

Abstract
Osteogenic differentiation, the process by which bone marrow mesenchymal stem/stromal (a.k.a. skeletal stem) cells and osteoprogenitors form osteoblasts, is a critical event for bone formation during development, fracture repair, and tissue maintenance. Extra cellular and intracellular signaling pathways triggering osteogenic differentiation are relatively well known; however, the ensuing change in cell energy metabolism is less clearly defined. We and others have previously reported activation of mitochondria during osteogenic differentiation. To further elucidate the involved bioenergetic mechanisms and triggers, we tested the effect of osteogenic media containing ascorbate and β-glycerol phosphate, or various osteogenic hormones and growth factors on energy metabolism in long bone (ST2)- and calvarial bone (MC3T3-E1)-derived osteoprogenitors. We show that osteogenic media and differentiation factors, Wnt3a and BMP2, stimulate mitochondrial oxidative phosphorylation (OxPhos) with little effect on glycolysis. The activation of OxPhos occurs acutely, suggesting a metabolic signaling change rather than protein expression change. To this end, we found that the observed mitochondrial activation is Akt dependent. Akt is activated by osteogenic media, Wnt3a, and BMP2, leading to increased phosphorylation of various mitochondrial Akt targets, a phenomenon known to stimulate OxPhos. In sum, our data provide comprehensive analysis of cellular bioenergetics during osteoinduction in cells of two different origins (mesenchyme vs neural crest) and identify Wnt3a and BMP2 as physiological stimulators of mitochondrial respiration through Akt activation.
Keywords: mitochondria, osteoblast, differentiation, Akt, Wnt, BMP2
Introduction
Mitochondria are dynamic organelles central to cellular energy production, metabolite synthesis, ion homeostasis, reactive oxygen species generation and scavenging, epigenetic signaling, and apoptotic signaling in nearly every cell type [1–3]. The current understanding of stem cell bioenergetics holds that following differentiation, cell reliance on oxidative phosphorylation (OxPhos) increases over glycolysis. Indeed, increasing the activity of glycolytic pathways is a common mechanism to induce stemness in cells in vivo and in induced pluripotent stem cells that revert to glycolysis to become more stem like. Therefore, the importance of mitochondria in cell types classically considered to be glycolytic such as those of stem cell niches is a nascent field of research [4]. The most obvious hypothesis for this metabolic transition is the ∼16-fold increased energetic efficiency of OxPhos compared to glycolysis in ATP synthesis allowing for heightened rates of cellular activity to meet the demands of differentiation. Alternatively, mitochondria are central hubs of cellular second messengers and epigenetic signals [5–7].
Bone marrow mesenchymal stem/stromal cells (BMSCs, a.k.a. skeletal stem cells) are somatic multipotent progenitors capable of differentiation into bone-forming osteoblasts (OBs), cartilage-forming chondrocytes, and fat-forming marrow adipocytes [8]. The heightened energy demand from the OB, compared to the other lineages, is due to high levels of collagen, bone matrix protein, biosynthesis followed by mineral deposition [9].
Recent work by our group and others demonstrate that OxPhos is important in osteogenically differentiating BMSCs and their progeny, OB and osteocytes, as measured by an increase in oxygen consumption [10–12]. Currently, it is unclear what role metabolic plasticity plays in OB differentiation and development. Increases in OxPhos in BMSCs [10,12,13] and calvarial OBs [11] have been reported; however, there remains the unanswered possibility that the observed increase in oxygen consumption is a result of decreased coupling efficiency or increases in non-mitochondrial oxygen consumption. There is also the possibility that long bone and calvarial bone OB metabolism are biologically distinct [14]. Conversely, there are reports that OxPhos is not activated during osteogenesis in long bone-derived cell lines [15], calvarial-derived cell lines [16], and primary BMSCs [17]; however, these studies describe changes in oxygen consumption under experimental conditions, which contain pyruvate, supraphysiological levels of glucose, or both. As pyruvate can feed directly into the mitochondrial citric acid cycle and high glucose is a powerful driver of the Crabtree effect, these conditions could bypass any constraint that specifically affects OxPhos.
The metabolic plasticity of mitochondria allows for the heterogeneous mix of substrates classically used in cell media and mitochondrial buffers: glucose, pyruvate, and glutamine to substitute as carbon entry points in the Krebs cycle [11,18]. Glutamine is converted to α-ketoglutarate to enter the Krebs cycle through the process of glutaminolysis and is used by OBs as an alternate fuel source [19]. Finally, bioenergetic characterization of the specific cause of cellular changes in oxygen consumption by mitochondrial versus non-mitochondrial respiration, respiration directed toward ATP synthesis, and uncoupled respiration [20] is often overlooked in these assays. We have previously demonstrated that under the most physiologically relevant cell culture conditions, OxPhos is indeed activated in BMSCs incubated in osteogenic media composed of 50 μg/mL ascorbate and 2.5 mM β-glycerol phosphate in the presence of low (ie, physiological 5 mM) glucose [12]. We found that this process is not accompanied by major gene expression or mitochondrial mass changes, but is accompanied by changes in mitochondrial dynamics. Concurrent work by the Kowaltowski group [21] reported similar results. The trigger(s) and upstream signaling for the observed mitochondrial changes during osteoinduction remain unclear and are the focus of this work.
In this study, we comprehensively explore the link between mitochondrial activity and osteogenic differentiation of two distinct long bone- or calvarial-derived osteoprogenitor cell lines (ST2 and MC3T3-E1, respectively), applying either commonly used, but artificial osteogenic media components, or various physiologically relevant osteoinduction factors. Our data demonstrate that acute stimulation of mitochondrial oxygen consumption by osteogenic media, Wnt3a, or BMP2 upon osteoinduction is a common feature in both long bone- and calvarial-derived osteoprogenitors. We further show that this mitochondrial activation is completely dependent on activation of Akt. Intriguingly, we observe that both the time course and degree of change in mitochondrial parameters are distinct depending on the osteogenic origin and mechanism of osteoinduction.
Materials and Methods
Cell culture
Mouse long bone-derived ST2 cells were a gift from Dr. Clifford Rosen. Mouse calvarial bone-derived MC3T3-E1 cells were acquired from ATCC. Cells were expanded in sterile conditions and maintained in a 37°C incubator at 5% CO2 in αMEM media (Gibco A10490–01) containing physiologic 1 g/L (ie, 5 mM) glucose, 0.3 g/L (ie, 1 mM) L-glutamine, ribonucleosides (0.01 g/L each), deoxyribonucleosides (0.01 g/L each), no ascorbic acid, 10% FBS (Gibco 10437–028) heat inactivated for 30 min at 55°C, and 0.1% Penn/Strep (Gibco 15140–122). Cells were maintained at passage numbers <20 as recommended by the supplier and from previous handling experience.
Osteoinduction and detection by cell staining
The addition of 50 μg/mL ascorbate (Sigma A4544) and 2.5 mM β-glycerol phosphate (USB Corp Cleveland, OH, 21655), 25 ng/mL BMP2 (R&D systems 355-BM-050/CF), 25 ng/mL Wnt3a (R&D systems 5036-WN-010), 5 ng/mL IGF1 (Sigma 13769–50UG), 0.2 ng/mL TGFβ (R&D systems 240-B), or 1 ng/mL PTH aa1–34 (R&D systems 3011/1) to αMEM-induced osteogenesis. To confirm osteogenesis, cells were stained with OB-specific ALP-specific stain (Thermo NBT/BCIP 1-step 34042) and with 0.5% crystal violet (CV) stain (Sigma C3886) to determine total cell count as previously described [13]. For metabolic inhibitory studies, cells were incubated with 0.1 μg/mL oligomycin (Oligo, Sigma 75351), 0.1 μM antimycin A (AA, Sigma A8674), 0.1 μM rotenone (ROT; Sigma R-8875), or 0.2 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, Sigma C2920) for 48 h before osteoinduction, and inhibitors were present during the entire time course of induction. For Akt signaling studies, cells were incubated with 10 μM Akt1/2 inhibitor (Sigma A6730) for 24 h before osteoinduction (or as indicated for time course studies) and were present during the period of osteoinduction.
Metabolic profiling
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured using Seahorse XF96 (Seahorse Bioscience). Cells were plated on Seahorse 96-well plates 48 h before the experiment at a density of 15,000 cells/well. Cells were incubated in media containing control, OBM, BMP2, or Wnt3a to induce osteogenesis for 1, 6, or 12 h. Immediately before the experiment, medium was replaced with unbuffered DMEM (Sigma D5030) containing 1 mM glutamine (Gibco 25030–081), 5 mM glucose (Sigma G8270), appropriate osteogenic inducer, and no pyruvate (pH 7.4). A baseline measurement of OCR and ECAR was taken, and then an inhibitory analysis was performed using serial injections of 1 μg/mL Oligo and 2 μM FCCP, and simultaneous addition of 1 μM each of AA and ROT. A parallel experiment was run with the following injections of Oligo+FCCP followed by 15 mM 2-deoxyglucose (Sigma D-3179). The following OxPhos and glycolytic indexes were calculated: basal respiration (OCRpre-Oligo − OCRpost-AntA), ATP-linked respiration (OCRpre-Oligo − OCRpost-Oligo), spare respiratory capacity (OCRpost-FCCP − OCRpre-Oligo), proton leak (OCRpost-Oligo − OCRpost-AA/ROT), basal glycolysis (ECARpre-Oligo), and glycolytic flux (ECARpost-Oligo/FCCP – ECAR pre-Oligo/FCCP).
Real-time RT-PCR
Total RNA was isolated using the RNeasy kit (Qiagen 74106) and reverse transcribed into cDNA using the qScript cDNA synthesis kit (Quanta 95048–500). cDNA was subjected to real-time RT-PCR. The primer pairs used for genes of interest are outlined in Table 1. Real-time RT-PCR was performed in the RotorGene system (Qiagen) using SYBR Green (Quanta 95072–012). The expression of genes of interest was normalized to expression of beta-2 microglobulin (B2m).
Table 1.
Primers Used in Real-Time RT-PCR
| Primer | Sequence 5′-3′ | Tm |
|---|---|---|
| Mouse beta-2 microglobulin (B2m) forward | AATGGGAAGCCGAACATAC | 59.9 |
| Mouse beta-2 microglobulin (B2m) reverse | CCATACTGGCATGCTTAACT | 60.1 |
| Mouse bone sialoprotein (Ibsp) forward | AATGGAGACGGCGATAGT | 60.2 |
| Mouse bone sialoprotein (Ibsp) reverse | GAGTGCCGCTAACTCAAA | 59.6 |
| Mouse osteocalcin (Bglap) forward | GACCTCACAGATGCCAAG | 59.5 |
| Mouse osteocalcin (Bglap) reverse | CAAGCCATACTGGTCTGATAG | 59.7 |
Western blotting
Cells were lysed with lysis buffer containing protease inhibitors and subjected to 4–12% SDS-PAGE followed by transfer to polyvinylidene difluoride membranes and blocking in 5% dry milk. For Akt detection, blots were probed with total Akt antibody (Cell Signaling 9272S) at a concentration of 1:2,000. For phosphorylated Akt detection, blots were probed with mouse monoclonal phospho serine 473 Akt (Cell Signaling 4060S) at a concentration of 1:1,000. Akt substrates were detected using p-Akt Substrate RXXS*/T* (Cell Signaling 110B7E) at 1:2,000. Loading controls were detected using antibodies against 1:2,000 Mitofusin 1 (Abcam Ab57602) and 1:20,000 β-actin (Invitrogen Cat-15). Following wash of primary antibody, blots were incubated with either horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Bio-Rad 170–6516) or HRP-conjugated goat anti-rabbit antibody at a concentration of 1:5,000. Antibody detection was developed with West Femto Substrate (Thermo Scientific 34095). Densitometry was performed using Fiji software “gel analysis” plug-in.
Mitochondrial isolation
Cells were grown on 15 cm plates until confluency and treated for 48 h with osteogenic media. Cells were rapidly transferred to cold PBS containing phosphatase inhibitors (cell signaling 5872S) by scraping (all following steps contain phosphatase inhibitors). Cells were spun down at 3,500 rpm and then resuspended in MannPrep buffer (mannitol at 195 mM, sucrose at 65 mM, HEPES at 2 mM, KH2PO4 at 0.05 mM, EGTA at 0.05 mM, and MgCl2 at 0.01 mM; pH 7.4 at 4°C). Cells were broken up by homogenizing them in glass homogenizer with Teflon pestle and 35 passes. Crude precipitate and nuclear material were removed by spin at 3,500 rpm for 5 min at 4°C in benchtop centrifuge. Supernatant was removed and spun at 12,000 rpm for 9 min at 4°C in benchtop centrifuge to pellet mitochondria. Crude mitochondrial pellet was resuspended in 0.02 mL protein lysis buffer containing phosphatase inhibitors. Mitochondrial protein content was assayed using BCA protein assay kit (Pierce 23227). Total protein labeling was performed using No-Stain™ Protein Labeling Reagent (Invitrogen A4449) as per the manufacturer's protocol
Statistical analysis
Mean values and standard deviation were calculated, and the statistical significance (P < 0.05) was established using either Student's t-test when two variables were compared or one-way analysis of variance (ANOVA) with post hoc Bonferroni correction when more than two variables were compared based on normal spread of our data as indicated in the figure legends. Principle component analysis (PCA) and heat map were performed using open access ClustVis software [22,23]. Biologic replicates are used to refer to experiments performed using different frozen stocks of cells, whereas technical replicates are used to refer to experiments repeated using the same cell stock.
Results
Long bone-derived ST2 and calvarial bone-derived MC3T3-E1 osteoprogenitors require mitochondrial activation in osteogenic media
ST2 and MC3T3-E1 are mouse-derived osteoprogenitor cell lines that do not have high alkaline phosphatase (ALP) or mineralization activities in nondifferentiating growth media, but become strongly positive for ALP after 2 days and for mineralization after 7 days of incubation in osteogenic media [24,25]. These changes are concomitant with gene expression for early and late markers of osteogenic differentiation as measured by real-time RT-PCR and ALP staining (Fig. 1A, B). As changes in mitochondrial activity are gaining attention as a hallmark for cellular reprogramming [26], we first asked whether osteogenic differentiation required mitochondrial activity.
FIG. 1.

Bone-derived cell differentiation in osteogenic media activates mitochondria. (A) Top: schematic representation of osteoprogenitor cell differentiation to OBs. ST2 and MC3T3-E1 cells represent osteoprogenitor stage cells of the long bone and calvarial bone, respectively (cell images generated using BioRender software). Bottom: schematic of time course. Cells grown to confluency (white bars) are then treated with osteoblast differentiation media (OBM, black bars) for 0, 2, or 7 days before measurements are taken (red arrows). (B) Expression of mRNA levels of OB markers Ibsp and Bglap relative to control B2m increases with differentiation. Control CV staining and OB marker ALP for same time points are shown below. Seahorse traces from (C) ECAR and (D) OCR for ST2 (triangles) and MC3T3-E1 (squares) cells with and without osteoinduction (empty and filled shapes, respectively). Arrows in Figures (C, D) indicate time point of injection of metabolic inhibitors: (antimycin A: AA, rotenone: ROT, oligomycin: Oligo, and carbonyl cyanide p-trifluoromethoxyphenylhydrazone: FCCP). Data are mean ± SD [n = 5 for qPCR (biologic replicates)], n = 15 Seahorse (three biologic replicates of five technical replicates). Staining images are representative of three biologic replicates. *P < 0.05 versus uninduced control as determined by ANOVA and confirmed by post hoc Bonferroni correction. ALP, alkaline phosphatase; OCR, oxygen consumption rate; ECAR, extracellular acidification rate; CV, crystal violet; OB, osteoblast. Color images are available online.
ST2 and MC3T3-E1 cells were incubated with low doses (1/10th required for complete inhibition) of various metabolic inhibitors (0.1 μg/mL oligomycin (Oligo), 0.1 μM rotenone (ROT), or 0.1 μM antimycin A (AA)) for 48 h in nondifferentiating growth media and then induced to undergo osteogenesis by the addition of ascorbate and β-glycerol phosphate (ie, osteoblast media: OBM) (Supplementary Fig. S1A). Following 48 h of low-dose mitochondrial inhibition in nondifferentiating media, we observed no change in cell quality as assayed by CV stain (Supplementary Fig. S1, “day 0”). However, following 4 days of osteoinduction by OBM, cells pretreated with mitochondrial inhibitors not only showed negative for ALP but also stained negative for crystal violet, whereas control cells showed robust staining (Supplementary Fig. S1, “day 4”). This suggests that mild mitochondrial inhibition of the electron transport chain or of ATP synthesis is not tolerated by cells actively undergoing differentiation along the OB lineage (Supplementary Fig. S1, “+AA, +ROT, +Oligo). This demonstrates the importance for mitochondrial respiratory activity as a common requirement for osteoprogenitors at the point of initiating the osteogenic program.
Osteogenic induction acutely activates mitochondrial bioenergetic parameters
Given that cells showed sensitivity to mitochondrial perturbation only upon osteoinduction, we asked whether mitochondrial activation was an early event during osteogenic differentiation. Moreover, previously published studies analyzed the effect of osteogenic differentiation on mitochondria only at conventional time points postosteoinduction (ie, 7 and 14 days); thus, the immediate bioenergetic responses remain unclear [12,27]. We therefore used the XF96 Seahorse Flux Bioanalyzer to measure ST2 and MC3T3-E1 cell OCR and ECAR under standard and osteogenic conditions following 1, 6, or 12 h of induction. This method allows for real-time monitoring of cellular OCR as a secondary measure of mitochondrial respiration and ECAR as a secondary measure of glycolysis. The serial incorporation of specific metabolic inhibitors to block mitochondrial ATP production (Oligo), uncouple mitochondrial respiration (FCCP), inhibit the election transport chain (Antimycin A+Rotenone, AA/Rot), or to inhibit glycolysis (hexokinase competitive inhibitor 2-Deoxyglucose, 2DG) allows for the calculation of aerobic and anaerobic contributions to cellular metabolism (see Methods section, Fig. 2A, B).
FIG. 2.

Bioenergetics in osteoprogenitor ST2 and MC3T3-E1 cell lines are acutely responsive to osteoinduction. (A) Specific metabolic contributions to OCR in ST2 cells were calculated as basal OCR (baseline minus AA/Rot), ATP production (baseline minus Oligo), respiratory capacity (FCCP minus baseline), proton leak (Oligo minus AA/Rot). Specific metabolic contributions to ECAR calculated as basal ECAR (baseline minus 2-DG) and glycolytic reserve (Oligo/FCCP minus baseline). (B) Specific metabolic contributions to OCR in MCT3T-E1 cells were calculated as in (A). (C, D) Energy map showing baseline (B: black outlined) and maximal (U: red outlined) OCR and ECAR following OBM addition as fold change over uninduced ST2 (C) or MC3T3-E1 (D) cells. The four quadrants represent cell metabolism attributable to “Quiescent” (lower left), “Aerobic” (upper left), “Energetic” (upper right) or “Glycolytic” (lower right) energy usage. Blue- and orange- colored ellipses represent the metabolic space covered by uninduced and induced cells, respectively. Inset shows the slope of each line generated from the xy-scatter plot. Slopes >1 indicate metabolic shifts toward aerobic respiration and slopes <1 indicated metabolic shifts toward glycolysis. Data are mean ± SD [n = 15 (three biologic replicates of five technical replicates)]. *P < 0.05 versus uninduced control as determined by ANOVA and confirmed by post hoc Bonferroni correction. Color images are available online.
As previously reported for BMSCs, undifferentiated ST2 and MC3T3-E1 osteoprogenitor cell lines relied marginally on both OxPhos and anaerobic metabolism (Fig. 1C, D white shapes) [12]. However, cellular respiration of both cell types was significantly and acutely increased upon addition of osteogenic media, in as little as 1 h (data not shown), and was sufficient to maximally stimulate mitochondrial OCR in ST2 and MC3T3-E1 cells after 12 h (Fig. 1D). This is in contrast to ECAR, which was not uniformly increased in both cell types and only reached significance in ST2 cells at baseline, and one measurement postinjection of Oligo+FCCP. Importantly, there was no change observed in non-mitochondrial OCR, indicating that changes in OCR are mitochondrial in origin.
Osteoinduction differentially stimulates OCR and ECAR in ST2 and MC3T3-E1 cells
Analysis of changes in cellular OCR and ECAR following the addition of metabolic inhibitors allows for the calculation of specific mitochondrial parameters (basal OCR, ATP-linked OCR, respiratory capacity, and proton leak), and of glycolytic parameters (basal glycolysis and glycolytic reserve) as described in the Methods above. The induction of osteogenic differentiation in ST2 and MC3T3-E1 cells resulted in a small, but significant increase in mitochondrial engagement as measured by basal respiration without significantly increasing basal glycolytic rates (Fig. 2A, B). ST2 cells rapidly induced doubling in both ATP-linked respiration and mitochondrial respiratory capacity and moderate, but significant increase in glycolytic capacity (Fig. 2A). These data indicate a heightened flux of metabolic processes providing substrates for glycolysis and the mitochondrial electron transport chain in osteoinduced cells.
Interestingly, the kinetics of osteoinduction in ST2 cells was markedly faster than those of MC3T3-E1 cells with majority of the increase in OCR present after 1 h of induction (data not shown). MC3T3-E1 cells showed progressive increases in OCR parameters with the largest increase occurring after 12 h. While ST2 cells demonstrated a more rapid response to OBM, MC3T3-E1 cells' maximal response was larger (Fig. 2B). Basal and maximal rates for both OCR and ECAR can be plotted against one another to establish a cell's “energy map.” This representation allows for comparison of shifts in metabolic programs with increased aerobic respiration appearing as a positive shift along the y-axis and increased anaerobic respiration appearing as a positive shift along the x-axis. Positive shifts along both axes represent a heightened energetic state, whereas negative shift along either axis indicates a quiescent state. Both ST2 and MC3T3-E1 cells show that osteoinduction rapidly activated an energetic phenotype (Fig. 2C, D). Measuring the slope of a line drawn between the basal and maximal rates shows a slope greater than 1 for all osteoinduced cells, indicating a bias toward aerobic respiration as the main driver of the heightened energetic state (Fig. 2C, D inset). Taken together, these data demonstrate that these osteoprogenitor cells acutely respond to osteoinduction by increasing mitochondrial bioenergetics. In addition, these data suggest that osteoprogenitors from different lineages may have distinct metabolic profiles following osteoinduction.
Biologically relevant osteoinducers, Wnt3a and BMP2, acutely activate mitochondrial bioenergetic parameters
Incubation of cells with ascorbic acid and β-glycerol phosphate is an accepted in vitro method of osteoinduction; however, it is not representative of physiologic induction. We therefore asked whether any cytokine or hormone known to promote osteogenic differentiation reproduced the acute response of OBM described in Fig. 2. We observed that incubation of cells with IGF1, TGFβ, or PTH peptide (aa1–34) for 12 h had no impact on the acute response of cells' energetic state. In contrast, both BMP2 and Wnt3a strongly induced aerobic activation (Supplementary Fig. S2). Upon close examination, we observe that, this increase in OCR matched the profile we observed in OBM, that is, stimulation of basal OCR and mitochondrial respiratory capacity, all significantly increased following addition of BMP2 (Fig. 3) or Wnt3a (Fig. 4). However, in contrast to OBM, ATP-linked OCR did not change following BMP2 or Wnt3a stimulation (Figs. 3 and 4).
FIG. 3.

Bioenergetics in osteoprogenitor ST2 and MC3T3-E1 cell lines are acutely responsive to BMP2. (A) Specific metabolic contributions to OCR in ST2 cells were calculated as basal OCR (baseline minus AA/Rot), ATP production (baseline minus Oligo), respiratory capacity (FCCP minus baseline), and proton leak (Oligo minus AA/Rot). Specific metabolic contributions to ECAR were calculated as basal ECAR (baseline minus 2-DG) and glycolytic reserve (Oligo/FCCP minus baseline). (B) Specific metabolic contributions to OCR in MCT3T-E1 cells were calculated as in panel (A). (C, D) Energy map showing baseline (B: black outlined) and maximal (U: red outlined) OCR and ECAR following OBM addition as fold change over uninduced ST2 (C) or MC3T3-E1 (D) cells. The four quadrants represent cell metabolism attributable to “Quiescent” (lower left), “Aerobic” (upper left), “Energetic” (upper right), or “Glycolytic” (lower right) energy usage. Blue- and orange-colored ellipses represent the metabolic space covered by uninduced and induced cells, respectively. Inset shows the slope of each line generated from the xy-scatter plot. Slopes >1 indicate metabolic shifts toward aerobic respiration and slopes <1 indicated metabolic shifts toward glycolysis. Data are mean ± SD [n = 15 (three biologic replicates of five technical replicates)]. *P < 0.05 versus uninduced control as determined by ANOVA and confirmed by post hoc Bonferroni correction. Color images are available online.
FIG. 4.

Bioenergetics in osteoprogenitor ST2 and MC3T3-E1 cell lines is acutely responsive to Wnt3a. (A) Specific metabolic contributions to OCR in ST2 cells were calculated as basal OCR (baseline minus AA/Rot), ATP production (baseline minus Oligo), respiratory capacity (FCCP minus baseline), and proton leak (Oligo minus AA/Rot). Specific metabolic contributions to ECAR were calculated as basal ECAR (baseline minus 2-DG) and glycolytic reserve (Oligo/FCCP minus baseline). (B) Specific metabolic contributions to OCR in MCT3T-E1 cells were calculated as in (A). (C, D) Energy map showing baseline (B: black outlined) and maximal (U: red outlined) OCR and ECAR following OBM addition as fold change over uninduced ST2 (C) or MC3T3-E1 (D) cells. The four quadrants represent cell metabolism attributable to “Quiescent” (lower left), “Aerobic” (upper left), “Energetic” (upper right) or “Glycolytic” (lower right) energy usage. Blue- and orange-colored ellipses represent the metabolic space covered by uninduced and induced cells, respectively. Inset shows the slope of each line generated from the xy-scatter plot. Slopes >1 indicate metabolic shifts toward aerobic respiration and slopes <1 indicated metabolic shifts toward glycolysis. Data are mean ± SD [n = 15 (three biologic replicates of five technical replicates)]. *P < 0.05 versus uninduced control as determined by ANOVA and confirmed by post hoc Bonferroni correction. Color images are available online.
In addition, we observed that ST2 cells continue to display increased metabolic response when compared to MC3T3-E1 cells as measured by their time to maximum response to osteoinduction, while MC3T3-E1 cells display greater maximal increase (data not shown). Finally, when looking at ECAR, whereas osteoinduction by OBM only significantly stimulated basal glycolysis, both BMP2 and Wnt3a showed significant stimulation of glycolytic reserve. Taken together, these data indicate that mitochondrial activation as measured by stimulation of membrane potential and oxygen consumption appears to be a universal component of early osteoinduction, but increased ATP-linked OCR, aka increased OxPhos, is not an early component of this activation by BMP2 and Wnt3a.
We next asked whether these datasets could be used to identify a distinct pattern of energetic activation during osteoinduction. Using unbiased PCA, we compared the changes in mitochondrial and glycolytic parameters over the three time point tests (1, 6, and 12 h) and in response to OBM, BMP2, and Wnt3a to determine whether ST2 and MC3T3-E1 cells did indeed have distinct metabolic profiles following osteoinduction (Supplementary Fig. S3A). This analysis revealed partially overlapping yet diverging metabolic spaces occupied by each cell type. Unbiased heatmap clustering of these metabolic rates confirmed that activation of OCR is a common response to osteoinduction as the 12-h postinduction data sets cluster together and have the greatest increase in OCR (Supplementary Fig. S3B). In addition, cell-specific (ST2 and MC3T3-E1) responses showed the next greatest impact on cluster formation, with the method of osteoinduction having the least impact on how the data separated.
Activation of Mitochondria requires Akt signaling
Mitochondrial activation can be the result of multiple signals and pathways, including bulk mitochondrial biogenesis, changes in cellular ratios of ATP/ADP, NAD+/NADH, calcium, metabolic intermediates of glycolysis and the Krebs' cycle, and mitochondrial fusion/fission dynamics. The observed acute time scales of mitochondrial activation suggest that the response is mediated through changes in signaling pathways. One such signaling pathway implicated in both osteogenic differentiation [28] and regulation of mitochondrial function [29] is Akt. We therefore asked whether we could detect differences in Akt phosphorylation status following acute osteoinduction. Western blotting revealed increased Akt phosphorylation and thus activation in less than 1 h after addition of OBM, BMP2, or Wnt3a to induce the osteogenic program in ST2 and MC3T3-E1 cells (Fig. 5A).
FIG. 5.

Osteoinduction activates Akt and increases phosphorylation of mitochondrial proteins. (A) Western blots of phospho-Akt and total Akt protein following 5-, 15-, 30-, or 60-min incubation with OBM (left), BMP2 (center), or Wnt3a (right). (B) (top) Western blot using anti-Akt phosphorylation sequence (RXXSp/Tp) to detect Akt target proteins in mitochondrial protein extracts following 48 h of osteoinduction. Quantification of total lane intensity shown below expressed as fold change over control lane. (bottom) No-Stain™ Protein Labeling Reagent used as total protein loading control. (C) Western blot of phospho-Akt and total Akt protein following incubation with pan 1/2/3 Akt inhibitor (iAkt) for 24 h, and then OBM (left), BMP2 (center), or Wnt3a (right) addition for 12 h for ST2 (C) and MC3T3-E1 (D) cells. Western blots and stains are representative images from n = 3 technical replicates. *P < 0.05 versus uninduced control as determined by t-test.”
Akt is known to have multiple phosphorylation targets in mitochondria [30]. To check if Akt phosphorylated mitochondrial targets, we isolated mitochondria and performed western blotting of mitochondrial protein extracts from ST2 or MC3T3-E1 cells using anti-Akt phosphorylation sequence (RXXSp/Tp) antibody. The assay showed that mitochondria from both cell types contained increased levels of Akt-dependent phosphorylation following osteoinduction compared to mitochondria from uninduced cells (Fig. 5B). MC3T3-E1 mitochondria uniquely showed an increase in the phosphorylation of a protein ∼35 kDa, whereas numerous bands were observed to increase in ST2 cells at molecular weights of ∼100, 80, 60, 55, 50 and 30 kDa. It is worth noting that Akt-dependent phosphorylation of mitochondrial proteins of molecular weights 100, 80, and 50 was described previously in mouse isogenic hepatocytes [31] and these proteins were identified as leucine-rich PPR motif-containing protein (100 kDa), aconitase (80 kDa), and glutamate dehydrogenase 1, aldehyde dehydrogenase, and ATP synthase subunit α and subunit β (all present within the 50 kDa band).
While BMP2 appeared to have the strongest impact on increased band intensity, all three methods of osteoinduction showed increases over control (Fig. 5B). Such Akt-mediated phosphorylation of various mitochondrial targets is well documented to stimulate mitochondrial OxPhos activity [30,32]. We next asked whether an Akt inhibitor could block the increased mitochondrial OCR observed in cells incubated in OBM. Incubation of cells with a pan-Akt inhibitor strongly abrogated Akt activation, as evidenced by decreased phosphorylated-Akt signal (Fig. 5C, D). In addition, mitochondrial activation in both ST2 and MC3T3-E1 cells following osteoinduction was completely and consistently blocked in the presence of the pan-Akt inhibitor (Fig. 6A). Finally, Akt inhibition prevented ST2 and MC3T3-E1 differentiation by OBM, and Wnt3a as evidenced by decreased ALP staining. Akt inhibition also prevented differentiation of ST2 cells by BMP2 and partially decreased MC3T3-E1 response to BMP2 (Fig. 6B, C).
FIG. 6.

Activation of mitochondrial spare capacity by osteoinduction requires active AKT. (A) Energy map of ST2 (top) and MC3T3-E1 (bottom) showing baseline (B: black outlined) and maximal (U: red outlined) OCR and ECAR of cells incubated with Akt inhibitor (iAkt) for 24 h and then induced using OBM (left), BMP2 (center), or Wnt3a (right) addition. Data represented as fold change over uninduced control cells. (B, C) ALP stain for cells treated with or without iAkt for 24 h before OB induction with OBM, BMP2, or Wnt3a for 4 days. Seahorse n = 15. Data are mean ± SD (n = 15). Statistical significance of conditions versus uninduced control was determined by ANOVA and confirmed by post hoc Bonferroni correction. No significance was found. Color images are available online.
Taken together, these data indicate that all three methods of osteoinduction converge on Akt at the signaling level leading to Akt stimulatory effect on mitochondria. This demonstrates that osteoinduction acts through Akt to promote mitochondrial bioenergetics. Taken together, our data demonstrate a model in which osteoinduction both through standard ascorbate/BGP treatment and physiologic signaling through BMP2 or Wnt3a results in activation of Akt, and increased Akt-mediated phosphorylation of mitochondrial proteins. Concurrently, osteoinduced cells exhibit an Akt-dependent increase in mitochondrial bioenergetic parameters, ultimately facilitating differentiation along the OB lineage (Fig. 7).
FIG. 7.
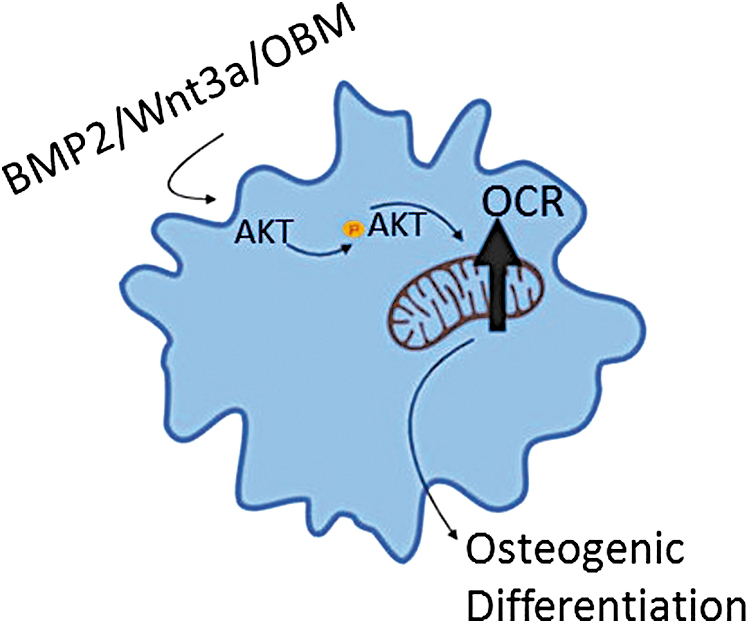
Schematic for Akt-dependent activation of mitochondrial function to induce osteogenic program. Standard acorbate/BGP treatment and physiologic signaling through BMP2 or Wnt3a result in activation of Akt, and increased Akt-mediated phosphorylation of mitochondrial proteins. Akt-dependent increase in mitochondrial bioenergetic parameters allows for differentiation along the osteoblast lineage. Image made using BioRender software. Color images are available online.
Discussion
The role of cellular metabolism during osteogenic differentiation is a topic of debate in the field. While it is routinely reported that various somatic stem cells rely on glycolysis for energy before differentiation and on the mitochondrial process of OxPhos during and after differentiation [4], the data regarding metabolism of osteoprogenitors remain controversial. Methodologic differences, including supraphysiological levels of glucose and glutamine, used in culture media to stimulate BMSC expansion have the potential to influence cell metabolism. The presence of extracellular pyruvate in metabolic assays could bypass any constraint that specifically affects OxPhos and artificially increases oxygen consumption in undifferentiated cells impeding the detection of change in OxPhos during differentiation. Furthermore, the presence of ascorbate in conventional media could prime osteogenic programming for activation and account for the alternate outcomes observed in reports that demonstrate increased OCR [10,11], and those that did not see changes on OCR [12,15,17].
In this study, our experimental model used cells grown in minimal media lacking ascorbate and containing physiologic (1 g/L = 5 mM) glucose and heat-inactivated FBS to best approximate physiologic conditions and to maintain cells in a true uninduced state. OCR and ECAR measures were performed in the absence of added pyruvate and with physiologic glucose (5 mM) and glutamine (1 mM). In addition, the inclusion of the glutaminase inhibitor BPTES did not impact OCR or ECAR levels in a separate experiment (data not shown), indicating that glutamine is not a significant fuel source in pre-OB cells in this acute time frame following osteoinduction.
Increased mitochondrial function can be a result of changes in the dynamic network of mitochondria within cells, changes in mitochondrial coupling, or activation of mitochondrial biogenesis. We chose time points on a short enough time scale to allow us to focus on changes in existing mitochondrial function rather than biogenesis of new mitochondrial protein machinery [33–35]. Using these techniques, we observed rapid and consistent increases in basal mitochondrial OCR, mitochondrial directed ECAR (ie, aerobic glycolysis), and respiratory capacity in both ST2 and MC3T3-E1 cells following addition of OBM, BMP2, or Wnt3a. However, the method of osteoinduction resulted in distinct changes in different metabolic parameters. OBM caused an increase in both ATP production and proton leak, while BMP2 and Wnt3a did not. This observation indicates that activation of OCR is a universal feature of osteoinduction; however, physiologic induction does not require a proportional increase in mitochondrially derived ATP.
Interestingly, while increase in proton leak was significant for MC3T3-E1 cells, it was not in ST2 cells. Activation of mitochondrial OCR associated with increased proton leak has implications for increased oxidative stress as well as a physiologic role in thermogenesis, glucose sensing, or maintaining carbon flux despite low ATP demand to facilitate nutrient supply to protein and nucleic acid synthesis [36]. We also note that ST2 cells responded to osteoinduction rapidly, reaching 90% of their maximal OCR values in the first hour postinduction. In contrast, MC3T3-E1 cells responded less rapidly, requiring 12 h to reach 90% of maximum.
Finally, ST2 cells' dynamic range (∼2-fold) for changes in OCR was consistently smaller than that observed in MC3T3-E1 cells (∼3-fold) following induction. Taken together, ST2 and MC3T3-E1 cells appear to occupy distinct metabolic spaces both preosteoinduction and postosteoinduction, despite responding to similar activators of induction. In addition, BMP2 and Wnt3a trigger metabolic activation of these cells with subtle differences when compared with the response to OBM. The activation of mitochondrial parameters early in osteoinduction is essential and these cells are unable to tolerate even mild inhibition of mitochondrial electron transport chain complexes or ATP synthase during this transition (Fig. 1C, D). Interestingly, mild uncoupling using FCCP did not negatively impact these cells (data not shown). Mild uncoupling has been investigated as a therapeutic strategy in improving metabolic health and could be acting by a similar mechanism in osteoinduction as well [37]. Further investigation is required to determine the specific metabolic requirements of early OB differentiation.
A number of signaling pathways, including Wnt3a and BMP2, are known to regulate osteoinduction of BMSCs into OBs [38]. Our data demonstrate that Wnt3a and BMP2 can trigger an acute activation of mitochondrial OCR and interventions that block mitochondrial activation prevent osteoinduction (Figs. 1 and 6). This agrees with the understanding of Wnt3a/β-catenin and BMP signaling as early signaling mechanisms during stem cell commitment stage [39]. In addition, posttranslational modification of β-catenin modulates its activity. Active Wnt signaling initiates hypophosphorylation of β-catenin and decreased ubiquitination resulting in stabilization of the Wnt protein and ultimately the activation of expression of canonical Wnt target genes. Phosphorylation promotes β-catenin degradation and is in competition with acetylation for amino acids in Wnt, which impact the ubiquitination status of the protein. [40]. Acetyl CoA (Ac-CoA) is a mitochondrially generated substrate for protein acetylation reactions by acetyltransferases. Thus, mitochondrially derived Ac-CoA may provide a link between cellular bioenergetic and differentiation pathways. Also, acetylation has been shown to stabilize and promote activities of other key osteogenic factors, Runx2 and Osterix [41,42].
Osteoinduction has been previously reported to involve both Wnt/Akt and Bmp/Akt signaling axis; however, these reports did not investigate mitochondrial parameters [43,44]. In addition, Akt has been shown separately to be a stimulator of mitochondrial function and an upstream regulator of the formation of mitochondria-associated membranes [29,32]. Our data indicate Akt signaling as absolutely essential for the acute stimulation of mitochondrial OCR during the process of Wnt- and BMP-mediated osteoinduction.
It is interesting to note that, while both ST2 and MC3T3-E1 cells had similar responses in our hands, their specific time to maximal response and the overall degree of mitochondrial activation were different. ST2 cells showed a rapid and large activation, while MC3T3-E1 cells demonstrated a slower and more modest activation. As these cell lines are derived from osteoprogenitor cells of different embryonic origins, this difference in metabolic activity is not surprising and has also been observed in previous studies [14]. Mice have been a staple of biologic research for decades and are useful models for bone development, repair, and osteoporosis; however, metabolic processes differ between mice and humans [45,46]. Of particular importance is the difference in metabolic rate between mice and humans, the former having a basal metabolic rate nearly seven times greater than the later [45,47]. This often translates to experiments in mice requiring higher concentrations of biologic agents to achieve the same physiologic outcome in humans.
In the context of bone, there are several documented differences. Mouse skeletal growth continues after the animal reaches sexual maturity, whereas in humans, peak bone mass aligns with sexual maturation [48]. Furthermore, in mice, new bone growth at the growth plates persists into adulthood, in contrast to humans whose growth plates seal during puberty [49]. Bone metabolism is also distinct as the turnover rate of cancellous bone in mice is measured to be 0.7% compared to 0.1% in humans [46,50,51]. Complete remodeling of bone in mice occurs in 2 weeks, whereas humans require up to 9 months [46,51,52]. Finally, the accessibility of internal regions of bone to osteoprogenitors is structurally distinct. Mice lack a true haversian system. This system, which provides enhanced delivery of OBs and osteoclasts to bone surfaces through blood vessels in humans, is lacking in Refs. 53–55. As this study focuses on the acute metabolic changes in bone-derived osteoprogenitor cells from mouse cell lines, it will be interesting to test whether similar results are generated using human-derived cells.
In conclusion, we found that (i) activation of mitochondrial OxPhos is achieved rapidly following induction of the osteogenic program in both long bone- and calvarial bone-derived osteoprogenitor cells; (ii) mitochondrial respiratory activity is required for successful transition into the osteogenic program; and (iii) activation of mitochondria using these methods is dependent upon active Akt signaling (Fig. 7). We, therefore suggest the scenario wherein the osteoprogenitor mitochondria are rapidly activated following Akt phosphorylation of mitochondrial targets allowing for the subsequent activation of the osteogenic program.
Supplementary Material
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
Funding was provided by the National Institute of Health grant to R.A.E. (R01 AR072601).
Supplementary Material
References
- 1. Nunnari J and Suomalainen A (2012). Mitochondria: in sickness and in health. Cell 148:1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McBride HM, Neuspiel M and Wasiak S (2006). Mitochondria: more than just a powerhouse. Curr Biol 16:R551. [DOI] [PubMed] [Google Scholar]
- 3. Schorr S and van der Laan M (2018). Integrative functions of the mitochondrial contact site and cristae organizing system. Semin Cell Dev Biol 76:191–200 [DOI] [PubMed] [Google Scholar]
- 4. Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA IV, Ramalho-Santos J, van Houten B and Schatten G (2011). Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6:e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sandhir R, Halder A and Sunkaria A (2017). Mitochondria as a centrally positioned hub in the innate immune response. Biochim Biophys Acta - Mol Basis Dis 1863:1090–1097 [DOI] [PubMed] [Google Scholar]
- 6. Cherry C, Thompson B, Saptarshi N, Wu J and Hoh J (2016). 2016: A “Mitochondria” Odyssey. Trends Mol Med 22:391–403 [DOI] [PubMed] [Google Scholar]
- 7. Vafai SB and Mootha VK (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491:374–383 [DOI] [PubMed] [Google Scholar]
- 8. Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ and Wang C-Y (2013). The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med 19:35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Motyl KJ, Guntur AR, Carvalho AL and Rosen CJ (2017). Energy Metabolism of Bone. Toxicol Pathol 45:887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen C-T, Shih Y-RV, Kuo TK, Lee OK and Wei Y-H (2008). Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 26:960–968 [DOI] [PubMed] [Google Scholar]
- 11. Guntur AR, Le PT, Farber CR and Rosen CJ (2014). Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 155:1589–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shum LC, White NS, Mills BN, de M Bentley KL and Eliseev RA (2016). Energy metabolism in mesenchymal stem cells during osteogenic differentiation. Stem Cells Dev 25:114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shares BH, Busch M, White N, Shum L and Eliseev RA (2018). Active mitochondria support osteogenic differentiation by stimulating-Catenin acetylation. J Biol Chem 293:16019–16027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quarto N, Wan DC, Kwan MD, Panetta NJ, Li S and Longaker MT (2010). Origin matters: differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones. J Bone Miner Res 25:1680–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW and Long F (2013). WNT-LRP5 signaling induces warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab 17:745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guntur AR, Gerencser AA, Le PT, DeMambro VE, Bornstein SA, Mookerjee SA, Maridas DE, Clemmons DE, Brand MD and Rosen CJ (2018). Osteoblast-like MC3T3–E1 Cells Prefer Glycolysis for ATP Production but Adipocyte-like 3T3–L1 cells prefer oxidative phosphorylation. J Bone Miner Res 33:1052–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pattappa G, Heywood HK, de Bruijn JD and Lee DA (2011). The metabolism of human mesenchymal stem cells during proliferation and differentiation. J Cell Physiol 226:2562–2570 [DOI] [PubMed] [Google Scholar]
- 18. Komarova SV, Ataullakhanov FI and Globus RK (2000). Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. Am J Physiol Cell Physiol 279:C1220–C1229 [DOI] [PubMed] [Google Scholar]
- 19. Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, Zhang H, Knudsen E, Zhang GF, Hilton MJ and Karner CM (2019). Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab 29:966–978.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ganz J, Kaslin J, Freudenreich D, Machate A, Geffarth M and Brand M (2011). Subdivisions of the adult zebrafish subpallium by molecular marker analysis Julia Ganz. J Comp Neurol 520:633–655 [DOI] [PubMed] [Google Scholar]
- 21. Forni MF, Peloggia J, Trudeau K, Shirihai O and Kowaltowski AJ (2016). Murine mesenchymal stem cell commitment to differentiation is regulated by mitochondrial dynamics. Stem Cells 34:743–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Metsalu T and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 43:W566–W570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jolliffe I (2014). Principal Component Analysis. In: Wiley StatsRef: Statistics Reference Online. John Wiley & Sons, Ltd, Chichester, United Kingdom [Google Scholar]
- 24. Otsuka E, Yamaguchi A, Hirose S and Hagiwara H (1999). Characterization of osteoblastic differentiation of stromal cell line ST2 that is induced by ascorbic acid. Am J Physiol Physiol 277:C132–C138 [DOI] [PubMed] [Google Scholar]
- 25. Wang D, Christensen K, Chawla K, Xiao G, Krebsbach PH and Franceschi RT (1999). Isolation and Characterization of MC3T3–E1 Vivo Differentiation/Mineralization Potential. J Bone Miner Res 14:893–903 [DOI] [PubMed] [Google Scholar]
- 26. Rastogi A, Joshi P, Contreras E and Gama V (2019). Remodeling of mitochondrial morphology and function: an emerging hallmark of cellular reprogramming. Cell Stress 3:181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kwon IK, Lee SC, Hwang YS and Heo JS (2015). Mitochondrial function contributes to oxysterol-induced osteogenic differentiation in mouse embryonic stem cells. Biochim Biophys Acta 1853:561–572 [DOI] [PubMed] [Google Scholar]
- 28. Raucci A, Bellosta P, Grassi R, Basilico C and Mansukhani A (2008). Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J Cell Physiol 215:442–451 [DOI] [PubMed] [Google Scholar]
- 29. Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N and Hall MN (2013). MTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A 110:12526–12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bijur GN and Jope RS (2003). Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem 87:1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li C, Li Y, He L, Agarwal AR, Zeng N, Cadenas E and Stiles BL (2013). PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic Biol Med 60:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen YH, Su CC, Deng W, Lock LF, Donovan PJ, Kayala MA, Baldi P, Lee HC, Chen Y and Wang PH (2019). Mitochondrial akt signaling modulated reprogramming of somatic cells. Sci Rep 9:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scarpulla RC. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638 [DOI] [PubMed] [Google Scholar]
- 34. Gottlieb RA and Bernstein D (2016). Mitochondrial remodeling: rearranging, recycling, and reprogramming. Cell Calcium 60:88–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilson-Fritch L, Burkart A, Bell G, Mendelson K, Leszyk J, Nicoloro S, Czech M and Corvera S (2003). Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol Cell Biol 23:1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Divakaruni AS and Brand MD (2011). The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 26:192–205 [DOI] [PubMed] [Google Scholar]
- 37. Cunha FM, Caldeira da Silva CC, Cerqueira FM and Kowaltowski AJ (2011). Mild mitochondrial uncoupling as a therapeutic strategy. Curr Drug Targets 12:783–789 [DOI] [PubMed] [Google Scholar]
- 38. Marie PJ. (2008). Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys 473:98–105 [DOI] [PubMed] [Google Scholar]
- 39. Valenti MT, Dalle Carbonare L and Mottes. M (2016). Osteogenic Differentiation in Healthy and Pathological Conditions. Int J Mol Sci 18: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao C, Xiao G and Hu J (2014). Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH, Ryoo HM, Choi JY, Yoshida M, et al. (2006). Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem 281:16502–16511 [DOI] [PubMed] [Google Scholar]
- 42. Lu J, Qu S, Yao B, Xu Y, Jin Y, Shi K, Shui Y, Pan S, Chen L and Ma C (2016). Osterix acetylation at K307 and K312 enhances its transcriptional activity and is required for osteoblast differentiation. Oncotarget 7:37471–37486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eiraku N, Chiba N, Nakamura T, Amir MS, Seong C, Ohnishi T, Kusuyama J, Noguchi K and Matsuguchi T (2019). BMP9 directly induces rapid GSK3-β phosphorylation in a Wnt-independent manner through class I PI3K-Akt axis in osteoblasts. FASEB J 33:12124–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ling L, Dombrowski C, Foong KM, Haupt LM, Stein GS, Nurcombe V, Van Wijnen AJ and Cool SM (2010). Synergism between Wnt3a and heparin enhances osteogenesis via a phosphoinositide 3-kinase/Akt/RUNX2 pathway. J Biol Chem 285:26233–26244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Demetrius L (2005). Of mice and men. EMBO Rep 6:S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jilka RL. (2013). The relevance of mouse models for investigating age-related bone loss in humans. J Gerontol A Biol Sci Med Sci 68:1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gillooly JF, Brown JH, West GB, Savage VM and Charnov EL (2001). Effects of size and temperature on metabolic rate. Science (80-) 293:2248–2251 [DOI] [PubMed] [Google Scholar]
- 48. Yuan R, Tsaih SW, Petkova SB, Evsikova CM, Xing S, Marion MA, Bogue MA, Mills KD, Peters LL, Bult CJ, Rosen CJ, Sundberg JP, Harrison DE, Churchill GA and Paigen B (2009). Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell 8:277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parfitt AM. (2002). Misconceptions (1): epiphyseal fusion causes cessation of growth. Bone 30:337–339 [DOI] [PubMed] [Google Scholar]
- 50. Jilka RL, O'Brien CA, Ali AA, Roberson PK, Weinstein RS and Manolagas SC (2009). Intermittent PTH stimulates periosteal bone formation by actions on post-mitotic preosteoblasts. Bone 44:275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weinstein RS, Jilka RL, Michael Parfitt A and Manolagas SC (1998). Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts end osteocytes by glucocorticoids potential mechanisms of their deleterious effects on bone. J Clin Invest 102:274–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Parfitt AM, Han ZH, Palnitkar S, Rao DS, Shih MS and Nelson D (1997). Effects of ethnicity and age or menopause on osteoblast function, bone mineralization, and osteoid accumulation in iliac bone. J Bone Miner Res 12:1864–1873 [DOI] [PubMed] [Google Scholar]
- 53. Lelovas PP, Xanthos TT, Thorma SE, Lyritis GP and Dontas IA (2008). The laboratory rat as an animal model for osteoporosis research. Comp Med 58:424–430 [PMC free article] [PubMed] [Google Scholar]
- 54. Parfitt AM. (1994). Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem 55:273–286 [DOI] [PubMed] [Google Scholar]
- 55. Schneider P, Krucker T, Meyer E, Ulmann-Schuler A, Weber B, Stampanoni M and Müller R (2009). Simultaneous 3D visualization and quantification of murine bone and bone vasculature using micro-computed tomography and vascular replica. Microsc Res Tech 72:690–701 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.