

SUMMARY
Alterations in chromatin remodeling genes have been increasingly implicated in human oncogenesis. Specifically, the biallelic inactivation of the SWI/SNF subunit SMARCB1 results in the emergence of extremely aggressive pediatric malignancies. Here, we developed embryonic mosaic mouse models of malignant rhabdoid tumors (MRT) that faithfully recapitulate the clinical-pathological features of the human disease. We demonstrated that SMARCB1-deficient malignancies exhibit dramatic activation of the unfolded protein response (UPR) and endoplasmic reticulum (ER) stress response via genetically intact MYC-p19ARF-p53 axis. As a consequence, these tumors display an exquisite sensitivity to agents inducing proteotoxic stress and inhibition of the autophagic machinery. In conclusion, our findings provide rationale for drug repositioning trials investigating combinations of agents targeting the UPR and autophagy in SMARCB1-deficient MRT.
Keywords: Rhabdoid tumors, embryonic mosaic GEM models, SMARCB1, MYC, p53, ER stress, autophagy
In Brief
By generating mouse models of SMARCB1-deficient malignant rhabdoid tumors (MRT), Carugo et al. find that loss of SMARCB1 activates the unfolded protein response, the ER stress response, and autophagy via a MYC–p19ARF–p53 axis, conferring sensitivity to proteasome and autophagy inhibitors in SMARB1-deficient MRT.
Graphical Abstract
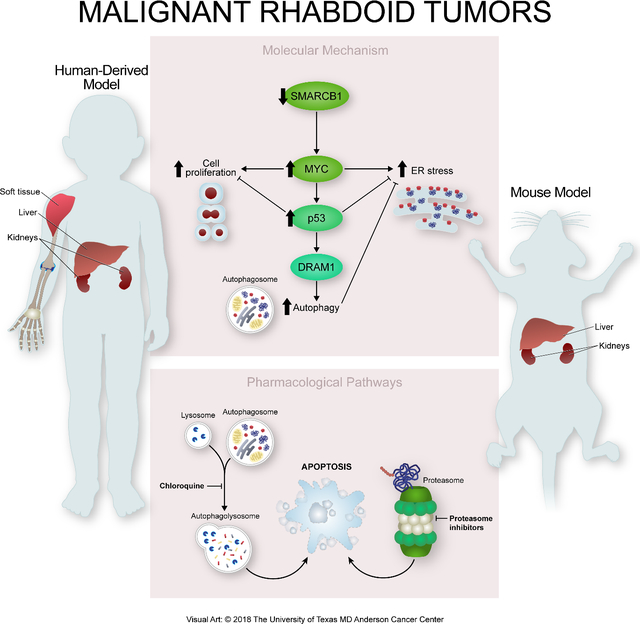
INTRODUCTION
The identification of genetic events affecting the SMARCB1 locus on chromosome 22 in malignant rhabdoid tumors (MRT) and atypical teratoid/rhabdoid tumors (AT/RT) provided the first evidence for involvement of the SWI/SNF chromatin remodeling complex in oncogenesis (Versteege et al., 1998). Since this original report of a causal association between SMARCB1 inactivation and pediatric rhabdoid tumors, the spectrum of neoplasms characterized by loss of this tumor suppressor gene has been expanded to include small cell undifferentiated hepatoblastomas (SCUD) and renal medullary carcinomas (RMC) (Trobaugh-Lotrario et al., 2009; Calderaro et al., 2016). Despite differences in anatomical distribution these malignancies share common features, including early age of onset, a prominent tendency to metastatic spread, and lack of response to current treatments, resulting in universally dismal prognoses (Reinhard et al., 2008; Oue et al., 2009; Buscariollo et al., 2012; Brennan et al., 2016; Ezekian et al., 2017). MRT and other SMARCB1-deficient pediatric neoplasms are considered orphan diseases and are currently managed with a combination of surgery, radiation therapy, and cytotoxic chemotherapy (Tekautz et al., 2005; Trobaugh-Lotrario et al., 2011). These approaches are marginally effective and can result in severe long-term disabilities and increased risk of secondary neoplasms for the few survivors (Dixon et al., 2018). To improve outcomes for these patients, a comprehensive understanding of the functional vulnerabilities of these diseases is needed to support the development of effective targeted therapeutic strategies.
From a biological standpoint, SMARCB1-deficient neoplasms display profound changes in their epigenetic architecture, resulting in the aberrant activation of transcriptional and metabolic programs that promote cell growth, cause deregulation of stem cell maintenance, and suppress terminal differentiation programs (Calderaro et al., 2016; Chun et al., 2016; Johann et al., 2016; Torchia et al., 2016). By contrast, these malignancies are characterized by remarkably few recurrent somatic events, suggesting that dysregulation of the SWI/SNF-dependent chromatin remodeling machinery is sufficient to reprogram normal embryonic and potentially adult cells into a malignant state (Kieran et al., 2012; Lee et al., 2012b; Lawrence et al., 2013). However, a lack of conditional genetic models of MRT has complicated the investigation of the molecular bases of malignant transformation and oncogenic dependencies associated with SMARCB1 loss in specific tissues. Here, we aimed to develop embryonic mosaic models of Smarcb1-null MRTs that faithfully recapitulate the clinical-pathological features of the human disease in order to further understand the biology of these malignancies, identify context specific vulnerabilities and develop more effective therapies.
RESULTS
Disruption of Smarcb1 in embryonic livers drives ER stress and autophagy
Clinical and genomic studies suggest that human MRT develop upon bi-allelic inactivation of SMARCB1 during early embryonic development, and, consequently, the vast majority of cases occur in infants in their first year of life (Bourdeaut et al., 2011). Consistently, leveraging a conditional murine model, Han et al. demonstrated that early embryonic neural progenitors are extremely sensitive to genetic ablation of Smarcb1, which results in AT/RT with high penetrance and short latency (Han et al., 2016). Similarly, genetic ablation of one copy of Smarcb1 predisposes to the development of soft-tissue MRT upon stochastic loss of heterozygosity (LOH) (Klochendler-Yeivin et al., 2000; Roberts et al., 2000). However, we have limited knowledge of the role of Smarcb1 in malignant transformation and tissue specification because of the early embryonic lethality in knockout mice (Roberts et al., 2000; Roberts et al., 2002; Gresh et al., 2005).
To investigate the functional consequences of Smarcb1 ablation during mouse organogenesis, we generated embryonic mosaic models of MRT of the liver (MRTL) by introducing a tissue-specific Cre recombinase expressed from the murine albumin promoter (Adx-A-Cre) via trans-uterine adenoviral injection in Smarcb1LoxP/LoxP embryos at embryonic day E12.5 (Figures 1A, S1A and S1B) (Mitchell et al., 2000). Genetic mosaicism enabled the study of malignant properties of Smarcb1-deficient cells by bypassing the early embryonic and perinatal lethality observed when classical knockout and conditional technologies are employed; it further allowed for the tissue-specific, time-restricted activation of a reporter and quantification of tumor burden and metastatic spread (Figures S1C and S1D). Specifically, the in utero mosaic Cre-mediated ablation of Smarcb1 targeted to E12.5 epithelial liver progenitor cells resulted in liver enlargement and invasive tumors with high propensity for celomatic and systemic dissemination in a subset of animals (Figures 1B–1E and S1E). Upon histopathological analysis, mice displayed liver hyperplasia with severe dysplastic and degenerative changes and disruption of normal liver architecture, livers also harbored focal lesions that faithfully recapitulated the histopathological and clinical features of MRT, including the presence of characteristic rhabdoid morphology, the diffuse infiltration of small, dyscohesive round cells expressing high levels of the mesenchymal marker vimentin, and high proliferative index (assessed by Ki67 immunostaining) (Figures S1F and S1G).
Figure 1. Genetic ablation of Smarcb1 in embryonic livers drives ER stress and autophagy.

A) Schematic display of the experimental procedure used to generate embryonic mosaic model of liver cancer. An adenoviral construct expressing the Cre recombinase under the liver-specific albumin promoter (Adx-A-Cre) was introduced in the liver of E12.5 Smarcb1LoxP/LoxP embryos by trans-uterine delivery. A LacZ expressing vector was used as negative control (Adx-LacZ). B) Top panels: representative coronal MRI sections from Smarcb1LoxP/LoxP mice transduced in utero with Adx-A-Cre (Cre) and Adx-LacZ (LacZ) vectors. Bottom panels: gross pathological appearance of liver lesions at necropsy. C) Kaplan-Meier survival analysis of Smarcb1LoxP/LoxP mice transduced in utero with different doses of Adx-A-Cre. Adx-LacZ transduced mice were used as controls. (Cre, 2×107 I.U.: n = 46; Cre, 2×106 I.U.: n = 31; LacZ, 2×107 I.U.: n = 25). D) Liver/body weight ratio in Smarcb1LoxP/LoxP mice transduced in utero with different doses of the Cre or the LacZ control (Cre, 2×106 I.U.: n = 9; Cre, 2×107 I.U. and LacZ, 2×107 I.U.: n = 10; all biological replicates). E) Relative representation of lesions according to histopathological grading from the experimental cohorts described above. F-G) Volcano plots showing the number of differentially expressed genes (F) and the expression of genes involved in cell cycle progression and regulation of protein metabolism (G) in Smarcb1-ablated tumors (Smarcb1Δ/Δ) and age matched LacZ control livers (Smarcb1+/+) (n = 5 biological replicates/group) (p < 0.01). H) Western blot analysis for markers of ER stress response and autophagy and loading control vinculin (VINC) in Smarcb1-deleted tumors (Smarcb1Δ/Δ) and LacZ transduced normal control livers (Smarcb1+/+). I) Ultrastructural analysis: transmission electron microscopy (TEM) of Smarcb1 deficient, proficient and restored cells. Top panels: representative TEM sections from Smarcb1 ablated tumors and normal control livers. Middle panels: representative TEM sections from Smarcb1 ablated and restored cells transduced with a lentiviral vector expressing the SMARCB1 coding sequence under a doxycycline (Dox) inducible promoter. Bottom panels: E12.5 embryonic liver progenitors cultures were transduced with an adenoviral Cre recombinase or LacZ control. Red arrowheads: autophagic vacuoles. Size bars: 4 μm. J) Quantification of vesicular structures in cultured Smarcb1LoxP/LoxP E12.5 ELP transduced with Cre or LacZ (n = 10 biological replicates/group). Cells were analyzed 96 hr after viral transduction. K) Quantification of vesicular structures in short term cultures established from Smarcb1 deficient MRT transduced with the pLIX-iSMARCB1 lentiviral system and assigned to doxycycline or vehicle treatment (n = 10 biological replicates/group). Cells were analyzed 96 hr after induction. L) UB-GFP quenching study in E12.5 Smarcb1LoxP/LoxP embryonic liver progenitors cultures transduced with Cre or LacZ. Cycloheximide (Chx) treated cells were used as negative controls, MG132 was used as a GFP rescue control in Cre transduced cells. M) LC3-GFP quenching study in E12.5 Smarcb1LoxP/LoxP embryonic liver progenitors transduced with an adenoviral Cre recombinase or LacZ control. Activation of autophagy was assessed by FACS analysis as a decrease in the number of GFP+ cells. Chx treated cells were used as negative controls, and chloroquine was used as a GFP rescue control in Cre transduced cells. Pharmacological treatments in L and M were started 12 hr before analysis. Extinction of the UB-GFP and LC3-GFP signal were assessed by FACS analysis 96 hr post infection. ** p < 0.01, *** p < 0.001, **** p < 0.0001 by unpaired two-tailed t-test (D, J, K) and Mantel-Cox test (C). Box-plots: Box and lines mark quartiles, ends of lines represent minimum and maximum. See also Figure S1 and Table S1.
To uncover the oncogenic networks activated upon Smarcb1 ablation, we performed a microarray analaysis to compare the transcriptome of Smarcb1-deficient lesions with age-matched Adx-LacZ control livers and identified 7759 differentially expressed genes (Figure 1F). Consistent with transcriptomic profiles of human MRT, GSEA pathway analysis revealed robust activation of multiple oncogenic networks, including pathways regulating cell-cycle progression (Figure S1H and Table S1) (Subramanian et al., 2005). In addition, we identified profound transcriptional changes in genes regulating protein translation and ribosome biogenesis, along with activation of cellular programs required for the maintenance of proteostasis and the UPR (Figures 1G, S1H and Table S1). Supportive of the transcriptomic data, Western blot analysis and immunohistochemical characterization of Smarcb1-deficient tumors confirmed dramatic up-regulation of proteins involved in the ER stress response and autophagy (Figures 1H and S1I). Transmission electron microscopy studies of murine tumors revealed signs of ER stress and autophagy including ER swelling, altered ER-ribosomes interface, accumulation of insoluble protein aggregates and reticulophagy (Figures 1I and S1K). In line with these evidences, acute ex vivo ablation of Smarcb1 in E12.5 embryonic liver progenitors (ELP) short term cultures resulted in a dramatic upregulation of ER stress markers and induction of autophagy as assessed by phagosome counts (Figures 1I, 1J, S1J and S1K). Similarly, restoration of SMARCB1 in tumor derived cultures leveraging a lentiviral based doxycycline inducible system (pLIX-iSMARCB1) resulted in a substantial suppression of ER stress markers and autophagy (Figures 1I, 1K, S1J and S1K). All together those results suggest that the ER stress response is tightly regulated by SMARCB1, rather than being an epiphenomenon of malignant transformation.
The prominent autophagic response and increased protein turnover were further confirmed ex vivo through GFP quenching assays using lentiviral-based reporter systems expressing LC3-GFP and Ub-GFP fusion constructs transduced into E12.5 embryonic liver progenitor cells isolated from Smarcb1LoxP/LoxP mice. Cre-mediated Smarcb1 ablation resulted in the reduction of the GFP signals by flow cytometry that was completely rescued upon treatment with the inhibitor of autophagy chloroquine (CQ) and the proteasome inhibitor MG132 (Figures 1L and 1M), indicative of enhanced autophagic flux and global protein turnover upon loss of Smarcb1 (Dantuma et al., 2000; Fung et al., 2008). Together, transcriptomic, functional, and protein analyses suggest activation of cellular programs involved in protein disposal and adaptation to UPR stress in Smarcb1-deficient MRT models.
Perturbation of UPR and autophagy induces regression of Smarcb1-deficient tumors
We hypothesized that the perturbation of proteostasis in Smarcb1-deficient tumors may be exploited therapeutically. Specifically, we speculated that suppression of autophagy may cooperate with pharmacological inhibition of the proteasome to promote tumor regression. To test this, we generated an embryonic mosaic model of MRT carrying a Flpo-inducible latent shRNA against Atg7, an essential autophagy gene, allowing for time-restricted suppression of autophagy upon tamoxifen treatment (R26L/F-SΔ/Δ-pLSM5-shAtg7) (Figures 2A and 2B) (Genovese et al., 2017). Tumor-bearing mice were monitored by luciferase imaging and assigned to treatment with tamoxifen, the proteasome inhibitors bortezomib or ixazomib, or a combination of tamoxifen with a proteasome inhibitor. Remarkably, while either monotherapy regimen resulted in a partial suppression of tumor growth and in a significant, yet transient, increase in survival, genetic suppression of autophagy along with inhibition of the proteasome resulted in complete tumor regression, as assessed by bioluminescence imaging and prolonged disease remissions (Figures 2C, 2D, S2A and S2B). Similar responses were confirmed using combinations of bortezomib/ixazomib/NVP-AUY-922 (HSP90 inhibitor) plus chloroquine further substantiating that perturbation of UPR and autophagy results in dramatic tumor regression in both primary tumors and secondary transplants upon induction of a prominent apoptotic response (Figures 2C, 2E–2H and S2C–S2E). To further support our findings, we generated secondary orthotopic transplants from advanced murine tumors that recurred after a first-line treatment with ifosfamide (Ifo) plus etoposide (VP16) (Figure 2I) to determine whether these relapsed tumors might respond to treatment with proteotoxic agents. While re-challenge with Ifo + VP16 showed marginal anti-tumor benefits, treatment with both of the proteotoxic combinations tested resulted in a dramatic apoptotic response and improved survival (Figures 2J and 2K).
Figure 2. Pharmacological perturbation of UPR and suppression of autophagy induce tumor regression in murine MRTL.

A) Schematic model of experimental design for tumor maintenance studies. A bicistronic lentiviral vector expressing the Cre recombinase under the KRT19 promoter and a latent Flpo-inducible shRNA specific for Atg7 (pLSM5-shAtg7) was introduced in the liver of Rosa26LSL-Luc/FlpoERT2-Smarcb1LoxP/LoxP (R26L/F-SΔ/Δ) E12.5 embryos by trans-uterine injection to generate R26L/F-SΔ/Δ-pLSM5-shAtg7 mice. Tumor bearing mice were treated with tamoxifen to induce the Flpo-mediated activation of the latent shRNA. B) Western blot analysis of ATG7 levels in ex vivo cultures generated from the model in (A). Cell lysates were obtained after 96 hr of treatment with 4-OHT or vehicle. C) Assessment of tumor burden at necropsy in R26L/F-SΔ/Δ-pLSM5-shAtg7 mice treated with bortezomib, ixazomib, tamoxifen, chloroquine or the reported combinations. D-E) Kaplan-Meier survival analysis of tumor bearing mice assigned to single or combination treatments with ixazomib and tamoxifen ([D] vehicle: n = 13, tamoxifen: n = 23, ixazomib: n = 14, ixazomib + tamoxifen: n = 11) or ixazomib and chloroquine ([E] vehicle: n = 14, chloroquine: n = 14, ixazomib: n = 14, ixazomib + chloroquine: n = 10). F) Evaluation of apoptotic response in R26L/F-SΔ/Δ-pLSM5-shAtg7 tumor bearing mice treated for 7 days with the reported drugs. Apoptosis was assessed by Cleaved Caspase-3 (CC3) immunostaining. G) Quantification of apoptotic response in the experimental arms described above (n = 4 biological replicates/group). The percentage of apoptotic cells was expressed as CC3 positive area/total area of the section. Box and lines mark quartiles, ends of lines represent minimum and maximum. H) Kaplan-Meier survival analysis of Rag2−/− mice orthotopically transplanted with R26L/F-SΔ/Δ-pLSM5-shAtg7 tumors and assigned to single agents or combination treatments with ixazomib, bortezomib, NVP-AUY-922, and chloroquine (n = 7/group; vehicle and chloroquine: n = 14). I) Schematic model of experimental workflow for second line therapy. Orthotopic transplants of MRTL were treated with a combination of ifosfamide and etoposide (VP16) and monitored until tumor recurrence. Secondary transplants generated from terminal mice were assigned to second line therapy with the original drug combination or the bortezomib/chloroquine or NVP-AUY-922/chloroquine combinations. J) Left panel: Kaplan-Meier survival analysis for mice treated with first line chemotherapy regimen (n = 5/group). Right panel: Kaplan-Meier analysis of survival of secondary orthotopic transplants assigned to rescue therapy with the reported combinations (n = 9/group). K) Immunophenotypic assessment of apoptosis (Cleaved Caspase-3) in the treatment groups described above. Tumors were harvested for analysis after 7 days of treatment. ** p < 0.01, *** p < 0.001, **** p < 0.0001 by Mantel-Cox test (D, E, H, J) and by unpaired two-tailed t-test (G). Size bars: 100 μm. Veh: vehicle, Bort: bortezomib, Ixa: ixazomib, Tx: tamoxifen, 4-OHT: 4-hydroxytamoxifen, Ifo: ifosfamide, AUY922: NVP-AUY-922. See also Figure S2.
To determine whether our observations are relevant to other organ sites, we developed a murine transplantation model of MRT of the kidney (MRTK) that faithfully recapitulates the aggressive clinical behavior, as well as the pathological and immunophenotypic characteristics of human kidney disease (Figures S2F–S2I). As observed in MRTL, ER stress markers phospho-EIF2A, BIP and PDIA6 were elevated in murine MRTK on immunohistochemical analysis (Figure S2I), indicating activation of the ER stress response.
Tumors were also highly sensitive to treatment with combinations of bortezomib plus CQ or NVP-AUY-922 plus CQ, both inducing apoptotic cell death and suppressing tumor growth (Figures S2J and S2K). Collectively, data from MRTL and MRTK models support that the perturbation of proteostasis achieved either by blocking the proteasome or the protein folding machinery promotes tumor regression in the context of the impaired autophagic response in Smarcb1-deficient cells.
Additionally, we queried in vitro drug-response databases (https://portals.broadinstitute.org/ccle, https://www.cancerrxgene.org/translation/Drug) to identify trends relevant to human MRT, and we identified a profound sensitivity of SMARCB1 deficient cell lines (G401, G402, KYM-1) to the ER stress-inducing drugs elesclomol and thapsigargin (Figures 3A, 3B and S3A) (Barretina et al., 2012; Yang et al., 2013; Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer, 2015). We further analyzed the levels of ER stress markers in pediatric MRT and RMC tissues. In all cases (n = 40), immunohistochemical staining showed accumulation of markers of the ER stress response (Figures 3C and 3D). Furthermore, in vitro treatments of human MRT and RMC cell lines with single-agent NVP-AUY-922, bortezomib, ixazomib or chlorquine confirmed the sensitivity to inhibition of proteostasis or autophagy (Figures 3E and S3B–S3D). In vivo, treatment of animals harboring orthotopic transplants of human MRTK (G401) or RMC (RMC2C) cell line-derived models with bortezomib plus chloroquine or NVP-AUY-922 plus chloroquine resulted in a massive apoptotic response and a significant increase in survival (Figures 3F, 3G, S3E and S3F). Taken together, our data support a therapeutic opportunity to treat SMARCB1-deficient tumors with drugs targeting the proteasome and autophagy.
Figure 3. Perturbation of UPR and sensitivity to proteotoxic stress are hallmarks of human MRT.

A) Dot plot displaying the copy number and mRNA expression values of SMARCB1 in the CCLE collection of human cell lines (https://portals.broadinstitute.org/ccle). B) Waterfall plots displaying the sensitivity to the ER stress inducer elesclomol in the CCLE cell line panel (https://www.cancerrxgene.org/translation/Drug). Myeloma cells were used as positive controls. C) Immunohistochemical analysis for the expression levels of the established markers of ER stress response phospho-EIF2A, BIP and PDIA6 in human MRT and RMC samples. H&E: hematoxylin-eosin. D) Quantification of the expression levels of ER stress response markers described in (C). The levels of ER stress markers were expressed as percentage of stained area/total area of the section. Normal adjacent tissue was used as control (tumors [T]: n = 40, normal [N]: n = 20, phospho-EIF2A normal: n = 22). E) Radar plots showing the dose-responses (measured as colony formation) of 4 human SMARCB1-deficient cell lines (3 MRTs: G401, G402, A204; 1 RMC: RMC2C) treated with the reported drugs. F-G) Kaplan-Meier survival analysis of NSG mice orthotopically transplanted with human RMC (RMC2C, [F]) and MRT (G401, [G]) cell lines and assigned to treatment with the reported agents and combinations (n = 5 for vehicle control and monotherapies, n = 7 for combinations). Pooled data are presented as mean ± the standard deviation of biological replicates. * p < 0.05, ** p < 0.01, *** p < 0.001 by Mantel-Cox test (F and G) and unpaired two-tailed t-test (D). Size bars: 100 μm. See also Figure S3.
p53 is a master regulator of UPR and autophagy in SMARCB1-deficient tumors
To understand cellular reprogramming in Smarcb1-deficient tumors, we conducted transcriptomic analysis of Cre-induced Smarcb1-null liver tumors and LacZ controls identifying significant upregulation of p53 pathways involved in the regulation of protein metabolism, ER stress adaptation, and autophagy in Smarcb1-null tumors compared to controls (Figures 4A, 4B and Table S1). These evidences suggest a role of p53 in the regulation of proteostasis in the context of Smarcb1 loss (Buckbinder et al., 1995; Crighton et al., 2006; Budanov and Karin, 2008; Yoon et al., 2009; Venneti et al., 2011). These results were further supported by Western blot analysis, where we observed increased levels of canonical p53 target proteins involved in the UPR response and autophagy (Figure 4C). Moreover, functional studies leveraging a lentiviral-based, luciferase reporter system (pLV-p53R-Cre), which enabled visualization of p53 activity in vivo by bioluminescence imaging, were able to confirm p53 activation in intact animals. Specifically, trans-uterine delivery of the reporter construct in E12.5 embryos resulted in a dramatic induction of luciferase activity in Smarcb1-ablated liver compared to wild-type controls (Figure 4D). In line with these findings, GSEA analysis of transcriptomic profiles of human MRT and normal samples, confirmed significant enrichment for genes involved in the UPR, as well as activation of the p19ARF-p53 pathway in tumors (Figure S4A and Table S2). Accordingly, nuclear accumulation of p53 was observed in SMARCB1-deficient human MRT and RMC specimens, suggesting baseline activation of the p53 pathway in human disease (Figures 4E and S4B). Of note, review of published human genomic data of MRT, along with other pediatric malignancies, failed to identify any somatic mutation in the TP53 locus (Figure 4F) (Chun et al., 2016; Johann et al., 2016) (https://pecan.stjude.cloud) (Zhou et al., 2016), suggesting that the p53 checkpoint is preserved and active and may have a role in the maintenance of cellular homeostasis in the context of SMARCB1 deficiency (Chun et al., 2016) (http://ocg.cancer.gov/programs/target).
Figure 4. SMARCB1 loss drives autophagy through a MYC-p53 axis.

A) Volcano plot highlighting the dysregulation of cell cycle and p53 pathways in Smarcb1-ablated tumors. B) GSEA analysis for enrichment of p53 and MYC pathway targets in Smarcb1Δ/Δ tumors versus Smarcb1+/+ livers (n = 5 biological replicates/group). C) Western blot dissection of the MYC-p53 axis involved in cell cycle regulation and modulation of UPR and autophagy. D) Top panel: schematic showing the pLV-p53R-Cre bicistronic lentiviral reporter which allows for the in vivo, tissue-restricted expression of the Cre recombinase and visualization of p53 activity through a dual bioluminescence/fluorescence reporter. Bottom panels: activation of the luciferase reporter in Smarcb1LoxP/LoxP (SΔ/Δ) mice transduced at E12.5 and imaged at P90. A conditional KrasLSL-G12D (KG12D) allele was used as positive control. Trp53LoxP/LoxP (PΔ/Δ) and Trp53LSL-R172H/+ (PR172H) mice were used as negative controls. WT: wild-type. RLU: relative luminescence units. E) Quantification of MYC and p53 levels in human MRT and RMC samples (p53 normal: n = 20, p53 tumors: n = 40, MYC normal: n = 12, MYC tumor: n = 25). The levels of the nuclear proteins were expressed as percentage of stained nuclei/total nuclei in the section. F) Bar plots demonstrating the relative representation of somatic mutations in TP53 across pediatric human cancers (MRT: Malignat rhabdoid tumor, AML: Acute myeloid leukemia, NBL: Neuroblastoma, ATRT: Atypical teratoid rhabdoid tumor, T-ALL: T cell acute lymphoblastic leukemia, EPD: Ependymoma, CPC: Choroid plexus carcinoma, MB: Medulloblastoma, B-ALL: B cell acute lymphoblastic leukemia, EWS: Ewing’s sarcoma, RHB: Rhabdosarcoma, WT: Wilms tumor, HGG: High-grade glioma, ACT: Adrenocortical carcinoma, OS: Osteosarcoma) from different published studies (https://pecan.stjude.cloud) (Chun et al., 2016; Johann et al., 2016). G) Schematic model of experimental design to generate Myc hypomorphic tumors. A bicistronic lentiviral vector carrying the Cre recombinase under the KRT19 promoter and a constitutive shRNA specific for Myc (pLSM4-shMyc) or pLSM4-shCtrl were introduced in the liver of Rosa26LSL-Luc/+-Smarcb1LoxP/LoxP (R26L/+-SΔ/Δ) E12.5 embryos by trans-uterine injection. H) Histopathological, immunohistochemical, and ultrastructural characterization of tumors generated as described in (G). I) Western blot analysis of tumors generated as described in (G). J-K) Flow cytometry analysis (J) and quantification (K) of protein biosynthesis through OPP (O-propargyl-puromycin) incorporation analysis in tumor-derived cultures generated as described in (G) (n = 4/group). L) Kaplan-Meier survival analysis of mice transplanted orthotopically with Myc proficient (shCtrl) and hypomorphic (shMyc) tumors and assigned to treatment with ixazomib or vehicle (n = 10/group). M) Analysis of apoptotic response by Cleaved Caspase-3 immunostaining in the experimental groups described above. Tumors were harvested for analysis after 10 days of treatment. **** p < 0.0001, NS = not significant by Mantel-Cox test (L) and unpaired two-tailed t-test (E and K). Pooled data are presented as mean ± the standard deviation of biological (E) and technical replicates (K). Size bars: 100 μm unless otherwise specified in panel. See also Figure S4 and Table S2.
Similar to p53, MYC target genes were transcriptionally upregulated in Smarcb1-deficient murine liver tumors (Figure 4B), and we observed accumulation of MYC protein in human MRT tissues and murine experimental models upon ablation of Smarcb1 (Figures 4E, S4B and S4C). Similarly, restoration of SMARCB1 levels in murine MRT resulted in a robust suppression of the levels of p53 and MYC (Figure S4D). Consistently, genetic studies leveraging a tissue-specific, conditional lentiviral-based RNAi approach in Smarcb1-deficient mice demonstrated that MYC is required for the global increase in protein biosynthesis to sustain cancer cell proliferation, as assessed by OPP (O-propargyl-puromycin) incorporation experiments, and acts as a major activator of downstream p53 signaling (Figures 4G–4K). In vivo, attenuation of MYC activity via RNAi yielded more indolent tumors compared to controls (Figure 4L). Accordingly, MYC hypomorphic tumors did not respond to treatment with the proteasome inhibitor ixazomib (Figures 4L and 4M), further supporting the hypothesis that MYC promotes an anabolic state that induces an adaptive ER stress response and increased sensitivity to pharmacological perturbation of proteostasis through proteasome inhibition.
To further investigate the functional role of p53 in MRT, we crossed the Smarcb1 conditional strain with a Flpo-inducible, conditional Trp53 allele (Trp53Frt) and an ubiquitous, tamoxifen-inducible Flpo-ER recombinase (Rosa26FlpoERT2) to generate Rosa26FlpoERT2/LSL-Gfp-Smarcb1LoxP/LoxP-Trp53Frt/Frt (R26F/G-SΔ/Δ-PF/F) animals (Figure 5A). E12.5 R26F/G-SΔ/Δ-PF/F embryos were transduced with the pLV-p53R-Cre lentiviral vector or the adenoviral Adx-A-Cre by trans-uterine injection and monitored post-natally for tumor formation. Subsequently, established tumors were transplanted in Rag2−/− mice and assigned to vehicle or tamoxifen treatment to assess the role of Trp53 ablation in the context of Smarcb1-deficiency (Figure 5A). Acute ablation of Trp53 in vivo resulted in suppression of p53 activity, as assessed functionally by a dramatic drop in luciferase reporter activity in pLV-p53R-Cre transduced animals and confirmed via immunohistochemical analysis of p53 levels in excised lesions (Figure 5B). In addition, consistent with our previous findings, Western blot and immunohistochemical analysis confirmed dysregulation of proteostasis and autophagy upon Trp53 ablation in vivo and in short-term cultures established from R26F/G-SΔ/Δ-PF/F tumors (Figures S5A and S5B). Functionally, these findings were supported by OPP incorporation studies, demonstrating a significant increase in the global levels of protein synthesis (Figures 5C and 5D), and by TEM analysis of 4-hydroxytamoxifen treated short-term cultures revealing a robust suppression of autophagy (Figure 5E).
Figure 5. Trp53 ablation drives a significant increase in protein biosynthesis and suppression of autophagy in Smarcb1-deficient tumors.

A) Schematic model showing the experimental workflow. E12.5 Rosa26FlpoERT2/LSL-Gfp-Smarcb1LoxP/LoxP-Trp53Frt/Frt (R26F/G-SΔ/Δ-PF/F) were transduced with the pLV-p53R-Cre lentiviral reporter or the Adx-A-Cre adenoviral vector. Secondary orthotopic transplants were generated in Rag2−/− recipients and assigned to vehicle or tamoxifen treatment to establish Trp53 deficient and proficient isogenic lines. Transplanted mice were treated with tamoxifen or vehicle control one day after surgery. B) Top panels: luciferase imaging showing the activity of the pLV-p53R-Cre lentiviral reporter in vivo upon Trp53 ablation as indicated in (A). Bottom panels: immunohistochemical analysis of p53 levels in tumors described above. C-D) FACS analysis (C) and quantification (D) of OPP incorporation in short-term cultures established from R26F/+-SΔ/Δ-PF/F tumors (n = 5/group). Cells were assessed for OPP incorporation 96 hr post treatment with 4-OHT or vehicle. E) Representative TEM sections from short-term cultures established from R26F/+-SΔ/Δ-PF/F tumors. Red arrowheads: autophagic vacuoles. F-G) In vitro evaluation of cell viability in short-term cultures established from R26F/+-SΔ/Δ-PF/F tumors after 72 hr of treatment with different concentrations of NVP-AUY-922 (F) or ixazomib (G). H) Quantification of colony formation assays from short-term cultures established from R26F/+-SΔ/Δ-PF/F tumors (n = 3/group). The number of colonies was expressed as a percentage of the vehicle treated controls. Pharmacological studies in F, G, H were started 96 hr after treatment with 4-OHT to ablate Trp53 or vehicle control. I) Kaplan-Meier analysis of survival in Rag2−/− mice orthotopically transplanted with R26F/G-SΔ/Δ-PF/F tumors and assigned to treatment with tamoxifen, ixazomib or the combination (n = 10/group). Tamoxifen treatment was started one day after transplantation. Pharmacological treatments were started 7 days after transplantation. J) Representative sections of orthotopic MRTL transplants of R26F/G-SΔ/Δ-PF/F tumors (induced using Adx-A-Cre) treated with vehicle and tamoxifen alone or in combination with ixazomib. Top panels: hematoxylin-eosin staining. Middle panels: co-immunofluorescence staining for GFP (tumor cells) and Cleaved Caspase-3 (apoptotic cells). Bottom panels: gross fluorescence visualization of liver tumors. Tumors were harvested for analysis after 10 days of pharmacological treatment. K) Representative images of colony formation assays from short-term cultures established from R26F/+-SΔ/Δ-PF/F tumors treated with vehicle or 4-OHT alone or combined with VP16 or chloroquine. Pharmacological studies were started 96 hr after treatment with 4-OHT to ablate Trp53 or vehicle control. L-M) Quantification of colonies from triplicate experiments of cultures treated with chloroquine (L) or VP16 (M) as described in K (n = 3/group). The number of colonies was expressed as a percentage of the vehicle treated controls. N) Kaplan-Meier analysis of survival in Rag2−/− mice orthotopically transplanted with R26F/G-SΔ/Δ-PF/F tumors and assigned to the treatment regimens reported in the panel (n = 20 for vehicle and tamoxifen controls, n = 10 for other groups). O) Representative sections of orthotopic transplants of R26F/G-SΔ/Δ-PF/F tumors (induced using Adx-A-Cre) treated with vehicle and tamoxifen alone or in combination with chloroquine. Top panels: hematoxylin-eosin staining. Middle panels: co-immunofluorescence staining for GFP (tumor cells) and Cleaved Caspase-3 (apoptotic). Bottom panels: gross fluorescence visualization of liver tumors. Tumors were harvested for analysis after 10 days of treatment with ixazomib. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, NS = not significant by unpaired two-tailed t-test (D, H, L, M) and Mantel-Cox test (I, N). Pooled data are presented as mean ± the standard deviation of technical replicates. Size bars: 100 μm unless otherwise specified in panel. See also Figure S5.
As predicted, genetic ablation of Trp53 in Smarcb1-deficient tumor-derived cultures resulted in enhanced in vitro sensitivity to NVP-AUY-922 and ixazomib and a prominent impairment of clonogenic growth (Figures 5F–5H and S5C). In line with these evidences, the tamoxifen-mediated in vivo ablation of Trp53 resulted in a significant increase in survival and a more pronounced apoptotic response upon treatment with ixazomib (Figures 5I and 5J). Accordingly, Trp53 loss conferred an increased resistance to the alkylating agents ifosfamide and VP16 and in the desensitization to chloroquine both in vitro and in vivo (Figures 5K–5O and S5D), suggesting that the adaptive induction of autophagy in response to the UPR is mostly dependent on a proficient p53 checkpoint. The requirement for a functional p19ARF-MDM2-p53 pathway was further supported by our observation that the suppression of p19ARF (Cdkn2a) through a conditional lentiviral-based RNAi approach, resulted in increased protein synthesis and an attenuated autophagic response (Figures S5E–S5K). The role of p53 in coordinating cell survival in response to perturbation of proteostasis in SMARCB1-deficient tumors was further validated in human MRT and RMC cell lines via RNAi-mediated knockdown experiments, where a significant increase in sensitivity to the proteasome inhibitor ixazomib upon TP53 suppression was readily appreciated (Figures S5L–S5N). Additional pharmacological studies with the MDM2 inhibitor idasanutlin demonstrated that, in Smarcb1-deficient tumors, induction of the p19ARF-MDM2-p53 pathway decreased tumorigenic potential but dramatically induced autophagy in a p53 dependent manner, as demonstrated by phenotypic rescue upon genetic ablation of Trp53 (Figures 6A–6D and S6A). Accordingly, treatment with idasanutlin in orthotopic transplants of murine MRT did not provide additional survival benefits to ixazomib monotherapy, however, pharmacological suppression of the p53-mediated autophagic response with chloroquine significantly prolonged survival (Figure S6B). The central role of p53 in the cellular response to proteotoxic stress through autophagy was further demonstrated by genetic ablation of Trp53 (Figure S6C).
Figure 6. p53 activates autophagy through DRAM1 in Smarcb1-deficient tumors.

A) Schematic model of the experimental workflow to demonstrate a proficient p53 check-point in murine MRTL. E12.5 Rosa26LSL-Gfp/+-Smarcb1LoxP/LoxP-Trp53+/+ (R26G/+-SΔ/Δ-P+/+) embryos were transduced with the Adx-A-Cre construct and assigned to vehicle or treatment with the MDM2 inhibitor idasanutlin at postnatal day 30. Daily dosage was continued for 3 weeks. Rosa26LSL-Gfp/+-Smarcb1LoxP/LoxP-Trp53LoxP/LoxP (R26G/+-SΔ/Δ-PΔ/Δ) were used as controls. B) Top panels: representative coronal T2-weighted MRI sections of tumor-bearing mice treated as described in (A). Bottom panels: histomorphological and immunohistochemical characterization of lesions harvested from Trp53 deficient and proficient tumor-bearing mice. Cell proliferation was assessed by Ki67 immunostaining. Apoptosis was assessed by Cleaved Caspase-3 immunostaining. Tumors were harvested after 10 days of treatment with idasanutlin. C) Kaplan-Meier analysis of survival in mice receiving oral idasanutlin or vehicle (R26G/+-SΔ/Δ-P+/+ + vehicle: n = 15, R26G/+-SΔ/Δ-P+/+ + idasanutlin: n = 17, R26G/+-SΔ/Δ-PΔ/Δ + vehicle: n = 12, R26G/+-SΔ/Δ-PΔ/Δ + idasanutlin: n = 15). D) Representative TEM sections from Trp53 proficient and deficient MRTs. E) Representative hematoxylin-eosin staining and co-immunofluorescence staining for GFP (tumor cells) and Cleaved Caspase-3 (apoptosis) in control and Dram1-silenced R26G/+-SΔ/Δ orthotopic transplants in Rag2−/− recipient mice treated with ixazomib and vehicle. Tumors were harvested for analysis after 10 days of treatment. F-G) Kaplan-Meier analysis of survival in control and Dram1-silenced R26G/+-SΔ/Δ orthotopic transplants in Rag2−/− recipient mice. Experimental cohorts were assigned to treatment with vehicle and ixazomib (n = 10/group) (F) or bortezomib (shCtrl + vehicle: n = 20, other groups: n = 10) (G). H) Schematic model showing the integrated function of MYC-p53-DRAM1 axis in the regulation of proteostasis within the SMARCB1 oncogenic network. *** p < 0.001, **** p < 0.0001, NS = not significant by Mantel-Cox test (C, F, G). Size bars: 100 μm unless otherwise specified in panel. Ida: idasanutlin. See also Figure S6.
Our data suggest that the specific perturbation of the proteostatic arm of the p53 network may generate context-specific vulnerabilities, while preserving its tumor-suppressive functions. To validate this hypothesis, we used RNAi to genetically suppress Dram1, a master regulator of autophagic flux and lysosome biogenesis involved in the p53-dependent autophagic response (Crighton et al., 2006). While Dram1 knockdown alone had limited effects on tumor growth in orthotopic transplants of Smarcb1-deficient MRTL, it potently synergized with the proteasome inhibitors bortezomib and ixazomib, resulting in a dramatic apoptotic response and prolonged survival when compared to shCtrl-transduced and vehicle-treated tumors (Figures 6E–6G and S6D–S6H). Collectively, our experiments predict a model wherein disruption of Smarcb1 functions activates diverse oncogenic pathways and induces UPR stress. In this context, p53 activation behaves as a pleiotropic regulator of cellular homeostasis, concurrently regulating cell-cycle progression and survival, as well as protein biosynthesis and disposal through autophagy (Figure 6H). In sum, our findings suggest that p53 has a context-specific, pro-survival role in SMARCB1-deficient cells that can be exploited therapeutically and should be taken into account when designing therapeutic regimens for patients with SMARCB1-deficient tumors.
High levels of the IAP protein BIRC5 promote tolerance to apoptosis in SMARCB1-deficient tumors
Our data suggest a seemingly contradictory role of p53 in the context of SMARCB1-deficient MRT biology, such that robust activation of p53 signaling induces pro-apoptotic programs and simultaneously protects cells from proteotoxic cell death (Figures 7A, 7B and S7A; Table S1 and S2). Seminal work from Paek et al. elegantly demonstrated that the fate of TP53-proficient cells upon apoptotic stimulus is dependent on levels of inhibitor of apoptosis (IAP) proteins and the dynamics of accumulation of IAPs and p53 (Paek et al., 2016). In this scenario, high levels of IAPs can suppress apoptosis at the level of effector caspases downstream of both extrinsic and intrinsic pathways. Indeed, expression analysis of SMARCB1-deficient murine and human tumors revealed dramatic upregulation of the IAP protein BIRC5 (Figures 7B, S7B and S7C), and experimental manipulation of SMARCB1 levels resulted in acute changes in BIRC5 protein expression (Figures S7D–S7F), suggesting that the Birc5 gene is tightly regulated by SMARCB1. To assess whether BIRC5 is required for tumor maintenance, we generated a conditional R26L/F-SΔ/Δ-pLSM5-shBirc5 model (Figure 7C) to allow for the tamoxifen-induced, Flpo-mediated acute ablation of Birc5 in primary experimental rhabdoid tumors. Birc5 ablation resulted in a dramatic induction of apoptosis (assessed by Cleaved Caspase-3 immunostaining) and complete tumor regression (Figures 7D, 7E and S7G–S7J). Accordingly, pharmacological treatment of human-derived models of MRT and RMC with the BIRC5 inhibitor YM155 confirmed a critical role of BIRC5 in the suppression of apoptosis in a system where sustained p53 activity is required for metabolic homeostasis (Figures 7F and 7G). Thus, the robust activation of p53 signaling is tolerated in a specific cellular context where execution of apoptosis is partially impaired through the upregulation of the IAP protein BIRC5.
Figure 7. Increased apoptotic threshold through BIRC5 mediates control of effector caspases in MRT.

A) GSEA analysis for enrichrment of the apoptotic program in murine Smarcb1-ablated MRT versus age-matched LacZ controls (n = 5/group). B) Immunohistochemical analysis of markers of apoptosis in Smarcb1-deficient tumors and Smarcb1-proficient controls. C) Schematic model of experimental design to knockdown Birc5 in vivo. A bicistronic lentiviral vector carrying the Cre recombinase under the KRT19 promoter and a latent, Flpo-inducible, shRNA specific for Birc5 or shCtrl vector were introduced in the liver of Rosa26LSL-Luc/FlpoERT2-Smarcb1LoxP/LoxP (R26L/F-SΔ/Δ) E12.5 embryos by trans-uterine injection to generate the R26L/F-SΔ/Δ-pLSM5-shBirc5-1, R26L/F-SΔ/Δ-pLSM5-shBirc5-2 and R26L/F-SΔ/Δ-pLSM5-shCtrl models. Mice were monitored weekly for signs of tumors by palpation and bioluminescence imaging. D) Representative luminescence images displaying the acute effect of the genetic suppression of Birc5 in murine MRT with 2 independent shRNA. shCtrl-transduced mice were used as controls. Images were acquired 3 weeks from the beginning of the treatment and the induction of the shRNAs. E) Representative co-immunofluorescence staining for Luciferase (LUC) (tumor cells) and Cleaved Caspase-3 in Birc5-silenced tumors harvested from experimental cohorts treated with vehicle or tamoxifen for 10 days. F) Quantification by flow cytometry of apoptotic responses (Propidium Iodide/Annexin V) in human-derived MRT (G401, G402 and A204) and RMC (RMC2C) models upon treatment with BIRC5 inhibitor YM155 for 72 hr (n = 3). Data are presented as mean ± the standard deviation of technical replicates. G) Schematic showing the interplay between BIRC5, p53 and ER stress in balancing cell survival and cell death in SMARCB1-deficient tumors. ** p < 0.01, *** p < 0.001, **** p < 0.0001, NS = not significant by unpaired two-tailed-t-test (F). Size bars: 100 μm unless otherwise specified in panel. See also Figure S7.
DISCUSSION
SMARCB1 inactivation defines a class of highly lethal diseases, the current standard of care for MRT and AT/RT is a combination of surgery, high dose alkylating agents and radiotherapy in patients 3-years old or older (Tekautz et al., 2005). Such approach, while able to induce temporary remissions, is often burdened with severe side effects including long-term developmental impairment and genotoxic hazard affecting the quality of life of the few long-term survivors and associated to an increased risk of secondary neoplasms (Oeffinger and Hudson, 2004). We leveraged embryonic mosaic GEM models of MRT and found that disruption of Smarcb1 during embryonic development results in a robust engagement of the UPR, ER stress response, and autophagy. These findings were confirmed in a murine transplantation model of MRTK and in human cell lines. Further, using pharmacological and genetic tools, we determined that interference with the cellular proteostatic machinery is highly lethal in Smarcb1-deficient tumors. Mechanistically, we demonstrated that the MYC-p19ARF-MDM2-p53 tumor suppressive axis has a central role in modulating proteostasis through the regulation of autophagy. Our data suggest that engagement of the UPR may be a byproduct of a MYC-induced hypermetabolic state. In this biological context, activation of the p53 checkpoint exerts a survival benefit by modulating protein translation and disposal through autophagy. Intriguingly, this phenomenon could explain the absence of TP53 mutations in MRT, in spite of significant engagement of the pathway (Chun et al., 2016; Johann et al., 2016).
Overall our evidence strongly supports a dual function of p53 in SMARCB1-deficient cells. In the context of tumor initiation, p53 signaling plays an antiproliferative and tumor suppressive role, while the pathway orchestrates an adaptive response during tumor maintenance under metabolic stress (Isakoff et al., 2005). Our preclinical studies and interrogation of clinical datasets provide the rationale for a therapeutic approach to exploit the “collateral proteopathy” characteristic of SMARCB1-deficient malignancies through the repositioning of drugs that have already shown acceptable toxicity profiles (Richardson et al., 2003; Ruiz-Irastorza et al., 2010; Kumar et al., 2014). Furthermore, our findings suggest that treatment with cytotoxic agents that induce the p53 checkpoint may result in clonal selection of malignant populations with an increased tolerance to proteotoxic stress and increased dependence on the autophagic machinery for their survival, highlighting a potential opportunity to combine standard-of-care with drugs targeting autophagy and the proteasome to yield durable responses.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Giannicola Genovese (GGenovese@mdanderson.org).
Experimental Models and Subject Details
Mouse strains and GEMM nomenclature.
Rosa26LSL-Luc, KrasLSL-G12D and Trp53LSL-R172H mice were generated by Dr. Tyler Jacks and obtained through the Jackson Laboratory (Jackson et al., 2001; Olive et al., 2004; Cheung et al., 2008). The Rosa26LSL-TdT was generated in Hongkui Zeng’s lab and obtained through the Jackson lab (Madisen et al., 2010). The Rosa26LSL-Cas9-Gfp strain was kindly donated by Dr. Feng Zhang (Platt et al., 2014). The Smarcb1LoxP strain was provided by Dr. Charles Roberts (Roberts et al., 2002). The Pax8Cre strain was generated by Dr. Meinrad Busslinger and obtained through the Jackson Laboratory (Bouchard et al., 2002). The Trp53LoxP strain generated by Dr. Anton Berns and donated by Dr. Ronald DePinho (Marino et al., 2000). The Rosa26FlpoERT2 was generated by Dr. Alexandra Joyner and obtained from the Jackson Laboratory (Lao et al., 2012). Trp53Frt animals were donated by Dr. David Kirsch (Lee et al., 2012a). Rag2−/− and NSG mice were obtained through the Jackson Laboratory. R26L/F-SΔ/Δ: Rosa26LSL-Luc/FlpoERT2-Smarcb1LoxP/LoxP. R26L/+-SΔ/Δ: Rosa26LSL-Luc/+-Smarcb1LoxP/LoxP. R26F/G-SΔ/Δ-PF/F: Rosa26FlpoERT2/LSL-Gfp-Smarcb1LoxP/LoxP-Trp53Frt/Frt. R26F/+-SΔ/Δ-PF/F: Rosa26FlpoERT2/+-Smarcb1LoxP/LoxP-Trp53Frt/Frt. R26G/+-SΔ/Δ-P+/+: Rosa26LSL-Gfp/+-Smarcb1LoxP/LoxP-Trp53+/+. R26G/+-SΔ/Δ-PΔ/Δ: Rosa26LSL-Gfp/+-Smarcb1LoxP/LoxP-Trp53LoxP/LoxP. R26F/+-SΔ/Δ: Rosa26FlpoERT2/+-Smarcb1LoxP/LoxP. R26G/+: Rosa26LSL-Gfp/+. R26L/+: Rosa26LSL-Luc/+. R26G/+-SΔ/Δ: Rosa26LSL-Gfp/+-Smarcb1LoxP/LoxP. PC/+-R26T/+-SΔ/Δ: Pax8Cre/+-Rosa26LSL-TdT/+-Smarcb1LoxP/LoxP. R26F/G-SΔ/Δ: Rosa26FlpoERT2/LSL-Gfp-Smarcb1LoxP/LoxP. Correct genotype was determined by PCR analysis and gel electrophoresis at birth and at death. No sex bias was introduced during the generation of experimental cohorts. Strains were kept in a mixed C57BL/6 and 129Sv/Jae background. Littermates of the same sex were assigned randomly to experimental arms. Trans uterine injections were performed in E12.5 pregnant female mice that were monitored carefully through pregnancy and delivery. For transplantation studies 6–9 weeks old mice were used. All animal studies and procedures were approved by the UTMDACC Institutional Animal Care and Use Committee. All experiments were conform to the relevant regulatory standards.
Human cell lines.
G401 (isolated from a male patient), G402 (isolated from a female patient) and A204 (isolated from a female patient) cells were obtained from ATCC, fingerprinted (according with the UTMDACC Institute for Applied Cancer Science internal protocols) and cultured in McCoy’s 5a (Thermo Fisher Scientific) supplemented with 10% FBS (Sigma Aldrich). SMARCB1 levels were assessed by Western blot analysis in all cell lines. RMC2C were isolated from a 35 year old African American male, affected by a SMARCB1-negative renal medullary carcinoma undergoing surgery at UTMDACC. The cell line were generated from a primary, treatment naive tumor arising from the left kidney and cultured in MEM (Thermo Fischer Scientific) supplemented with 1% penicillin–streptomycin (Thermo Fisher Scientific), 10% FCS (Sigma-Aldrich), 1% MEM non-essential amino acid (Thermo Fisher Scientific), 1% Insulin-Transferrin-Selenium (ScienceCell) and 5 μg/ml EGF (Millipore). SMARCB1 loss was confirmed by Western blot and immunohistochemical analysis. Cells were cultured at 37°C in a humidified atmosphere wi th 5% CO2. All cell lines were tested for Mycoplasma contamination (Venor™ GeM Mycoplasma Detection Kit, PCR-based, Sigma-Aldrich) according to manufacture’s instructions.
Patient-derived samples.
Patient-derived samples were obtained from patients who provided informed consent under an Institutional Review Board (IRB).
Methods
Adenoviral lentiviral vectors and other plasmids.
The pLSM5 vector was generated as previously described (Genovese et al., 2017). To generate pLSM4 construct, a synthetic cassette (Geneart, Life Technologies) containing the U6 promoter and the Cre recombinase sequence under the human KERATIN 19 promoter flanked by two Frt sites (XbaI-Frt-U6-HpaI-XhoI-KRT19-Cre-Frt-WPRE-KpnI) was cloned into the XbaI/KpnI sites of the pLSM5 vector. The shRNA oligos were cloned as previously described (Genovese et al., 2017). To generate the pLV-p53R-Cre, a XbaI-EcoRI-KRT19-Cre-KpnI synthetic cassette (Geneart, Life Technologies) was cloned into the XbaI-KpnI site of the pLMS5 vector. The p53RE-Luc-2A-Gfp-WPRE cassette was PCR amplified from the pGF-p53-mCMV-EF1α-Puro Lentivector (SBI) and inserted in XbaI-EcoRI site. All the constructs were verified by restriction digestion and sequencing. The pMSCV-LoxP-dsRed-LoxP-eGFP-Puro-WPRE vector was used for virus titration in HEK293 cells, provided by Dr. Hans Clevers (Koo et al., 2012) and obtained through Addgene. Adx-A-Cre adenoviral construct: the murine Albumin promoter was PCR amplified from the ALB-GFP vector and cloned into the I-CeuI/BamHI sites of the ICPI-CS-iCreP2A-tdTomato vector to generate the ICPI-Alb-iCreP2A-tdTomato construct. The Cre sequence was amplified from the pLSM5 construct and cloned into the BamHI/EcoRI sites of the ICPI-Alb-iCreP2A-tdTomato to generate the ICPI-Alb-Cre vector. The Alb-Cre-SV40-pA fragment was liberated by restriction digestion and cloned into the I-CeuI and pi-SceI sites of the Adx vector to generate the Adx-A-Cre construct. The Adx, ICPI-CS-iCreP2A-tdTomato and Adx-LacZ vectors were kindly provided by Dr. Oka Kazuhiro. The ALB-GFP vector was generated by Snorri Thorgeirsson and acquired through Addgene (Heo et al., 2006). The pBABE-Puro-LC3-GFP vector was generated in the Debnath lab and provided by Dr. Andrea Viale (Fung et al., 2008). The pUb-GFP vector was generated by cloning the Ub-GFP ORF into the pLenti6/UBC by LR recombination. The Ub-GFP ORF was generated by Dr. Nico Dantuma and obtained through Addgene (Dantuma et al., 2000). The pLIX-iSMARCB1 and pLIX-iLacZ constructs were generated by introducing the SMARCB1 and LacZ CDS in to the pLIX403 vector by Gateway recombination. The pLIX403 vector was generated in David Root’s lab and obtained through Addgene.
Virus preparation.
Infectious viral particles were produced using helper plasmids psPAX2 and pMD2.G obtained through Addgene as previously described (Genovese et al., 2017). Briefly, 293T cells were cultured in DMEM (Thermo Fisher Scientific) containing 10% FBS, 1% penicillin–streptomycin, caffeine 4 mM (Sigma-Aldrich) and transfected using Polyethylenimine (PEI) (Sigma-Aldrich). Virus-containing supernatant was collected 48–72 hr after transfection, spun at 3000 rpm for 10 min and filtered through 0.45 μm low protein binding filters (Corning). High-titer preps were obtained by multiple rounds of ultracentrifugation at 23.000 RPM for 2 hr each. Viral titer was quantified in HEK293T cells stably transduced with a Cre-inducible Gfp reporter. High titer adenoviral preps were produced using the Adeno-X purification kit (Takara) according to manufacturer’s instructions. Viral titer was measured using the Adeno-X Rapid Titer Kit (Takara) following manufacturer’s instructions.
Surgical procedures.
Adenoviral and lentiviral particles were resuspended in PBS (Thermo Fisher Scientific). E12.5 pregnant females were anesthetized using isoflurane (Henry Schein Animal Health) and put in the Trendelenburg position. Shaved skin was disinfected with betadine (Dynarex) and ethanol (Covidien). 1-cm incisions were performed through the skin/subcutaneous and muscular/peritoneal layers on the midline in the lower abdominal quadrant. Uterine horns were sequentially exposed and embryos counted. 3 μl of viral solution was introduced into the embryonic liver by trans uterine delivery. Uterine horns were carefully repositioned into the abdominal cavity. The muscular/peritoneal planes were closed individually by continuous absorbable sutures. The skin/subcutaneous planes were closed using metal clips. Analgesia was achieved with buprenorphine (0.1 mg/Kg BID) (Par Parmaceutical). Mothers were monitored carefully for signs of distress through pregnancy and during delivery. Transduced pups were monitored twice/week for signs of liver dysfunction/abdominal distension. Liver transplantations experiments were described elsewhere (Zender et al., 2006; Winslow et al., 2011). Kidney transplantation studies: mice were anesthetized using isoflurane. Shaved skin was disinfected with betadine and ethanol. 1-cm incisions were performed on the left flank through the skin/subcutaneous and muscular layers, the layers retracted and the kidney exposed. 2×105 cells were resuspended in a 2:1 solution of OPTI-MEM (Thermo Fisher Scientific) and matrigel (BD Biosciences) and transplanted into the kidney parenchyma of 6–9 weeks old mice in a single injection (25 μl). Hemostasis was controlled with a bipolar cautery (Bioseb). The muscular plane was closed with continuous absorbable sutures. The skin/subcutaneous planes were closed with metal clips (Word precision instrument). Analgesia was achieved with buprenorphine (0.1 mg/Kg BID). Mice were monitored twice/week for sign of tumor growth by manual palpation and bioluminescence imaging when appropiate. Human lines were transplanted in NSG mice, murine cells were transplanted in Rag2−/− mice. Viral titer and embryonic/perinatal survival were assessed by counting the numbers of live transduced pups over the number of injected embryos. Transduction efficiency was confirmed by fluorescence analysis leveraging the Rosa26LSL-Gfp reporter.
Flow cytometry, and protein synthesis ex vivo.
Tumor-derived cells and primary lines were cultured in vitro for <5 passages before experiments. The autophagic flux and the protein turnover were assessed by measuring the mean fluorescence of Smarcb1 proficient and ablated E12.5 liver progenitors transduced with lentiviral constructs expressing the LC3-GFP and UB-GFP, respectively. Cycloheximide (20 μM), chloroquine (50 μM) (Sigma-Aldrich) and MG132 (5 μM) (EMD Millipore) treated cells were used as controls. Protein synthetic rate was assessed as previously described using the Click-iT® Plus OPP Alexa Fluor® 594 or 488 Protein Synthesis Assay Kit (Thermo Fisher Scientific) according to manufacturer’s instructions (Genovese et al., 2017). Briefly, the fraction of positive cells and the intensity of incorporation were assessed by FACS analysis. Cells cultured in the presence of cyclohexemide were used as negative controls. Gating strategies to exclude aggregates were employed. After staining, samples were acquired using a BD FACS Canto II flow cytometer. Data were analyzed by FlowJo (Tree Star).
Annexin V-FITC/PI double-staining.
Human rhabdoid cell lines were pre-seeded (3×105 cells) in 10 cm plate (Falcon BD) for 24 hr and then treated with NVP-AUY-922, bortezomib (LC Laboratories), chloroquine, ixazomib (Selleckchem) and YM155 (Proactive Molecular Research) at the respective IC50 (see Table S3). After 72 hr, cells were collected and stained according to manufactorer’s kit with annexin V-FITC and PI (Thermo Fisher Scientific). Stained samples were acquired using a Beckman Coulters Gallios flow cytometer. Data were analyzed by FlowJo. Experiments were repeated at least in triplicate.
Tumor cells isolation and culture.
Isolation and culture of embryonic liver progenitors were performed according to Zender et al. (Zender et al., 2006). Ex vivo cultures from primary tumor explants were generated by mechanical dissociation and incubation for 1 hr at 37°C with a solution of Collagenase IV/Dispase (4 mg/ml) (Invitrogen). Cells derived from primary mouse tumors were kept in culture for less than 5 passages. Cells were plated on collagen IV coated plates (Corning) and cultured in DMEM (Lonza) supplemented with 2 mM glutamine (Invitrogen), 15% FBS (Lonza), 40 ng/ml hEGF (PeproTech), 20 ng/ml hFGF (PeproTech), 5μg/ml h-Insulin (Roche), 40 ng/ml HGF (Sigma-Aldrich), 1% penicillin–streptomycin. Embryonic renal epithelial cells were isolated and briefly expanded ex vivo according to a protocol modified from Tanigawa et al. (Tanigawa et al., 2016). Briefly, E15 kidneys were pulled, digested at 37°C for 20 min in a 0.25% Coll agenase A (w/v) (Sigma-Aldrich), 1% Pancreatin digest (w/v) solution (Sigma-Aldrich). Cells were plated on Recombinant Laminin-511E8 (BioLamina) coated plates and cultured in DMEM/F12 (Life Technologies) supplemented with triiodothyronine (Sigma-Aldrich), hydrocortisone (Sigma-Aldrich), Insulin-Transferrin-Selenium, 10 ng/ml TGF-α (Peprotech), and 50 ng/ml FGF2 (Peprotech)/FGF9 (R&D Systems), 1 μM CHIR99021 (Wako), 2.5 μM DAPT (Merck Millipore), 5 ng/ml BMP7 (R&D Systems), 5 ng/ml mouse LIF (Millipore), and 10 μM Y27632 (Wako). Medium was changed every 24 hr for 3–4 days until the next passage. Cell were briefly expanded in vitro before transplantation in Rag2−/− mice.
In vitro treatments.
For drug treatments, low passages tumor cells were collected, washed, digested with trypsin and counted in presence of trypan blue to exclude dead cells (Countless, Invitrogen). 18 hr after plating cells were incubated with chloroquine (50 μM), 4-hydroxytamoxifen (250 nM) (Sigma-Aldrich), NVP-AUY-922 (5 nM), ixazomib (100 nM) and VP16 (5 μM) (Sigma-Aldrich). Colonies were stained with crystal violet (Sigma-Aldrich) and manually counted. In vitro assessment of p53 signaling activation and induction of autophagy was assessed after 24 hr of treatment with idasanutlin (250 nM). The drug response in colony formation assay and/or cell viability assay were performed seeding 1×103 cells/well in 6-well plates (Falcon BD) or 96-well plates (clear bottom black Corning) respectively. After 24 hr pre-seeding, cells were treated with different drugs at the selected concentrations. Colony forming assay: upon 10–15 days of drug inhibition, colonies were washed with PBS and stained with 1 mL of 0.1% crystal violet/20% ethanol in each well for 1 hr at room temperature. After washing with dH2O for three times, the cells were air-dried and then scanned. 2 ml of 10% acetic acid (Sigma-Aldrich) were added in each well and gently shaken for 30 min at room temperature. Absorbance were read at 590 nm. Cell viability assay: after 72 hr of drug treatment cell viability was evaluated by ATP measurement with CellTiter–Glo, according to manufacturer’s instructions (CellTiter-Glo® 2.0, Promega). All in vitro experiments were repeated at least in triplicate.
In vivo studies and treatment schedules.
For pre-clinical studies, NVP-AUY-922 was administered i. p. (intraperitoneally) at 75 mg/kg every other day for 21 days; bortezomib was administered i. v. (intravenous) at 1 mg/kg twice per week for 3 weeks; chloroquine was administered i. p. daily at 60 mg/kg for 3 weeks; ifosfamide (Sigma-Aldrich) was administered i. p. 90 mg/kg on days 1, 2, 3; VP16 was administered i. p. at 12 mg/kg on days 1, 2, 3; idasanutlin (Selleckchem) was administered daily by oral gavage at 30 mg/kg for 20 days. Ixazomib was administrated by oral gavage at 11 mg/kg twice per week for 3 weeks. Tamoxifen was administered i. p. every other day for a total of 5 injections as a 15 mg/ml solution in corn oil (100 μl) (Sigma-Aldrich), NVP-AUY-922 was dissolved 10% DMSO (Sigma-Aldrich)/25% water (Sigma-Aldrich)/65% PEG 400 (Sigma-Aldrich), idasanutlin was dissolved in a 1% Klucel LF (Ashland)/0.1% Tween 80 (Sigma-Aldrich) solution, ixazomib was dissolved in 5% 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich), bortezomib, chloroquine, ifosfamide and VP16 were re-suspended in 0.9% saline solution.
MRI and Ivis Imaging.
The procedure has been described elsewhere (Genovese et al., 2017). A 4.7T Bruker Biospec (BrukerBioSpin), equipped with 35 mm innerdiameter volume coil and 6 cm inner-diameter gradients, was used for imaging the animals. A fast acquisition with relaxation enhancement (RARE) sequence with TR/TE of 2,000/38 ms, matrix size 256× 192, 0.75 mm slice thickness, 0.25 mm slice gap, 4× 3 cm FOV, 101 kHz bandwidth, 3 NEX was used for acquired in coronal and axial geometries a multi-slice T2-weighted images. To reduce the respiratory motion the axial scan sequences were gated. IVIS-100 imaging system was used for the detection of luciferase activity. Mice were injected with d-luciferin bioluminescence substrate (Perkin Elmer), according to manufacturer’s instructions, and imaged 5 min after the injection. In ex vivo experiments mice were sacrificed and tissues harvested 5 min after luciferin injection. The living Image 4.3 software (Perkin Elmer) was used for analysis of the images after acquisition.
For ex vivo fluorescence imaging animals were sacrificed and imaged using a Leica MZ125 Stereo Zoom microscope and a DFC450C camera.
Immunohistochemistry and immunofluorescence.
Immunohistochemistry and immunofluorescence were performed as previously described (Genovese et al., 2017). Briefly, tumor samples were fixed in 4% formaldehyde (Fisher Scientific) for 24 hr at room temperature, moved in 70% ethanol (Fisher Scientific) for 48 hr, and embedded in paraffin (Leica ASP300S). After cutting (Leica RM2235), baking and de-paraffinization, slides were treated with Citrate Buffer solution (Electron Microscopy Sciences) according to manufacturer’s instructions. For immunohistochemistry staining, endogenous peroxidases were inactivated in a solution of 3% hydrogen peroxide (Sigma-Aldrich) for 20 min. Non-specific signals were blocked for 1 hr using 10% FBS and 5% BSA (Sigma-Aldrich). Tumor samples were stained with primary antibodies for 12 hr at 4°C. HRP-conjugated secondary antibodies (ImmPress, Vector Lab) and Nova RED peroxidase substrate (Vector Lab) were used for detection. Images were captured using a Nikon EclipseTi microscope and a Nikon DS-Fi1 digital camera. For immunofluorescence studies, Alexa488 and 555 conjugated secondary antibodies (Molecular Probes) were used. Images were acquired with a Hamamatsu C11440 digital camera, on a wide-field Nikon EclipseTi microscope.
Transmission electron microscopy.
The procedure was been described elsewhere (Genovese et al., 2017) and was performed at the UTMDACC High Resolution Electron Microscopy Facility. Briefly, after 1 hr of fixation with a solution containing 3% glutaraldehyde, 2% paraformaldehyde in 0.1 M cacodylate buffer at pH 7.3 the samples were washed and treated with 0.1% Millipore-filtered cacodylate-buffered tannic acid, post-fixed with 1% buffered osmium tetroxide for 30 min, stained and bloc with 1% Millipore-filtered uranyl acetate. Increasing concentrations of ethanol were used to dehydrated the samples that were subsequently infiltrated and embedded in LX-112 medium. Polymerization was induced at 60°C for 2 days. A leica Ultracut microtome was use d to cut ultra-thin sections that were stained with uranyl acetate and lead citrate in a Leica EM Stainer. Sections were scanned using a JEM 1010 transmission electron microscope (JEOL) at an accelerating voltage of 80 kV. Digital images were acquired using an AMT Imaging System (Advanced Microscopy Techniques Corp). Quantification of autophagy was performed by manually counting the number of vacuoles per cell.
Western blotting.
Western blot was performed as previously described (Genovese et al., 2017). Briefly, protein lysates were resolved on 5–15% gradient pre cast polyacrylamide SDS gels (Bio-Rad) and transferred onto PVDF membranes (Bio-Rad) according to manufacturer’s instructions. To reduce non-specific signal membranes were blocked in 3% BSA or Odyssey blocking buffer (Licor). Membranes were incubated with indicated primary antibodies overnight at 4°C, washed in TBST [10 mmol/L Tris, p H 8.0, 150 mmol/L NaCl (Fischer Scientific), 0.5% Tween 20 (Fisher Scientific)] buffer and probed with HRP-conjugated secondary antibodies (Cell Signaling Technology) at room temperature for 1 hr. The detection of bands was carried out upon chemiluminescence reaction (Denville Scientific) followed by film exposure (Phenix Research Products).
Expression profiling and data analysis.
RNA and DNA were isolated using RNeasy Mini Kit and DNeasy Blood and Tissue Kit (Qiagen) according to manufacturer’s instructions. Microarray Core Facility at the UTMDACC performed the gene expression profiling on Gene Chip Mouse Genome 430 2.0 Array (Thermo Scientific). RMA (robust multi-array average) methods were used with default options (background correction, quantile normalization, and log transformation) to normalize raw data from batches using R/Bioconductor’s affymetrix package (12925520) (Gautier et al., 2004) and analyzed with GSEA as previously described (Genovese et al., 2017).
Differential Expression:
Human MRT and matched normal expression data were obtained from the TARGET database (http://ocg.cancer.gov/programs/target). Human and murine expression data were analized using the limma package in R (Ritchie et al., 2015) at a p value threshold of 0.01. Pathway analysis was conducted using the Broad Institute’s Gene Set Enrichment Analysis (GSEA) tool (Mootha et al., 2003; Subramanian et al., 2005). Similarly, differential expression and pathway analyses were conducted on microarray data, and volcano plots were generated using GSEA gene sets.
Mutational Analysis:
Mutation rates for the TP53 gene for various pediatric cancers were obtained from the St. Jude’s Pediatric Cancer (PeCan) database (https://pecan.stjude.cloud) (Zhou et al., 2016). TP53 mutational data in rhabdoid tumors and in ATRTs were obtained from recent publications (Chun et al., 2016; Johann et al., 2016).
Quantification and statistical analysis.
Quantitative analysis was performed using ImageJ according to the providers’ instructions. Differences in mean between 2 groups were calculated using a two-tailed Student’s t-test. Results from survival and incidence experiments were analyzed with a log-rank (Mantel–Cox) test and expressed as Kaplan–Meier curves. Statistical analyses were performed using GraphPad Prism 7. Group size was established based on preliminary experimental results. For in vivo studies mice were assigned randomly to experimental arms. No statistical methods were used to establish sample size. Analysis of outcome was not performed in a blinded manner. The number of samples and replicates that were analyzed are included in the figure legends.
Data and software availability
The accession number for the microarray data reported in this paper is EMBL-EBI: E-MTAB-7497
Supplementary Material
Table S1. Gene set enrichment analysis in murine MRT samples, related to Figure 1.
Table S2. Gene set enrichment analysis in human MRT samples, related to Figure 4.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-AKT | Cell Signaling Technology | Cat#9272, RRID:AB_329827 |
| Rabbit monoclonal anti-AMPKα (D63G4) | Cell Signaling Technology | Cat#5832, RRID:AB_10624867 |
| Rabbit monoclonal anti-ATF4 (D4B8) | Cell Signaling Technology | Cat#11815 RRID:AB_2616025 |
| Rabbit monoclonal anti-Atg7 (D12B11) | Cell Signaling Technology | Cat#8558, RRID:AB_10831194 |
| Mouse monoclonal anti-beta-Actin (clone AC-74) | Sigma-Aldrich | Cat#A2228, RRID:AB_476697 |
| Mouse monoclonal anti-BAF47 | BD Bioscience | Cat#612111, RRID:AB_2191717 |
| Rabbit monoclonal anti-BIP (C50B12) | Cell Signaling Technology | Cat#3177, RRID:AB_2119845 |
| Rabbit monoclonal anti-c-Jun (60A8) | Cell Signaling Technology | Cat#9165, RRID:AB_2130165 |
| Mouse monoclonal anti-c-Myc (9E10) | Santa Cruz Biotechnology | Cat#sc-40, RRID:AB_627268 |
| Rabbit polyclonal anti-c-Myc | Proteintech Group | Cat#10828–1-AP, RRID:AB_2148585 |
| Mouse monoclonal anti-CHOP (L63F7) | Cell Signaling Technology | Cat#2895, RRID:AB_2089254 |
| Mouse monoclonal anti-Cip1 | BD Biosciences | Cat#610233, RRID:AB_397628 |
| Rabbit monoclonal anti-Cleaved Caspase-3 (Asp175) (5A1E) | Cell Signaling Technology | Cat#9664, RRID:AB_2070042 |
| Rabbit monoclonal anti-Cleaved PARP (Asp214) (D64E10) XP | Cell Signaling Technology | Cat#5625, RRID:AB_10699459 |
| Rabbit polyclonal anti-DRAM1 | LifeSpan | Cat#LS-B2726–50, RRID:AB_1932638 |
| Rabbit monoclonal anti-eIF2alpha (D7D3) XP | Cell Signaling Technology | Cat#5324, RRID:AB_10692650 |
| Rabbit polyclonal anti-EIF2AK2 | Proteintech Group | Cat#18244–1-AP, RRID:AB_2246451 |
| Rabbit polyclonal anti-EIF2AK2/PKR | LifeSpan | Cat#LS-B563, RRID:AB_2095733 |
| Goat polyclonal anti-Firefly Luciferase | LifeSpan | Cat#LS-C147186–500, RRID:AB_11134935 |
| Chicken polyclonal anti-GFP | Abcam | Cat#ab13970, RRID:AB_300798 |
| Rabbit monoclonal anti-GFP (D5.1) | Cell Signaling Technology | Cat#2956, RRID:AB_1196615 |
| Rabbit monoclonal anti-HSP90 (C45G5) | Cell Signaling Technology | Cat#4877, RRID:AB_2233307 |
| Mouse monoclonal anti-IGFBP3 (B-5) | Santa Cruz Biotechnology | Cat#sc-365936, RRID:AB_10917037 |
| Rabbit monoclonal anti-IRE1α (14C10) | Cell Signaling Technology | Cat#3294, RRID:AB_823545 |
| Rabbit monoclonal anti-Ki-67 (SP6) | Thermo Fisher Scientific | Cat#MA5–14520, RRID:AB_10979488 |
| Rabbit monoclonal anti-LC3A/B (D3U4C) XP | Cell Signaling Technology | Cat#12741, RRID:AB_2617131 |
| Mouse monoclonal anti-MDM2 (SMP14) | Santa Cruz Biotechnology | Cat#sc-965, RRID:AB_627920 |
| Rat monoclonal anti-p19 ARF (5-C3–1) | Santa Cruz Biotechnology | Cat#sc-32748, RRID:AB_628071 |
| Mouse monoclonal anti-p53 (1C12) | Cell Signaling Technology | Cat#2524, RRID:AB_331743 |
| Mouse monoclonal anti-p53 (DO-7) | Cell Signaling Technology | Cat#48818, RRID:AB_2713958 |
| Rabbit monoclonal anti-p53 (7F5) | Cell Signaling Technology | Cat#2527, RRID:AB_331211 |
| Rabbit monoclonal anti-p70 S6 Kinase (49D7) | Cell Signaling Technology | Cat#2708, RRID:AB_390722 |
| Rabbit polyclonal anti-PDIA6 | Proteintech Group | Cat#18233–1-AP, RRID:AB_10805765 |
| Rabbit monoclonal anti-PERK (D11A8) | Cell Signaling Technology | Cat#5683, RRID:AB_10831515 |
| Rabbit monoclonal anti-phospho-AKT (Ser473) (D9E) XP | Cell Signaling Technology | Cat#4060, RRID:AB_2315049 |
| Rabbit monoclonal anti-phpspho-AMPKα (Thr172) (40H9) | Cell Signaling Technology | Cat#2535, RRID:AB_331250 |
| Rabbit monoclonal anti-phospho-eIF2alpha (Ser51) (D9G8) XP | Cell Signaling Technology | Cat#3398, RRID:AB_2096481 |
| Rabbit monoclonal anti-phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) XP | Cell Signaling Technology | Cat#4858, RRID:AB_916156 |
| Mouse monoclonal anti-phospho-p70 S6 Kinase (Thr389) (1A5) | Cell Signaling Technology | Cat#9206, RRID:AB_331790 |
| Rabbit monoclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) (81E11) | Cell Signaling Technology | Cat#4668, RRID:AB_2307320 |
| Rabbit polyclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) | Cell Signaling Technology | Cat#9251, RRID:AB_331659 |
| Rabbit monoclonal anti-phospho-4E-BP1 (Thr37/Thr46) (236B4) | Cell Signaling Technology | Cat#2855, RRID:AB_560835 |
| Mouse monoclonal anti-S6 Ribosomal Protein (54D2) | Cell Signaling Technology | Cat#2317, RRID:AB_2238583 |
| Rabbit polyclonal anti-SAPK/JNK | Cell Signaling Technology | Cat#9252, RRID:AB_2250373 |
| Rabbit monoclonal anti-SESN2 (EPR18907) | Abcam | Cat#ab178518, RRID:AB_2716805 |
| Rabbit polyclonal anti-SESN2 | Proteintech Group | Cat#21346–1-AP, RRID:AB_10733238 |
| Rabbit polyclonal anti-SMARCB1 | Sigma-Aldrich | Cat#HPA018248, RRID: AB_1851162 |
| Rabbit monoclonal anti-Survivin (71G4B7) | Cell Signaling Technology | Cat#2808, RRID:AB_2063948 |
| Rabbit polyclonal anti-TP53 | Proteintech Group | Cat#21891–1-AP, RRID:AB_10896826 |
| Mouse monoclonal anti-Ubiquitin (P4D1) | Cell Signaling Technology | Cat#3936, RRID:AB_10839120 |
| Rabbit monoclonal anti-Vimentin (D21H3) XP | Cell Signaling Technology | Cat#5741, RRID:AB_10695459 |
| Mouse monoclonal anti-Vimentin (RV202) | Abcam | Cat#ab8978, RRID:AB_306907 |
| Rabbit monoclonal anti-Vinculin (E1E9V) XP | Cell Signaling Technology | Cat#13901, RRID:AB_2728768 |
| Rabbit monoclonal anti-XBP-1s (D2C1F) | Cell Signaling Technology | Cat#12782, RRID:AB_2687943 |
| Rabbit monoclonal anti-4E-BP1 (53H11) | Cell Signaling Technology | Cat#9644, RRID:AB_2097841 |
| Bacterial and Virus Strains | ||
| psPAX2 packaging vector | Didier Trono | Addgene Cat#12260 |
| pMD2.G packaging vector | Didier Trono | Addgene Cat#12259 |
| Biological Samples | ||
| Copy number and mRNA expression values of SMARCB1 in human cell lines | Cancer Cell Lines Encyclopedia | https://portals.broadinstitute.org/ccle |
| Human MRT and normal samples | TARGET database | http://ocg.cancer.gov/programs/target |
| Mutation Signatures | PeCan database | https://pecan.stjude.cloud |
| Mutation Signatures for MRT and ATRT | Chun, H. J. E. et al., 2016; Johann, P. D. et al., 2016 | N/A |
| Rhabdoid FFPE | Dinesh Rakheja (dinesh.rakheja@utsouthwestern.edu) | N/A |
| Rhabdoid FFPE | Dolores Lopez-Terrada (dhterrad@texaschildrenshospital.org) | N/A |
| Rhabdoid FFPE | Liren Li (lilr@sysucc.org.cn) | N/A |
| RMC FFPE | George J. Netto (gnetto1@jhmi.edu) | N/A |
| RMC FFPE | Priya Rao (prao@mdanderson.org) | N/A |
| RMC2C cell line | Jose A. Karam (JAKaram@mdanderson.org) and Nizar M. Tannir (ntannir@mdanderson.org) | N/A |
| Sensitivity to the ER stress in human cell lines | Genomics of Drug Sensitivity in Cancer | https://www.cancerrxgene.org/translation/Drug |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bortezomib | LC Laboratories | Cat#B-1408 |
| Chloroquine diphosphate salt | Sigma-Aldrich | Cat#C6628 |
| Cycloheximide solution | Sigma-Aldrich | Cat#C4859 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891 |
| Etoposide | Sigma-Aldrich | Cat#E1383 |
| Idasanutlin (RG-7388) | Selleckchem | Cat#S7205 |
| Ifosfamide | Sigma-Aldrich | Cat#I4909 |
| Ixazomib Citrate (MLN9708) | Selleckchem | Cat#S2181 |
| MG-132 | EMD Millipore | Cat#474787 |
| NVP-AUY922 | LC Laboratories | Cat#N-5300 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| YM-155 | Proactive Molecular Research | Chemical Number P06–10194 CAS_RN 781661–94-7 |
| (Z)-4-Hydroxytamoxifen | Sigma-Aldrich | Cat#H7904 |
| Critical Commercial Assays | ||
| Adeno-X™ Maxi Purification Kit | Takara Bio Inc | Cat#631533 |
| Adeno-X™ Rapid Titer Kit | Takara Bio Inc | Cat#632250 |
| CellTiter-Glo® 2.0 Assay | Promega | Cat#G9243 |
| Click-iT™ Plus OPP Alexa Fluor™ 488 Protein Synthesis Assay Kit | Thermo Fisher Scientific | Cat#C10456 |
| Click-iT™ Plus OPP Alexa Fluor™ 594 Protein Synthesis Assay Kit | Thermo Fisher Scientific | Cat#C10457 |
| Dead Cell Apoptosis Kit with Annexin V FITC and PI, for flow cytometry | Thermo Fisher Scientific | Cat#V13242 |
| DNeasy Blood & Tissue Kit (250) | Qiagen | Cat#69506 |
| GeneChip™ Mouse Genome 430 2.0 Array | Thermo Fisher Scientific | Cat#900496 |
| Mouse on Mouse (M.O.M.™) Elite Peroxidase Kit | Vector Laboratories | Cat#PK-2200 |
| RNeasy Mini Kit (250) | Qiagen | Cat#74106 |
| Venor™ GeM Mycoplasma Detection Kit, PCR-based | Sigma Aldrich | Cat#MP0025 |
| XenoLight D-Luciferin-K+ Salt Bioluminescent Substrate | PerkinElmer | Cat#122799 |
| Deposited Data | ||
| Microarray data | EMBL-EBI | E-MTAB-7497 |
| Experimental Models: Cell Lines | ||
| A204 | ATCC | Cat#HTB-82, RRID:CVCL_1058 |
| HEK293T | ATCC | Cat#CRL-3216, RRID:CVCL_0063 |
| G401 | ATCC | Cat#CRL-1441, RRID:CVCL_0270 |
| G-402 | ATCC | Cat#CRL-1440, RRID:CVCL_1221 |
| RMC2C | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Rosa26LSL-Luc: B6.129S4-Gt(ROSA)26Sortm3(CAG-luc)Tyj/J | The Jackson Laboratory | JAX:009044; RRID:IMSR_JAX:009044 |
| Mouse: KRASLSL/G12D: B6.129S4-Krastm4Tyj/J | The Jackson Laboratory | JAX:008179; RRID:IMSR_JAX:008179 |
| Mouse: Trp53LSL-R172H: 129S-Trp53tm2Tyj/J | The Jackson Laboratory | JAX:008652; RRID:IMSR_JAX:008652 |
| Mouse: Rosa26LSL-TdT: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX:007909; RRID:IMSR_JAX:007909 |
| Mouse: Rosa26LSL-Cas9-Gfp | Platt et al., 2014 | N/A |
| Mouse: Smarcb1LoxP | Roberts et al., 2002 | N/A |
| Mouse: Pax8Cre: B6.129P2(Cg)-Pax8tm1.1(cre)Mbu/J | The Jackson Laboratory | JAX:028196; RRID:IMSR_JAX:028196 |
| Mouse: Trp53LoxP | Marino et al., 2000 | N/A |
| Mouse: Rosa26FlpoERT2: STOCK Gt(ROSA)26Sortm3(CAG-flpo/ERT2)Alj/J | The Jackson Laboratory | JAX:018906; RRID:IMSR_JAX:018906 |
| Mouse: Trp53Frt | Lee et al., 2012a | N/A |
| Mouse: Rag2−/−: B6(Cg)-Rag2tm1.1Cgn/J | The Jackson Laboratory | JAX:008449; RRID:IMSR_JAX:008449 |
| Mouse: NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX:005557; RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| shRNA Atg7 seed sequence: 5’-GCCAACATCCCTGGATACAAG-3’ |
This paper | N/A |
| shRNA Birc5–1 seed sequence: 5’-CAAAGACTACCCGTCAGTCAA-3’ |
This paper | N/A |
| shRNA Birc5–2 seed sequence: 5’-GAAGAACTAACCGTCAGTGAA-3’ |
This paper | N/A |
| shRNA CDKN2A seed sequences: 5’-GCTGGGTGGTCTTTGTGTA-3’ |
This paper | N/A |
| shRNA DRAM1–1 | Sigma Aldrich | Cat#SHCLNG-NM_027878, TRCN0000329081 |
| shRNA DRAM1–2 | Sigma Aldrich | Cat#SHCLNG-NM_027878, TRCN0000329023 |
| shRNA Myc seed sequence: 5’-TGGAGATGATGACCGAGTTAC-3’ |
This paper | N/A |
| shRNA TP53–1 | Sigma Aldrich | Cat#SHCLNG-NM_000546, TRCN0000003753 |
| shRNA TP53–2 | Sigma Aldrich | Cat#SHCLNG-NM_000546, TRCN0000003755 |
| Recombinant DNA | ||
| Adx | Oka Kazuhiro | N/A |
| Adx-A-Cre | This paper | N/A |
| Adx-LacZ | Oka Kazuhiro | N/A |
| ICPI-CS-iCreP2A-tdTomato | Oka Kazuhiro | N/A |
| pALB-GFP | Snorri Thorgeirsson (Heo et al., 2006) | Addgene Cat#55759 |
| pBABEpuro GFP-LC3 | Jayanta Debnath (Fung et al., 2008) | Addgene Cat#22405 |
| pGF-p53-mCMV-EF1α-Puro Lentivector | SBI | Cat#TR200pa-p |
| pLenti6/UbC/V5-DEST™ Gateway™ Vector | Thermo Fisher Scientific | Cat#V49910 |
| pLIX-iLacZ | This paper | N/A |
| pLIX-iSMARCB1 | This paper | N/A |
| pLIX_403 | David Root | Addgene Cat#41395 |
| pMSCV-loxP-dsRed-loxp-eGFP-Puro-WPRE | Hans Clevers (Koo et al., 2012) | Addgene Cat#32702 |
| pLSM4 | This paper | N/A |
| pLSM5 | Genovese et al., 2017 | N/A |
| pUb-GFP | This paper | N/A |
| pLV-p53R-Cre | This paper | N/A |
| Ub-M-GFP | Nico Dantuma (Dantuma et al., 2000) | Addgene Cat#11938 |
| Software and Algorithms | ||
| FlowJo_V10 | FlowJo LLC | www.flowjo.com |
| Gene Set Enrichment Analysis (GSEA) | Subramanian et al., 2005; Mootha et al., 2003 | http://software.broadinstitute.org/gsea/index.jsp |
| GraphPad Prism 7 | GraphPad Software | www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Linear Models for Microarray Data (limma package) | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Living Image 4.3 software | PerkinElmer | http://www.perkinelmer.com |
| Methods for Affymetrix Oligonucleotide Arrays (R/Bioconductor’s affy package (12925520)) | Bioconductor | http://bioconductor.org/packages/affy/ |
| RStudio version 1.0.143 with R version 3.4.3 | R-project | www.r-project.org/ |
| Other | ||
SIGNIFICANCE.
This work describes immediately testable clinical hypotheses to treat highly recalcitrant diseases affecting children and young adults. Our study sheds light on the crucial role of the tumor suppressor gene, TP53, in the regulation of global protein metabolism and in the adaptation to ER stress in the context of SMARCB1 loss. Our findings have profound implications regarding the current standard-of-care cytotoxic drug regimen prescribed to patients with MRTs: specifically predicting that cytotoxic chemotherapeutics may promote a rapid adaptation to proteotoxic stress through a p53-dependent, autophagic response. In this context, rational combinations with inhibitors of autophagy or cycles of cytotoxic and proteotoxic combinations may result in durable remissions for a class of aggressive pediatric malignancies.
Highlights.
Smarcb1-deficient mosaic GEM models recapitulate malignant rhabdoid tumors (MRT)
Autophagy and UPR are essential adaptive mechanisms to proteotoxic stress in MRT
The MYC-p53 axis regulates proteostasis in SMARCB1-deficient cells
Autophagy and proteasome inhibitors achieve durable responses in MRT models.
ACKNOWLEDGEMENTS
We thank Drs. Philip Jones, Joe Marszalek and Carlo Toniatti for discussions and suggestions. We thank Dr. Angela Deem for stylistic editing. We thank Dr. Papia Ghosh and Claudia Penati for the continuous support. We thank Alessia Petrocchi, Sahil Seth, Dr. Emilia Di Francesco and members of the Draetta lab for discussions and reagents. We thank Dr. Kenneth Dunner, Jr. and the High Resolution Electron Microscopy Facility at MDACC; the MDACC Small Animals Imaging Facility, the UTMDACC Microarray Core Facility, the MDACC Department of Veterinary Medicine, the UTMDACC Flow Facility and the UT MDACC Creative Communications.
Dr. Pavlos Msaouel was supported by a Conquer Cancer Foundation Young Investigator Award, and by a Kidney Cancer Association Young Investigator Award. Dr. Giulio Draetta was supported by the Sheikh Ahmed Bin Zayed Al Nahyan Center for Pancreatic Cancer Grant and the Pancreatic Cancer Action Network Translational Research Grant. Dr. Alessandro Carugo was supported by the FIRC-AIRC fellowship. Dr. Federica Carbone was supported by the AIRC fellowship. Dr.Nizar M. Tannir was supported by the Ransom Horne Jr. Professorship for Cancer Research. Dr. Genovese was supported by the Barbara Massie Memorial Fund and the CPRIT Research Grant RP170722.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M, Souabni A, Mandler M, Neubuser A, and Busslinger M (2002). Nephric lineage specification by Pax2 and Pax8. Genes Dev 16, 2958–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F, Andre N, Stephan JL, Perel Y, Oberlin O, et al. (2011). Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17, 31–38. [DOI] [PubMed] [Google Scholar]
- Brennan B, De Salvo GL, Orbach D, De Paoli A, Kelsey A, Mudry P, Francotte N, Van Noesel M, Bisogno G, Casanova M, and Ferrari A (2016). Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur J Cancer 60, 69–82. [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, and Kley N (1995). Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 377, 646–649. [DOI] [PubMed] [Google Scholar]
- Budanov AV, and Karin M (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscariollo DL, Park HS, Roberts KB, and Yu JB (2012). Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118, 4212–4219. [DOI] [PubMed] [Google Scholar]
- Calderaro J, Masliah-Planchon J, Richer W, Maillot L, Maille P, Mansuy L, Bastien C, de la Taille A, Boussion H, Charpy C, et al. (2016). Balanced Translocations Disrupting SMARCB1 Are Hallmark Recurrent Genetic Alterations in Renal Medullary Carcinomas. Eur Urol 69, 1055–1061. [DOI] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia C, and Genomics of Drug Sensitivity in Cancer C (2015). Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AF, Dupage MJ, Dong HK, Chen J, and Jacks T (2008). Regulated expression of a tumor-associated antigen reveals multiple levels of T-cell tolerance in a mouse model of lung cancer. Cancer Res 68, 9459–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HJ, Lim EL, Heravi-Moussavi A, Saberi S, Mungall KL, Bilenky M, Carles A, Tse K, Shlafman I, Zhu K, et al. (2016). Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 29, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, and Ryan KM (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134. [DOI] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, and Masucci MG (2000). Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol 18, 538–543. [DOI] [PubMed] [Google Scholar]
- Dixon SB, Bjornard KL, Alberts NM, Armstrong GT, Brinkman TM, Chemaitilly W, Ehrhardt MJ, Fernandez-Pineda I, Force LM, Gibson TM, et al. (2018). Factors influencing risk-based care of the childhood cancer survivor in the 21st century. CA Cancer J Clin 68, 133–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezekian B, Englum B, Gilmore BF, Nag UP, Kim J, Leraas HJ, Routh JC, Rice HE, and Tracy ET (2017). Renal medullary carcinoma: A national analysis of 159 patients. Pediatr Blood Cancer 64. [DOI] [PubMed] [Google Scholar]
- Fung C, Lock R, Gao S, Salas E, and Debnath J (2008). Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 19, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, and Irizarry RA (2004). affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. [DOI] [PubMed] [Google Scholar]
- Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, Nezi L, Corti D, Minelli R, Pettazzoni P, et al. (2017). Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 542, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresh L, Bourachot B, Reimann A, Guigas B, Fiette L, Garbay S, Muchardt C, Hue L, Pontoglio M, Yaniv M, and Klochendler-Yeivin A (2005). The SWI/SNF chromatin-remodeling complex subunit SNF5 is essential for hepatocyte differentiation. EMBO J 24, 3313–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ZY, Richer W, Freneaux P, Chauvin C, Lucchesi C, Guillemot D, Grison C, Lequin D, Pierron G, Masliah-Planchon J, et al. (2016). The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun 7, 10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J, Factor VM, Uren T, Takahama Y, Lee JS, Major M, Feinstone SM, and Thorgeirsson SS (2006). Hepatic precursors derived from murine embryonic stem cells contribute to regeneration of injured liver. Hepatology 44, 1478–1486. [DOI] [PubMed] [Google Scholar]
- Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, Wang X, Biegel JA, Pomeroy SL, Mesirov JP, and Roberts CW (2005). Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U S A 102, 17745–17750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DT, Sturm D, Hermann C, Segura Wang M, et al. (2016). Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29, 379–393. [DOI] [PubMed] [Google Scholar]
- Kieran MW, Roberts CW, Chi SN, Ligon KL, Rich BE, Macconaill LE, Garraway LA, and Biegel JA (2012). Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer 59, 1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, and Yaniv M (2000). The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep 1, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BK, Stange DE, Sato T, Karthaus W, Farin HF, Huch M, van Es JH, and Clevers H (2012). Controlled gene expression in primary Lgr5 organoid cultures. Nature methods 9, 81–83. [DOI] [PubMed] [Google Scholar]
- Kumar SK, Bensinger WI, Zimmerman TM, Reeder CB, Berenson JR, Berg D, Hui AM, Gupta N, Di Bacco A, Yu J, et al. (2014). Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood 124, 1047–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao Z, Raju GP, Bai CB, and Joyner AL (2012). MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep 2, 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CL, Moding EJ, Huang X, Li Y, Woodlief LZ, Rodrigues RC, Ma Y, and Kirsch DG (2012a). Generation of primary tumors with Flp recombinase in FRT-flanked p53 mice. Dis Model Mech 5, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, et al. (2012b). A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Mitchell M, Jerebtsova M, Batshaw ML, Newman K, and Ye X (2000). Long-term gene transfer to mouse fetuses with recombinant adenovirus and adeno-associated virus (AAV) vectors. Gene Ther 7, 1986–1992. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Oeffinger KC, and Hudson MM (2004). Long-term complications following childhood and adolescent cancer: foundations for providing risk-based health care for survivors. CA Cancer J Clin 54, 208–236. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, and Jacks T (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Oue T, Fukuzawa M, Okita H, Mugishima H, Horie H, Hata J, Saito M, Nozaki M, Chin M, Nakadate H, et al. (2009). Outcome of pediatric renal tumor treated using the Japan Wilms Tumor Study-1 (JWiTS-1) protocol: a report from the JWiTS group. Pediatr Surg Int 25, 923–929. [DOI] [PubMed] [Google Scholar]
- Paek AL, Liu JC, Loewer A, Forrester WC, and Lahav G (2016). Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 165, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard H, Reinert J, Beier R, Furtwangler R, Alkasser M, Rutkowski S, Fruhwald M, Koscielniak E, Leuschner I, Kaatsch P, and Graf N (2008). Rhabdoid tumors in children: prognostic factors in 70 patients diagnosed in Germany. Oncol Rep 19, 819–823. [PubMed] [Google Scholar]
- Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, et al. (2003). A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348, 2609–2617. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, and Orkin SH (2000). Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proceedings of the National Academy of Sciences of the United States of America 97, 13796–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Leroux MM, Fleming MD, and Orkin SH (2002). Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2, 415–425. [DOI] [PubMed] [Google Scholar]
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, and Khamashta MA (2010). Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 69, 20–28. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa S, Taguchi A, Sharma N, Perantoni AO, and Nishinakamura R (2016). Selective In Vitro Propagation of Nephron Progenitors Derived from Embryos and Pluripotent Stem Cells. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, Krasin M, Dalton J, Hale G, Kun LE, et al. (2005). Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23, 1491–1499. [DOI] [PubMed] [Google Scholar]
- Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, Norman JD, Guilhamon P, Garzia L, Agamez NR, et al. (2016). Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 30, 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobaugh-Lotrario AD, Finegold MJ, and Feusner JH (2011). Rhabdoid tumors of the liver: rare, aggressive, and poorly responsive to standard cytotoxic chemotherapy. Pediatr Blood Cancer 57, 423–428. [DOI] [PubMed] [Google Scholar]
- Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, Gore L, and Feusner JH (2009). Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer 52, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Le P, Martinez D, Eaton KW, Shyam N, Jordan-Sciutto KL, Pawel B, Biegel JA, and Judkins AR (2011). p16INK4A and p14ARF tumor suppressor pathways are deregulated in malignant rhabdoid tumors. J Neuropathol Exp Neurol 70, 596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, and Delattre O (1998). Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206. [DOI] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. (2011). Suppression of lung adenocarcinoma progression by Nkx2–1. Nature 473, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. (2013). Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41, D955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon CH, Lee ES, Lim DS, and Bae YS (2009). PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc Natl Acad Sci U S A 106, 7852–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, et al. (2006). Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, Li Y, Zhang Z, Rusch MC, Parker M, et al. (2016). Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 48, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene set enrichment analysis in murine MRT samples, related to Figure 1.
Table S2. Gene set enrichment analysis in human MRT samples, related to Figure 4.
Data Availability Statement
The accession number for the microarray data reported in this paper is EMBL-EBI: E-MTAB-7497