

Abstract
An accurate in vitro model of human adipose tissue could assist in the study of adipocyte function and allow for better tools for screening new therapeutic compounds. Cell culture models on two-dimensional surfaces fall short of mimicking the three-dimensional in vivo adipose environment, while three-dimensional culture models are often unable to support long-term cell culture due, in part, to insufficient mass transport. Microfluidic systems have been explored for adipose tissue models. However, current systems have primarily focused on 2D cultured adipocytes. In this work, a 3D human adipose microtissue was engineered within a microfluidic system. Human adipose-derived stem cells (ADSCs) were used as the cell source for generating differentiated adipocytes. The ADSCs differentiated within the microfluidic system formed a dense lipid-loaded mass with the expression of adipose tissue genetic markers. Engineered adipose tissue showed a decreased adiponectin secretion and increased free fatty acid secretion with increasing shear stress. Adipogenesis markers were downregulated with increasing shear stress. Overall, this microfluidic system enables the on-chip differentiation and development of a functional 3D human adipose microtissue supported by the interstitial flow. This system could potentially serve as a platform for in vitro drug testing for adipose tissue-related diseases.
Graphical Abstract
This work presents a microfluidic system for the engineering and analysis of 3D human adipose tissue under controlled flow.
Adipose tissue plays a central role in regulating energy expenditure and glucose homeostasis through both organ and systemic level function1. Excess accumulation of subcutaneous and visceral fat due to positive energy balance leads to obesity, a critical health care issue2. Obesity is associated with hyperglycemia and insulin resistance3 and various forms of cancer, including colorectal, breast, endometrial, renal cell, oesophageal, pancreatic, and prostate cancers4,5. Meanwhile, alterations in adipose tissue function with subsequent adipokine secretion contribute to various pathological conditions6,7. Due to its involvement in a vast array of physiological processes, adipose tissue has tremendous potential as a drug target8. A deeper understanding of adipose tissue development, function, and regulating mechanisms are needed for developing therapeutic strategies to treat obesity and related metabolic complications.
Two-dimensional (2D) cell culture models have been used extensively to study adipocyte differentiation and function9. For example, studies performed on the repression of PPARγ function models confirmed that PPARγ is necessary and sufficient for adipogenesis10. Compared to 2D culture, three-dimensional (3D) in vitro culture models can better mimic the complex in vivo physiological environment of adipose tissue11 and have been shown to have improved adipogenic differentiation12,13. Adipocytes have been cultured or differentiated in 3D using natural biomaterials such as fibrin14, collagen15, silk16, and hyaluronic acid scaffold17, or synthetic biomaterials such as polyglycolic acid18 and poly(ethylene glycol) hydrogels19. 3D tissue growth requires the effective transport of nutrition to the cells and the removal of waste. Compared to static 3D adipose tissue models, dynamic conditions created by orbital shakers and bioreactors have shown increased triglyceride accumulation, leptin secretion and enabled prolonged adipogenic differentiation16,18,20. Although these dynamic models realized better mass transport over static models, a more controlled flow through the engineered tissue is needed for accurate analysis of adipose tissue differentiation and function.
Microfluidic systems permit small scale cell culture with a constant flow that can represent in vivo physiological mass transport conditions21. 3T3-L1 preadipocytes, murine primary preadipocytes, and human adipose-derived stem cells (ADSCs) have been cultured and differentiated in various microfluidic devices to study adipokine secretions, glycerol secretions, non-esterified fatty acid secretions, as well as mammalian target of rapamycin (mTOR) regulations22-26. To protect adipocytes from direct fluidic shear stress, researchers have developed dual-layer microfluidic devices in which porous membranes separate cell culture chambers from fluid flow26-28. In most of the systems, preadipocytes were either differentiated on glass coverslips and later sealed into the microfluidic device or seeded to surface-coated microfluidic chambers before differentiation, making the systems essentially 2D models. Efforts have been made in recent years to incorporate 3D adipose tissue into microfluidic systems. For example, Godwin et al. integrated a reservoir into a passively operated microfluidic device to culture primary murine adipocytes suspended and entrapped in collagen hydrogel21. Zambon et al. developed a microfluidic platform to study glucose uptake and insulin sensitivity profiles of human adipose tissue biopsy samples under physiopathological stimulations29. In current models, the adipose tissues were isolated from lab animals or taken from a human donor through liposuction. Using an adipose tissue biopsy in the microfluidic systems has a few drawbacks. First, due to the inherent buoyancy of adipose tissue, it is difficult to trap or anchor the biopsy into microfluidic devices30. Second, the process of integrating the tissue into the microfluidic system could cause deterioration of the tissue and lead to lipid leakage and structure deformation, rendering the microfluidic analysis inaccurate31. Furthermore, interstitial flow plays a significant role in tissue morphogenesis and function32. Shear stress induced by interstitial level flow has been shown to affect tissue microenvironment modification33, stem cell differentiation34, and tumor cell invasion35. Using fluorescence recovery after photobleaching, Chary et al. measured the interstitial flow velocities to be in the range of 0 – 2.0 μm/s36. In current 3D adipose tissue microfluidic systems, the flow was designed to go around instead of perfusing through the tissue, making it difficult to control interstitial flow, evaluate flow conditions, or study adipocyte function under physiological level interstitial flow.
In this paper, we present a microfluidic system that enables on-chip differentiation of stem cells into a 3D adipose microtissue and subsequently analyze adipocyte function under physiological level interstitial flow conditions. The microfluidic device has five connected cell culture chambers in which the 3D tissue resides, flanked by two side channels that deliver medium to the cell culture chambers. The design strategy of flanking 3D constructs with medium channels has been proved to enable the control over mass transport in 3D culture37,38, and has been successfully used to develop different microfluidic models, including microvascular networks39,40, blood-brain barrier41, microtumors42, and cancer cell extravasation43, et al. Fibrin was used in this study as the 3D scaffold since it has been shown to support adipogenic differentiation44. Human ADSCs were suspended in fibrinogen and thrombin solution and then seeded into the cell culture chambers to polymerize into cell/fibrin 3D construct. Adipogenic medium was applied through side channels. After the adipose tissue was formed, interstitial flow was applied to the tissue to study adipocyte function. To our knowledge, this is the first report on 3D adipose tissue differentiation and formation in a microfluidic device and adipose tissue response under physiological level interstitial shear stress stimulation.
Methods
Fabrication of microfluidic device
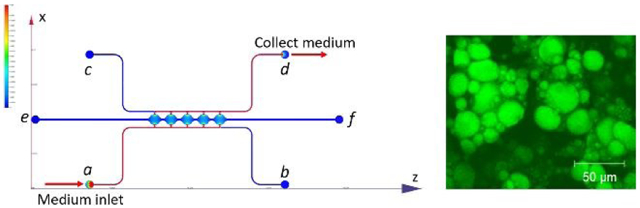
The microfluidic device consists of two side channels (port a-b, c-d) separated by 5 cell culture chambers (Figure 1A). The cell culture chambers are 1 mm in diameter and 200 μm in height. The side channels are connected on either side of the cell culture chambers via connecting pores with a width of 50 μm and a height of 100 μm (Figure S1A). The curved cell culture chambers were designed to resemble fat lobules since a morphology resemblance of in vivo tissue could be beneficial in the development of in vitro tissue models45. For on-chip 3D culture, ADSCs suspended in fibrin gel were loaded through port e. The side-connecting pores (100 μm × 50 μm) were designed to be smaller than the chamber-connecting channels (200 μm × 150 μm) to ensure that the fibrin could advance from port e to port f without bursting into the side channels46. Side channels (a-b, c-d) have a height and width of 100 μm. Reservoirs or syringe pumps can be applied to the ports to control the direction and magnitude of the convective flow through the cell culture chambers.
Figure 1. Microfluidic system design and assembly.

(A) Microfluidic device consists of two fluidic channels (port a-b, c-d) separated by 5 cell culture chambers (e-f). (B) Assembly of the microfluidic device. (C) Assembled microfluidic device with four reservoirs. (D) Dimensions of the cell culture chamber and fluidic channels measured from microscopic image.
The mold for making the microfluidic devices was manufactured using a computer numeric control (CNC) milling machine (Haaz VF2, Haas Automation, Inc.) at the Illinois Institute of Technology Idea Shop Prototyping Lab. The material used for the mold was polyoxymethylene (POM), also known as Delrin. POM is an engineering thermoplastic used in precision parts requiring high stiffness, low friction, and excellent dimensional stability47. The microfluidic mold was milled on a POM block with a length of 40 mm and a width of 25 mm (Figure S1B). The microfluidic device was subsequently fabricated using polydimethylsiloxane (PDMS) soft lithography. First, a PDMS precursor solution was formed by mixing prepolymer with curing agent at a 10:1 ratio (Sylgard 184, Dow Corning, Midland, MI) and poured onto the mold. After degassing in a vacuum desiccator for 2 hours, the PDMS was cured for 4 hours at 65°C. After peeling the PDMS slab from the mold, inlet/outlet ports were punched through using a 0.75 mm biopsy punch (World precision instruments, Sarasota, FL). The thickness of the PDMS slab was ~5 mm. To assemble the microfluidic device, a 1-mm-thick PDMS sheet was first bonded to a glass slide after plasma treatment in a plasma cleaner (PDC-32G, Harrick Plasma, Ithaca, NY). The PDMS slab was then bonded to the PDMS sheet after plasma treatment, forming closed channels and cell culture chambers, as shown in Figure 1B. After each bonding, pieces were placed in an oven at 65°C for 10-15 minutes. The assembled PDMS device was sterilized in an autoclave for 30 minutes at 110°C before loading cells.
Medium reservoirs and syringe pumps (NE-300, New Era Pump Systems Inc.) are both used to provide interstitial flow through the 3D tissue construct in the cell culture chambers. For reservoir setup, plastic cryogenic vials (Thermo scientific) with bottoms removed were used as reservoirs and bonded over the ports of fluidic channels (a, b, c, and d) on the assembled microfluidic device using a PDMS precursor solution. Interstitial flow through cell culture chambers was driven by hydraulic pressure from medium height difference in reservoirs on two sides. Since medium height can be changed easily, for all microfluidic devices, reservoirs were used at the beginning of culture. Medium height was switched on the two sides every 3 days to alter interstitial flow direction to promote uniform differentiation of the cells. To study the influence of shear stress, the cells were first cultured using static reservoirs for 3 weeks and then switched to syringe pump setup with different flow rates. For syringe pump setup, ports b and c were blocked. Medium was introduced to the system from port a, perfused through the cell culture chambers, and evacuated from port d. Tygon tubing (0.01-inch ID, 0.03-inch OD) was used for medium infusion.
Simulation modeling
Autodesk CFD 2018 (Autodesk Inc.) was used for simulating fluid dynamics within the microfluidic device. The fluid in the channels was set as cell culture medium with a viscosity of 0.94 centipoises48. The material in the cell culture chambers was set as resistance material with a permeability of 1.5×10−13 m2 based on previous studies with fibrin49. To model the flow in reservoir setup, ports a and b were set to have a pressure of 10 mm H2O, ports c and d were set to have a pressure of 5 mm H2O, creating a 5 mm H2O hydraulic pressure difference between two sides of the cell culture chambers. To model the flow in the syringe pump setup, port a was set as medium inlet with a constant volumetric flow rate, port d was set as medium outlet with a gauge pressure of 0 Pa. Flow through porous fibrin was considered incompressible for both setups.
Cell culture and differentiation
Human adipose-derived stromal cells (ADSCs) were purchased from Lonza (PT-5006, Batch 0000535975). The donor for the cells was a 33-year-old male with a body mass index (BMI) of 25. Cells were cultured in ADSC Growth Medium (ADSCGM, PT-4505, Lonza) containing 10% fetal bovine serum (FBS).
For 3D on-chip culture, cells were loaded into microfluidic devices in a fibrin gel. Briefly, when ADSC reached 90% confluence in flasks, the cells were washed with phosphate buffered saline (PBS), dissociated with TrypLE™ Express (12605036, Gibco), and counted. ADSCs were suspended in 10 mg/mL fibrinogen and 10 U/mL thrombin solution and loaded into the microfluidic device with a pipette through cell inlet port e with steady pressure until the solution reached the cell outlet port f. Both the cell inlet and outlet were subsequently blocked with filter tips (EP0030077512, Eppendorf). The device was then incubated in 37°C humidified incubator for 30 minutes to ensure full fibrin polymerization. After fibrin hydrogel was formed, ADSCGM was introduced into both fluidic channels (a-b, c-d). Starting from one port (for example, port a or port c), medium was pipetted slowly by applying steady pressure until the air in the channel was expelled and medium reaches the other port (for example, port b or port d) and forms a small bubble of media. Additional medium was added to the reservoirs. A fluid height difference was maintained in the four reservoirs to allow the medium to perfuse across the cell culture chambers. The caps of the reservoirs were screwed loosely to allow free gas exchange. The cell-loaded microfluidic devices were incubated in a 37°C humidified incubator with 5% CO2.
To induce adipogenic differentiation, the cells were given ADSCGM supplemented with 5 μg/mL insulin, 1 nM 3,3’,5-triiodoL-thyronine (T3), 2 μg/mL dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine (IBMX). After 5 days of induction, the cells were given ADSCGM supplemented with 5 μg/mL insulin and 1 nM T3 for the rest of the cell culture.
Staining
A Live/Dead viability/cytotoxicity kit (L3224, Invitrogen) was used to evaluate cell viability within the microfluidic devices. ADSCs were loaded to microfluidic devices at a density of 5 million/mL and cultured in ADSCGM. Cell viability was tested at 2 hours, 2 days, 4 days, and 6 days after loading following the manufacturer’s protocol. Briefly, ADSCGM supplemented with Live/Dead staining reagents calcein AM (green fluorescent dye stains live cells, 494 nm) and ethidium homodimer-1 (red fluorescent dye stains dead cells, 528 nm) was added to reservoirs on one side of cell culture chamber with a height of 10 mm. The reservoirs on the other side were added with ADSCGM with a height of 5 mm, creating a hydraulic pressure to allow staining reagents to flow across the cell culture chambers. After 1 hour of incubation, samples were imaged using confocal microscopy (Axiovert 200M, Carl Zeiss Microimaging, Inc., Thornwood, NY). Result images were analyzed using customized MATLAB scripts (MathWorks, Inc). The live cell ratio was calculated by dividing the live cell area by total cell area.
To confirm the formation of lipid droplets in differentiated cells, samples were evaluated with BODIPY staining. Cells were fixed by flowing 10% Buffered Formalin (SF100-4, Fisher Scientific) through the microfluidic chambers overnight. Formalin was removed by flowing PBS through the chambers. BODIPY 493/503 (4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a- diaza-s-indacene) was added to PBS at 0.01 mg/mL and flew through the chambers. After staining, excess BODIPY was washed away with PBS. Images acquired from the confocal microscope were quantified using Image J and custom MATLAB scripts in terms of BODIPY staining area and lipid droplet size.
Gene expression analysis
For gene analysis, after defined culture times, the microfluidic devices were cut open to access the cells in the chambers. The reservoirs were removed from the microfluidic devices, after which the PDMS slab was cut using a No.11 surgical scalpel blade to expose the cell culture chambers. The RNA was extracted using RNeasy Mini kit (74104, Qiagen) following the manufacturer’s instructions. The purity and concentration of the RNA were determined using Nanodrop 2000 (Thermo Scientific). The cDNA was synthesized with Qscript (Quanta Biosciences). Reverse transcription polymerase chain reaction (RT-PCR) was performed using SYBR green on CFX Connect Real-Time System (Bio-Rad, Hercules, California). Primers are shown in Table 1. Melt curve analysis was used to assess primer specificity. GAPDH-1 was used as the housekeeping gene. Gene expression levels were evaluated by the 2−ΔΔCT Method50.
Table 1.
RT-PCR primer sequences
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| GAPDH 1 | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
| ADIPOQ | TGCTGGGAGCTGTTCTACTG | TACTCCGGTTTCACCGATGTC |
| PLIN1 | TGGAAACTGAGGAGAACAAG | ATGTCACAGCCGAGATG |
| PPARG | CAGTGGGGATGTCTCATAA | CTTTTGGCATACTCTGTGAT |
| AP2 | TGGTGGTGGAATGCGTCAT | GGTCAACGTCCCTTGGCTTA |
| CEBPa | CTTGTGCCTTGGAAATGCAA | GCTGTAGCCTCGGGAAGGA |
| IL6 | AGACAGCCACTCACCTCTTCAG | TTCTGCCAGTGCCTCTTTGCTG |
| IL1B | CCACAGACCTTCCAGGAGAATG | GTGCAGTTCAGTGATCGTACAGG |
Adipokine and FFA secretion
Medium collected from the microfluidic devices was analyzed for adipokines and free fatty acid (FFA) secretion. Briefly, after adipogenic differentiation of ADSCs in the reservoir setup, medium reservoirs were removed from the microfluidic devices, revealing inlet and outlet ports and switched to the syringe pump setup. The medium was then collected in a centrifuge tube and stored at −20°C until further analysis. Adiponectin and leptin secretion from differentiated adipocytes were analyzed using enzyme-linked immunosorbent assay (ELISA) kits (Thermo Fisher Scientific) according to the manufacturer’s instructions. Results of the ELISA assay were obtained using a plate reader (SpectraMax M2, Molecular Devices) and analyzed using GraphPad Prism (GraphPad Software, LLC.). FFA secretion was tested using EnzyChrom™ Free Fatty Acid Assay Kit (EFFA-100) according to the manufacturer’s instructions.
Fatty acid uptake
The metabolic function of the differentiated adipocytes was evaluated by fatty acid uptake. After 3 weeks of differentiation, 1μM 4,4-difluoro-5-methyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoic acid (BODIPY D-3823, Molecular Probes) was added to the medium in reservoirs on one side of cell culture chambers and incubated in 37°C humidified incubator. BODIPY D-3823 is a fluorescently labeled fatty acid analog that undergoes native-like transport and metabolism in cells51,52. After incubation, the cells were imaged over time using fluorescent microscopy and signal intensity was quantified using customized MATLAB scripts.
Scanning electron microscopy (SEM)
The structure of the developed adipose microtissue was evaluated using scanning electron microscopy (SEM). First, the microtissue was fixed by flowing 10% buffered formalin through the microfluidic chambers overnight. The next day, formalin was removed by flowing PBS through the chambers. Then, reservoirs were removed from the microfluidic devices, after which PDMS slab was cut open to expose the adipose tissue residing in the cell culture chambers. The samples were post-fixed with 1% aqueous osmium tetraoxide for 30 minutes, dehydrated in a series of increasing concentration of ethanol solution (50%, 70%, 85%, 95%, and 100% ethanol), washed in hexamethyldisilazane (HMDS), and air-dried in a chemical hood. The dried samples were placed on PELCO Tabs (Ted Pella Inc.) and imaged using a scanning electron microscope (Phenom ProX).
Statistics
One-way analysis of variance (ANOVA) followed by Tukey’s posttest was used for comparison in different data groups. In all cases, p < 0.05 was considered statistically significant.
Results
Characterization and simulation modeling of the microfluidic device
The POM mold manufactured from the CNC machine was used directly to make PDMS slab for the microfluidic device (Figure S1C). An example of assembled microfluidic devices with the reservoir setup is shown in Figure 1C. A red dye was perfused through the microchannels in the assembled device to test for leakage and channel dimensions. As shown in the left image in Figure 1D, the side channel was measured to have a width of ~100 μm, and the connection between the side channels and the cell culture chambers was measured to have a width of ~50 μm. PDMS slab was also cut open to show the cross-section of the device. As shown in the right image in Figure 1D, the side channel was measured to have a height of ~100 μm, and the central chamber was measured to have a height of ~200 μm. The measurements from microscope images confirmed that the manufactured microfluidic device accurately created the structures from the design.
Flow in the cell culture chambers was simulated for both the syringe pump and reservoir setups. With the differentiation of adipocytes in the cell culture chambers, the structure of the microtissue changes and is difficult to predict. To simplify the simulation, this model was based on a fibrin-loaded microfluidic device without cells. This simulation can provide an estimation for the levels of flow rate and shear stress. Figure 2A is an illustration of the medium flow direction and velocity distribution in both the side channels and central chambers using the syringe pump setup. Medium was introduced from port a at different volumetric flow rates (1, 4, and 10 μl/hour) and collected from port d. The upper image in Figure 2B shows the colormap of the flow velocity distribution in cell culture chambers with 1 μl/hour medium input. The velocity is lower than 1 μm/s in most regions of the cell culture chambers but increases rapidly near the connecting pores at the two ends of each chamber. The area close to the opening of the pores can be excluded from the average flow calculation since it has a small number of cells34,39. The lower image in Figure 2B shows the colormap of the flow velocity distribution with the area near the connecting pores omitted. Simulation results show that this area (effective region) covers 90.16% of the total cell culture chamber area and has an average flow velocity of 0.324 μm/s. Meanwhile, the flow velocity distributions are similar for all five chambers regardless of location in the chain. The effective region covers around 90% (90.32%, 90.16%, 90.11%, 89.95% and 90.26% for chambers 1-5, respectively) of each chamber, with average flow velocities of 0.319 μm/s, 0.323 μm/s, 0.332 μm/s, 0.327 μm/s and 0.317 μm/s for the five chambers, respectively. For the same effective region, 4 μl/hour and 10 μl/hour medium inputs resulted in average flow velocities of 1.29 μm/s and 3.24 μm/s, respectively. Similarly, for the reservoir setup, the average flow velocity generated from 5 mm H2O hydraulic pressure was 1.08 μm/s. Figure 2C shows flow velocity colormap in the central chamber cross-section at different syringe pump volumetric flow rates. In all three conditions, velocity changes slower near the center of the chambers compare to the two sides of the chambers. Velocity in the X direction (red line in the upper image) is shown in Figure 2D. In accordance with Figure 2C, graphs in Figure 2D show flat lines near the center of the chambers and an increase in slop near two sides of the chambers. When medium flow into the center of the chamber, velocities drop as the chambers get wider. When medium flow out of the chamber, velocities rise as the chambers get narrower. Meanwhile, in all three cases, hydrostatic pressure drops in the direction of the flow (Figure 2E). Figure 2F shows top view flow velocity in the five chambers at different syringe pump volumetric flow rates. Velocity in the Z direction (red line in the upper image) is shown in Figure 2G. Velocities in all three conditions show similar shapes with different magnitude. Velocities are highest at the centers of each chamber and the lowest between two chambers. The hydrostatic pressure in the Z-direction is relatively constant for each case (Figure 2H).
Figure 2. Simulation modeling results from the microfluidic device.

(A) Overview of flow direction and velocity distribution in both the side channels and central chambers using the syringe pump setup. (B) Colormap of the flow velocity distribution in cell culture chambers with 1 μl/hour medium input. (C) Flow velocities in the central chamber cross-section at different syringe pump volumetric flow rates. (D) Velocity in the X-direction in cell culture chambers. (E) Hydrostatic pressure in the X-direction. (F) Top view flow velocity in the five chambers at different syringe pump volumetric flow rates. (G) Velocity in the Z-direction in cell culture chambers. (H) Hydrostatic pressure in the Z-direction across the 5 cell culture chambers.
To estimate the shear stress induced by interstitial flow, the following equation developed by Wang et al. and Shi et al.53,54 was applied:
| (1) |
where τ is the interstitial flow-induced shear stress, μ is the viscosity of the flow medium, v is the flow velocity, and Kp is the permeability of the 3D material. Using this equation, the average shear stresses in the chambers generated from volumetric flow rates of 1 μl/hour, 4 μl/hour, and 10 μl/hour were estimated to be 0.00079 Pa, 0.00313 Pa, and 0.00786 Pa, respectively. Similarly, shear stress generated from reservoirs was 0.00262 Pa. Shear stresses generated by the microfluidic system fall within the physiological range and can be used to analyze cell response at in vivo interstitial level flow34,36.
Live/Dead assay
Once the cell culture chambers are loaded with fibrin, the two side channels would be separated by the hydrogel. Survival of the cells in the fibrin hydrogel relies on the delivery of nutrients from the two side channels. To visualize the perfusion of medium from one side channel to the other, medium containing fluorescein isothiocyanate-dextran (25 μg/mL, FD70S, Sigma) was added to one side channel with higher hydraulic pressure and allow to perfuse through the fibrin in the cell culture chamber (Figure S2). The dextran was evenly distributed in the chamber after 2 hours. Despite the results from the simulation that the area between chambers has a very low flow rate, that area does not show an obvious weaker fluorescent signal compared to the center of the chamber. Since the length of the chambers is small (~ 1mm) and that fibrin scaffold has a porous structure, medium was diffusing between the chambers and hence could provide nutrients to the cells that reside there.
To test cell survival in the microfluidic device, ADSCs (5 million/mL) were suspended in fibrinogen (10 mg/mL) and thrombin (10 U/mL) solution and loaded to the cell culture chambers. Cells were distributed homogeneously and started to spread and migrate after loaded into the chambers. As shown in Figure 3, cell Live/Dead assay at 2 hours after loading showed that cells could survive the loading process with a 99.9% live cell percentage. Cell viability in the chambers after 2, 4, and 6 days were 98.7%, 97.6%, and 97.9%, indicating that cells could stay alive in the fibrin gel in the microfluidic device. Live/Dead staining reagents were added to one side and perfused through cell culture chambers as interstitial flow. Cells close to the side with staining solution were in contact with reagents for a longer time and hence had a stronger staining signal (green). Overall, the live/dead assay indicated that this microfluidic device could maintain cell viability.
Figure 3. Live/Dead assay of ADSCs in the microfluidic device.

(A) Cell viability at 2 hours, 2 days, 4 days, and 6 days after loading to cell culture chambers. (B) Live cell ratio at different time points. Values are means ± SD, n = 10 or 14. One-way ANOVA followed by Tukey’s multiple comparisons test was performed.
On-chip differentiation
To test lipid formation, ADSCs were loaded into the microfluidic device and given the adipogenic medium. After differentiation, cells were stained with BODIPY. After one week of differentiation, small lipid droplets could be observed within the cells (Figure 4A). The lipid droplets increased in size through 3 weeks of differentiation. Quantification of the BODIPY staining area (Figure 4B) and lipid droplet size (Figure 4C) indicated that intracellular lipid in the cells significantly increased from week 1 to week 3, suggesting continued adipogenesis and lipid loading during this time. A reconstructed 3D confocal image of adipocytes within the microfluidic device is shown in Figure S3.
Figure 4. ADSCs differentiation within the microfluidic device.

(A) Brightfield and BODIPY staining images of cells at week 1 through week 3. (B) BODIPY staining quantification showed increased lipid loading through the course of 3 weeks. Values are means ± SD, n = 9 or 10. (C) Lipid droplet size at different time points during adipogenic differentiation. Values are means ± SD, n = 5. (D) Gene expression of adipocytes in microfluidic chambers at different time points. Values are means ± SD, n = 4 or 5. (E) Gene expression of adipocytes in microfluidic chambers with different densities. Values are means ± SD, n = 3 to 5. For all datasets, one-way ANOVA followed by Tukey’s multiple comparisons test was performed. *denotes significant difference (p < 0.05) between the marked groups.
The expression of adipogenic markers was also examined at different time points. ADSCs were loaded at 10 million/mL into the microfluidic devices and differentiated for 1 to 3 weeks before the microtissues were harvested for RT-PCR (Figure 4D). ADIPOQ expressions at week 2 and week 3 were significantly upregulated compared to week 1 cells, suggesting increased ipocyte maturity over time. CEBPA expression at week 3 was significantly upregulated compared to week 1 cells, suggesting faster lipid droplet accumulation55. Although not statistically significant, the PPARγ expression showed upregulation during the differentiation of the cells. Overall, RT-PCR data indicated increased adipogenesis during the 3 weeks of differentiation.
To study the influence of cell density on adipogenesis, ADSCs were loaded at 5 million/mL, 10 million/mL, and 20 million/mL densities and differentiated for 3 weeks before being extracted for RT-PCR (Figure 4E). ADIPOQ expression was shown to be highest at 20 million/mL. This result was consistent with a study in 2D in which a higher density of human mesenchymal stem cells favored adipogenesis56. Although the higher density is preferable for adipogenesis, the increased cell density might lead to uneven development within the cell culture chambers, as shown in Figure S4.
Microtissue developed in the microfluidic devices for 4 weeks were characterized by both BODIPY staining and SEM. Figure 5A shows the BODIPY staining (left column, scale bar: 200 μm) and SEM (right column, scale bar: 300 μm) images of the adipose tissue at different densities (5 million/mL, 10 million/mL, and 20 million/mL) after 4 weeks of differentiation. During the dehydration process for SEM analysis, the microtissues went through volume shrinkage due to the high-water content of the fibrin hydrogel. At 5 million/mL ADSCs density, the tissue scattered into smaller pieces. At 10 million/mL and 20 million/mL ADSCs densities, the tissues showed less shrinkage and bigger pieces, indicating higher volume taken up by adipocytes and ECM secreted by the adipocytes. Overall, SEM images confirmed the formation of 3D adipose tissue within the microfluidic chambers. Figure 5B shows BODIPY staining quantification of the microtissues at different seeding densities. Lipid staining percent area increases with higher initial seeding density. Figure 5C shows the quantification of lipid droplet size. Cells seeded at 5 million/mL showed a bigger average lipid droplet size than other groups, suggesting that a lower density in the chamber provides bigger room for the cells to expand. Figure 5D shows the lipid content of the adipocytes in the microfluidic device (left) and native adipose tissue57 (right). Lipid diameter from cells in the engineered adipose tissue is approaching that of native adipose tissue.
Figure 5. BODIPY staining and SEM imaging of adipose tissue in the microfluidic device.

(A) BODIPY staining (left column, scale bar: 200 μm) and SEM images (right column, scale bar: 300 μm) of adipose tissue formed in the microfluidic devices at 4 weeks under three different cell densities. (B) BODIPY stain percent area of adipose tissue under different cell densities. (C) Lipid droplet size at different cell densities. (D) Lipid content of the adipocytes in microfluidic device (left) and of native adipose tissue (right, modified from Aubin, K et al. 2015). For all datasets, values are means ± SD, n = 5. One-way ANOVA followed by Tukey’s multiple comparisons test was performed. *denotes significant difference (p < 0.05) between the marked groups.
Fatty acid uptake
The uptake of fatty acids in adipocytes is important for energy storage and lipid distribution. We used a fluorescently labeled fatty acid analog to visualize fatty acid uptake of the differentiated adipocytes in the microfluidic device. The fatty acid analog was applied to one side channel at a concentration of 1 μM/mL in medium. The active fatty acid uptake was verified in Figure S5. Figure 6A shows fluorescent images of the chamber after 4, 8, 24, 48, 72, and 96 hours of incubation. Figure 6B shows a brightfield image of the cell chamber. The fluorescent fatty acid was initially taken up near the inlet side channel after a few hours but over days reached the opposite side of the chamber. A possible explanation for the significant lag time is that adipocytes near the inlet side channel were taking up the fatty acid in the medium relatively fast so that the fatty acid concentration was dropping as the medium was perfused through the cell culture chamber. Figure 6C shows the comparison of fatty acid uptake at different cell densities. For all groups, the fluorescent signal intensity increased with time, suggesting continued fatty acid uptake. Although there was not a significant difference, the signal intensity from the group with 20 million/mL cells had a lower average value than the other groups. This may suggest that higher cell density leads to slower medium perfusion or faster uptake of fatty acid near the inlet side channel.
Figure 6. Adipose tissue fatty acid uptake in the microfluidic device.

(A) Fluorescent images of fatty acid uptake at different time points. (B) Bright-field image of the cell culture chamber (scale bar: 200 μm). (C) Quantification of fatty acid uptake at different ADSCs densities. Values are means ± SD, n = 5. One-way ANOVA followed by Tukey’s multiple comparisons test was performed.
Effects of shear stress
To study the effect of interstitial flow on the adipocytes in the microfluidic device, adipocytes were exposed to different flow rates for 5 days after 3 weeks of differentiation. Volumetric flow rates of 1, 4, and 10 μl/hour from the syringe pumps which, based on modeling studies, generated average interstitial flow velocities of 0.324 μm/s, 1.29 μm/s, and 3.24 μm/s, respectively were applied to the microfluidic devices. The flow-through medium was collected to test for adipokine secretion and free fatty acid secretion. The cells in the microfluidic chambers were then extracted for RT-PCR. ELISA results showed that adiponectin secretion was significantly reduced at 4 μl/hour and 10 μl/hour compared to the 1 μl/hour flow rate (Figure 7A). Leptin secretion was not significantly different among the groups (Figure 7B). Meanwhile, Free fatty acid secretion increased significantly with increased flow rate (Figure 7C), suggesting higher levels of lipolysis at higher shear stresses. As shown in Figure 7D, ADIPOQ, CEBPA, PPARG, PLIN1, AP2, and IL1B genes were all downregulated with increased flow rate, indicating that adipocytes favor a low shear stress environment. IL6 gene was upregulated with an increased flow rate.
Figure 7. Adipose tissue response to different shear stresses.
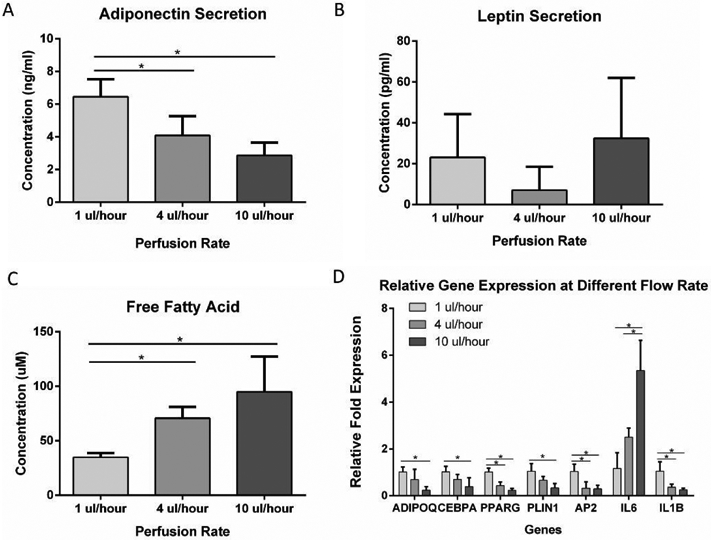
(A) Adiponectin secretion from adipose tissue in the microfluidic device at different perfusion rates. (B) Leptin secretion from adipose tissue in the microfluidic device at different perfusion rates. (C) Free fatty acid secretion from adipose tissue in the microfluidic device at different perfusion rates. (D) Gene expression of adipose tissue in the microfluidic device in response to different perfusion rates. For all datasets, values are means ± SD, n = 5. One-way ANOVA followed by Tukey’s multiple comparisons test was performed. *denotes significant difference (p < 0.05) between the marked groups.
Discussion
The effort for establishing in vitro cell culture model that can faithfully represent the in vivo environment has led to the development of enormous tissue-on-chip models. As an essential endocrine organ, adipose tissue has been engineered into different microfluidic devices to study adipocyte function or interaction with other tissues. For example, in Kongsuphol et al.’s design, two cell culture compartments were designed to host adipocytes and immune cells respectively58. Human preadipocytes were cultured on the 2D surface of the lower compartment until confluence before the differentiation medium was applied to start adipogenesis for 14 days. Similarly, in Liu et al.’s paper to construct an adipose model for immune-metabolic analysis in type II diabetes, two compartments separated by micro-channel arrays were used to culture adipocytes and immune cells 59. Pre-adipocytes were also cultured and differentiated on a 2D surface in the compartment. After a few days of differentiation, cells in the adipocyte compartment showed detachment from the surface. In Zhu et al.’s model, pre-adipocytes were differentiated for 14 days before co-cultured with macrophages in the same compartment 60. Compared to the currently published models, our system focuses on the establishment and study of a 3D adipose microtissue in which the adipocytes were differentiated in the presence of ECM, in this case, fibrin. The difference between 2D and 3D culture of human adipocytes has been well characterized recently15. Cells in 3D culture showed faster adipogenesis, higher leptin secretion, and upregulation of multiple adipogenic genes compared to 2D culture. To provide interstitial flow to the cells in the 3D environment, both reservoirs and syringe pumps were used. Reservoirs were used at the beginning of the adipogenic differentiation to generate uniformly differentiated cells. Syringe pumps were then used to provide more controlled interstitial flow and facilitate addition assays. Figure S6 shows that adiponectin expression is significantly upregulated in 3D microfluidic devices compared with 2D culture in well-plates. Supplementary videos showed a distinct difference in the morphology of adipocytes cultured in 2D and 3D (Video S1 and Video S2). Cells cultured in 2D spread on the surface and exhibited limited cell-cell contact. Cells cultured in chambers showed clusters of cells. The clusters were present in multiple focal planes, indicating that they were in a 3D environment within the fibrin gel.
Using the microfluidic system, we realized the on-chip engineering of 3D human adipose tissue and analyzed the response of adipose tissue to interstitial shear stress. Human ADSCs were differentiated toward adipocytes in the microfluidic chambers to form a 3D human adipose microtissue. The upregulation of ADIPOQ and C/EBPα in cells in the microfluidic chambers from 1 week to 3 weeks showed increased adipocyte maturity during differentiation. Expressions of adipogenic markers were not significantly different between week 2 and week 3 cells, suggesting the adipocytes reached maturity after 2 weeks of differentiation. The higher density of ADSCs showed upregulated ADIPOQ expression, suggesting a high density of ADSCs was favorable for adipogenesis.
In the body, interstitial shear stress is induced by extravascular fluid flowing through the extracellular matrix. At normal physiological conditions, the interstitial shear stress is estimated to be lower than 0.01 Pa34,36. Current studies on the effect of flow on adipogenesis are either on 2D surfaces or resulted from applying much higher fluid shear stress than interstitial levels. The influence of interstitial levels of shear stress on 3D cultured adipocytes has been investigated for the first time in this work. Data shows that even at low levels of interstitial flow, adipose tissue showed decreased adiponectin secretion and increased lipolysis in response to increased interstitial shear stress. RT-PCR analysis showed that ADIPOQ, CEBPA, PPARG, PLIN1, and AP2 genes were downregulated with increased shear stress. ADIPOQ modulates numerous pathways related to fat storage and metabolism through the secretion of adiponectin, a protein hormone that can regulate insulin sensitivity and fatty acid breakdown61. During maturation, differentiating adipocytes accumulate lipid droplets through the expression of CEBPA and PPARG gene55. Perilipin 1, encoded by the PLIN1 gene, is the most abundant lipid droplet-associated protein in adipocytes and is essential for the mobilization of lipids in adipose tissue62. Adipocyte Protein 2 (aP2) is closely linked to both metabolic and inflammatory processes through modulating lipid-sensitive pathways in adipocytes63. IL6, a gene that has been shown to respond to the change of shear stress64-66, was upregulated with the increases of interstitial flow. The downregulation of multiple adipogenic markers in response to increased shear stress suggests that even the interstitial level of flow can have inhibitory effects on adipogenesis.
Shear stress has been shown to have varied effects on different cell types. There has been evidence that interstitial fluid flow may act as a signal of blood pressure and blood flow rate and stimulate vascular smooth muscle cells53. Shear stress has significant effects in modifying the tissue microenvironment. Studies have shown that fibroblasts in vitro began to align the collagen matrix after stimulation with high levels of interstitial flow and cells then reorient in the direction of the newly realigned matrix33. Interstitial flow can directly promote tumor cell invasion through autologous chemotaxis, and indirectly support tumor invasion via biophysical cues from stromal cells 35. Shear stress induced by the interstitial level of flow promoted osteogenic differentiation of mesenchymal stem cells (MSCs)34. Adipogenesis is easily affected by mechanical stimuli such as substrate stiffness19,67, compressive forces68, and shear stress69. PPARγ2 and C/EBPα expressions are shown to be inhibited by fluid shear stress during adipocyte maturation, leading to reduced lipid accumulation69. The inhibition of adipogenesis and promotion of osteogenesis from sheer stress is in accordance with the discovery that osteogenesis and adipogenesis are reciprocally controlled by ERK/MAP kinase phosphorylation of Runx2 and PPARγ transcription factors70. Due to the micro-scale of the microfluidic chambers, proteins collected from the system were not sufficient for western blot analysis. There have been reports of validating on-chip hASCs adipogenesis at the protein level by using off-chip cultured cells25. However, off-chip culture often could not represent the sheer stress that the cells experience in the micro-environment in the microfluidic chamber. Currently, we are exploring new assays to facilitate more accurate protein analysis in microscale culture systems71.
Conclusions
This work presented a microfluidic system that enables the engineering and analysis of 3D human adipose tissue under controlled flow. The engineered adipose microtissue resembles the function and morphology of native adipose tissue. By controlling interstitial flow in the 3D micro-tissue, this work explored adipose tissue response under physiological level interstitial shear stress. This model could potentially serve as an in vitro drug testing platform for adipose tissue-related diseases.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Patrick Karnia at the Illinois Institute of Technology Idea Shop Prototyping Lab for assistance with microfluidic mold CNC machining, and Dr. Carlo U. Segre’s lab for assistance with SEM imaging. This research was funded by the National Institute of Health (5R01EB020604-02).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Luo L and Liu M, J. Endocrinol, 2016, 231, R77–R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prieto-Hontoria PL, Pérez-Matute P, Fernández-Galilea M, Bustos M, Martínez JA and Moreno-Aliaga MJ, Biochim. Biophys. Acta - Bioenerg, 2011, 1807, 664–678. [DOI] [PubMed] [Google Scholar]
- 3.Rosen ED and Spiegelman BM, Nature, 2006, 444, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Percik R and Stumvoll M, Exp. Clin. Endocrinol. Diabetes, 2009, 117, 563–566. [DOI] [PubMed] [Google Scholar]
- 5.Ungefroren H, Gieseler F, Fliedner S and Lehnert H, Horm. Mol. Biol. Clin. Investig, 2015, 21, 5–15. [DOI] [PubMed] [Google Scholar]
- 6.Jung UJ and Choi MS, Int. J. Mol. Sci, 2014, 15, 6184–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao H, J. Endocrinol, 2014, 220, T47–T59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nawrocki AR and Scherer PE, Drug Discov. Today, 2005, 10, 1219–1230. [DOI] [PubMed] [Google Scholar]
- 9.Lee MJ, Wu YY and Fried SK, Obesity, 2012, 20, 2334–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farmer SR, Cell Metab, 2006, 4, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W and Chen CS, Cell Stem Cell, 2009, 5, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girandon L, Kregar-Velikonja N, Bozikov K and Barlic A, Folia Biol. (Praha), 2011, 57, 47–56. [PubMed] [Google Scholar]
- 13.Stacey DH, Hanson SE, Lahvis G, Gutowski KA and Masters KS, Tissue Eng. Part A, 2009, 15, 3389–3399. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y. Il, Kim HI, Choi MY, Son SH, Seo MJ, Seo JY, Jang WH, Youn YC, Choi KJ, Cheong SH and Shelby J, J. Cell. Physiol, 2010, 224, 807–816. [DOI] [PubMed] [Google Scholar]
- 15.Louis F, Kitano S, Mano JF and Matsusaki M, Acta Biomater., 2018, 84, 194–207. [DOI] [PubMed] [Google Scholar]
- 16.Abbott RD, Raja WK, Wang RY, Stinson JA, Glettig DL, Burke KA and Kaplan DL, Methods, 2015, 84, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korurer E, Kenar H, Doger E and Karaoz E, J. Biomed. Mater. Res. Part A, 2014, 102, 2220–2229. [DOI] [PubMed] [Google Scholar]
- 18.Fischbach C, Seufert J, Staiger H, Hacker M, Neubauer M, Göpferich A and Blunk T, Tissue Eng., 2004, 10, 215–229. [DOI] [PubMed] [Google Scholar]
- 19.Vaicik MK, Morse M, Blagajcevic A, Rios J, Larson JC, Yang F, Cohen RN, Papavasiliou G and Brey EM, J. Mater. Chem. B, 2015, 3, 7903–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerlach JC, Lin Y-C, Brayfield CA, Minteer DM, Li H, Rubin JP and Marra KG, Tissue Eng. Part C-Methods, 2012, 18, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godwin LA, Brooks JC, Hoepfner LD, Wanders D, Judd RL and Easley CJ, Analyst, 2015, 140, 1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai N, Sims JK, Jeon NL and Lee K, Tissue Eng. Part C-Methods, 2012, 18, 958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark AM, Sousa KM, Jennings C, MacDougald OA and Kennedy RT, Anal. Chem, 2009, 81, 2350–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dugan CE, Grinias JP, Parlee SD, El-Azzouny M, Evans CR and Kennedy RT, Anal. Bioanal. Chem, 2017, 409, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, Schneider N, Platen A, Mitra I, Blazek M, Zengerle R, Schule R and Meier M, Proc Natl Acad Sci U S A, 2016. 113, E4143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanataweethum N, Zelaya A, Yang F, Cohen RN, Brey EM and Bhushan A, Biotechnol. Bioeng, 2018, 115, 1979–1987. [DOI] [PubMed] [Google Scholar]
- 27.Loskill P, Sezhian T, Tharp KM, Lee-Montiel FT, Jeeawoody S, Reese WM, Zushin PJH, Stahl A and Healy KE, Lab Chip, 2017, 17, 1645–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Kongsuphol P, Gourikutty SBN and Ramadan Q, Biomed. Microdevices, 2017, 19, 1–10. [DOI] [PubMed] [Google Scholar]
- 29.Zambon A, Zoso A, Gagliano O, Magrofuoco E, Fadini GP, Avogaro A, Foletto M, Quake S and Elvassore N, Anal. Chem, 2015, 87, 6535–6543. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Brooks JC, Hu J, Ford KI and Easley CJ, Lab Chip, 2017, 17, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eto H, Suga H, Matsumoto D, Inoue K, Aoi N, Kato H, Araki J and Yoshimura K, Plast. Reconstr. Surg, 2009, 124, 1087–1097. [DOI] [PubMed] [Google Scholar]
- 32.Swartz MA and Fleury ME, Annu. Rev. Biomed. Eng, 2007, 9, 229–256. [DOI] [PubMed] [Google Scholar]
- 33.Ng CP and Swartz MA, Ann. Biomed. Eng, 2006, 34, 446–454. [DOI] [PubMed] [Google Scholar]
- 34.Kim KM, Choi YJ, Hwang JH, Kim AR, Cho HJ, Hwang ES, Park JY, Lee SH and Hong JH, PLoS One,, DOI: 10.1371/journal.pone.0092427. [DOI] [Google Scholar]
- 35.Shieh AC and Swartz MA, Phys. Biol, , DOI: 10.1088/1478-3975/8/1/015012. [DOI] [PubMed] [Google Scholar]
- 36.Chary SR and Jain RK, Proc. Natl. Acad. Sci, 1989, 86, 5385–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moya ML, Hsu YH, Lee AP, Hughes CCW and George SC, Tissue Eng. Part C-Methods, 2013, 19, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu Y-H, Moya ML, Abiri P, Hughes CCW, George SC and Lee AP, Lab Chip, 2013, 13, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu Y-H, Moya ML, Hughes CCW, George SC and Lee AP, Lab Chip, 2013, 13, 2990–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Lee H, Chung M and Jeon NL, Lab Chip, 2013, 13, 1489–1500. [DOI] [PubMed] [Google Scholar]
- 41.Campisi M, Shin Y, Osaki T, Hajal C, Chiono V and Kamm RD, Biomaterials, 2018, 180, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sobrino A, Phan DTT, Datta R, Wang XL, Hachey SJ, Romero-Lopez M, Gratton E, Lee AP, George SC and Hughes CCW, Sci. Rep, , DOI: 10.1038/srep31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M and Kamm RD, Proc. Natl. Acad. Sci, 2015, 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verseijden F, Sluijs SJP, van Neck JW, Hofer SOP, Hovius SER and van Osch G, Cell Transplant., 2012, 21, 2283–2297. [DOI] [PubMed] [Google Scholar]
- 45.Ho C-T, Lin R-Z, Chen R-J, Chin C-K, Gong S-E, Chang H-Y, Peng H-L, Hsu L, Yew T-R, Chang S-F and Liu C-H, Lab Chip, 2013, 13, 3578–3587. [DOI] [PubMed] [Google Scholar]
- 46.Huang CP, Lu J, Seon H, Lee AP, Flanagan LA, Kim H-Y, Putnam AJ and Jeon NL, Lab Chip, 2009, 9, 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammer CF, Koch TA and Whitney JF, J. Appl. Polym. Sci, 1959, 1, 169–178. [Google Scholar]
- 48.Fröhlich E, Bonstingl G, Höfler A, Meindl C, Leitinger G, Pieber TR and Roblegg E, Toxicol. Vitr, 2013, 27, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang XL, Phan DTT, Sobrino A, George SC, Hughes CCW and Lee AP, Lab Chip, 2016, 16, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Livak KJ and Schmittgen TD, Methods, 2001, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 51.Bai J and Pagano RE, Biochemistry, 1997, 36, 8840–8848. [DOI] [PubMed] [Google Scholar]
- 52.Kasurinen J, Biochem. Biophys. Res. Commun, 1992, 187, 1594–1601. [DOI] [PubMed] [Google Scholar]
- 53.Wang S and Tarbell JM, Arterioscler. Thromb. Vasc. Biol, 2000, 20, 2220–2225. [DOI] [PubMed] [Google Scholar]
- 54.Shi Z-D, Ji X-Y, Qazi H and Tarbell JM, Am. J. Physiol. Circ. Physiol, 2009, 297, H1225–H1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang Q-Q, Zhang J-W and Daniel Lane M, Biochem. Biophys. Res. Commun, 2004, 318, 213–218. [DOI] [PubMed] [Google Scholar]
- 56.McBeath R, Pirone DM, Nelson CM, Bhadriraju K and Chen CS, Dev. Cell, 2004, 6, 483–495. [DOI] [PubMed] [Google Scholar]
- 57.Aubin K, Safoine M, Proulx M, Audet-Casgrain MA, Cote JF, Tetu FA, Roy A and Fradette J, PLoS One, , DOI: 10.1371/journal.pone.0137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kongsuphol P, Gupta S, Liu Y, Bhuvanendran Nair Gourikutty S, Biswas SK and Ramadan Q, Sci. Rep, 2019, 9, 4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Kongsuphol P, Chiam SY, Zhang QX, Gourikutty SBN, Saha S, Biswas SK and Ramadan Q, Lab Chip, 2019, 19, 241–253. [DOI] [PubMed] [Google Scholar]
- 60.Zhu J, He J, Verano M, Brimmo AT, Glia A, Qasaimeh MA, Chen P, Aleman JO and Chen W, Lab Chip, 2018, 18, 3550–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diez JJ and Iglesias P, Mini-Reviews Med. Chem, 2010, 10, 856–869. [DOI] [PubMed] [Google Scholar]
- 62.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ and Londos C, J. Biol. Chem, 1991, 266, 11341–11346. [PubMed] [Google Scholar]
- 63.Furuhashi M HG, Nat. Rev. Drug Discov, 2010, 7, 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.V Sterpetti A, Cucina A, Morena AR, Di Donna S, D’Angelo LS, Cavallaro A and Stipa S, Surgery, 1993, 114, 911–914. [PubMed] [Google Scholar]
- 65.Wang P, Zhu F, Lee NH and Konstantopoulos K, J. Biol. Chem, 2010, 285, 24793–24804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitajima M, Miyazawa M, Ueda M and Kitajima M, Gastroenterology, 2000, 118, A903. [Google Scholar]
- 67.Zhao W, Li X, Liu X, Zhang N and Wen X, Mater. Sci. Eng. C, 2014, 40, 316–323. [DOI] [PubMed] [Google Scholar]
- 68.Hossain MG, Iwata T, Mizusawa N, Shima SWN, Okutsu T, Ishimoto K and Yoshimoto K, J. Biosci. Bioeng, 2010, 109, 297–303. [DOI] [PubMed] [Google Scholar]
- 69.Choi J, Lee SY, Yoo YM and Kim CH, Cell Biochem. Biophys, 2017, 75, 87–94. [DOI] [PubMed] [Google Scholar]
- 70.Ge C, Cawthorn WP, Li Y, Zhao G, MacDougald OA and Franceschi RT, J. Cell. Physiol, 2016, 231, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christenson C, Baryeh K, Ahadian S, Nasiri R, Dokmeci MR, Goudie M, Khademhosseini A and Ye JY, in Label-free Biomedical Imaging and Sensing (LBIS) 2020, eds. Shaked NT and Hayden O, SPIE, 2020, vol. 176, p. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.