

Abstract
The mitochondrial F1Fo ATP synthase is one of the most abundant proteins of the mitochondrial inner membrane, which catalyzes the final step of oxidative phosphorylation to synthesize ATP from ADP and Pi. ATP synthase uses the electrochemical gradient of protons (ΔμH+) across the mitochondrial inner membrane to synthesize ATP. Under certain pathophysiological conditions, ATP synthase can run in reverse to hydrolyze ATP and build the necessary ΔμH+ across the mitochondrial inner membrane. Tight coupling between these two processes, proton translocation and ATP synthesis, is achieved by the unique rotational mechanism of ATP synthase and is necessary for efficient cellular metabolism and cell survival. The uncoupling of these processes, dissipation of mitochondrial inner membrane potential, elevated levels of ROS, low matrix content of ATP in combination with other cellular malfunction trigger the opening of the mitochondrial permeability transition pore in the mitochondrial inner membrane. In this review we will discuss the new role of ATP synthase beyond oxidative phosphorylation. We will highlight its function as a unique regulator of cell life and death and as a key target in mitochondria-mediated neurodegeneration and neuroprotection.
Introduction
Mitochondria originated from endosymbiotic alphaproteobacteria more than one billion years ago (Roger et al., 2017). Motility is one of the many features that mitochondria inherited from their ancestors: mitochondria are extremely dynamic organelles; they can move long distances based on cellular energy requirements. The regulation of mitochondrial motility, as a response to different environmental and metabolic changes is crucial for maintaining energy homeostasis to support cellular function. Neurons are especially susceptible to disruptions in mitochondrial motility and quality control mechanisms. Quality control processes include removal of damaged mitochondria by mitophagy and dynamic processes of mitochondrial fusion and fission. Recent studies indicate that dysregulation of mitochondrial dynamics and mitochondrial dysfunction comprise an important underlying cause of aging, different neurological and degenerative diseases, highlighting further the central role of mitochondria in cell death. Mitochondria regulate cell death through Bax/Bak-mediated mitochondrial outer membrane permeabilization (MOMP), which can lead to caspase-dependent apoptosis or caspase-independent cell death (Chipuk et al., 2008; Finucane et al., 1999), as well as by another major mechanism termed mitochondrial permeability transition (mPT). mPT is traditionally defined as a phenomenon associated with the sudden change in the mitochondrial inner membrane permeability to different solutes with a size of <1.5 kDa (Haworth and Hunter, 1979; Hunter and Haworth, 1979a, b; Hunter et al., 1976). The mPT causes osmotic dysregulation of mitochondria leading to mitochondrial swelling, rupture of the outer mitochondrial membrane and cell death (Haworth and Hunter, 1979; Hunter and Haworth, 1979a, b; Hunter et al., 1976). These events are linked to the opening of a mitochondrial inner membrane channel, the so-called mitochondrial permeability transition pore (mPTP) (Kinnally et al., 1989; Lohret et al., 1996; Petronilli et al., 1989; Szabo and Zoratti, 1992; Zoratti and Szabo, 1995; Zorov et al., 1992). mPTP is a high conductance, voltage-gated, non-selective protein channel of controversial molecular content (Kinnally et al., 1989; Lohret et al., 1996; Petronilli et al., 1989; Szabo and Zoratti, 1992; Zoratti and Szabo, 1995; Zorov et al., 1992). A high level of matrix Ca2+ was shown to be one of the main factors to induce mPTP opening, along with inorganic phosphate, CypD and oxidants (Bernardi, 1992, 1996; Bernardi et al., 1992; Carraro and Bernardi, 2016; Petronilli et al., 1994; Szabo et al., 1992). mPTP opening can be inhibited by other divalent and trivalent cations, such as, Mg2+, Ba2+, Gd3+, as well as by H+ and adenine nucleotides (Bernardi, 1996; Mnatsakanyan et al., 2019; Nicolli et al., 1993; Szabo et al., 1992).
The exact structure and molecular composition of mPTP has been extensively studied and various mitochondrial proteins, the adenine nucleotide translocator (ANT) (Halestrap and Brenner, 2003), the voltage-dependent anion channel (VDAC) (Szabo et al., 1993; Szabo and Zoratti, 1993), the phosphate carrier (PiC) (Kwong et al., 2014) and the translocator protein (TSPO) (Chelli et al., 2001; Li et al., 2007; Pastorino et al., 1994) were suggested to constitute the channel of mPTP. However, further studies revealed the important role of these proteins as channel regulators rather than as a key structural element of mPTP (Baines et al., 2007; Gutierrez-Aguilar et al., 2014; Kokoszka et al., 2004; Sileikyte et al., 2014). The mitochondrial ATP synthase dimers (Carraro et al., 2018; Carraro et al., 2014; Giorgio et al., 2013; Guo et al., 2019; Urbani et al., 2019) or c-subunit ring alone (Alavian et al., 2014; Azarashvili et al., 2014; Bonora et al., 2013; Mnatsakanyan et al., 2019; Morciano et al., 2018; Neginskaya et al., 2019; Pavlov et al., 2005) were suggested and still remain strong candidates for housing the mPTP channel according to several recent studies. Here we will discuss the role of ATP synthase c-subunit leak channel (ACLC) in human health and disease, in mitochondrial homeostasis and remodeling, synaptic plasticity and metabolic efficiency of neurons, brain ischemia and excitotoxicity, neurodegenerative disease and most importantly as a therapeutic target in mitochondria-mediated neuroprotection.
The structure, oligomeric state of ATP synthase and its arrangement in the mitochondrial inner membrane
The overall structure of ATP synthase is highly conserved in all unicellular and multicellular organisms. ATP synthase is composed of two multi-subunit subcomplexes, F1 and FO, which drive ATP synthesis and hydrolysis in a unique and efficient manner, by using the rotation of their own subunits (Abrahams et al., 1994; Menz et al., 2001). The mitochondrial ATP synthase has by far the most complex structure and subunit composition compared with its simplest versions found in bacteria. The FO subdomain consists of subunits a, b, c, d, F6, e, f, g, A6L, 6.8PL, DAPIT (diabetes-associated protein in insulin sensitive tissues) and OSCP (oligomycin sensitivity-conferring protein) (Kuot et al., 2019; Zhou et al., 2015). The c-subunit ring is completely embedded in the membrane and has 8 copies in mammalian mitochondria (Watt et al., 2010). The F1 subdomain consists of subunits α, β, γ, δ, ε and the regulatory protein IF1, which are located in the mitochondrial matrix. The three copies of α and β subunits alternate with each other to form the hexameric catalytic head of F1, which does not rotate during ATP synthase catalysis (Menz et al., 2001). Subunits γ, δ and ε form the central stalk of ATP synthase by connecting F1 with FO. The central stalk rotates along with the c-subunit ring during ATP synthesis or hydrolysis. FO subunits b, d, F6 and OSCP form the peripheral stator stalk of ATP synthase by connecting the F1 α3β3γ complex with the a subunit of FO (Zhou et al., 2015). The additional function of the peripheral stator stalk is to provide the elasticity necessary for efficient coupling of F1 and FO motors (Murphy et al., 2019).
When the electron transport chain complexes are fully functional to build the ΔμH+, the downhill flow of protons through FO drives the rotation of c-subunit ring and the consequent rotation of γδε-subunits of F1 to synthesize ATP. During the reverse reaction, ATP hydrolysis in β catalytic centers of F1 powers the rotation of γδε-subunits and the subsequent rotation of the c-subunit ring to drive proton translocation across the membrane (Nicholls, 2013). Cellular conditions inducing the reverse catalytic reaction of ATP synthase will be discussed in later sections of this review.
Recent advances in structural biology, especially in the fields of cryo-electron microscopy and cryo-electron tomography have significantly increased our understanding of ATP synthase structure and its oligomeric state. The high-resolution structure of mitochondrial ATP synthase dimers from green alga Polytomella sp, determined by single-particle electron cryomicroscopy at 6.2 A° resolution revealed a most unexpected horizontal location of a-subunits (Allegretti et al., 2015). This structure provides unique insights into the proton translocation mechanism through FO and how the passage of protons through the membrane drives ATP production (Allegretti et al., 2015).
Another recent structure of mitochondrial ATP synthase from porcine heart shows the high-resolution structure of ATP synthase tetramer in its inhibited state with IF1 molecules bound to it (Gu et al., 2019). Interestingly, c-subunit lumen in this structure was filled by a 40 amino acid-long alpha-helical protein instead of lipids or detergents as previously reported (Matthies et al., 2009; Meier et al., 2001; Oberfeld et al., 2006). The authors assigned 6.8PL proteolipid subunit of ATP synthase to this density although further studies are required to confirm the identity of this protein. According to this structure, the C-termini of the four FO e subunits are bent toward the four c-subunit rings in the tetramer, which allows them to interact with the C-terminal ends of the 6.8PL helices occupying the central pore of the four c-rings (Gu et al., 2019). This interaction between subunits c, 6.8PL and e might be important for preventing dissipation of membrane potential under normal physiological conditions by keeping the ACLC closed or substantially reducing the probability of its opening.
Cryo-electron tomographic analysis of mitochondria revealed the morphology of mitochondrial inner membrane cristae and the in situ oligomeric state of ATP synthase (Davies et al., 2012; Davies et al., 2014). Recent reports have shown that ATP synthase molecules are organized in rows of dimers (Blum et al., 2019). Dimeric conformation of ATP synthase is thought to be essential for the formation of the lamellar morphology of the inner membrane cristae (Blum et al., 2019; Davies et al., 2012). ATP synthase FO subunits e and g are proposed to be crucial for causing the curvature of cristae because a mutant strain of yeast lacking subunit e or g also lacks the normal lamellar shape of the cristae and has balloon-shaped cristae instead (Davies et al., 2012). The role of ATP synthase in shaping the inner membrane cristae and controlling mitochondrial fission and fusion processes will be discussed in later sections.
The role of ATP synthase in human health and disease
A. ATP synthase c-subunit leak channel as a main contributor to mPT
Mitochondrial F1FO ATP synthase was suggested to house the channel of mPTP according to several recent studies (Alavian et al., 2014; Azarashvili et al., 2014; Bonora et al., 2013; Carraro et al., 2018; Carraro et al., 2014; Giorgio et al., 2013; Guo et al., 2019; Mnatsakanyan et al., 2019; Morciano et al., 2018; Neginskaya et al., 2019; Pavlov et al., 2005; Urbani et al., 2019). Bernardi’s laboratory first described the specific interaction of ATP synthase OSCP subunit with the peptidyl-prolyl cis-trans isomerase cyclophilin D (CypD) (Giorgio et al., 2009), a well-known regulator of mPTP (Baines et al., 2005; Karch and Molkentin, 2014), which allowed them to hypothesize that ATP synthase is a key player in mPTP formation. Numerous studies performed in Bernardi’s group suggested that the channel is formed by the ATP synthase dimers and subunits e and g, required for ATP synthase dimerization, are also crucial for forming the channel (Carraro et al., 2018; Carraro et al., 2014; Giorgio et al., 2013; Guo et al., 2019; Urbani et al., 2019). The experiments with yeast mutant strains without subunits e and g displayed a remarkable resistance to channel opening (Carraro et al., 2018; Carraro et al., 2014). Additionally, channel formation was observed in lipid bilayer recordings of purified reconstituted ATP synthase tetramers and dimers but not in monomers isolated from bovine and porcine hearts, indicating that ATP synthase dimerization is required for pore formation (Giorgio et al., 2013; Urbani et al., 2019).
Our studies have recently shown that ATP synthase monomer forms a high, multi-conductance channel with the biophysical characteristics of mPTP, suggesting that the channel is not located between e and g subunits (Mnatsakanyan et al., 2019). We reported that the channel resides in the membrane embedded c-subunit ring (Alavian et al., 2014). Several other reports suggest that the c-subunit comprises the pore (Alavian et al., 2014; Azarashvili et al., 2014; Bonora et al., 2013; Mnatsakanyan et al., 2019; Morciano et al., 2018; Neginskaya et al., 2019; Pavlov et al., 2005). The c-subunit was found as the main component of the chloroform extract of mitochondrial membranes from rat liver; the channel activity of this extract closely resembled the behavior of the mPTP (Azarashvili et al., 2014; Pavlov et al., 2005). We found that the affinity purified c-subunit indeed forms a large, voltage-sensitive, multi-conductance channel in patch-clamp recordings (Alavian et al., 2014). However, the activity of the c-subunit channel was not sensitive to the well-known mPTP regulators, cyclosporine A (CsA) and Ca2+, since these regulators bind to ATP synthase F1 subunits or FO peripheral stalk subunits located in the matrix (Alavian et al., 2014). Depletion of the c-subunit attenuated Ca2+-induced depolarization of the inner mitochondrial membrane of primary hippocampal neurons, while its overexpression sensitized neurons to cell death. We also reported that c-subunit ring undergoes measurable conformational changes by enlarging its size upon activation of the channel (Alavian et al., 2014). Additionally, mutations that loosen the packing of the ring and expand its diameter led to significantly increased conductance compared to WT (Alavian et al., 2014). These mutations also sensitized cells to death. We also reported that CypD/Ca2+-mediated dissociation of ATP synthase F1 subunits from FO is required for unmasking of the c-subunit ring and initiation of its channel conductance (Alavian et al., 2014). Further evidence that F1 becomes uncoupled from FO and leads to uncoupling of ATP hydrolysis from H+ pumping in the presence of high Ca2+ was reported recently by another group (Nesci et al., 2017). Interestingly, age-dependent decrease in F1 subunit level compared to Fo, reducing the F1/FO ratio, have been reported in rat brain and heart mitochondria (Guerrieri et al., 1992a; Guerrieri et al., 1992b).
However, the important role of the c-subunit ring in mPTP formation has been questioned in a recent study (He et al., 2017). The ATP synthase c-subunit knockout HAP1-A12 cells were shown to have the same sensitivity of the mPT to calcium as wild-type (WT) cells in calcium retention capacity (CRC) experiments (He et al., 2017). This allowed the authors to rule out the possibility that ATP synthase c-subunit ring could form the channel of mPT (He et al., 2017). The CRC experiment, however, may only indicate a loss of membrane potential; it is not a direct measure of a channel activity, nor does it assess mPT-induced swelling (He et al., 2017; Mansson et al., 2007; Morota et al., 2013). Patch-clamp recordings, which are a direct measure of channel activity revealed that these same c-subunit knockout mitoplasts (mitochondria lacking the outer membrane) from HAP1-A12 cells lack the high conductance CsA-sensitive mPTP channel activity recorded in WT cells (Neginskaya et al., 2019), indicating the prominent role of c-subunit ring in forming the mPTP channel.
We have discussed all the recent concepts and controversies regarding ACLC, as well as proposed new models describing the physiological gating of ACLC and its pathological openings in our recent publication (Mnatsakanyan and Jonas, 2020) and, partially, in the later sections of this review.
B. Role of ATP synthase in mitochondrial homeostasis and cristae remodeling
More than a century ago it was described that organelles later understood to be mitochondria undergo shape changes rapidly and in response to signals (Chan, 2012). Mitochondrial structural alterations occur during cell stress and death but are also relevant to normal metabolism, cell growth, differentiation, aging and senescence. Mitochondrial quality control is maintained by the mitophagy of defective mitochondria and by the dynamic processes of mitochondrial fusion (the joining of two or more mitochondria) and fission (division), identifying these processes as important targets in neuroprotection.
Mitochondrial fission was found to be carried out by a family of proteins first identified for their role in endocytosis, the dynamin super-family (De Camilli et al., 1995). This family is comprised proteins that hydrolyze GTP to fuse or separate membranes. Drp1 was first described in yeast (DNM1, encoding the protein Dnm1p) (Gammie et al., 1995) and subsequently found to regulate mitochondrial shape and distribution (Bleazard et al., 1999; Otsuga et al., 1998) (Baker et al., 2019; Chan, 2006, 2012; Flippo and Strack, 2017; Smirnova et al., 2001). In neurons, Drp1 regulates Ca2+ dependent shape change and movement arrest necessary for synaptic signaling (Hollenbeck, 2005; Rintoul et al., 2003) and for cell death. It is required for normal brain and synapse development and for normal synaptic growth and plasticity (Ishihara et al., 2009). (Berman et al., 2009a; Guo et al., 2005; Li et al., 2008; Verstreken et al., 2005).
Mitochondrial fusion is brought about by several proteins, but key are the mitofusins (1 and 2) and optic atrophy protein 1 (OPA1) (Giacomello et al., 2020). Mitofusin 1 (Mfn1) interacts with OPA1 to bring about fusion on the outer mitochondrial membrane. Mfn2 also tethers mitochondria to other organelles (de Brito and Scorrano, 2008); mutations in Mfn cause Charcot-Marie-Tooth disease, a sensory motor neuropathy (Giacomello et al., 2020). OPA1 loss or mutation leads to genetic diseases including optic nerve atrophy (Giacomello et al., 2020). Upregulation of OPA1 in mice protects against muscular atrophy, ischemic damage in the heart and brain and hepatocyte death, since OPA-1 prevents the release of pro-apoptotic signals (Civiletto et al., 2015; Varanita et al., 2015).
Morphological changes in isolated mitochondria occur during metabolic changes in in vitro experiments. Healthy mitochondria switch between an “orthodox” configuration, in the absence of substrate or ADP, to a “condensed” configuration when performing electron transport and producing ATP (Hackenbrock, 1966). In the orthodox configuration, the matrix is electron light and the cristae appear small and disconnected from each other or the inner boundary membrane. The inner boundary membrane also appears tightly juxtaposed to the outer membrane (Fig. 1). In the condensed configuration, the matrix takes up less of the space of the outer limiting membrane even though the overall volume of the mitochondria is not changed. The matrix becomes exceedingly dense while the cristae spaces enlarge and appear interconnected and connected to an enlarged space between the outer membrane and the inner boundary membrane (Fig. 1). Often one or more enlarged cristae spaces appear within the center of the mitochondria. These changes occur within seconds of the supply of substrate and ADP to mitochondria (Hackenbrock, 1966) and are truly a remarkable example of extreme metabolic plasticity linked to structural changes of mitochondria (Fig. 1).
Figure 1.
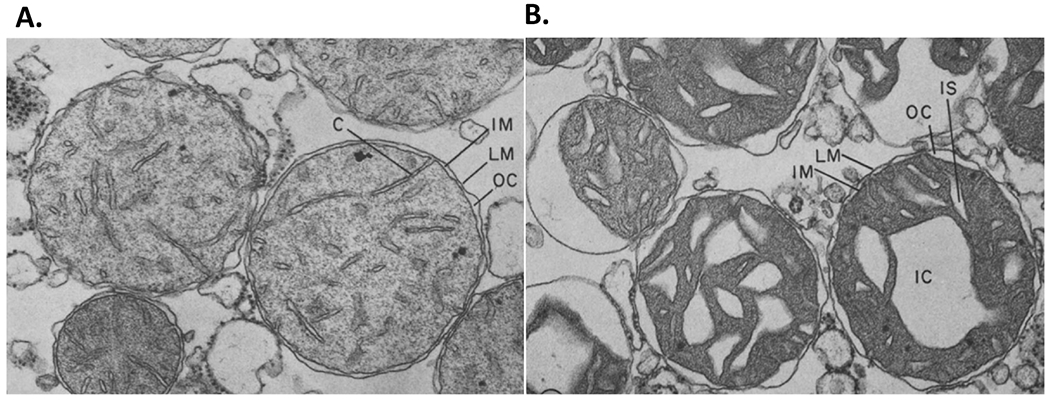
The interplay between metabolic plasticity and structural reorganization of mitochondria. Mitochondria switch between an A. orthodox configuration when they have no substrate or ADP, to a B. condensed folded arrangement of cristae while they perform electron transport and produce ATP. These changes occur within seconds of the supply of substrate and ADP to mitochondria (Hackenbrock, 1966). The orthodox configuration of cristae is characterized by the electron light matrix; the cristae appear small and disconnected from each other, and from the inner boundary membrane. In the condensed configuration, the matrix becomes exceedingly dense while the cristae spaces enlarge and appear interconnected and connected to an enlarged space between the outer membrane and the inner boundary membrane. Cristae, C, limiting membrane, LM; inner membrane, IM; outer compartment OC, intracristal space, IS. x 50,000. (Reproduced from Hackenbrock, 1966, by permission of Journal of Cell Biology).
The observed rapid structural changes linked to ATP production raise questions about the underlying molecular mechanisms. Cristae remodeling requires forces driven by the dynamin-like proteins and adapter proteins including OPA-1 and ATP synthase. OPA-1 is a master regulator of the mitochondrial contact site and cristae organizing system (MICOS), in that it participates in opening of the cristae junction (Glytsou et al.,2016) (Fig. 2). MICOS is composed of MIC10, MIC12, MIC19 MIC25, MIC26, MIC27 (also known as MOMA1 and APOOL) and MIC60 (also known as mitofilin); these are subdivided into two independent protein assemblies, MIC27–MIC10–MIC12 and MIC60–MIC19, linked via MIC19 (Friedman et al., 2015). MIC60 is an essential component of the complex; its depletion leads to loss of normal cristae morphology and disconnection of the cristae from the inner boundary membrane. MIC60 KO cells also have abnormalities in oxidative phosphorylation and mitochondrial RNA translation (John et al., 2005; Rabl et al., 2009; Zerbes et al., 2012), implicating MIC60 in organization of electron transport and ATP production. Overexpression of OPA-1 tightens the cristae junction and removal of OPA-1 dissolves it. The inner membrane-anchored form of OPA1 (L-OPA1, long-OPA1) can be cleaved by proteases, generating transmembrane-free, short form of OPA1 (S-OPA1) (Ishihara et al., 2006). Many enzymes participate in L-OPA-1 cleavage, including OMA-1, which may create an imbalance between long and short forms favoring mitochondrial fission and causing mitochondrial and cellular injury (Baker et al., 2014). The S-OPA1 was thought to be incompetent in mitochondrial fusion and energetic maintenance for a long time, however, a recent study suggested that S-OPA1 maintains energetic activity (Anand et al., 2014; Lee et al., 2017). Cells lacking OPA1 not only show highly fragmented mitochondria, but also widened and dysfunctional mitochondrial cristae, and an increased susceptibility to cell death (Frezza et al., 2006; Olichon et al., 2003). Mild overexpression of OPA1 enhances mitochondrial supercomplex formation and respiration efficiency by increasing cristae tightness (Cogliati et al., 2013). In contrast, OPA1-KO cells show no or very few cristae and are unable to support the maturation of the ATP synthase: a significant amount of F1 can be detected without assembly into the mature ATP synthase complex (Lee et al., 2017).
Figure 2.
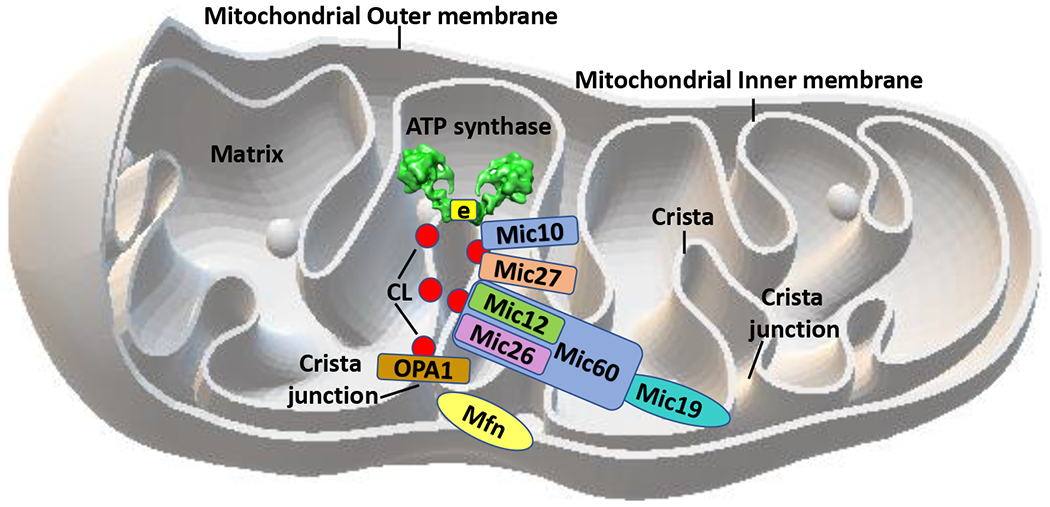
Cristae architecture and interplay between OPA1, MICOS-complex and the F1FO ATP synthase. Schematic representation of OPA1, MICOS subunits and ATP synthase in the mitochondrial inner membrane: Mic10 interacts with ATP synthase and its e-subunit, while Mic27 was shown to be an important bridge mediating the interaction between Mic10/ATP synthase and the remaining MICOS complex, Mic60-Mic19-Mic26-Mic12. The cristae shape is formed by OPA-1 and MICOS interactions at the cristae junction and ATP synthase structural organization at the cristae tip, so the complexes work together, at opposite ends of the cristae to form its normal architecture. MICOS and the ATP synthase together with cardiolipin (CL, red circles) and other membrane lipids support the normal formation and plasticity of the cristae structure. CL interacts with MICOS, OPA-1, ATP synthase and with the other members of the electron transport chain. The 3D model of mitochondria from Microsoft Paint 3D was used.
ATP synthase is crucial for cristae modeling (Paumard et al., 2002a; Paumard et al., 2002b; Quintana-Cabrera et al., 2018). Dimerization of the synthase and the further organization of dimers into rows forms the shape and optimal function of the cristae tips (Davies et al., 2012; Davies et al., 2011). Loss of ATP synthase dimers through depletion of membrane embedded FO subunits e and g, that are implicated in assisting in oligomerization, causes a flattening of the cristae tips opposite to the cristae junction with the inner boundary membrane, ballooning the cristae (Davies et al., 2012). OPA1 enhances the stability of ATP synthase oligomeric structures when the mitochondria are under stress (Quintana-Cabrera et al., 2018). Studies show that ATP synthase interacts directly with MICOS component MIC10 (Rampelt et al., 2017), implicating either a different location for at least part of the ATP synthase or high mobility of ATP synthase monomers in the cristae membrane. Chemical crosslink of Mic10 with ATP synthase subunit e and Mic10 to Mic27 in yeast mitochondria has been observed (Eydt et al., 2017) . Therefore, Mic27 was shown to be an important bridge mediating the interaction between Mic10/ATP synthase and the remaining MICOS complex, Mic60-Mic19-Mic26-Mic12 (Eydt et al., 2017) (Fig. 2).
MIC60/MIC19 form a different subcomplex from Mic27/Mic10/Mic12. This latter complex is selectively dependent on the presence of mtDNA, suggesting a critical role of respiratory complexes as a whole, but not any specific respiratory complex, in the assembly and/or stabilization of this MICOS subcomplex (Friedman et al., 2015). In yeast, Fcj1, the homologue of MIC60, assists in decreasing higher order oligomers of F1FO ATP synthase, allowing the formation of the appropriate dimerization, but subunits e and g of the ATP synthase are required for normal cristae tip formation (Rabl et al., 2009). Therefore, the cristae shape is likely formed by OPA-1 and MICOS interactions at the cristae junction and ATP synthase structural organization at the cristae tip or edge, so the complexes work together but at opposite ends of the cristae to form its normal architecture (Rabl et al., 2009). The data suggest that MICOS and the ATP synthase together with cardiolipin and other membrane lipids support the normal formation and plasticity of the cristae structure (Friedman et al., 2015; Wollweber et al., 2017) (Fig. 2).
Cardiolipin (CL) is a major lipid membrane component of the acute angle at the cristae junctions and at the ATP synthase edge. CL is necessary for interactions between inner and outer membranes. The mitochondrial fusion and fission, as well as protein transport are all dependent on CL. CL interacts with MICOS, OPA-1, ATP synthase and with the other members of the electron transport chain. CL is highly acidic. It has a glycerol head group that is esterified to two phosphatidylglyceride backbone fragments, combined with four acyl chains. This gives cardiolipin its dimeric shape, resulting in a conical structure that creates the acute bends (Paradies et al., 2014). CL is almost exclusively associated with membranes designed to produce ATP through electron transport activities (mitochondrial and bacterial membranes); it affects the efficiency and stability of respiratory enzyme supercomplexes (Paradies et al., 2014). Most of the major respiratory enzymes, metabolite carriers and ion transporters of the inner membrane are dependent on CL. CL also contributes to activity of the metabolite transporter proteins by providing a binding site to clamp the transporters in position. CL is critical for oligomerization of ATP synthase multimeric complexes either by direct interaction with ATP synthase or by producing the proper membrane curvature. CL and ATP synthase interaction may reduce the free energy of the extreme membrane curvature to stabilize high-curvature folds (Acehan et al., 2011). These properties affect cristae morphology and increase energy use and production efficiency. Cardiolipin-bound cytochrome c (cyt c) acts to perform H2O2-dependent CL peroxidation, essential for the release of cyt c during apoptosis. Alterations in CL structure or amount are associated with mitochondrial dysfunction in hypo and hyperthyroid states (Paradies and Ruggiero, 1990; Paradies et al., 1991), heart ischemia–reperfusion (Paradies et al., 1999), nonalcoholic fatty liver disease (Petrosillo et al., 2007), diabetes (Han et al., 2005), Barth syndrome (Schlame and Ren, 2006) and aging (Paradies et al., 2011; Semba et al., 2019).
CL likely assists in the formation of supercomplexes of the respiratory chain enzymes (Acin-Perez et al., 2008; Wittig et al., 2006). Evidence suggests that CL improves electron transfer efficiency by tightening the interaction of the supramolecular assemblies, including improving the efficiency of oxygen consumption and metabolite flux (Acin-Perez et al., 2008; Genova and Lenaz, 2013, 2014), enhancing efficiency of electron/proton exchange by as much as 35% (Claypool, 2009). These assemblies also reduce ROS production and increase the capacity for ATP production. CL has been suggested to use the phosphate head groups as a proton trap, restricting movement of pumped protons, the buffering of which may provide a structural basis for mitochondrial membrane potential (ΔΨ) by increasing the voltage component of the mitochondrial membrane potential. This also may provide an avenue for supply of protons to the ATP synthase (Haines and Dencher, 2002). CL could locally contribute to driving directional ATP export and ADP import through the ADP/ATP carrier (Klingenberg, 2009).
The assembly and disassembly of the complexes is dynamic and subject to change depending on physiological and pathological conditions (Beutner et al., 2014; Pfeiffer et al., 2003; Zhang et al., 2002). Therefore fusion, fission and structural changes to the cristae are all highly dependent on CL.
C. Role of mPTP in cristae remodeling during apoptosis
Understanding cristae remodeling during apoptosis has helped to gain an understanding of the normal remodeling that accompanies physiological alterations in metabolism. Mitochondrially mediated apoptosis is a well-orchestrated event beginning with translocation of tBID’s (truncated BID, BH-3 only Bcl-2 family member) to the mitochondrial outer membrane. tBID’s activate Bax or Bak oligomerization to release cyt c from mitochondria through a Bax/Bak dependent channel. Cyt c activates the apoptosome including caspase 9 and APAF-1, which then activate downstream caspases initiating the breakdown of cellular and nuclear components (Adams and Cory, 2007; Hardwick and Youle, 2009). The release of almost 100% of mitochondrial cyt c occurs during these events, raising the question of how the intracristal cyt c gets out without obvious matrix swelling, since only about 16% of cyt c is available for release from the intermembrane space (Scorrano et al., 2002). Later it was shown that a reorganization of the cristae occurs to release the majority of cyt c (Giacomello et al., 2020; Scorrano et al., 2002). The original studies showed that tBID-induced apoptotic signaling induced partial mitochondrial membrane depolarization and release of calcein (a matrix-entrapped fluorescent dye). Both processes were inhibited by CsA but did not cause appreciable swelling, indicating reversible activation of the mPTP during apoptotic cyt c release. CsA in the presence of tBID still resulted in 17% cyt c release, suggesting that CsA could not inhibit the release of cyt c from the intermembrane space but could block its release from the intracristal compartments. Therefore, the major portion of cyt c is contained in the intracristal space and this cyt c is not available for release unless mPTP opens. After loosening its anchor from the intracristal location, another action is required to release cyt c from the outer membrane and this proved to be Bax/Bak oligomerization since the CsA sensitive membrane depolarization and calcein release still occurred in Bax/Bak KO cells but cyt c release was abrogated (Scorrano et al., 2002). These studies confirmed that the release of cyt c during apoptosis is made up of two events: A cyt c mobilization event dependent on tBID and mPTP and a separate releasing event dependent on tBID activation of Bax/Bak in the outer membrane, since Bax/Bak deficient mitochondria were able to redistribute, but not release, cyt c.
The cyt c releasing event involves a major reorganization of the cristae, as noted in electron micrographs (Mannella, 2006). Especially important is that the connection to the intermembrane space, the crista junction, widens during apoptosis to about 5x its normal width, indicating that this is the size most likely needed for cyt c release (Mannella, 2006).
Studies of the ER-associated BH3 only protein BIK (BCL-2 interacting killer) indicated that BIK, similarly to tBID, activates cristae remodeling. These rapid changes in cristae morphology are dependent on BIK’s BH3 domain, Ca2+ uptake by mitochondria, the mPTP, and functional Drp1 (Germain et al., 2005). The order of events is that calcium enters the mitochondria from the ER through the MCU, it activates the mPTP and this induces cristae membrane remodeling at the same time that Ca2+ localizes Drp1 to the outer membrane and activates calcineurin to activate Drp1 GTPase activity (Germain et al., 2005). The mechanism of how calcium-dependent Drp1 activity causes cristae membrane remodeling is not understood but may involve Drp1 interactions with inner membrane proteins at the adjacent cristae junction.
During cell death Drp1 dependent remodeling requires high calcium. In contrast, the cristae remodeling that occurs during normal ATP production does not require calcium but only substrate and ADP (Hackenbrock, 1966). New studies using super resolution imaging of fluorescently labeled cristae indicate that rapid physiological changes in cristae morphology occur almost constantly even in resting cells (Kondadi et al., 2020). Therefore, rapid changes in structure brought on by normal variation in daily physiological metabolic events are the norm. The key feature observed in the early studies was cristae enlargement, albeit in the presence of abnormally high ADP/ATP ratio, probably causing a very rapid shift to ADP dependent oxidative phosphorylation (state III) (Hackenbrock, 1966). By analogy with apoptosis and cyt c release, ATP and other metabolite release through the cristae junction would be the most likely reason for the drastic remodeling of the cristae and matrix compartment during normal state III respiration. The large change in volume of the intracristal space occurring during these events is likely to be brought on by osmotic changes, particularly K+ influx into the crista compartment (Kaasik et al., 2007). The ions (and water) may derive from two sources: The first could be an enlarged connection formed at the crista junction with the inner boundary membrane. The second is uptake of K+ from the matrix, since the matrix rapidly transforms into an extremely electron dense compartment and shares the same volume, fixed by the outer membrane, with the enlarged cristae during this time. MICOS and OPA-1 must participate in the rapid changes in fusion with the inner boundary membrane but the mechanisms of K+ flux from the matrix at this time are completely unknown. These could involve the mPTP operating in its physiological mode, particularly as it was observed that the tBID induced, CsA sensitive changes that cause cyt c release appear not to require changes in total mitochondrial volume (Scorrano et al., 2002).
ATP synthase as a key target in mitochondria-mediated neurodegeneration: Reduced F1/FO ratio in neurodegenerative disease
Recent studies indicate that mitochondrial changes are a driving force, rather than a consequence, of the aging process and neurodegeneration. Abnormal accumulation of amyloid-β protein (Aβ) was found not only in the mitochondria of transgenic mouse models of AD that overexpress Aβ (Dragicevic et al., 2010; Manczak et al., 2006) but also was reported to interact directly with ATP synthase subunits and cause detrimental changes to its structure and function (Beck et al., 2016). Alzheimer’s Disease (AD) is one of the most commonly occurring neurodegenerative diseases. AD pathology causes synaptic dysfunction followed by the death of brain cells, leading to significant tissue shrinkage affecting memory, attention, speech and behavior (Cadonic et al., 2015; Selkoe, 2011). High levels of reactive oxygen species produced in mitochondria cause oxidative damage and redox imbalance, impairing mitochondrial function (Beal, 2005; Trushina and McMurray, 2007). Such changes were observed in human pathological samples of early AD (Cenini et al., 2019; Du et al., 2012; Du and Yan, 2010a, b; Nunomura et al., 2001). Interestingly, the α subunit of the mitochondrial ATP-synthase was reported as the most common lipoxidized protein in the entorhinal cortex of all AD cases at stages I/II of the disease (Terni et al., 2010). Additionally, it was reported that ATP synthase α subunit accumulates in the cytosol either alone or associated with tau aggregates of neurofibrillary tangles in degenerating neurons of AD (Sergeant et al., 2003). In the brains of AD individuals and in an AD mouse model, a decrease in ATP synthase OSCP subunit level was observed (Beck et al., 2016) and OSCP was shown to be a binding site for the aggregated protein Aβ (Beck et al., 2016). Decreases in OSCP levels were pronounced especially in neuronal mitochondria (Beck et al., 2016). Furthermore, increased F1 dissociation from FO and significantly high free F1 levels were reported in neuronal mitochondria in the AD mouse (Beck et al., 2016). The loss of OSCP and reduced F1 levels led to reduced ATP production, elevated oxidative stress and activated mPT and these abnormalities were alleviated in the mouse by OSCP overexpression (Beck et al., 2016).
Similarly, we have now found that loss of the ATP synthase F1 β subunit and reduction in F1/FO ratio occurs in a DJ-1 deficient, Parkinson’s Disease (PD) mouse model (Chen et al., 2019). DJ-1 is a molecular chaperone and peptidase C56 family protein with known and uncharacterized cellular functions (Bonifati et al., 2003). Familial PD protein DJ-1 mutations are linked to early onset PD. DJ-1 mutant animals show increased sensitivity to neuronal toxins, and in different species DJ-1 is required for normal life span, motor function, and neuronal resistance to oxidative damage (Biosa et al., 2017; Flao et al., 2010; Malgieri and Eliezer, 2008). DJ-1 translocates to mitochondria from the cytosol in response to mitochondrial stress (Canet-Aviles et al., 2004; Junn et al., 2009; Weinert et al., 2019) and defects in DJ-1 alter mitochondrial morphology and function (Larsen et al., 2011). We have found that DJ-1 binds directly to the F1FO ATP synthase β subunit and this interaction decreases mitochondrial uncoupling and enhances ATP production efficiency (Chen et al., 2019). In contrast, DJ-1 deficient mitochondria demonstrate a leaky mitochondrial inner membrane and the leak is produced by the ATP synthase (Chen et al., 2019; Hao et al., 2010). The abnormalities of the ATP synthase in DJ-1−/− animals are associated with reduced enzymatic function, reduced ATP levels and impaired neurite extension in isolated dopaminergic neurons, suggesting that normal ATP synthase function is critical for dopaminergic neuron growth (Chen et al., 2019). Levels of the ATP synthase β subunit are low in aged DJ-1−/− mouse brain and in patient-derived cell lines even though levels of the c-subunit are normal, suggesting that mutant DJ-1-induced degenerative disease is associated with a reduction in F1/FO ratio with resultant ACLC formation.
α-synuclein is the most abundant protein found in Lewy bodies, the pathological hallmark of Parkinson’s disease. Under physiological conditions monomeric α-synuclein interacts with ATP synthase α subunit and enhances ATP synthase activity and mitochondrial function (Ludtmann et al., 2016). However, α-synuclein aggregation generates β sheet-rich oligomers that localize to the mitochondria in close proximity to several mitochondrial proteins including complex I and ATP synthase. These oligomers interact with the ATP synthase β subunit, induce its selective oxidation and mitochondrial lipid peroxidation, and increase the probability of mPTP opening, triggering mitochondrial swelling and cell death (Ludtmann et al., 2018). Therefore, transition of α-synuclein from its monomeric to oligomeric state switches its role from physiological to pathological leading to mitochondrial dysfunction in Parkinson’s disease through a direct interaction of pathological α-synuclein oligomers with the ATP synthase. This highlights the role of ATP synthase in PD pathology.
ATP synthase as a key target in mitochondria-mediated neuroprotection
A. ATP synthase leak channel as a regulator of synaptic plasticity and metabolic efficiency in neurons
We have reported that overexpression of the anti-apoptotic protein Bcl-xL in neurons markedly increases ATP production, while lowering oxygen consumption (Alavian et al., 2011). This contradiction was resolved when we found that Bcl-xL is bound directly to the ATP synthase β subunit (Alavian et al., 2011; Chen et al., 2011), where it increases ATP synthase activity rate and reduces inner mitochondrial membrane leak, enhancing the coupling and efficiency of oxidative phosphorylation. Bcl-xL inhibitors ABT-737 (Alavian et al., 2011) and WEHI-539, induce ATP synthase c-subunit related inner membrane leak formation by inhibiting the binding of Bcl-xL to the ATP synthase β subunit. Bcl-xL depletion causes energy loss and instability of the mitochondrial membrane potential (Chen et al., 2011). Bcl-xL KO causes embryonic lethality just before E12.5 (Motoyama et al., 1995), but conditional KO mice lacking Bcl-xL in neuronal progenitors survive, albeit with a highly malformed cortex and severe loss of neurite growth and synaptogenesis (Chen et al., 2011).
By increasing the metabolic efficiency of neurons Bcl-xL contributes to enhancing synaptic growth and plasticity through regulating mitochondrial biogenesis. Bcl-xL increases the numbers of presynaptic vesicle clusters (Li et al., 2008), synapses and mitochondria. Bcl-xL coordinates with the mitochondrial fission protein, Drp-1 and the biogenesis factor PGC-1 alpha to achieve mitochondrial positioning, followed by mitochondrial fission to supply the enlarged synapses with newly minted mitochondria (Berman et al., 2009a, b). Bcl-xL and Drp-1 work together directly on synaptic vesicles to bring about activity dependent vesicle pool refilling within the first few minutes after high frequency action potential firing (Li et al., 2013). Despite its important anti-apoptotic functions, Bcl-xL can be cleaved to ΔN-Bcl-xL, an N-terminal, pro-apoptotic product during ischemia or excitotoxicity. The role of Bcl-xL in modulating the ACLC activity during excitotoxic conditions will be discussed in the next section.
B. Modulation of ATP synthase leak channel activity in ischemia-triggered excitotoxic conditions
Oxygen and nutrient deprivation during acute stroke causes energy crisis in neurons and cell death by necrotic mechanisms (Moskowitz et al., 2010). Depolarization of neuronal plasma membranes and failure of active transporters result in accumulation of high glutamate levels in synaptic terminals (Wang et al., 2003), creating glutamate excitotoxicity, an important cause of cell death during ischemia. Subsequent transport of calcium into the intracellular compartment and the mitochondria, results in opening of the mPTP, mitochondrial swelling, rupture of the mitochondrial outer membrane and oxidant stress (Arundine and Tymianski, 2004; Nicholls, 2004, 2008). The opening of mPTP is less dramatic and possibly reversible in the area of brain tissue that has partial blood supply, the penumbra (Astrup et al., 1981; Moskowitz et al., 2010), while it can be acute in other cases. It is during this period in the peri-infarct territory that programmed cell death mechanisms are activated. These mechanisms follow a path determined by genetically encoded molecular players carrying out predetermined roles through protein/protein and protein/membrane interactions (Adams and Cory, 2007; Broughton et al., 2009; Park and Jonas, 2017).
The Bcl-2 family proteins are critical in regulating neuronal death in the at-risk penumbra. Pro-death Bcl-2 family members, Bax and Bak, produce outer mitochondrial membrane permeabilization, leading to cyt c release and activation of downstream signaling pathways, including activation of enzymes known as cell-destructive caspases (Adams and Cory, 2007; Hardwick et al., 2012; Hardwick and Youle, 2009). The death-inducing functions of the pro-apoptotic versions of Bcl-2 family proteins are opposed by those of the anti-death proteins such as Bcl-2, Bcl-xL, Mcl-1 and others. Small molecule inhibitors of the Bcl-2 proteins such as ABT-737 and others were developed to bind into a groove in the BH1-3 domains and prevent the interaction of the anti-apoptotic proteins with the pro-apoptotic proteins, releasing Bax and Bak to permeabilize mitochondrial membranes. These powerful and selective reagents are useful for their chemotherapeutic activity (Chipuk et al., 2008; Oltersdorf et al., 2005)
The BH4-domain-containing anti-apoptotic proteins such as Bcl-xL carry out important survival functions in the brain, but during ischemia or excitotoxicity, Bcl-xL is cleaved to ΔN-Bcl-xL, an N-terminal, cleavage product (Fujita et al., 1998; Miyawaki et al., 2008; Ofengeim et al., 2012) and it activates large conductance mitochondrial channel activity (Hickman et al., 2008; Jonas et al., 2004; Ofengeim et al., 2012) similar to that of oligomerized Bax (Jonas et al., 2005a). Blocking formation of ΔN-Bcl-xL or the mitochondrial channel activity of ΔN-Bcl-xL with the small molecule inhibitor ABT-737 rescues neurons from ischemic death (Hickman et al., 2008; Ofengeim et al., 2012; Park and Jonas, 2017; Park et al., 2017). ABT-737 also blocks synaptic depression produced by hypoxia or by injection of recombinant ΔN-Bcl-xL protein into the presynaptic terminal (Hickman et al., 2008; Jonas et al., 2004; Jonas et al., 2005b). A mouse expressing a cleavage resistant form of Bcl-xL is protected from ischemic brain damage (Ofengeim et al., 2012).
In addition to its known outer mitochondrial membrane functions (to release cyt c and activate apoptosis), (Ofengeim et al., 2012) ΔN-Bcl-xL, similarly to full-length Bcl-xL, functions at the mitochondrial inner membrane where it exacerbates the effects of glutamate excitotoxicity on mitochondrial depolarization, cyt c release, caspase activation, loss of ATP production and death (Park et al., 2017). These effects are prevented by low dose ABT-737, which preferentially binds to ΔN-Bcl-xL overfull length Bcl-xL, the latter of which requires higher concentrations to cause mitochondrial dysfunction in neurons (Park et al., 2017). ΔN-Bcl-xL depolarizes the mitochondrial inner membrane most likely by opening the ACLC. It is localized to the mitochondrial inner membrane by immunocytochemistry and by membrane fractionation studies. In neurons challenged with excitotoxicity, ΔN-Bcl-xL overexpression causes cell death prevented by the relative depletion of ATP synthase c-subunit with shRNA (Park et al., 2017). Neuronal dendritic and axonal processes are highly sensitive to metabolic perturbations. Depletion of c-subunit prevents ΔN-Bcl-xL-induced neurite dysfunction and cell death by modulating ACLC activity.
C. ATP synthase leak channel as a target in ischemia/reperfusion injury
One of the leading causes of death in the United States is ischemia/reperfusion (I/R) resulting from stroke or myocardial infarction (Benjamin et al., 2018). During I/R injury tissue damage and cell death occur first in response to the initial ischemic insult and then later by the subsequent reperfusion. Severe reduction in ATP levels and intracellular pH during prolonged ischemia are associated with hypoxia-induced inhibition of oxidative phosphorylation and lactate accumulation due to enhanced glycolysis. The ATP- dependent ion transport mechanisms (Na+/K+ ATPase etc.) become dysfunctional under these conditions, causing neurons to depolarize and release excessive amount of glutamate into the synaptic cleft, initiating excitotoxic injury (Chomova and Zitnanova, 2016; Kalogeris et al., 2012; Mehta et al., 2007; Nakka et al., 2008). The glutamate overflow activates post-synaptic NMDA and AMPA receptors leading to intracellular and mitochondrial calcium overload and mitochondrial swelling. The conditions after reperfusion, including elevated levels of reactive oxygen species and phosphate and increased intracellular pH orchestrate the opening of the mPTP, which is a critical factor for inducing both apoptotic and necrotic cell death during I/R injury (Halestrap et al., 2004; Kalogeris et al., 2012) (Fig. 3). Increasing evidence suggests that mPTP is a promising target for neuro and cardioprotection during I/R injury.
Figure 3.
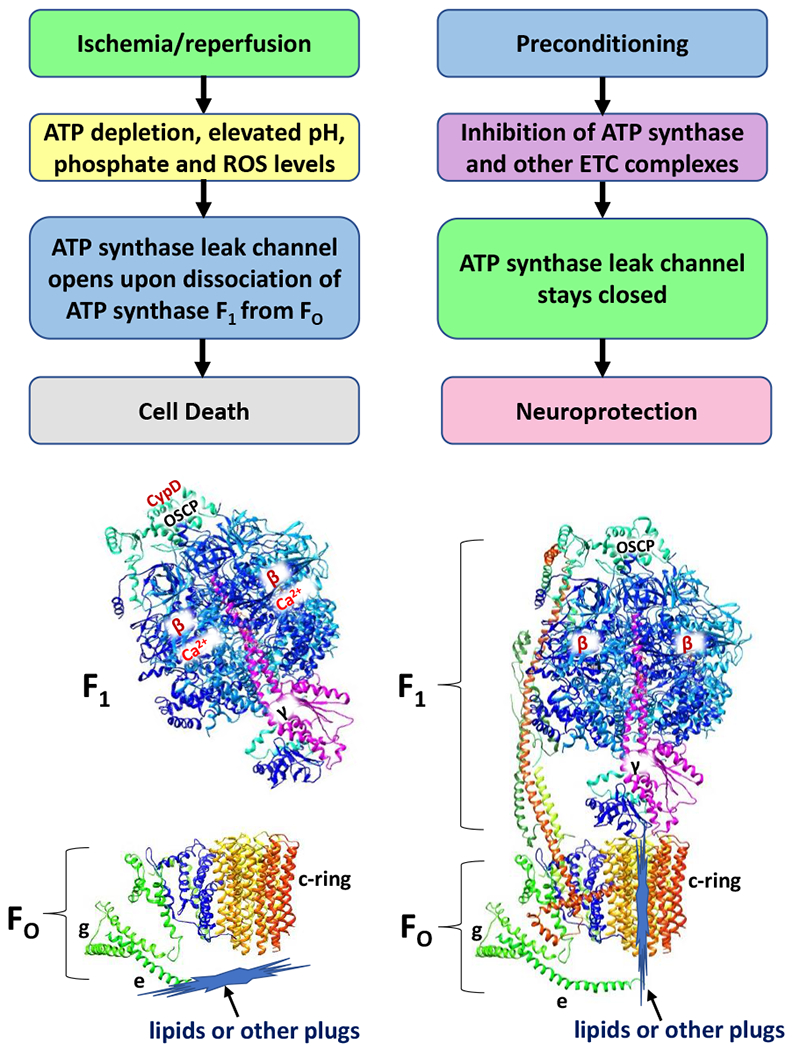
ATP depletion, elevated pH, phosphate, and ROS levels after ischemia/reperfusion injury trigger the opening of the mPTP and induce cell death. The opening of ACLC occurs upon non-reversible dissociation of F1 subcomplex from FO (Alavian et al., 2014), due to the binding of Ca2+ to ATP synthase β subunits, CypD to OSCP and upon removal of lipid or other plugs from the c-subunit lumen. The ischemic preconditioning, stimulated by the inhibitors of ATP synthase and of other mitochondrial electron transport chain complexes prevent the opening of ACLC and thus increase the brain’s tolerance against ischemic cell death, highlighting the role of mPTP as a promising target for neuro and cardioprotection during I/R injury. ATP synthase subunits are drawn as ribbon representations (modified PDB ID code: 6J5I) (Gu et al., 2019).
Ischemic preconditioning is found to be a promising therapy for increasing the brain’s tolerance against ischemic cell death. A short, non-damaging ischemic episode, preceding sustained ischemia, stimulated by different therapeutic conditions or pharmacological agents proves useful in different animal models and in in vitro cell cultures (Kitagawa et al., 1991; Murry et al., 1986; Schurr et al., 1986). However, the underlying molecular mechanisms mediating ischemic preconditioning protection are not fully understood.
One of the protective mechanisms was reported to be related to ATP synthase inhibition during preconditioning (Vuorinen et al., 1995). A brief global ischemic episode used for preconditioning of the rat heart myocardium caused ATP synthase inhibition that persisted throughout the initial reperfusion period, resulting in a better final recovery of the preconditioned hearts (Vuorinen et al., 1995). Under normoxic conditions mitochondrial ATP synthase drives ATP synthesis. However, as the H+ gradient drops under hypoxic conditions the ATP synthase begins to run in reverse, contributing to mitochondrial membrane potential generation by ATP hydrolysis; this process depletes cellular ATP content and activates mPTP opening (Galkin et al., 2009). The intrinsic inhibitor protein IF1 binds to ATP synthase at low pH (under ischemic conditions) and prevents the reverse reaction of ATP hydrolysis and the ATP wastage, thus IF1 is reported to be protective against ischemic injury (Campanella et al., 2008). Meanwhile, increased pH level (under reperfusion conditions) impairs IF1 interaction with ATP synthase allowing ATP hydrolysis to occur freely (Cabezon et al., 2000a; Cabezon et al., 2000b; Campanella et al., 2008). Interestingly, lack of IF1 induces a non-thyroidal hypermetabolic syndrome in humans known as Luft’s disease (DiMauro et al., 1976; Yamada and Huzel, 1992). It is a rare mitochondrial disease, characterized by altered mitochondrial morphology (abnormally large mitochondria with packed cristae) and uncoupled mitochondrial respiration with high basal ATP hydrolysis activity. Furthermore, the rate of energy-dependent calcium uptake by isolated mitochondria is normal, but the amount and retention of accumulated calcium is much reduced suggesting that mPTP opening participates in the disease phenotype. These findings highlight the key role of ATP synthase and IF1 in mitochondrial physiology and in cell survival during ischemia.
Inhibitors of ATP synthase and of other mitochondrial electron transport chain complexes have been reported to cause preconditioning-like effects (Arbelaez-Quintero and Palacios, 2017; Della-Morte et al., 2009; Vuorinen et al., 1995) (Fig. 3). Oligomycin, which inhibits ATP synthase through its Fo domain, preserves mitochondrial ATP content and protects against or postpones cell death during ischemia. Although a 5-minute preischemic infusion of oligomycin caused initial decline in the cellular energy state due to inhibition of ATP synthesis, it significantly reduced further decreases in ATP levels during the subsequent ischemic period (Vuorinen et al., 1995). Consequently, preservation of mitochondrial ATP allows neurons to survive the long-lasting hypoxia and energy deprivation that occur during an ischemic episode. Although mPTP channel activity was not investigated in this study, we suggest that the preconditioning period of ischemia, as well as oligomycin-induced ATP synthase inhibition can lead to the closure of the mPTP channel to increase the bioenergetic efficiency of neurons (Fig. 3). This is in agreement with our recent observation of oligomycin inhibition/closure of the ACLC activity in in vitro patch-clamp recordings of porcine ATP synthase reconstituted liposomes (Mnatsakanyan et al., 2019).
In recent studies, mild hypoxic conditions in general were reported to protect cells and animal models from defects in mitochondrial electron transport chain (Ferrari et al., 2017; Jain et al., 2019; Jain et al., 2016). In these studies, oxygen deprivation was shown to activate the hypoxic response, an evolutionarily conserved adaptive mechanism allowing mammals to survive under limited oxygen conditions by switching their metabolism from oxidative phosphorylation to glycolysis (Jain et al., 2016). Neurons prefer to use mitochondrial oxidative phosphorylation over glycolysis to meet the high energy needs of the brain. Due to such reliance on oxidative phosphorylation mitochondria are critical to neuronal activities and mitochondrial dysfunction becomes a main cause of neurodegeneration. A genome-wide Cas9-mediated screening of ~18,000 genes for identification of protective factors during mitochondrial disease conditions revealed the importance of the Von Hippel-Lindau (VHL) factor, a key regulator of the hypoxic response pathway, as the most effective genetic suppressor. Activation of the hypoxia response by either genetic or pharmacological interventions protects cultured cells or zebrafish models of mitochondrial dysfunction (Jain et al., 2016). Additionally, chronic hypoxia (11% O2) improves the disease phenotype in the Ndufs4 KO mouse model of the pediatric mitochondrial disease Leigh syndrome, the most common mitochondrial disease in children (Jain et al., 2016). The Ndufs4 gene encodes subunit 4 of complex I, the deficiency of which is the most frequent biochemical cause of Leigh syndrome. Chronic hypoxia is also reported not only to prevent but to reverse brain lesions in mice with advanced neuropathology in a mouse model of Leigh syndrome (Ferrari et al., 2017; Jain et al., 2019).
Another compound, which acts as an electron transport chain inhibitor with beneficial effects in I/R injury and in Alzheimer’s disease is metformin, a common drug used to treat type 2 diabetes (Arbelaez-Quintero and Palacios, 2017; Yin et al., 2011). Metformin is a mild inhibitor of complex I. It inhibits respiration leading to activation of AMP-activated protein kinase (AMPK) and resulting in beneficial metabolic effects (Adak et al., 2018). Furthermore, metformin and other biguanides may inhibit ATP synthase activity (Bridges et al., 2014). However, their direct interaction sites with ATP synthase and the inhibitory mechanisms are currently unknown.
One of the most promising pharmacological agents used in preconditioning is resveratrol, a natural polyphenol and antibiotic found in grapes, berries and their products, such as wines (Della-Morte et al., 2009; Khoury et al., 2016; Khoury et al., 2019; Raval et al., 2006; Salehi et al., 2018). Resveratrol was shown to have neuroprotective effects in cerebral ischemia and other models of neurological disease, such as Parkinson’s, Alzheimer’s and Huntington’s (Kursvietiene et al., 2016; Salehi et al., 2018). The neuroprotective effect of resveratrol was first reported to be through the activation of Sirtuin 1 (SIRT1), which is an NAD-dependent deacetylase that removes acetyl groups from various proteins contributing to cellular regulation (Ghosh, 2008; Howitz et al., 2003; Lagouge et al., 2006). SIRT1 activates the PGC1-alpha and ERR-alpha transcription factors, which are important metabolic regulators playing a central role in transcriptional control of mitochondrial biogenesis and respiratory function (Lagouge et al., 2006; Scarpulla, 2011). SIRT1 also inhibits NF-κB-regulated inflammation related gene expression and thus extends lifespan and delays aging in mice (Lawrence, 2009). Other studies suggest that the effect of resveratrol on SIRT1 is indirect in that it may be due to the inhibition of mitochondrial respiration, which increases the AMP/ATP ratio and thus activates AMPK, the upstream activator of SIRT1 (Kulkarni and Canto, 2015).
Resveratrol preconditioning was reported to significantly reduce uncoupling protein 2 (UCP2) levels in rat brain by 35% and increase the ATP synthesis ratio per oxygen molecule (ADP/O ratio) in hippocampal mitochondria suggesting that resveratrol preconditioning increases overall metabolic and bioenergetic efficiency of mitochondria (Della-Morte et al., 2009).
In further studies, a long window of cerebral ischemic tolerance that lasts for two-weeks, mediated by resveratrol preconditioning, was reported in mice (Khoury et al., 2019). RNA-seq experiments of the preconditioned mouse cortex revealed downregulation of more than 100 genes involved in transcription, synaptic signaling, and neurotransmission. Meanwhile, the overexpression of genes involved in pyruvate uptake, TCA cycle, and oxidative phosphorylation was observed, along with increased ATP levels and mitochondrial abundance. However, both basal and maximal OCR were significantly reduced, suggesting reduced inner membrane leak and more efficient respiration (Khoury et al., 2019). This could be explained partially by the overexpression of the anti-apoptotic protein Bcl-2 and decreased expression of the pro-apoptotic proteins Bax, caspase-3 and cleaved caspase-3 observed during resveratrol preconditioning, overall resulting in the attenuation of ischemia-induced cell death (Liu et al., 2016). We have discussed the role of Bcl-xL in closing the ACLC and increasing the bioenergetic efficiency of neurons in the previous sections. However, other regulators of the ACLC in resveratrol preconditioning are yet to be investigated.
Resveratrol and other related polyphenols, quercetin and piceatannol bind directly to mitochondrial ATP synthase (Gledhill et al., 2007). Resveratrol interacts with specific residues of α, β and γ subunits by binding to the hydrophobic pocket between the C-terminal tip of the γ subunit and the βTP subunit (Gledhill et al., 2007) (Fig. 4). Resveratrol binding prevents both ATP synthesis and hydrolysis activities by blocking the rotation of γ subunit in mammalian ATP synthase (Gledhill et al., 2007). Investigation of resveratrol effects on regulation of ACLC activity will help us understand if neuroprotective effects of resveratrol can also be explained by its direct interaction with the mitochondrial ATP synthase.
Figure 4.
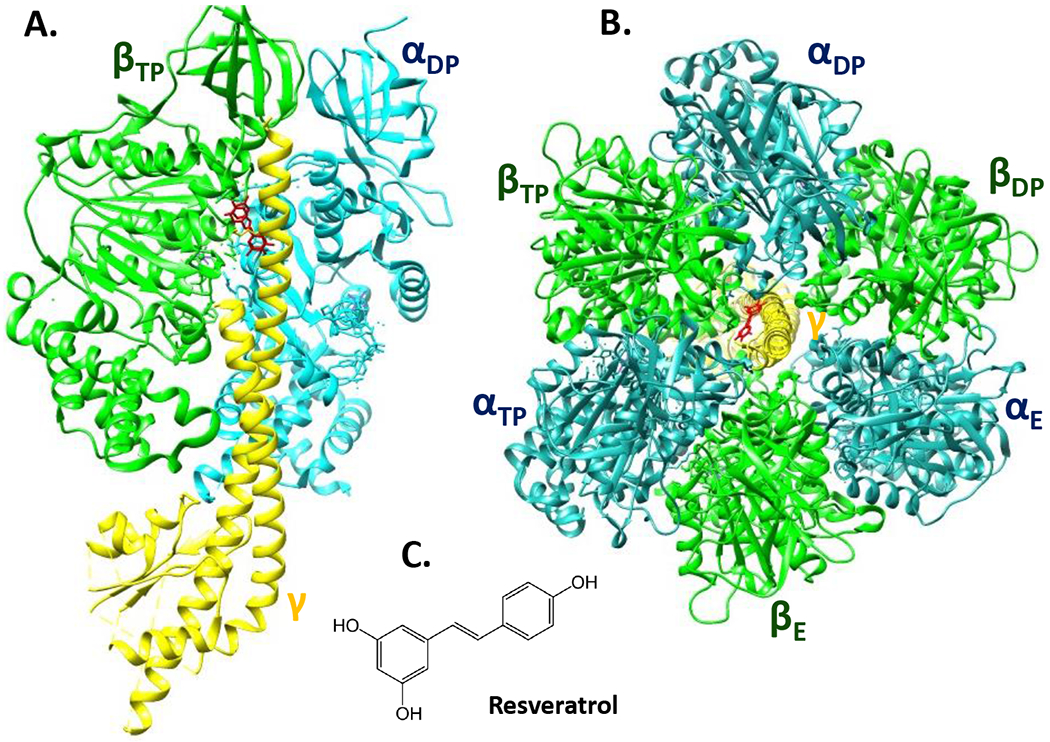
The structure of bovine heart mitochondrial F1-ATPase in 2.3-Å resolution showing the site of binding of resveratrol. PDB ID: 2JIZ (Gledhill et al., 2007). The α, β, γ subunits are blue, green, yellow respectively, and resveratrol is red. A. Side view of F1-ATPase with resveratrol bound in a pocket between βTP and γ subunits. For clarity, the other two α and β subunits were removed. B. Top view of F1-ATPase showing the binding site of resveratrol between the βTP and γ subunits. C. Chemical structure of resveratrol.
In contrast to its neuroprotective effects, resveratrol displays anti-cancer properties by inducing cell growth arrest or triggering apoptotic cell death signals in tumors. In contrast to neurons, cancer cells have different energetic needs; they exhibit an altered metabolism by switching from oxidative phosphorylation to aerobic glycolysis, known as the Warburg effect. Resveratrol exposure reverses Warburg metabolic phenotype in colon cancer cells, by causing a metabolic shift from glycolysis to oxidative phosphorylation; resveratrol slows down cell proliferation without inducing massive cell death in this study (Saunier et al., 2017). Taken together these studies suggest that resveratrol-mediated mitochondrial changes, its neuroprotective effects in ischemia and neurodegeneration and apoptotic effects in cancer cells depend on the metabolic phenotype of the cell type and more specifically on its bioenergetic profile.
Concluding remarks
F1Fo ATP synthase has always been considered as a crucial component of the mitochondrial electron transport chain since it synthesizes ATP during oxidative phosphorylation to meet cellular energy needs. The importance of F1Fo ATP synthase in cell physiology was further highlighted by the recent findings that it houses the cell death channel of mitochondrial permeability transition. In this review we have discussed the new role of ATP synthase beyond oxidative phosphorylation. We have described the physiological function of ACLC in enhancing the metabolic efficiency and synaptic plasticity of neurons, as well as its function in mitochondrial remodeling and shaping cristae morphology. We have highlighted the function of ATP synthase as a unique regulator of cell life and death in ischemia-reperfusion injury and in neurodegenerative disease, such as Alzheimer’s and Parkinson’s. We suggested that the ATP depletion, elevated pH, phosphate and ROS levels after ischemia/reperfusion injury trigger the opening of ACLC upon non-reversible dissociation of F1 subcomplex from FO, and upon removal of lipid or other plugs from the c-subunit lumen (Fig. 3). We described that the ischemic preconditioning, stimulated by the inhibitors of ATP synthase and of other mitochondrial electron transport chain complexes prevent the opening of ACLC and thus increase the brain’s tolerance against ischemic cell death, highlighting the role of ATP synthase as a promising target for neuro and cardioprotection during I/R injury.
Future structural and functional studies of ACLC will make it an irreplaceable target for the treatment of degenerative and metabolic diseases.
Acknowledgments
Work was supported by NIH Grants NS045876, NS112706, NS081746 (to E.A.J.), NIA Grant K01AG054734 (to N.M.). Correspondence should be addressed to Nelli Mnatsakanyan (nelli.mnatsakanyan@yale.edu)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- Abrahams JP, Leslie AG, Lutter R, Walker JE, 1994. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628. [DOI] [PubMed] [Google Scholar]
- Acehan D, Malhotra A, Xu Y, Ren M, Stokes DL, Schlame M, 2011. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J 100, 2184–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA, 2008. Respiratory active mitochondrial supercomplexes. Mol Cell 32, 529–539. [DOI] [PubMed] [Google Scholar]
- Adak T, Samadi A, Unal AZ, Sabuncuoglu S, 2018. A reappraisal on metformin. Regul Toxicol Pharmacol 92, 324–332. [DOI] [PubMed] [Google Scholar]
- Adams JM, Cory S, 2007. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr., Jonas EA, 2014. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 111, 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA, 2011. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol 13, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Klusch N, Mills DJ, Vonck J, Kuhlbrandt W, Davies KM, 2015. Horizontal membrane-intrinsic alpha-helices in the stator a-subunit of an F-type ATP synthase. Nature 521, 237–240. [DOI] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T, 2014. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbelaez-Quintero I, Palacios M, 2017. To Use or Not to Use Metformin in Cerebral Ischemia: A Review of the Application of Metformin in Stroke Rodents. Stroke Res Treat 2017, 9756429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M, 2004. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cellular & Molecular Life Sciences 61, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup J, Siesjo BK, Symon L, 1981. Thresholds in cerebral ischemia - the ischemic penumbra. Stroke 12, 723–725. [DOI] [PubMed] [Google Scholar]
- Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J, Saris NE, 2014. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 55, 69–77. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death.[see comment]. Nature 434, 658–662. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD, 2007. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T, 2014. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J 33, 578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N, Patel J, Khacho M, 2019. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 49, 259–268. [DOI] [PubMed] [Google Scholar]
- Beal MF, 2005. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58, 495–505. [DOI] [PubMed] [Google Scholar]
- Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H, 2016. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun 7, 11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Council on, E., Prevention Statistics, C., Stroke Statistics, S., 2018. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 137, e67–e492. [DOI] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM, 2009a. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. Journal of Cell Biology 184, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM, 2009b. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol 184, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, 1992. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem 267, 8834–8839. [PubMed] [Google Scholar]
- Bernardi P, 1996. The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim Biophys Acta 1275, 5–9. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zoratti M, 1992. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem 267, 2934–2939. [PubMed] [Google Scholar]
- Beutner G, Eliseev RA, Porter GA Jr., 2014. Initiation of electron transport chain activity in the embryonic heart coincides with the activation of mitochondrial complex 1 and the formation of supercomplexes. PLoS One 9, e113330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biosa A, Sandrelli F, Beltramini M, Greggio E, Bubacco L, Bisaglia M, 2017. Recent findings on the physiological function of DJ-1: Beyond Parkinson’s disease. Neurobiol Dis 108, 65–72. [DOI] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM, 1999. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol 1, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum TB, Hahn A, Meier T, Davies KM, Kuhlbrandt W, 2019. Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P, 2003. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P, 2013. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12, 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges HR, Jones AJ, Pollak MN, Hirst J, 2014. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 462, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG, 2009. Apoptotic mechanisms after cerebral ischemia. Stroke 40, e331–339. [DOI] [PubMed] [Google Scholar]
- Cabezon E, Arechaga I, Jonathan P, Butler G, Walker JE, 2000a. Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1. J Biol Chem 275, 28353–28355. [DOI] [PubMed] [Google Scholar]
- Cabezon E, Butler PJ, Runswick MJ, Walker JE, 2000b. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J Biol Chem 275, 25460–25464. [DOI] [PubMed] [Google Scholar]
- Cadonic C, Sabbir MG, Albensi BC, 2015. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol Neurobiol. [DOI] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR, 2008. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab 8, 13–25. [DOI] [PubMed] [Google Scholar]
- Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR, 2004. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A 101, 9103–9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro M, Bernardi P, 2016. Calcium and reactive oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast. Cell Calcium 60, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro M, Checchetto V, Sartori G, Kucharczyk R, di Rago JP, Minervini G, Franchin C, Arrigoni G, Giorgio V, Petronilli V, Tosatto SCE, Lippe G, Szabo I, Bernardi P, 2018. High-Conductance Channel Formation in Yeast Mitochondria is Mediated by F-ATP Synthase e and g Subunits. Cell Physiol Biochem 50, 1840–1855. [DOI] [PubMed] [Google Scholar]
- Carraro M, Giorgio V, Sileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabo I, Bernardi P, 2014. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 289, 15980–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenini G, Lloret A, Cascella R, 2019. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid Med Cell Longev 2019, 2105607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC, 2006. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22, 79–99. [DOI] [PubMed] [Google Scholar]
- Chan DC, 2012. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46, 265–287. [DOI] [PubMed] [Google Scholar]
- Chelli B, Falleni A, Salvetti F, Gremigni V, Lucacchini A, Martini C, 2001. Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem Pharmacol 61, 695–705. [DOI] [PubMed] [Google Scholar]
- Chen R, Park HA, Mnatsakanyan N, Niu Y, Licznerski P, Wu J, Miranda P, Graham M, Tang J, Boon AJW, Cossu G, Mandemakers W, Bonifati V, Smith PJS, Alavian KN, Jonas EA, 2019. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis 10, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM, 2011. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. Journal of Cell Biology 195, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Fisher JC, Dillon CP, Kriwacki RW, Kuwana T, Green DR, 2008. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proceedings of the National Academy of Sciences of the United States of America 105, 20327–20332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomova M, Zitnanova I, 2016. Look into brain energy crisis and membrane pathophysiology in ischemia and reperfusion. Stress 19, 341–348. [DOI] [PubMed] [Google Scholar]
- Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, Zeviani M, 2015. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 21, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool SM, 2009. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim Biophys Acta 1788, 2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V , Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L, 2013. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Anselmi C, Wittig I, Faraldo-Gomez JD, Kuhlbrandt W, 2012. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci U S A 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Daum B, Gold VA, Muhleip AW, Brandt T, Blum TB, Mills DJ, Kuhlbrandt W, 2014. Visualization of ATP synthase dimers in mitochondria by electron cryo-tomography. J Vis Exp, 51228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kuhlbrandt W , 2011. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci U S A 108, 14121–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L, 2008. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Takei K, McPherson PS, 1995. The function of dynamin in endocytosis. Curr Opin Neurobiol 5, 559–565. [DOI] [PubMed] [Google Scholar]
- Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA, 2009. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 159, 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Bonilla E, Lee CP, Schotland DL, Scarpa A, Conn H Jr., Chance B, 1976. Luft’s disease. Further biochemical and ultrastructural studies of skeletal muscle in the second case. J Neurol Sci 27, 217–232. [DOI] [PubMed] [Google Scholar]
- Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC, 2010. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis 20 Suppl 2, S535–550. [DOI] [PubMed] [Google Scholar]
- Du H, Guo L, Yan SS, 2012. Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid Redox Signal 16, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Yan SS, 2010a. Mitochondrial medicine for neurodegenerative diseases. Int J Biochem Cell Biol 42, 560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Yan SS, 2010b. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim Biophys Acta 1802, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eydt K, Davies KM, Behrendt C, Wittig I, Reichert AS, 2017. Cristae architecture is determined by an interplay of the MICOS complex and the F1FO ATP synthase via Mic27 and Mic10. Microb Cell 4, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK, Zapol WM, 2017. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci U S A 114, E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR, 1999. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem 274, 2225–2233. [DOI] [PubMed] [Google Scholar]
- Flippo KH, Strack S, 2017. Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci 130, 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L, 2006. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Mourier A, Yamada J, McCaffery JM, Nunnari J, 2015. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Nagahashi A, Nagashima K, Rokudai S, Tsuruo T, 1998. Acceleration of apoptotic cell death after the cleavage of Bcl-XL protein by caspase-3-like proteases. Oncogene 17, 1295–1304. [DOI] [PubMed] [Google Scholar]
- Galkin A, Abramov AY, Frakich N, Duchen MR, Moncada S, 2009. Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury? J Biol Chem 284, 36055–36061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammie AE, Kurihara LJ, Vallee RB, Rose MD, 1995. DNM1, a dynamin-related gene, participates in endosomal trafficking in yeast. J Cell Biol 130, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genova ML, Lenaz G, 2013. A critical appraisal of the role of respiratory supercomplexes in mitochondria. Biol Chem 394, 631–639. [DOI] [PubMed] [Google Scholar]
- Genova ML, Lenaz G, 2014. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta 1837, 427–443. [DOI] [PubMed] [Google Scholar]
- Germain M, Mathai JP, McBride HM, Shore GC, 2005. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J 24, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh HS, 2008. The anti-aging, metabolism potential of SIRT1. Curr Opin Investig Drugs 9, 1095–1102. [PubMed] [Google Scholar]
- Giacomello M, Pyakurel A, Glytsou C, Scorrano L, 2020. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21, 204–224. [DOI] [PubMed] [Google Scholar]
- Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G, 2009. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 284, 33982–33988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P, 2013. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 110, 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gledhill JR, Montgomery MG, Leslie AG, Walker JE, 2007. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci U S A 104, 13632–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glytsou C, Calvo E, Cogliati S, Mehrotra A, Anastasia I, Rigoni G, Raimondi A, Shintani N, Loureiro M, Vazquez J, Pellegrini L, Enriquez JA, Scorrano L, Soriano ME, 2016. Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep 17, 3024–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Zhang L, Zong S, Guo R, Liu T, Yi J, Wang P, Zhuo W, Yang M, 2019. Cryo-EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1. Science 364, 1068–1075. [DOI] [PubMed] [Google Scholar]
- Guerrieri F, Capozza G, Kalous M, Papa S, 1992a. Age-related changes of mitochondrial F0F1 ATP synthase. Ann N Y Acad Sci 671, 395–402. [DOI] [PubMed] [Google Scholar]
- Guerrieri F, Capozza G, Kalous M, Zanotti F, Drahota Z, Papa S, 1992b. Age-dependent changes in the mitochondrial F0F1 ATP synthase. Arch Gerontol Geriatr 14, 299–308. [DOI] [PubMed] [Google Scholar]
- Guo L, Carraro M, Carrer A, Minervini G, Urbani A, Masgras I, Tosatto SCE, Szabo I, Bernardi P, Lippe G, 2019. Arg-8 of yeast subunit e contributes to the stability of F-ATP synthase dimers and to the generation of the full-conductance mitochondrial megachannel. J Biol Chem 294, 10987–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE, 2005. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP, 2014. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J Mol Cell Cardiol 72, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR, 1966. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 30, 269–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines TH, Dencher NA, 2002. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett 528, 35–39. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Brenner C, 2003. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 10, 1507–1525. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Javadov SA, 2004. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res 61, 372–385. [DOI] [PubMed] [Google Scholar]
- Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW, 2005. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 44, 16684–16694. [DOI] [PubMed] [Google Scholar]
- Hao LY, Giasson BI, Bonini NM, 2010. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proceedings of the National Academy of Sciences of the United States of America 107, 9747–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Chen YB, Jonas EA, 2012. Multipolar functions of BCL-2 proteins link energetics to apoptosis. Trends Cell Biol 22, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Youle RJ, 2009. SnapShot: BCL-2 proteins. Cell 138, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR, 1979. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195, 460–467. [DOI] [PubMed] [Google Scholar]
- He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE, 2017. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A 114, 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA, 2008. Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. Journal of Neurophysiology 99, 1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, 2005. Mitochondria and neurotransmission: evacuating the synapse. Neuron 47, 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA, 2003. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, 1979a. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 195, 453–459. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, 1979b. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys 195, 468–477. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, Southard JH, 1976. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem 251, 5069–5077. [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K, 2006. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25, 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K, 2009. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11, 958–966. [DOI] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goldberger O, Marutani E, Wojtkiewicz GR, Ast T, Wang H, Schleifer G, Stepanova A, Brepoels K, Schoonjans L, Carmeliet P, Galkin A, Ichinose F, Zapol WM, Mootha VK, 2019. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab 30, 824–832 e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM, Mootha VK, 2016. Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J, 2005. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 16, 1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hardwick JM, Kaczmarek LK, 2005a. Actions of BAX on mitochondrial channel activity and on synaptic transmission. Antioxidants & Redox Signaling 7, 1092–1100. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, Ivanovska I, Basanez G, Kinnally KW, Zimmerberg J, Hardwick JM, Kaczmarek LK, 2004. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proceedings of the National Academy of Sciences of the United States of America 101, 13590–13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Hardwick JM, Kaczmarek LK, 2005b. Exposure to hypoxia rapidly induces mitochondrial channel activity within a living synapse. Journal of Biological Chemistry 280, 4491–4497. [DOI] [PubMed] [Google Scholar]
- Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM, 2009. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J Neurosci Res 87, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, Safiulina D, Zharkovsky A, Veksler V, 2007. Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol 292, C157–163. [DOI] [PubMed] [Google Scholar]
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ, 2012. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298, 229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch J, Molkentin JD, 2014. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 111, 10396–10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury N, Koronowski KB, Perez-Pinzon MA, 2016. Long-term window of ischemic tolerance: An evolutionarily conserved form of metabolic plasticity regulated by epigenetic modifications? J Neurol Neuromedicine 1, 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury N, Xu J, Stegelmann SD, Jackson CW, Koronowski KB, Dave KR, Young JI, Perez-Pinzon MA, 2019. Resveratrol Preconditioning Induces Genomic and Metabolic Adaptations within the Long-Term Window of Cerebral Ischemic Tolerance Leading to Bioenergetic Efficiency. Mol Neurobiol 56, 4549–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnally KW, Campo ML, Tedeschi H, 1989. Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr 21, 497–506. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R, Ueda H, Handa N, Kimura K, Kamada T, 1991. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res 561, 203–211. [DOI] [PubMed] [Google Scholar]
- Klingenberg M, 2009. Cardiolipin and mitochondrial carriers. Biochim Biophys Acta 1788, 2048–2058. [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC, 2004. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondadi AK, Anand R, Hansch S, Urbach J, Zobel T, Wolf DM, Segawa M, Liesa M, Shirihai OS, Weidtkamp-Peters S, Reichert AS, 2020. Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. EMBO Rep 21, e49776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni SS, Canto C, 2015. The molecular targets of resveratrol. Biochim Biophys Acta 1852, 1114–1123. [DOI] [PubMed] [Google Scholar]
- Kuot A, Ronci M, Mills R, Klebe S, Snibson G, Wiffen S, Loh R, Corbett M, Zhou T, Chataway T, Burdon KP, Craig JE, Urbani A, Sharma S, 2019. Reduced expression of apolipoprotein E and immunoglobulin heavy constant gamma 1 proteins in Fuchs endothelial corneal dystrophy. Clin Exp Ophthalmol. [DOI] [PubMed] [Google Scholar]
- Kursvietiene L, Staneviciene I, Mongirdiene A, Bernatoniene J, 2016. Multiplicity of effects and health benefits of resveratrol. Medicina (Kaunas) 52, 148–155. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD, 2014. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ 21, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J, 2006. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122. [DOI] [PubMed] [Google Scholar]
- Larsen NJ, Ambrosi G, Mullett SJ, Berman SB, Hinkle DA, 2011. DJ-1 knock-down impairs astrocyte mitochondrial function. Neuroscience 196, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T, 2009. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1, a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Smith SB, Yoon Y, 2017. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem 292, 7115–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P, Licznerski P, Graham M, Uo T, Guo J, Rahner C, Duman RS, Morrison RS, Jonas EA, 2013. A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol 15, 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA, 2008. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America 105, 2169–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang J, Zeng Y, 2007. Peripheral benzodiazepine receptor ligand, PK11195 induces mitochondria cytochrome c release and dissipation of mitochondria potential via induction of mitochondria permeability transition. Eur J Pharmacol 560, 117–122. [DOI] [PubMed] [Google Scholar]
- Liu S, Sun J, Li Y, 2016. The Neuroprotective Effects of Resveratrol Preconditioning in Transient Global Cerebral Ischemia-Reperfusion in Mice. Turk Neurosurg 26, 550–555. [DOI] [PubMed] [Google Scholar]
- Lohret TA, Murphy RC, Drgon T, Kinnally KW, 1996. Activity of the mitochondrial multiple conductance channel is independent of the adenine nucleotide translocator. J Biol Chem 271, 4846–4849. [DOI] [PubMed] [Google Scholar]
- Ludtmann MH, Angelova PR, Ninkina NN, Gandhi S, Buchman VL, Abramov AY, 2016. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J Neurosci 36, 10510–10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, Al-Menhali AS, Ranasinghe RT, Whiten DR, Yapom R, Dolt KS, Devine MJ, Gissen P, Kunath T, Jaganjac M, Pavlov EV, Klenerman D, Abramov AY, Gandhi S, 2018. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun 9, 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malgieri G, Eliezer D, 2008. Structural effects of Parkinson’s disease linked DJ-1 mutations. Protein Sci 17, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH, 2006. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15, 1437–1449. [DOI] [PubMed] [Google Scholar]
- Mannella CA, 2006. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta 1762, 140–147. [DOI] [PubMed] [Google Scholar]
- Mansson R, Hansson MJ, Morota S, Uchino H, Ekdahl CT, Elmer E, 2007. Re-evaluation of mitochondrial permeability transition as a primary neuroprotective target of minocycline. Neurobiol Dis 25, 198–205. [DOI] [PubMed] [Google Scholar]
- Matthies D, Preiss L, Klyszejko AL, Muller DJ, Cook GM, Vonck J, Meier T, 2009. The c13 ring from a thermoalkaliphilic ATP synthase reveals an extended diameter due to a special structural region. J Mol Biol 388, 611–618. [DOI] [PubMed] [Google Scholar]
- Mehta SL, Manhas N, Raghubir R, 2007. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54, 34–66. [DOI] [PubMed] [Google Scholar]
- Meier T, Matthey U, Henzen F, Dimroth P, Muller DJ, 2001. The central plug in the reconstituted undecameric c cylinder of a bacterial ATP synthase consists of phospholipids. FEBS Lett 505, 353–356. [DOI] [PubMed] [Google Scholar]
- Menz RI, Walker JE, Leslie AG, 2001. Structure of bovine mitochondrial F(1)-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell 106, 331–341. [DOI] [PubMed] [Google Scholar]
- Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S, Bonanni L, Bennett MV, Zukin RS, Jonas EA, 2008. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America 105, 4892–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ, Jonas EA, 2019. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat Commun 10, 5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morciano G, Preti D, Pedriali G, Aquila G, Missiroli S, Fantinati A, Caroccia N, Pacifico S, Bonora M, Talarico A, Morganti C, Rizzo P, Ferrari R, Wieckowski MR, Campo G, Giorgi C, Trapella C, Pinton P, 2018. Discovery of Novel 1,3,8-Triazaspiro[4.5]decane Derivatives That Target the c Subunit of F1/FO-Adenosine Triphosphate (ATP) Synthase for the Treatment of Reperfusion Damage in Myocardial Infarction. J Med Chem 61, 7131–7143. [DOI] [PubMed] [Google Scholar]
- Morota S, Manolopoulos T, Eyjolfsson A, Kimblad PO, Wierup P, Metzsch C, Blomquist S, Hansson MJ, 2013. Functional and pharmacological characteristics of permeability transition in isolated human heart mitochondria. PLoS One 8, e67747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C, 2010. The science of stroke: Mechanisms in search of treatments. Neuron 67, 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. , 1995. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267, 1506–1510. [DOI] [PubMed] [Google Scholar]
- Murphy BJ, Klusch N, Langer J, Mills DJ, Yildiz O, Kuhlbrandt W, 2019. Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1-Fo coupling. Science 364. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA, 1986. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. [DOI] [PubMed] [Google Scholar]
- Nakka VP, Gusain A, Mehta SL, Raghubir R, 2008. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol Neurobiol 37, 7–38. [DOI] [PubMed] [Google Scholar]
- Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, Pavlov EV, 2019. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep 26, 11–17 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesci S, Trombetti F, Ventrella V, Pirini M, Pagliarani A, 2017. Kinetic properties of the mitochondrial F1FO-ATPase activity elicited by Ca(2+) in replacement of Mg(2). Biochimie 140, 73–81. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, 2004. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Current Molecular Medicine 4, 149–177. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, 2008. Oxidative stress and energy crises in neuronal dysfunction. Annals of the New York Academy of Sciences 1147, 53–60. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ, 2013. The Chemiosmotic Proton Circuit in Isolated Organelles: Theory and Practice, Bioenergetics, Fourth edition, p. 53–87.
- Nicolli A, Petronilli V, Bernardi P, 1993. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry 32, 4461–4465. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA, 2001. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60, 759–767. [DOI] [PubMed] [Google Scholar]
- Oberfeld B, Brunner J, Dimroth P, 2006. Phospholipids occupy the internal lumen of the c ring of the ATP synthase of Escherichia coli. Biochemistry 45, 1841–1851. [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, Alavian KN, Pontarelli F, Roelofs BA, Hickman JA, Hardwick JM, Zukin RS, Jonas EA, 2012. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci 15, 574–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G, 2003. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278, 7743–7746. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, 2005. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. [DOI] [PubMed] [Google Scholar]
- Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, Shaw JM, 1998. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol 143, 333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G, 2014. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta 1837, 408–417. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Paradies V, Ruggiero FM, 2011. Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int 58, 447–457. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM, 1999. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic Biol Med 27, 42–50. [DOI] [PubMed] [Google Scholar]
- Paradies G, Ruggiero FM, 1990. Enhanced activity of the tricarboxylate carrier and modification of lipids in hepatic mitochondria from hyperthyroid rats. Arch Biochem Biophys 278, 425–430. [DOI] [PubMed] [Google Scholar]
- Paradies G, Ruggiero FM, Dinoi P, 1991. The influence of hypothyroidism on the transport of phosphate and on the lipid composition in rat-liver mitochondria. Biochim Biophys Acta 1070, 180–186. [DOI] [PubMed] [Google Scholar]
- Park HA, Jonas EA, 2017. DeltaN-Bcl-xL, a therapeutic target for neuroprotection. Neural Regen Res 12, 1791–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HA, Licznerski P, Mnatsakanyan N, Niu Y, Sacchetti S, Wu J, Polster BM, Alavian KN, Jonas EA, 2017. Inhibition of Bcl-xL prevents pro-death actions of DeltaN-Bcl-xL at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ 24, 1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Simbula G, Gilfor E, Hoek JB, Farber JL, 1994. Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J Biol Chem 269, 31041–31046. [PubMed] [Google Scholar]
- Paumard P, Arselin G, Vaillier J, Chaignepain S, Bathany K, Schmitter JM, Brethes D, Velours J, 2002a. Two ATP synthases can be linked through subunits i in the inner mitochondrial membrane of Saccharomyces cerevisiae. Biochemistry 41, 10390–10396. [DOI] [PubMed] [Google Scholar]
- Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brethes D, di Rago JP, Velours J, 2002b. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J 21, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov E, Zakharian E, Bladen C, Diao CT, Grimbly C, Reusch RN, French RJ, 2005. A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys J 88, 2614–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P, 1994. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem 269, 16638–16642. [PubMed] [Google Scholar]
- Petronilli V, Szabo I, Zoratti M, 1989. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett 259, 137–143. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G, 2007. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta 1767, 1260–1267. [DOI] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H, 2003. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278, 52873–52880. [DOI] [PubMed] [Google Scholar]
- Quintana-Cabrera R, Quirin C, Glytsou C, Corrado M, Urbani A, Pellattiero A, Calvo E, Vazquez J , Enriquez JA, Gerle C, Soriano ME, Bernardi P, Scorrano L, 2018. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat Commun 9, 3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl R, Soubannier V, Scholz R, Vogel F, Mendl N, Vasiljev-Neumeyer A, Korner C, Jagasia R, Keil T, Baumeister W, Cyrklaff M, Neupert W, Reichert AS, 2009. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J Cell Biol 185, 1047–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelt H, Bohnert M, Zerbes RM, Horvath SE, Warscheid B, Pfanner N, van der Laan M, 2017. Mic10, a Core Subunit of the Mitochondrial Contact Site and Cristae Organizing System, Interacts with the Dimeric F1Fo-ATP Synthase. J Mol Biol 429, 1162–1170. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Perez-Pinzon MA, 2006. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab 26, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ, 2003. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23, 7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger AJ, Munoz-Gomez SA, Kamikawa R, 2017. The Origin and Diversification of Mitochondria. Curr Biol 27, R1177–R1192. [DOI] [PubMed] [Google Scholar]
- Salehi B, Mishra AP, Nigam M, Sener B, Kilic M, Sharifi-Rad M, Fokou PVT, Martins N, Sharifi-Rad J, 2018. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunier E, Antonio S, Regazzetti A, Auzeil N, Laprevote O, Shay JW, Coumoul X, Barouki R, Benelli C, Huc L, Bortoli S, 2017. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci Rep 7, 6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC, 2011. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Ren M, 2006. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett 580, 5450–5455. [DOI] [PubMed] [Google Scholar]
- Schurr A, Reid KH, Tseng MT, West C, Rigor BM, 1986. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res 374, 244–248. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ, 2002. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Developmental Cell 2, 55–67. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2011. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med 17, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Semba RD, Moaddel R, Zhang P, Ramsden CE, Ferrucci L, 2019. Tetra-linoleoyl cardiolipin depletion plays a major role in the pathogenesis of sarcopenia. Med Hypotheses 127, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Wattez A, Galvan-valencia M, Ghestem A, David JP, Lemoine J, Sautiere PE, Dachary J, Mazat JP, Michalski JC, Velours J, Mena-Lopez R, Delacourte A, 2003. Association of ATP synthase alpha-chain with neurofibrillary degeneration in Alzheimer’s disease. Neuroscience 117, 293–303. [DOI] [PubMed] [Google Scholar]
- Sileikyte J, Blachly-Dyson E, Sewell R, Carpi A, Menabo R, Di Lisa F, Ricchelli F, Bernardi P, Forte M, 2014. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J Biol Chem 289, 13769–13781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM, 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Molecular Biology of the Cell 12, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Bernardi P, Zoratti M, 1992. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem 267, 2940–2946. [PubMed] [Google Scholar]
- Szabo I, De Pinto V, Zoratti M, 1993. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett 330, 206–210. [DOI] [PubMed] [Google Scholar]
- Szabo I, Zoratti M, 1992. The mitochondrial megachannel is the permeability transition pore. J Bioenerg Biomembr 24, 111–117. [DOI] [PubMed] [Google Scholar]
- Szabo I, Zoratti M, 1993. The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett 330, 201–205. [DOI] [PubMed] [Google Scholar]
- Terni B, Boada J, Portero-Otin M, Pamplona R, Ferrer I, 2010. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol 20, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, McMurray CT, 2007. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145, 1233–1248. [DOI] [PubMed] [Google Scholar]
- Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, Abe K, Maeda S, Shinzawa-Itoh K, Bogers JFM, McMillan DGG, Gerle C, Szabo I, Bernardi P, 2019. Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat Commun 10, 4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P, Viscomi C, Zeviani M, Di Lisa F, Mongillo M, Sandri M, Scorrano L, 2015. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21, 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ, 2005. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. [DOI] [PubMed] [Google Scholar]
- Vuorinen K, Ylitalo K, Peuhkurinen K, Raatikainen P, Ala-Rami A, Hassinen IE, 1995. Mechanisms of ischemic preconditioning in rat myocardium. Roles of adenosine, cellular energy state, and mitochondrial F1F0-ATPase. Circulation 91, 2810–2818. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Xiao AY, Sheline C, Hyrc K, Yang A, Goldberg MP, Choi DW, Yu SP, 2003. Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. Journal of Cell Science 116, 2099–2110. [DOI] [PubMed] [Google Scholar]
- Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE, 2010. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci U S A 107, 16823–16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert M, Millet A, Jonas EA, Alavian KN, 2019. The mitochondrial metabolic function of DJ-1 is modulated by 14-3-3beta. FASEB J 33, 8925–8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Carrozzo R, Santorelli FM, Schagger H, 2006. Supercomplexes and subcomplexes of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1757, 1066–1072. [DOI] [PubMed] [Google Scholar]
- Wollweber F, von der Malsburg K, van der Laan M, 2017. Mitochondrial contact site and cristae organizing system: A central player in membrane shaping and crosstalk. Biochim Biophys Acta Mol Cell Res 1864, 1481–1489. [DOI] [PubMed] [Google Scholar]
- Yamada EW, Huzel NJ, 1992. Distribution of the ATPase inhibitor proteins of mitochondria in mammalian tissues including fibroblasts from a patient with Luft’s disease. Biochim Biophys Acta 1139, 143–147. [DOI] [PubMed] [Google Scholar]
- Yin M, van der Horst IC, van Melle JP, Qian C, van Gilst WH, Sillje HH, de Boer RA, 2011. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am J Physiol Heart Circ Physiol 301, H459–468. [DOI] [PubMed] [Google Scholar]
- Zerbes RM, van der Klei IJ, Veenhuis M, Pfanner N, van der Laan M, Bohnert M, 2012. Mitofilin complexes: conserved organizers of mitochondrial membrane architecture. Biol Chem 393, 1247–1261. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W, 2002. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277, 43553–43556. [DOI] [PubMed] [Google Scholar]
- Zhou A, Rohou A, Schep DG, Bason JV, Montgomery MG, Walker JE, Grigorieff N, Rubinstein JL, 2015. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife 4, e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I, 1995. The mitochondrial permeability transition. Biochim Biophys Acta 1241, 139–176. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Kinnally KW, Perini S, Tedeschi H, 1992. Multiple conductance levels in rat heart inner mitochondrial membranes studied by patch clamping. Biochim Biophys Acta 1105, 263–270. [DOI] [PubMed] [Google Scholar]