

Summary
Immunotherapy has achieved notable success in tumor treatment, but it is restricted to a small number of patients due to multiple immunosuppressive pathways in the tumor microenvironment. Here, we present a step-by-step protocol to prepare functional cellular nanovesicles from HEK293-FT cells displaying PD1 and TRAIL. TRAIL specifically induces immunogenic cancer cell death to initiate an immune response, and ectogenic PD1 blocks the PD1/PDL1 checkpoint signal to reactivate anergic tumor-specific CD8+ T cells.
For complete details on the use and execution of this protocol, please refer to Wu et al. (2020).
Subject areas: Cell membrane, Cancer, Immunology
Graphical Abstract
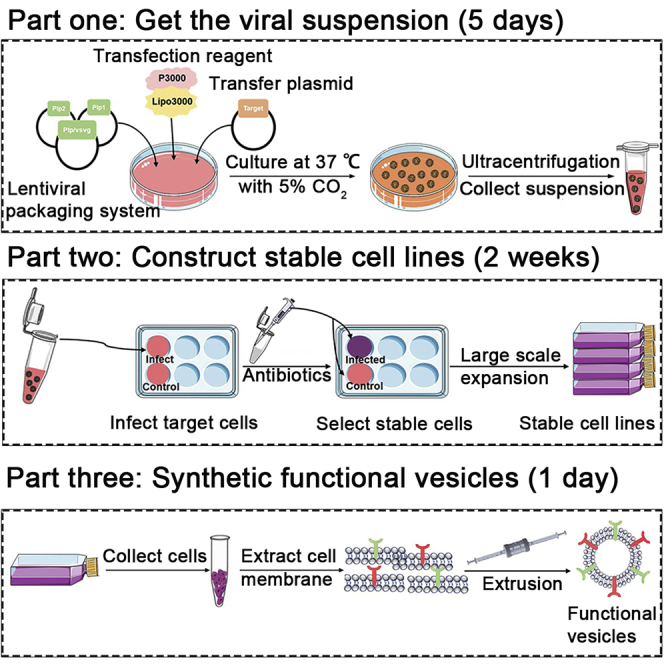
Highlights
-
•
Established stable HEK293 FT cells expressing PD1 and TRAIL
-
•
Prepare functional cellular nanovesicles (FCNVs) from stable HEK293 FT cells
-
•
FCNVs can boost antitumor response by the cooperative roles of PD1 and TRAIL
Immunotherapy has achieved notable success in tumor treatment, but it is restricted to a small number of patients due to multiple immunosuppressive pathways in the tumor microenvironment. Here, we present a step-by-step protocol to prepare functional cellular nanovesicles from HEK293-FT cells displaying PD1 and TRAIL. TRAIL specifically induces immunogenic cancer cell death to initiate an immune response, and ectogenic PD1 blocks the PD1/PDL1 checkpoint signal to reactivate anergic tumor-specific CD8+ T cells.
Before you begin
Design plasmids
Timing: 1 day
-
1.
Download the target gene sequence from NCBI Gene database. For this protocol, we use house mouse PD1 (Programmed cell death 1, Pdcd1) (Tang et al., 2018), TRAIL (Tumor necrosis factor superfamily member 10, Tnfsf10) (von Karstedt et al., 2017), and their Accession are NC_000067.7 and NC_000069.7, respectively. See Figure 1.
-
2.Integrate target gene sequence with Kozak sequence, linker sequence and fluorescent protein sequence (Figure 2).
-
a.Acquire Kozak sequence on the website (https://baike.sogou.com/v7771534.htm)
-
b.Determine the Linker sequence according to experimental needs (Chen et al., 2013). For this protocol, we choose the GS linker to connect the target gene sequence and the fluorescent protein sequence.Note: According to their functions in the construction of recombinant fusion proteins, peptide linkers are generally classified into three categories: flexible linkers, rigid linkers, and in vivo cleavable linkers. Among these linkers, flexible linkers (usually peptides rich in small or hydrophilic amino acids) could provide a certain degree of flexibility between the joined domains to allow interaction or increase spatial separation between the domains, while rigid linkers (such as helical structures and peptides rich in Prolines) could keep the distance between the joined domains to maintain the stability and bioactivity of the domains. And in vivo cleavable linkers are commonly used to generate commensurable proteins, which are physically separated from each other to prevent any potential interaction and steric hindrance. Particularly in this protocol, in order to make sure that the target protein and fluorescent protein are constantly fused to each other to facilitate the imaging, meanwhile to increase spatial separation to minimize steric hindrance, the most commonly used flexible linker consisting of Glycine and Serine (abbreviated as “GS” linker) is chosen as the peptide linker. In addition to act as a flexible linker used in fusion proteins, GS linker also can be used in single-chain variable fragment (scFv) to link the variable regions of heavy and light chain of the complete antibody, or optimized to improve the oral efficacy of recombinant granulocyte colony-stimulating factor and transferrin fusion protein.
-
c.Download the fluorescent protein sequence as step 1 described from the NCBI Gene database. For this protocol, we select red fluorescent protein (mCherry, ADV78248.1) and green fluorescent protein (EGFP, NC_025025.1) as fluorescence tags.
-
d.Kozak sequence, Linker sequence, target gene sequence and fluorescent protein sequence are connected in order as shown in Figure 2, and the integrity of the integrated sequences is checked by Snapgene software.
-
e.Entrust the company (Sangon Biotech (Shanghai) Co., Ltd. https://www.sangon.com/) to synthesize the integrated sequences and insert them into the lentivirus vector plasmid (pCDH-CMV-MCS-EF1-Puro and pCDH-CMV-MCS-EF1-Blasticidin) through linking at the end of the C-terminal of the plasmid (Zhang et al., 2018a, 2018b).Alternatives: The pre-synthesized lentivirus vectors can also be homemade by the following steps. First, design the PCR primers to amplify the coding sequence of the target gene (remember to contain Kozak sequence, restriction enzyme cutting site and optimal bases for flanking the restriction site at the 5'end of the forward primer; for the reverse primer, stop codon should be removed to fuse the protein with GS linker and fluorescent protein). Second, the target gene fragment is generated by PCR amplification from cDNA template and purified by electrophoresis. Next, the PCR product and vector plasmid are respectively subjected to restriction enzyme digestion and purified by electrophoresis. The purified linear vector and the PCR fragment are ligated by T4 ligase to construct the interested plasmid, which is further imported into competent cells and dotted onto LB agar plates (plus antibiotic). The monoclonal colonies on the plate are selected and multiplied in LB medium for PCR identification, and the positive clones are sequenced. At last, the correct clone is expanded in large-scale and the plasmid is extracted with EndoFree Maxi Plasmid Kit following the manufacturer’s instructions.
-
a.
Figure 1.

The detailed target gene information from the NCBI database
Figure 2.

Schematic illustration of the integration and insertion of the sequence of interest
Plasmid amplification
After obtaining the above mentioned two plasmids (pCDH-CMV-PD1-mCherry-Puro, pCDH-CMV-TRAIL-EGFP-Blasticidin), high-quality plasmids were obtained by purification with EndoFree Maxi Plasmid Kit (TIANGEN, DP117, https://www.tiangen.com/?app.html).
-
3.
To prepare LB medium, dissolve 10 g tryptone, 5 g Yeast extract and 10 g NaCl in 1 L of ultrapure water (use magnetic stirrer, 500 rpm/min), aliquot to 200 mL and autoclave at 121°C for 15 min. After the medium is naturally cooled to 20°C–25°C, add antibiotics (containing ampicillin 100 μg/mL or kanamycin 50 μg/mL).
-
4.
Take out the Competent Cell (TransGen, CD501-02, https://www.transgen.com.cn/) from the −80°C refrigerator and leave on the ice for 15 min.
-
5.
Add 20 μL Competent Cell and 5 μL plasmids into a sterilized 1.5 mL Eppendorf (EP) tube, mix well, and place on ice for 0.5 h.
-
6.
The mixture is then placed on 42°C water bath for 30 s, then immediately placed on ice for 2 min.
-
7.
500 μL free-antibiotic LB medium is added to EP tube and shaken at 37°C for 40–60 min to restore normal growth of competent cell, using bench type thermostatic oscillator.
-
8.
The mixture is added to 200 mL LB medium containing the antibiotics and shaken at 37°C for 12–14 h to expand plasmids.
9. Note: Unless otherwise indicated, antibiotics are applied as: 100 μg/mL ampicillin and 50 μg/mL kanamycin. Plasmids are extracted from the microbial broth according to the instructions of the EndoFree Maxi Plasmid Kit (TIANGEN, DP117, https://www.tiangen.com/?app.html).
Pause point: The microbial broth can be stocked in 25% glycerol at −80°C for 1 year.
-
10.
The concentration of the extracted plasmids is detected by NanoDrop 2000/2000c Spectrophotometers (Thermo Fisher Scientific), and the sequence of plasmids is verified by biological company (BioSune (Shanghai) Co., Ltd. http://www.biosune.com/).
Note: A good quality and endotoxin free of plasmids are important for successful transfection in HEK293 FT cells.
Cell culture
-
11.
Prepare cell culture medium. Mix 500 mL of DMEM, 50 mL of FBS, 5 mL of Penicillin-Streptomycin solution (penicillin: 10,000 units/mL, streptomycin 10,000 mg/mL) in a sterilized environment.
-
12.Culture cells for transfection and infection
-
a.Thaw HEK293FT cells cryo-preserved in a liquid nitrogen freezer at 37°C for 5 min, add 10 mL of PBS to wash and centrifuge at 300 × g for 5 min. Aspirate the PBS.
-
b.Resuspend the cell pellet in 1 mL of culture medium as prepared in step 11, transfer the cells into T25 cell culture flask and then supplement with 4 mL of culture medium.
-
c.Place the cell culture flask in an incubator with 5% CO2 at 37°C.Note: After culture the cells in incubator for about 12 h, there are some dead cells floating in the medium, then the medium should be replaced by fresh one.
-
d.When cells reach 80%–90% confluence, aspirate the culture medium from the cell culture flask and rinse the cells once with 5 mL of PBS using an electronic pipette.
-
e.Add 0.5 mL trypsin-EDTA to the cells and place the cells into the incubator with 5% CO2 at 37°C incubator for 3 min.
-
f.Add 10 mL culture medium and detach the cells by slowly pipetting up and down, and count cell number under a microscope using a hemocytometer.
-
g.Transfer the culture medium into a T75 cell culture flask using an electronic pipette.
-
h.Place the cell culture flask in an incubator with 5% CO2 at 37°C. When cells have reached 80%–90% confluence, split cells 1:4 into another T75 cell culture flask by repeating steps d–f.
-
i.Continue to sub-culture cells, maintaining 80%–90% confluence, until ready for construction of stable cell lines expressing PD1 and TRAIL.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-PD1 | Abcam | Cat# ab214421 |
| Mouse monoclonal anti-TRAIL | Abcam | Cat# ab10516 |
| Goat anti-rabbit IgG (H+L), HRP conjugate | TransGen Biotech | Cat# HS101-01 |
| Goat anti-mouse IgG (H+L), HRP conjugate | TransGen Biotech | Cat# HS201-01 |
| Bacterial and virus strains | ||
| Trans1-T1 phage resistant chemically competent cell | TransGen Biotech | Cat# CD501-02 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Gibco | Cat# 11995500 |
| Opti-MEM | Gibco | Cat# 31985-LSG |
| FBS | PAN | Cat# P30-3302 |
| PBS | HyClone | Cat# SH30256.01 |
| Penicillin-streptomycin | Gibco | Cat# 15140122 |
| Ampicillin | Solarbio | Cat# YZ-1033000 Cas# 69-53-4 |
| Kanamycin | Solarbio | Cat# YZ-1355006 Cas# 5965-95-7 |
| Tryptone | OXOID | Cat# LP0042 |
| Yeast extract | OXOID | Cat# LP0021 |
| NaCl | Sinopharm | Cat# 73522260 Cas# 7647-14-5 |
| DiO | Beyotime | Cat# C1038 |
| DiI | Beyotime | Cat# C1036 |
| DAPI | Sigma-Aldrich | Cat# D9542 Cas# 28718-90-3 |
| PMSF | Beyotime | Cat# ST506 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Polybrene | Santa Cruz | Cat# SC-134220 |
| Paraformaldehyde, 4% | Solarbio | Cat# P1110 |
| RIPA lysis buffer | Servicebio | Cat#G2002 |
| Critical commercial assays | ||
| EndoFree Maxi Plasmid Kit | TIANGEN BIOTECH (BEIJING) | Cat# DP117 |
| Easy II Protein Quantitative Kit | TransGen | Cat# DQ111-01 |
| Membrane and Cytosol Protein Extraction Kit | Beyotime | Cat# P0033 |
| Lipofectamine 3000 Transfection Kit | Thermo Fisher | Cat# L3000015 |
| Mouse PD-1 Duo Set ELISA Kit | R&D Systems | Cat# DY1021 |
| Mouse TRAIL/TNFSF10 Duo Set ELISA Kit | R&D Systems | Cat# DY1121 |
| Experimental models: cell lines | ||
| Human: HEK293FT | ATCC | N/A |
| Recombinant DNA | ||
| Plasmid: pCDH-CMV-MCS-EF1-Puro | This paper | N/A |
| Plasmid: pCDH-CMV-MCS-EF1-Blasticidin | This paper | N/A |
| Plasmid: pCDH-CMV-PD1-mCherry-Puro | This paper | N/A |
| Plasmid: pCDH-CMV- TRAIL-EGFP-Blasticidin | This paper | N/A |
| Software and algorithms | ||
| Flowjo | Tree Star | 10.0 |
| Zen | Carl Zeiss | 2012 SP2(blue edition) |
| Other | ||
| Confocal dish | NEST | Cat# 801005 |
| 6-well plate | Costar | Cat# 3335 |
| 0.22 μm filter | Merck Millipore | Cat#PRO3683 |
| 15 mL centrifuge tubes | Corning | Cat# 430790 |
| 25 cm2 cell culture flasks | Corning | Cat# 430639 |
| 75 cm2 cell culture flasks | Corning | Cat# 430641 |
| Cell scraper | Thermo Fisher | Cat# 179693PK |
| CO2 incubator | Thermo Scientific | HERAcell150i |
| Thermostat water bath | Jinghong | DK-8D |
| Clean bench | AIRTECH | SW-CJ-2FD |
| Autoclave sterilizer | ZEALWAY | GR85DR |
| Milli-Q gradient system | Milli-Q Reference | Milli-Q Integral 3 |
| NanoDrop2000/2000c spectrophotometer | Thermo Scientific | ND2000LAPTOP |
| Ultracentrifuge | Beckman Coulter | Optima XPN-100 |
| Centrifuge | Thermo Fisher | Sorvall ST 16R |
| Dry bath | ZHISUN | Mk-20 |
| Avanti mini-syringe | Avanti | 610017-1Ea |
| Confocal laser scanning microscope | Zeiss | LSM780 |
| Flow cytometry | BD | FACSVerse |
| Fluorescence microscope | Zeiss | Axio Vert.A1 |
Materials and equipment
Preparation of 2× loading buffer
| Reagent | Amount |
|---|---|
| 1 M Tris-HCl (pH 6.8) | 1.25 mL |
| 10% SDS | 3.0 mL |
| Bromophenol Blue | 1.0 mg |
| Glycerin | 1 mL |
| β-Mercaptoethanol | 0.5 mL |
Step-by-step method details
Construction of stable cell lines expressing PD1 and TRAIL
When the plasmids containing PD1 or TRAIL sequence and HEK293FT cells are ready, the genetic engineered cells are constructed according to the overexpression protocol which involves plasmid transfection, antibiotic selection to construct stable cell lines (Elegheert et al., 2018). The procedure includes two parts. In the first part (steps 1–9), the HEK293FT cell lines are transfected using Lipofectamine 3000 kit (Thermo Fisher, L3000015), and lentivirus supernatant are collected. In the second part, according to the resistance gene in the target gene plasmid, the corresponding antibiotic is added to screen the infected cells, then large-scale expanded (steps 10–13) (Chaudhary et al., 2012).
-
1.
Inoculate the parent HEK293FT cells in T75 culture flask in advance, and proceed to the next step after the cell density reaches 70%–80%.
-
2.
Prepare reagents for gene transfection. Prepare a 15 mL centrifuge tube (numbered A) and a 1.5 mL EP tube (numbered B). Transfer 1 mL Opti-MEM medium (Gibco, 31985-LSG, https://www.thermofisher.com/us/en/home/brands/gibco.html?CID=cn-ebz-sem-sgk-gene-Gibco-aBrand-pc-subb-082920-abcd-00000009FD973428) and 40 μL Lipo 3000 in Tube A, mix them by using gentle pipette motion. Tube B contains target gene plasmid (10 μg), lentivirus packaging systems (plp1 plasmid: 7.5 μg; plp2 plasmid: 3 μg; plp/vsvg plasmid: 4 μg) and P3000 (40 μL), which are dissolved in 1 mL Opti-MEM medium (Gibco, 31985-LSG,https://www.thermofisher.com/us/en/home/brands/gibco.html?CID=cn-ebz-sem-sgk-gene-Gibco-aBrand-pc-subb-082920-abcd-00000009FD973428). See Table 1 for the description of all reagent for transfection. Then, set tube A and B aside for 5 min.
-
3.
Transfer the solution in Tube B to Tube A, mix them by using gentle pipette motion, and let the mixture stand for 20 min.
-
4.
Add the above mixture to the cell culture flask, and lentiviral supernatant are collected after 24 h to 72 h of incubation.
Note: mCherry tag and EGFP tag inserted in the target gene plasmid provide the fluorescent signals to evaluate the gene transfection efficiency. Under fluorescence microscope, when the percent of fluorescence cells reach above 60% (Figure 3), and the cell maintain normal morphology, it is suitable to collect the supernatant. If the transfection efficiency is low, See Troubleshooting: Problem 1.
Table 1.
The detailed description of transfection reagent
| Amount | |
|---|---|
| Reagent | |
| Opti-MEM | 2 mL |
| Lipo 3000 | 40 μL |
| P3000 | 40 μL |
| Plasmid | |
| Plp1 | 7.5 μg |
| Plp2 | 3.0 μg |
| Plp/vsvg | 4.0 μg |
| Target gene plasmid (PD1 or TRAIL ) | 10.0 μg |
Figure 3.

Fluorescence images of HEK293 FT cells after transfection for 24 h
Scale bar, 100 μm.
CRITICAL: Collect the supernatant and add fresh culture medium. Try to operate along the side wall of the culture flask, gently and slowly, and avoid direct contact with the cells.
-
5.
Filter the supernatant containing lentivirus by passing it though a 0.22 μm filter to remove large cell fragments.
-
6.
Transfer the sample to a sterilized ultracentrifugation tube. Place the centrifugation tube into a pre-cooled centrifuge. Counterbalance when needed.
-
7.Program the ultracentrifuge run using the following settings:
-
a.Rotor type: SW 32.1 Ti Rotor
-
b.Speed: 76,800 × g
-
c.Time: 90 min
-
d.Temperature: 4°C
-
e.Acceleration: 0 (MAX)
-
f.Deceleration: 0 (MAX)
-
a.
-
8.
Start the ultracentrifuge.
-
9.
After centrifugation, the supernatant is gently discarded, and 1 mL of fresh culture medium is added to resuspend the virus, then aliquot into 3–5 tubes (virus titer: 106–107 IFU/mL), and stored at −80°C for future use.
-
10.
Seed 1 mL of the cell suspension of HEK293FT cells (containing 1 × 105 cells) in a 6-well cell culture plate, place the plate in an incubator with 5% CO2 at 37°C overnight.
Note: Incubation time can be adjusted and terminated when 70%–80% cell confluence reach.
-
11.
The cell culture is replaced by fresh medium supplemented with a tube of virus solution (containing 106–107 IFU) and 5 μg/mL polybrene reagent to infect the cells. If the infection efficiency not high, see Troubleshooting: Problem 2. After 2 or 3 days of culture, add puromycin or blasticidin to the cell for selecting PD-1 or TRAIL stably expression cells, respectively. The parent cells without lentiviral infection are used as control.
Note: The concentration of puromycin or blasticidin should be adjusted above the lethal doses that kill nearly all the parent cells. If the high dose antibiotics also fail to screen, see Troubleshooting: Problem 3.
-
12.
Cells in the infected group are screened for 1–2 weeks until the percent of fluorescence cells reached above 95%, proved that the stably expression cells are successfully screened.
Note: Formation of puromycin or blasticidin resistance cells suggests the successful infection of lentivirus.
-
13.
The stably expression cells are large-scale expanded.
Note: When cells have reached 80%–90% confluence, split cells 1:3 onto another 6-well plate with fresh culture medium.
Characterization of stable cell lines
The constructed stable cell lines are characterized by confocal laser scanning microscopy observation (Dong et al., 2019), western blot assay (Ohno et al., 2013) and flow cytometry assay (Koh et al., 2017) to confirm the target protein is well expressed and localized onto the cell membrane.
-
14.Confocal microscopy observation
-
a.Cells are seeded in a confocal dish at a density of 1 × 105 per well. Place the dish in an incubator with 5% CO2 at 37°C until they completely adhered.
-
b.Wash cells three times with PBS, and fix them with 4% paraformaldehyde for 10–15 min
-
c.Cell nucleus is stained with DAPI for 10 min, washed three times with PBS.
-
d.Cell membrane is stained with DiO or DiI for 15 min, washed three times with PBS, and observed under a confocal laser scanning microscope (Zeiss LSM780). See Figure 4.
-
a.
-
15. Western blot assay
-
a.Collect cells into a 1.5 mL EP tube by using cell scraper (Thermo Fisher Scientific), when the cells reach 70%–80% confluence in T75 cell culture flask.
-
b.The cells are lysed on ice with RIPA lysis buffer for 30 min. After lysis, the cells are centrifuged at 4°C for 30 min (13,000 × g), and the supernatant is collected for BCA protein quantification, using EasyⅡ Protein Quantitative Kit (TransGen, DQ111-01, https://www.transgen.com.cn/).
-
c.According to the BCA protein quantification result, the sample is diluted to 4 mg/mL and mixed evenly with 2× loading buffer (volume ratio 1:1), and the mixed sample is boiled at 100°C for 10 min. For details on preparation of 2× loading buffer, see the Materials and equipment.
-
d.The prepared sample is subjected to polyacrylamide gel electrophoresis for protein separation. Select β-actin protein as the internal reference protein for comparison. And then, transfer to the NC membrane by membrane transfer. Afterwards, the NC membrane is placed in 5% milk and blocked at 20°C–25°C for 2 h, and washed with TBST buffer (100 mM Tris-HCl (pH=8.0), 1.5 mM NaCl, 0.1% Tween 20) for 3 times, 10 min per time. After that, add the primary antibody (dilution rate, 1:1,000), See Key resources table, and incubate for 12–16 h at 4°C.
-
e.The next day, recycle the primary antibody and wash the membrane with TBST buffer for 3 times with 15 min per time. Afterwards, add the secondary antibody and incubate for 1 h at 20°C–25°C. Then recycle the secondary antibody (dilution rate, 1:5,000), See Key resources table, and wash the membrane with TBST buffer for 3 times with 10 min per time. Preparation the ECL and imaging on the gel imager (ChemiDoc MP) (Figure 5).
-
a.
-
16.
Flow cytometer assay
Figure 4.

Confocal images of HEK293 FT cells stably expressing mouse PD1
Figure 5.

Western blot assay to confirm the expression of PD1 (indicated as PD1+) and TRAIL (indicated as TRAIL+) in 293FT cells
Actin was used as an internal reference.
Above prepared 105 ∼5 × 105 cells are collected, centrifuged at 20°C–25°C for 5 min (300 × g) and resuspended in PBS. The fluorescent protein signal was analyzed by flow cytometry (BD, FACSVerse) (Figure 6).
Figure 6.

The representative flow cytometric analysis of HEK293 FT cells before and after transfected with PD 1 and TRAIL, respectively
Cell membrane extraction
-
17.
When cells have reached 80%–90% confluence, remove medium and add 1 mL trypsin to digest cells. Harvest cells from plate and centrifuge cells at 600 × g for 5 min (Kim et al., 2017).
-
18.
Discard the supernatant, collect the cell precipitation and extract the cell membrane with a cell membrane extraction kit according to the manufacture’s instruction (Beyotime, P0033, https://www.beyotime.com/index.htm).
-
19.
Determine the protein content in the extracted membrane by BCA kit, and then store the membrane in −80°C refrigerator for future use.
Note: The storage time of the extracted membrane should be no more than 1 month in −80°C refrigerator.
Synthesize the functional vesicles
An Avanti mini-syringe extruder (Figures 7 and 8) is used to prepare cell membrane vesicles using the mechanical force imposed by the extruding process to disrupt the membrane structure and enable it to reform into nano-scale vesicles (Fang et al., 2014).
-
20.Assemble the mini-syringe extruder
-
a.Place the 2 Internal Membrane Supports on a flat test bed with the O-rings vertically upward
-
b.Pre-wet 2 Filter Supports with DI water, or buffer, and place over orifice of Internal Membrane Supports. The Filter Supports should adhere to the Teflon orifice inside the O-ring inner scope.
-
c.Insert the Internal Membrane Support into the Extruder Outer.
-
d.Place 1 Polycarbonate Membrane in the Extruder Outer Casing over the Filter Support and O-ring.Note: Do not install the blue paper disks which separate the polycarbonate membranes in packing box.
-
e.Pre-wet a second pair of Filter Supports with DI water, or buffer, and place over orifice of remaining Internal Membrane Support.
-
f.Carefully place the second Internal Membrane Support into the casing (O-ring facing down).
-
g.Place the Retainer Nut on the threaded end of the Extruder Outer Casing and tighten. Tighten the Retainer Nut by hand just until it is finger tight; do not use a wrench.Note: Autoclaving the Teflon inserts is not recommended as slight distortion may occur (∼0.002 mm). The use of ethylene oxide or UV irradiation is suggested as an alternative. The syringes should be sterilized with ethanol.
-
a.
Figure 7.

The Avanti mini-syringe extruder
Figure 8.

Schematic illustration of the parts of the Avanti mini-syringe extruder
Figure 9.

Installation process for the Avanti mini-syringe extruder
(A and B) Place two Internal Membrane Support on a flat test bed with the O-rings vertically upward.
(C) Insert one Internal Membrane Support into the Extruder Outer Casing.
(D) Place one pre-wetted Filter Support over orifice of the Internal Membrane Support in the Extruder Outer Casing.
(E) Place one Polycarbonate Membrane over the Filter Support Support and O-ring in the Extruder Outer Casing.
(F) Place the second pair of pre-wetted Filter Support over orifice of the other Internal Membrane Support.
(G) Carefully place the second Internal Membrane Support into the Extruder Outer Casing (O-ring facing down).
(H) Place the Retainer Nut on the threaded end of the Extruder Outer Casing and tighten.
(I) Place the empty gas-tight syringe (with plunger is set to zero) into the one end of the mini-extruder.
-
21.
Add the cell membrane solution into one of the gas-tight syringes and carefully place into one end of the mini-extruder (Figure 10).
Figure 10.

Schematic illustration of drawing the sample by syringe
Note: To reduce the dead volume and facilitate extrusion, pre-wet the extruder parts by passing a syringe full of buffer through the extruder.
-
22.
Place the empty gas-tight syringe into the other end of the mini-extruder. Make sure the empty syringe plunger is set to zero; the syringe will fill automatically as the cell membrane is extruded through the polycarbonate membranes.
-
23.
Gently push the plunger of the filled syringe until the cell membrane solution is completely transferred to the alternate syringe (Figure 11).
-
24.
Gently push the plunger of the alternate syringe to transfer the solution back to the original syringe.
Figure 11.

Schematic illustration of extrusion operation
Repeat above steps 23 and 24. Total of 11 passes through the 1,000 nm, 400 nm, 200 nm, and 100 nm polycarbonate membranes, respectively. If the FCNVs fail to maintain their biological functions, see Troubleshooting: Problem 4.
Note: The final extrusion should fill the alternate syringe. This is to reduce the chances of contamination with larger particles or foreign material.
-
25.
After the final extrusion, inject the obtained solution into a clean sample vial.
The characterization of FCNVs
-
26.
The size distribution of FCNVs is measured by dynamic light scattering (DLS) method through Zetasizer NanoZS system (Malvern Instruments, Southborough, MA, USA). Therein, the size measurement is performed in Size module with 200 s equilibration time using DTS0012 cuvette. The ultrastructure, integrity and morphology of FCNVs are evaluated by transmission electron microscopy (TEM). Therein, the sample is dropped onto a Cu-grid that covered with carbon film, and dried for 2 h. Subsequently, the sample is stained by 50 μL of tungstophosphoric acid for 5 min, and the redundant solvent was removed blotters. After drying again, the morphology was observed and photographed using JEM-1400Plus transmission electron microscopy (JEOL, Japan). The specific ratio and content of TRAIL and PD1 in FCNVs are determined by ELISA (R&D Systems, DY1121 for TRAIL and DY1021 for PD1, https://www.rndsystems.com/cn).
Assessment of immunotherapy of FCNVs
The TRAIL component on FCNVs could initiate the immune responses by specifically inducing immunogenic cancer cell death, which could be further strengthened by collaborating with presented ectogenic PD1 proteins as the checkpoint blockade to reactivate the anergic tumor-specific CTLs. The corresponding function of FCNVs can be evaluated both in vitro and in vivo.
-
27.
In vitro immune checkpoint inhibition of FCNVs: naive mouse CD8+ T cells (CTLs) are first isolated from the spleen of BALB/c mice, followed by CD3/CD28 antibody stimulation to obtain activated CD8+ T cells. Subsequently, cancer cells with various treatments, such as blocking PDL1 on cell membranes by our FCNVs or PDL1 antibody, are added to the activated CD8+ T cells to check the cytotoxic activity by using Annexin V-FITC and PI cell apoptosis assay.
-
28.
In vitro immunogenic cancer cell death (ICD) induced by FCNVs: Anticancer effect of TRAIL proteins on FCNVs can be assessed by Annexin V-FITC and PI cell apoptosis assay, and the danger-associated molecular patterns (DAMPs) like CRT proteins released from dead cancer cells are checked by immunofluorescent staining. Finally, the release of DAMPs together with tumor-associated antigens (TAAs) from dead cells are co-incubated with borrow-derived dendritic cells (BMDCs) to check whether they can promote the maturation of BMDCs though dual staining for CD80 and CD86, the typical markers on the surface of maturated DCs.
-
29.
In vivo antitumor effect of FCNVs: The comprehensive immuno-modulating ability and robust antitumor immunity of our FCNVs can be demonstrated through evaluating tumor growth regression, metastasis prevention, distal and immune-memory effects, and long-term survival benefits.
All of the above corresponding results can be found in our recent iScience publication (Wu et al., 2020).
Expected outcomes
Following this protocol, we prepared functional cellular nanovesicles (FCNVs) with surface presenting PD1 and TRAIL. We advise to perform the particle and protein characterization to qualitatively and quantitatively assess the FCNVs, respectively. ELISA determination of PD1 and TRAIL content on FCNVs is necessary. Immunoprecipitation (IP) assay verifies the orientation of functional proteins on FCNVs. The size distribution is measured by dynamic light scattering (DLS) methods. FCNVs ultrastructure, integrity and morphology are evaluated by transmission electron microscopy (TEM). For detailed instructions on how to perform FCNVs characterization and the corresponding results can be found in our recent iScience publication (Wu et al., 2020).
We demonstrated the ability of this protocol to prepare FCNVs with high purity, which is a prerequisite to obtain reliable data for antitumor immune responses. For full details, we refer to (Wu et al., 2020). Theoretically, in addition to PD1 and TRAIL, other functional proteins decorated cellular nanovesicles can be also obtained by our protocols. Furthermore, the inner core of FCNVs can be used to encapsulated other functional components, for example, catalase is loaded into nanovesicles to overcome tumor hypoxia after producing O2, in turn to increase infiltration of effector T cells while deplete immunosuppressive cells in tumor (Wu et al., 2020).
Limitations
While this protocol was developed to obtain the functional cellular nanovesicles with multiple immuno-modulating ability to elicit robust and systematic antitumor immunity, the procedure is labor intensive and low-throughput, thus keep the same quality FCNVs from batch to batch and large-scale production might be an obstacle for pharmaceutical translation. Moreover, some functional proteins on FCNVs have right orientation (outwards), but some presented inwards which certainly can not play the right function in immunotherapy after co-extrusion, but in our current protocol it is very difficult to precisely control the orientation of functional proteins on the surface of FCNVs.
Troubleshooting
Problem 1
Why the efficiency of gene transfection is not high?
Potential solution
There are many factors affecting the transfection efficiency, such as the state of the cells, the quality of the transfection reagent and the purity of the plasmid. Therefore, the cells should be handled in a good condition, the transfection reagent free of quality problems, and the plasmid has high purity and the sequence has been verified before transfection.
Problem 2
Why the efficiency of infected target cells is not high?
Potential solution
We found that the transfection efficiency and the viral titer may directly influent the infection efficiency. Make sure the transfect efficiency more than 80% after 48 h of transfection. And the viral titer should reach above 106 IFU/mL to ensure better infection efficiency. Meanwhile, the storage time of collected virus supernatant at 4°C should not exceed 1 week. Also, if the efficiency of infection is still very low and you may choose secondary infection.
Problem 3
Why the antibiotics fail to screen even at high dose?
Potential solution
Make sure the resistant drugs are within its warranty and properly stored. Then, determine whether the target cells already have the corresponding resistance gene. Alternatively, flow cytometry can also be used to select stably expression cells instead of drug screening.
Problem 4
How to obtain homogeneous FCNVs with uniform morphology while maintain their biological functions?
Potential solution
We advise to mechanically harvest stable cells from cell culture flask by cell scraper rather than trypsin digestion to avoid potential harms to the functional proteins on the cell membranes including PD1 and TRAIL. Another suggestion to maintain the biological function is that the extracted membrane should not be lyophilized for storage. To improve the homogeneity of FCNVs, the repeats of extruding process in steps 23 and 24 can be appropriately increased.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Xiaolong Liu (xiaoloong.liu@gmail.com).
Materials availability
This study did not generate new unique materials.
Data and code availability
This study did not generate any unique datasets or code.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 61727823), the joint research projects of Health and Education Commission of Fujian Province (grant no. 2019-WJ-20), the Joint Funds for the innovation of science and Technology, Fujian province (grant no. 2019Y9046), and the Scientific Foundation of Fuzhou Municipal Health commission (grant nos. 2020-S-wt7 and 2020-S-wp6).
Author contributions
P.Y. and D.Z. wrote and prepared the manuscript. C.Z., M.W., and X.L. edited and helped in section organization.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100324.
Contributor Information
Ming Wu, Email: wmmj0419@163.com.
Xiaolong Liu, Email: xiaoloong.liu@gmail.com.
References
- Chaudhary S., Pak J.E., Gruswitz F., Sharma V., Stroud R.M. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat. Protoc. 2012;7:453–466. doi: 10.1038/nprot.2011.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Zaro J.L., Shen W. Fusion protein linkers: property, design and functionality. Adv. Drug Deliver. Rev. 2013;65:1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Gao J., Zhang C.Y., Hayworth C., Frank M., Wang Z. Neutrophil membrane-derived nanovesicles alleviate inflammation to protect mouse brain injury from ischemic stroke. ACS Nano. 2019;13:1272–1283. doi: 10.1021/acsnano.8b06572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elegheert J., Behiels E., Bishop B., Scott S., Woolley R.E., Griffiths S.C., Byrne E.F.X., Chang V.T., Stuart D.I., Jones E.Y. Lentiviral transduction of mammalian cells for fast, scalable, and high-level production of soluble and membrane proteins. Nat. Protoc. 2018;13:2991–3017. doi: 10.1038/s41596-018-0075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R., Hu C., Luk B., Gao W., Copp J., Tai Y., O'Connor D.E., Zhang L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Han C., Jo W., Kang S., Cho S., Jeong D., Gho Y.S., Park J. Cell-engineered nanovesicle as a surrogate inducer of contact-dependent stimuli. Adv. Healthc. Mater. 2017;6:1700381. doi: 10.1002/adhm.201700381. [DOI] [PubMed] [Google Scholar]
- Koh E., Lee E., Nam G., Hong Y., Cho E., Yang Y., Kim I. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials. 2017;121:121–129. doi: 10.1016/j.biomaterials.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., Yu J.X., Hubbard-Lucey V.M., Neftelinov S.T., Hodge J.P., Lin Y. The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2018;17:854–855. doi: 10.1038/nrd.2018.210. [DOI] [PubMed] [Google Scholar]
- von Karstedt S., Montinaro A., Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer. 2017;17:352–366. doi: 10.1038/nrc.2017.28. [DOI] [PubMed] [Google Scholar]
- Wu M., Zheng D., Zhang D., Yu P., Peng L., Chen F., Lin Z., Cai Z., Li J., Wei Z. Converting immune cold into hot by biosynthetic functional vesicles to boost systematic antitumor immunity. iScience. 2020;23:101341. doi: 10.1016/j.isci.2020.101341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Zhang L., Qin Z., Hua S., Guo Z., Chu C., Lin H., Zhang Y., Li W., Zhang X. Genetically engineered liposome-like nanovesicles as active targeted transport platform. Adv. Mater. 2018;30:1705350. doi: 10.1002/adma.201705350. [DOI] [PubMed] [Google Scholar]
- Zhang X., Wang C., Wang J., Hu Q., Langworthy B., Ye Y., Sun W., Lin J., Wang T., Fine J. PD-1 blockade cellular vesicles for cancer immunotherapy. Adv. Mater. 2018;30:1707112. doi: 10.1002/adma.201707112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.