glaucoma patients. To explore the possibility of an inherent IOP homeostatic mechanism, we experimentally elevated pressure and looked for a corrective response in perfused human anterior segment organ culture, the most accepted system for studying outflow regulation (Bradley et al., 2001). As shown schematically in figure 3, when we doubled the perfusion flow rate from 1x to 2x (2.5 to 5 μl/min), we observed an immediate doubling of the measured pressure, i.e. the outflow facility (~ flow/pressure) did not change. However, although we maintained the perfusion flow rate at 2x over the next few days, the pressure gradually returned to the previous level (Bradley et al., 2001). In response to the elevated pressure, the outflow resistance was adjusted down to restore pressure to normal levels. In the constant flow system, IOP homeostasis is seen as restoration of the original pressure although flow remains at 2x. To our knowledge, this is the first direct experimental evidence for the existence of a robust IOP homeostatic mechanism. Since the word IOP homeostasis is used casually throughout the literature, we felt it important to define the term precisely. We defined IOP homeostasis as a corrective response to sustained pressure deviations in which the outflow resistance is adjusted to restore the IOP to within the normal narrow acceptable range (Acott et al., 2014), rather than including any manipulation that just changes outflow facility.
Figure 3.
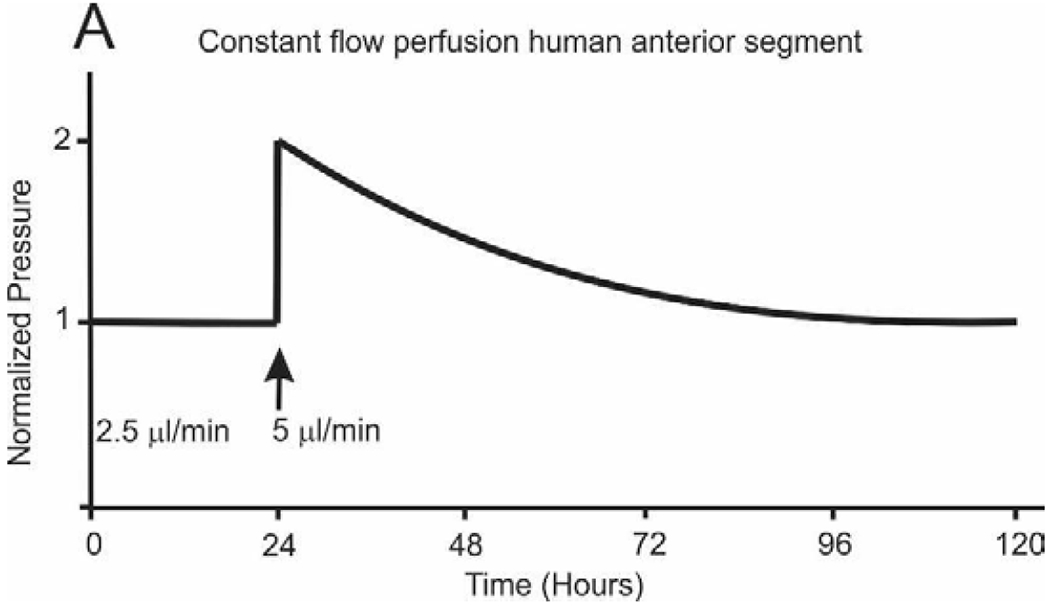
Idealized curve of IOP homeostatic response to 2x pressure challenge initiated by doubling flow rate. Perfusion stabilized at 1x and then flow doubled to 2x which doubles the pressure. Over several days, the outflow resistance is reduced and pressure returns. Modeled after study by Bradley, et. al., (Bradley et al., 2001).
The same response, reduction of the outflow resistance, is observed in constant pressure perfusion. When the pressure is doubled from 1x to 2x and the flow rate doubles immediately and then gradually increases over a few days as the outflow pathway attempts to correct the elevated pressure. Conversely, if the pressure is too low going from 1x (8.8 mm Hg) and drops to 0.5x (4.4 mm Hg) the flow rate immediately drops to 0.5x and then slowly declines further as the outflow resistance is gradually increased. Note that since the pressure is maintained at 4.4 mm Hg, the resistance adjustment does not correct the pressure and thus the outflow pathway continues to increase the resistance attempting unsuccessfully to correct the pressure.
Figure 4 shows actual data where normalized outflow facility is plotted for constant pressure perfusion studies in humans with n = 23 (Acott et al., 2014). These corrective responses to sustained pressure increases or decreases provide a mechanism for maintaining the IOP within a narrow, non-damaging range throughout life. Hence, most people do not have sustained pressure outside of the narrow acceptable range and consequently do not ever develop glaucoma (Acott et al., 2014; Acott et al., 2016). Of course, some that are maintained within this range do develop normal tension glaucoma for other reasons.
Figure 4.
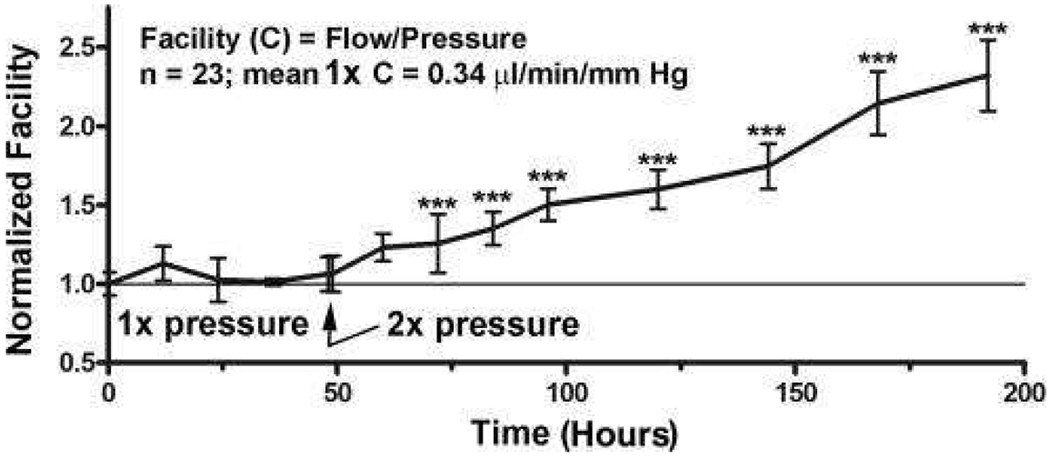
IOP homeostasis with constant pressure perfusion in human anterior segments (Acott et al., 2014; Acott et al., 2016). Perfusion at 1x pressure (8.8mm Hg) gives a normalized facility of 1.0 and doubling the pressure to 2x doubles the outflow immediately but the facility (~flow/pressure) does not change. However, over the next few days maintaining pressure at 2x, the resistance is decreased as the outflow system is attempting to compensate and the flow rate and facility increase. N=23 separate human anterior segments and significance was determined by one-way ANOVA with Dunnett’s Multiple Comparison Test.
4.2. Mechanical stretch/distortion sensing and MMP responses
With our initial testing of the IOP homeostasis hypothesis (Bradley et al., 2001), we observed that associated with the IOP homeostatic process there was an increase in MMP2 activity. Since the ECM is thought to be the source of much of the outflow resistance, increasing the level or activity of ECM degrading enzymes like MMP2 implies that this is an initiating step in the resistance adjustment process. In parallel with this, MMP14 was increased and tissue inhibitor of metalloproteinases, TIMP2, was decreased (Bradley et al., 2001). This suggested that MMPs initiated the resistance modulation that occurs in the IOP homeostatic response.
A likely conclusion is that the sustained pressure differences produce mechanical stretching or distortion of the JCT’s ECM, which could be sensed by JCT TM cells triggering the homeostatic response (Acott and Kelley, 2008). Increased MMP activity or expression would thus be the initiating step producing ECM degradation. This would be accompanied by more complete ECM turnover steps, including ECM fragment uptake, replacement ECM biosynthesis, and secretion to produce a slightly modified ECM with different outflow resistance. Mechanically stretching TM cells produced MMP2 and MMP14 increases with TIMP2 decreases (Bradley et al., 2001).
Since we had previously shown that adding or inducing the production of MMPs increased outflow facility, while inhibiting endogenous MMPs by several means decreased facility (Bradley et al., 1998), these changes are suggestive of important regulatory steps in response to mechanical stretching of TM cells. Mechanical stretching appears to serve as a surrogate or sensor for changes in IOP (Bradley et al., 2001).
4.3. Additional ECM turnover events with mechanical stretch
Of course, just increasing MMPs and initiating ECM degradation will not produce controlled changes in the ECM and the outflow resistance. In addition to the MMP-related ECM degradation effects, mechanical stretching triggers numerous ECM expression changes that will facilitate ECM turnover, producing a modified outflow resistance with different quantities, composition or organization of the outflow resistance ECM. Microarray studies of mechanical stretch responses of TM cells yield numerous changes in ECM component expression and composition (Vittal et al., 2005). There were also a number of alternative mRNA splice variations associated with mechanical stretching including versican, fibronectin, CD44, tenascin C and type XII collagen (Acott and Kelley, 2008; Keller et al., 2007; Vittal et al., 2005).
4.4. Signal transduction
Since mechanical stretch triggers both MMP activation and increases in levels, understanding the signal transduction involved is of importance (Bradley et al., 2003). If TM cells sense sustained pressure differentials as mechanical stretching or distortion, probably via integrins, the signal transduction is likely through downstream components. By using signal transduction inhibitors such as Wortmannin and Rapamycin and following phosphorylation of specific components, we unraveled the pathways involved in production of MMP2 and MMP14 (Figure 5; (Bradley et al., 2003)).
Figure 5.
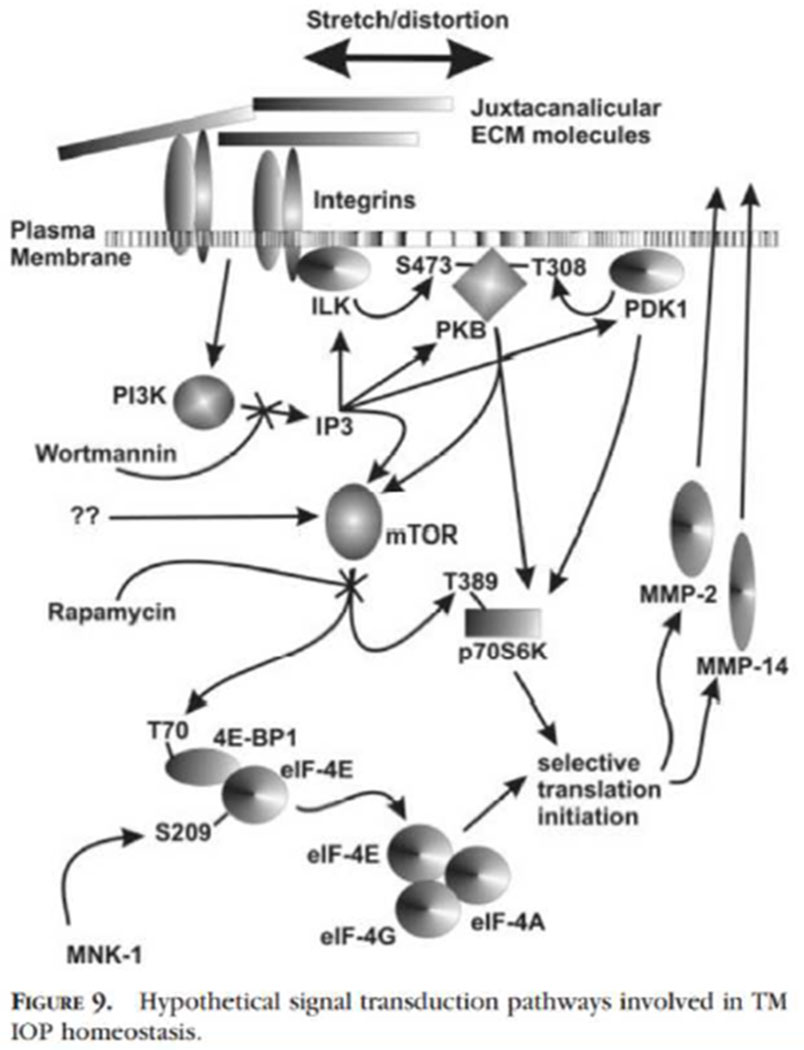
Signal transduction following mechanical stretch of TM cells producing increased translation of MMP2 and MMP14 (Bradley et al., 2003). Mechanical stretching triggers the PKB, PI3K, mTOR and p70S6K pathways and works through several downstream modulators to initiate MMP-2 and MMP-14 translation.
Integrin moves or changes within the plasma membrane triggering PI3K activity which increases IP3 and is blocked by Wortmannin. PI3K or ILK direct activation increases phosphorylations of PKB and PDK1, both of which normally affect phosphorylation of p70S6K. Rapamycin inhibition of mTOR activity on p70S6K or 4E-PP1/eIF-4E blocks specific steps in MMP2 and MMP14 selective protein translation and secretion. Several other complex changes are involved, such as facilitating cap protein binding, unmasking a strongly double-stranded inhibitory region upstream of MMP14 mRNA via an unfoldase, etc. This is presented in more detail in the original manuscript (Bradley et al., 2003).
4.5. Glaucoma, TM cellularity and reduced cellularity glaucoma model
Alvarado counted TM cells in TEM sections comparing normal aging and 36 age-matched glaucomatous eyes (Alvarado et al., 1984; Alvarado et al., 1981). He demonstrated a clear reduction in cellularity in glaucomatous eyes compared to age-matched normal eyes as shown in figure 6. Note that the TM cellularity within the entire meshwork area declines significantly with age, particularly within the first 10-20 years, but then slowly through the remainder of life (solid line and solid dots). Glaucomatous eyes (dashed line and open dots) decline approximately in parallel but more extensively.
Figure 6.
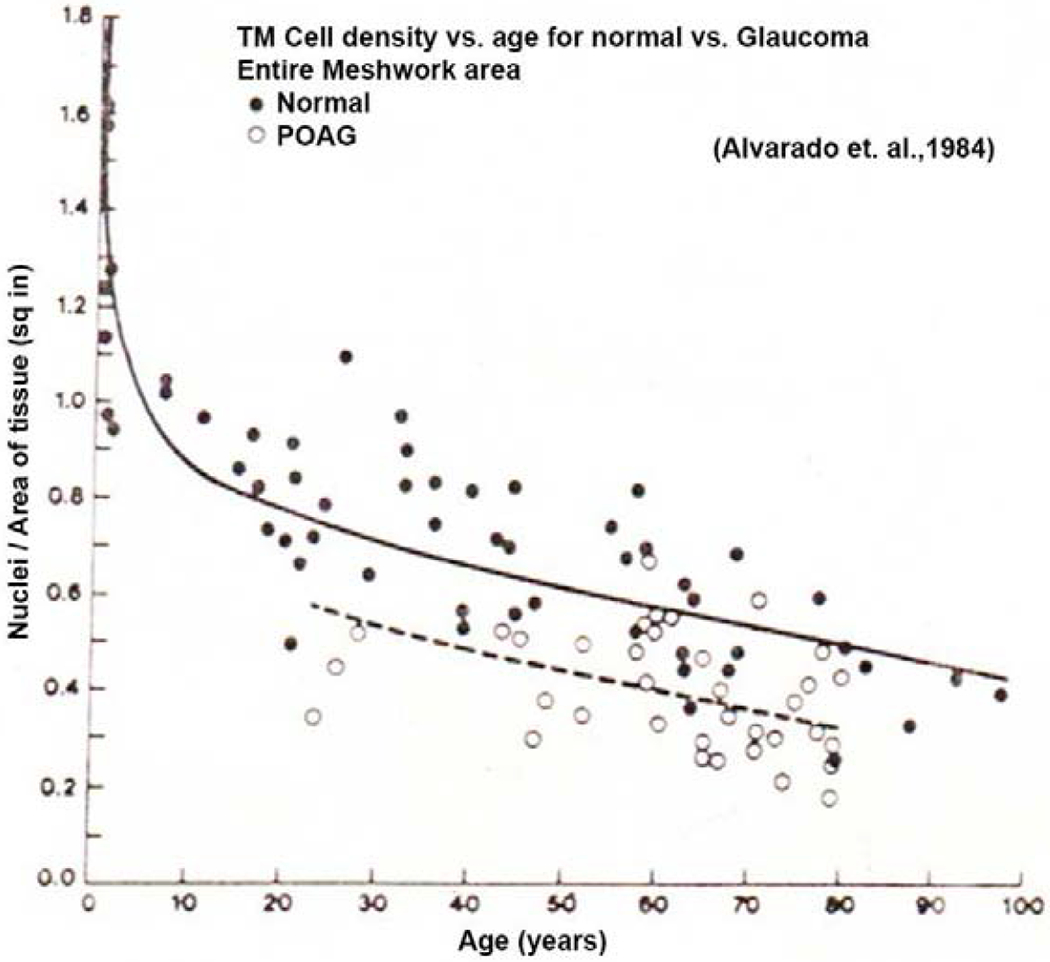
TM cell density as a function of age for normal and glaucomatous eyes (Alvarado et. al., 1984). N = 36 human eyes.
Of the 36 eyes examined, all but a few glaucomatous eyes exhibited comparatively reduced cellularity. Since there are currently well over 100 identified individual glaucoma risk alleles with none accounting for more than a few percent of glaucoma cases (Khawaja et al., 2018; Youngblood et al., 2019), it is safe to assume that all or nearly all of these eyes owe their glaucoma to a different gene variant. This suggests that one common feature of all or many types of glaucoma is this cell loss. At this stage, this is the only common feature or pathway that has been identified in spite of considerable attention being paid to this idea.
To experimentally evaluate the relevance of Alvarado’s observation, we developed a cell-loss glaucoma model using the detergent, saponin, in perfused human anterior segment organ culture (Abu-Hassan et al., 2015). Treatment with saponin produced an approximate 30% reduction in TM cellularity. This reduction did not affect outflow facility at 1x perfusion pressure (8.8mm Hg) and perfusion at 2x pressure produced an immediate doubling of outflow rate (Figure 7) in both treatment and vehicle control. Over the next few days, the vehicle control reduced the outflow resistance attempting to achieve IOP homeostasis, but the cell-depleted saponin treated anterior segments were unable to adjust the outflow resistance and thus the flow remained constant over the next few days. The normal IOP homeostatic response was lost (Abu-Hassan et al., 2015). This strongly suggests that the glaucomatous cell loss may be mechanistically relevant in glaucoma.
Figure 7.
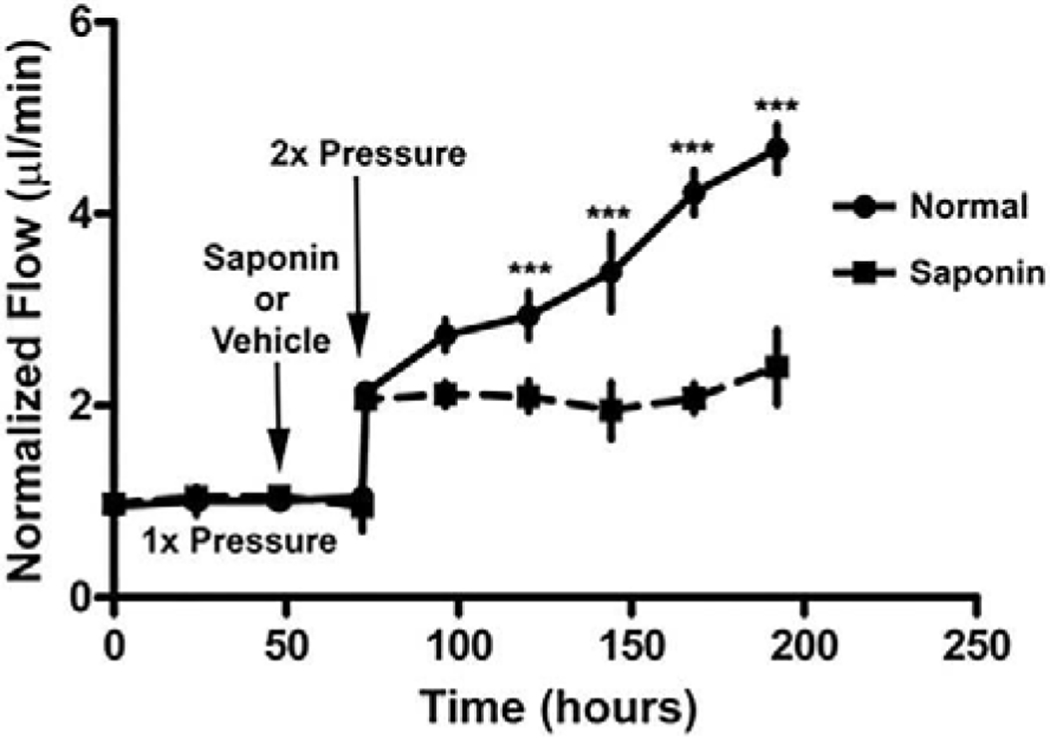
Saponin glaucoma cell loss model and IOP homeostatic effect (Abu-Hassan et al., 2015). Treatment with saponin which kills approximately 30% of TM cells, does not immediately change outflow facility at 1x or at 2x but does block the IOP homeostatic response to a pressure challenge over the next few days. N=8 normal and =17 saponin with one-way ANOVA for *** = P<0.001.
4.6. Cellular replacement and restoration of IOP homeostatic response to pressure challenge
To extend these studies, TM cells were transplanted into saponin-treated anterior segments (Abu-Hassan et al., 2015) as shown in figure 8. First the anterior segments were stabilized at 1x and then treated with saponin. In response to a 2x pressure challenge (the first 2x in the figure) no homeostatic resistance correction was observed. The perfusion pressure was then returned to 1x, HTM cells perfused in, and the flow stopped overnight to allow attachment. Next, a 2x pressure challenge was initiated and now the transplanted cells supported a strong IOP homeostatic resistance adjustment and the outflow facility increased with time. This further supports the mechanistic relevance of Alvarado’s initial observation on TM cellularity in glaucoma, since restoration of TM cellularity was able to restore function.
Figure 8.
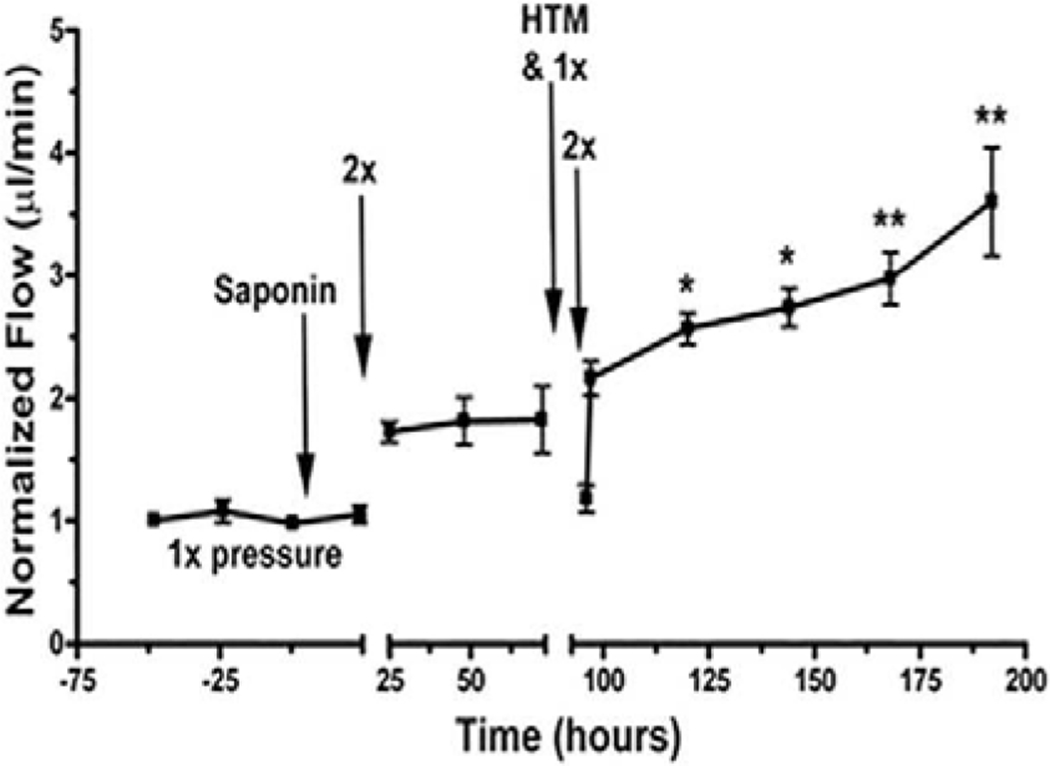
Restoration of IOP homeostatic response after transplantation of 300,000 TM cells to saponin model (Abu-Hassan et al., 2015). N=6 and one-way ANOVA where * = P<0.05 and ** = P<0.001.
With an eye to future therapeutic approaches to glaucoma, in a parallel study we differentiated induced pluripotent stem cells (iPSCs) into TM-like iPSCs and transplanted them into the saponin cell-depletion model (Abu-Hassan et al., 2015) (figure 9). This also restored the IOP homeostatic response to a 2x pressure challenge, suggesting that this would be an effective treatment for glaucoma. Several other cell types, including fibroblasts, undifferentiated iPSCs and HUVECs, were unable to restore this IOP homeostatic response.
Figure 9.
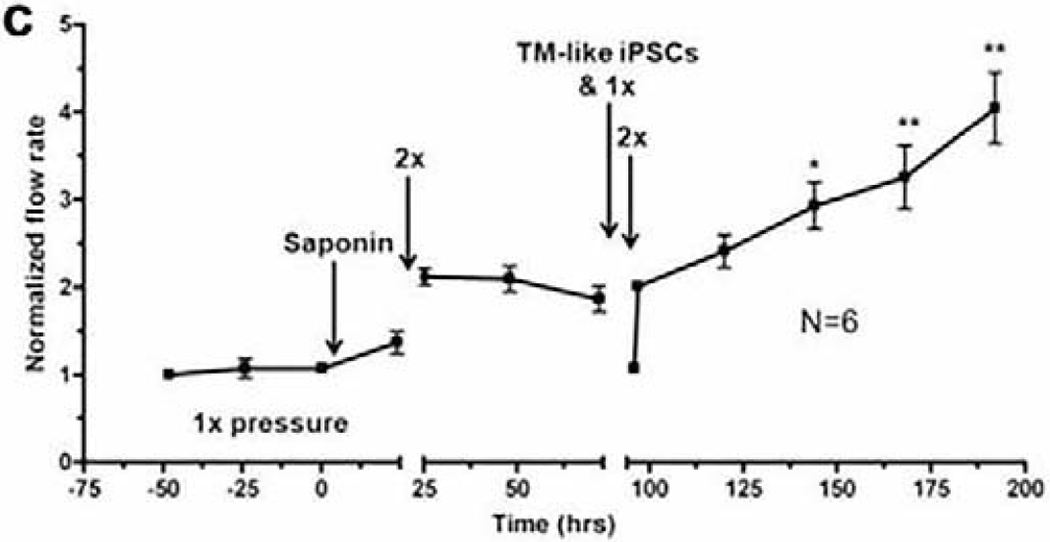
Restoration of IOP homeostatic response after transplantation of TM-like iPSCs (Abu-Hassan et al., 2015). TM-like iPSCs (300,000) restored IOP homeostatic response where N=6 and one-way ANOVA where * = P<0.05 and ** = P<0.001.
4.7. Glaucoma and IOP homeostasis
From the studies detailed above, one might expect that glaucomatous eyes would be unable to mount an IOP homeostatic response to a 2x pressure challenge. We conducted these studies and found that this is indeed the case (Raghunathan et al., 2018). Perfused human anterior segments from glaucomatous donors are unable to adjust the outflow resistance in response to a 2x pressure challenge, while anterior segments from normal eyes could. Instead, as indicated in figure 10, doubling the pressure doubled the flow, but over the next several days the resistance was not adjusted in response to the pressure challenge for the glaucomatous anterior segments. This further establishes the physiologic relevance of the original Alvarado observation of cell loss in glaucomatous eyes (Alvarado et al., 1984) and provides a direct differentiating characteristic for glaucomatous compared to normals.
Figure 10.
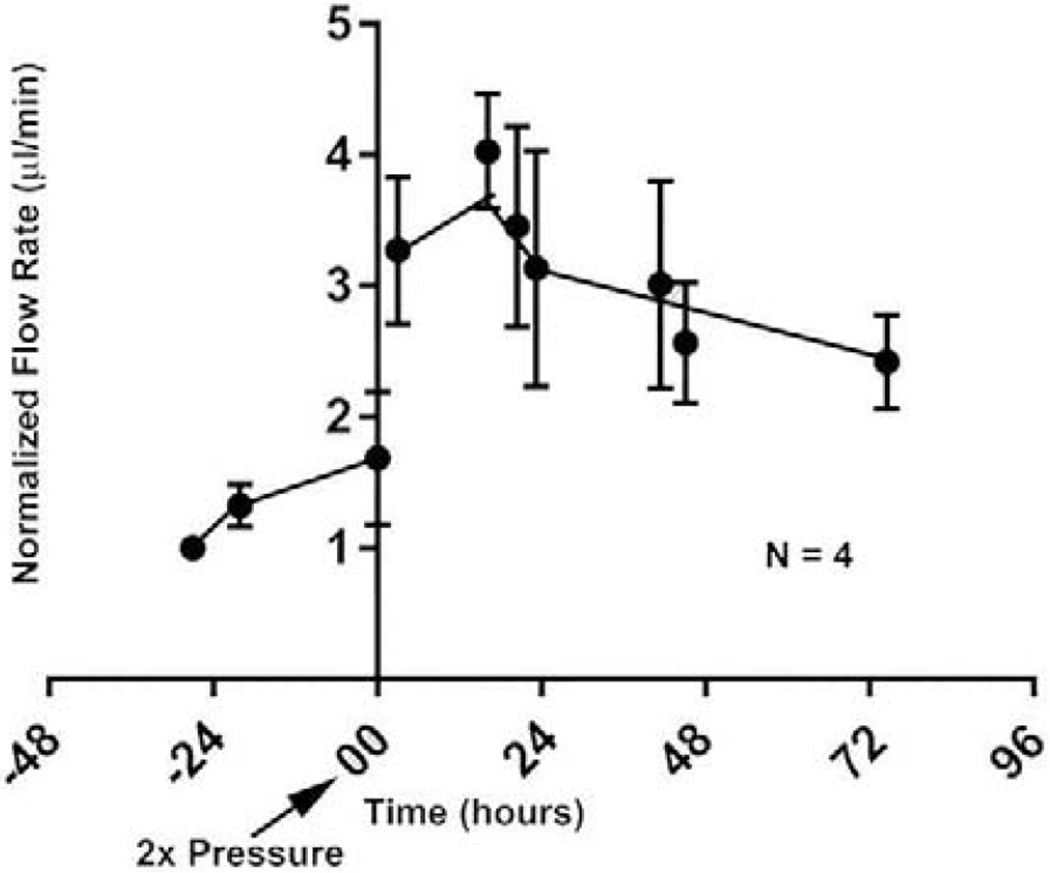
Loss of IOP homeostatic response by glaucomatous anterior segments (Raghunathan et al., 2018). N=4 glaucomatous eyes from 3 individuals and lack of significance was determined by one-way ANOVA with Dunnet’s Multiple Comparison Test.
4.8. ECM crosslinking and IOP homeostasis
Discrete tissues have distinct biomechanical properties, which are subject to change with aging and disease (Frantz et al., 2010). Biomechanical properties of the ECM depend not only on composition, but also on organization and post-translational modifications such as crosslinking (Cox and Erler, 2011). Enzyme-mediated crosslinking is primarily regulated by lysyl oxidase (LOX), lysyl oxidase-like (LOXL) family members, and the transglutaminase enzyme family. LOX family proteins catalyze the crosslinking of collagens and elastin through oxidative deamination of lysine and hydroxylysine residues, which stabilize collagen and elastin fibers in the ECM (Kagan and Li, 2003; Sethi et al., 2012). Human TM cells have been documented to express all members of the LOX family. Transglutaminase enzymes catalyze the posttranslational modification of proteins through the formation of isopeptide bonds resulting in crosslinking of ECM proteins including fibronectin, collagen, laminin and elastin (Beninati and Piacentini, 2004; Greenberg et al., 1991; Tovar-Vidales et al., 2011). Tissue transglutaminase 2 (TGM2) was expressed and active in both normal and glaucomatous TM tissues and cells, and the protein levels and enzyme activities are elevated in glaucomatous cells (Tovar-Vidales et al., 2008). In the TM, both TGM2 and LOX family protein expression were induced by TGFβ2, a cytokine overexpressed in glaucoma (Sethi et al., 2011; Tovar-Vidales et al., 2008; Welge-Lussen et al., 2000; Wordinger and Clark, 2014; Wordinger et al., 2014).
Crosslinking of ECM proteins can obstruct proteolytic breakdown, resulting in decreased ECM turnover, ECM accumulation and increased tissue stiffness (Levental et al., 2009). Previous studies found morphological changes in the ECM of glaucomatous TM tissues and suggested that the tissues are more crosslinked than those from normal age-matched individuals (Lutjen-Drecoll, 2005; Rohen et al., 1993). In addition to the apparent ultrastructural changes in the TM, the elastic modulus of glaucomatous TM tissue was found to be much higher than normal age-matched TM tissues (Last et al., 2011), as discussed below, suggesting that changes in the biomechanical properties of the TM may be directly related to the aqueous humor outflow resistance and IOP. Interestingly LOXL1 was significantly overexpressed in cell derived matrices of glaucomatous donors in comparison with non-glaucomatous (Raghunathan et al., 2018). Active LOX has been shown to stiffen tissues and can compromise their function (Levental et al., 2009), whereas reduction of LOX activity reduces tissue stiffness and prevents fibrosis (Georges et al., 2007; Levental et al., 2009).
To mimic altered stiffness in vitro, two agents, genipin or beta-aminopropionitrile (BAPN), were used to induce or inhibit, respectively, matrix cross-linking. The effects on outflow were then investigated in perfusion culture (Yang et al., 2016). Inhibiting collagen cross-linking increased outflow rates whereas induction of cross-linking produced opposite effects. These effects on outflow were correlated with the effects of the cross-linking agents on MMP levels and activity. By Western immunoblotting, genipin caused high MW complexes consisting of TIMP-2 and MMP-14, with reduced MMP-2 activity. Both agents also caused alterations in expression of ECM components. Thus, altering cross-linking appears to significantly affect remodeling of TM ECM components of the outflow resistance. This is consistent with the paradigm that the stiffer the ECM, the lower the aqueous outflow facility through the TM (Yang et al., 2016).
5. Outflow segmentation
5.1. Segmentation
Aqueous humor outflow through the conventional outflow pathway has long been observed to show a non-uniform pattern of flow around the circumference of the eye. This has been shown in studies of both normal and glaucomatous human eyes using tracers of different composition and size (Buller and Johnson, 1994; Chang et al., 2014; de Kater et al., 1989; Ethier and Chan, 2001; Hann et al., 2005; Hann and Fautsch, 2009; Keller et al., 2011; Vranka et al., 2015). This segmental distribution is demonstrated by the presence of relatively high (HF), moderate or intermediate (MF or IF), and low-to-no flow (LF) regions of the TM, and SC. There are also structures of the distal outflow pathway, i.e. beyond SCE inner wall including the collector channels, that may contribute significantly to the outflow resistance (Cha et al., 2016). In addition to human eyes, non-uniform or segmental patterns of aqueous outflow have been demonstrated in monkey, mouse, porcine and bovine eyes (Battista et al., 2008; Lu et al., 2008; Swaminathan et al., 2014). Pigmentation within the TM was originally suggested to be a marker for preferential aqueous outflow (Gottanka et al., 2001). However, few studies have established a direct correlation. Pigmented regions were not shown to differ in TM cell number or ultrastructural characteristics when compared with non-pigmented regions in the same eye (Johnson et al., 1989). Interestingly, recent studies have identified morphological differences coincident with regions of non-uniform outflow. Collector channels have been associated with areas of higher outflow (Cha et al., 2016; Hann and Fautsch, 2009), although this does not appear to be a strict correlation. In addition, SCE cells along the inner wall have trans-endothelial pores (border or B-pores and intracellular or I-pores) that allow fluid flow through or between inner wall cells (Bill and Svedbergh, 1972; Ethier, 2002; Ethier et al., 1998; Johnson and Erickson, 2000; Johnson et al., 1990; Tamm, 2009). A recent study demonstrated a positive correlation of B-pores with high flow regions of the JCT and the inner wall SCE (Braakman et al., 2015), suggesting that the presence of pores could indicate active flow regions and possibly influence localized outflow.
5.2. Segmentation patterns
In addition to morphological differences observed in segmental regions of the TM and inner wall SCE, we have investigated the molecular differences between high or active-outflow regions (HF) and low-to-no outflow regions (LF). Our previous study characterized segmental flow regions of the TM using a variety of fluorescently-labeled microspheres, of both different sizes and chemical modifications, perfused into anterior segments to label flow regions (Vranka et al., 2015). Carboxy- and amine-modified 200nm fluospheres localized to similar areas along the TM suggesting the existence of diverse binding sites within common fluid flow areas. We also showed that the distribution of the fluorescent tracers varied on both a macro- and micro-scale. The macro-scale regions, which are mm sized, distribute unevenly and roughly to 1/3rd each of HF, intermediate or medium flow, and LF. The micro-scale regions appear to occur with a periodic frequency of approximately 50 – 100 μm (Vranka et al., 2015). This micro-scale variation in human trabecular meshwork has also been shown by others (Chang et al., 2014).
5.3. Cells from HF/LF
To facilitate understanding of the innate molecular differences between TM cells in high or active outflow regions and low or no outflow regions, we have established primary TM cell lines from these respective regions of TM tissues from multiple donor eyes. Our intention was to determine whether these cells show differences in cell growth, differentiation, proliferation, and morphology. Our preliminary data show no differences between HF and LF TM cells in these respects. We are also in the process of measuring ECM gene and protein expression levels between cultured HF and LF TM cells. Our initial studies have identified a small number of ECM genes that appear to be upregulated in LF TM cells relative to HF TM cells from the same donor (Staverosky et al, manuscript in review). Since we see relatively few differences between the HF and LF TM cells, we hypothesize that the substrates upon which the cells are grown will likely exert an influence on the cells. Our previous data in TM tissues suggests that the ECM has a strong influence on the resident TM cells in situ. We are currently investigating the influence of substrates of varying stiffness and composition on the HF and LF TM cells from multiple donors.
5.4. Molecules and segmentation
The underlying molecular basis for how the segmental pattern arises and its ultimate consequences on establishing and maintaining outflow resistance is poorly understood. We have hypothesized that the molecular composition of ECM molecules within the HF regions may show some important differences from that of the LF regions. Initial studies investigating molecular differences between segmental outflow regions identified several ECM genes and proteins enriched in HF or LF regions from perfused human anterior segments using quantitative PCR arrays (Vranka et al., 2015). In this study we found a number of collagen and metalloproteinase genes that were differentially expressed, as well as some integrins and matricellular proteins. In addition, MMP enzyme activity was measured from HF and LF TM regions extracted from perfused anterior segments, but these did not show any difference between the segmental flow regions. In an unpublished immunohistochemical study, PKB phosphorylation on T308 was undetectable in the TM/SCE of HF and LF regions after 24 hours at 1x perfusion pressure and in the LF region at 2x perfusion pressure. However, phosphorylation was very pronounced in the SCE/JCT region of HF regions after perfusion at 2x pressure.
5.4.1. Segmentation and versican
Our prior study showed that total versican is inversely correlated with flow, i.e. high total versican in LF regions and low total versican in HF regions. However, isoforms V1 and V2 levels were low in LF regions and high in HF regions from TM tissues (Keller et al., 2011). Presumably, different concentrations of CS GAGs per volume would impact fluid permeability. In addition, orientation, extension or collapse and organization of versican isoforms and how they are bound on HA strands or interact with other binding partners would impact fluid movement within the segments (Gard et al., 1993).
5.4.2. Segmentation and mRNA levels
In addition to measuring protein levels in segmental flow regions, we also investigated gene expression levels of ECM genes from TM tissues (Vranka and Acott, 2017). In this study we identified different subsets of ECM genes that were differentially expressed in segmental regions of the TM from anterior segments that were perfused at either physiologic (1x), or at elevated (2x), pressure for 48 hours. This work suggests that a limited subset of ECM genes is differentially regulated in segmental flow regions of the TM in response to elevated pressure.
5.4.3. Segmentation and protein levels
Our later study used a quantitative proteomics approach to identify specific ECM proteins that are up- and down-regulated in the HF and LF regions of the TM (Vranka et al., 2018). The most dramatic differences measured in this same study identified a specific group of ECM proteins to be upregulated in response to elevated (2x) pressure in HF and LF regions suggesting a key role in modulating the normal homeostatic response to elevated pressure (Vranka et al., 2018). This study also measured the biomechanical properties of the HF and LF regions of the TM tissues after perfusion ex vivo at either physiologic or elevated pressure. LF regions were more than twice as stiff as the HF regions, and this difference was more pronounced at elevated perfusion pressure, indicating an important difference in biomechanical response to pressure.
5.4.4. Pressure elevation and expression
As mentioned earlier, elevated pressure induces a response in the TM. This has been established previously using primary cultured TM cells and subjecting them to stretch for various time points. Key ECM genes were shown to be up- or down-regulated at different time points in response to sustained stretch (Vittal et al., 2005). Our recent studies have been the first to measure molecular differences in segmental flow regions in response to sustained elevated pressure (Vranka et al., 2017; Vranka and Acott, 2017). Numerous ECM components are differentially regulated in response to a sustained 2x pressure challenge. We are in the process of conducting manipulation studies to determine which of these are most relevant to the IOP homeostatic response.
5.5. Regional segmental half life
Some questions that remained unanswered were: 1) how long-lived are the individual segmental flow regions; 2) are these regions affected by perfusion at elevated pressure; and 3) what is the function of LF regions, which do not seem to have an apparent function? We recently investigated these questions using perfusion organ culture and introducing two different colored fluorescent labels one week apart. We compared the relative distributions of the HF, intermediate flow (IF or MF) and LF regions when anterior segments were perfused for 1 week at physiologic (1x) pressure with those that were perfused for 1 week at elevated (2x) pressure. Segmental flow regions of the TM are relatively stable after 7 days of perfusion at physiologic pressure using an ex vivo human organ culture system. Relative amounts of IF or MF regions are significantly increased after perfusion at elevated pressure. These dynamic changes in segmental outflow regions appear to be an important part of the normal IOP homeostatic response to elevated pressure (Vranka et al., 2020).
5.6. Glaucoma and segmentation
Until recently, no studies had been done to experimentally determine the involvement of segmental outflow in glaucoma. This is in spite of the fact that many groups have shown the presence of ultrastructural abnormalities in the ECM of the TM and the inner wall of SCE in glaucoma eyes (Lutjen-Drecoll 1981, 1999, 2005; Rohen 1981 Tektas 2009; Furuyoshi et al 1997). In addition, the biomechanical properties are altered in glaucoma, where the TM was shown to be 20-fold stiffer than age-matched normal TM tissues (Last et al, 2011) as detailed in section 6.1.1.later. Our recent work aimed to measure the relative amounts of segmental flow regions in glaucoma eyes relative to those in normal eyes (Raghunathan et al 2018; Vranka et al 2018). When we assessed the relative portion of HF and LF regions in glaucomatous eyes, we found that there is a distinctive increase in the amount of LF regions compared to the amount in normal eyes (Raghunathan et al., 2018). Thus, in glaucomatous eyes, not only are there fewer HF or active-flowing regions of the TM, but the LF or non-flowing regions were much more prevalent than in normal tissues. This observation is likely to contribute to the development of glaucoma whereby the active outflow is restricted to smaller regions of the TM.
Interestingly, we also measured the number of cells in segmental regions of the TM and found no differences in cell numbers between the HF and LF regions of normal eyes. There also were no differences between the HF and LF regions of glaucomatous eyes. In contrast, we saw a significant decrease in the cell numbers in segmental regions of glaucomatous eyes compared to the corresponding regions in normal eyes, which is in good agreement with previous studies showing a decrease in cellularity in the TM of glaucoma eyes (Abu-Hassan et al., 2015; Alvarado et al., 1984; Alvarado et al., 1981; Grierson and Howes, 1987). Thus, this decreased cellularity appears to be a general effect of glaucoma and does not preferentially occur in particular segmental regions of the TM.
6. Biomechanics
6.1. Tissue mechanics
6.1.1. Total TM tissue
Changes in TM ECM composition, organization, and remodeling may be due to multiple factors such as ageing, secretion of cytokines, stress responses or subtle genetic disbalances. Because of their critical role in regulating outflow resistance, TM JCT cells are adept at sensing and responding to pressure differences and are accustomed to pressure fluctuations which are likely to be interpreted by the cells as constant yet dynamic changes in strain. The outflow resistance is adjusted many, many times throughout life in response to homeostatic challenges, which, may result in the ECM turnover process drifting further and further away from its optimal state. This process is thought to be distinct from the regular ocular pulse and accompanying IOP fluctuations along the cardiac cycle that is extensively covered elsewhere in the literature (Carreon et al., 2016; Johnstone, 2004; Li et al., 2013; Sun et al., 2015; Xin et al., 2017). These IOP changes have been shown to be dynamic by continuous IOP monitoring in nonhuman primates (Downs, 2015). In any case, it is conceivable that when TM cells are unable to respond correctly to an IOP homeostatic challenge due to such this drift, they progress towards a pathologic state where eventually their ECM becomes disorganized. This alters the mechanical properties and flow state of the tissue. It is therefore clear that characterization of both healthy and diseased ECM pliability and compliance is of fundamental interest. Indeed, numerous studies have measured the elastic moduli of the human TM using several engineering techniques (Table 1). While most of the studies rely upon excision of the tissue and subjecting it to compressive or tensile loading ex vivo, a few have performed advanced imaging coupled with finite element modeling (FEM) to evaluate the biomechanical properties. Thus, the room for methods development and growth in the area is extensive. Here we summarize some of the key findings of our and others’ studies.
Table 1.
Elastic moduli of TM reported using a variety of methods and species
| Study | Technique | Tissue Species | Elastic Modulus | Comments |
|---|---|---|---|---|
| (Last et al., 2011) | AFM | Human | Normal: 4.0 ± 2.2 kPa Glaucomatous: 80.8 ± 32.5 kPa | Mathematical modeling correlated increased moduli (JCT side up, 1μm sphere) to flow resistance. |
| (Yuan et al., 2011) | AFM | Porcine | 1.38 ± 0.47 kPa | |
| (Camras et al., 2014; Camras et al., 2012) | Tensile | Human | Normal: 51.5 ± 13.6 MPa Glaucoma: 12.5 ± 1.4 MPa | Did not correlate with postmortem time, age, facility or cellularity. |
| (Camras et al., 2014; Camras et al., 2012) | Tensile | Porcine | Normal: 2.49 ± 1.5 MPa | |
| (Li et al., 2014b) | SD-OCT & mathematical model for SC deformation | Mice | Naïve: 2.16 kPa +GFP: 2.20 - 2.82 kPa +BMP2: 3.46 kPa – 5.01 kPa |
GFP or BMP overexpressing mice were imaged. Modulus was calculated as a function of cross-sectional area of SC relative to that with no pressure difference across the TM. Mathematical models described in (Johnson and Kamm, 1983; Li et al., 2014a) |
| (Huang et al., 2015) | AFM | Rat | 162 ± 1.2 Pa | Measured on uveal side, validated with mathematical model for non-Hookean materials |
| (Raghunathan et al., 2015) | AFM | Rabbit | Control: 1.03 ± 0.55 kPa Dexamethasone: 3.89 ± 2.55 kPa |
3 weeks, b.i.d. 0.1% Dexamethasone topical |
| (Johnson et al., 2015) | Inverse FEM | Human | Normal: 128 kPa | Beam-bending analytical model for a linearly elastic material with simplified geometry. |
| (Wang et al., 2017) | Inverse FEM | Human | Normal: 70 ± 20 Kpa Glaucoma: 98 ± 19 kPa |
|
| AFM | Normal: 1.37 ± 0.56 kPa 2.75 ± 1.19 kPa |
Measure on the Corneoscleral/Uveal meshwork side. | ||
| (Pant et al., 2017) | Inverse FEM | Human | Normal: 5.75 kPa | Applied a neo-Hookean solid model for nearly incompressible materials. |
| (Raghunathan et al., 2017) | AFM | Non-human primate | Set 1 Control: 3.31 ± 0.32 kPa Set 2 Control: 2.63 ± 0.14 kPa Set 1 ExGl: 0.46 ± 0.036 kPa Set 2 ExGl: 0.15 ± 0.014 kPa |
Mechanics determined in unlasered eyes (control), and unlasered portions of lasered eye (ExGl) |
| (Vranka et al., 2018) | AFM | Human | Normal: kPa HF1X: 3.05±0.86 HF2X (24 Hr): 1.31±0.59 HF2X (72Hr): 2.49±0.56 |
|
| (Raghunathan et al., 2018) | AFM | Human | Glaucoma: HF: 1.86 ± 0.77 kPa IF: 2.98 ± 1.85 kPa LF: 76.55 ± 24.4 kPa |
|
| (Wang et al., 2018) | AFM | Mouse | Control: 1.99 ± 0.91 Dexamethasone: 2.38 ± 1.31 kPa |
Mouse eyes perfused, cryofixed, measurements done on sections. |
| (Li et al., 2019) | Inverse FEM / OCT | Mouse | Control: 29 kPa Dexamethasone: 69 kPa |
measured by factoring loads exerted on SC/JCT/TM regions, pressure within lumen of SC, and forces and moments contributed by the iris |
| (Vahabikashi et al., 2019a) | AFM | Human | Normal: 7.62 ± 1.43 kPa Glaucoma: 11.67 ± 2.26 kPa |
10 μm sphere |
| Normal: 11.83 ± 1.33 kPa Glaucoma: 53.91 ± 17.77 kPa |
1μm sphere |
Abbreviations: AFM = atomic force microscopy, JCT = juxtacanalicular trabecular meshwork, OCT = optical coherence tomography, SD-OCT = spectral domain OCT, FEM = finite element modeling, SC = Schlemm’s canal, kPa = kilo Pascals, MPa = mega Pascals, HF = High flow, LF = Low flow, IF = Intermediate flow.
6.1.2. Methods and measurements of normal and glaucomatous tissues
Various studies have documented the elastic modulus of the human trabecular meshwork from both non-glaucomatous and glaucomatous donors. These measurements have been obtained using various methods that are now commonplace in the realm of biomechanics; and the choice of method is dependent on various factors. Nevertheless, the most widely employed technique has been atomic force microscopy (AFM) that enables the quantitation of local changes in tissue biomechanics. However, even with the usage of AFM the studies vary between each other on how a sample was prepared, choice of indenter diameter, location of indentation, depth of indentation, and type and parameters of cantilevers used. For example, the very first study to report definitive alterations in the biomechanical properties of the human TM in glaucoma utilized AFM (Last et al., 2011). The TM tissue was removed and probed, SCE side up, using a relatively small spherical indenter. The apparent elastic modulus of glaucomatous TM was reported to be 20-fold greater than that of age-matched normal TM, suggesting an important link between tissue compliance and the disease state. We also demonstrated that the modulus varied with location along the length of tissue, although segmental outflow regions were not identified or considered.
Since then, AFM has been used to report TM moduli in porcine (Yuan et al., 2011), rabbit (Raghunathan et al., 2015), rat (Huang et al., 2015) or mice (Wang et al., 2017; Wang et al., 2018) tissues. Interestingly, AFM in mouse tissues (Wang et al., 2017; Wang et al., 2018) was performed using sagittal cryosections, and demonstrated for the first time a small but significant correlation between TM modulus and outflow resistance, but not IOP, with dexamethasone treatment; however, discrepancies continue to exist regarding the site or indenter geometry used for these measurements. In an attempt to further reconcile the differences in values reported, at least using AFM, and to further identify the specific region where there may be increased modulus at the TM/SC tissue Vahabikashi et al (Vahabikashi et al., 2017) determined the elastic modulus of both SCE and their substrate in normal and glaucomatous human eyes using various tip size and indentation depth. This provided a new view of the depth-dependent stiffness in the tissue as assessed by several approaches. Measurements were obtained by indenting the SCE side similarly to the technique reported by Last et al (Last et al., 2011). They report that the elastic moduli of SCE cells in glaucomatous tissues were elevated compared with SCE cells in normal tissue using a 10 μm spherical bead tip. With the same bead diameter for the indenter they found no difference in matrix mechanics underlying SCE cells. This is unsurprising considering the large diameter used for moduli measurements over relatively small indentation depths. It is generally accepted that the radius of the indenter should be 1/0th that of the indentation depth in order to apply Hertzian/Sneddon models for estimation of the elastic modulus. The relationship between indenter diameter, indentation depth, and mechanical properties of materials have been recognized and describing their relationship is out of the scope of this review. Further, when a smaller diameter bead was used (1 μm), the values of elastic moduli reported for matrix mechanics underlying SCE cells were comparable to those found by Last et al (Last et al., 2011). Accompanying the biomechanical changes, Vahabikashi et al (Vahabikashi et al., 2019a) showed elevated moduli values to correlate with reduced outflow facility in glaucomatous eyes. This is consistent with another recent study from our group (Raghunathan et al., 2018) where we document that glaucomatous eyes with stiffer TMs not only have increased resistance, but also exhibit a loss of the IOP homeostatic response in aqueous outflow facility. Interestingly, as applies to section 7, both groups’ data are compatible with the biomechanical outcome that the outflow resistance, as impacted by glaucoma, resides within around 1 μm of the inner wall of SCE. In terms of probe shape and size, another recent analysis suggests that sharp probes have some intrinsic advantages in measuring the compliance of this region, although careful assumptions and data fitting can overcome this advantage (Vahabikashi et al., 2019b).
Besides AFM, elastic moduli of the TM has also been investigated using the conventional uniaxial tensile testing technique in human and porcine (Camras et al., 2014; Camras et al., 2012) tissues. Another technique that has been used in engineering widely, and in characterizing mechanical properties of tissues in silico is finite element modeling (FEM). For many of these studies, advanced OCT was used to image the irido-corneal angle, and then using FEM, the moduli were estimated. Wang et al (Wang et al., 2017) showed that the TM stiffness is higher in glaucomatous patients with outflow facility, in both normal and glaucomatous human eyes, correlating inversely with TM stiffness (Wang et al., 2017). More recently, using a pseudo-2D FEM geometry on integrated OCT and histology images, the elastic modulus of the TM in mouse eyes in vivo was measured by factoring loads exerted on SCE/JCT/TM regions, pressure within lumen of SC, and as well as forces and moments contributed by the iris (Li et al., 2019). Such comprehensive in situ analyses revealed TM stiffness in steroid induced eyes to be 69 kPa against 29 kPa in control eyes. Further, the study showed for the first time that the SCE was more resistant to collapse at elevated IOPs, reflecting increased TM stiffness with steroid treatment (Li et al., 2019).
Here we would like to emphasize that different techniques will yield different numerical values for elastic moduli. Tensile moduli values of human and porcine TM were orders of magnitude greater, for example in megapascals (MPa), compared with reports using the AFM and FEM measures, where moduli values for human TM were in the order of kilopascals (kPa),. Tensile measurements would stretch the bulk of the tissue along the axis of the orientation of the fibers and indention measurements would be localized to specific regions of the TM (nano-micron scale) and against the orientation of the collagen and elastin fibers of the TM. On the other hand, finite element modelling accounts for mathematically breaking a bulk tissue into smaller elements on which boundary conditions are applied for estimating moduli. Further, sample preparation techniques, hydration methods, indenter geometry, clamping force for tensile measurements, the magnitude, and the rate at which the tensile or compressive forces are applied would all dictate the estimates determined for the elastic moduli. Thus, every method that one uses comes with its own set of assumptions, advantages, and challenges that ought to be carefully considered.
6.1.3. Biomechanics of segmental flow regions
While variations in the elastic modulus along the TM are suggestive of segmental outflow, evidence to substantiate this was missing until recently. We found in normal eyes (Vranka et al., 2018), where IOP homeostatic response was maintained, that the elastic modulus of LF regions was 2.3-fold larger than HF regions at physiological (1x) pressure. After 2x pressure elevation for 24 or 72 hours, LF regions were 7.4-fold or 3.5-fold larger than HF regions, respectively. Also, changes in elastic moduli were seen comparing the same flow regions across different pressure conditions, albeit to a smaller extent. Elastic moduli of HF regions were 2.3- and 1.2-fold smaller at 24 and 72 hours of 2x pressure, respectively, than those at 1x pressure (Vranka et al., 2018). Collectively, these data demonstrated a compensatory effect in HF regions but a deleterious positive feedback response in LF regions whereby at elevated pressures, HF regions got softer, and LF regions became stiffer within 24 hours before attempting to normalize at 72 hours. This is consistent with the homeostatic response where with increased pressure, normalized facility increases with time. Such a compensatory response in HF regions becoming softer is not inexplicable. We showed that unlasered regions of the TM in experimental glaucoma (ExGl) in non-human primates (NHPs) were softer compared with control unlasered NHPs (3.3 ± 0.32 kPa). This suggests that eyes (non-diseased) have the capacity to compensate for chronic IOP elevation in ExGl by altering the composition and subsequent mechanical properties of the ECM in the JCT region to compensate for reduced outflow facility (Raghunathan et al., 2017), although homeostasis may still be disrupted. In a follow up study (Raghunathan et al., 2018), we compared elastic moduli in segmental flow regions of glaucomatous eyes where homeostatic response was lacking. We noted that LF regions of the glaucomatous TM exhibited an 8- to 10-fold increase in elastic moduli compared to LF regions of non-glaucomatous TM. Concurrently, a 50% decrease was observed in the moduli of the HF regions of glaucomatous TMs, when compared with HF regions of non-glaucomatous TM, although it did not reach statistical significance due to a relatively small number of biological replicates. This was accompanied by a relative increase in the number of LF regions accompanying a loss of HF regions. This, however, is consistent with our ExGl NHP data, where smaller HF regions in glaucomatous TMs are even more compliant to maintain IOP at acceptable levels.
6.2. Cellular biomechanics
6.2.1. Variability in measurement methods and conditions
Multiple studies have determined the elastic moduli of TM or SCE cells using an AFM. However, differences in the indentation conditions or indenter geometry are inconsistent between investigators and, as such, direct comparisons of ‘E’ values are difficult. Nevertheless, all studies demonstrate similar effects in that cells treated with glaucomatous stimuli, such as TGFβ, or the corticosteroid dexamethasone (Dex), or stiff substrates, all of which induce a pro-fibrotic phenotype in TM cells, exhibit a larger elastic modulus than control cells.
6.2.2. Human TM cells
We demonstrated that primary human TM cells, when cultured on glass surfaces were stiffer when treated with 100 nM Dex than control cells (Control: 2.82 ± 1.01 kPa vs Dex: 5.87 ± 2.93 kPa)(Raghunathan et al., 2015). Furthermore, we showed that replicative senescence in TM cells is associated with cell stiffening (Morgan et al., 2015a; Wang et al., 2017) and with Wnt inhibition. Indeed, Wnt inhibition has also been observed with glaucomatous stimuli (e.g Dex or TGFβ) (Mao et al., 2012a; Mao et al., 2012b; Raghunathan et al., 2015; Wang et al., 2008; Webber et al., 2018; Webber et al., 2016), and is associated with elevations in IOP (Pang et al., 2015; Wang et al., 2008; Webber et al., 2016) and cell stiffening (Morgan et al., 2015a, b). More recently, we demonstrate activation of the Wnt pathway, after chronic Wnt inhibition in normal TM cells softens them, suggesting at least in normal cells that the phenotype may be reversible (Dhamodaran et al., 2020). TM cell response to actin disrupting drugs has also been observed to be substratum stiffness dependent (McKee et al., 2011). Briefly, in control conditions, untreated cells cultured on 4 kPa and 90 kPa hydrogels were softer than when cultured on glass substrates. After treatment for 90 minutes with 0.2 μM Latrunculin-B, significant actin re-polymerization and increased cell stiffness was observed on glass (GPa; 11.0 ± 2.3 kPa) and stiff (90 kPa; 4.0 ±1.8 kPa) hydrogels, but this was not observed on soft (4 kPa; 2.3 ± 0.2 kPa) hydrogels. In addition, 270 minutes after treatment, cell stiffness in all groups returned to basal levels.
6.2.3. Human SCE cells
Similar substratum dependent effects were also observed in contractile SCE cells, where glaucomatous SCE cells appear to be hyper-responsive to substrate stiffness (Overby et al., 2014b; Stamer et al., 2015). Both normal and glaucomatous SCE cells were observed to stiffen when cultured on stiffer hydrogels. Simultaneously, glaucomatous SCE cell stiffness was attributed to subcortical stiffness using a spherical AFM probe, while sharp tips resulted in no significant differences. Impact of indenting tip geometry on SCE cell stiffness was demonstrated by Vargas-Pinto et al (Vargas-Pinto et al., 2013). Their data suggested that sharp tips are good for measuring mechanics at the cell cortex, while spherical or rounded tips are better for subcortical measurements. (Spherical tip: 0.7150 kPa in HUVEC and 0.9450 kPa in SCE cells; Sharp tips: 3.2350 kPa in HUVEC; 6.6751 kPa in SCE cells). The authors also demonstrated tip-geometry dependent changes in elastic moduli when treated with Latrunculin-A. Latrunculin-A greatly reduced moduli for sharp and rounded tips and also reduced the ratio of the values measured with a sharp tip as compared to a rounded tip. Latrunculin-A loaded polymer micelles were also observed to reduce SCE stiffness and morphology more recently (Stack et al., 2018). Agreeing with these in vitro data are the recent findings of SCE cells stiffness in situ in excised tissue; glaucomatous SC cells were stiffer than non-glaucomatous SCE cells (Vahabikashi et al., 2019a; Vahabikashi et al., 2019b).
6.2.4. Implications of cell stiffness in glaucoma
Gene/protein expression changes have been observed to correlate with changes in substratum compliance and cell mechanics. Alterations in cytoskeletal protein expression, cellular response to drugs, or mechanotransduction have all been reported. However, a direct causal relationship between an altered cellular phenotype or gene/protein expression and cell biomechanics is difficult to determine without unintended consequences in a biological system. Furthermore, whether a stiffer cell translates to altered contractility via increased traction forces exerted on substrates remains to be shown. Also, direct ECM remodeling via altered cellular biomechanics has also not been demonstrated. Nevertheless, we quantified and compared the expression levels of the versican isoforms in human TM cells cultured from normal and glaucomatous donor eyes (Figure 11). While levels of total versican were not significantly different between normal and glaucomatous TM cells, the V1 versican isoform, was significantly upregulated in the glaucoma TM cells as compared to the normal TM cells. As V1 is the predominant versican isoform in the TM, this difference is striking and likely to have functional significance with regards to the function of versican in the TM. It is important to recognize that dysregulation in the contractility of the TM/SC tissue to impair aqueous humor outflow may be mediated by localized traction forces exerted by TM cells locally. To the best of our knowledge, very few studies have documented changes in traction forces exerted by TM/SCE cells.
Fig. 11.
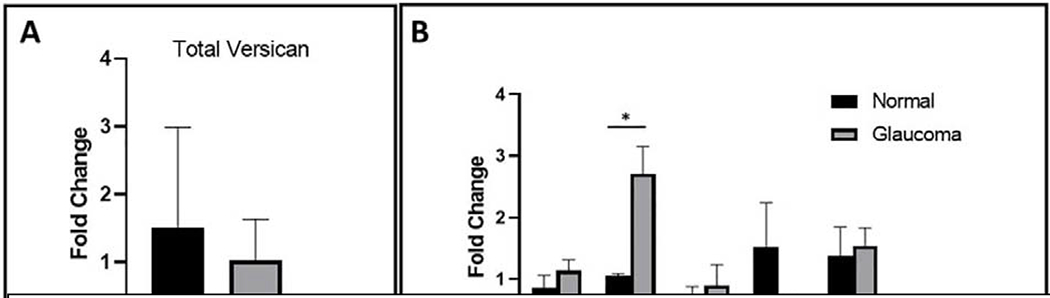
Versican isoform mRNA levels in normal and glaucoma TM cells. Total RNA was isolated from primary cultured human TM cells from normal (n = 3) or glaucomatous (n = 4) donors. (A) Expression levels of total versican and (B) of each versican isoforms (V0-V4) were measured using quantitative RT-PCR. Fold change of each isoform was normalized to total levels of versican. Statistical significance was determined using unpaired two-tailed t-tests, where p<0.05 was considered significant (“*”) (Raghunathan et al., 2018).
6.3. Stiffness of cell derived matrices
While there is increasing acceptance that TM/SCE tissue and cells are stiffer with glaucomatous stimuli, the role of the matrix in dictating the mechanics was unclear. Our group thus optimized the conditions to obtain TM cell derived matrices and quantified the indentation moduli of ECM. Initially we demonstrated that ECM derived from primary human TM cells treated with TGFβ3 (0.98 ± 0.13 kPa) were stiffer than those deposited by control cells (0.26 ± 0.12 kPa) (Raghunathan et al., 2015). Subsequently, we showed that both treated and untreated glaucomatous cells deposit an ECM stiffer than non-glaucomatous cells, and that this stiffness is further elevated in the presence of Dex [NTM ECM: Control: 0.37 ± 0.26 kPa ; Dex: 1.35 ± 0.45 kPa (Raghunathan et al., 2015) GTM ECM: Control: 1.49 ± 0.68 kPa ; Dex: 2.78 ± 1.21 kPa (Raghunathan et al., 2018). In the aforementioned studies, we documented substantial differences in the composition, morphology, and organization of the matrix deposited by the cells. Furthermore, we demonstrated that when freshly cultured normal TM cells are re-plated on these cell derived matrices, marked changes in cell biomechanics are observed. Thus, it is clear that manipulating the matrix upon which TM cells grow has a direct effect on TM cell behavior.
6.4. Effect of stiffness on gene/protein expression in TM cells
In an early study (Russell et al., 2008), it was shown that anisotropic nanopatterned surfaces containing biomimetic length scale features influenced human TM cellular behavior. Importantly, it was demonstrated that mRNA anc protein levels of myocilin were significantly higher when cells were cultured on nanopatterned surfaces. This suggests that suppressed myocilin expression, as observed in cells plated on flat tissue culture plastic or glass substrates, is likely an artifact of a non-physiologic culture environment lacking relevant biophysical cues. Subsequently, Wood et al (Wood et al., 2011) showed that cell attachment, migration and proliferation rates were significantly lower on softer hydrogels whose elastic moduli mimic those of healthy TM tissue. In other studies (Raghunathan et al., 2013), we also established that mechanotransducers, YAP/TAZ, were inversely regulated by substratum stiffness; YAP was upregulated on softer substrates while canonical Wnt modulators, TAZ and sFRP-1, were overexpressed on stiffer hydrogels. In addition, cells were more responsive to steroid induced expression of MYOC and Angptl7 on stiffer substrates. McKee et al (McKee et al., 2011) showed that HTM cell mechanics correlated with substratum stiffness with no additional treatment. However, a substratum stiffness and time-dependent increase in TM cell elastic modulus was observed upon withdrawal of the latrunculin-B, an actin disruptor. Further, it was observed that treatment of TM cells with Latrunculin-B dramatically downregulated both SPARC (secreted protein acidic and cysteine-rich) and myocilin on hydrogels with stiffness mimicking glaucomatous tissue, while, cells grown on TCP expressed greater or similar amounts of SPARC and myocilin mRNA after Latrunculin-B treatment (Thomasy et al., 2012). Thus, if drugs were screened in cells cultured in an environment that is far removed from the native microenvironment, we may obtain artifactual results that would skew our ability to identify drugs that may not be effectively translatable to in vivo models.
We have also observed stiffness-mediated changes in ECM proteins expressed by human TM cells. The effect of stiffness on versican isoform expression is demonstrated when normal TM cells were cultured in hydrogels with varying stiffness (Figure 12). The soft hydrogels (2–5 kPa) mimic normal TM tissue stiffness, whereas the 75 kPa stiffness mimics the stiffness of glaucoma TM (Last et al., 2011; Raghunathan et al., 2018; Schlunck et al., 2008). The V1 isoform predominates on the stiffest substrates (tissue culture plastic); however, on soft substrates (2–5 kPa), the V1 isoform expression is highly reduced. With increasing substrate stiffness V1 expression is increased. Conversely, the V2 isoform is more highly expressed in soft substrates and is reduced as the substrate becomes more rigid. Thus, increased stiffness, as is observed in glaucomatous TM tissue, not only affects synthesis of ECM proteins, but can also affect expression of alternative isoforms of ECM proteins.
Figure 12.
Human TM cells grown for 48 hours on hydrogels of various stiffnesses (2-75 kPa). Conditioned serum-free media was subject to SDS-PAGE and Western immunoblots and probed with a versican monoclonal antibody (12C5; Developmental Studies hybridoma Bank, University of Iowa, Iowa). Densitometry was used to quantitate each of the gel bands and the relative fluorescent units (RFUs) were averaged. The line in the plots shows the median with N=6 technical replicates from 4 normal TM cell strains. TCP = tissue culture plastic; TCP + FNC = tissue culture plastic coated with fibronectin-collagen mixture that was used to coat the hydrogels. Isoforms V1 and V2 are shown.
Altogether, it is clear that mechanistic studies in vitro need to be performed on better model systems that simulate the multi-faceted and complex properties of the extracellular matrix in vivo. Engineering environments that incorporate stiffness, topography, composition, and cellcell communication are thus essential.
7. Outflow resistance
7.1. Resistance localization and open spaces
As discussed above, the TM and JCT regions contain many relatively large “open spaces”. It has been estimated that one opening approximately 12 μm in diameter would be sufficient to completely account for the passage of aqueous through the outflow pathway (McEwen, 1958). In electron micrographs, there are many open spaces this large or larger in the JCT, which is thought to be the site of the outflow resistance. The intertrabecular spaces between trabecular beams are relatively huge and are not thought to contribute directly to the outflow resistance. See example in figure 13 where (A) is a TEM and (B) is a quick-freeze deep-etch SEM (Gong et al., 2002), both showing significant open spaces (OS), although the latter definitely shows less than the former. The basal lamina (BL), Schlemm’s canal (SC) and giant vacuoles (V), are shown.
Figure 13.
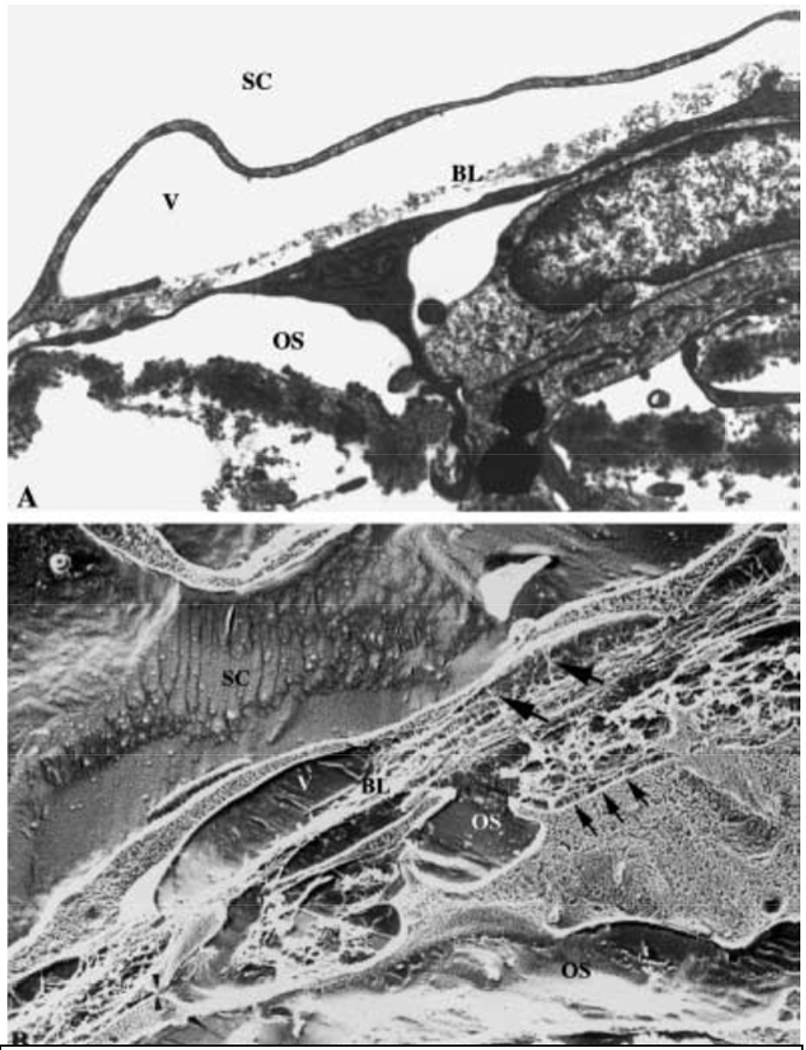
TEM (A) and quick-freeze deep-etch SEM (B) of SCE and deep JCT (Gong et al., 2002). SC is Schlemm’s canal, V is giant vacuole, BL is basal lamina, OS is open space. Small arrows point to JCT-SCE connections, larger arrows point to SCE-ECM connections and arrowheads show JCT-JCT connections.
7.2. Hypothesis - Resistance localization to 1-2 μm region beneath inner wall SCE
Eyes were perfusion fixed after being perfused at 10 or 25mm Hg (Hann and Fautsch, 2009) and the JCT region is shown above the red line (see the two images in figure 14).
Figure 14.
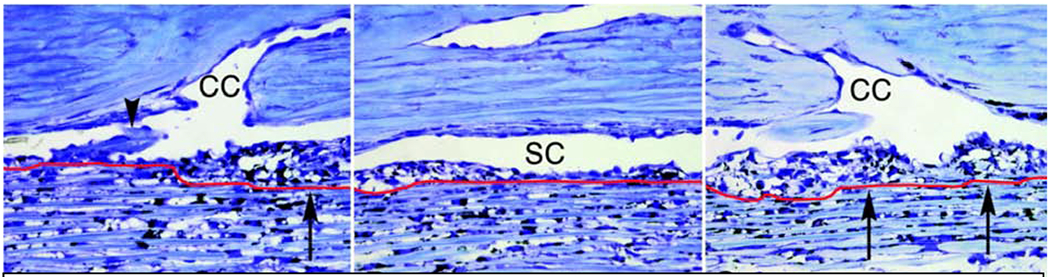
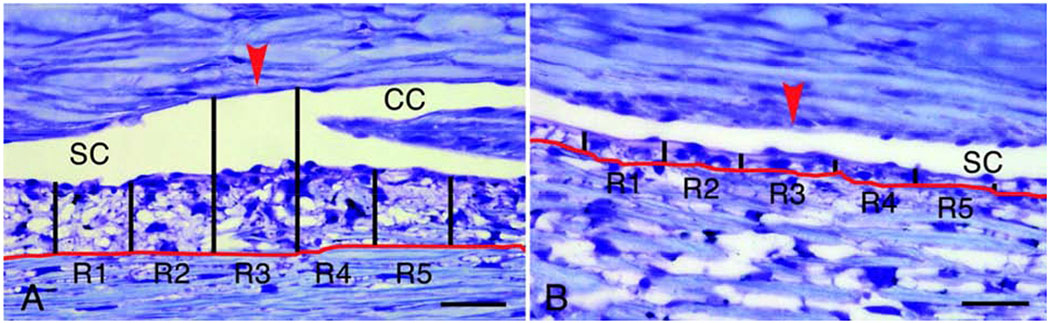
TM/SCE perfusion fixed at 10 (upper left) and 25mm Hg (other panels above and below) showing JCT region above the red line (Hann and Fautsch, 2009). Black lines and R#s in figure below were for their computations, CC is collector channel and SC is Schlemm’s canal, scale bar is 50 microns above and 20 micron below, black arrows show expanded JCT region.
Looking at these images, it is apparent that at either pressure, particularly at the higher pressure, the SCE is pushed out into SC and the open spaces within the JCT region are more frequent and dramatically enlarged, particularly near the collector channels. These enlarged open spaces are many microns in diameter. Note that beyond these open spaces, there is a very thin line of SCE and its basement membrane. The enlarged open spaces must be upstream from the resistance and thus the resistance must be within this 1-2 μm region, which includes the SCE cells themselves and their greater basement membrane. This appears to be clear evidence that the outflow resistance is not in the greater JCT, but actually only in the deepest 1-2 μm of it. Localizing the resistance to this very thin region dramatically changes our thinking about the nature of and molecular contributors to the resistance. The resistance is not the ECM of the total JCT, but just this thin SCE greater basement membrane.
7.3. What is in this 1-2 μm region?
A quick-freeze deep-etch SEM, in the left panel of figure 15, provides a physical image of this potential resistance region (Gong et al., 2002). This is a region showing continuous basement membrane and no discontinuities, a situation that will be discussed later in section 7.5.. As indicated in the schematic on the right of this image (figure 15), just beneath the SCE cell is the lamina rara, which in this case is approximately 125 nm thick and is fairly open with just a few strands reaching across it, hence its name. This should include primarily laminin tails and some integrin external domains. The next region is the lamina densa, which is very dense and approximately 500 nm thick, containing the laminin heads, collagen IV, nidogen, perlecan and other basement membrane proteins. This relatively dense region is a likely candidate for most of the outflow resistance.
Figure 15.
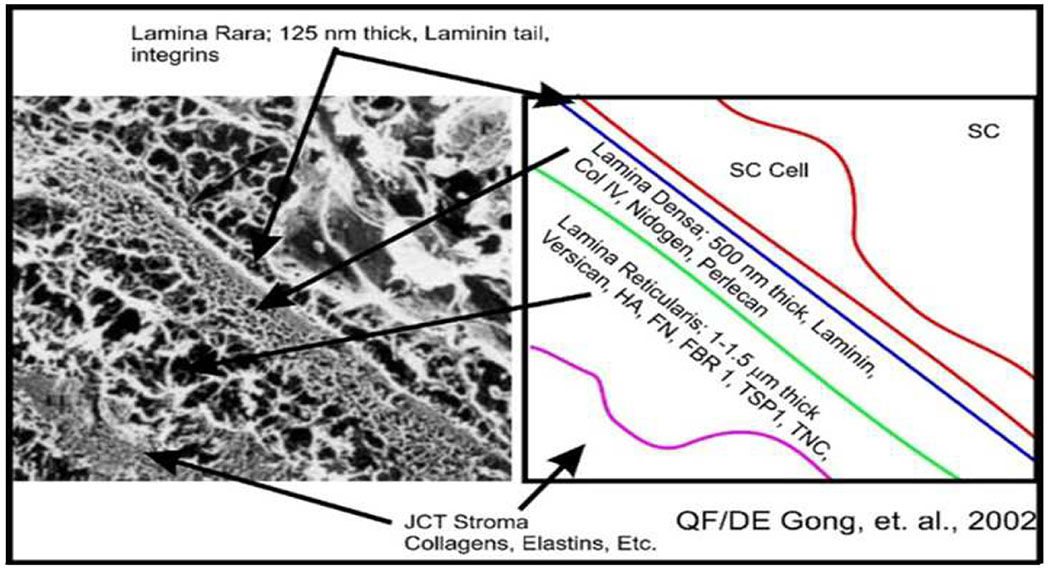
Quick-freeze deep-etch SEM of continuous SCE BM region on the left (Gong et al., 2002) with our diagrammatic characteristics on the right. Smaller two-headed arrow in SEM shows inner and outer SCE plasma membrane and arrows point to regions. Col is collagen, HA is hyaluronan, FN is fibronectin, FBR is fibrillin, TSP is thrombospondin and TNC is tenascin C.
The next region, the lamina reticularis, is 1-1.5 mm thick and should contain other basement membrane and general ECM components, such as versican, other proteoglycans, fibronectin, hyaluronan, fibrillin 1, thrombospondin 1, tenascin C, etc. This region is more amorphous with moderate sized apparent open areas and fibrillar material. Beyond that at over 2 mm from the SCE is the stromal ECM region containing structural collagens such as I and III, elastic fibers and numerous other ECM components and extending 5–15 mm beyond the lamina reticularis.
Based on other tissues, the lamina rara and lamina densa will be synthesized primarily by the SCE cells and the lamina reticularis will be primarily synthesized by the JCT cells. Compared to many other tissues, this is a thick basement membrane (Thomsen et al., 2017a; Thomsen et al., 2017b). However, some others are even thicker, e.g. bronchial epithelial basement membranes can be 5–8 microns (Watanabe et al., 1997). Separately, there is some evidence that the JCT cells themselves make their own basement membranes (Fuchshofer et al., 2006).
7.4. Additional aspects of the JCT/SCE organization
Recent studies using 3D reconstructions from serial block-face SEM imaging provide additional information about the tissue and cells (Lai et al., 2019). The SCE cell inner wall monolayer includes many strong tight and adherens junctional connections between the SCE cells. In addition, TM cells have many projections onto the basal surface of the inner wall SCE cells. In immersion fixed tissue, there was an average of 132 (range 46–237) JCT cell or ECM connections to the basal surface of each SCE cell, while fixation at 15mm Hg gave an average of 29 (range 6–76) JCT cell or ECM connections to each SCE cell. For just JCT cell connections to each SCE cell, the means were 50 and 14 connections, at 0 or 15mm Hg fixation pressure, respectively. This has significant implications for the outflow resistance, but our focus here will be on the potential for the JCT cell projections to be involved in focal SCE basement membrane turnover.
7.5. Basement membrane discontinuities
The SCE basement membrane, particularly the lamina densa, has long been recognized to have regions of discontinuity (Gong et al., 2002; Johnson, 2006; Lutjen-Drecoll, 1999). This is particularly noticeable TEMs, which dramatically under-visualize GAGs. One study found no difference in JCT ECM or SCE basement membranes in normal and glaucomatous eyes (Hann et al., 2001). Studies have found an association between SCE pores and SCE basement membrane (BM) discontinuities, suggesting that the discontinuities may have some function in outflow regulation. In addition, dexamethasone treatment increased SCE BM thickness and enhanced continuity, proportionally to reduced outflow facility (Lai et al., 2017; Lai et al., 2019; Overby et al., 2014a). These and other studies can be interpreted to support a role for modulation of these discontinuities as a component of outflow resistance modulation, as proposed in our hypothetical model. They also highlight the difficulty in studying this process.
7.6. Basal lamina assembly
This has been studied in other tissues and a self-assembly process has been proposed and studied extensively (Yurchenco and Patton, 2009). As shown diagrammatically in figure 16, taken with permission from this manuscript, laminin tails attach to specific integrins and self-assemble into a lattice stabilized and organized by interactions between the laminin arms and heads. Next, type IV collagen self-assembles into a chicken wire-like structure via interactions of their non-collagenous domains. Nidogen then links laminin to collagen IV, stabilizing these structures. Other key basal lamina proteins, such as the proteoglycans perlecan and agrin, then integrate into the structure.
Figure 16.
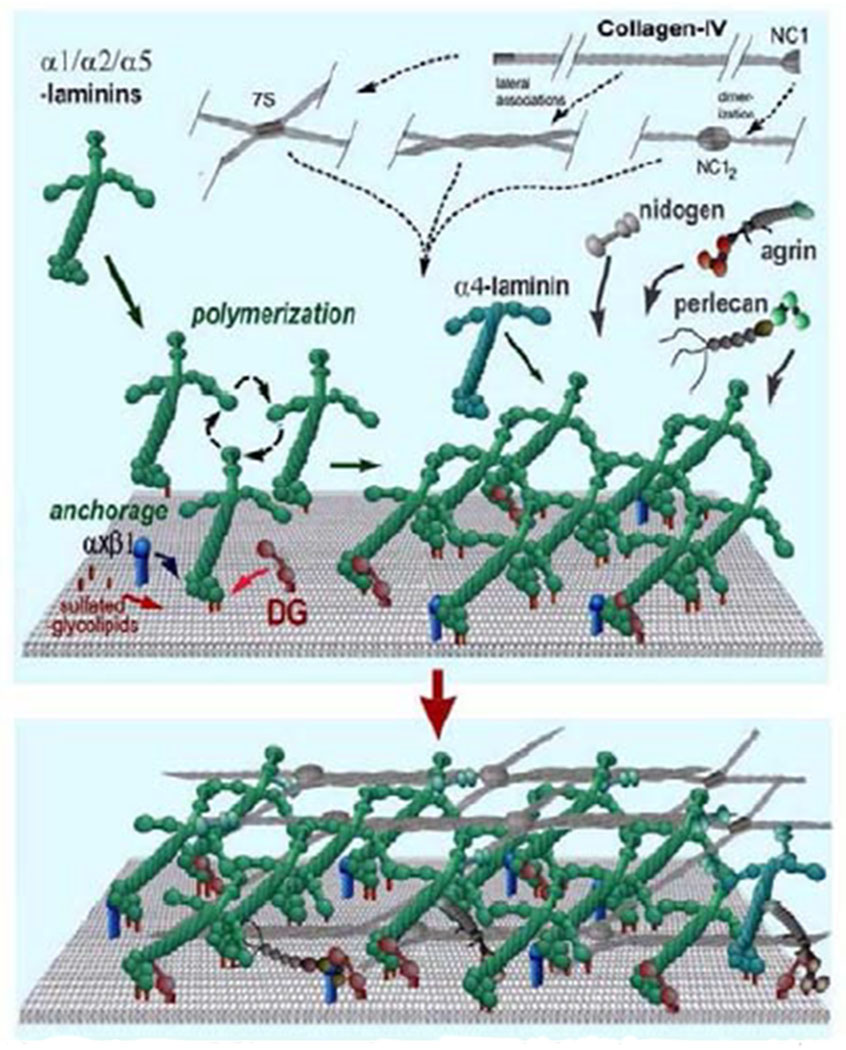
Basal lamina assembly (Yurchenco and Patton, 2009).
7.7. Focal regulated ECM turnover and PILS
As discussed earlier, PILS are sites of focal MMPs, particularly MMP14 and MMP2, and a variety of other components commonly involved in ECM turnover (Aga et al., 2008). These focal highly-controlled ECM turnover sites are thus integrated centrally into our model providing a discrete method of introducing basement membrane discontinuities exactly where desired and of precisely the size needed.
7.8. Hypothetical dynamic aqueous humor resistance model
Based on our and others’ data, we have developed a dynamic hypothetical model of how SCE basement membrane remodeling and discontinuities could regulate the outflow resistance. We know that ongoing ECM turnover is required to maintain the outflow resistance (Bradley et al., 1998) and that ECM turnover is tightly associated with changes in the resistance (Bradley et al., 1998). It appears that the IOP homeostatic response to sustained pressure deviation involves dynamic modifications of the outflow resistance. Modulation of SCE basement membrane may be primary to outflow facility regulation.
In this model, we have included changes in several major components and likely modulators as indicated in the key (figure 17). Initially, as seen in figure 18, JCT cells are attached to the basal surface of the inner wall SCE, the lamina densa is continuous as in the QF-DE SEM, figure 15 earlier. A laminin lattice is bound to SCE cellular integrins and interacts via nidogen with the chicken-wire collagen IV lattice. Perlecan proteoglycans are dispersed over this structure. Versican proteoglycans with numerous chondroitin sulfate GAG side chains are dispersed throughout the lamina reticularis binding to hyaluronan chains and interacting with thrombospondin and other ECM macromolecules. Because of the dense and continuous nature of the lamina densa, flow is highly restricted in this region. No giant vacuoles or pores are apparent in the SCE.
Figure 17.
Component key for figures 18–22. The JCT cells are blueish and the SCE cell lies on top of the image with its roundish nucleus.
Figure 18.
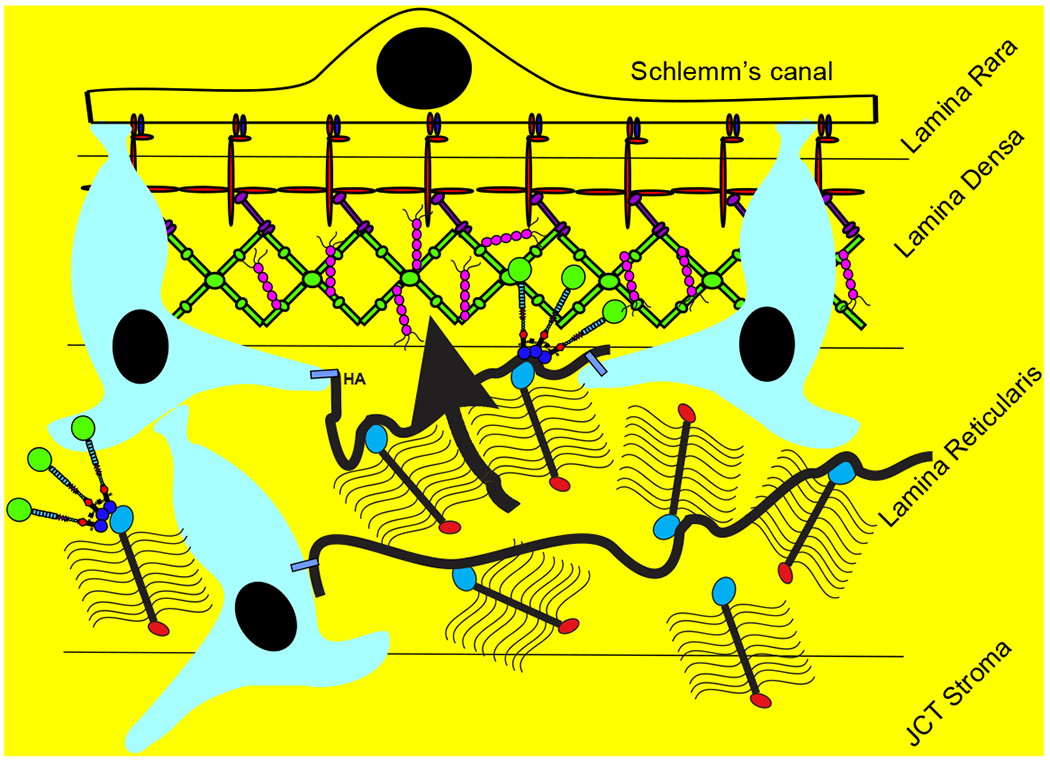
Resistance model with continuous basement membrane (lamina densa) and flow restricted.
In figure 19, the JCT cells, via their ECMs, sense a sustained pressure deviation and respond by relocating PILS with MMP2 and MMP14 to a site on the JCT cell that is adjacent to an area of the lamina densa. These MMPs on the PILS degrade a small region of the lamina densa and perhaps the lamina rara, providing a potential flow channel.
Figure 19.
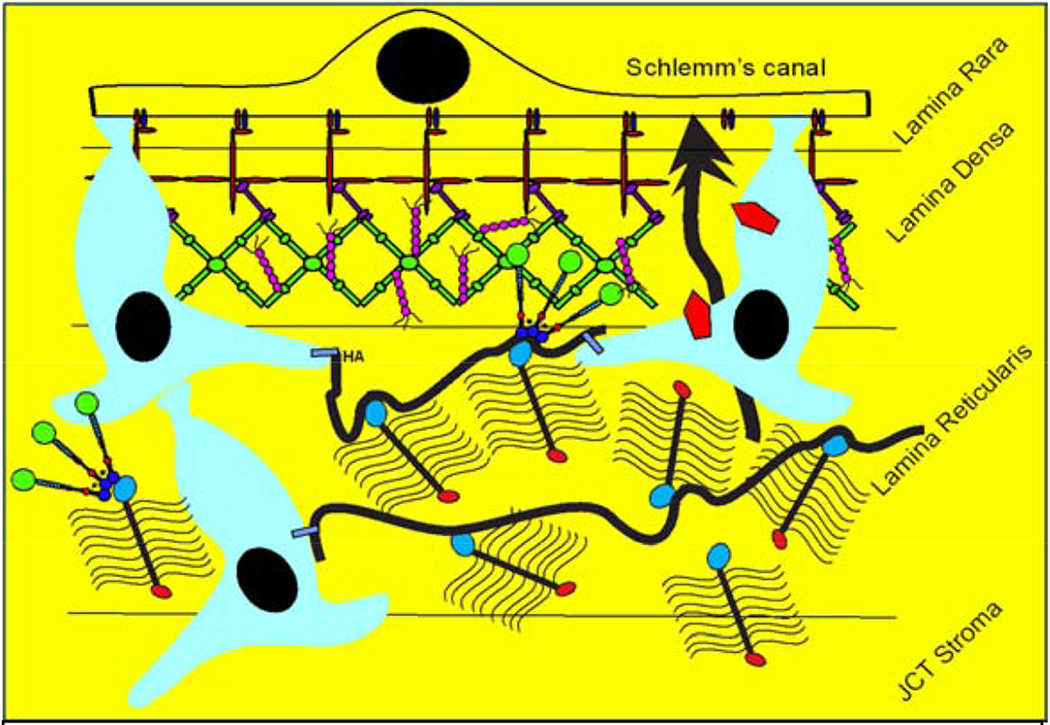
Resistance model with JCT ECM sensing sustained pressure and relocating PILS (red) into position to begin degrading a very focal region of the basal lamina.
Presumably, the degradation is primarily of laminin and collagen IV with some activity on other BM components. The flow channel that this creates will likely be less than 1 mm in diameter but will provide access for aqueous humor to pass through it and reach the basal surface of the SCE cell. The degraded region could be 1 μm or it could be even 1/10th that size but would be sufficient to allow fluid to reach the SCE cell.
In figure 20, the fluid pressure on the basal surface of the SCE produces a giant vacuole and then a pore forms within it, allowing passage of aqueous humor through the cell and into SC.
Figure 20.
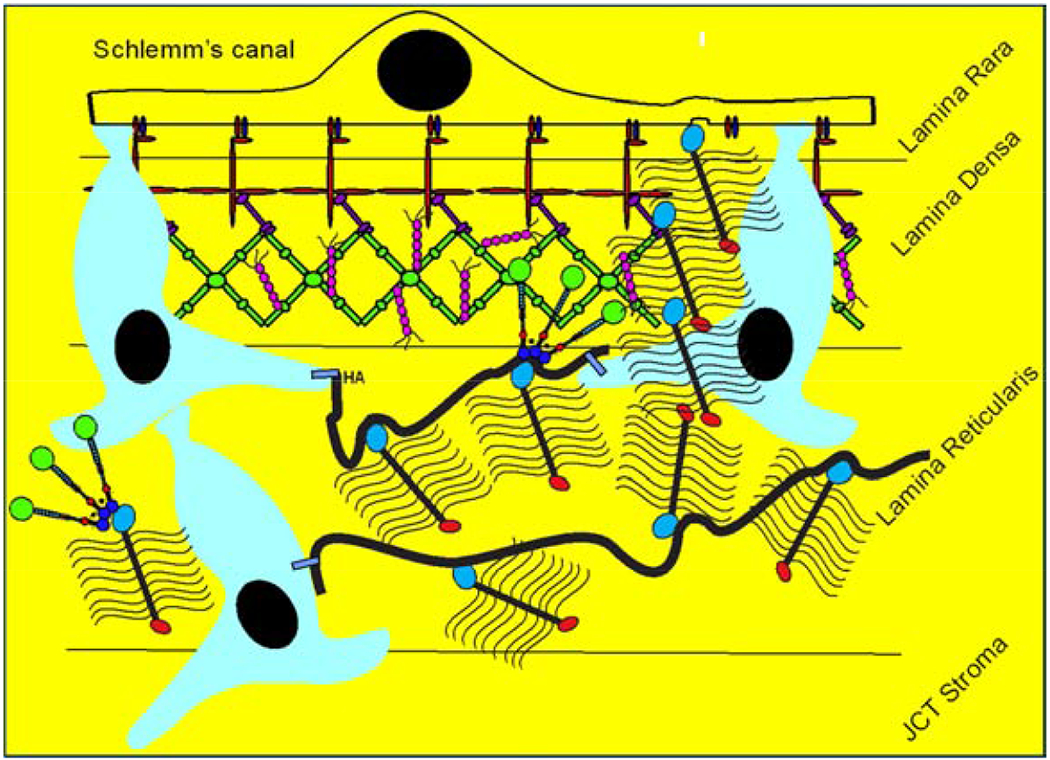
Degraded discontinuous basal lamina allows flow through and a giant vacuole with a pore is forming in the SCE, allowing fluid flow.
In figure 21, to control this fluid flow and maintain the IOP, versican molecules, either newly synthesized or currently present, are shifted by this JCT cell into the discontinuity to restrict flow. The numerous versican GAG side chains are extremely space filling and water trapping and by filling the discontinuity, can easily block or control the amount of fluid that passes. We previously showed, as have others, that versican’s chondroitin sulfate GAG sidechains can modulate flow through pores quite effectively in a concentration-dependent manner (Gard et al., 1993).
Figure 21.
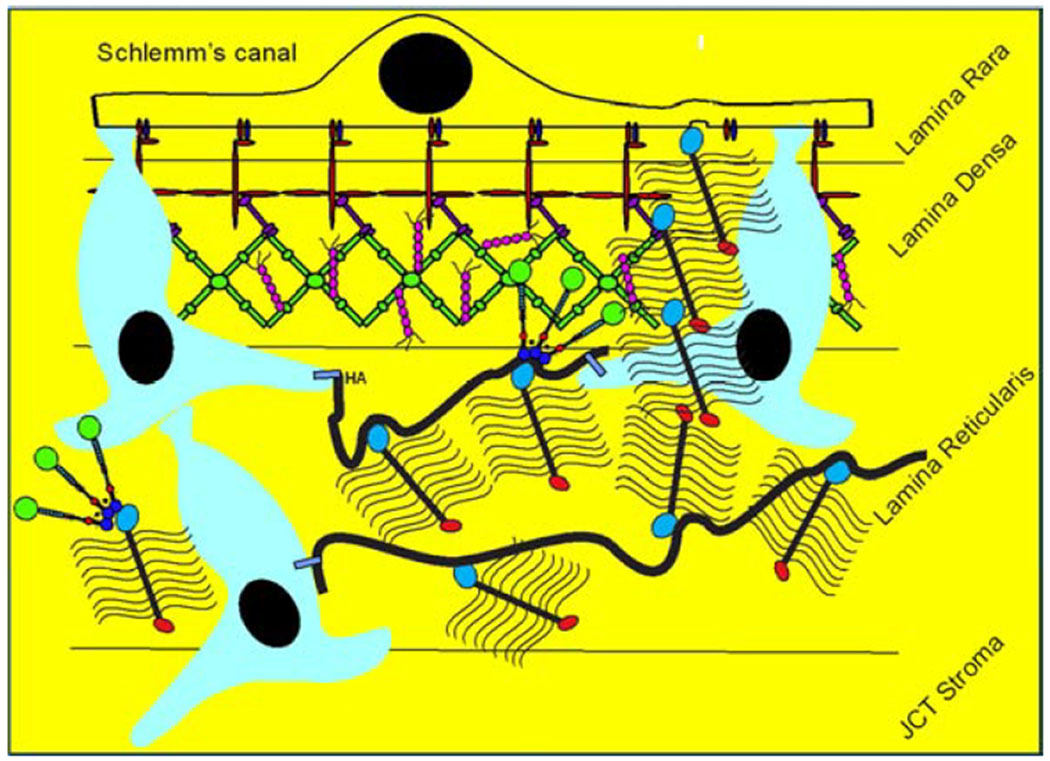
To control or restrain aqueous flow, the JCT cell moves or secretes new versican into the discontinuity and restricts or controls fluid passage by modulating position or concentrations of GAG chains.
This would allow the JCT cell to transfer the versican around or to replace it with alternatively spliced isoforms with more or less GAG side chains, effectively modulating the functional concentration of GAG charges within this discontinuity and thus manipulate the resistance to flow. This could be done dynamically by the JCT cell, perhaps in concert with the SCE cell, since the JCT-SCE cell contact is extremely likely to serve a direct signaling or communication role as well. The length of versican V0 and V1 proteoglycan cores as assessed by rotary shadow are approximately 400 nm and 300-350 nm (Morgelin et al., 1989; Perissinotto et al., 2000). The GAG side chains are up to 58 nm long. Thus, the V0 isoform would be around 400 nm long and 120-130 nm in diameter with the many GAGs sticking out on all sides. The V1 isoform would be similar, but only 300-350 nm long. In addition, the length and density of GAG chains is variable and regulatable, providing additional regulatory flow control. Based on this, one or several versican molecules would completely fill a 0.1 to 1 μm BM discontinuity. Versican is often bound to chains of Hyaluronin and to other ECM proteins, so it is easy to imagine the JCT cell filling the discontinuity with a few versican molecules. Separately, as mentioned above, we have shown that changing salt concentrations or type collapses or expands the GAG sidechains (Gard et al., 1993), which would add another mode of regulating the flow through the versican-plugged discontinuities. Using preexisting versican molecules, this could be relatively rapid or using freshly synthesized versican molecules, this could take hours or longer.
In figure 22 the SCE and or JCT cell refill the discontinuity with basal lamina molecules and restore the continuous and resistant to flow basal lamina. The timing of the initial formation of the discontinuity is likely fairly rapid after the sustained pressure increase, i.e. hours to a day or so. The "plugging” of the discontinuity with versican can be expected to occur in similar time frames, i.e. a few minutes to hours after the discontinuity is formed. However, the amount of time that an open discontinuity exists with versican filling it and modulating the flow is not clear. This could occur in a few days or could take weeks or longer.
Figure 22.
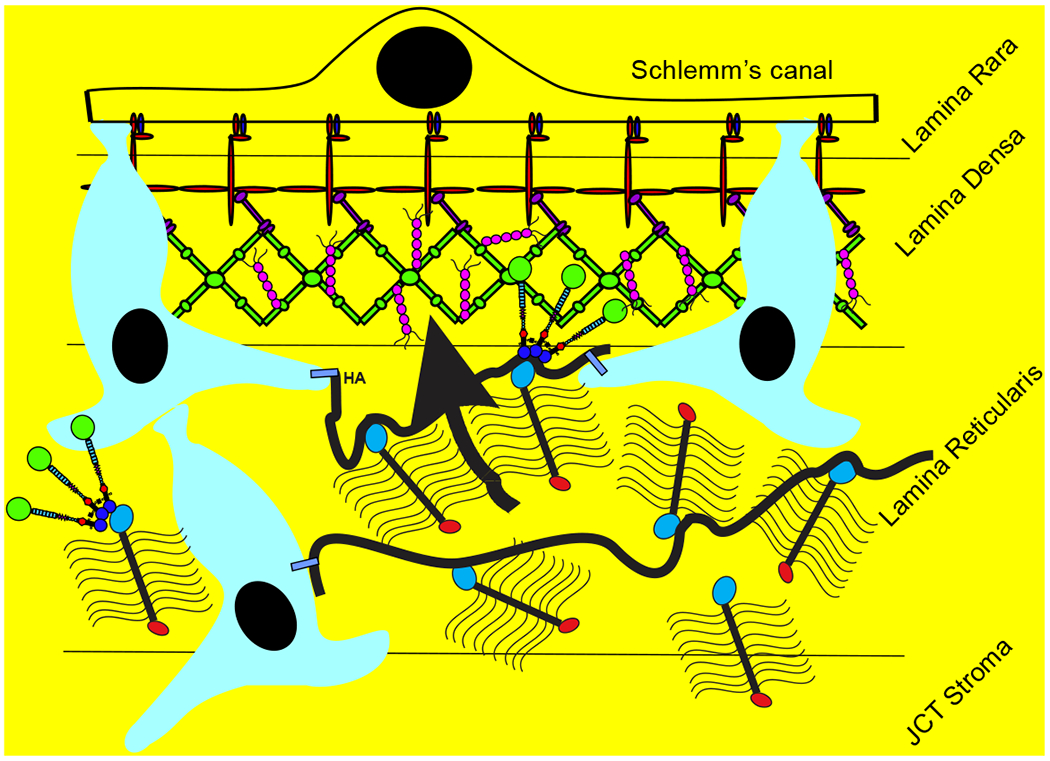
Basement membrane components are restored and the basement membrane is continuous again and obstructs flow.
Another point is that SEM or TEM imaging cannot tell if the discontinuity is full of versican GAG side chains or not, since they are fairly "invisible” by these methods. GAGs collapse dramatically in response to the harsh preparation conditions of these methods and do not allow visualization. The possibility that a discontinuity, apparently open, could exist for long periods of time in this model, is compatible with observations that the SCE basal lamina is often visualized in EM to have many discontinuities.
7.9. Images that support this hypothetical model
We have conducted several studies that provide evidence supportive of this hypothetical mode. In figure 23, red FluoSpheres (200nm and amino-derivatized; labeled FS594) were perfused into a human anterior segment in organ culture in our tissue perfusion unit at 1x pressure (8.8mm Hg) to label the segmental regions. A frontal section through a moderately high flow region is shown with each label separately in panels and then below merged. Note the two arrows pointing to versican spots (green) that coincide with the red flow marker and with an open space in the type IV collagen in grey. When merged, the type IV collagen discontinuity contains a red FluoSphere, which shows modest yellow colocalization with the versican. This image suggests that versican is filling a basal lamina collagen IV discontinuity and that flow is passing through it. If the versican is at a level of allowing some fluid passage, marked by the red FluoSphere (FS594), but otherwise filling the collagen IV discontinuity, this would support our model.
Figure 23.
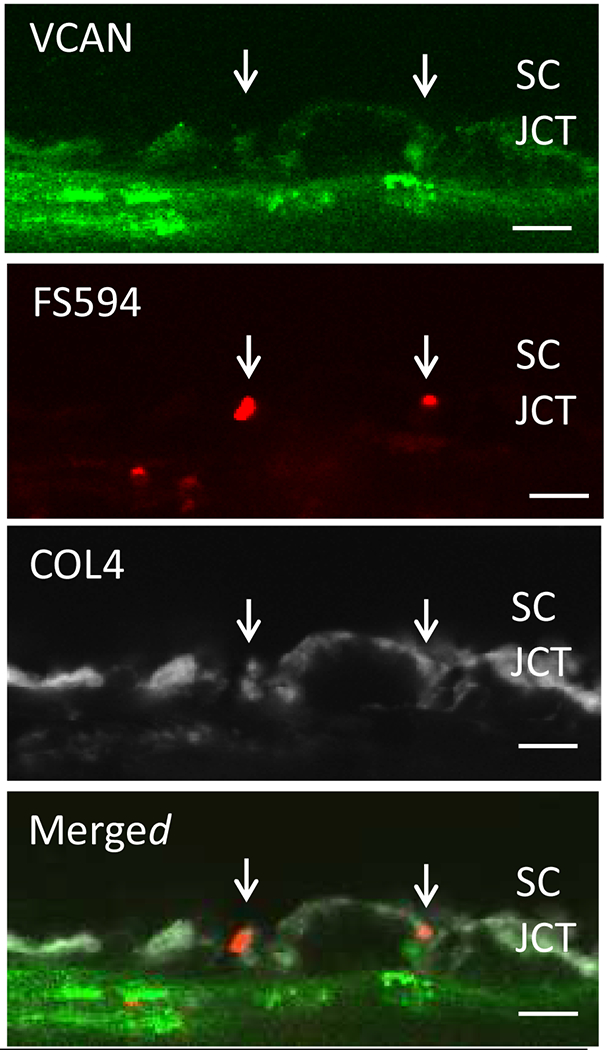
Colocalization of flow channel (red FluoSpheres) and versican (green) in collagen IV discontinuity (grey). Cropped and zoomed with Imaris software in HF region. Bar = 8 μm.
In figure 24, versican (green) colocalizes with apparent discontinuities in type IV collagen (grey) providing a similar case of versican in a potential basal lamina discontinuity (again marked by the arrows).
Figure 24.
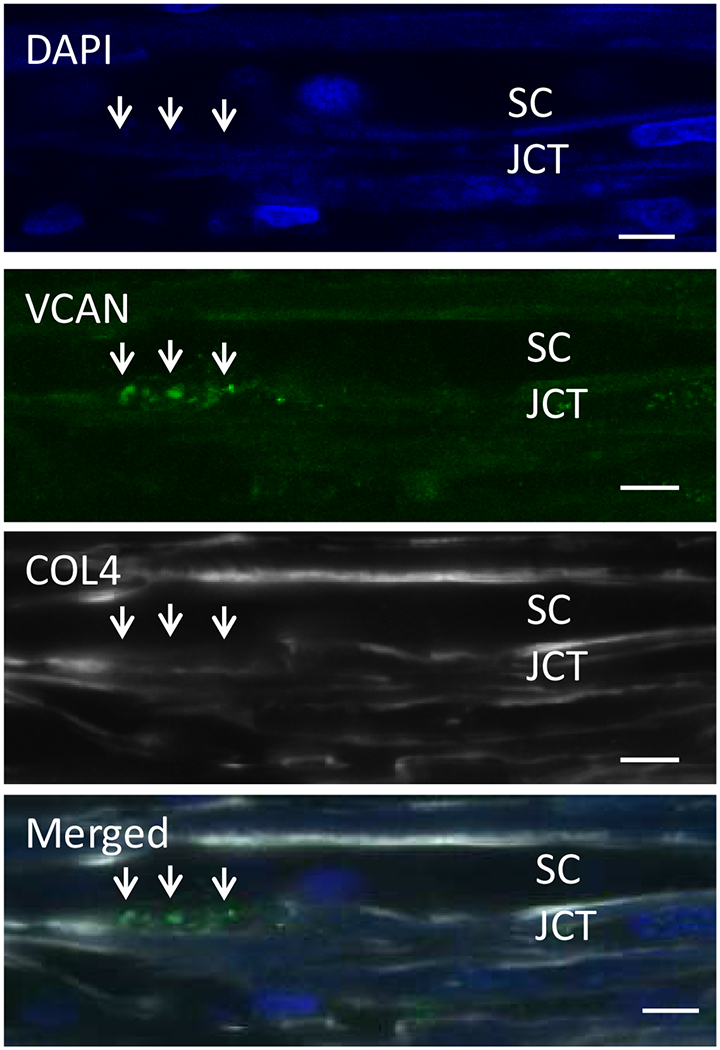
Colocalization of versican (green) with collagen IV discontinuities (grey) in apparent basal lamina discontinuities. Bar = 8 μm.
These images are compatible with our model, but do not establish it. It is noteworthy that supportive images are available, but direct evidence for our model is not as easily found. The next step is to conduct direct component manipulations and use more precise discontinuity identification approaches to test the hypothesis.
8. Future directions and conclusions
We and others have made considerable progress over the years with various aspects of the molecular and cellular nature and the regulation of the aqueous humor outflow resistance. TM and SCE cells, particularly those from glaucomatous individuals are more clearly understood and new methodologies are rapidly extending their sub-types and phenotypic characteristics. The key role and many molecular details of ECM turnover in IOP regulation have been increasingly elucidated. The identification and characterization of a robust IOP homeostatic response, which may be the reason many people do not develop glaucoma, will provide strong leads in the future for glaucoma therapies that actually address the underlying issue. i.e. the loss of IOP homeostatic capability in glaucoma. TM cell loss appears to be a key component of glaucoma and TM cell replacement can restore the lost IOP homeostatic response to pressure challenges in a cell-loss glaucoma model. This has significant potential to provide novel glaucoma therapies. Demonstration that some glaucomatous eyes cannot mount an IOP homeostatic response to a pressure challenge may indicate that this is a primary characteristic of this disease. The differences between biomechanical properties of the normal and glaucomatous outflow pathway is notable and understanding more clearly how this relates to outflow properties is of particular interest. More fully understanding the molecular causes of the differences in biomechanical properties in normal and glaucomatous eyes and the relationship to outflow segmentation will be of considerable future use. Further elucidating outflow segmentation and the molecular causes and consequences associated with it seems to be at the center of outflow regulation differences between normal and glaucomatous individuals. Actually identifying the specific molecules that comprise the outflow resistance and determining their exact location and functions within the outflow pathway seems possible now and will be of upmost importance, since this appears to be at the heart of glaucomatous loss of IOP homeostatic function. Our current hypothesis of the molecules that form the resistance, how they are manipulated to generate basement membrane discontinuities, and how the discontinuities are modulated to control the outflow resistance in real time is potentially important in IOP regulation and glaucoma. However, as should be blatantly clear from the details presented above, there is still a significant amount of information that remains to be elucidated.
Acknowledgments
9. Funding Support
NIH/NEI grants EY030238, EY025721, EY026048, EY021800, EY019643, EY003279, EY008247, P30 EY010572, Lewis Rudin Glaucoma Prize and Research to Prevent Blindness Foundation grant to the Casey Eye Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Acott: none; Vranka: none; Keller, none; Raghunathan: none; Kelley: none.
Author statement
The authors believe they are in compliance with the “Ethics in publishing” guidelines
The authors have no financial conflicts of interest
The authors have avoided redundant or concurrent publications, although portions of the work described have been published and are indicated as such and some figures will require permission from the original publication and we will obtain that before publication
The authors have attempted to use inclusive language without using race, sex, culture, his/her, etc.
Author contribution is indicated in the full manuscript
The first author contributed more and all other authors contributed equally and are all to be considered equivalent second authors
Funding sources are listed
We would prefer to publish this as Open Access, if possible
10. References
- Abu-Hassan DW, Li X, Ryan EI, Acott TS, Kelley MJ, 2015. Induced pluripotent stem cells restore function in a human cell loss model of open-angle glaucoma. Stem cells 33, 751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acott TS, Kelley MJ, 2008. Extracellular matrix in the trabecular meshwork (Review). Exp Eye Res 86, 543–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acott TS, Kelley MJ, Keller KE, Vranka JA, Abu-Hassan DW, Li X, Aga M, Bradley JM, 2014. Intraocular pressure homeostasis: maintaining balance in a high-pressure environment. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics 30, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acott TS, Kelley MJ, Keller KE, Vranka JA, Abu-Hassan DW, Li X, Aga M, Bradley JM, 2016. IOP Homeostasis - Why most people do not ever develop glaucoma, in: Knepper PA, Samples JR (Eds.), Glaucoma Research and Clinical Advances 2016-2018. Kugler Publications, Amsterdam, The Netherlands, pp. 87–98. [Google Scholar]
- Acott TS, Kingsley PD, Samples JR, Van Buskirk EM, 1988. Human trabecular meshwork organ culture: Morphology and glycosaminoglycan synthesis. Investigative Ophthalmology Visual Science 29, 90–100. [PubMed] [Google Scholar]
- Acott TS, Samples JR, Bradley JMB, Bacon DR, Bylsma SS, Van Buskirk EM, 1989. Trabecular repopulation by anterior trabecular meshwork cells after laser trabeculoplasty. American Journal Ophthalmology 107, 1–6. [DOI] [PubMed] [Google Scholar]
- Acott TS, Westcott M, Passo MS, Van Buskirk EM, 1985. Trabecular meshwork glycosaminoglycans in human and cynomolgus monkey eye. Investigative Ophthalmology Visual Science 26, 1320–1329. [PubMed] [Google Scholar]
- Aga M, Bradley J, Keller K, Kelley M, Acott T, 2008. Specialized Podosome- or Invadopodia-like Structures (PILS) for Focal Trabecular Meshwork Extracellular Matrix Turnover. Invest Ophthalmol Vis Sci 49, 5353–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aga M, Bradley JM, Wanchu R, Yang YF, Acott TS, Keller KE, 2014. Differential effects of caveolin-1 and -2 knockdown on aqueous outflow and altered extracellular matrix turnover in caveolin-silenced trabecular meshwork cells. Invest Ophthalmol Vis Sci 55, 5497–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JP, Acott TS, 2001. Involvement of protein kinase C in TNFa regulation of trabecular matrix metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci 42, 2831–2838. [PubMed] [Google Scholar]
- Alexander JP, Acott TS, 2003. Involvement of the Erk-MAP kinase pathway in TNFα regulation of trabecular metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci 44, 164–169. [DOI] [PubMed] [Google Scholar]
- Alvarado J, Murphy C, Juster R, 1984. Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology 91, 564–579. [DOI] [PubMed] [Google Scholar]
- Alvarado J, Murphy C, Polansky J, Juster R, 1981. Age-related changes in trabecular meshwork cellularity. Invest Ophthalmol Vis Sci 21, 714–727. [PubMed] [Google Scholar]
- Anderson DR, 1981. The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. Trans Am Ophthalmol Soc 79, 458–485. [PMC free article] [PubMed] [Google Scholar]
- Bárány E, 1953. In vitro studies of the resistance to flow through the angle of the anterior chamber. Acta Soc Med Upsal 59, 260–276. [PubMed] [Google Scholar]
- Battista SA, Lu Z, Hofmann S, Freddo T, Overby DR, Gong H, 2008. Reduction of the available area for aqueous humor outflow and increase in meshwork herniations into collector channels following acute IOP elevation in bovine eyes. Invest Ophthalmol Vis Sci 49, 5346–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beninati S, Piacentini M, 2004. The transglutaminase family: an overview: minireview article. Amino Acids 26, 367–372. [DOI] [PubMed] [Google Scholar]
- Bermudez JY, Montecchi-Palmer M, Mao W, Clark AF, 2017. Cross-linked actin networks (CLANs) in glaucoma. Exp Eye Res 159, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A, Svedbergh B, 1972. Scanning electronmicroscopic studies of the trabecular meshwork and the canal of Schlemm: An attempt to localize the main resistance to ouflow of aqueous humor in man. Acta ophthalmologica 1972, 295–320. [DOI] [PubMed] [Google Scholar]
- Borras T, 2003. Gene expression in the trabecular meshwork and the influence of intraocular pressure. Prog Retin Eye Res 22, 435–463. [DOI] [PubMed] [Google Scholar]
- Braakman ST, Read AT, Chan DW, Ethier CR, Overby DR, 2015. Colocalization of outflow segmentation and pores along the inner wall of Schlemm’s canal. Exp Eye Res 130, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JM, Kelley MJ, Rose A, Acott TS, 2003. Signaling pathways used in trabecular matrix metalloproteinase response to mechanical stretch. Invest Ophthalmol Vis Sci 44, 5174–5181. [DOI] [PubMed] [Google Scholar]
- Bradley JMB, Anderssohn AM, Colvis CM, Parshley DE, Zhu XH, Ruddat MS, Samples JR, Acott TS, 2000. Mediation of laser trabeculoplasty-induced matrix metalloproteinase expression by IL-1β and TNFα. Invest Ophthalmol Vis Sci 41, 422–430. [PubMed] [Google Scholar]
- Bradley JMB, Kelley MJ, Zhu XH, Anderssohn AM, Alexander JP, Acott TS, 2001. Effects of mechanical stretching on trabecular matrix metalloproteinases. Invest Ophthalmol Vis Sci 42, 1505–1513. [PubMed] [Google Scholar]
- Bradley JMB, Vranka JA, Colvis CM, Conger DM, Alexander JP, Fisk AS, Samples JR, Acott TS, 1998. Effects of matrix metalloproteinase activity on outflow in perfused human organ culture. Invest Ophthalmol Vis Sci 39, 2649–2658. [PubMed] [Google Scholar]
- Brubaker RF, 1970. The measurement of pseudofacility and true facility by constant pressure perfusion in the normal rhesus monkey eye. Investigative Ophthalmology 9, 42–52. [PubMed] [Google Scholar]
- Buller C, Johnson D, 1994. Segmental variability of the trabecular meshwork in normal and glaucomatous eyes. Invest Ophthalmol Vis Sci 35, 3841–3851. [PubMed] [Google Scholar]
- Camras LJ, Stamer DW, Epstein D, Gonzalez P, Yuan F, 2014. Erratum in: Differential effects of trabecular meshwork stiffness on outflow facility in normal human and porcine eyes. Invest Ophthalmol Vis Sci 55, 2316. [DOI] [PubMed] [Google Scholar]
- Camras LJ, Stamer WD, Epstein D, Gonzalez P, Yuan F, 2012. Differential effects of trabecular meshwork stiffness on outflow facility in normal human and porcine eyes. Invest Ophthalmol Vis Sci 53, 5242–5250. [DOI] [PubMed] [Google Scholar]
- Carreon T, van der Merwe E, Fellman RL, Johnstone M, Bhattacharya SK, 2016. Aqueous outflow - A continuum from trabecular meshwork to episcleral veins. Prog Retin Eye Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha ED, Xu J, Gong L, Gong H, 2016. Variations in active outflow along the trabecular outflow pathway. Exp Eye Res 146, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Folz SJ, Laryea SN, Overby DR, 2014. Multi-scale analysis of segmental outflow patterns in human trabecular meshwork with changing intraocular pressure. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics 30, 213–223. [DOI] [PubMed] [Google Scholar]
- Clark AF, Brotchie D, Read AT, Hellberg P, English-Wright S, Pang IH, Ethier CR, Grierson I, 2005. Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton 60, 83–95. [DOI] [PubMed] [Google Scholar]
- Clark AF, Miggans ST, Wilson K, Browder S, McCartney MD, 1995. Cytoskeletal changes in cultured human glaucoma trabecular meshwork cells. Journal Glaucoma 4, 183–188. [PubMed] [Google Scholar]
- Clark AF, Wilson K, McCartney MD, Miggans ST, Kunkle M, Howe W, 1994. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Investigative Ophthalmology Visual Science 35, 281–294. [PubMed] [Google Scholar]
- Covin Y, Laroche D, MMG O, 2014. The Societal Costs of Blindness From Uncontrolled Glaucoma, Glaucoma Today. [Google Scholar]
- Cox TR, Erler JT, 2011. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech 4, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Tamm ER, 2004. Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. BioEssays : news and reviews in molecular, cellular and developmental biology 26, 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DM, Sowinski S, 2008. Membrane nanotubes: dynamic long-distance connections between animal cells. Nat Rev Mol Cell Biol 9, 431–436. [DOI] [PubMed] [Google Scholar]
- De Groef L, Van Hove I, Dekeyster E, Stalmans I, Moons L, 2013. MMPs in the trabecular meshwork: promising targets for future glaucoma therapies? Invest Ophthalmol Vis Sci 54, 7756–7763. [DOI] [PubMed] [Google Scholar]
- de Kater AW, Melamed S, Epstein DL, 1989. Patterns of aqueous humor outflow in glaucomatous and nonglaucomatous human eyes. A tracer study using cationized ferritin. Arch Ophthalmol 107, 572–576. [DOI] [PubMed] [Google Scholar]
- Dhamodaran K, Baidouri H, Sandoval L, Raghunathan V, 2020. Wnt Activation After Inhibition Restores Trabecular Meshwork Cells Toward a Normal Phenotype. Invest Ophthalmol Vis Sci 61, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JC, 2015. IOP telemetry in the nonhuman primate. Exp Eye Res 141, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethier CR, 2002. The inner wall of Schlemm’s canal. Exp Eye Res 74, 161–172. [DOI] [PubMed] [Google Scholar]
- Ethier CR, Chan D-H, 2001. Cationic ferritin changes outflow facility in human eyes whereas anionic ferritin does not. Invest Ophthalmol Vis Sci 42, 1795–1802. [PubMed] [Google Scholar]
- Ethier CR, Coloma FM, Sit AJ, Johnson M, 1998. Two pore types in the inner-wall endothelium of Schlemm’s canal. Invest Ophthalmol Vis Sci 39, 2041–2048. [PubMed] [Google Scholar]
- Ethier CR, Kamm RD, Palaszewski BA, Johnson MC, Richardson TM, 1986. Calculations of flow resistance in the juxtacanalicular meshwork. Investigative Ophthalmology & Visual Science 27, 1741–1750. [PubMed] [Google Scholar]
- Filla MS, Schwinn MK, Nosie AK, Clark RW, Peters DM, 2011. Dexamethasone-associated cross-linked actin network formation in human trabecular meshwork cells involves beta3 integrin signaling. Invest Ophthalmol Vis Sci 52, 2952–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois J, 1975. The importance of the mucopolysaccharides in intraocular pressure regulation. Investigative Ophthalmology 14, 173–176. [PubMed] [Google Scholar]
- Frantz C, Stewart KM, Weaver VM, 2010. The extracellular matrix at a glance. J Cell Sci 123, 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DS, Wolfs RC, O’Colmain BJ, Klein BE, Taylor HR, West S, Leske MC, Mitchell P, Congdon N, Kempen J, Eye Diseases Prevalence Research, G., 2004. Prevalence of open-angle glaucoma among adults in the United States. Arch Ophthalmol 122, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchshofer R, Welge-Lussen U, Lutjen-Drecoll E, Birke M, 2006. Biochemical and morphological analysis of basement membrane component expression in corneoscleral and cribriform human trabecular meshwork cells. Invest Ophthalmol Vis Sci 47, 794–801. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Inoue T, Inoue-Mochita M, Tanihara H, 2016. Live cell imaging of actin dynamics in dexamethasone-treated porcine trabecular meshwork cells. Exp Eye Res 145, 393–400. [DOI] [PubMed] [Google Scholar]
- Gard TL, Van Buskirk EM, Acott TS, 1993. Ionic modulation of flow resistance in an immobilized proteoglycan model of the trabecular meshwork. J Glaucoma 2, 183–192. [PubMed] [Google Scholar]
- Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, Janmey PA, Furth EE, Wells RG, 2007. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol 293, G1147–1154. [DOI] [PubMed] [Google Scholar]
- Gong H, Ruberti J, Overby D, Johnson M, Freddo TF, 2002. A new view of the human trabecular meshwork using quick-freeze, deep-etch electron microscopy. Exp Eye Res 75, 347–358. [PubMed] [Google Scholar]
- Gottanka J, Johnson DH, Martus P, Lutjen-Drecoll E, 2001. Beta-adrenertic blocker therapy and the trabecular meshwork. Graefes Arch Clin Exp Ophthalmol 239, 138–144. [DOI] [PubMed] [Google Scholar]
- Grant WM, 1956. Facility of flow through the trabecular meshwork. Arch Ophthalmol 54, 245–248. [DOI] [PubMed] [Google Scholar]
- Grant WM, 1958. Further studies on facility of flow through the trabecular meshwork. Archives Ophthalmology 60, 523–533. [DOI] [PubMed] [Google Scholar]
- Greenberg CS, Birckbichler PJ, Rice RH, 1991. Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. FASEB J 5, 3071–3077. [DOI] [PubMed] [Google Scholar]
- Grierson I, Howes RC, 1987. Age-related depletion of the cell population in the human trabecular meshwork. Eye 1 (Pt 2), 204–210. [DOI] [PubMed] [Google Scholar]
- Grierson I, Lee WR, 1974. Junctions between the cells of the trabecular meshwork. Albrecht Von Graefes Arch Klin Exp Ophthalmol 192, 89–104. [DOI] [PubMed] [Google Scholar]
- Grierson I, Lee WR, Abraham S, Howes RC, 1978. Associations between the cells of the walls of Schlemm’s canal. Albrecht Von Graefes Arch Klin Exp Ophthalmol 208, 33–47. [DOI] [PubMed] [Google Scholar]
- Gu X, Reagan AM, McClellan ME, Elliott MH, 2017. Caveolins and caveolae in ocular physiology and pathophysiology. Prog Retin Eye Res 56, 84–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann CR, Bahler CK, Johnson DH, 2005. Cationic ferritin and segmental flow through the trabecular meshwork. Invest Ophthalmol Vis Sci 46, 1–7. [DOI] [PubMed] [Google Scholar]
- Hann CR, Fautsch MP, 2009. Preferential fluid flow in the human trabecular meshwork near collector channels. Invest Ophthalmol Vis Sci 50, 1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann CR, Springett MJ, Wang X, Johnson DH, 2001. Ultrastructural localization of collagen IV, fibronectin, and laminin in the trabecular meshwork of normal and glaucomatous eyes. Ophthalmic Res 33, 314–324. [DOI] [PubMed] [Google Scholar]
- Hoare MJ, Grierson I, Brotchie D, Pollock N, Cracknell K, Clark AF, 2009. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Invest Ophthalmol Vis Sci 50, 1255–1263. [DOI] [PubMed] [Google Scholar]
- Hogan MJ, Alvarado JA, Weddell JE, 1971. Histology of the human eye. An atlas and textbook. W. B. Saunders Company, Philadelphia. [Google Scholar]
- Hosseini M, Rose AY, Song K, Bohan C, Alexander JP, Kelley MJ, Acott TS, 2006. IL-1 and TNF induction of matrix metalloproteinase-3 by c-Jun N-terminal kinase in trabecular meshwork. Invest Ophthalmol Vis Sci 47, 1469–1476. [DOI] [PubMed] [Google Scholar]
- Huang J, Camras LJ, Yuan F, 2015. Mechanical analysis of rat trabecular meshwork. Soft Matter 11, 2857–2865. [DOI] [PubMed] [Google Scholar]
- Hubbard W, Johnson M, Gong H, Gabelt B, Peterson J, Sawhney R, Freddo T, Kaufman P, 1997. Intraocular pressure and outflow facility are unchanged following acute and chronic intracameral chondroitinase ABC and hyaluronidase in monkeys. Exp Eye Res 65, 177–190. [DOI] [PubMed] [Google Scholar]
- Jain A, Zode G, Kasetti RB, Ran FA, Yan W, Sharma TP, Bugge K, Searby CC, Fingert JH, Zhang F, Clark AF, Sheffield VC, 2017. CRISPR-Cas9-based treatment of myocilin-associated glaucoma. Proc Natl Acad Sci U S A 114, 11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DH, Richardson TM, Epstein DL, 1989. Trabecular meshwork recovery after phagocytic challenge. Curr Eye Res 8, 1121–1130. [DOI] [PubMed] [Google Scholar]
- Johnson M, 2006. ‘What controls aqueous humour outflow resistance?’. Exp Eye Res 82, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Erickson K, 2000. Mechanisms and routes of aqueous humor drainage., in: Albert D, Jakobiec F (Eds.), Principles and Practice of Ophthalmology, 2nd ed. W.B. Saunders, Philadelphia, pp. 2577–2595. [Google Scholar]
- Johnson M, Johnson D, Kamm RD, de Kater A, Epstein D, 1990. The filtration characteristics of the aqueous outflow system. Exp Eye Res 50, 407–418. [DOI] [PubMed] [Google Scholar]
- Johnson M, Schuman JS, Kagemann L, 2015. Trabecular meshwork stiffness in the living human eye, ARVO, p. 3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MC, Kamm RD, 1983. The role of Schlemm’s canal in aqueous outflow from the human eye. Investigative Ophthalmology Visual Science 24, 320–325. [PubMed] [Google Scholar]
- Johnstone MA, 2004. The aqueous outflow system as a mechanical pump: evidence from examination of tissue and aqueous movement in human and non-human primates. J Glaucoma 13, 421–438. [DOI] [PubMed] [Google Scholar]
- Kagan HM, Li W, 2003. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem 88, 660–672. [DOI] [PubMed] [Google Scholar]
- Kaufmann C, Bachmann LM, Robert YC, Thiel MA, 2006. Ocular pulse amplitude in healthy subjects as measured by dynamic contour tonometry. Arch Ophthalmol 124, 1104–1108. [DOI] [PubMed] [Google Scholar]
- Keller K, Aga M, Bradley J, Kelley M, Acott T, 2009a. Extracellular matrix turnover and outflow resistance. Exp Eye Res 88, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller K, Bradley JM, Acott TS, 2009b. Differential effects of ADAMTSs -1, -4, and -5 in the Trabecular Meshwork. Invest Ophthalmol Vis Sci 50, 5769–5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Acott TS, 2013. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. Journal of ocular biology 1, 3. [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Bradley JM, Kelley MJ, Acott TS, 2008. Effects of modifiers of glycosaminoglycan biosynthesis on outflow facillity in perfusion culture. Invest Ophthalmol Vis Sci 49, 2495–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Bradley JM, Sun YY, Yang YF, Acott TS, 2017. Tunneling Nanotubes are Novel Cellular Structures That Communicate Signals Between Trabecular Meshwork Cells. Invest Ophthalmol Vis Sci 58, 5298–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Bradley JM, Vranka JA, Acott TS, 2011. Segmental versican expression in the trabecular meshwork and involvement in outflow facility. Invest Ophthalmol Vis Sci 52, 5049–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Kelley MJ, Acott TS, 2007. Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci 48, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Kelley MJ, Rose A, Song K, Lystrup B, Samples JW, Acott TS, 2007a. p38 MAP Kinase Pathway and Stromelysin Regulation in Trabecular Meshwork Cells. Invest Ophthalmol Vis Sci 48, 3126–3137. [DOI] [PubMed] [Google Scholar]
- Kelley MJ, Rose AY, Song K, Chen Y, Bradley JM, Rookhuizen D, Acott TS, 2007b. Synergism of TNF and IL-1 in the induction of matrix metalloproteinase-3 in trabecular meshwork. Invest Ophthalmol Vis Sci 48, 2634–2643. [DOI] [PubMed] [Google Scholar]
- Khawaja AP, Cooke Bailey JN, Wareham NJ, Scott RA, Simcoe M, Igo RP Jr., Song YE, Wojciechowski R, Cheng CY, Khaw PT, Pasquale LR, Haines JL, Foster PJ, Wiggs JL, Hammond CJ, Hysi PG, Eye UKB, Vision C, Consortium N, 2018. Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nature genetics 50, 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T, Sato H, Okada A, Ohuchi E, Imai K, Okada Y, Seiki M, 1998. TIMP-2 promotes activation of progelatinase A by membrane-type 1 matrix metalloproteinase immobilized on agarose beads. J Biol Chem 273, 16098–16103. [DOI] [PubMed] [Google Scholar]
- Kischel P, Waltregny D, Dumont B, Turtoi A, Greffe Y, Kirsch S, De Pauw E, Castronovo V, 2010. Versican overexpression in human breast cancer lesions: known and new isoforms for stromal tumor targeting. Int J Cancer 126, 640–650. [DOI] [PubMed] [Google Scholar]
- Kizhatil K, Ryan M, Marchant JK, Henrich S, John SW, 2014. Schlemm’s canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process. PLoS biology 12, e1001912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper PA, Farbman AI, Telser AG, 1984. Exogenous hyaluronidases and degradation of hyaluronic acid in the rabbit eye. Investigative Ophthalmology & Visual Science 25, 286–293. [PubMed] [Google Scholar]
- Kuchtey J, Kunkel J, Esson D, Sapienza JS, Ward DA, Plummer CE, Gelatt KN, Kuchtey RW, 2013. Screening ADAMTS10 in dog populations supports Gly661Arg as the glaucoma-causing variant in beagles. Invest Ophthalmol Vis Sci 54, 1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchtey J, Olson LM, Rinkoski T, Mackay EO, Iverson TM, Gelatt KN, Haines JL, Kuchtey RW, 2011. Mapping of the disease locus and identification of ADAMTS10 as a candidate gene in a canine model of primary open angle glaucoma. PLoS genetics 7, e1001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Su Y, Huang D, Gong H, 2017. The role of cellular connections in Schlemm’s canal endothelial cells in regulating segmental aqueous outflow., Assoc Res Vis Ophthalmol, pp. 1077–B0365. [Google Scholar]
- Lai J, Su Y, Swain DL, Huang D, Getchevski D, Gong H, 2019. The Role of Schlemm’s Canal Endothelium Cellular Connectivity in Giant Vacuole Formation: A 3D Electron Microscopy Study. Invest Ophthalmol Vis Sci 60, 1630–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Last JA, Pan T, Ding Y, Reilly CM, Keller K, Acott TS, Fautsch MP, Murphy CJ, Russell P, 2011. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci 52, 2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM, 2009. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Farsiu S, Chiu SJ, Gonzalez P, Lutjen-Drecoll E, Overby DR, Stamer WD, 2014a. Pilocarpine-induced dilation of Schlemm’s canal and prevention of lumen collapse at elevated intraocular pressures in living mice visualized by OCT. Invest Ophthalmol Vis Sci 55, 3737–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Farsiu S, Qiu J, Dixon A, Song C, McKinnon SJ, Yuan F, Gonzalez P, Stamer WD, 2014b. Disease progression in iridocorneal angle tissues of BMP2-induced ocular hypertensive mice with optical coherence tomography. Mol Vis 20, 1695–1709. [PMC free article] [PubMed] [Google Scholar]
- Li G, Lee C, Agrahari V, Wang K, Navarro I, Sherwood JM, Crews K, Farsiu S, Gonzalez P, Lin CW, Mitra AK, Ethier CR, Stamer WD, 2019. In vivo measurement of trabecular meshwork stiffness in a corticosteroid-induced ocular hypertensive mouse model. Proc Natl Acad Sci U S A 116, 1714–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Shen TT, Johnstone M, Wang RK, 2013. Pulsatile motion of the trabecular meshwork in healthy human subjects quantified by phase-sensitive optical coherence tomography. Biomed Opt Express 4, 2051–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Kopp P, 2005. Podosomes at a glance. J Cell Sci 118, 2079–2082. [DOI] [PubMed] [Google Scholar]
- Lu Z, Overby DR, Scott PA, Freddo TF, Gong H, 2008. The mechanism of increasing outflow facility by rho-kinase inhibition with Y-27632 in bovine eyes. Exp Eye Res 86, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusthaus J, Goldberg I, 2019. Current management of glaucoma. Med J Aust 210, 180–187. [DOI] [PubMed] [Google Scholar]
- Lutjen-Drecoll E, 1999. Functional morphology of the trabecular meshwork in primate eyes. Prog Retin Eye Res 18, 91–119. [DOI] [PubMed] [Google Scholar]
- Lutjen-Drecoll E, 2005. Morphological changes in glaucomatous eyes and the role of TGFbeta2 for the pathogenesis of the disease. Exp Eye Res 81, 1–4. [DOI] [PubMed] [Google Scholar]
- Maatta M, Tervahartiala T, Harju M, Airaksinen J, Autio-Harmainen H, Sorsa T, 2005. Matrix metalloproteinases and their tissue inhibitors in aqueous humor of patients with primary open-angle glaucoma, exfoliation syndrome, and exfoliation glaucoma. J Glaucoma 14, 64–69. [DOI] [PubMed] [Google Scholar]
- Maepea O, Bill A, 1992. Pressures in the juxtacanalicular tissue and Schlemm’s canal in monkeys. Experimental Eye Research 54, 879–883. [DOI] [PubMed] [Google Scholar]
- Mao W, Millar JC, Wang WH, Silverman SM, Liu Y, Wordinger RJ, Rubin JS, Pang IH, Clark AF, 2012a. Existence of the canonical Wnt signaling pathway in the human trabecular meshwork. Invest Ophthalmol Vis Sci 53, 7043–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao W, Rubin JS, Anoruo N, Wordinger RJ, Clark AF, 2012b. SFRP1 promoter methylation and expression in human trabecular meshwork cells. Exp Eye Res 97, 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen WK, 1958. Application of Poiseuille’s law to aqueous outflow. Archives of Ophthalmology 60, 290–294. [DOI] [PubMed] [Google Scholar]
- McKee CT, Wood JA, Shah NM, Fischer ME, Reilly CM, Murphy CJ, Russell P, 2011. The effect of biophysical attributes of the ocular trabecular meshwork associated with glaucoma on the cell response to therapeutic agents. Biomaterials 32, 2417–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead TJ, Apte SS, 2018. ADAMTS proteins in human disorders. Matrix Biol 71-72, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JT, Raghunathan VK, Chang YR, Murphy CJ, Russell P, 2015a. The intrinsic stiffness of human trabecular meshwork cells increases with senescence. Oncotarget 6, 15362–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JT, Raghunathan VK, Chang YR, Murphy CJ, Russell P, 2015b. Wnt inhibition induces persistent increases in intrinsic stiffness of human trabecular meshwork cells. Exp Eye Res 132, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgelin M, Paulsson M, Malmstrom A, Heinegard D, 1989. Shared and distinct structural features of interstitial proteoglycans from different bovine tissues revealed by electron microscopy. J Biol Chem 264, 12080–12090. [PubMed] [Google Scholar]
- Muir KW, Lee PP, 2011. Glaucoma medication adherence: room for improvement in both performance and measurement. Arch Ophthalmol 129, 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan J, Crosbie DE, Cassidy PS, Sherwood JM, Flugel-Koch C, Lutjen-Drecoll E, Humphries MM, Reina-Torres E, Wallace D, Kiang AS, Campbell M, Stamer WD, Overby DR, O’Brien C, Tam LCS, Humphries P, 2017. Therapeutic potential of AAV-mediated MMP-3 secretion from corneal endothelium in treating glaucoma. Hum Mol Genet 26, 1230–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly S, Pollock N, Currie L, Paraoan L, Clark AF, Grierson I, 2011. Inducers of cross-linked actin networks in trabecular meshwork cells. Invest Ophthalmol Vis Sci 52, 7316–7324. [DOI] [PubMed] [Google Scholar]
- Overby DR, Bertrand J, Tektas OY, Boussommier-Calleja A, Schicht M, Ethier CR, Woodward DF, Stamer WD, Lutjen-Drecoll E, 2014a. Ultrastructural changes associated with dexamethasone-induced ocular hypertension in mice. Invest Ophthalmol Vis Sci 55, 4922–4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overby DR, Zhou EH, Vargas-Pinto R, Pedrigi RM, Fuchshofer R, Braakman ST, Gupta R, Perkumas KM, Sherwood JM, Vahabikashi A, Dang Q, Kim JH, Ethier CR, Stamer WD, Fredberg JJ, Johnson M, 2014b. Altered mechanobiology of Schlemm’s canal endothelial cells in glaucoma. Proc Natl Acad Sci U S A 111, 13876–13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang IH, Millar JC, Clark AF, 2015. Elevation of intraocular pressure in rodents using viral vectors targeting the trabecular meshwork. Exp Eye Res 141, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant AD, Kagemann L, Schuman JS, Sigal IA, Amini R, 2017. An imaged-based inverse finite element method to determine in-vivo mechanical properties of the human trabecular meshwork. J Model Ophthalmol 1, 100–111. [PMC free article] [PubMed] [Google Scholar]
- Perissinotto D, Iacopetti P, Bellina I, Doliana R, Colombatti A, Pettway Z, Bronner-Fraser M, Shinomura T, Kimata K, Morgelin M, Lofberg J, Perris R, 2000. Avian neural crest cell migration is diversely regulated by the two major hyaluronan-binding proteoglycans PG-M/versican and aggrecan. Development 127, 2823–2842. [DOI] [PubMed] [Google Scholar]
- Quigley HA, 2011. Glaucoma. Lancet 377, 1367–1377. [DOI] [PubMed] [Google Scholar]
- Raghunathan V, Eaton JS, Christian BJ, Morgan JT, Ver Hoeve JN, Yang CC, Gong H, Rasmussen CA, Miller PE, Russell P, Nork TM, Murphy CJ, 2017. Biomechanical, ultrastructural, and electrophysiological characterization of the non-human primate experimental glaucoma model. Scientific reports 7, 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan VK, Benoit J, Kasetti R, Zode G, Salemi M, Phinney BS, Keller KE, Staverosky JA, Murphy CJ, Acott T, Vranka J, 2018. Glaucomatous cell derived matrices differentially modulate non-glaucomatous trabecular meshwork cellular behavior. Acta biomaterialia 71, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan VK, Morgan JT, Dreier B, Reilly CM, Thomasy SM, Wood JA, Ly I, Tuyen BC, Hughbanks M, Murphy CJ, Russell P, 2013. Role of substratum stiffness in modulating genes associated with extracellular matrix and mechanotransducers YAP and TAZ. Invest Ophthalmol Vis Sci 54, 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan VK, Morgan JT, Park SA, Weber D, Phinney BS, Murphy CJ, Russell P, 2015. Dexamethasone Stiffens Trabecular Meshwork, Trabecular Meshwork Cells, and Matrix. Invest Ophthalmol Vis Sci 56, 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohen JW, Lutjen-Drecoll E, Flugel C, Meyer M, Grierson I, 1993. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG). Exp Eye Res 56, 683–692. [DOI] [PubMed] [Google Scholar]
- Russell P, Gasiorowski JZ, Nealy PF, Murphy CJ, 2008. Response of human trabecular meshwork cells to topographic cues on the nanoscale level. Invest Ophthalmol Vis Sci 49, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfarazi M, Child A, Stoilova D, Brice G, Desai T, Trifan OC, Poinoosawmy D, Crick RP, 1998. Localization of the fourth locus (GLC1E) for adult-onset primary open-angle glaucoma to the 10p15-p14 region. Am J Hum Genet 62, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaguchi S, Yue B, Yeh P, Tso M, 1992. Effects of intracameral injection of chondroitinase ABC in vivo. Arch Ophthalmol 110, 110–117. [DOI] [PubMed] [Google Scholar]
- Schlotzer-Schrehardt U, Lommatzsch J, Kuchle M, Konstas AG, Naumann GO, 2003. Matrix metalloproteinases and their inhibitors in aqueous humor of patients with pseudoexfoliation syndrome/glaucoma and primary open-angle glaucoma. Invest Ophthalmol Vis Sci 44, 1117–1125. [DOI] [PubMed] [Google Scholar]
- Schlunck G, Han H, Wecker T, Kampik D, Meyer-ter-Vehn T, Grehn F, 2008. Substrate rigidity modulates cell matrix interactions and protein expression in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 49, 262–269. [DOI] [PubMed] [Google Scholar]
- Schuman JS, Chang W, Wang N, de Kater AW, Allingham RR, 1999. Excimer laser effects on outflow facility and outflow pathway morphology. Invest Ophthalmol Vis Sci 40, 1676–1680. [PubMed] [Google Scholar]
- Sethi A, Jain A, Zode GS, Wordinger RJ, Clark AF, 2011. Role of TGFbeta/Smad signaling in gremlin induction of human trabecular meshwork extracellular matrix proteins. Invest Ophthalmol Vis Sci 52, 5251–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A, Wordinger RJ, Clark AF, 2012. Focus on molecules: lysyl oxidase. Exp Eye Res 104, 97–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowden JC, 2007. Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond) 21, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Spiga MG, Borras T, 2010. Development of a gene therapy virus with a glucocorticoid-inducible MMP1 for the treatment of steroid glaucoma. Invest Ophthalmol Vis Sci 51, 3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack T, Vahabikashi A, Johnson M, Scott E, 2018. Modulation of Schlemm’s canal endothelial cell stiffness via latrunculin loaded block copolymer micelles. Journal of biomedical materials research. Part A 106, 1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer WD, Acott TS, 2012. Current understanding of conventional outflow dysfunction in glaucoma. Curr Opin Ophthalmol 23, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer WD, Braakman ST, Zhou EH, Ethier CR, Fredberg JJ, Overby DR, Johnson M, 2015. Biomechanics of Schlemm’s canal endothelium and intraocular pressure reduction. Prog Retin Eye Res 44, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer WD, Clark AF, 2017. The many faces of the trabecular meshwork cell. Exp Eye Res 158, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilova D, Child A, Trifan OC, Crick RP, Coakes RL, Sarfarazi M, 1996. Localization of a locus (GLC1B) for adult-onset primary open angle glaucoma to the 2cen-q13 region. Genomics 36, 142–150. [DOI] [PubMed] [Google Scholar]
- Stone EM, Fingert JH, Alward WLM, Nguyen TD, Polansky JR, Sunden SLF, Nishimura D, Clark AF, Nystuen A, Hichols BE, Mackey DA, Ritch R, Kalenak JW, Craven ER, Sheffield VC, 1997. Identification of a gene that causes primary open angle glaucoma. Science 275, 668–670. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhu Q, Guo P, Zhang Y, Tighe S, Zhu Y, 2019a. Trabecular meshwork cells are a valuable resource for cellular therapy of glaucoma. J Cell Mol Med 23, 1678–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YC, Li P, Johnstone M, Wang RK, Shen TT, 2015. Pulsatile motion of trabecular meshwork in a patient with iris cyst by phase-sensitive optical coherence tomography: a case report. Quant Imaging Med Surg 5, 171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YY, Yang YF, Keller KE, 2019b. Myosin-X Silencing in the Trabecular Meshwork Suggests a Role for Tunneling Nanotubes in Outflow Regulation. Invest Ophthalmol Vis Sci 60, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan SS, Oh DJ, Kang MH, Rhee DJ, 2014. Aqueous outflow: segmental and distal flow. Journal of cataract and refractive surgery 40, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm ER, 2009. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res 88, 648–655. [DOI] [PubMed] [Google Scholar]
- Thomasy SM, Wood JA, Kass PH, Murphy CJ, Russell P, 2012. Substratum stiffness and latrunculin B regulate matrix gene and protein expression in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 53, 952–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T, 2017a. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J Neurochem 140, 741–754. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, Routhe LJ, Moos T, 2017b. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 37, 3300–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar-Vidales T, Clark AF, Wordinger RJ, 2011. Focus on molecules: transglutaminase 2. Exp Eye Res 93, 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar-Vidales T, Roque R, Clark AF, Wordinger RJ, 2008. Tissue transglutaminase expression and activity in normal and glaucomatous human trabecular meshwork cells and tissues. Invest Ophthalmol Vis Sci 49, 622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong TN, Li H, Hong YK, Chen L, 2014. Novel characterization and live imaging of Schlemm’s canal expressing Prox-1. PloS one 9, e98245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahabikashi A, Dong B, Ahang HF, Johnson M, 2017. In-sity stiffness characterization of the human inner wall endothelium of Schlemm’s canal and its substrate, ISER/BrightFocus 2017 Glaucoma Symposium, Atlanga, GA, p. 141. [Google Scholar]
- Vahabikashi A, Gelman A, Dong B, Gong L, Cha EDK, Schimmel M, Tamm ER, Perkumas K, Stamer WD, Sun C, Zhang HF, Gong H, Johnson M, 2019a. Increased stiffness and flow resistance of the inner wall of Schlemm’s canal in glaucomatous human eyes. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahabikashi A, Park CY, Perkumas K, Zhang Z, Deurloo EK, Wu H, Weitz DA, Stamer WD, Goldman RD, Fredberg JJ, Johnson M, 2019b. Probe Sensitivity to Cortical versus Intracellular Cytoskeletal Network Stiffness. Biophys J 116, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buskirk EM, Brett J, 1978. The canine eye: in vitro dissolution of the barriers to aqueous outflow. Invest Ophthalmol Vis Sci 17, 258–271. [PubMed] [Google Scholar]
- Van Buskirk EM, Cioffi GA, 1992. Glaucomatous optic neuropathy. Am J Ophthalmol 113, 447–452. [DOI] [PubMed] [Google Scholar]
- Vargas-Pinto R, Gong H, Vahabikashi A, Johnson M, 2013. The effect of the endothelial cell cortex on atomic force microscopy measurements. Biophys J 105, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SS, Cooke Bailey JN, Lucas A, Bradford Y, Linneman JG, Hauser MA, Pasquale LR, Peissig PL, Brilliant MH, McCarty CA, Haines JL, Wiggs JL, Vrabec TR, Tromp G, Ritchie MD, e, M.N., Consortium, N., 2016. Epistatic Gene-Based Interaction Analyses for Glaucoma in eMERGE and NEIGHBOR Consortium. PLoS genetics 12, e1006186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS, 2005. Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci 46, 2857–2868. [DOI] [PubMed] [Google Scholar]
- Vranka J, Yang YF, Sun YY, Staverosky J, Acott T, Keller K, 2017. Expression of alternative versican isoforms in human trabecular meshwork cells and tissues., ISER/BrightFocus 2017 Glaucoma Symposium, Atlanta, GA, p. #57. [Google Scholar]
- Vranka JA, Acott TS, 2017. Pressure-induced expression changes in segmental flow regions of the human trabecular meshwork. Exp Eye Res 158, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranka JA, Bradley JM, Yang YF, Keller KE, Acott TS, 2015. Mapping molecular differences and extracellular matrix gene expression in segmental outflow pathways of the human ocular trabecular meshwork. PloS one 10, e0122483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranka JA, Staverosky JA, Raghunathan V, Acott TS, 2020. Elevated pressure influences relative distribution of segmental regions of the trabecular meshwork. Exp Eye Res 190, 107888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranka JA, Staverosky JA, Reddy AP, Wilmarth PA, David LL, Acott TS, Russell P, Raghunathan VK, 2018. Biomechanical Rigidity and Quantitative Proteomics Analysis of Segmental Regions of the Trabecular Meshwork at Physiologic and Elevated Pressures. Invest Ophthalmol Vis Sci 59, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Johnstone MA, Xin C, Song S, Padilla S, Vranka JA, Acott TS, Zhou K, Schwaner SA, Wang RK, Sulchek T, Ethier CR, 2017. Estimating Human Trabecular Meshwork Stiffness by Numerical Modeling and Advanced OCT Imaging. Invest Ophthalmol Vis Sci 58, 4809–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li G, Read AT, Navarro I, Mitra AK, Stamer WD, Sulchek T, Ethier CR, 2018. The relationship between outflow resistance and trabecular meshwork stiffness in mice. Scientific reports 8, 5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WH, McNatt LG, Pang IH, Millar JC, Hellberg PE, Hellberg MH, Steely HT, Rubin JS, Fingert JH, Sheffield VC, Stone EM, Clark AF, 2008. Increased expression of the WNT antagonist sFRP-1 in glaucoma elevates intraocular pressure. J Clin Invest 118, 1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Senju S, Toyoshima H, Yoshida M, 1997. Thickness of the basement membrane of bronchial epithelial cells in lung diseases as determined by transbronchial biopsy. Respir Med 91, 406–410. [DOI] [PubMed] [Google Scholar]
- Webber HC, Bermudez JY, Millar JC, Mao W, Clark AF, 2018. The Role of Wnt/beta-Catenin Signaling and K-Cadherin in the Regulation of Intraocular Pressure. Invest Ophthalmol Vis Sci 59, 1454–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber HC, Bermudez JY, Sethi A, Clark AF, Mao W, 2016. Crosstalk between TGFbeta and Wnt signaling pathways in the human trabecular meshwork. Exp Eye Res 148, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL, Martin KR, 2016. Primary open-angle glaucoma. Nat Rev Dis Primers 2, 16067. [DOI] [PubMed] [Google Scholar]
- Welge-Lussen U, May CA, Lutjen-Drecoll E, 2000. Induction of tissue transglutaminase in the trabecular meshwork by TGF-beta1 and TGF-beta2. Invest Ophthalmol Vis Sci 41, 2229–2238. [PubMed] [Google Scholar]
- Wight TN, 2002. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol 14, 617–623. [DOI] [PubMed] [Google Scholar]
- Wight TN, 2017. Provisional matrix: A role for versican and hyaluronan. Matrix Biol 60-61, 38–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight TN, Heinegard DK, Hascall VC, 1991. Proteoglycans. Structure and function, in: Hay ED (Ed.), Cell Biology of Extracellular Matrix, 2 ed. Plenum Press, New York, pp. 45–78. [Google Scholar]
- Wight TN, Mecham RP, 1987. Biology of Proteoglycans, 1st ed. Academic Press, New York. [Google Scholar]
- Wirtz MK, Bradley JM, Xu H, Domreis J, Nobis CA, Truesdale AT, Samples JR, Van Buskirk EM, Acott TS, 1997a. Proteoglycan expression by human trabecular meshworks. Curr Eye Res 16, 412–421. [DOI] [PubMed] [Google Scholar]
- Wirtz MK, Samples JR, Kramer PL, Rust K, Topinka JR, Yount J, Koler RD, Acott TS, 1997b. Mapping a gene for adult-onset primary open-angle glaucoma to chromosome 3q. Am J Human Genet 60, 296–304. [PMC free article] [PubMed] [Google Scholar]
- Wirtz MK, Samples JR, Rust K, Lie J, Nordling L, Schilling K, Acott TS, Kramer PL, 1999. GLC1F, a new primary open-angle glaucoma locus, maps to 7q35-q36. Arch Ophthalmol 117, 237–241. [DOI] [PubMed] [Google Scholar]
- Wood JA, McKee CT, Thomasy SM, Fischer ME, Shah NM, Murphy CJ, Russell P, 2011. Substratum compliance regulates human trabecular meshwork cell behaviors and response to latrunculin B. Invest Ophthalmol Vis Sci 52, 9298–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wordinger RJ, Clark AF, 2014. Lysyl oxidases in the trabecular meshwork. J Glaucoma 23, S55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wordinger RJ, Sharma T, Clark AF, 2014. The role of TGF-beta2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics 30, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin C, Wang RK, Song S, Shen T, Wen J, Martin E, Jiang Y, Padilla S, Johnstone M, 2017. Aqueous outflow regulation: Optical coherence tomography implicates pressure-dependent tissue motion. Exp Eye Res 158, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YF, Sun YY, Acott TS, Keller KE, 2016. Effects of induction and inhibition of matrix crosslinking on remodeling of the aqueous outflow resistance by ocular trabecular meshwork cells. Scientific reports 6, 30505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood H, Hauser MA, Liu Y, 2019. Update on the genetics of primary open-angle glaucoma. Exp Eye Res 188, 107795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Camras LJ, Gonzalez P, 2011. Trabecular meshwork stiffness in ex vivo perfused porcine eyes, ARVO, p. 6693. [Google Scholar]
- Yurchenco PD, Patton BL, 2009. Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des 15, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ognibene CM, Clark AF, Yorio T, 2007. Dexamethasone inhibition of trabecular meshwork cell phagocytosis and its modulation by glucocorticoid receptor beta. Exp Eye Res 84, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Russell P, 2005. Versican splice variants in human trabecular meshwork and ciliary muscle. Mol Vis 11, 603–608. [PubMed] [Google Scholar]
- Zimmerman GA, Lorant DE, McIntyre TM, Prescott SM, 1993. Juxtacrine intercellular signaling: another way to do it. Am J Respir Cell Mol Biol 9, 573–577. [DOI] [PubMed] [Google Scholar]