

Abstract
The mosquito body hosts highly diverse microbes, which influence different physiological traits of both larvae and adults. The composition of adult mosquito microbiota is tightly linked to that of larvae, which are aquatic and feed on organic detritus, algae and prokaryotic microorganisms present in their breeding sites. Unraveling the ecological features of larval habitats that shape the structure of bacterial communities and their interactions with the mosquito host is still a poorly investigated topic in the Asian tiger mosquito Aedes albopictus, a highly invasive species that is vector of numerous arboviruses, including Dengue, Chikungunya, and Zika viruses. In this study, we investigated the composition of the bacterial community present in the water from a natural larval breeding site in which we separately reared wild-collected larvae and hatched eggs of the Foshan reference laboratory strain. Using sequence analysis of bacterial 16S rRNA gene amplicons, we comparatively analyzed the microbiota of the larvae and that of adult mosquitoes, deriving information about the relative impact of the breeding site water on shaping mosquito microbiota. We observed a higher bacterial diversity in breeding site water than in larvae or adults, irrespective of the origin of the sample. Moreover, larvae displayed a significantly different and most diversified microbial community than newly emerged adults, which appeared to be dominated by Proteobacteria. The microbiota of breeding site water significantly increased its diversity over time, suggesting the presence of a dynamic interaction among bacterial communities, breeding sites and mosquito hosts. The analysis of Wolbachia prevalence in adults from Foshan and five additional strains with different geographic origins confirmed the described pattern of dual wAlbA and wAlbB strain infection. However, differences in Wolbachia prevalence were detected, with one strain from La Reunion Island showing up to 18% uninfected individuals. These findings contribute in further understanding the dynamic interactions between the ecology of larval habitats and the structure of host microbiota, as well as providing additional information relative to the patterns of Wolbachia infection.
Keywords: breeding site, water, larvae, 16S rRNA gene, Wolbachia
Introduction
The microbiota of Aedes mosquitoes is known to play a significant role in host physiology, including egg production, blood digestion (Gaio Ade et al., 2011; Coon et al., 2016a), immunity regulation (Xi et al., 2008), host–pathogen interaction and vector competence (Ramirez et al., 2012; Charan et al., 2013; Goncalves et al., 2014; Dickson et al., 2017; Souza-Neto et al., 2019).
The microbiota of both laboratory and wild Aedes mosquitoes has been investigated and shown to be dominated by Proteobacteria (Wang et al., 2018). However, despite the overall composition of the microbiota was similar between laboratory-reared and wild mosquitoes (David et al., 2016), the diversity of midgut bacterial communities was found to be higher in field-caught mosquitoes (Osei-Poku et al., 2012). In Aedes aegypti, different bacterial communities were detected between domestic and sylvatic habitats (Dickson et al., 2017). Moreover, when Ae. aegypti populations from the field were reared in the laboratory, they were shown to display a similar midgut microbiota (Dickson et al., 2018). These findings indicate the importance of the environment in shaping mosquito microbiota.
Mosquitoes are holometabolous insects with larval and adult stages occupying different ecological niches, thus exploiting different resources. Larvae develop in aquatic habitats, while adults are terrestrial. Larval stages acquire their symbionts primarily through feeding in their breeding site water, with their microbiota representing a subset of the bacteria found in the water (Coon et al., 2014, 2016b; Dada et al., 2014; Dickson et al., 2017; Wang et al., 2018). Adults can introduce bacteria through feeding on nectar (Merritt et al., 1992), imbibing water from their breeding sites at emergence, as occurring in Anopheles gambiae, or trans-stadially from larval gut bacterial during metamorphosis (Lindh et al., 2008). Thus, larval and adult stages are not independent from each other and biotic and abiotic features of the larval environment can influence adult microbiota.
The Asian tiger mosquito Aedes albopictus (Skuse, 1894) is a highly invasive species of growing public health concern due to its ability to transmit at least 22 arboviruses, including Chikungunya, Dengue, and Zika viruses (ISSG Invasive Species Specialist Group, 2020; Lwande et al., 2020). The worldwide expansion of this species was favored both by anthropogenic factors such as increased mobility and trades, and biological characteristics of this species, such as its ability to undergo photoperiodic diapause and use natural and artificial breeding sites. A number of studies have described the microbiota of Ae. albopictus in recent years, including the prevalence of Wolbachia (Zouache et al., 2009; Chouaia et al., 2010; Minard et al., 2013; Valiente Moro et al., 2013; Minard et al., 2014, 2015; Yadav et al., 2015, 2016; Coon et al., 2016b; Park et al., 2016; Ahmad et al., 2017; Muturi et al., 2017; Mancini et al., 2018; Rosso et al., 2018; Thongsripong et al., 2018; Wang et al., 2018; Guegan et al., 2018; Hu et al., 2020; Mancini et al., 2020).
Wolbachia is a genus of Gram-negative bacteria infecting about 40% of arthropod species (Zug and Hammerstein, 2012) and almost 30% of mosquito species (Ricci et al., 2002). Wolbachia is transovarially transmitted and is able to exert a number of reproductive manipulations, such as cytoplasmic incompatibility, that can be exploited for the control of mosquito populations (Floate et al., 2006; Bourtzis et al., 2014). Moreover, the generation of novel Wolbachia transinfections in Ae. aegypti showed to have an impact on host susceptibility for several pathogens (see for example Moreira et al., 2009; Bian et al., 2010; Walker et al., 2011; van den Hurk et al., 2012; Frentiu et al., 2014; Ye et al., 2015; Ant et al., 2018). Aedes albopictus transinfected with the wMel Wolbachia strain is unable to transmit Dengue (Blagrove et al., 2012) or Chikungunya (Blagrove et al., 2013) viruses in laboratory assays. Aedes albopictus is naturally superinfected with two Wolbachia strains, wAlbA and wAlbB (Dutton and Sinkins, 2004). Thus, to develop effective Wolbachia-based strategies for controlling Ae. albopictus, it is essential to assess the stability and population invasion potential for Wolbachia newly generated infections. Because this is most likely linked to Wolbachia inter-strain interactions (Ant and Sinkins, 2018), it important to determine wAlbA and wAlbB prevalence across Ae. albopictus populations and strains.
In Ae. albopictus, most studies focused on the characterization of adult endosymbionts, and little is known about larval microbiota and how it can shape adult biological traits. Additionally, the complexity of the interactions between the mosquito host and the bacteria in breeding sites and the relationships between Wolbachia infection and other components of the microbiota are still poorly understood.
In this exploratory study, we aimed at expanding the current understanding of the dynamic interactions between the ecology of larval habitats and the structure of host microbiota, as well as the patterns of Wolbachia infection. To do so, we addressed three main questions: (i) to what extent is the breeding site microbiota affecting endosymbiont community assemblage in larval and adult Ae. albopictus? (ii) Is the bacterial community of the breeding site water changing over time during mosquito development? (iii) What is the frequency of Wolbachia in adult samples reared as larvae in the wild-collected water?
To answer these questions, we compared bacterial community composition among (i) water from natural larval breeding sites, (ii) mosquito larvae, and (iii) adult individuals. In water from a natural breeding site, we reared both wild-collected larvae and larvae of the Foshan reference strain to derive information about the relative impact of the environment and the genetic background of mosquitoes on shaping mosquito microbiota. Finally, we analyzed Wolbachia presence in Foshan and five additional laboratory strains with different geographic origins.
Materials and Methods
Water Sampling
Water from Ae. albopictus larval breeding sites was collected in a private garden in Crema, Italy (45°21′51.2′′N 9°40′57.7′′E), which was an accessible site characterized by high population density, at the end of August 2018, when climate conditions are optimal for mosquito development. Domestic collection of water, eggs, and larvae originated from two plastic buckets of a maximum volume of 500 ml that were placed in the same garden next to ornamental plants. Environmental water collected in these buckets derived from dew and rain and was monitored for its level and clarity every day for 4 days. No sedimentary layer was observed. Water and eggs/larvae were then collected in sterile 50-ml Falcon tubes and transferred to the insectary of the University of Pavia, Italy. The collected water was divided into three aliquots and used as follows: (i) an aliquot of 200 ml was brought to the ZooPlant laboratory in Milano, Italy, to characterize its microbial composition (samples hereafter called W Start); (ii) 300 ml were aliquoted in two different pans (150 ml/pan) and daily monitored for natural larval hatching in the Pavia insectary (samples named CR); (iii) 600 ml were used to hatch eggs from the Foshan strain (Chen et al., 2015; Palatini et al., 2020), after having verified the absence of wild eggs using a stereomicroscope. The Foshan strain is an established laboratory colony derived from wild mosquitoes from South-East China and reared in the Pavia insectary since 2013 (Palatini et al., 2017). A total of 400 Foshan eggs were hatched, in batches of 100, each in 150 ml of breeding site water (samples named FO). To avoid overcrowding, the amount of water was determined based on the average hatching rate of the Foshan strain in our insectary conditions. No food was added in any pan to allow larvae to grow based on the nutrients present in the breeding site water.
For both the CR and FO samples, the developing individuals were collected separately as fourth instar larvae (in pools of eight individuals) and adults (individually collected the day of their emergence). In the case of adult collections, pupae developed in the larval rearing pans were individually transferred to cups containing 10 ml of the same wild water until adult eclosion. When mosquito development was completed and all adults had eclosed, the remaining water from each rearing pan was transferred into sterile 50 ml Falcon tubes for the analysis of the microbiota (samples hereafter called W End).
All mosquito life stages were maintained in the Pavia insectary at 28°C and 80% RH, with a photoperiod of 12:12 hrs light:dark.
Chemical Analyses of Water Samples
For each water sample, total nitrogen (N_tot), nitrites (NO2), nitrates (NO3), ammoniacal nitrogen (NH4-N), and phosphate (P_tot) were measured with a spectrophotometer (Spectroquant Pharo 300, Merck). Moreover, pH and conductivity were also recorded. Analyses were performed using the following kits, according to the manufacturer’s instructions: Ammonium Test Photometric Method NH4-N, Nitrate Test Photometric Method NO3-N, Nitrite Test Photometric Method NO2-N, Total Nitrogen Test Photometric Method, Phosphate Test Photometric Method PO4-P, Merck Spectroquant®.
Sample Pre-processing and DNA Extraction
Genomic DNA was extracted from (i) water samples at the two time-points described above; (ii) pooled larval (L) samples; (iii) individual adult (A) mosquitoes collected immediately after eclosion.
Water samples were processed according to a modified version of a previously described protocol (Bruno et al., 2017a). First water samples were filtered by a serial vertical (orthogonal) filtration using nitrocellulose membrane filters with a pore size of 8 μm, followed by a filtration with a membrane with pores of 3 μm. Vacuum was generated by a vacuum pump (ME 2 NT VacuubrandTM) connected to a filtering apparatus. The filtrate was collected and concentrated using tangential flow filtration (TFF) to recover as much biological material as possible. The TFF system involved a peristaltic pump (Masterflex L/S Economy Drive), Tygon® tubing, sterile reservoirs and filtration modules. The used tangential flow filter was a VivaFlow® 200 cassette (Sartorius) made of polyethersulfone (PES) with a nominal pore rating of 10000 MWCO and a surface area of 200 cm2. The system was scaled up with an additional unit connected in parallel to increase the filtration surface area and the flow speed. The TFF system was run at a transmembrane pressure of 1.5 bar. The initial water samples were concentrated to a final retentate volume of 100 mL, which was further reduced to 1 mL through the Vivaspin® 20 ultrafiltration unit (Sartorius) made of PES with a nominal pore rating of 10000 MWCO. The final volume was allowed to be adsorbed onto a membrane filter before DNA extraction.
All tubing, tubing connections, and containers were sterilized with sodium hypochlorite or autoclaved prior to each experiment and among samples. Every step was conducted in the laminar flow cabinet in a pre-amplification dedicated laboratory.
Environmental DNA was extracted separately from each of the filters obtained by water filtration using DNeasy® PowerSoil® Kit (Qiagen) with the QIAcube (Qiagen) automated system, following the manufacturer protocol. DNA was eluted in 75 μL of warmed (40°C) elution buffer, to increase DNA concentration. DNA extraction negative controls were included.
A total of 25 Ae. albopictus samples were processed, namely 11 larval pools (four CR and seven FO samples, respectively) and 14 individual adults (five CR, of which four males and one female; nine FO, of which six males and three females, respectively). The finding that sequencing pools of six mosquitoes allowed to capture a level of bacterial diversity comparable to that of single mosquitoes (Bennett et al., 2019), together with our aim to achieve the maximum information level, prompted us to process larvae and adults differently. Larval samples were treated as pools of eight individuals, similarly to other studies (e.g., Wang et al., 2018) to minimize the biological variability of the microbiota among individuals and, at the same time, avoid overcycling in amplicon PCR. Individual adults were also used to obtain data related to Wolbachia infection at the single-mosquito level. Prior to DNA extraction from both larval pools and adults, each individual was surface-washed twice with 1X PBS, after washing in ethanol 70% (following Seabourn et al., 2020). DNA extraction was performed using the Wizard Genomic DNA Purification Kit (Promega) following the manufacturer’s instructions. DNA extraction negative controls were included.
All the procedures were carried out in a laminar flow cabinet, in order to avoid contamination with exogenous DNA and inter-samples contamination, and in separate rooms for the pre- and post- amplification steps, with dedicated personal protective equipment.
16S Metagenomic Sequencing of Mosquito and Water Samples
Illumina MiSeq 16S libraries were generated following the standard protocol “16S Metagenomic Sequencing Library Preparation, Part # 15044223 Rev B.” Amplicon PCR was performed using PCR primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) with Illumina library adaptors, for 25 cycles of amplification. DNA extraction negative controls (Bianco_1EXT, Bianco_2EXT) and amplicon PCR negative controls were included in library preparation. Libraries quantification through TapeStation 4100 (Agilent) showed no amplicon signal for amplicon PCR negative controls, which were excluded from the sequencing. In the case of Ae. albopictus samples, amplicon sequencing was run by Macrogen, Inc. using an Illumina MiSeq platform and the Herculase II Fusion DNA Polymerase Nextera XT Index Kit V2. Water samples were sequenced by the Center for Translational Genomics and Bioinformatics – San Raffaele Scientific Institute (Milan, Italy) with Illumina MiSeq 2 × 300 paired-end chemistry (MiSeq Reagent Kit v3).
Illumina Data Processing and Analyses of the Microbiota Composition
The raw paired-end FASTQ reads were imported into the Quantitative Insights Into Microbial Ecology 2 program (QIIME2, ver. 2017.9.01) (Caporaso et al., 2010) and demultiplexed using native plugin. The Divisive Amplicon Denoising Algorithm 2 (DADA2) (Callahan et al., 2016) was used to quality filter, trim, denoise, and mergepair the data and remove chimeric sequences.
The resulting Amplicon Sequence Variants (ASVs) with less than a 50x coverage were discarded from further analyses. The classification of the obtained ASVs was run using the feature-classifier plugin implemented in QIIME2 against the SILVA SSU non-redundant database (138 release), adopting a consensus confidence threshold of 0.8. The analysis on the bacterial diversity as well as the corresponding figures were done using the phyloseq R package (McMurdie and Holmes, 2013).
Any reads assigned to Wolbachia were filtered using the filtered taxa plugin of QIIME2.
Microbiota diversity was described in terms of within (alpha) and between (beta) sample diversities. The Shannon index and Observed Features alpha diversity metrics were calculated to estimate the variation of bacterial diversity in the water, larvae and adult samples. Values were compared using the pairwise Kruskal–Wallis test.
To explore the bacterial diversity of our samples, we used a relative abundance bar plot to show phyla and family distribution between our three sample types (water, larvae, and adults). The distribution of the 100 most abundant ASVs was studied with a heatmap plot. Both visualizations were obtained with the phyloseq R package.
Rarefaction is used to subsample and calculate distances among samples. Beta diversity was estimated with quantitative distance metrics using the diversity QIIME2 plugin based on the rarefied dataset with a sampling depth of 10,000 sequences. Samples with lower depth were automatically discarded from beta diversity analysis.
We estimated the unweighted UniFrac and Bray–Curtis dissimilarity indexes by sampling 10,000 reads per sample (Lozupone et al., 2007). Statistical significance among groups, including sampling site and developmental stage, was determined by a permutation-based ANOVA (PerMANOVA) test using ADONIS (Anderson, 2005) and a 999 permutation-based UniFrac distance metrics. PerMANOVA Pairwise contrast was performed by the beta-group-significance command of diversity plugin. The structure of microbial communities was explored by Non-Multidimensional Scaling (NMDS), an ordination approach (Kruskal, 1964). Sequences representative of each community were aligned with MAFFT and used for phylogenetic reconstruction in FastTree (Price et al., 2010).
The list of bacterial genera, as derived from ASVs, in each sample was compared using Venn diagrams following two criteria. We considered ASVs showing more than 50 reads and occurring in at least two tested samples as ‘general’ ASVs. Then, among these ASVs, we looked for those occurring in at least 70% of all tested samples; we considered these ASVs as the “conserved” mosquito microbiota. This analysis allowed us to explore the flux of bacterial symbionts acquired from the water environment by the mosquito larvae and maintained until the adult stage. Venn diagrams were created using an online tool1.
qPCR Amplification of 16S rRNA Gene in Water Samples
Quantitative Real Time PCR (qPCR) assays were performed targeting the 16S rRNA gene to verify the sensitivity of our approach in detecting the microbiota of water samples and to compare relative bacterial abundances between W Start and W End, according to a protocol previously described (Bruno et al., 2017a). Water samples, samples deriving from TFF filtrate, DNA extraction negative controls and amplification negative controls were tested in triplicates.
Cycling conditions adopted were as follows: an initial denaturation at 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 s and annealing-elongation at 55°C for 1 min. A final dissociation stage was performed. Amplification reaction consisted of 5.0 μl SsoFast EvaGreen Supermix with Low ROX (Bio-Rad S.r.l., Segrate, Milan, Italy), 0.1 μl each 10 μmol l–1 primer solution, 2 μl DNA sample, and 2.8 μl of Milli-Q water. Assays were performed on an AB 7500 thermocycler (Applied Biosystem) and results analyzed as previously described (Bruno et al., 2017a).
Estimates of Wolbachia Prevalence by PCR
To obtain a qualitative validation of Wolbachia infection status in the 25 adult samples used for metagenomic analysis, PCR reactions for Wolbachia detection were performed in 15 μl reaction volumes consisting of 7.5 μl DreamTaq Green Master Mix (Thermo Fisher Scientific, Eugene, OR, United States), 4 μl of autoclaved Milli-Q water, 1 μl primers (10 μM), and 1.5 μl of DNA (∼20 ng). The PCR cycling conditions were as follows: 3 min at 95°C for the initial denaturation step, followed by 35 cycles of 1 min at 95°C, 1 m at 55°C, 1 m at 72°C and 10 min at 72°C for the final extension. The published wsp primers were used (Zhou et al., 1998): 328F (5′-CCAGCAGATACTATTGCG-3′) and 691R (5′-AAAAATTAAACGCTACTCCA-3′) for the detection of wAlbA, and 183F (5′-AAGGAACCGAAGTTCATG-3′) and 691R for wAlbB. PCRs reactions were run on the same samples also using the primer set Aealbo18S_F1 (5′-TGCCATGGATGCTTTCATTA-3′) and Aealbo18S_R1 (5′-GTACAAAGGGCAGGGACGTA-3′) to test for DNA quality. All PCR products were visualized on 1% agarose gels and sequenced.
Moreover, the presence of Wolbachia was assessed by PCRs in Foshan (generation 37) and five additional long-established laboratory strains deriving from Canton (China, G20), Recife (Brasil, G25), Tapachula (Mexico, G31), Tampon (La Reunion, G26), and Crema (Italy, G33). These strains were selected for several reasons. First, to compare Wolbachia prevalence estimated through metagenomics in CR and FO samples (reared in wild-collected water) with estimates of Wolbachia presence in the Foshan and Crema laboratory strains. The Crema strain was established in 2016 from mosquito eggs collected in the same breeding site used in this study. Second, to determine whether insectary rearing for several generations have led to a 100% Wolbachia frequency in adult individuals.
Results
The microbiota of breeding site water (W), larval (L), and adult (A) Ae. albopictus samples was examined by sequencing of the bacterial 16S rRNA gene. Sequences of a total of 37 libraries (i.e., 10 water samples; four CR and seven FO larval samples; five CR and nine FO adults, respectively; two DNA extraction negative controls) resulted in 8,191,150 million sequence reads, ranging from 14,277 to 264,318 (with an average of 126,018). No reads were reported in negative controls. Total number of reads per sample before and after Wolbachia-associated reads removal is reported in Supplementary Table S1. Identified ASVs per sample ranged between 19 and 3410 (Supplementary Table S2).
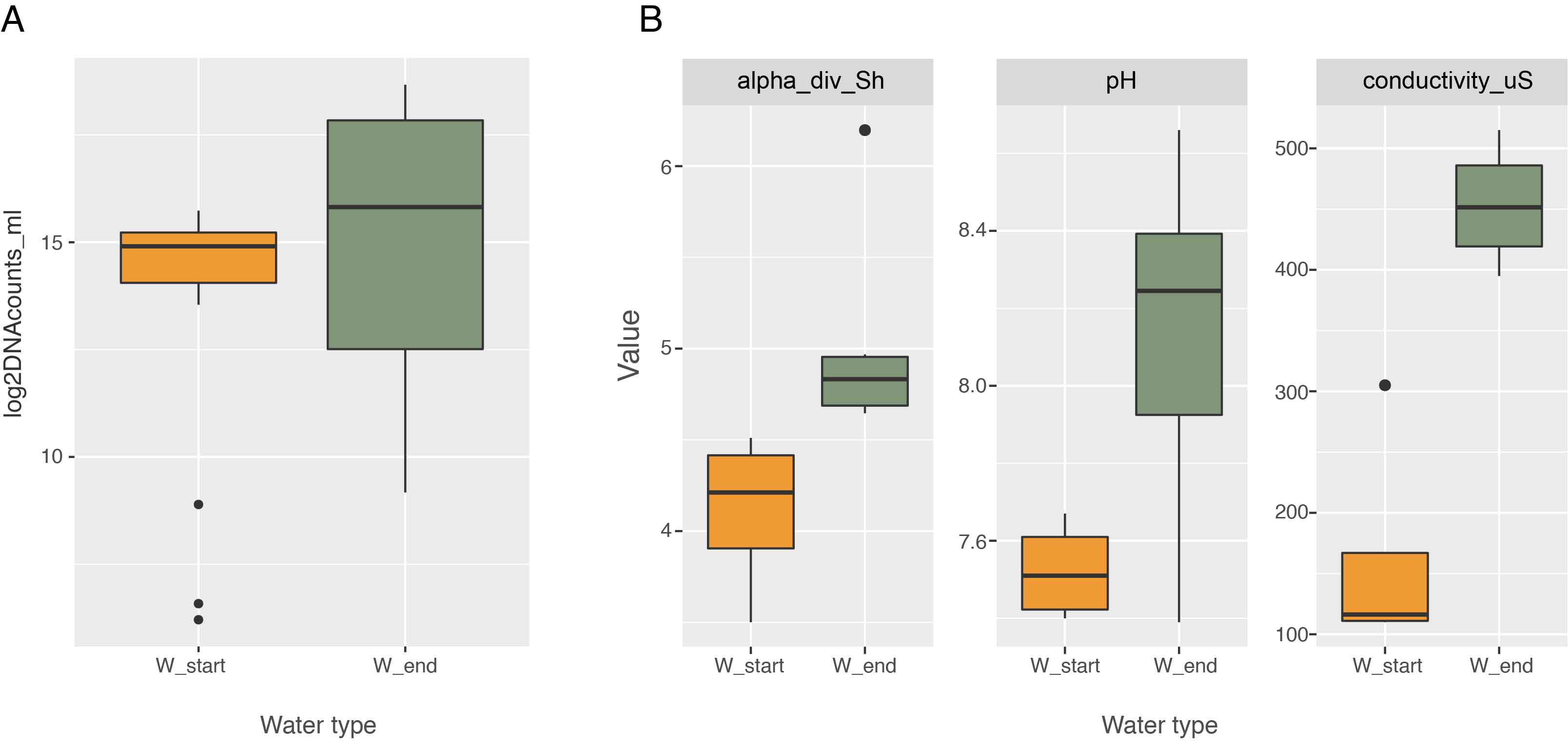
W End samples showed higher variability in the number of ASVs than W Start samples; larval samples showed a much higher variability than adults, irrespective of their origin (Figure 1). These differences are statistically supported, as shown by the results of the Kruskal–Wallis test (Table 1). Quantity of bacterial DNA in water samples was also estimated by qPCR (Supplementary Figure S1A and Supplementary Table S3) confirming a significantly higher number of bacterial DNA copies in W End in comparison to W Start (ANOVA, Tukey post hoc test: p < 0.05).
FIGURE 1.

Alpha diversity indices (richness and Shannon). Box plots of alpha diversity values distribution for both indices (richness and Shannon diversity) of the water (W Start, orange; W End, light green), larval (CR, golden; FO, blue), and adult (CR, coral; FO, dark green) samples are shown. The line inside each box represents the median value. Outliers are shown as dots.
TABLE 1.
Pairwise comparison (Kruskal–Wallis test) of diversity indices between water, larval, and adult samples.
| Water | Crema | Foshan | ||||
| W Start | W End | L_CR | A_CR | L_FO | ||
| Water | W End | 6.54 (0.010) | ||||
| Crema | L_CR | 0.083 (0.773) | 3.682 (0.05) | |||
| A_CR | 6.00 (0.014) | 7.50 (0.006) | 6.00 (0.014) | |||
| Foshan | L_FO | 4.321 (0.038) | 9.00 (0.003) | 2.893 (0.089) | 7.180 (0.007) | |
| A_FO | 7.384 (0.007) | 9.60 (0.02) | 5.654 (0.017) | 1.371 (0.242) | 1.929 (0.165) |
H and p-values (in brackets) are shown. Significant comparisons (P < 0.05) are indicated in bold. The number of samples within each group were: W Start (n = 4). W End (n = 6), L_CR (n = 4), L_FO (n = 7), A_CR (n = 5), A_FO (n = 9).
Water chemistry at mosquito breeding sites has been shown to have a key impact on mosquito survival and abundance (Chen et al., 2009; Rajesh et al., 2013; Onchuru et al., 2016). Additionally, specific physical and chemical parameters (i.e., oxygen and conductivity) were found to be associated with microbiota composition in Ae. aegypti (Hery et al., 2021). Thus, in an attempt to provide a qualitative analysis that could support the metagenomic results, a set of physical and chemical parameters were measured for both W Start and W End samples. Both pH and conductivity significantly increased in W End samples (t-test, P < 0.05) (Supplementary Figure S1B and Supplementary Table S4). Moreover, nitrates, ammoniacal nitrogen and phosphate values were higher (at least > 2.5 folds) in W End than in W Start, providing further support to the presence of differences between the two considered samples, despite measurements were often close to detection limits (Supplementary Table S4).
Microbiota Composition and Distribution
A total of 28 bacterial phyla and 262 families were identified across all samples. Taxonomic analysis showed that most of the sequences are associated with the phylum Proteobacteria (59.7%), followed by Bacteroidota (16.4%), Firmicutes (3.7%), and Spirochaetota (3.6%).
In W Start samples, the main bacterial phyla were Proteobacteria (>95%), followed by Bacteroidota (0.5%) and Verrucomicrobiota (0.3%). In addition to these phyla, in W End samples we also identified Patescibacteria, Dependentiae, Cyanobacteria, and Acidobacteria (Figure 2A). The trend of increasing diversity from W Start to W End samples is also evident at the family taxonomic level (Figure 2B). Proteobacteria and Bacteroidota dominated the microbiota of larvae; the bacterial community of adults displayed a much lower level of diversity, with Proteobacteria representing almost the totality of the microbiota of adult mosquitoes (95% in A_CR and 71% in A_FO) (Supplementary Table S5).
FIGURE 2.

Relative abundance bar plot of bacteria at phylum (A) and family (B) taxonomy level in water, larval, and adult samples from wild-collected and laboratory mosquitoes. Each bar represents the proportion of sequencing reads (relative abundance expressed as percentage) assigned to a given bacterial taxon. Only the 50 most abundant Amplicon Sequence Variants were considered and assigned to corresponding taxa. The legend lists the most abundant taxonomic categories (9 in A and 29 in B). The number of samples analyzed for each category is reported in parentheses.
The trend of decreasing ASVs variability from water to adult samples is also depicted in a heatmap generated using the 100 most abundant ASVs assigned at the taxonomic level of Family (unweighted UniFrac distance) (Figure 3). The heatmap shows that the 100 most abundant ASVs are differently distributed in the water, larvae, and adult samples. Differences are evident also between W Start and W End, as well as between CR and FO samples, independently of the developmental state. At the family taxonomic level, W Start includes Sphingobacteriaceae, Spirosomaceae, Chitinophagaceae and, to a lesser extent, Nocardiaceae. W End samples include Cellvibrionaceae, Burkholderiaceae, Caulobacteraceae, Planococcaceae, Cytophagaceae, Blastocatellaceae, and several unculturable unidentified bacteria. Larval samples are characterized by Weeksellaceae, Spirosomaceae and Chitinophagaceae (phylum Bacteroidetes), Spirochaetaceae (phylum Spirochaetes), Sphingomonadaceae and Rhodobacteraceae (phylum Proteobacteria) (Figures 2B, 3).
FIGURE 3.

Heatmap showing the relative abundance of the components of the microbiota in the breeding site water, larvae, and adult samples from laboratory and wild-collected mosquitoes. The distribution of the 100 most abundant Amplicon Sequence Variants was explored in each analyzed sample with a heatmap plot based on rarefied tables. Each sample is shown on the X-axis; W refers to water samples, L and A to larvae and adult samples, respectively. Start and End refers to water collected at the moment of mosquito collection and after all adults had emerged, respectively. FO and CR refer to mosquitoes of the Foshan laboratory strain or collected in the wild, respectively. Heatmap colors (from dark to light blue) indicate increasing abundance of each microbiota component. The heatmap was generated with the phyloseq R package.
Wild and laboratory larval samples differentiated by the presence of Verrucomicrobiaceae, Chitinophagaceae, Pirellulaceae, Cellvibrionaceae, and Terrimicrobiaceae exclusively in laboratory samples. The only bacterial family uniquely present in wild larval samples was Microbacteriaceae, with the Herbiconiux, Leifsonia, and Leucobacter genera.
In addition to bacteria already known to be part of the microbiota of Aedes mosquitoes, such as Sphingomonas and Chryseobacterium (see Scolari et al., 2019 for a review), in Foshan larval samples we also identified bacteria of the Paenibacillus genus, which were previously associated only with Ae. aegypti (Scolari et al., 2019). In Crema samples, genera reported for both Ae. aegypti and Ae. albopictus, such as Bacillus, Pseudomonas, and Escherichia–Shigella, were also found.
In the case of adult samples, the families Verrucomicrobiaceae, Acetobacteracea, Planococcaceae, Weeksellaceae, Crocinitomicaceae, Cytophagaceaea, Corynebacteriaceae, Blastocatellaceae, and Bryobacteriaceae were typical of the Foshan laboratory samples, with Weeksellaceae and Planococcaceae being particularly abundant.
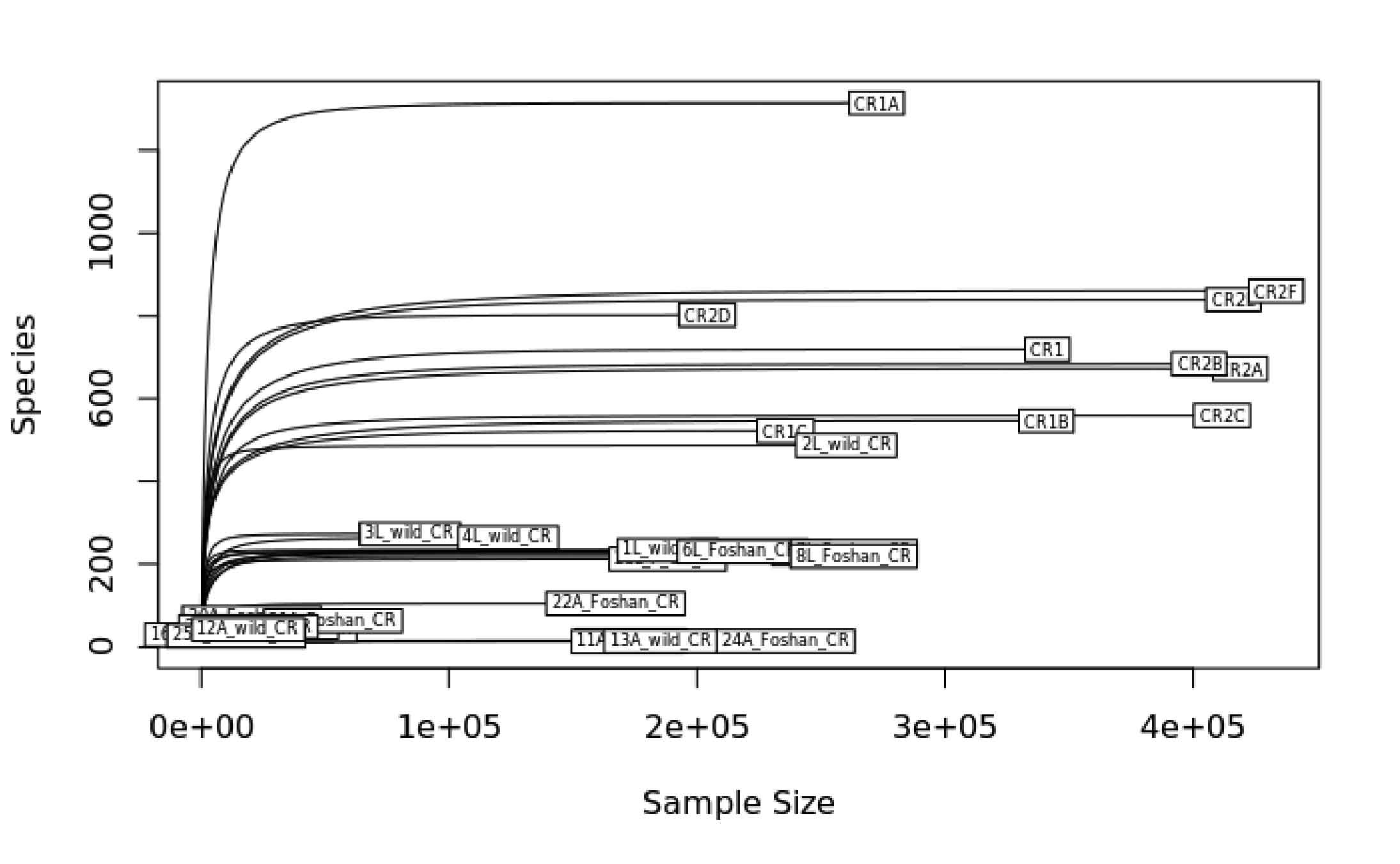
Based on these results, beta diversity metrics were computed to further explore differences among samples. For more reliable results we used a rarefaction process to resample the data. Obtained rarefaction curves are shown in Supplementary Figure S2.
Samples separated into clusters in a non-metric multidimensional scaling (NMDS) ordination plot (Figure 4). Using pairwise weighted UniFrac distance matrix, the obtained stress value was <0.1. W Start and W End samples clustered together, although into two identifiable groups. All larval samples clustered together, suggesting a limited contribution of the genetic background in shaping larval microbiota. On the opposite, adult samples showed greater intra and inter sample variability, with a portion of them not being clearly separated from the larvae. PerMANOVA pairwise comparison based on the unweighted UniFrac distance metrics revealed statistically significant differences among the groups (Table 2).
FIGURE 4.

Non-metric multidimensional scaling (NMDS) of water, larval and adult samples. Colors in the bidimensional NMDS plot are used according to the different sample origin as shown in the legend. Start and End refers to water collected at the moment of mosquito collection and after all adults had emerged, respectively. FO and CR refer to mosquitoes of the Foshan laboratory strain or collected in the wild, respectively.
TABLE 2.
PerMANOVA pairwise comparison based on unweighted UniFrac distance metrics between water, larval, and adult samples.
| Water | Crema | Foshan | ||||
| W Start | W End | L_CR | A_CR | L_FO | ||
| Water | W End | 2.884 (0.006) | ||||
| Crema | L_CR | 2.718 (0.023) | 3.497 (0.007) | |||
| A_CR | 3.230 (0.013) | 4.540 (0.003) | 2.421 (0.048) | |||
| Foshan | L_FO | 3.700 (0.008) | 4.870 (0.001) | 1.330 (0.094) | 3.279 (0.002) | |
| A_FO | 3.760 (0.003) | 5.621 (0.001) | 2.483 (0.002) | 1.537 (0.163) | 2.9351 (0.001) |
Pseudo-F and p-values (in brackets) are shown. Significant comparisons (P < 0.05) are indicated in bold. The number of samples within each group were: W Start (n = 4), W End (n = 6), L_CR (n = 4), L_FO (n = 7), A_CR (n = 5), A_FO (n = 9).
Sample-Unique Bacteria
The intersections among the list of bacterial genera found in analyzed samples were calculated to identify the components of the microbiota that are unique of each sample (Figure 5). As expected according to the previously described data, the number of genera shared between larvae and water samples were higher than those shared between larvae and adults, both in the case of Crema and Foshan samples (Figure 5). Wild larvae unique genera included Dietzia, Blautia, and Leifsonia; in wild adults, unique genera were Tepidimonas, Cloacibacterium, Haemophilus, and Neisseria. An unidentified uncultured bacterium from the family Rhodobacteraceae was found to be unique of Foshan larvae; Dietzia, Meiothermus, Porphyromonas, Blautia, Micrococcus, and Fructobacillus were the unique genera in Foshan adults.
FIGURE 5.

Number of unique bacterial genera in breeding site water (W), larval (L), and adult (A) samples from wild (CR) or laboratory (FO) samples. Venn diagrams show the number of shared genera in (A) Crema (CR) and (B) Foshan (FO) samples with respect to what observed in breeding site water collected at the beginning (start) or after all adults had emerged (end). The Venn diagrams were created using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
When focusing on comparison between W Start and W End, and in both larvae and adults between FO and CR samples, the most striking result was the number of ASVs uniquely detected in adults of Foshan, and including genera such as Prosthecobacter, Solimonas, Ancylobacter, Anaerococcus, Schlesneria, and Micrococcus (Supplementary Figure S3).
Conserved Microbiota
We define “conserved” microbiota the bacterial genera detected in at least 70% of all our mosquito samples. Mosquito conserved microbiota includes 102 genera (Figure 6 and Supplementary Tables S6, S7). A total of 81 genera were also detected in water samples (i.e., W Start), emphasizing the role of the breeding site in shaping mosquito microbiota. Among the bacteria absent in water samples (both W Start and W end), Vulcaniibacterium was the only present in both larvae and adults, of both CR and FO samples. Escherichia–Shigella was instead found only in CR adult samples. Several unclassified bacteria were found to be conserved in larval samples and/or adults, absent from the W Start samples but detected in the W End samples (Supplementary Table S7).
FIGURE 6.

Number of unique bacterial genera from the conserved microbiota of larval (L) and adult (A) samples from wild (CR) or laboratory (FO) mosquitoes. Venn diagrams show the number of conserved genera in (A) adults and larvae from Crema (CR) and Foshan (FO), (B) within CR mosquito samples, and (C) within FO mosquito samples. The Venn diagrams were created using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Wolbachia Abundance Impacts Microbial Richness in Ae. albopictus Adults
Shannon diversity index was calculated for each sample resulting into two groups. One group included five samples (11A_wild_CR, 13A_wild_CR, 22A_FO, 10A_Wild_CR, and 24A_FO) with Shannon diversity index < 1.5; all the other samples had a Shannon diversity index higher or equal to 2 (Supplementary Figure S4). Interestingly, samples from the first group had lower frequency of Wolbachia reads (ranging from 0% in 11A_wild_CR and 13A_wild_CR to 41.2% in 22A_FO) than samples of the second group, thus we called these two groups “Wol_low” and “Wol_high,” respectively, and we compared their alpha diversity after excluding Wolbachia reads. Wol_high samples displayed a significantly higher number of ASVs than Wol_low samples (t-test, P < 0.05) (Figure 7).
FIGURE 7.

Alpha diversity indices (richness and Shannon) in samples with low (Wol_low, in orange) or high (Wol_high, in blue) Wolbachia abundance. Wol_low grouped five samples (11A_wild_CR, 13A_wild_CR, 22A_FO, 10A_Wild_CR, and 24A_FO), which had individually a Shannon diversity index < 1.5, as shown in Supplementary Figure S4, and a frequency of Wolbachia reads up to 41.2%. Wol_high grouped all the other samples, which had a frequency of Wolbachia reads > 86.8% and an individual Shannon diversity index > 2 (Supplementary Figure S4). The line inside each box represents the median value. Outliers are shown as dots. Wol_high samples displayed a significantly higher number of ASVs than Wol_low samples (t-test, P < 0.05).
Wolbachia Presence in Different Laboratory Strains
The absence of Wolbachia in four adult samples prompted us to further investigate Wolbachia prevalence in six long-established laboratory strains. The prevalence of wAlbA and wAlbB strains was also determined. In all mosquito strains, both wAlbA and wAlbB were detected. However, while Wolbachia was present in all tested individuals in Foshan, Canton, and Recife strains, some individuals were shown to not carry Wolbachia in Crema, Tapachula, and Tampon (Table 3). The percentage of Wolbachia-negative mosquitoes ranged between 3% in Crema to 18% in Tampon. Absence was more prevalent for wAlbA than wAlbB. Patterns of Wolbachia infections were also evaluated in relation to mosquito sex (Table 3). Dual infection was more common in females than in males in Foshan, Canton, and Recife strains, while in Crema and Tampon, males had the highest values of infection. Furthermore, wAlbA-only infection was never detected in males, while wAlbB-only infection appeared to be mostly limited to males.
TABLE 3.
Wolbachia prevalence in six Ae. albopictus laboratory strains.
| Wolbachia prevalence (%) | ||||||
| Strain (generation) | N. samples | wAlbA | wAlbB | wAlbA + wAlbB | Total Wolbachia+ | Total Wolbachia – |
| Foshan (G37) | 40 | 0 | 22 | 78 | 100 | 0 |
| (22M, 18F) | (0M, 0F) | (22M, 0F) | (33M, 45F) | |||
| Canton (G20) | 40 | 0 | 10 | 90 | 100 | 0 |
| (20M, 20F) | (0M, 0F) | (10M, 0F) | (40M, 50F) | |||
| Recife (G25) | 40 | 5 | 10 | 85 | 100 | 0 |
| (20M, 20F) | (0M, 5F) | (10M, 0F) | (40M, 45F) | |||
| Crema (G33) | 40 | 0 | 0 | 97 | 97 | 3 |
| (20M, 20F) | (0M, 0F) | (0M, 0F) | (50M, 47F) | (50M, 47F) | (0M, 3F) | |
| Tampon (G26) | 40 | 5 | 5 | 72 | 82 | 18 |
| (20M, 20F) | (0M, 5F) | (2.5M, 2.5F) | (42M, 30F) | (45M, 37F) | (5M, 13F) | |
| Tapachula (G31) | 40 | 5 | 22 | 65 | 92 | 8 |
Patterns of infection in relation to the sex were evaluated in positive samples for Foshan, Canton, Recife, Crema, and Tampon. M, males; F, females.
Discussion
Aedes albopictus Larvae Acquire Microbiota From Breeding Site Water
The analysis of the diversity of bacterial communities showed that Ae. albopictus larvae contain a subset of the ASVs present in breeding site water, supporting the idea that larvae are colonized by a fraction of the bacteria ingested through feeding (Minard et al., 2013; Coon et al., 2016b; Strand, 2018). Higher ASVs richness in water than in mosquito samples has been already described in Aedes spp. (Dada et al., 2014; Dickson et al., 2017; Wang et al., 2018; Alfano et al., 2019). Among the bacterial genera present in the breeding site water and acquired by both CR and FO larvae, it is worth mentioning Chryseobacterium, which is known to localize in the larval gut (see Scolari et al., 2019 for a review). We also observed differences between CR and FO larvae in bacterial genera shared with W Start samples, although all larval samples grouped together in the NMDS plot, suggesting a limited contribution of the genetic background in shaping larval microbiota. For example, Foshan larval samples shared with W Start bacteria such as Roseococcus, Paenibacillus, and Ferruginibacter, which were not detected in Crema larvae. The microbiota of Crema larvae showed Bacillus, which was previously detected in Ae. aegypti (Koneman et al., 1992) and Ae. albopictus field-collected larvae (Coon et al., 2016b), and Ensifer, Sphingobium, and Aeromicrobium.
Microbial Community of Breeding Site Water Changes Over Time
Despite the intrinsic impossibility to provide a comparison with respect to breeding site prior to mosquito oviposition (i.e., a ‘natural’ control, characterized by total absence of mosquito eggs and larvae), the comparison between W Start and W End water samples suggests the presence of a dynamic interaction between larvae and their breeding site. Such a difference was also detected by qPCR amplification of the 16S rRNA gene, which confirmed a significantly higher number of bacterial DNA copies in W End in comparison to W Start. Although unculturable candidate phyla are likely present in our water samples, the procedure of relying on 16S rRNA quantification to infer bacterial load in breeding site water and mosquito samples is routinely used (e.g., Charan et al., 2013; Wang et al., 2018).
Future studies comprising water samples collected in breeding sites before mosquito oviposition will provide important data to clarify the impact of mosquito larvae on shaping the water bacterial communities. Such studies will require the possibility to robustly predict the likelihood of a water collection to become a mosquito breeding site. Moreover, additional studies comprising water controls collected at breeding sites and maintained without larval samples over time to cover the mosquito developmental window will provide further ground to investigate the actual modifications induced by larval development to the water microbial communities.
Mosquitoes are able to modify their aquatic habitat through larval feeding and excretion of bacteria from the digestive tract, an effect that can be magnified in small volume containers (Coon et al., 2016b), as adopted in our experiments. Mosquitoes are impacting their breeding sites through excretion of relatively high levels of ammonium (NH4+) from the larval anal papillae, as shown in Ae. aegypti (Donini and O’Donnell, 2005; Weihrauch et al., 2012). The chemical analysis of the W End samples is in line with this finding, since we detected fourfold higher levels of ammonia than in the W Start samples.
Competition among larvae may impact breeding site microenvironment and consequently affect the composition of the water microbiota over time. Competition driven by excreted chemicals has indeed been shown to occur in Ae. aegypti (Bédhomme et al., 2005). Larval feeding and excretion in the breeding water may contribute to developing the optimal conditions for the growing of bacteria that, in the initial water samples, could not find the ideal environment for proliferation and may thus have gone undetected. The composition of detritus-associated microfauna is indeed known to be altered over time as a consequence of larval grazing in Aedes triseriatus (Walker et al., 2010). Moreover, similarly to what occurs for Wolbachia, which is transmitted vertically from mother to offspring via the egg cytoplasm (Werren, 1997), other bacteria can be vertically transmitted (Moran et al., 2008). This can be the case of certain bacterial genera identified in our study that are not present in the water at the breeding sites, thus not being acquired through feeding. For example, Escherichia–Shigella was found to colonize ovaries of An. gambiae (Mancini et al., 2018). In Ae. albopictus, the reported wide dominance of Wolbachia in the ovaries (94% according to Mancini et al., 2018), could have masked the identification of other bacteria, which may have an importance in the biology of the species. Further studies aimed at localizing the larval- and/or adult-specific bacteria in mosquito tissues will be essential to clarify this aspect.
When considering the composition of the W End samples, it is worth mentioning the presence of Patescibacteria, which were previously shown to be associated with different developmental stages of Ae. albopictus (Qing et al., 2020) and adult Culex nigripalpus females (Duguma et al., 2019). Patescibacteria are prevalent in water environments (Tian et al., 2020), especially in groundwater (Bruno et al., 2017b; Schwab et al., 2017), and due to a reduced genome and consequent limited biosynthetic capabilities, the presence of members of this phylum may depend on nutrient uptake from other members (autotroph) of the microbial community (Brown et al., 2015; Herrmann et al., 2019). Members of this phylum have been associated with oligotrophic environments (Herrmann et al., 2019). This feature could explain the presence of members of this phylum in our water, since we allowed the larvae to grow solely based on the nutrients present in the water collected at the breeding site, which may have become depleted as a consequence of feeding. Another phylum that characterized W End samples is Dependentiae that, similarly to Patescibacteria, are widespread across different environments, including wastewater (McLean et al., 2013; Yeoh et al., 2015), and have limited metabolic capacities, suggesting their dependence on other aquatic autotrophic and heterotrophic microorganisms (Deeg et al., 2019). The abundance of Cyanobacteria is in general not surprising, since these bacteria are known to be present in mosquito breeding sites and have been previously isolated from mosquito guts (Thiery et al., 1991; Vazquez-Martinez et al., 2002). Members of this phylum are particularly abundant in environments with high phosphate concentrations (Roldán and Ramírez, 2008), which is the case of the water collected at the end of our experiments. Moreover, the growth of cyanobacteria has been shown to be favored by increase concentrations of nitrates and ammonia (Kim et al., 2017), which in our experiments could be related to mosquito development. Acidobacteria have been previously found in mosquito breeding sites (Onchuru et al., 2016), as well as adult individuals [e.g., Culex nigripalpus (Duguma et al., 2019), An. coluzzii (Mancini et al., 2018), An. gambiae (Mwadondo et al., 2017), An. stephensi (Kalappa et al., 2018), but also Ae. albopictus (Wang et al., 2018)].
We also identified bacteria, such as Nocardiaceae, that are shared between W Start samples and all tested mosquito samples (i.e., both CR and FO larvae and adults) but they are absent in the W End sample. This pattern may indicate that members of this family are acquired and digested. In triatomines, Nocardiaceae play the role of nutritional symbionts able to provide the host with essential nutrients (Salcedo-Porras et al., 2020). Conversely, Burkholderiaceae were present in larval and adult individuals, as well as the W End, but not in W Start. Burkholderiaceae were previously found to be associated with Ae. albopictus breeding sites (Shelomi, 2019) as well as adults (Seabourn et al., 2020). Similarly, larval and pupal samples from Ae. koreicus were found to be dominated by this bacterial family (Alfano et al., 2019).
The presence of mosquito larvae has been previously shown to have different impacts on the composition of the bacterial community of breeding site water. For example, microcosmos-based experiments involving Cx. restuans and Ae. triseriatus showed that larvae affect the composition of the bacterial community in breeding sites and reduce bacterial abundance, diversity and richness (Walker et al., 1991; Muturi et al., 2020); a similar trend was seen in natural breeding sites in tree holes for Ae. triseriatus (Walker et al., 1991). These results contrast with those obtained by Kaufman et al. (1999) in the same mosquito species, in a microcosms-based set-up, as they found that larval presence increase total bacterial numbers, similarly to the increased bacterial abundance detected in water columns in small container habitats reported for the pitcher plant mosquito Wyeomyia smithii (Heard, 1994; Cochran-Stafira and von Ende, 1998).
Larvae can contribute to the development of enriched and more anoxic conditions, which favor the growth of facultative anaerobe bacteria.
Newly Emerged Ae. albopictus Adults Display a Simplified Microbiota
The data obtained in this study show a decrease in microbiota complexity from larval to adult stage. This trend, evident in both wild and laboratory samples, is most likely due to the dramatic changes the gut epithelium undergoes during metamorphosis. In particular, the formation of two meconial peritrophic matrices (MPM1 and MPM2) was shown to contribute to Ae. aegypti adult midgut sterilization by sequestering microorganisms ingested during the larval stage, which are excreted after emergence (Moll et al., 2001; Moncayo et al., 2005). This process is not fully understood in Ae. albopictus.
After such loss of components, adults shape a new microbiota and diet plays a key role. Mosquito hosts, such as plants and animals, are a source of bacteria, as well as viruses and other microorganisms. Indeed, it is known that the midgut of the two sexes harbors different microbial communities: the microbiota of females is typical of blood feeding insects, and it is mostly colonized by Gammaproteobacteria; males, instead, display a midgut mainly colonized by Firmicutes (Minard et al., 2013).
Developmental Stage Affects Microbiota Composition
Our results suggest that the developmental stage influences microbiota composition in mosquitoes. Only one genus, i.e., Vulcaniibacterium, was found to be shared by both Foshan and Crema samples. This bacterium has been previously found in the gut of both sexes of Bactrocera oleae (Koskinioti et al., 2019) but it was never detected in a mosquito species so far.
Microbacteriaceae, the only family found to be uniquely present in wild CR larval samples, was previously isolated from Ae. albopictus larvae and reported to be absent from the other developmental stages (Yadav et al., 2016). Similarly, in Ae. aegypti, Microbacteriaceae were found to be lost during metamorphosis (Frankel-Bricker et al., 2020). In our work, we confirmed the larval-specificity of this bacterial family in Ae. albopictus. While Herbiconiux was previously identified in Ae. aegypti larvae (Hery et al., 2021), and it is present also in our wild sampled larvae, Leifsonia was found in Ae. albopictus for the first time. This bacterium has been identified in the breeding site water of A. darlingi in Brazil, and in the midgut of the sandfly Leishmania major (Louradour et al., 2017), but to the extent of our knowledge it was never identified in Ae. albopictus before. The role of this bacterium in the microbiota of the tiger mosquito will require further investigation, extending the sampling to other natural breeding sites. Leucobacer was previously detected in the larvae of Ae. aegypti and were rare in breeding site water and nearly absent in adults (Coon et al., 2014), mirroring our results in Ae. albopictus.
Wolbachia Abundance Contributes to Bacterial Community Structure
In addition to the effects of the breeding site environment, also interactions between members of the microbiota within a host can induce changes in the structure of the microbial communities and in the relative abundance of its components (Brinker et al., 2019). Recent studies began to show that the history of Wolbachia colonization has an impact on the physiological changes mediated by this bacterium in the host, with effects on the resident microbiota. After a stable transinfection, Wolbachia was shown to lead to a decrease in microbial diversity in Ae. aegypti (Audsley et al., 2018). This decrease in microbial diversity has been suggested to be mediated by immune system modulation, resource competition and the pH (Simhadri et al., 2017; Audsley et al., 2018). Wolbachia-mediated immune regulation appears to be lost in hosts developing long-term co-evolutionary relationships with this bacterium (Shi et al., 2018). Differently from what occurs in Ae. aegypti, wAlbA and wAlbB are native Wolbachia infections of Ae. albopictus with a long history of co-association (Dutton and Sinkins, 2004). Accordingly, here we show that Ae. albopictus adults displaying a high Wolbachia prevalence have a more diverse microbiota than mosquitoes with no or low Wolbachia reads. Our study did not focus on investigating whether and to what extent Wolbachia prevalence is linked to features of water at mosquito breeding sites. Further studies involving an extended sample size are needed to confirm results from our exploratory study as well as to further clarify the complex interactions between mosquito host, the environment and the different members of the bacterial communities residing in different tissues and organs, as mentioned above. The identification of Wolbachia also in somatic tissues of insects, including mosquitoes (see Pietri et al., 2016 for a review), led to formulate the hypothesis that this bacterium can also be acquired from the environment and/or host sharing, as suggested for ants and triatomines (Espino et al., 2009; Andersen et al., 2012; Frost et al., 2014).
Wolbachia Prevalence in Laboratory Strains Varies
This study contributed to further support that Wolbachia is highly prevalent in Ae. albopictus. However, we found that in one strain from La Reunion Island (i.e., Tampon), the prevalence was particularly low with respect to what detected in the other five strains and previously reported (Noor Afizah et al., 2015). Moreover, in the Crema strain derived from eggs collected in 2016 in the same site used for this study, we found that 3% of the mosquitoes were uninfected. Given that Wolbachia is transmitted vertically and this strain has been reared in our insectary for 33 generations, this result was unexpected and provides ground for further analyses.
Our results also confirmed the prevalence of dual Wolbachia infection in Ae. albopictus strains derived from different geographic populations. In the field, the prevalence of double infection by Wolbachia has been reported to be over 99.41% (Kittayapong et al., 2002a) and in Korean populations more than 98.8% (Park et al., 2016). We found dual infection ranging from 65 to 97%. As reported by previous studies, dual infection is mainly present in females (Joanne et al., 2015; Noor Afizah et al., 2015; Ahmad et al., 2017), but in two of the strains we analyzed, namely Crema and Tampon, both Wolbachia were present with higher frequency in males. A previous study that investigated wAlbA density in wild-sampled Ae. albopictus males from two Italian localities in the Central and Southern regions (i.e., Central Italy: Crevalcore, Bologna; Southern Italy: Anguillara Sabazia, Rome) showed that wAlbA titer was very low in about half of the collected males from both sites (Calvitti et al., 2015), in agreement with the findings of Tortosa et al. (2010). However, density of both Wolbachia strains was confirmed to be impacted by adult age, population geographic origin and environmental conditions such as temperature and food availability in larval breeding sites (Calvitti et al., 2015).
Our data suggest that an extensive survey of Wolbachia prevalence in wild populations is extremely important not only to better understand the role of this bacterium in contributing to shape the microbial community in Ae. albopictus, but also to provide essential basic-biology information to inform current and future mosquito control programs based on the Incompatible Insect Technique approach, especially given that cytoplasmic incompatibility level has been correlated with wAlbA density (Calvitti et al., 2015).
Data Availability Statement
The datasets generated for this study can be found in the European Nucleotide Archive (ENA) under the BioProject: PRJEB41031. The codes used for our analyses are available at the following link: https://gitlab.com/anna.sandionigi/metamosquito.
Author Contributions
MB and MCas conceived and designed the experiment. FS, AS, MCar, and AB performed the experiments. FS, AS, and AB analyzed the data. FS, AS, AB, and MB wrote the manuscript. All the authors provided critical comments on the manuscript, read and approved the final version of the manuscript.
Conflict of Interest
AS is employed by the company Quantia Consulting srl. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Jayme Souza-Neto (São Paulo State University) for helpful discussion.
Funding. This research was funded by a European Research Council Consolidator Grant (ERC-CoG) under the European Union’s Horizon 2020 Programme (Grant Number ERC-CoG 682394) to MB, the Italian Ministry of Education, University and Research FARE-MIUR project R1623HZAH5 to MB, the Human Frontier Science Program Research Grant RGP0007/2017 to MB, and the Italian Ministry of Education, University and Research (MIUR): Dipartimenti Eccellenza Program (2018–2022) Department of Biology and Biotechnology “L. Spallanzani,” University of Pavia, and the Italian Ministry of Education, University and Research project ‘Sistemi Alimentari e Sviluppo Sostenibile-tra ricerca e processi internazionali e africani’ (H42F16002450001) to MCas.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.624170/full#supplementary-material
Water samples analyses. (A) 16S rDNA-based bacterial quantification in water samples. On the X-axis, water samples are reported, colored by sampling period (W Start – orange – and W End – green – refers to water collected at the moment of mosquito collection and after all adults had emerged, respectively). Values are expressed as log2(DNA counts)/mL. (B) Physical analysis of W Start and W End water samples. Shannon alpha diversity index, pH, and conductivity values are shown. Both pH and conductivity significantly increased in W End samples (t-test, P < 0.05).
{kind=link}
Rarefaction curves used to discover all the microbial ‘species’/ASVs in each sample where Wolbachia reads were removed.
{kind=link}
Bacterial genera shared between breeding site water, and wild and laboratory larvae and adults. Venn diagrams show the number of genera shared in (A) water (W Start vs. W End), (B) larval (CR vs. FO), and (C) adult (CR vs. FO) samples.
{kind=link}
Alpha diversity indices of adult individuals. Alpha diversity (Shannon) values distribution for adults (CR and FO) samples are shown. Each sample is indicated with a different color.
{kind=link}
Number of sequencing reads per sample before and after removal of Wolbachia-associated reads. DNA extraction negative controls are included. Samples with no Wolbachia reads are indicated in bold.
List of ASVs identified in each larval, adult, and water sample.
Bacteria quantification through qPCR assays.
Physio-chemical analyses of water samples.
Proportion of sequencing reads assigned to a given bacterial taxon at the phylum and family level in water, larval, and adult samples. Only 50 most abundant ASVs were considered.
List of bacterial genera identified in the conserved microbiota for each larval, adult, and water sample.
List of unique and conserved bacterial genera from the microbiota of larvae and adults from wild or laboratory samples.
References
- Ahmad N. A., Vythilingam I., Lim Y. A. L., Zabari N. Z. A. M., Lee H. L. (2017). Detection of Wolbachia in Aedes albopictus and their effects on chikungunya virus. Am. J. Trop. Med. Hyg. 96 148–156. 10.4269/ajtmh.16-0516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano N., Tagliapietra V., Rosso F., Manica M., Arnoldi D., Pindo M., et al. (2019). Changes in microbiota across developmental stages of Aedes koreicus, an invasive mosquito vector in Europe: indications for microbiota-based control strategies. Front. Microbiol. 10:2832. 10.3389/fmicb.2019.02832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen S. B., Boye M., Nash D. R., Boomsma J. J. (2012). Dynamic Wolbachia prevalence in Acromyrmex leaf-cutting ants: potential for a nutritional symbiosis. J. Evol. Biol. 25 1340–1350. 10.1111/j.1420-9101.2012.02521.x [DOI] [PubMed] [Google Scholar]
- Anderson M. J. (2005). Permanova: A FORTRAN Computer Program for Permutational Multivariate Analysis of Variance. Auckland: University of Auckland. [Google Scholar]
- Ant T. H., Sinkins S. P. (2018). A Wolbachia triple-strain infection generates self-incompatibility in Aedes albopictus and transmission instability in Aedes aegypti. Parasit. Vectors 11:295. 10.1186/s13071-018-2870-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ant T. H., Herd C. S., Geoghegan V., Hoffmann A. A., Sinkins S. P. (2018). The Wolbachia strain wAu provides highly efficient virus transmission blocking in Aedes aegypti. PLoS Pathog. 14:e1006815. 10.1371/journal.ppat.1006815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audsley M. D., Seleznev A., Joubert D. A., Woolfit M., O’Neill S. L., McGraw E. A. (2018). Wolbachia infection alters the relative abundance of resident bacteria in adult Aedes aegypti mosquitoes, but not larvae. Mol. Ecol. 27 297–309. 10.1111/mec.14436 [DOI] [PubMed] [Google Scholar]
- Bédhomme S., Agnew P., Sidbore C., Michalakis Y. (2005). Pollution by conspecifics as a component of intraspecific competition among Aedes aegypti larvae. Ecol. Entomol. 30 1–7. 10.1111/j.0307-6946.2005.00665.x [DOI] [Google Scholar]
- Bennett K. L., Gómez-Martínez C., Chin Y., Saltonstall K., McMillan W. O., Rovira J. R., et al. (2019). Dynamics and diversity of bacteria associated with the disease vectors Aedes aegypti and Aedes albopictus. Sci. Rep. 9:12160. 10.1038/s41598-019-48414-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian G., Xu Y., Lu P., Xie Y., Xi Z. (2010). The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog. 6:e1000833. 10.1371/journal.ppat.1000833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagrove M. S., Arias-Goeta C., Di Genua C., Failloux A. B., Sinkins S. P. (2013). A Wolbachia wMel transinfection in Aedes albopictus is not detrimental to host fitness and inhibits chikungunya virus. PLoS Negl. Trop. Dis. 7:e2152. 10.1371/journal.pntd.0002152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagrove M. S., Arias-Goeta C., Failloux A. B., Sinkins S. P. (2012). Wolbachia strain wMel induces cytoplasmic incompatibility and blocks dengue transmission in Aedes albopictus. Proc. Natl. Acad. Sci. U.S.A. 109 255–260. 10.1073/pnas.1112021108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtzis K., Dobson S. L., Xi Z., Rasgon J. L., Calvitti M., Moreira L. A., et al. (2014). Harnessing mosquito-Wolbachia symbiosis for vector and disease control. Acta Trop. 132 S150–S163. 10.1016/j.actatropica.2013.11.004 [DOI] [PubMed] [Google Scholar]
- Brinker P., Fontaine M. C., Beukeboom L. W., Falcao Salles J. (2019). Host, symbionts, and the microbiome: the missing tripartite interaction. Trends Microbiol. 27 480–488. 10.1016/j.tim.2019.02.002 [DOI] [PubMed] [Google Scholar]
- Brown C. T., Hug L. A., Thomas B. C., Sharon I., Castelle C. J., Singh A., et al. (2015). Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523 208–211. 10.1038/nature14486 [DOI] [PubMed] [Google Scholar]
- Bruno A., Sandionigi A., Galimberti A., Siani E., Labra M., Cocuzza C., et al. (2017a). One step forwards for the routine use of high-throughput DNA sequencing in environmental monitoring. An efficient and standardizable method to maximize the detection of environmental bacteria. Microbiologyopen 6:e00421. 10.1002/mbo3.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno A., Sandionigi A., Rizzi E., Bernasconi M., Vicario S., Galimberti A., et al. (2017b). Exploring the under-investigated “microbial dark matter” of drinking water treatment plants. Sci. Rep. 7:44350. 10.1038/srep44350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J., Holmes S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvitti M., Marini F., Desiderio A., Puggioli A., Moretti R. (2015). Wolbachia density and cytoplasmic incompatibility in Aedes albopictus: concerns with using artificial Wolbachia infection as a vector suppression tool. PLoS One 10:e0121813. 10.1371/journal.pone.0121813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charan S. S., Pawar K. D., Severson D. W., Patole M. S., Shouche Y. S. (2013). Comparative analysis of midgut bacterial communities of Aedes aegypti mosquito strains varying in vector competence to dengue virus. Parasitol. Res. 112 2627–2637. 10.1007/s00436-013-3428-x [DOI] [PubMed] [Google Scholar]
- Chen C. D., Lee H. L., Stella-Wong S. P., Lau K. W., Sofian-Azirun M. (2009). Container survey of mosquito breeding sites in a university campus in Kuala Lumpur, Malaysia. Dengue Bull. 33 187–193. [Google Scholar]
- Chen X. G., Jiang X. T., Gu J. B., Xu M. (2015). Genome sequence of the Asian Tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc. Natl. Acad. Sci. U.S.A. 112 E5907–E5915. 10.1073/pnas.1516410112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouaia B., Rossi P., Montagna M., Ricci I., Crotti E., Damiani C., et al. (2010). Molecular evidence for multiple infections as revealed by typing of Asaia bacterial symbionts of four mosquito species. Appl. Environ. Microbiol. 76 7444–7450. 10.1128/AEM.01747-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran-Stafira D. L., von Ende C. N. (1998). Integrating bacteria into food webs: studies with Sarracenia purpurea inquilines. Ecology 79 880–898. [Google Scholar]
- Coon K. L., Brown M. R., Strand M. R. (2016a). Gut bacteria differentially affect egg production in the anautogenous mosquito Aedes aegypti and facultatively autogenous mosquito Aedes atropalpus (Diptera: Culicidae). Parasit. Vectors 9:375. 10.1186/s13071-016-1660-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon K. L., Brown M. R., Strand M. R. (2016b). Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol. Ecol. 25 5806–5826. 10.1111/mec.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon K. L., Vogel K. J., Brown M. R., Strand M. R. (2014). Mosquitoes rely on their gut microbiota for development. Mol. Ecol. 23 2727–2739. 10.1111/mec.12771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dada N., Jumas-Bilak E., Manguin S., Seidu R., Stenstrom T. A., Overgaard H. J. (2014). Comparative assessment of the bacterial communities associated with Aedes aegypti larvae and water from domestic water storage containers. Parasit. Vectors 7:391. 10.1186/1756-3305-7-391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M. R., dos Santos L. M. B., Vicente A. C. P., Maciel-de-Freitas R. (2016). Effects of environment, dietary regime and ageing on the dengue vector microbiota: evidence of a core microbiota throughout Aedes aegypti lifespan. Mem. Inst. Oswaldo Cruz 111 577–587. 10.1590/0074-02760160238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeg C. M., Zimmer M. M., George E. E., Husnik F., Keeling P. J., Suttle C. A. (2019). Chromulinavorax destructans, a pathogen of microzooplankton that provides a window into the enigmatic candidate phylum Dependentiae. PLoS Pathog. 15:e1007801. 10.1371/journal.ppat.1007801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson L. B., Ghozlane A., Volant S., Bouchier C., Ma L., Vega-Rua A., et al. (2018). Diverse laboratory colonies of Aedes aegypti harbor the same adult midgut bacterial microbiome. Parasit. Vectors 11:207. 10.1186/s13071-018-2780-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson L. B., Jiolle D., Minard G., Moltini-Conclois I., Volant S., Ghozlane A., et al. (2017). Carryover effects of larval exposure to different environmental bacteria drive adult trait variation in a mosquito vector. Sci. Adv. 3:e1700585. 10.1126/sciadv.1700585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donini A., O’Donnell M. J. (2005). Analysis of Na+, Cl-, K+, H+ and NH4+ concentration gradients adjacent to the surface of anal papillae of the mosquito Aedes aegypti: application of self-referencing ion-selective microelectrodes. J. Exp. Biol. 208 603–610. 10.1242/jeb.01422 [DOI] [PubMed] [Google Scholar]
- Duguma D., Hall M. W., Smartt C. T., Debboun M., Neufeld J. D. (2019). Microbiota variations in Culex nigripalpus disease vector mosquito of West Nile virus and Saint Louis Encephalitis from different geographic origins. PeerJ 6:e6168. 10.7717/peerj.6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton T. J., Sinkins S. P. (2004). Strain-specific quantification of Wolbachia density in Aedes albopictus and effects of larval rearing conditions. Insect Mol. Biol. 13 317–322. 10.1111/j.0962-1075.2004.00490.x [DOI] [PubMed] [Google Scholar]
- Espino C. I., Gómez T., González G., do Santos M. F., Solano J., Sousa O., et al. (2009). Detection of Wolbachia bacteria in multiple organs and feces of the triatomine insect Rhodnius pallescens (Hemiptera, Reduviidae). Appl. Environ. Microbiol. 75 547–550. 10.1128/AEM.01665-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floate K. D., Kyei-Poku G. K., Coghlin P. C. (2006). Overview and relevance of Wolbachia bacteria in biocontrol research. Biocontrol Sci. Technol. 16 767–788. 10.1080/09583150600699606 [DOI] [Google Scholar]
- Frankel-Bricker J., Buerki S., Feris K. P., White M. M. (2020). Influences of a prolific gut fungus (Zancudomyces culisetae) on larval and adult mosquito (Aedes aegypti)-associated microbiota. Appl. Environ. Microbiol. 86:e2334-19. 10.1128/AEM.02334-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frentiu F. D., Zakir T., Walker T., Popovici J., Pyke A. T., van den Hurk A., et al. (2014). Limited dengue virus replication in field-collected Aedes aegypti mosquitoes infected with Wolbachia. PLoS Negl. Trop. Dis. 8:e2688. 10.1371/journal.pntd.0002688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C. L., Pollock S. W., Smith J. E., Hughes W. O. H. (2014). Wolbachia in the flesh: symbiont intensities in germ-line and somatic tissues challenge the conventional view of Wolbachia transmission routes. PLoS One 9:e95122. 10.1371/journal.pone.0095122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaio Ade O., Gusmao D. S., Santos A. V., Berbert-Molina M. A., Pimenta P. F., Lemos F. J. (2011). Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae) (L.). Parasit. Vectors 4:105. 10.1186/1756-3305-4-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves C. M., Melo F. F., Bezerra J. M. T., Chaves B. A., Silva B. M., Silva L. D., et al. (2014). Distinct variation in vector competence among nine field populations of Aedes aegypti from a Brazilian dengue-endemic risk city. Parasit. Vectors 7:320. 10.1186/1756-3305-7-320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guegan M., Minard G., Tran F. H., Tran Van V., Dubost A., Valiente Moro C. (2018). Short-term impacts of anthropogenic stressors on Aedes albopictus mosquito vector microbiota. FEMS Microbiol. Ecol. 94:fiy188. 10.1093/femsec/fiy188 [DOI] [PubMed] [Google Scholar]
- Heard S. B. (1994). Pitcher-plant midges and mosquitos – a processing chain commensalism. Ecology 75 1647–1660. 10.2307/1939625 [DOI] [Google Scholar]
- Herrmann M., Wegner C. E., Taubert M., Geesink P., Lehmann K., Yan L. J., et al. (2019). Predominance of cand. Patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Front. Microbiol. 10:1407. 10.3389/fmicb.2019.01407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hery L., Guidez A., Durand A. A., Delannay C., Normandeau-Guimond J., Reynaud Y., et al. (2021). Natural variation in physicochemical profiles and bacterial communities associated with Aedes aegypti breeding sites and larvae on Guadeloupe and French Guiana. Microb. Ecol. 81, 93–109. 10.1007/s00248-020-01544-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. P., Xi Z. Y., Liu X. B., Wang J., Guo Y. H., Ren D. S., et al. (2020). Identification and molecular characterization of Wolbachia strains in natural populations of Aedes albopictus in China. Parasit. Vectors 13:28. 10.1186/s13071-020-3899-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISSG Invasive Species Specialist Group (2020). Global Invasive Species Database. Available online at: http://www.iucngisd.org/gisd/100_worst.php (accessed October 29, 2020). [Google Scholar]
- Joanne S., Vythilingam I., Yugavathy N., Leong C. S., Wong M. L., AbuBakar S. (2015). Distribution and dynamics of Wolbachia infection in Malaysian Aedes albopictus. Acta Trop. 148 38–45. 10.1016/j.actatropica.2015.04.003 [DOI] [PubMed] [Google Scholar]
- Kalappa D. M., Subramani P. A., Basavanna S. K., Ghosh S. K., Sundaramurthy V., Uragayala S., et al. (2018). Influence of midgut microbiota in Anopheles stephensi on Plasmodium berghei infections. Malaria J. 17:385. 10.1186/s12936-018-2535-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman M. G., Walker E. D., Smith T. W., Merritt R. W., Klug M. J. (1999). Effects of larval mosquitoes (Aedes triseriatus) and stemflow on microbial community dynamics in container habitats? Appl. Environ. Microbiol. 65 2661–2673. 10.1128/AEM.65.6.2661-2673.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Jo B. Y., Kim H. S. (2017). Effect of different concentrations and ratios of ammonium, nitrate, and phosphate on growth of the blue-green alga (cyanobacterium) Microcystis aeruginosa isolated from the Nakdong River, Korea. Algae 32 275–284. 10.4490/algae.2017.32.10.23 [DOI] [Google Scholar]
- Kittayapong P., Baimai V., O’Neill S. L. (2002a). Field prevalence of Wolbachia in the mosquito vector Aedes albopictus. Am. J. Trop. Med. Hyg. 66 108–111. 10.4269/ajtmh.2002.66.108 [DOI] [PubMed] [Google Scholar]
- Kittayapong P., Baisley K. J., Sharpe R. G., Baimai V., O’Neill S. L. (2002b). Maternal transmission efficiency of Wolbachia superinfections in Aedes albopictus populations in Thailand. Am. J. Trop. Med. Hyg. 66 103–107. 10.4269/ajtmh.2002.66.103 [DOI] [PubMed] [Google Scholar]
- Koneman E. W., Allen S. D., Janda W. M., Schreckenberger P. C., Winn W. C. (1992). Color Atlas and Textbook of Diagnostic Microbiology. Philadelphia, PA: Lippincott Williams & Wilkins. [Google Scholar]
- Koskinioti P., Ras E., Augustinos A. A., Tsiamis G., Beukeboom L. W., Caceres C., et al. (2019). The effects of geographic origin and antibiotic treatment on the gut symbiotic communities of Bactrocera oleae populations. Entomol. Exp. Appl. 167 197–208. 10.1111/eea.12764 [DOI] [Google Scholar]
- Kruskal J. B. (1964). Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29 1–27. 10.1007/BF02289565 [DOI] [Google Scholar]
- Lindh J. M., Borg-Karlson A. K., Faye I. (2008). Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop. 107 242–250. 10.1016/j.actatropica.2008.06.008 [DOI] [PubMed] [Google Scholar]
- Louradour I., Monteiro C. C., Inbar E., Ghosh K., Merkhofer R., Lawyer P., et al. (2017). The midgut microbiota plays an essential role in sand fly vector competence for Leishmania major. Cell. Microbiol. 19:e12755. 10.1111/cmi.12755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C. A., Hamady M., Kelley S. T., Knight R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73 1576–1585. 10.1128/AEM.01996-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lwande O. W., Obanda V., Lindstrom A., Ahlm C., Evander M., Naslund J., et al. (2020). Globe-trotting Aedes aegypti and Aedes albopictus: risk factors for arbovirus pandemics. Vector Borne Zoonotic Dis. 20 71–81. 10.1089/vbz.2019.2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M. V., Damiani C., Accoti A., Tallarita M., Nunzi E., Cappelli A., et al. (2018). Estimating bacteria diversity in different organs of nine species of mosquito by next generation sequencing. BMC Microbiol. 18:126. 10.1186/s12866-018-1266-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M. V., Herd C. S., Ant T. H., Murdochy S. M., Sinkins S. P. (2020). Wolbachia strain wAu efficiently blocks arbovirus transmission in Aedes albopictus. PLoS Negl. Trop. Dis. 14:e0007926. 10.1371/journal.pntd.0007926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean J. S., Lombardo M. J., Badger J. H., Edlund A., Novotny M., Yee-Greenbaum J., et al. (2013). Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc. Natl. Acad. Sci. U.S.A. 110 E2390–E2399. 10.1073/pnas.1219809110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie P. J., Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt R. W., Dadd R. H., Walker E. D. (1992). Feeding behavior, natural food, and nutritional relationships of larval mosquitoes. Annu. Rev. Entomol. 37 349–376. 10.1146/annurev.en.37.010192.002025 [DOI] [PubMed] [Google Scholar]
- Minard G., Tran F. H., Dubost A., Tran-Van V., Mavingui P., Moro C. V. (2014). Pyrosequencing 16S rRNA genes of bacteria associated with wild tiger mosquito Aedes albopictus: a pilot study. Front. Cell. Infect. Microbiol. 4:59. 10.3389/fcimb.2014.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minard G., Tran F. H., Raharimalala F. N., Hellard E., Ravelonandro P., Mavingui P., et al. (2013). Prevalence, genomic and metabolic profiles of Acinetobacter and Asaia associated with field-caught Aedes albopictus from Madagascar. FEMS Microbiol. Ecol. 83 63–73. 10.1111/j.1574-6941.2012.01455.x [DOI] [PubMed] [Google Scholar]
- Minard G., Tran F. H., Van V. T., Goubert C., Bellet C., Lambert G., et al. (2015). French invasive Asian tiger mosquito populations harbor reduced bacterial microbiota and genetic diversity compared to Vietnamese autochthonous relatives. Front. Microbiol. 6:970. 10.3389/fmicb.2015.00970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll R. M., Romoser W. S., Modrzakowski M. C., Moncayo A. C., Lerdthusnee K. (2001). Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J. Med. Entomol. 38 29–32. 10.1603/0022-2585-38.1.29 [DOI] [PubMed] [Google Scholar]
- Moncayo A. C., Lerdthusnee K., Leon R., Robich R. M., Romoser W. S. (2005). Meconial peritrophic matrix structure, formation, and meconial degeneration in mosquito pupae/pharate adults: histological and ultrastructural aspects. J. Med. Entomol. 42 939–944. 10.1093/jmedent/42.6.939 [DOI] [PubMed] [Google Scholar]
- Moran N. A., McCutcheon J. P., Nakabachi A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42 165–190. 10.1146/annurev.genet.41.110306.130119 [DOI] [PubMed] [Google Scholar]
- Moreira L. A., Iturbe-Ormaetxe I., Jeffery J. A., Lu G., Pyke A. T., Hedges L. M., et al. (2009). A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and plasmodium. Cell 139 1268–1278. 10.1016/j.cell.2009.11.042 [DOI] [PubMed] [Google Scholar]
- Muturi E. J., Dunlap C., Caceres C. E. (2020). Microbial communities of container aquatic habitats shift in response to Culex restuans larvae. FEMS Microbiol. Ecol. 96:fiaa112. 10.1093/femsec/fiaa112 [DOI] [PubMed] [Google Scholar]
- Muturi E. J., Ramirez J. L., Rooney A. P., Kim C. H. (2017). Comparative analysis of gut microbiota of mosquito communities in central Illinois. PLoS Negl. Trop. Dis. 11:e0005377. 10.1371/journal.pntd.0005377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwadondo E. M., Ghilamicael A., Alakonya A. E., Kasili R. W. (2017). Midgut bacterial diversity analysis of laboratory reared and wild Anopheles gambiae and Culex quinquefasciatus mosquitoes in Kenya. Afr. J. Microbiol. Res. 11 1171–1183. 10.5897/AJMR2016.8256 [DOI] [Google Scholar]
- Noor Afizah A., Roziah A., Nazni W. A., Lee H. L. (2015). Detection of Wolbachia from field collected Aedes albopictus Skuse in Malaysia. Indian J. Med. Res. 142 205–210. 10.4103/0971-5916.164259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onchuru T. O., Ajamma Y. U., Burugu M., Kaltenpoth M., Masiga D., Villinger J. (2016). Chemical parameters and bacterial communities associated with larval habitats of Anopheles, Culex and Aedes mosquitoes (Diptera: Culicidae) in western Kenya. Int. J. Trop. Insect Sci. 36 146–160. 10.1017/S1742758416000096 [DOI] [Google Scholar]
- Osei-Poku J., Mbogo C. M., Palmer W. J., Jiggins F. M. (2012). Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 21 5138–5150. 10.1111/j.1365-294X.2012.05759.x [DOI] [PubMed] [Google Scholar]
- Palatini U., Masri R. A., Cosme L. V., Koren S., Thibaud-Nissen F., Biedler J. K., et al. (2020). Improved reference genome of the arboviral vector Aedes albopictus. Genome Biol. 21:215. 10.1186/s13059-020-02141-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatini U., Miesen P., Carballar-Lejarazu R., Ometto L., Rizzo E., Tu Z., et al. (2017). Comparative genomics shows that viral integrations are abundant and express piRNAs in the arboviral vectors Aedes aegypti and Aedes albopictus. BMC Genomics 18:512. 10.1186/s12864-017-3903-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. H., Lim H., Kim H., Lee W. G., Roh J. Y., Park M. Y., et al. (2016). High prevalence of Wolbachia infection in Korean populations of Aedes albopictus (Diptera: Culicidae). J. Asia Pac. Entomol. 19 191–194. 10.1016/j.aspen.2015.12.014 [DOI] [Google Scholar]
- Pietri J. E., DeBruhl H., Sullivan W. (2016). The rich somatic life of Wolbachia. Microbiologyopen 5 923–936. 10.1002/mbo3.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing W., Zhijing X., Guangfu Y., Fengxia M., Qiyong L., Zhong Z., et al. (2020). Variation in the microbiota across different developmental stages of Aedes albopictus is affected by ampicillin exposure. MicrobiologyOpen 9 1162–1174. 10.1002/mbo3.1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh K., Dhanasekaran D., Tyagi B. K. (2013). Survey of container breeding mosquito larvae (Dengue vector) in Tiruchirappalli district, Tamil Nadu, India. J. Entomol. Zool. 1 88–91. [Google Scholar]
- Ramirez J. L., Souza-Neto J. A., Torres Cosme R., Rovira J., Ortiz A., Pascale J. M., et al. (2012). Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS Negl. Trop. Dis. 6:e1561. 10.1371/journal.pntd.0001561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci I., Cancrini G., Gabrielli S., D’amelio S., Favia G. (2002). Searching for Wolbachia (Rickettsiales: Rickettsiaceae) in mosquitoes (Diptera: Culicidae): large polymerase chain reaction survey and new identifications. J. Med. Entomol. 39 562–567. 10.1603/0022-2585-39.4.562 [DOI] [PubMed] [Google Scholar]
- Roldán G., Ramírez J. J. (2008). Fundamentos de Limnologia Neotropical. Medellín: Universidad de Antioquia. [Google Scholar]
- Rosso F., Tagliapietra V., Albanese D., Pindo M., Baldacchino F., Arnoldi D., et al. (2018). Reduced diversity of gut microbiota in two Aedes mosquitoes species in areas of recent invasion. Sci. Rep. 8:16091. 10.1038/s41598-018-34640-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo-Porras N., Umana-Diaz C., de Oliveira Barbosa Bitencourt R., Lowenberger C. (2020). The role of bacterial symbionts in triatomines: an evolutionary perspective. Microorganisms 8:1438. 10.3390/microorganisms8091438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab V. F., Herrmann M., Roth V. N., Gleixner G., Lehmann R., Pohnert G., et al. (2017). Functional diversity of microbial communities in pristine aquifers inferred by PLFA- and sequencing-based approaches. Biogeosciences 14 2697–2714. 10.5194/bg-14-2697-2017 [DOI] [Google Scholar]
- Scolari F., Casiraghi M., Bonizzoni M. (2019). Aedes spp. and their microbiota: a review. Front. Microbiol. 10:2036. 10.3389/fmicb.2019.02036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabourn P., Spafford H., Yoneishi N., Medeiros M. (2020). The Aedes albopictus (Diptera: Culicidae) microbiome varies spatially and with Ascogregarine infection. PLoS Negl. Trop. Dis. 14:e0008615. 10.1371/journal.pntd.0008615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelomi M. (2019). Bacterial and eukaryote microbiomes of mosquito habitats in dengue-endemic southern Taiwan. J. Asia Pac. Entomol. 22 471–480. 10.1016/j.aspen.2019.02.011 [DOI] [Google Scholar]
- Shi M., White V. L., Schlub T., Eden J. S., Hoffmann A. A., Holmes E. C. (2018). No detectable effect of Wolbachia wMel on the prevalence and abundance of the RNA virome of Drosophila melanogaster. Proc. R. Soc. B. 285:20181165. 10.1098/rspb.2018.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simhadri R. K., Fast E. M., Guo R., Schultz M. J., Vaisman N., Ortiz L., et al. (2017). The gut commensal microbiome of Drosophila melanogaster is modified by the endosymbiont Wolbachia. mSphere 2:e00287-17. 10.1128/mSphere.00287-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza-Neto J. A., Powell J. R., Bonizzoni M. (2019). Aedes aegypti vector competence studies: a review. Infect. Genet. Evol. 67 191–209. 10.1016/j.meegid.2018.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand M. R. (2018). Composition and functional roles of the gut microbiota in mosquitoes. Curr. Opin. Insect Sci. 28 59–65. 10.1016/j.cois.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery I., Nicolas L., Rippka R., Demarsac N. T. (1991). Selection of cyanobacteria isolated from mosquito breeding sites as a potential food source for mosquito larvae. Appl. Environ. Microbiol. 57 1354–1359. 10.1128/AEM.57.5.1354-1359.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongsripong P., Chandler J. A., Green A. B., Kittayapong P., Wilcox B. A., Kapan D. D., et al. (2018). Mosquito vector-associated microbiota: metabarcoding bacteria and eukaryotic symbionts across habitat types in Thailand endemic for dengue and other arthropod-borne diseases. Ecol. Evol. 8 1352–1368. 10.1002/ece3.3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R. M., Ning D. L., He Z. L., Zhang P., Spencer S. J., Gao S. H., et al. (2020). Small and mighty: adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 8:51. 10.1186/s40168-020-00825-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortosa P., Charlat S., Labbé P., Dehecq J. S., Barré H., Weill M. (2010). Wolbachia age-sex-specific density in Aedes albopictus: a host evolutionary response to cytoplasmic incompatibility? PLoS One 5:e9700. 10.1371/journal.pone.0009700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente Moro C., Tran F. H., Raharimalala F. N., Ravelonandro P., Mavingui P. (2013). Diversity of culturable bacteria including Pantoea in wild mosquito Aedes albopictus. BMC Microbiol. 13:70. 10.1186/1471-2180-13-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk A. F., Hall-Mendelin S., Pyke A. T., Frentiu F. D., McElroy K., Day A., et al. (2012). Impact of Wolbachia on infection with chikungunya and yellow fever viruses in the mosquito vector Aedes aegypti. PLoS Negl. Trop. Dis. 6:e1892. 10.1371/journal.pntd.0001892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Martinez M. G., Rodriguez M. H., Arredondo-Jimenez J. I., Mendez-Sanchez J. D., Bond-Compean J. G., Gold-Morgan M. (2002). Cyanobacteria associated with Anopheles albimanus (Diptera : Culicidae) larval habitats in southern Mexico. J. Med. Entomol. 39 825–832. 10.1603/0022-2585-39.6.825 [DOI] [PubMed] [Google Scholar]
- Walker E. D., Kaufman M. G., Merritt R. W. (2010). An acute trophic cascade among microorganisms in the tree hole ecosystem following removal of omnivorous mosquito larvae. Community Ecol. 11 171–178. 10.1556/ComEc.11.2010.2.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker E. D., Lawson D. L., Merritt R. W., Morgan W. T., Klug M. J. (1991). Nutrient dynamics, bacterial populations, and mosquito productivity in tree hole ecosystems and microcosms. Ecology 72 1529–1546. 10.2307/1940953 [DOI] [Google Scholar]
- Walker T., Johnson P. H., Moreira L. A., Iturbe-Ormaetxe I., Frentiu F. D., McMeniman C. J., et al. (2011). The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature 476 450–453. 10.1038/nature10355 [DOI] [PubMed] [Google Scholar]
- Wang X. M., Liu T., Wu Y., Zhong D. B., Zhou G. F., Su X. H., et al. (2018). Bacterial microbiota assemblage in Aedes albopictus mosquitoes and its impacts on larval development. Mol. Ecol. 27 2972–2985. 10.1111/mec.14732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihrauch D., Donini A., O’Donnell M. J. (2012). Ammonia transport by terrestrial and aquatic insects. J. Insect Physiol. 58 473–487. 10.1016/j.jinsphys.2011.11.005 [DOI] [PubMed] [Google Scholar]
- Werren J. H. (1997). Biology of Wolbachia. Annu. Rev. Entomol. 42 587–609. 10.1146/annurev.ento.42.1.587 [DOI] [PubMed] [Google Scholar]
- Xi Z. Y., Ramirez J. L., Dimopoulos G. (2008). The Aedes aegypti Toll pathway controls dengue virus infection. PLoS Pathog. 4:e1000098. 10.1371/journal.ppat.1000098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav K. K., Bora A., Datta S., Chandel K., Gogoi H. K., Prasad G. B., et al. (2015). Molecular characterization of midgut microbiota of Aedes albopictus and Aedes aegypti from Arunachal Pradesh, India. Parasit. Vectors 8:641. 10.1186/s13071-015-1252-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav K. K., Datta S., Naglot A., Bora A., Hmuaka V., Bhagyawant S., et al. (2016). Diversity of cultivable midgut microbiota at different stages of the Asian Tiger Mosquito, Aedes albopictus from Tezpur, India. PLoS One 11:e0167409. 10.1371/journal.pone.0167409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y. H., Carrasco A. M., Frentiu F. D., Chenoweth S. F., Beebe N. W., van den Hurk A. F., et al. (2015). Wolbachia reduces the transmission potential of dengue-infected Aedes aegypti. PLoS Negl. Trop. Dis. 9:e0003894. 10.1371/journal.pntd.0003894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh Y. K., Sekiguchi Y., Parks D. H., Hugenholtz P. (2015). Comparative genomics of candidate phylum TM6 suggests that parasitism is widespread and ancestral in this lineage. Mol. Biol. Evol. 33 915–927. 10.1093/molbev/msv281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Rousset F., O’Neil S. (1998). Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc. Biol. Sci. 265 509–515. 10.1098/rspb.1998.0324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouache K., Voronin D., Tran-Van V., Mousson L., Failloux A. B., Mavingui P. (2009). Persistent Wolbachia and cultivable bacteria infection in the reproductive and somatic tissues of the mosquito vector Aedes albopictus. PLoS One 4:e6388. 10.1371/journal.pone.0006388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zug R., Hammerstein P. (2012). Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One 7:e38544. 10.1371/journal.pone.0038544 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Water samples analyses. (A) 16S rDNA-based bacterial quantification in water samples. On the X-axis, water samples are reported, colored by sampling period (W Start – orange – and W End – green – refers to water collected at the moment of mosquito collection and after all adults had emerged, respectively). Values are expressed as log2(DNA counts)/mL. (B) Physical analysis of W Start and W End water samples. Shannon alpha diversity index, pH, and conductivity values are shown. Both pH and conductivity significantly increased in W End samples (t-test, P < 0.05).
Rarefaction curves used to discover all the microbial ‘species’/ASVs in each sample where Wolbachia reads were removed.
Bacterial genera shared between breeding site water, and wild and laboratory larvae and adults. Venn diagrams show the number of genera shared in (A) water (W Start vs. W End), (B) larval (CR vs. FO), and (C) adult (CR vs. FO) samples.
Alpha diversity indices of adult individuals. Alpha diversity (Shannon) values distribution for adults (CR and FO) samples are shown. Each sample is indicated with a different color.
Number of sequencing reads per sample before and after removal of Wolbachia-associated reads. DNA extraction negative controls are included. Samples with no Wolbachia reads are indicated in bold.
List of ASVs identified in each larval, adult, and water sample.
Bacteria quantification through qPCR assays.
Physio-chemical analyses of water samples.
Proportion of sequencing reads assigned to a given bacterial taxon at the phylum and family level in water, larval, and adult samples. Only 50 most abundant ASVs were considered.
List of bacterial genera identified in the conserved microbiota for each larval, adult, and water sample.
List of unique and conserved bacterial genera from the microbiota of larvae and adults from wild or laboratory samples.
Data Availability Statement
The datasets generated for this study can be found in the European Nucleotide Archive (ENA) under the BioProject: PRJEB41031. The codes used for our analyses are available at the following link: https://gitlab.com/anna.sandionigi/metamosquito.