

Abstract
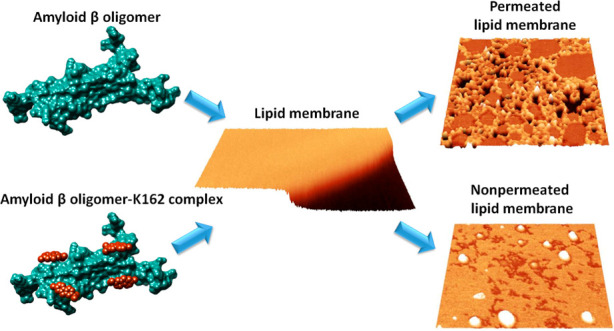
Alzheimer’s disease (AD) is characterized by progressive neurodegeneration associated with amyloid β (Aβ) peptide aggregation. The aggregation of Aβ monomers (AβMs) leads to the formation of Aβ oligomers (AβOs), the neurotoxic Aβ form, capable of permeating the cell membrane. Here, we investigated the effect of a fluorene-based active drug candidate, named K162, on both Aβ aggregation and AβO toxicity toward the bilayer lipid membrane (BLM). Electrochemical impedance spectroscopy (EIS), atomic force microscopy (AFM), and molecular dynamics (MD) were employed to show that K162 inhibits AβOs-induced BLM permeation, thus preserving BLM integrity. In the presence of K162, only shallow defects on the BLM surface were formed. Apparently, K162 modifies Aβ aggregation by bypassing the formation of toxic AβOs, and only nontoxic AβMs, dimers (AβDs), and fibrils (AβFs) are produced. Unlike other Aβ toxicity inhibitors, K162 preserves neurologically beneficial AβMs. This unique K162 inhibition mechanism provides an alternative AD therapeutic strategy that could be explored in the future.
Keywords: Alzheimer’s disease, amyloid β, membrane permeation, toxicity inhibition, amyloid β aggregation, atomic force microscopy
1. Introduction
Alzheimer’s disease (AD), the most prevalent type of dementia,1 is a fatal, neurodegenerative disorder that leads to a cognitive impairment such as memory loss, communication difficulties, and personality changes. Pathology of AD is associated with misfolding of amyloid β (Aβ) peptide and tau protein, and their aggregation into amyloid plaques and neurofibrillary tangles, respectively, the two hallmarks of AD.
Aβ aggregates via nucleation-dependent polymerization in which Aβ monomers (AβMs) associate into Aβ oligomers (AβOs) and then Aβ fibrils (AβFs).2−4 Recent studies consider AβOs to be the most toxic form, while AβMs and AβFs are considered nontoxic.5−7 AβOs can permeate the cell membrane,8−12 and once incorporated into the cell, AβOs can cause learning and cognition deficiency,13,14 deterioration of synapses,15,16 leakage of lysosomal enzymes,17 inhibition of mitochondrial activity,18 increased production of reactive oxygen species,19 and neuroinflammation.20,21
Aβ inhibitors act either by (i) stimulating AβMs aggregation into nontoxic off-pathway oligomers with nonamyloidogenic conformation,22−24 (ii) binding to fibril surface, thus preventing AβOs formation via secondary nucleation,25,26 or (iii) accelerating Aβ aggregation, thus decreasing the lifetime of toxic AβOs, preventing their growth, and stimulating the formation of nontoxic AβFs.27−29 In all these therapeutic strategies, AβMs are consumed. AβMs stimulate brain development,30 positively contribute to differentiation and proliferation of neural progenitor cells31 and human neural stem cells,32 enhance survival of neurons,33 and protect neurons from excitotoxic cell death.34 Therefore, the disadvantage of previously developed therapeutic strategies is that with the consumption of AβMs, many beneficial physiological abilities of AβMs are lost.
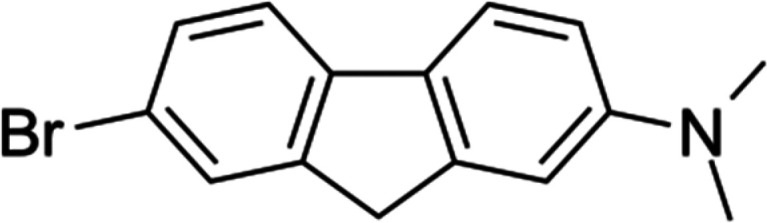
A fluorene-based compound known as K162 or K01–162 (Scheme 1) decreases AβOs toxicity in vivo.35 Evidently, K162 penetrates the blood-brain barrier, inhibits AβOs binding to synapses, and decreases amyloid load inside MC65 cells and the hippocampus of 5xFAD mice brain. However, the mechanism of these K162 in vivo effects is not described.
Scheme 1. Structural Formula of K162 (C15H14BrN).
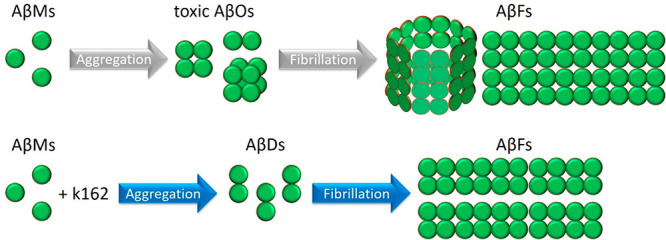
Here, we reveal that K162 prevents AβOs-induced bilayer lipid membrane (BLM) poration by altering the Aβ aggregation pathway. In the K162-modified Aβ aggregation, AβMs dimerize. Then, these Aβ dimers (AβDs) do not oligomerize but fibrillate. This way, the formation of membrane-permeating AβOs is bypassed. Unlike other amyloid inhibitors, K162 preserves the neurologically beneficial AβMs.
2. Results and Discussion
K162 Inhibits BLM Permeation by AβOs–EIS Studies
Electrochemical impedance spectroscopy (EIS) measures the impedance of the electrode coated with BLM. This BLM is an insulating layer on the electrode surface. When the BLM integrity is compromised, the electrode impedance changes because of the ion transfer from the electrolyte solution bulk to the electrode surface. For highly capacitive systems, like a nonpermeated phospholipid bilayer, the impedance vs frequency curve slope does not change, while the phase angle vs frequency curve displays a plateau at a phase angle ∼90° in a low-frequency region.36,37 These EIS features are observed for the BLM in the absence of AβOs and K162 (Figures S1a and S1b). For the BLM in the presence of AβOs (BLM-AβOs), the impedance vs frequency curve displays the small “kink-like” feature below ∼1 Hz (black curve in Figure 1a). The corresponding phase angle vs frequency curve exhibits the minimum with the lowest phase angle value (∼74°) at ∼0.4 Hz (black curve in Figure 1b). These results indicate the BLM permeation by AβOs, in agreement with our previous study.38 To test whether K162 inhibits this permeation, the preincorporation method was used. That is, K162 was mixed with AβMs and then allowed to interact with them for 24 h at 4 °C. Next, lipid vesicles were mixed with the Aβ-K162 solution at room temperature by 10-min sonication. Finally, the 1-thio-β-d-glucose (Tg)-modified Au(111) electrode was immersed in the K162-Aβ-lipid mixture to allow for sample depositing overnight (for details, see the Materials and Methods section).
Figure 1.
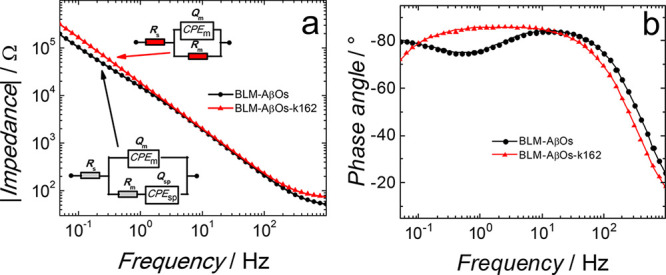
(a) Impedance and (b) phase angle as a function of frequency for (black curve) fBLM-AβOs and (red curve) fBLM-AβOs-K162 in the PBS (0.01 M phosphate buffer, 0.0027 M KCl, and 0.137 M NaCl, pH = 7.4) solution at 0 V vs SCE. Symbols and curves of the same colors represent experimental data and results of fitting of parameters of the equivalent electrical circuits, shown as insets in Panel (a), to the EIS data, respectively, for the same measurement at a single potential. Rs and Rm – solution and membrane resistance, respectively; CPEm and CPEsp – constant-phase element for the membrane and submembrane (spacer) region, respectively.
For that sample, the impedance vs frequency curve does not display the “kinklike” feature (red curve in Figure 1a). Moreover, there is no minimum in the phase angle vs frequency curve. Instead, there is a plateau with a phase angle of ∼85° (red curve in Figure 1b). These EIS features indicate that BLM-AβOs-K162 is not permeated, and AβOs are inactive toward BLM in the presence of K162. Possibly, K162 either interacts with AβMs, thus preventing them from forming toxic AβOs, or K162 interacts with toxic AβOs and converts them into a nontoxic form.
The external addition procedure was utilized to test the second possibility. That is, K162 was added to a solution of already formed AβOs and then allowed to interact with AβOs for 24 h at 4 °C. Next, lipid vesicles were mixed with the AβOs-K162 solution at room temperature by 10-min sonication. Finally, the Tg-modified Au(111) electrode was immersed in the K162-Aβ-lipid mixture to allow for sample depositing overnight (for details, see the Materials and Methods section). In this case, both the “step-like” feature in the impedance vs frequency curve (Figure S1c) and the minimum in the phase angle vs frequency curve (Figure S1d) are absent. These EIS results indicate that K162 effectively inhibits the toxicity of preformed AβOs.
Equivalent electric circuits, shown as insets in Figure 1a, were fitted to the EIS data to obtain information about membrane capacitance, Qm, and resistance, Rm, and their changes in the presence of toxins.38,39 Results of the EIS fitting are shown in Table S1. As expected, Qm is the lowest for BLM, i.e., ∼3.72 μF cm–2. The addition of AβOs to BLM resulted in an increase of Qm to ∼11.57 μF cm–2, in agreement with our previous study.38 The preincorporation or external addition of K162 does not change Qm, i.e., Qm is ∼11.79 μF cm–2 and ∼10.19 μF cm–2, respectively. A decrease in Rm is an excellent indication of membrane permeation. As expected, Rm is the highest for BLM, equaling ∼7.25 MΩ cm2. The addition of AβOs to BLM (BLM-AβOs) resulted in an ∼100-fold decrease in Rm, i.e., to ∼0.079 MΩ cm2. This result supports the conclusion that BLM is permeated by AβOs, in agreement with our previous study.38 Moreover, when K162 is preincorporated or externally added, Rm is ∼0.954 and 3.29 MΩ cm2, respectively. That is, Rm is decreased much less than that for the nonpermeated BLM. The difference in the Rm change between the K162 added externally and preincorporated into Aβ solution is negligible (∼2 MΩ cm2) and can be considered comparable. Such experimental variations are demonstrated in Figure S2, which shows Rm for BLM in the absence of Aβ and K162 determined from two independent measurements. Overall, the EIS results indicate that K162 inhibits the BLM permeation by AβOs.
K162 Inhibits the BLM Permeation by AβOs–AFM Studies
EIS is a technique that provides average information about the sample. High-resolution atomic force microscopy (AFM) imaging was used to complement EIS results by providing molecular-level information about the Aβ interaction with BLM in the K162 absence and presence. In the absence of both AβOs and K162, the BLM morphology is typical for lipid bilayers, i.e., it is an ∼6 nm thick film with a smooth surface (Figures 2a and 2d). Interestingly, a higher resolution image (inset in Figure 2a) shows that even though BLM is in a gel phase only, it is not homogeneous; it consists of small domains. These domains, also known as lipid clusters, were observed in cholesterol-containing ternary40 and quaternary41 lipid bilayer mixtures42,43 (detailed discussion on this part is provided in the SI).
Figure 2.

AFM topography images of (a) BLM, (b) BLM in the presence of AβOs, and (c) BLM in the presence of AβOs and K162 in the PBS (0.01 M phosphate buffer, 0.0027 M KCl, and 0.137 M NaCl, pH = 7.4) solution at room temperature. Insets in Panels (a) and (b) show higher-resolution AFM images of the BLM in the absence of AβO, and the pores and AβO clusters formed in the BLM-AβOs, respectively. (d) Cross-sectional profile across the line in Panel (a) showing the BLM thickness. (e) Cross-sectional profile across the line in the inset in Panel (b) showing the pore depth and the membrane-protruding AβOs cluster’s height. (f) Cross-sectional profile across line 1 in Panel (c) displaying the depth of scratches and globular structures’ height. The inset in Panel (f) shows the cross-sectional profile along line 2 in Panel (c), displaying the height of the AβOs cluster located on top of the BLM-AβOs-K162. The height in all AFM images is scaled using the lowest point of the image as a reference.
Figure 2b shows the BLM morphology in the presence of AβOs (BLM-AβOs), significantly different than that of BLM in the AβOs absence. In the presence of AβOs, a network of domains consisting of pores surrounded by AβO clusters is formed. This network occupies ∼40% of the total surface area of the membrane. The pore depth is in the range of 3 to 4.5 nm, with the most populated pore depth of ∼3.9 nm (Figures 2e and S4a). Our previous study showed that AβOs induce conformational changes of lipid acyl chains by increasing the number of gauche conformers characteristic for melted lipid acyl chains.38 The thickness of the BLM with melted acyl chains, i.e., in the liquid crystalline phase, is ∼5 nm (Figure S3). Therefore, the pore depth distribution (Figure S4a) shows that AβOs entirely compromised the integrity of the ∼5 nm thick BLM (Figure 2b). The AβO clusters with an equivalent disk radius of ∼5 nm (Figure S4b) protrude ∼2–3 nm from the membrane surface (Figure 2e).
Figure 2c shows the morphology of BLM in the presence of both AβOs and K162 (BLM-AβOs-K162). The presence of K162 prevents AβOs from forming the network of domains with pores passing through the entire BLM. Instead, three distinctive features are visible on the BLM surface, i.e., (i) defects of irregular shapes (indicated with green arrow 1′), globular particles (purple arrow 2′), and AβO clusters similar to those shown in Figure 2a (red arrow 3′). The defects are ∼0.7–1.2 nm deep (Figures 2f and S4c), and they occupy ∼7% of the BLM surface area. These defects are very shallow compared to the pores in BLM-AβO (Figures 2e and S4a). The globular structures with a height of ∼4–6 nm (Figure 2f) occupy ∼3% of the BLM surface area. The (∼2–3)-nm thick AβO clusters (inset in Figure 2f), similar to those for BLM-AβOs in the absence of K162 (Figure 2b), also appear in the K162 presence, indicating that K162 does not inhibit AβOs clustering on the BLM surface. The lack of pores in the presence of K162 clearly indicates that the resulting clusters do not permeate BLM, thus confirming the EIS results. In the presence of K162, there is no network of AβO clusters (Figure 2c), seen in the absence of K162 (Figure 2b). Instead, only a few separate AβO clusters and many large globular structures, mostly located in irregularly shaped, shallow defects, are visible (Figure 2c). This observation indicates that the K162 molecules break the AβO cluster network and stimulate individual AβO clusters to shrink into globular structures. During this shrinking, AβO clusters leave defects on the BLM surface like imprints showing their location on the membrane surface before the shrinking. The formation of globular structures only in the K162 presence supports this explanation. Moreover, these structures are higher than AβO clusters (Figure 2c and inset in Figure 2f), as expected, if the globular structure is formed by a mass accumulation of laterally long AβO clusters. Furthermore, the shape and radius of the defects (Figure S4d) are very similar to those of AβO clusters formed in the absence of K162 (Figure S4b), supporting the hypothesis that the defects are imprints of AβO clusters that shrank and diffused away from the BLM surface. This hypothesis could also account for the K162-induced inhibition of AβOs binding to synapses.35
K162 contains aromatic rings (Scheme 1), making the molecule very hydrophobic. Hence, K162 could potentially incorporate into the membrane and alter its structure. Therefore, the morphology of BLM exposed to K162 only (without Aβ) was studied by AFM. Both morphology and thickness of the membrane in the absence (Figure 2a) and presence of K162 (Figure S5) are similar, indicating that K162 does not affect the membrane structure at the concentration used in the present research. Moreover, no particles were adsorbed on the membrane or mica surface.
K162 Influence on the Aβ Aggregation in Solution–AFM Studies
The EIS and AFM results show that K162 inhibits BLM permeation by AβOs, formed after 24 h of Aβ aggregation. To understand this inhibition mechanism, we examined the K162 influence on the Aβ aggregation in the absence of lipids. Figure 3a shows the AFM image of Aβ molecules deposited on mica from a freshly prepared Aβ solution (0 h of aggregation) in the absence of K162. Globular structures are only visible. The most populated globules (∼47%) with a height of ∼0.3 nm correspond to AβMs, while the remaining globules constitute a minor population of small AβOs (Figure 3g and Table S2). These results are in excellent agreement with the literature.44 After 24 h of Aβ aggregation in the absence of K162 (Figure 3b), the AβM population significantly decreases from ∼47% to ∼19%, and two types of globular AβOs, with their respective heights of ∼1.2 and ∼2.4 nm, are formed (Figure 3h and Table S2). Considering the height of AβMs, one can infer that the two populations of AβOs correspond to tetramers and octamers, respectively. The Aβ aggregation occurs through the so-called nucleated conversion mechanism.45,46 This mechanism indicates the growth of AβOs by their stacking on top of each other, i.e., two AβM molecules stack to produce an Aβ dimer (AβD), then two AβD molecules stack to produce a tetramer, etc. This mechanism explains why AβMs, AβDs, tetramers, and octamers dominate over trimers, pentamers, hexamers, etc.
Figure 3.

AFM topography images of Aβ forms produced after (a, d) 0, (b, e) 24, and (c, f) 48 h of Aβ aggregation in the (a–c) absence and (d–f) presence of K162 in the PBS (0.01 M phosphate buffer, 0.0027 M KCl, and 0.137 M NaCl, pH = 7.4) solution at 4 °C. The corresponding height distributions of Aβ forms produced after (g, j) 0, (h, k) 24, and (i, l) 48 h of Aβ aggregation in the (g, h, i) absence and (j, k, l) presence of K162. Histograms corresponding to monomers, tetramers, and octamers of Aβ are colored in blue, red, and green, respectively.
In the presence of K162, a freshly prepared Aβ solution contains a slightly higher population of AβMs (∼58%) and a lower population of AβOs (Figures 3d and 3j) in comparison to those found in the K162 absence (Figures 3a and 3g). After 24 h of the Aβ aggregation in the presence of K162, only globular structures are formed (Figure 3e) at the expense of the AβMs population, which decreased from ∼58% to ∼44%. This decrease is significantly lower than that in the absence of K162 (Figures 3k vs3h). That is, after 24-h Aβ aggregation, 60 and 25% of the monomers aggregated, forming oligomers in the absence and presence of K162, respectively (Figures 3h and 3k and Table S2). Consequently, the population of AβOs formed in the presence of K162 is lower than that in the K162 absence. Moreover, the height distribution of AβOs formed in the presence of K162 significantly differs from that observed in the K162 absence. That is, tetramers dominate over the other forms of AβOs, and the number of octamers is negligible compared to AβOs distribution in the K162 absence (Table S2). These results suggest that K162 substantially preserves AβMs and inhibits their aggregation into membrane-permeating AβOs like tetramers, octamers, etc.
So far, we showed that K162 suppressed the formation of AβOs during the first 24 h of Aβ aggregation. However, it is unclear whether K162 only delays or effectively inhibits the formation of toxic AβOs. In the former case, the BLM damage would be delayed but unavoidable, making K162 an ineffective therapeutic. Therefore, the Aβ aggregation in the absence and presence of K162 was monitored by AFM for 48 h. After 48 h of Aβ aggregation in the absence of K162, both globular and elongated structures are formed (Figures 3c and S6a). The AβMs population decreased significantly (Figure 3i) compared to that observed after 24 h of Aβ aggregation (Figure 3h). Moreover, the most dominant AβOs are tetramers, while the octamer population decreased (Table S2). It might be surprising that globular Aβ aggregates formed after 48 h of aggregation (Figure 3i) are smaller than those formed after 24 h of aggregation (Figure 3h). However, a recent study showed that AβOs rather dissociate than grow into larger forms,47 even though AβFs are formed later along the aggregation pathway. In the K162 absence, most elongated structures assume a ringlike shape, while a minor population has an elongated shape typical for AβFs (Figure 3c). The mechanism of the ringlike shape formation is not elucidated yet. Presumably, only AβOs of a certain molecular weight can adopt the ringlike shape. Because all AβOs are transiently stable, some will assemble into the ringlike shape, while others will aggregate into higher-order AβOs. The K162 occupies hydrophobic residues of ring-forming Aβ oligomers, thus preventing their assembling into the ringlike shape. The elongated structures’ cross-sectional profile showed that their height is ∼1.2 nm (Figure S6c), identical to the Aβ tetramers’ height, indicating that these structures are formed lateral assembly of Aβ tetramers.
In the presence of K162, both globules and AβFs are formed after 48 h of aggregates (Figures 3f and S6b), similarly as in the K162 absence (Figures 3c and S6a). However, the height distribution differs significantly between the two cases. In the K162 presence, AβMs and AβDs represent a substantial part of the Aβ aggregates (Figure 3l and Table S2). Interestingly, in the presence of K162, there are no ringlike structures. The cross-sectional profiles show that AβFs, formed in the presence of K162 (Figures 3f and S6d), are much longer and by ∼50% thinner than the AβFs formed in the K162 absence (Figures 3c and S6c). The height of the AβFs formed in the presence of K162 indicates that they are composed of Aβ dimers (AβDs), not tetramers, as it is observed in the K162 absence.
K162 Influence on the Aβ Aggregation in Solution–MD Simulations
Molecular dynamics (MD) simulations were performed to gain further insight into the K162 interaction with Aβ. Two different Aβ structures were used for the MD calculations. AβMs and AβDs are represented by the Aβ42 that assumes a so-called β-hairpin structure with an antiparallel β-sheet arrangement.48 This structure is typical for the prefibrillar Aβ forms, i.e., misfolded AβMs and toxic AβOs.49,50 For AβFs, the structure of Aβ42 fibrils with in-register parallel β-sheet architecture, composed of AβDs, was used.51 The in-register parallel β-sheet is typical for AβFs.51−54 Interactions of these structures with K162 are shown in Figure 4.
Figure 4.
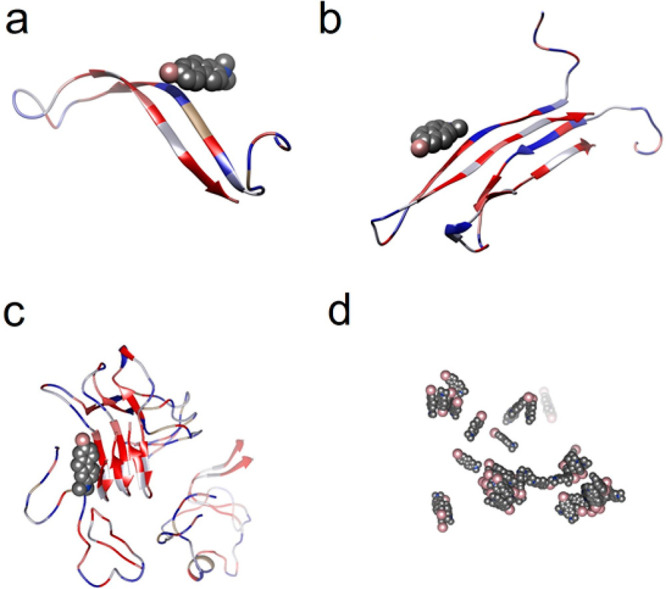
Molecular dynamics modeled examples of configurations of the K162 complexes with (a) AβMs, (b) AβDs, (c) AβFs, and (d) K162. K162 molecules are represented as spheres and colored by the type of element, i.e., carbon is gray, bromine is pink, and nitrogen is blue. All Aβ forms are shown as ribbon structures, and their residues are colored by lipophilicity, i.e., hydrophilic residues are blue, neutral residues are white, and hydrophobic residues are red.
MD identifies the binding sites and energies of the K162-Aβ interactions, thus providing information about the K162 affinity toward AβMs, AβDs, and AβFs and its influence on Aβ aggregation. MD simulations show that K162 binds to all Aβ forms, i.e., AβMs, AβDs, and AβFs, but it also aggregates itself (Figure 4). The dispersive and hydrophobic interactions, mostly π–π stacking of aromatic rings, are responsible for the favorable attractive interactions between K162 molecule. Moreover, they allow for partial K162 dehydration and contact-pair formation. In the K162-AβM complex (Figure 4a), K162 interacts with hydrophilic residues, i.e., 13–16, of AβM55 via its Br terminal. In contrast to AβM, K162 binds to hydrophobic residues of AβDs and AβFs (Figure 4b and 4c, respectively) via its hydrophobic aromatic ring. The difference between K162-AβDs and K162-AβFs interactions is that K162 interacts with both the middle part (residues 16–18) and the C-terminal side (residues 31–35) of AβDs, while it binds only to the C-terminal part (residues 30–42) of AβFs.
The binding energy values for all Aβ-Aβ interactions in the K162 absence are negative, indicating a high affinity of Aβ to aggregate (Table S3). On the other hand, the positive binding energies of all Aβ-Aβ interactions in the K162 presence suggest that once the K162 binds to any Aβ form, its further aggregation is energetically unfavorable. However, AFM imaging shows that despite the K162 presence, the Aβ aggregates are formed even at a K162 concentration 10-fold higher than that of Aβ (the K162:Aβ ratio is 10:1). K162 self-aggregates (Figure 4d), thus suggesting that not all K162 molecules bind to Aβ. Therefore, Aβ aggregation is not entirely prevented because the K162 self-aggregation competes with the K162-Aβ interaction.
Moreover, K162 does not inhibit the aggregation of all Aβ forms equally because it does not bind to all of them with the same preference. The binding energy values for all K162-Aβ interactions indicate that the affinity of K162 increases in the order of K162 < AβFs < AβMs < AβDs (Table S3). This different affinity can be explained by considering the structural changes of all Aβ forms during Aβ aggregation. The Aβ aggregates in the following order: (i) aggregation of misfolded AβMs to AβOs rich in antiparallel β-sheets,48−50,56 (ii) conversion of AβOs to fibrillar seeds with in-register parallel β-sheets, and (iii) lateral assembly of fibril seeds to AβFs with in-register parallel β-sheets.51−54 During Aβ aggregation, the hydrophobic residues of Aβ adopt β-sheets conformation and thus are the primary residues driving the Aβ aggregation.55 As Aβ aggregation proceeds, hydrophobic residues get buried more efficiently in the Aβ aggregates interior and thus are less exposed and less accessible for K162.
An AFM study shows that a high amount of AβMs is preserved after 24 h of Aβ aggregation (Figure 3k and Table S2). However, both AβDs and AβFs are formed after 48 h of Aβ aggregation (Figure 3l and Table S2). That is because K162 binds to the hydrophilic residues but not to aggregation-relevant hydrophobic residues of the AβMs (Figure 4a). Therefore, K162 inhibits but does not entirely prevent AβM aggregation.
After 24 h of Aβ aggregation in the K162 presence, a population of globular AβOs, larger than AβDs, is minor (Figure 3k and Table S2). Hence, K162 inhibits AβD oligomerization. After 48 h of Aβ aggregation in the K162 presence, AβDs and AβFs, composed of AβDs, dominate (Figures 3f, 3l, and S6b). Apparently, some AβDs manage to convert to fibril seeds and fibrillate in the presence of K162. Although K162 binds to aggregation-relevant hydrophobic residues of both AβDs and fibril seeds/AβFs (Figures 4b and 4c), the hydrophobic residues of AβFs are more deeply buried, thus remaining less accessible to K162. Therefore, the K162-AβD interaction is energetically more favorable than the K162-AβF interaction (Table S3). Hence, K162 more effectively prevents AβD oligomerization than fibrillation of AβD converted to fibril seeds.
Long AβFs are observed in the K162 presence (Figures 3f and S6b), indicating that K162 cannot compromise them. The in-register parallel β-sheet arrangement in fibril seeds/AβFs makes them very robust and stable, giving them a strength comparable to steel and mechanical stiffness comparable to that of silk57 that is significantly higher than that of AβOs.58
Our results explain why only AβMs and AβDs were observed inside neurons.35 The only remaining kind of Aβ aggregates formed in the K162 presence is nontoxic AβFs, which adsorb on the BLM surface without destroying it,10,11 and thus they cannot insert into neurons.35 Importantly, AβFs are widely known as nontoxic because they are inactive toward various biosystems like lipid vesicles,5 PC12 cells,6 glial cells in CGC cultures and macrophage J774 cells,7 MC65 cells, U18666A-treated neurons, and Tg6799 5xFAD mice.35 Moreover, our findings explain the influence of K162 on the formation of nontoxic AβDs. In general, hydrophobic residues of AβOs are essential for their toxicity. The higher the surface hydrophobicity of AβOs, the higher their toxicity.59−62 The interaction of AβO hydrophobic residues with BLM’s hydrophobic core leads to the BLM permeabilization.11,38 Our results show that K162 occupies the toxicity-relevant hydrophobic residues of AβDs, thus inhibiting the BLM permeation by these AβOs.
3. Conclusions
We have demonstrated that K162 inhibits BLM poration by AβOs. The EIS results showed that BLM integrity was preserved in the presence of AβOs and K162. This observation was confirmed by AFM imaging, showing no pores in the membrane typically formed by AβOs. The BLM protection from AβOs by K162 results from K162 binding to hydrophobic residues of Aβ aggregates. These residues are relevant not only for AβOs toxicity but also for Aβ aggregation. Once K162 binds to Aβ, its further aggregation is unfavorable. However, under the conditions used in the present study, K162 cannot entirely prevent aggregation of all Aβ forms present in the solution because of competitive K162 self-aggregation. Therefore, K162 inhibits AβMs aggregation, prevents AβDs oligomerization, but allows partial AβDs fibrillation (Scheme 2). As a result, nontoxic Aβ forms, i.e., AβMs, AβDs, and AβFs, are only formed in the presence of K162. This behavior is not observed for Aβ alone, where Aβ aggregation leads to the formation of high-molecular-weight toxic oligomers. These results show that K162 affects the Aβ aggregation pathways. This way, the production of BLM-permeating AβOs is bypassed. Unlike other Aβ toxicity inhibitors, K162 preserves neurologically beneficial AβMs. However, it remains to be elucidated whether K162-bound AβMs retain their beneficial neurological abilities. Even if they do not, the present findings describe a unique Aβ toxicity inhibition mechanism that may inspire the production of a novel type of AD therapeutics.
Scheme 2. Aβ Aggregation Pathways in the Absence (Grey Arrows) and Presence (Blue Arrows) of K162.
4. Materials and Methods
Aβ Peptide Preparation
Lyophilized amyloid β (1–42) peptide was purchased from rPeptide (Watkinsville, USA) and Bachem (Bubendorf, Switzerland). Its purity was high, as evidenced by MS analysis, reported in our previous work,38 showing molecular mass identical to that expected for the Aβ monomer. The peptide solution was prepared by following the previously developed protocol63 with slight modifications introduced in our previous studies.11,38 Briefly, Aβ was first dissolved to reach the 0.5-mg mL–1 concentration in trifluoroacetic acid (TFA) from Sigma-Aldrich by 5-min vortexing to remove pre-existing Aβ aggregates. Then, TFA was removed under an Ar stream, leaving the peptide film on the glass vial wall. Next, the peptide film was dissolved at the 0.5 mg mL–1 concentration in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) from Sigma-Aldrich by 5-min vortexing. Subsequently, HFIP was evaporated under an Ar stream, leaving the Aβ film on the glass vial wall. The dissolution of this Aβ film in HFIP and subsequent removal of HFIP were repeated once more. Next, the Aβ film was dissolved at the 0.25 mg mL–1 concentration in HFIP by 5-min vortexing. The Aβ solution was divided into 20 aliquots, each containing 50 μg of the peptide per centrifuge tube. The tubes were covered with Kimtech wipes to protect them from contamination and then left under the fume hood overnight to allow for HFIP evaporation. Next, residual HFIP was removed under decreased pressure in a desiccator for 1 h, and the resulting transparent peptide films were stored in the freezer at −20 °C. A single aliquot was used for each experiment. The peptide film was resuspended in 20 μL of dimethyl sulfoxide (DMSO) from Sigma-Aldrich. The Aβ aggregation was initiated by diluting the Aβ/DMSO solution to 50 μg mL–1 Aβ concentration with the 0.01 M phosphate buffer saline (PBS) solution. The PBS solution was prepared by dissolving PBS tablets from Sigma-Aldrich in 200 mL of Milli Q water, 18.2 MΩ cm, thus obtaining the 0.01 M phosphate buffer, 0.0027 M KCl, and 0.137 M NaCl (pH = 7.4) solution. Initially, the 50 μg mL–1 Aβ solution contained AβMs. The AβMs solution was then immediately stored in the fridge at 4 °C to aggregate for 24 h. The Aβ aggregation rate is lower at low temperatures.64 Therefore, storing AβMs solution at 4 °C for 24 h allowed us to obtain a solution of small, BLM-permeating AβOs, as we have previously shown.11,38
Incorporation of K162 into Aβ Solution
A powder of K162 (Scheme 1) from Sigma-Aldrich (No. 200487) was dissolved at 5 mM concentration in DMSO to make a K162 stock solution. Then, K162 was incorporated into Aβ solutions in two ways. In one, named the preincorporation protocol, the 20 μL of K162/DMSO stock solution was used to resuspend the 50-μg Aβ aliquot, thus obtaining the AβMs/K162/DMSO solution. This solution was then diluted to the 50-μg mL–1 Aβ concentration and subsequently allowed to aggregate in the fridge at 4 °C for 24 h, thus providing the same aggregation conditions as those in the drug absence. In the other way, named the external addition protocol, first, 50 μg of AβMs, subsequently dissolved in 20 μL of DMSO and 980 μL of PBS, was allowed to aggregate for 24 h at 4 °C. Next, the AβOs solution was mixed with the 20 μL of K162/DMSO stock solution. The final AβOs/K162 solution was stored again at 4 °C for 24 h. In this way, conditions of the K162 interaction with both AβMs and AβOs were identical. The concentration of K162 and Aβ in the Aβ-K162 mixture was 100 and 10 μM, respectively, resulting in the drug-to-peptide ratio, in all samples, of 10:1 (v:v).
Lipid Vesicle Preparation
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol (Chol), porcine brain sphingomyelin (SM), and ovine brain monosialoganglioside (GM1), purchased from Sigma-Aldrich, were used without further purification. Structural formulas of the lipids used are shown elsewhere.38 DSPE was dissolved in the chloroform:methanol (9:1, v:v) mixed solvent solution at 50 °C. Other lipids were dissolved in chloroform at room temperature. The lipid stock solutions were stored in the freezer at −20 °C. An aliquot of each lipid stock solution was transferred to a glass vial, and the final solution contained 1 mg of lipids. This mixed lipid solution contained 50% DSPE, 15% DPPC, 25% Chol, 8% SM, and 2% GM1 by weight. This lipid composition was used in our previous study38 to mimic aged lipid rafts formed in the cells’ membrane from the human frontal cortex found in AD patients’ brains.65 The solvents were evaporated under an Ar stream accompanied by vortexing to produce a lipid film on the glass vial’s bottom. Then, this film was resuspended in the PBS solution to reach the 1 mg mL–1 lipid concentration. Finally, lipid vesicles were formed after a 20-min sonication of the lipid solution at 45 °C using ultrasonic cleaner Sonorex Digiplus DL 102 H from Bandelin (Berlin, Germany).
In all experiments, the lipid vesicle and AβOs solutions (either without or with K162) were mixed to reach the peptide-to-lipid mass ratio of 1:20. Then, the mixture was sonicated for 10 min at room temperature and used immediately afterward.
Electrochemical Impedance Spectroscopy (EIS) Measurements
The vesicle fusion method was used to prepare a BLM.66 A single-crystal Au(111) electrode (surface area of 0.7854 cm2) was used as the working electrode for electrochemical measurements. Before use, this electrode was pretreated according to the previously developed procedure.67 Briefly, the electrode was rinsed with Milli-Q water and then flame annealed using a Bunsen burner. After cooling down to room temperature, the electrode was immersed in a 0.4 mg mL–1 1-thio-β-d-glucose (Tg) solution from Sigma-Aldrich for 5 h. That way, a self-assembled monolayer of Tg (SAM-Tg) was formed on the gold surface. The SAM-Tg provides a hydrophilic cushion layer that enhances vesicle fusion and relieves stress imposed by the gold substrate on the BLM.68 Moreover, it provides a water-rich layer underneath the lipid bilayer, thus mimicking the natural cell membrane environment. After SAM-Tg formation, the electrode was rinsed with Milli-Q water and then immersed in the lipid vesicle solution (either containing or not containing AβOs and K162) overnight. Finally, the electrodes were withdrawn from the solutions, and then the excess of the solution was gently removed with a Kimtech wipe.
The all-glass three-electrode cell was used for all electrochemical measurements. An Au(111), Au wire, and saturated calomel electrode (SCE) were used as the working, counter, and reference electrode, respectively. Before each measurement, the solution was purged for 30 min with an Ar stream for deaeration. During the experiments, an Ar cushion was flowing over the solution. The Au(111) electrode coated with the BLM was assembled in the electrochemical cell in the hanging meniscus configuration. That is, the electrode was slowly pushed down vertically toward the electrolyte solution until it touched it. Then, it was raised until a meniscus between the electrode surface and the electrolyte was formed.
Electrochemical impedance spectroscopy (EIS) measurements were conducted using VSP electrochemical interface (Biologic). The EIS spectra were acquired in the potential range of 0.3 to −0.4 V vs SCE. During EIS measurements, an excitation sinusoidal voltage signal of the amplitude of 10 mV was applied, and the spectra were recorded in the frequency range of 103 to 0.05 Hz. Equivalent electric circuits were fitted to the EIS data using ZView software (Scribner Associates Inc.).
Atomic Force Microscopy (AFM) Imaging
The BLM samples were imaged with AFM in the PeakForce quantitative nanomechanical mapping (PF-QNM) mode using a MultiMode 8 system (Bruker) equipped with an E scanner. The system was turned on and allowed to equilibrate for at least 30 min before each experiment. The BL-AC40TS (Olympus) and RTESPA300 (Bruker) cantilevers with a spring constant of 0.1 and 40 N m–1, and the resonance frequency of 50 and 300 kHz, respectively, were used for sample imaging in liquid and air, respectively. The AFM cantilevers were cleaned by consecutive immersing in a detergent bath, 2-propanol, and Milli-Q water for 10 min. Next, the cantilevers were ozonized in the UVC-1014 UV ozone cleaner (Nanobioanalytics, Berlin, Germany) for 10 min. The cantilevers were calibrated using the thermal tune method. The tip radius was determined by imaging the Ti roughness sample (Bruker) routinely used for tip radius determination.69,70 The V1 grade mica disks (Ted Pella, Inc.) were mounted on metallic disks using an adhesive tape. Next, mica was cleaned in ethanol and then in Milli-Q water. After drying with an Ar stream, its top layer was piled off using an adhesive tape, resulting in a clean and atomically flat surface. The samples were immediately deposited on the freshly cleaned mica surface.
The PF-QNM in a fluid mode was used to study the morphology of BLM, BLM-AβOs, and BLM-AβOs-K162 in the PBS solution (pH = 7.4) at 21 °C. Before the imaging, the fluid cell and AFM accessories were cleaned in a detergent bath, followed by sequential rinsing with ethanol and then Milli-Q water. A 30-μL aliquot of the lipid vesicle solution (either without or with AβOs and K162) was deposited on a freshly cleaved mica substrate and then left for 45 min to form BLM on the substrate surface. Finally, the sample was rinsed with Milli-Q water filtered through a Whatman syringe filter (GE Healthcare Life Sciences) of 0.02 μm porosity and then mounted for AFM imaging.
The PF-QNM in air mode was used for monitoring Aβ aggregation in the absence and presence of K162. A freshly prepared AβMs solution (either without or with K162) was deposited on a freshly cleaved mica substrate. After 5 min of deposition, the sample was rinsed with filtered Milli-Q water, dried with a gentle stream of Ar, and subsequently mounted for AFM imaging. The imaging was performed at 21 °C.
All AFM images were processed and analyzed using Gwyddion software.71
Molecular Dynamics (MD) Simulations
To gain molecular-level insight into the energetics of the Aβ interaction with K162, we carried out molecular dynamics simulations and postprocessed obtained trajectories using the Molecular Mechanics Poisson–Boltzmann Surface Area (MMPBSA)72 method. The AβM and AβD were constructed using the PDB:6RHY structure.48 The AβF structure was prepared using the PDB:2NAO structure.51 First, we simulated mixtures of AβMs, AβDs, or AβFs with K162 in the molar ratio of Aβ:K162 equal 1:10. We modeled solvent explicitly using the OPC3 water model,73 which provides a realistic description of solution dielectric properties and responses.74 The MD simulation protocol consisted of a sequence of the following steps: (i) steepest descent optimization of initial configuration, (ii) heating to room temperature, (iii) density optimization, and (iv) 50 ns production run within the isothermal–isobaric ensemble. The simulations were carried out under ambient conditions with the temperature controlled by stochastic Langevin thermostat (collision frequency γ = 2 ps–1) and pressure controlled by Berendsen barostat (coupling constant τ = 1 ps). Second, we extracted the bounded complexes of K162 with AβM, AβD, or AβF from the last configuration in the mixture production runs. These complexes were immersed in bulk water and then simulated for another 50 ns. Finally, the single-complex trajectories were postprocessed using the MMPBSA72 method to estimate the K162-Aβ binding energies. The interaction parameters for Aβ were assigned based on the residue-partitioning and connectivity using the Amber ff14SB75 force field. The interaction model for K162 was developed using the Density Function Theory (theory level B3LYP/6-311++G**). This procedure included three steps: i.e., (i) geometry optimization, (ii) Merz–Singh–Kollman76 electrostatic potential partitioning into partial charges, and (iii) assignment of short-range interaction parameters and bonding terms from the GAFF77 force-field library. The first-principle calculations were carried out using NWChem78 and Gaussian79 packages; the MD simulations were carried out using both Amber80 and Gromacs81 packages.
Acknowledgments
This research was supported by receiving funding from the Polish National Science Centre, grant No. OPUS12 2016/23B/ST4/02791, awarded to P.P. The research activity of D.M. was supported by funds from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 711859 and by financial resources for science in the years 2017–2021 awarded by the Polish Ministry of Science and Higher Education for the implementation of an international cofinanced project. P.Z. acknowledges support from the US Department of Energy (DOE) Chemical Sciences, Geosciences, and Biosciences Division under Contract DE-AC02-05CH11231. The collaboration between the Polish Academy of Sciences and the Lawrence Berkeley National Laboratory was supported by grant NCN Sonata-Bis (DEC-2016/22/E/ST4/00446). J.L. acknowledges support of Natural Sciences and Engineering Research Council of Canada (NSERC) grant (RG-03958).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00754.
Figure S1, impedance and phase angle as function of frequency for BLM and BLM-AβOs-K162; Figure S2, two independent measurements of membrane resistance as function of applied potential in PBS; Figure S3, AFM image and corresponding cross-sectional profile of BLM; Figure S4, depth of pores and equivalent disk radius of AβO clusters surrounding pores in BLM-AβOs; Figure S5, AFM topography image and corresponding cross-sectional profile of BLM in presence of K162 in PBS; Figure S6, AFM imaged topography of elongated Aβ aggregates formed in absence and resence of K162 after 48 h of Aβ aggregation; Table S1, numerical results of equivalent electric circuits fittings to the EIS data; Table S2, population of different Aβ forms during Aβ aggregation; and Table S3, binding energies of interactions of Aβ with K162 (PDF)
Author Contributions
P.P. designed the study and managed the project. D.M. conducted the electrochemical and AFM measurements. P.Z. performed molecular dynamics simulations. D.M. and M.M. prepared the samples. D.M. conducted the data processing. D.M., I.S.P., R.N., and P.P. analyzed the data. D.M. and P.P. drafted the main text of the manuscript. D.M., W.K., J.L., and P.P. wrote the manuscript. All authors have given their final approval for the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Scheltens P.; Blennow K.; Breteler M. M. B.; de Strooper B.; Frisoni G. B.; Salloway S.; Van der Flier W. M. (2016) Alzheimer’s Disease. Lancet 388 (10043), 505–517. 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Cukalevski R.; Michaels T. C. T.; Šarić A.; Törnquist M.; Vendruscolo M.; Dobson C. M.; Buell A. K.; Knowles T. P. J.; Linse S. (2018) Distinct Thermodynamic Signatures of Oligomer Generation in the Aggregation of the Amyloid-β Peptide. Nat. Chem. 10 (5), 523–531. 10.1038/s41557-018-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield S. M.; Lashuel H. A. (2010) Amyloidogenic Protein-Membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem., Int. Ed. 49 (33), 5628–5654. 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- Arosio P.; Knowles T. P. J.; Linse S. (2015) On the Lag Phase in Amyloid Fibril Formation. Phys. Chem. Chem. Phys. 17 (12), 7606–7618. 10.1039/C4CP05563B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagmeier P.; De S.; Wirthensohn D. C.; Lee S. F.; Vincke C.; Muyldermans S.; Knowles T. P. J.; Gandhi S.; Dobson C. M.; Klenerman D. (2017) Ultrasensitive Measurement of Ca 2+ Influx into Lipid Vesicles Induced by Protein Aggregates. Angew. Chem., Int. Ed. 56 (27), 7750–7754. 10.1002/anie.201700966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. (2009) Structure-Neurotoxicity Relationships of Amyloid β-Protein Oligomers. Proc. Natl. Acad. Sci. U. S. A. 106 (35), 14745–14750. 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizas P.; Budvytyte R.; Morkuniene R.; Moldovan R.; Broccio M.; Lösche M.; Niaura G.; Valincius G.; Borutaite V. (2010) Size-Dependent Neurotoxicity of β-Amyloid Oligomers. Arch. Biochem. Biophys. 496 (2), 84–92. 10.1016/j.abb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N.; Rojas E.; Pollard H. B. (1993) Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. U. S. A. 90 (2), 567–571. 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist A.; Doudevski I.; Lin H.; Azimova R.; Ng D.; Frangione B.; Kagan B.; Ghiso J.; Lal R. (2005) Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc. Natl. Acad. Sci. U. S. A. 102 (30), 10427–10432. 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode D. C.; Freeley M.; Nield J.; Palma M.; Viles J. H. (2019) Amyloid-β Oligomers Have a Profound Detergent-like Effect on Lipid Membrane Bilayers, Imaged by Atomic Force and Electron Microscopy. J. Biol. Chem. 294 (19), 7566–7572. 10.1074/jbc.AC118.007195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrdenovic D.; Majewska M.; Pieta I. S.; Bernatowicz P.; Nowakowski R.; Kutner W.; Lipkowski J.; Pieta P. (2019) Size-Dependent Interaction of Amyloid β Oligomers with Brain Total Lipid Extract Bilayer—Fibrillation Versus Membrane Destruction. Langmuir 35 (36), 11940–11949. 10.1021/acs.langmuir.9b01645. [DOI] [PubMed] [Google Scholar]
- Delgado D. A.; Doherty K.; Cheng Q.; Kim H.; Xu D.; Dong H.; Grewer C.; Qiang W. (2016) Distinct Membrane Disruption Pathways Are Induced by 40-Residue β-Amyloid Peptides. J. Biol. Chem. 291 (23), 12233–12244. 10.1074/jbc.M116.720656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. (2008) Amyloid-β Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 14 (8), 837–842. 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Donald J. M.; Savva G. M.; Brayne C.; Welzel A. T.; Forster G.; Shankar G. M.; Selkoe D. J.; Ince P. G.; Walsh D. M. (2010) The Presence of Sodium Dodecyl Sulphate-Stable Aβ Dimers Is Strongly Associated with Alzheimer-Type Dementia. Brain 133 (5), 1328–1341. 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar G. M.; Bloodgood B. L.; Townsend M.; Walsh D. M.; Selkoe D. J.; Sabatini B. L. (2007) Natural Oligomers of the Alzheimer Amyloid- Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 27 (11), 2866–2875. 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor P. N.; Buniel M. C.; Furlow P. W.; Sanz Clemente A.; Velasco P. T.; Wood M.; Viola K. L.; Klein W. L. (2007) A Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 27 (4), 796–807. 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G.; Jäättelä M. (2005) Lysosomes and Autophagy in Cell Death Control. Nat. Rev. Cancer 5 (11), 886–897. 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- Rosales-Corral S.; Acuna-Castroviejo D.; Tan D. X.; López-Armas G.; Cruz-Ramos J.; Munoz R.; Melnikov V. G.; Manchester L. C.; Reiter R. J. (2012) Accumulation of Exogenous Amyloid- Beta Peptide in Hippocampal Mitochondria Causes Their Dysfunction: A Protective Role for Melatonin. Oxid. Med. Cell. Longevity 2012, 1–15. 10.1155/2012/843649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez-Prieto M.; Velasco A.; Tabernero A.; Medina J. M. (2018) Endocytosis and Transcytosis of Amyloid-β Peptides by Astrocytes: A Possible Mechanism for Amyloid-β Clearance in Alzheimer’s Disease. J. Alzheimer's Dis. 65 (4), 1109–1124. 10.3233/JAD-180332. [DOI] [PubMed] [Google Scholar]
- Tomiyama T.; Matsuyama S.; Iso H.; Umeda T.; Takuma H.; Ohnishi K.; Ishibashi K.; Teraoka R.; Sakama N.; Yamashita T.; Nishitsuji K.; Ito K.; Shimada H.; Lambert M. P.; Klein W. L.; Mori H. (2010) A Mouse Model of Amyloid Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss In Vivo. J. Neurosci. 30 (14), 4845–4856. 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti M. T.; Bruno M. A.; Ducatenzeiler A.; Klein W. L.; Cuello A. C. (2012) Intracellular Aβ-Oligomers and Early Inflammation in a Model of Alzheimer’s Disease. Neurobiol. Aging 33 (7), 1329–1342. 10.1016/j.neurobiolaging.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Thapa A.; Jett S. D.; Chi E. Y. (2016) Curcumin Attenuates Amyloid-β Aggregate Toxicity and Modulates Amyloid-β Aggregation Pathway. ACS Chem. Neurosci. 7 (1), 56–68. 10.1021/acschemneuro.5b00214. [DOI] [PubMed] [Google Scholar]
- Lopez del Amo J. M.; Fink U.; Dasari M.; Grelle G.; Wanker E. E.; Bieschke J.; Reif B. (2012) Structural Properties of EGCG-Induced, Nontoxic Alzheimer’s Disease Aβ Oligomers. J. Mol. Biol. 421 (4–5), 517–524. 10.1016/j.jmb.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. (2008) EGCG Redirects Amyloidogenic Polypeptides into Unstructured, off-Pathway Oligomers. Nat. Struct. Mol. Biol. 15 (6), 558–566. 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Arosio P.; Presto J.; Kurudenkandy F. R.; Biverstål H.; Dolfe L.; Dunning C.; Yang X.; Frohm B.; Vendruscolo M.; Johansson J.; Dobson C. M.; Fisahn A.; Knowles T. P. J.; Linse S. (2015) A Molecular Chaperone Breaks the Catalytic Cycle That Generates Toxic Aβ Oligomers. Nat. Struct. Mol. Biol. 22 (3), 207–213. 10.1038/nsmb.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt T.; Łapińska U.; Kumita J. R.; Whiten D. R.; Klenerman D.; Wilson M. R.; Cohen S. I. A.; Linse S.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J.; Arosio P. (2019) Secondary Nucleation and Elongation Occur at Different Sites on Alzheimer’s Amyloid-β Aggregates. Sci. Adv. 5 (4), eaau3112 10.1126/sciadv.aau3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbocker R.; Chia S.; Ruggeri F. S.; Perni M.; Cascella R.; Heller G. T.; Meisl G.; Mannini B.; Habchi J.; Michaels T. C. T.; Challa P. K.; Ahn M.; Casford S. T.; Fernando N.; Xu C. K.; Kloss N. D.; Cohen S. I. A.; Kumita J. R.; Cecchi C.; Zasloff M.; Linse S.; Knowles T. P. J.; Chiti F.; Vendruscolo M.; Dobson C. M. (2019) Trodusquemine Enhances Aβ42 Aggregation but Suppresses Its Toxicity by Displacing Oligomers from Cell Membranes. Nat. Commun. 10 (1), 225. 10.1038/s41467-018-07699-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieschke J.; Herbst M.; Wiglenda T.; Friedrich R. P.; Boeddrich A.; Schiele F.; Kleckers D.; Lopez del Amo J. M.; Grüning B. A.; Wang Q.; Schmidt M. R.; Lurz R.; Anwyl R.; Schnoegl S.; Fändrich M.; Frank R. F.; Reif B.; Günther S.; Walsh D. M.; Wanker E. E. (2012) Small-Molecule Conversion of Toxic Oligomers to Nontoxic β-Sheet-Rich Amyloid Fibrils. Nat. Chem. Biol. 8 (1), 93–101. 10.1038/nchembio.719. [DOI] [PubMed] [Google Scholar]
- Yang J.; Dear A. J.; Yao Q.-Q.; Liu Z.; Dobson C. M.; Knowles T. P. J.; Wu S.; Perrett S. (2020) Amelioration of Aggregate Cytotoxicity by Catalytic Conversion of Protein Oligomers into Amyloid Fibrils. Nanoscale 12 (36), 18663–18672. 10.1039/D0NR01481H. [DOI] [PubMed] [Google Scholar]
- Bernabeu-Zornoza A.; Coronel R.; Palmer C.; Monteagudo M.; Zambrano A.; Liste I. (2019) Physiological and Pathological Effects of Amyloid-β Species in Neural Stem Cell Biology. Neural Regener. Res. 14 (12), 2035. 10.4103/1673-5374.262571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Dong C. (2009) Aβ40 Promotes Neuronal Cell Fate in Neural Progenitor Cells. Cell Death Differ. 16 (3), 386–394. 10.1038/cdd.2008.94. [DOI] [PubMed] [Google Scholar]
- Bernabeu-Zornoza A.; Coronel R.; Palmer C.; Calero M.; Martínez-Serrano A.; Cano E.; Zambrano A.; Liste I. (2019) Aβ42 Peptide Promotes Proliferation and Gliogenesis in Human Neural Stem Cells. Mol. Neurobiol. 56 (6), 4023–4036. 10.1007/s12035-018-1355-7. [DOI] [PubMed] [Google Scholar]
- Kim J.; Onstead L.; Randle S.; Price R.; Smithson L.; Zwizinski C.; Dickson D. W.; Golde T.; McGowan E. (2007) A 40 Inhibits Amyloid Deposition In Vivo. J. Neurosci. 27 (3), 627–633. 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida M. L.; Caraci F.; Pignataro B.; Cataldo S.; De Bona P.; Bruno V.; Molinaro G.; Pappalardo G.; Messina A.; Palmigiano A.; Garozzo D.; Nicoletti F.; Rizzarelli E.; Copani A. (2009) β-Amyloid Monomers Are Neuroprotective. J. Neurosci. 29 (34), 10582–10587. 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.-S.; Maezawa I.; Budamagunta M.; Rana S.; Shi A.; Vassar R.; Liu R.; Lam K. S.; Cheng R. H.; Hua D. H.; Voss J. C.; Jin L.-W. (2010) Candidate Anti-Aβ Fluorene Compounds Selected from Analogs of Amyloid Imaging Agents. Neurobiol. Aging 31 (10), 1690–1699. 10.1016/j.neurobiolaging.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z.; Shodiev M.; Leitch J. J.; Abbasi F.; Lipkowski J. (2018) Role of Transmembrane Potential and Defects on the Permeabilization of Lipid Bilayers by Alamethicin, an Ion-Channel-Forming Peptide. Langmuir 34 (21), 6249–6260. 10.1021/acs.langmuir.8b00928. [DOI] [PubMed] [Google Scholar]
- Su Z.; Ho D.; Merrill A. R.; Lipkowski J. (2019) In Situ Electrochemical and PM-IRRAS Studies of Colicin E1 Ion Channels in the Floating Bilayer Lipid Membrane. Langmuir 35 (25), 8452–8459. 10.1021/acs.langmuir.9b01251. [DOI] [PubMed] [Google Scholar]
- Mrdenovic D.; Su Z.; Kutner W.; Lipkowski J.; Pieta P. (2020) Alzheimer’s Disease-Related Amyloid β Peptide Causes Structural Disordering of Lipids and Changes the Electric Properties of a Floating Bilayer Lipid Membrane. Nanoscale Adv. 2 (8), 3467–3480. 10.1039/D0NA00292E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valincius G., and Mickevicius M. (2015) Tethered Phospholipid Bilayer Membranes. In Advances in Planar Lipid Bilayers and Liposomes, Vol. 21, pp 27–61, Elsevier Inc., 10.1016/bs.adplan.2015.01.003. [DOI] [Google Scholar]
- Heberle F. A.; Wu J.; Goh S. L.; Petruzielo R. S.; Feigenson G. W. (2010) Comparison of Three Ternary Lipid Bilayer Mixtures: FRET and ESR Reveal Nanodomains. Biophys. J. 99 (10), 3309–3318. 10.1016/j.bpj.2010.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konyakhina T. M.; Wu J.; Mastroianni J. D.; Heberle F. A.; Feigenson G. W. (2013) Phase Diagram of a 4-Component Lipid Mixture: DSPC/DOPC/POPC/Chol. Biochim. Biophys. Acta, Biomembr. 1828 (9), 2204–2214. 10.1016/j.bbamem.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle F. A.; Feigenson G. W. (2011) Phase Separation in Lipid Membranes. Cold Spring Harbor Perspect. Biol. 3 (4), a004630. 10.1101/cshperspect.a004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman D. G.; Feigenson G. W. (2015) Lipid Bilayers: Clusters, Domains and Phases. Essays Biochem. 57, 33–42. 10.1042/bse0570033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S.; Wirthensohn D. C.; Flagmeier P.; Hughes C.; Aprile F. A.; Ruggeri F. S.; Whiten D. R.; Emin D.; Xia Z.; Varela J. A.; Sormanni P.; Kundel F.; Knowles T. P. J.; Dobson C. M.; Bryant C.; Vendruscolo M.; Klenerman D. (2019) Different Soluble Aggregates of Aβ42 Can Give Rise to Cellular Toxicity through Different Mechanisms. Nat. Commun. 10 (1), 1541. 10.1038/s41467-019-09477-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z.; Aucoin D.; Davis J.; Van Nostrand W. E.; Smith S. O. (2015) Mechanism of Nucleated Conformational Conversion of Aβ42. Biochemistry 54 (27), 4197–4207. 10.1021/acs.biochem.5b00467. [DOI] [PubMed] [Google Scholar]
- Fu Z.; Aucoin D.; Ahmed M.; Ziliox M.; Van Nostrand W. E.; Smith S. O. (2014) Capping of Aβ42 Oligomers by Small Molecule Inhibitors. Biochemistry 53 (50), 7893–7903. 10.1021/bi500910b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dear A. J.; Michaels T. C. T.; Meisl G.; Klenerman D.; Wu S.; Perrett S.; Linse S.; Dobson C. M.; Knowles T. P. J. (2020) Kinetic Diversity of Amyloid Oligomers. Proc. Natl. Acad. Sci. U. S. A. 117 (22), 12087–12094. 10.1073/pnas.1922267117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciudad S.; Puig E.; Botzanowski T.; Meigooni M.; Arango A. S.; Do J.; Mayzel M.; Bayoumi M.; Chaignepain S.; Maglia G.; Cianferani S.; Orekhov V.; Tajkhorshid E.; Bardiaux B.; Carulla N. (2020) Aβ(1–42) Tetramer and Octamer Structures Reveal Edge Conductivity Pores as a Mechanism for Membrane Damage. Nat. Commun. 11 (1), 3014. 10.1038/s41467-020-16566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerf E.; Sarroukh R.; Tamamizu-Kato S.; Breydo L.; Derclaye S.; Dufrêne Y. F.; Narayanaswami V.; Goormaghtigh E.; Ruysschaert J.-M.; Raussens V. (2009) Antiparallel β-Sheet: A Signature Structure of the Oligomeric Amyloid β-Peptide. Biochem. J. 421 (3), 415–423. 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- Gu L.; Liu C.; Stroud J. C.; Ngo S.; Jiang L.; Guo Z. (2014) Antiparallel Triple-Strand Architecture for Prefibrillar Aβ42 Oligomers. J. Biol. Chem. 289 (39), 27300–27313. 10.1074/jbc.M114.569004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälti M. A.; Ravotti F.; Arai H.; Glabe C. G.; Wall J. S.; Böckmann A.; Güntert P.; Meier B. H.; Riek R. (2016) Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A. 113 (34), E4976–E4984. 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Gonnelli L.; Luchinat C.; Mao J.; Nesi A. (2011) A New Structural Model of Aβ 40 Fibrils. J. Am. Chem. Soc. 133 (40), 16013–16022. 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- Tycko R. (2011) Solid-State NMR Studies of Amyloid Fibril Structure. Annu. Rev. Phys. Chem. 62 (1), 279–299. 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin M. T.; Silvers R.; Ni Q. Z.; Can T. V.; Sergeyev I.; Rosay M.; Donovan K. J.; Michael B.; Wall J.; Linse S.; Griffin R. G. (2016) Atomic Resolution Structure of Monomorphic Aβ 42 Amyloid Fibrils. J. Am. Chem. Soc. 138 (30), 9663–9674. 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abelein A.; Abrahams J. P.; Danielsson J.; Gräslund A.; Jarvet J.; Luo J.; Tiiman A.; Wärmländer S. K. T. S. (2014) The Hairpin Conformation of the Amyloid β Peptide Is an Important Structural Motif along the Aggregation Pathway. JBIC, J. Biol. Inorg. Chem. 19 (4–5), 623–634. 10.1007/s00775-014-1131-8. [DOI] [PubMed] [Google Scholar]
- Tomaselli S.; Esposito V.; Vangone P.; van Nuland N. A. J.; Bonvin A. M. J. J.; Guerrini R.; Tancredi T.; Temussi P. A.; Picone D. (2006) The α-to-β Conformational Transition of Alzheimer’s Aβ-(1–42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of β Conformation Seeding. ChemBioChem 7 (2), 257–267. 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- Smith J. F.; Knowles T. P. J.; Dobson C. M.; MacPhee C. E.; Welland M. E. (2006) Characterization of the Nanoscale Properties of Individual Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A. 103 (43), 15806–15811. 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri F. S.; Adamcik J.; Jeong J. S.; Lashuel H. A.; Mezzenga R.; Dietler G. (2015) Influence of the β-Sheet Content on the Mechanical Properties of Aggregates during Amyloid Fibrillization. Angew. Chem., Int. Ed. 54 (8), 2462–2466. 10.1002/anie.201409050. [DOI] [PubMed] [Google Scholar]
- Mannini B.; Mulvihill E.; Sgromo C.; Cascella R.; Khodarahmi R.; Ramazzotti M.; Dobson C. M.; Cecchi C.; Chiti F. (2014) Toxicity of Protein Oligomers Is Rationalized by a Function Combining Size and Surface Hydrophobicity. ACS Chem. Biol. 9 (10), 2309–2317. 10.1021/cb500505m. [DOI] [PubMed] [Google Scholar]
- Bolognesi B.; Kumita J. R.; Barros T. P.; Esbjorner E. K.; Luheshi L. M.; Crowther D. C.; Wilson M. R.; Dobson C. M.; Favrin G.; Yerbury J. J. (2010) ANS Binding Reveals Common Features of Cytotoxic Amyloid Species. ACS Chem. Biol. 5 (8), 735–740. 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- Ladiwala A. R. A.; Litt J.; Kane R. S.; Aucoin D. S.; Smith S. O.; Ranjan S.; Davis J.; Van Nostrand W. E.; Tessier P. M. (2012) Conformational Differences between Two Amyloid β Oligomers of Similar Size and Dissimilar Toxicity. J. Biol. Chem. 287 (29), 24765–24773. 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R.; Akcan M.; Khondker A.; Rheinstädter M. C.; Bozelli J. C.; Epand R. M.; Huynh V.; Wylie R. G.; Boulton S.; Huang J.; Verschoor C. P.; Melacini G. (2019) Atomic Resolution Map of the Soluble Amyloid Beta Assembly Toxic Surfaces. Chem. Sci. 10 (24), 6072–6082. 10.1039/C9SC01331H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao S.-C.; Ma K.; Talafous J.; Orlando R.; Zagorski M. G. (1997) Trifluoroacetic Acid Pretreatment Reproducibly Disaggregates the Amyloid β-Peptide. Amyloid 4 (4), 240–252. 10.3109/13506129709003835. [DOI] [Google Scholar]
- Kusumoto Y.; Lomakin A.; Teplow D. B.; Benedek G. B. (1998) Temperature Dependence of Amyloid -Protein Fibrillization. Proc. Natl. Acad. Sci. U. S. A. 95 (21), 12277–12282. 10.1073/pnas.95.21.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz M.; Fabelo N.; Ferrer I.; Marín R. (2018) Lipid Raft Aging” in the Human Frontal Cortex during Nonpathological Aging: Gender Influences and Potential Implications in Alzheimer’s Disease. Neurobiol. Aging 67, 42–52. 10.1016/j.neurobiolaging.2018.02.022. [DOI] [PubMed] [Google Scholar]
- McConnell H. M.; Watts T. H.; Weis R. M.; Brian A. A. (1986) Supported Planar Membranes in Studies of Cell-Cell Recognition in the Immune System. Biochim. Biophys. Acta, Rev. Biomembr. 864 (1), 95–106. 10.1016/0304-4157(86)90016-X. [DOI] [PubMed] [Google Scholar]
- Richer J.; Lipkowski J. (1986) Measurement of Physical Adsorption of Neutral Organic Species at Solid Electrodes. J. Electrochem. Soc. 133 (1), 121–128. 10.1149/1.2108505. [DOI] [Google Scholar]
- Kycia A. H.; Wang J.; Merrill A. R.; Lipkowski J. (2011) Atomic Force Microscopy Studies of a Floating-Bilayer Lipid Membrane on a Au(111) Surface Modified with a Hydrophilic Monolayer. Langmuir 27 (17), 10867–10877. 10.1021/la2016269. [DOI] [PubMed] [Google Scholar]
- Hutter J. L.; Bechhoefer J. (1993) Calibration of Atomic-force Microscope Tips. Rev. Sci. Instrum. 64 (7), 1868–1873. 10.1063/1.1143970. [DOI] [Google Scholar]
- Lévy R.; Maaloum M. (2002) Measuring the Spring Constant of Atomic Force Microscope Cantilevers: Thermal Fluctuations and Other Methods. Nanotechnology 13 (1), 33–37. 10.1088/0957-4484/13/1/307. [DOI] [Google Scholar]
- Nečas D.; Klapetek P. (2012) Gwyddion: An Open-Source Software for SPM Data Analysis. Open Phys. 10 (1), 181–188. 10.2478/s11534-011-0096-2. [DOI] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. (2000) Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 33 (12), 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Izadi S.; Onufriev A. V. (2016) Accuracy Limit of Rigid 3-Point Water Models. J. Chem. Phys. 145 (7), 074501. 10.1063/1.4960175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzycki P.; Gilbert B. (2020) Temperature-Dependence of the Dielectric Relaxation of Water Using Non-Polarizable Water Models. Phys. Chem. Chem. Phys. 22 (3), 1011–1018. 10.1039/C9CP04578C. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. (2015) Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh U. C.; Kollman P. A. (1984) An Approach to Computing Electrostatic Charges for Molecules. J. Comput. Chem. 5 (2), 129–145. 10.1002/jcc.540050204. [DOI] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. (2004) Development and Testing of a General Amber Force Field. J. Comput. Chem. 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Aprà E.; Bylaska E. J.; de Jong W. A.; Govind N.; Kowalski K.; Straatsma T. P.; Valiev M.; van Dam H. J. J.; Alexeev Y.; Anchell J.; Anisimov V.; Aquino F. W.; Atta-Fynn R.; Autschbach J.; Bauman N. P.; Becca J. C.; Bernholdt D. E.; Bhaskaran-Nair K.; Bogatko S.; Borowski P.; Boschen J.; Brabec J.; Bruner A.; Cauët E.; Chen Y.; Chuev G. N.; Cramer C. J.; Daily J.; Deegan M. J. O.; Dunning T. H.; Dupuis M.; Dyall K. G.; Fann G. I.; Fischer S. A.; Fonari A.; Früchtl H.; Gagliardi L.; Garza J.; Gawande N.; Ghosh S.; Glaesemann K.; Götz A. W.; Hammond J.; Helms V.; Hermes E. D.; Hirao K.; Hirata S.; Jacquelin M.; Jensen L.; Johnson B. G.; Jónsson H.; Kendall R. A.; Klemm M.; Kobayashi R.; Konkov V.; Krishnamoorthy S.; Krishnan M.; Lin Z.; Lins R. D.; Littlefield R. J.; Logsdail A. J.; Lopata K.; Ma W.; Marenich A. V.; Martin del Campo J.; Mejia-Rodriguez D.; Moore J. E.; Mullin J. M.; Nakajima T.; Nascimento D. R.; Nichols J. A.; Nichols P. J.; Nieplocha J.; Otero-De-La-Roza A.; Palmer B.; Panyala A.; Pirojsirikul T.; Peng B.; Peverati R.; Pittner J.; Pollack L.; Richard R. M.; Sadayappan P.; Schatz G. C.; Shelton W. A.; Silverstein D. W.; Smith D. M. A.; Soares T. A.; Song D.; Swart M.; Taylor H. L.; Thomas G. S.; Tipparaju V.; Truhlar D. G.; Tsemekhman K.; Van Voorhis T.; Vázquez-Mayagoitia Á.; Verma P.; Villa O.; Vishnu A.; Vogiatzis K. D.; Wang D.; Weare J. H.; Williamson M. J.; Windus T. L.; Woliński K.; Wong A. T.; Wu Q.; Yang C.; Yu Q.; Zacharias M.; Zhang Z.; Zhao Y.; Harrison R. J. (2020) NWChem: Past, Present, and Future. J. Chem. Phys. 152 (18), 184102. 10.1063/5.0004997. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., and Fox D. J. (2016) Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT.
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. (2005) The Amber Biomolecular Simulation Programs. J. Comput. Chem. 26 (16), 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. (1995) GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 91 (1–3), 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.