

Abstract
Developing drugs for the central nervous system (CNS) requires fine chemical modifications, as a strict balance between size and lipophilicity is necessary to improve the permeability through the blood-brain barrier (BBB). In this context, morpholine and its analogues represent valuable heterocycles, due to their conformational and physicochemical properties. In fact, the presence of a weak basic nitrogen atom and of an oxygen atom at the opposite position provides a peculiar pKa value and a flexible conformation to the ring, thus allowing it to take part in several lipophilic–hydrophilic interactions, and to improve blood solubility and brain permeability of the overall structure. In CNS-active compounds, morpholines are used (1) to enhance the potency through molecular interactions, (2) to act as a scaffold directing the appendages in the correct position, and (3) to modulate pharmacokinetic/pharmacodynamic (PK/PD) properties. In this perspective, selected morpholine-containing CNS drug candidates are discussed to reveal the active pharmacophores accountable for the (1) modulation of receptors involved in mood disorders and pain, (2) bioactivity toward enzymes and receptors responsible for neurodegenerative diseases, and (3) inhibition of enzymes involved in the pathology of CNS tumors. The medicinal chemistry/pharmacological activity of morpholine derivatives is discussed, in the effort to highlight the importance of morpholine ring interactions in the active site of different targets, particularly reporting binding features retrieved from PDB data, when available.
Keywords: brain, blood-brain barrier, Alzheimer’s disease, Parkinson’s disease, cancer, enzyme inhibitor
Introduction
The development of treatments for central nervous system (CNS) disorders is accompanied by challenging tasks and difficulties, as compared to other therapeutic areas. In general, the chance for CNS drug candidates to reach the market is about 50% lower than that for other drug applications, and their development costs are 30% higher relative to most other areas of drug discovery.1 It is not surprising that several Big Pharma companies have reduced research in this area in recent years, despite the multibillion-dollar interest in developing drugs for diseases such as Alzheimer’s, Parkinson’s, depression, anxiety, schizophrenia, and stroke, that are of paramount importance in the context of an aging global population.2 The high cost and low approval rate of CNS drugs is mainly related to the fact that the majority of neuropsychiatric leads fail in the pricey Phase III of clinical trials. The reasons for the neuro-failures are numerous, ranging from the inadequate understanding of the biology behind CNS (brain and spinal cord) to the lack of reliable animal models, and also due to complications associated with the blood-brain barrier (BBB), a dynamic and selective membrane that restricts the flow to foreign agents, thus requiring fine chemical modifications of drug candidates. Generally, large size, high topological polar surface area, and a high degree of hydrogen bonding are not compatible with the passive diffusion of the molecules through the BBB,3 thus limiting the use of antibodies and peptides as orally administered CNS drugs.4−7 This is complicated in some patients with neurodegenerative conditions, where BBB is altered, thus affecting further drug permeability uptake.8 A balance between decreasing size and increasing lipophilicity is necessary to improve the penetrability of a drug,9,10 while simultaneously ensuring that P-glycoprotein-mediated efflux or plasma-mediated or fatty tissue-mediated drug sequestration are not activated.11 Thus, even though drugs affecting the CNS were the first to be discovered by humans thousands of years ago, there is still room for improving the exploration of the CNS-relevant chemical space.12 Among the many different N-containing heterocycles, morpholine represents one of the most useful scaffolds for the development of CNS drug candidates, thanks to its well-balanced lipophilic–hydrophilic profile, the reduced pKa value, and the chairlike flexible conformation. This heterocycle has been the object of a plethora of research studies, and several review papers have been reported regarding synthetic methodologies for its preparation,13−16 as well as the different biological activities displayed by compounds in which it is found.17−22 In fact, morpholine is used to modulate the pharmacokinetic/pharmacodynamic (PK/PD) properties of the overall structure, as the presence of a weak basic nitrogen at the opposite position of the oxygen atom provides to the ring a pKa value similar to the pH of blood, thus enhancing solubility and brain permeability. Also, it can be added to enhance the potency toward a target protein, as the oxygen atom can form hydrogen bonds, while the relatively electron-deficient ring can establish hydrophobic interactions.23 Finally, the flexible conformation resulting from the equilibrium between the chairlike and skew-boat topologies provides the optimal features for a suitable scaffold directing the appendages in the right position.24,25 Morpholine has also an improved CYP3A4 profile, prolonged bioavailability, and optimal clearance, being oxidized easily into nontoxic derivatives.17−22 A plethora of morpholine derivatives have already been present in the market for several decades (Figure 1). Just to give some examples, doxapram (1976),26 phendimetrazine (1979),27 moclobemide (1992),28 reboxetine (1997),29 and aprepitant (2003)30−32 are drugs with applications in several CNS diseases, mainly as anxiolytics and/or antidepressants.
Figure 1.

Approved morpholine-containing CNS drugs.
Several morpholine-containing pharmacophores have been identified as responsible for the selective interaction with specific molecular targets, such as aryl-morpholines interacting with PI3K kinase family33 or morpholine–azaindoles with cannabinoid receptors.34 They are structurally similar to endogenous neurotransmitters (as in the case of aryl-morpholines) and it has been demonstrated that they play a key role in improving the crossing of the blood-brain barrier. Although the biological activity of morpholine derivatives has been reviewed elsewhere,17−22 herein selected morpholine-containing CNS drug candidates are discussed to reveal the active pharmacophores accountable for the modulation of (1) receptors involved in mood disorders and pain, (2) enzymes and receptors responsible for neurodegenerative diseases, and (3) enzymes involved in the pathology of CNS tumors. The medicinal chemistry/pharmacological activity of morpholine derivatives is discussed, in the effort to highlight the importance of morpholine ring interactions in the active site of different targets, particularly reporting the binding features retrieved from PDB data, when available. The role of the morpholine ring is highlighted using different colors, depending on whether it acts as (1) interacting element (red), (2) scaffold (blue), or (3) PK/PD modulator (green) and, when appropriate, by comparing morpholine-containing compounds with analogues that lack this ring.
Biological Activity of Morpholine Derivatives in Mood Disorders and Pain
The human nervous system comprises millions of neurons, that communicate quickly between each other thanks to the action of a plethora of different receptors. Three different classes of receptors have been identified in the CNS, many of them involved in the pathological development of different disorders. Although their classification still needs to be clearly understood, neurotransmitter receptors can be divided into ionotropic receptors, ligand-gated ion channels that are excited by the binding of neurotransmitters activating the passage of different ions, and metabotropic receptors, often G protein-coupled receptors that respond to the binding of the neurotransmitter by activating a signal transduction pathway inside the cell. For some of the most important classes of neurotransmitter receptors involved in the regulation of mood disorders and pain control, morpholine derivatives are found to be able to desensitize or downregulate their mode of action, as they usually present structural similarity with their endogenous ligands (Figure 2). In some cases, a precise pharmacophore containing the morpholine ring can be identified, whereas in some other cases, the morpholine ring plays a key role by improving the overall pharmacokinetic profile and the brain permeability of the final compound.
Figure 2.
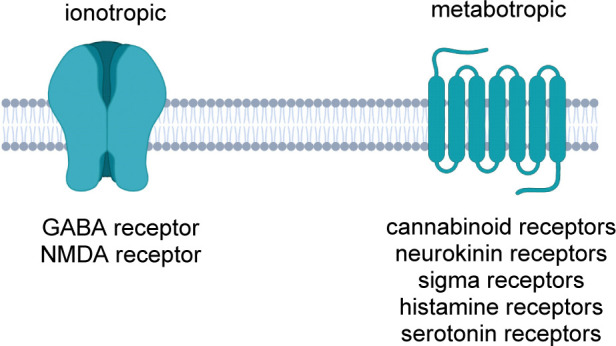
Neuroreceptors as molecular targets of morpholine-containing compounds.
Cannabinoid receptors interact endogenously with several different ligands, and their partial agonism is of interest for the development of therapeutics for pain management, inflammation, obesity, and substance abuse disorders.35,36 Of the many different compounds that can act as agonists toward the two main CB1 and CB2 receptors, aminoalkylindoles (AAI) are very active, such as pravadoline, discovered serendipitously in the 1990s,37,38 or compounds 1 and 2,39 although they do not share any obvious structural similarity with the classical cannabinoid (−)-Δ9-THC or with endogenous fatty acid ethanolamide ligands (such as anandamide) (Figure 3).40 The crystal structures of the human CB1 receptor41 and CB2 receptor42 have been resolved only in recent years and provided important insights for the development of selective CB2 receptor agonists. In fact, while the CB1 receptor is mainly found in the central nervous system and its inhibition can produce addictive effects, the selective targeting of the CB2 receptor, being found only in the peripheral immune system and in diseased brain cells, has a much more desirable therapeutic value for pain relief, anti-inflammatory activity, and inhibition of cough without addictive side effects.43−46 Recent studies have highlighted how the substitution pattern of the indole core plays a key role in the selectivity among the two receptors. In particular, bulky substituents at position 3 of the indole ring can increase the selectivity for the CB2 receptor, as in compound 1 (Figure 3b).39 The N-morpholinoethyl moiety occupies the deep hydrophobic pocket surrounded by conserved aromatic residues (Trp172, Tyr190, and Trp194) in the fourth transmembrane domain,34 which have been identified as crucial residues for ligand recognition,47 while the indole moiety creates arene–H interactions with F87. The introduction of a methoxy group at position 7 of the indole core, as in compound 2, results in a switch from agonist to antagonist activity for the CB2 receptor,42 possibly due to a different binding mode to the CB2 receptor. The presence of the methoxy group may force the rotation of the indole ring, producing the loss of the interaction with the crucial tryptophan residues (Figure 3b).
Figure 3.

Structure of endogenous cannabinoid receptor ligands, (−)-Δ9-THC and pravalodine. (b) Structure of a selective CB2 receptor agonist (compound 1) and antagonist (compound 2) and their interactions in the CB2 binding site (Adapted with permission from ref (39). Copyright (2017) American Chemical Society.).
The N-morpholinoethyl moiety is found to be important also for the modulation of other metabotropic receptors, such as sigma receptors (σ1R and σ2R)48 and serotonin receptors (5-hydroxytriptamine, 5-HT),49 molecular targets involved in the regulation of drug dependence, amnesia, depression, and cognition, and in the endocrine system modulation. A comprehensive overview of morpholine-containing antagonists of sigma and serotonin receptors is reported by Kouronakis,17 and two selected examples of them are reported in Figure 4.
Figure 4.

Structure of sigma and serotonin receptor antagonists containing the N-morpholinoethyl moiety.
MR-309 is a sigma receptor antagonist, where the presence of N-morpholinoethyl in the molecular structure has been balanced by the addition of a naphthyl group, to provide the desirable lipophilicity required for CNS permeability.50 MR-309 has a potential therapeutic application in those patients affected by spinal cord injury (SCI) that develop central neuropathic pain (CNP), by reducing the expression of extracellular pain mediators (TNF-α and IL-1β) and intracellular signaling cascades (ERK/pERK).51 As σ1R can regulate the N-methyl-d-aspartate receptor (NMDAR), that has been implicated in the treatment of ischemic stroke, this compound may be used also for the treatment of cerebral ischemia, as demonstrated by the reduction of cerebral infarct size and neurological deficits in mice with middle cerebral artery occlusion (MCAO) after intravenous treatment with MR-309.52 In addition, temanogrel (APD791) is a potent inverse agonist of the serotonin receptor, in which the N-morpholinoethyl moiety provides selectivity for the 5-HT2A isoform versus the 5-HT2B and 5-HT2c receptors (Figure 4).53 As 5-HT2A is involved in the reduction of thrombus formation, by inhibiting the inositol phosphate accumulation, temanogrel entered Phase I clinical trial as a potential antiplatelet agent.54,55
Another class of GPCR metabotropic receptors that play a key role in the CNS is the family of tachykinin or neurokinin receptors (NK1, NK2, and NK3), that are involved in a variety of conditions, including migraine, asthma, nausea, analgesia, inflammation, emesis, pruritus, and bronchoconstriction. The endogenous ligands of these receptors are Substance P (SP), and neurokinins A (NKA) and B (NKB), that are small peptides sharing the conserved C-terminal structural motif FxGLM-NH2.56,57 After 80 years of research around substance P antagonists (SPAs), a morpholine-containing compound (aprepitant) became the first approved oral drug for the treatment of chemotherapy-induced nausea and vomiting (CINV).30−32 The crystal structure of aprepitant complexed with the NK1 receptor has been reported (Figure 5),58 thus revealing the importance of the morpholine ring, acting as a scaffold able to direct the three interacting arms in the correct positions. Specifically, the 1H-1,2,4-triazol-5(4H)-one moiety is found establishing hydrogen bonds with Glu193, Trp184, and Gln165, the latter interacting also with morpholine, and the two substituted phenyl rings found interacting with Phe264 and Phe 268.
Figure 5.
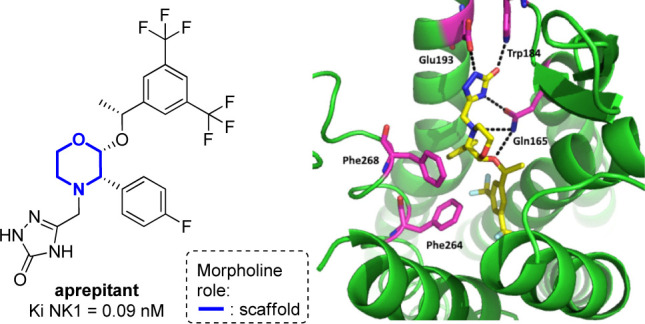
Crystal structure of the human neurokinin 1 receptor in complex with aprepitant (left) and detailed interactions with the receptor, viewed from the extracellular side above helix I (right, PDB: 6HLO).59
Histamine H3 receptor, regulating histamine levels in different areas of the CNS (cerebral cortex, striatum, and hypothalamus), is a molecular target with potential application for the treatment not only of allergic disorders, but also of schizophrenia, insomnia, and metabolic diseases, such as obesity and diabetes.60,61 Several different histamine H3 receptor modulators contain a morpholine ring, as this nucleus is often added to improve the pharmacokinetic properties of the overall compound, especially addressing the half-life and the distribution in CNS tissues. For example, the substitution of the piperidine ring with a morpholine in a series of (4-aminobutyn-1-yl)benzylamines H3 antagonists results in the improvement of potency and CNS druggability, as shown by JNJ-10181457 possessing a shorter plasma half-life and a higher ability to penetrate the BBB, with brain concentration values that roughly parallel those found in plasma (Figure 6).62
Figure 6.

Structure of a histamine H3 receptor antagonist in comparison with the morpholine-containing analogue JNJ-10181457.
Similarly, morpholine is contained in different mGlu2 (metabotropic Glutamate receptor 2) negative allosteric modulators (NAM) to increase their pharmacokinetic properties. mGlu2 is another metabotropic receptor that is broadly expressed in the CNS and represents an important therapeutic target for mood disorders (anxiety, depression, schizophrenia, addiction), as well as for neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD).63−65 First-generation mGlu2 NAMs were characterized by poor physiochemical properties (e.g., high lipophilicity, poor solubility) and very low CNS penetration (as shown by the value of rat brain:plasma partition ratios Kp, that is, 0.30 for compounds 4 and 5, Figure 7a). Recently, a novel mGlu2 NAM based on the thieno[3,2-b]pyridine-5-carboxamide core (compound 6) has been developed, showing that the introduction of 2,5-dimethylmorpholine allowed the compound to be highly CNS penetrant (Kp = 5.62), with moderate predicted hepatic clearance in microsomes and in vivo efficacy (Figure 7b).66
Figure 7.

(a) Structure of first-generation mGlu2 NAMs and (b) structure of a morpholine-containing compound able to modulate the mGlu2 receptor.
Morpholine chemotypes can be found also in antagonists or agonists of ionotropic neurotransmitter receptors, even though few examples are known in the literature. Even in this case, different roles for the morpholine nucleus can be evinced, namely, as a key interacting element or as a suitable appendage to better modulate the pharmacokinetic properties. For example, in the case of GABA (γ-aminobutyric acid) receptor, it has been recently reported how the introduction of morpholine to a 1,5-benzodiazepine derivative (such as clobazam) can increase the anxiolytic activity, by modulating the overall pharmacokinetic profile of compound 7 (Figure 8).67 On the contrary, in the case of NMDA (N-methyl-d-aspartate) receptor, a calcium channel target for achieving analgesic and antipyretic effects, the phencyclidine analogue 8 with agonist activity (Figure 8) possesses a morpholine instead of a methylamino group (as in ketamine), resulting in the morpholine moiety directly interacting with the receptor as a pharmacophoric element.68
Figure 8.

Structure of (a) compounds able to modulate ionotropic receptors (GABA and NMDA receptors), in comparison with (b) morpholine-containing analogues.
Biological Activity of Morpholine Derivatives in Neurodegenerative Diseases
Parkinson’s disease (PD) is a long-term progressive degenerative disorder that affects more than 10 million people worldwide, with a variety of clinical symptoms related to the motor system, such as shaking, rigidity, slowness of movement, and difficulty with walking.69 Although many pharmacological agents have been specifically developed over the past decades, there is still no cure for Parkinson’s disease, but only some treatments that can alleviate motor symptoms, even though with undesirable adverse effects. Thus, there is still a relevant and unmet clinical need for treatments that offer disease modification and that addresses symptoms resistant to levodopa.70 In this context, morpholine containing compounds represent powerful therapeutic agents, being able to target different receptors and enzymes involved in the pathological development of the disease. For example, mutations in LRRK2 (encoding leucine-rich repeat kinase 2) are the most common autosomal dominant mutations responsible for the development of either familial and sporadic PD, leading to an increase of LRRK2 kinase activity in neurons, mitochondrial DNA dysfunction, autophagy, inflammatory response, and apoptosis.71 Selective LRRK2 kinase inhibitors with favorable drug-like properties, such as MLi-2 (Figure 9a), may represent an alternative therapy for PD. This compound, possessing a 2,5-dimethylmorpholine ring, is able to inhibit LRRK2 with a IC50 of 0.76 nM and a 295-fold selectivity over 300 kinases, and showed good brain permeability and a good tolerance in different animal models, including MitoPark mice.72 Starting from the structure of MLi-2, compound 9 was recently developed and labeled with fluorine-18 (18F), in order to obtain a positron emission tomography (PET) tracer for the visualization of LRRK2 in the brain (Figure 9a).73 Other important molecular targets involved in PD are dopamine receptors.74,75 These GPCR receptors are classified in two different subtypes, D1-like receptors (D1 and D5) and D2-like receptors (D2, D3, and D4), that show a different distribution in the CNS system.74,75 D2 receptors are mainly found in basal ganglia neurons at the supraspinal level and are involved in adenylyl cyclase and phosphatidylinositol hydrolysis activity, as well as in the release of arachidonic acid. On the contrary, D3 receptors are present only in a few areas of the brain, such as the limbic system, which is associated with cognition and emotion. A dysregulation of either D2 and D3 receptors is found in a number of CNS disorders. Nevertheless, the selective inhibition of D3 over D2 is crucial, as an increase of D3 receptors was observed in PD-affected brains, while D2 receptors are not altered and provoke diverse side effects if activated.76 In this context, the N-morpholinopropyl moiety, as already seen for other metabotropic receptors, being functionalized with an aryl group in the 2-position has been widely used for the development of selective D3 receptor agonists, such as compound 10 that shows structural similarity to noradrenaline (Figure 9b).77 The N-morpholinopropyl moiety is also found in compound 12, a nuclear factor erythroid 2-related factor 2 (Nrf2)78 inhibitor able to attenuate the loss of dopaminergic neurons.79 Even in this case, previously developed lead compound 11 exhibited undesirable drug-like properties,80 and the introduction of a morpholine ring resulted in the improvement of the activity (EC50) and the CNS permeability (as proved by PAMPA-BBB assay) (Figure 9c).
Figure 9.

Structure of morpholine-containing compounds able to modulate molecular targets involved in Parkinson’s disease, such as (a) LRRK2 kinase, (b) dopamine receptors, and (c) Nrf2 transcription factor.
Alzheimer’s disease (AD) is the most common cause of dementia, with over 50 million affected people worldwide, a number that is expected to reach 152 million in 2050.81 Even though the molecular mechanisms behind Alzheimer’s pathogenesis is far from being completely unveiled, the amyloid cascade hypothesis remains one of the less debated.82−84 Several different molecular targets are involved in this process, and most of them have been studied for the development of novel therapeutic agents. For example, β-Secretase (BACE-1) is the enzyme that cleaves the extracellular domain of the Amyloid Precursor Protein (APP) into a N-terminal fragment C99, which is then processed by γ-secretase into the pathogenic neurotoxic peptides Aβ40 and Aβ42.85 Thus, either β- and γ-secretases have been devised as therapeutic targets of clear importance, although their inhibition proved to be challenging, in particular, for the development of compounds able to pass efficiently the BBB.86 Morpholines were shown to be important molecular scaffolds for the inhibition of BACE-1. For example, compound 13, a hydroxyethylamine-based peptide isostere, provided the right flexible architecture to fit perfectly into the active site of BACE-1, thus orienting its substituents toward the S1, S1′, and S2′ pockets, and showing capabilities for BBB entrance (Figure 10a).87 The synclinal relationship between the endocyclic oxygen and the first exocyclic carbon atoms was found to be the preferred conformation of morpholine in solution due to the exoanomeric effect. In this orientation, the methyl group of morpholine is oriented toward S1 (Figure 10b). The morpholine ring was also found to establish polar interactions with its nitrogen atom pointing toward the carbonyl group of Gly34, and with Thr72 on the flap region taking advantage of the oxygen atom.
Figure 10.
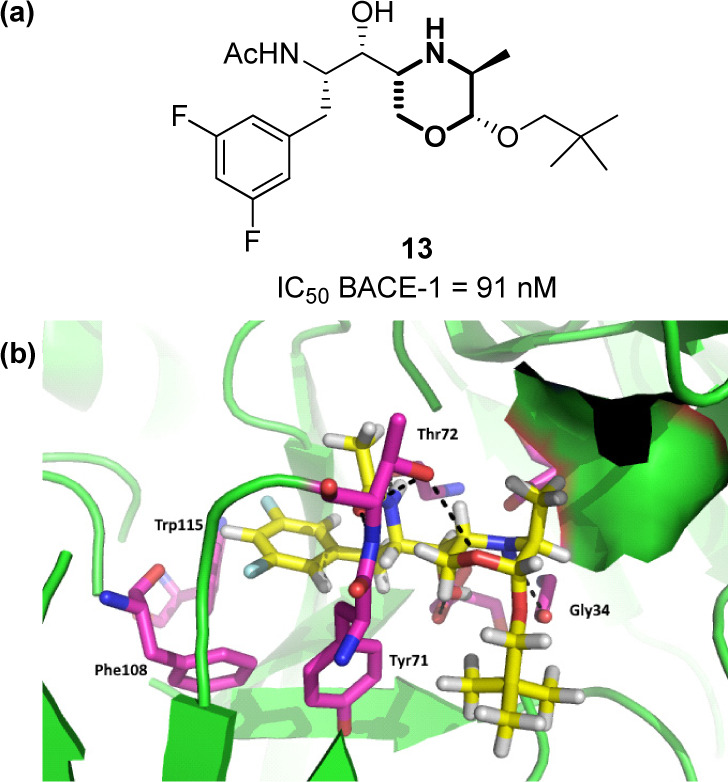
(a) Structure of compound 13, and (b) its interactions within the active site of BACE-1 (PDB: 6BFX).59
In addition, the morpholine moiety possessing the amidino group in position 3 (1,4-oxazin-3-amine) was found to be a crucial molecular platform for the inhibition of this enzyme. Just to give an example, compound 14, developed by Rambouts and co-workers, inhibits BACE-1 with IC50 of 22 nM (Figure 11a).88 The morpholine ring is important for presenting the two nitrogen atoms of the amidino group in the right position to interact with the catalytic aspartic acids (Asp32 and Asp228) and to address other interacting sites (i.e., the oxygen atom establishing a hydrogen bond with the OH group of Thr72 and with the NH group of Arg235). The long hydrophobic chain provided by the amide-tethered biaryl system can efficiently address the S1 and S3 pockets, with the NH of the amide bond interacting with Gly230, and the two aromatic rings establishing π-stacking interactions with Tyr71, Phe108, Trp76, and Trp115 (Figure 11a). An important issue in the development of BACE-1 inhibitors is the right optimization of the structure to allow good brain permeation through the BBB, and subsequent efforts of the same authors were directed through the reduction of the amidine pKa value to modulate the overall cLogP of the compounds. The authors found that the substitution of the chlorine atom with a CN group and the introduction of a fluorine atom at C-2 position can increase the inhibition potency as well as the CNS permeation (compound 15, Figure 11b).89 An additional challenge of BACE-1 inhibitors is to avoid the inhibition of hERG, which causes cardiotoxicity as a severe side effect.90 In this view, the authors demonstrated that the replacement of the 2-fluoro substituent by a CF3 group (compound 16) resulted in a reduced hERG inhibition, with an improved cardiovascular safety profile to allow the entrance of compound 16 into clinical trials (Figure 11b).89
Figure 11.
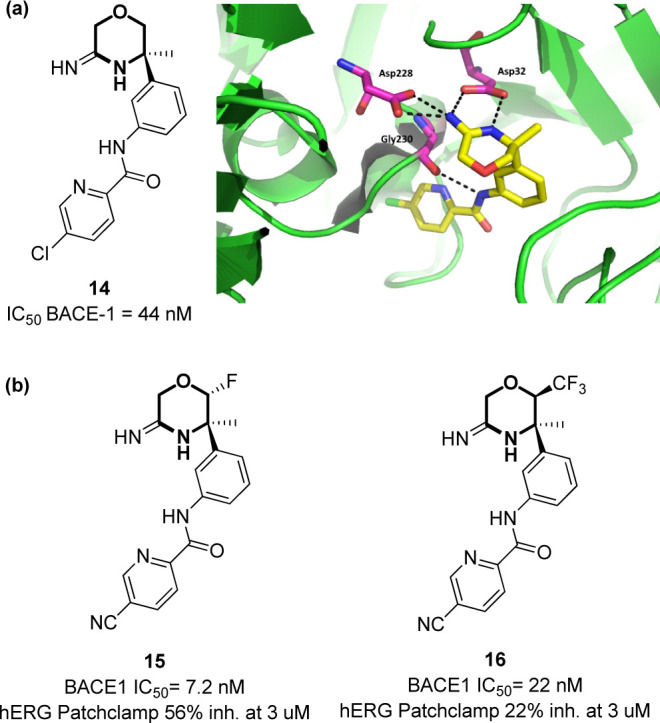
(a) Structure of compound 14 and key interactions within the active site of BACE-1 (PDB: 5CLM).59 (b) Lead optimization of compound 14 into compounds 15 and 16.
Our efforts in this field consist of the development of morpholine-containing peptidomimetics able to inhibit BACE-1. We recently reported how a bicyclic morpholine containing acetal scaffold decorated with a C=S bond was found to be active as a BACE-1 inhibitor (compound 17, Figure 12).91 Docking studies revealed the importance of the morpholine appendage for interacting with polar and apolar residues and of the sulfur atom for addressing the flap region.92,93 Successively, we developed C-2 substituted morpholine derivatives containing a thioamide group, identifying compound 18 with micromolar activity against BACE-1. According to molecular docking calculations, the binding mode of this compound was characterized by the thioamide moiety experiencing interactions with catalytic Asp228 and by the phenyl substituent interacting with aromatic residues on the flap region (Figure 12).94
Figure 12.

Structure of morpholine derivatives 17 and 18 developed as BACE-1 inhibitors.
Aβ40 and Aβ42 peptides are known to be the main toxic factors for neuronal injury and death in Alzheimer’s disease (AD), due to the ion-channel formation, free radical generation, and interaction with various proteins (e.g., apolipoprotein E and mitochondrial hydroxyacyl-CoA dehydrogenase).95 The presence of these peptides can be reduced by inhibiting γ-secretase, the enzyme that processes the N-terminal fragment C99 of the APP protein into these peptides.95 This enzyme can be inhibited by the 2,6-disubstituted morpholine N-arylsulfonamide 20, a compound able to reduce Aβ levels in Tg CRND8 mice (Figure 13a).96 The replacement of the piperidine scaffold with a morpholine ring in a previous series of compounds97 resulted in an overall improvement of potency and CYP3A4 liability (see compound 19 in Figure 13a). The introduction of morpholine was found beneficial also in a class of C-glucosyl chromen-4-ones, as the p-morpholinyl flavone 21 stood out for its ability to interact with Aβ42 peptides, reducing Aβ42-induced cell death in AD and neuroblastoma model systems (Figure 13b).98
Figure 13.

Structure of morpholine-containing compounds involved in the reduction of amyloid β (Aβ) peptides for the treatment of Alzheimer’s disease by inhibiting γ-secretase or reducing Aβ42-induced cell toxicity.
Finally, another molecular target that has been recently proposed due to its involvement in the amyloid cascade is δ-secretase, also known as asparagine endopeptidase (AEP) or legumain.99 This lysosomal cysteine proteinase cleaves both amyloid precursor protein (APP) and tau, two major pathogenic players in AD. APP is the peptide that is also demolished by β- and γ-secretase, while tau is cleaved into SET fragments, small peptides that inhibit the activity of protein phosphatase 2A (PP2A). Recently, a high-throughput screening to find δ-secretase inhibitors resulted in the identification of 7-morpholinobenzo[c][1,2,5]oxadiazol-4-amine (compound 22, Figure 14a) and a 2,4-dimorpholinoaniline (compound 23, Figure 14b) as nontoxic and potent δ-secretase inhibitors, able to reduce tau and APP cleavage in in vivo murine models.100 The crystal structure of δ-secretase in complex with these two compounds revealed the importance of morpholine for the interaction, both in the active site and in the regulatory allosteric exosite of δ-secretase. Specifically, the morpholine ring in compound 22 was found to be interacting within the S1 pocket with Cys189, Gly149, and His148 (Figure 14a), and the benzofurazan moiety interacting with Ser215 and Tyr217 of the S2 pocket, with fewer contact points as compared to those found in the interaction with the allosteric site. Compound 23, not possessing the benzofurazan core, was developed to increase the preference of the inhibitor for the active site and not for the allosteric one. As shown in Figure 14b, compound 23 is able not only to maintain the interaction with Cys189, Gly149, and His148 at the S1 site typical of the morpholine ring, but also to mimic the canonical P2–S2 and P3–S3 interactions found in the active site, with an increased binding affinity for δ-secretase.
Figure 14.
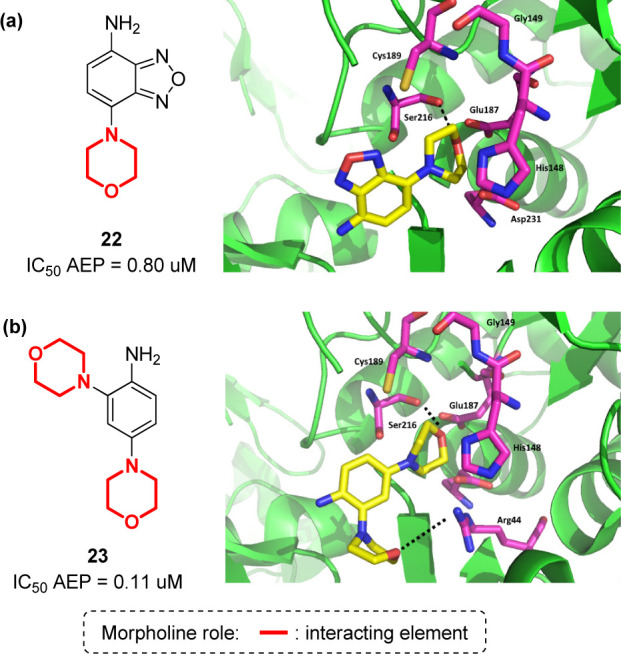
(a) Structure of compound 22 and its interaction in the active site of δ-secretase (PDB: 5LUB);59 (b) structure of compound 23 and its interaction in the active site of δ-secretase (PDB: 5LUA).59
Although not strictly related to the amyloid cascade hypothesis, there has been reported evidence regarding the involvement of acetylcholine, noreprinephrine, dopamine, and serotonin levels in the etiology of Alzheimer’s disease.101,102 Thus, acetylcholinesterase (AChE), monoamine oxidase (MAO-A and MAO-B), and the M1 acetylcholine receptor, responsible for the regulation of such biogenic amines in the CNS, are considered interesting therapeutic targets in AD drug discovery. The morpholine ring is frequently observed in acetylcholinesterase (AChE) and MAO inhibitors, thus confirming the importance of this heterocycle in providing useful interactions with different receptors.103 For example, compound 25 is a dual inhibitor of acetylcholinesterase (AChE) and monoamine oxidase B (MAO-B) with good BBB permeability, representing a promising lead compound for the prevention and treatment of AD (Figure 15b).104 Again, the replacement of the piperidine ring with morpholine resulted in a 100-fold enhancement of the activity toward MAO-B, as evinced by molecular modeling studies that revealed how the morpholine ring interacts in the entrance cavity, directing the coumarin scaffold in the substrate cavity. In addition, the benzomorpholine compound VU0486846 is able to activate the M1 subtype of muscarinic acetylcholine receptor through a positive allosteric modulation (PAM), showing a potential recovery of cognition in AD animal models (Figure 15b).105 This compound was developed by a scaffold hopping screening campaign, and showed good CNS penetration, with no classic cholinergic adverse events in the prefrontal cortex (PFC), as evinced by the absence of any interaction with the orthosteric acetylcholine (ACh) site and no seizure liability at high brain exposures.106
Figure 15.

Structure of (a) compounds able to modulate molecular targets (acetylcholinesterase enzyme (AChE), monoamine oxidase (MAO-A and MAO-B) and M1 acetylcholine receptor) involved in the regulation of several biogenic amines, in comparison with (b) morpholine-containing analogues.
Biological Activity of Morpholine Derivatives on CNS Tumors
The development of morpholine containing anticancer drugs with minimal or no side effects is an active field in drug discovery,17−22 and this is especially true in the case of CNS tumors, including astrocytoma, glioblastoma, medulloblastoma, ependymal, and meningeal tumors. Targeted therapy is one the most effective ways to treat those type of cancers, and especially for CNS tumors, major targets that can be addressed with morpholine-containing compounds are the kinases involved in cell cycle regulation, namely, phosphatidylinositol 3-kinase (PI3K)107,108 and the mammalian target of rapamycin (mTOR1 and mTOR2).109,110 In particular, the PI3K-mTOR pathway has attracted much attention to the scientific community involved in CNS drug discovery, as an overactivated PI3K/mTOR pathway has been related not only to aberrant cancer cell growth, but also to several neurodegenerative pathways.111,112 Thus, the development of brain penetrant dual pan-PI3K/mTOR inhibitors may enable the identification of novel agents for the treatment of brain metastasis and brain tumors. Many morpholine-containing PI3K inhibitors have been developed over the last decades,113−116,17,22 with a particular recurrence of morpholino-pyrimidine,113 morpholino-triazine,114 and morpholinopyrimidine-5-carbonitrile.115 Just to give some examples, NVP-BKM120 is a compound able to inhibit all four class I PI3K isoforms with 50-fold selectivity over the other protein kinases (Figure 16a).117 As shown by the crystal structure of this compound, one morpholine ring interacts with the hinge valine Val882 through its oxygen atom, a key interaction established by several PI3K inhibitors in the ATP-binding site. The second morpholine interacts with Thr887, though it was added as a double substitution to maintain the capacity of interacting with the key Val882, considering that morpholine is highly susceptible to in vivo oxidation. The NH2 group of the aminopyridine ring forms hydrogen bonds with the carboxylic group of aspartates Asp836, Asp841, and Asp964. Starting from this compound, Beaufils and co-workers replaced the pyrimidine ring with a triazine ring, developing PQR309 (also called bimiralisib), a compound able to inhibit all PI3K kinases of class I as well as mTOR kinase (Figure 16b).118 This compound is orally active, crosses the blood-brain barrier, and displays favorable pharmacokinetic parameters in several animal models, thus demonstrating its potential for the treatment of brain tumors or CNS metastasis. The crystal structure revealed the same interaction of one morpholine oxygen atom with Val882, with the other morpholine establishing a hydrogen bond with Lys890, and maintaining those interactions of the aminopyridine NH2 group with Asp964, whereas the triazine ring was found interacting through π–π stacking interactions with Phe961, Tyr867, and Phe965.
Figure 16.
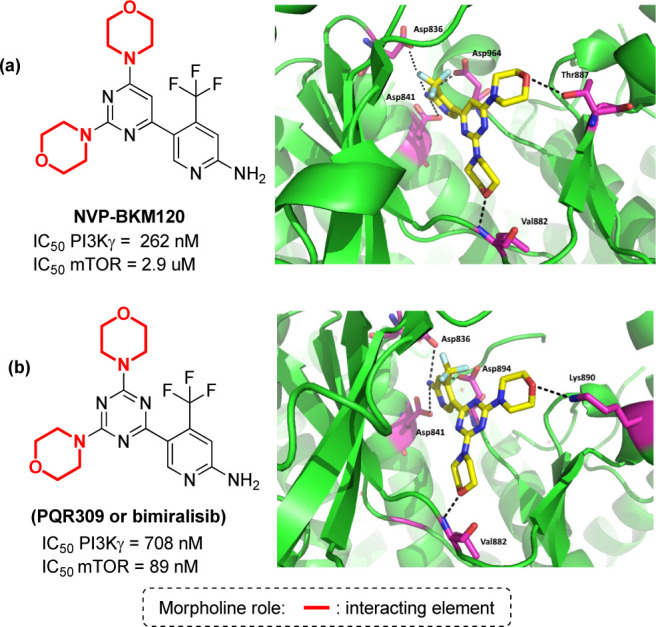
(a) Structure of compound NVP-BKM120 and its interaction pattern in the active site of PI3Kγ (PDB: 3SD5).59 (b) Structure of compound PQR309 and its interaction pattern in the active site of PI3Kγ (PDB: 5OQ4).59
Although the dual inhibition of PI3K and mTOR enzyme is of potential interest for the development of anticancer drugs, the selective inhibition of the mTOR complex over the PI3K family is of special relevance in the field of CNS drug discovery. In fact, for several dual inhibitors (including PQR309) it has been observed that when mTOR1 is inhibited, class IA PI3Ks are overactivated, resulting in a mTOR2-dependent increase in the PKB/Akt activity.119−121 Compounds targeting both mTOR1 and mTOR2, without significantly inhibiting PI3K family, are expected to provide a more efficient blockade of growth factors, as well as to display a beneficial effect in Alzheimer’s and Parkinson’s diseases,122,123 mostly because the inhibition of mTOR promotes the removal of toxic protein complexes by inducing autophagy. The crystal structure of mTOR has been reported by Yang and co-workers,124 in complex with PI-103, a morpholine-containing compound that is able to inhibit both mTOR and class I PI3K kinase (Figure 17). This study revealed that the active site of this enzyme is remarkably similar to canonical PI3K kinases. Thus, the hinge interaction with valine (here Val2240) is conserved, as well as the presence of several hydrophobic residues. Thus, PI-103 does not provide any selectivity for this catalytic cleft, as the morpholine ring makes the classical hydrogen bonds to the “hinge” Val2240 and Cys2243, while the m-phenol group binds to the inner pocket establishing two hydrogen bonds with Tyr2225 and Asp2195 side chains at the back of the cleft (Figure 17). In addition, the central aromatic scaffolds establish a π-stacking interaction with Trp2239.
Figure 17.
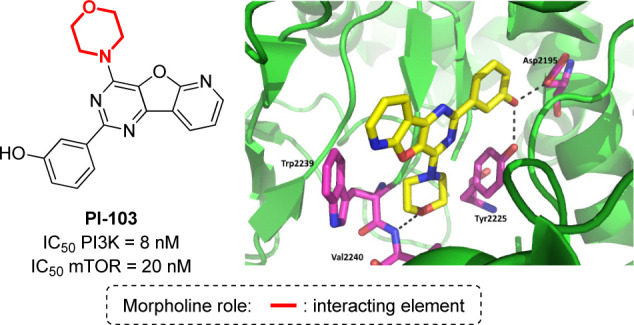
Crystal structure of PI-103 in complex with mTOR (PDB: 4JT6).59
As mTOR has a deeper pocket as compared to PI3K, the introduction of substituents on the morpholine ring, such as an ethylene bridge between positions 3 and 5 or a methyl group at position 3, have been studied in order to develop selective and highly brain penetrant mTOR kinase inhibitors.125 In particular, the introduction of the bridged morpholine moiety is often beneficial for CNS candidates, as they are effective in decreasing lipophilicity, partly because of an enhancement of the polar surface area resulting from conformational changes.24,126 For example, PQR620, containing two 3,5-bridged morpholines, inhibits mTOR kinase potently and selectively, and showed antitumor effects in vitro and in vivo, with promising benefits in CNS indications due to its brain/plasma distribution ratio.127 Similarly, compound 27, possessing two 3-methylmorpholines, is a potent, orally available, and specific ATP-competitive mTOR inhibitor with selectivity against all Class I PI3K isoforms and other kinases (Figure 18).128 In particular, the thiazolopyrimidine 27 was found to be brain-penetrant and active in neuronal cell-based models of mTOR hyperactivity, significantly improving the survival rate of mice with neuronal-specific ablation of the Tsc1 gene. Molecular docking studies revealed the importance of the pyrazole moiety of compound 27 in providing additional hydrogen-bonding interactions with catalytic Lys2187 and Asp2195 residues, in addition to the interaction of the morpholine oxygen atom with Val2240.
Figure 18.

Structure of morpholine-containing compounds able to selectively inhibit mTOR complex over the PI3K kinase family.
Conclusion
Developing drugs for the central nervous system (CNS) require fine chemical modifications, as a strict balance between size and lipophilicity is necessary to improve the permeability through the blood-brain barrier (BBB). In this context, morpholine and its analogues represent valuable heterocycles, due to their conformational and physicochemical properties. In fact, the presence of a weak basic nitrogen, at the opposite position of the oxygen atom, provide to the ring a peculiar pKa value and a flexible conformation, thus allowing it to take part in several lipophilic–hydrophilic interactions, and to improve blood solubility and brain permeability of the overall structure. Herein, the analysis of selected morpholine-containing CNS drug candidates revealed the active pharmacophores accountable for the modulation of different molecular targets, that have been subdivided in three classes: (1) receptors involved in mood disorders and pain; (2) enzymes and receptors responsible for neurodegenerative diseases; and (3) enzymes involved in the pathology of CNS tumors. This analysis allowed us to highlight the cases when morpholine contributes in the overall drug structure by (1) directly interacting with key residues in the binding site of the target, (2) having a scaffolding function to direct the appendages in the right position, or (3) improving the overall pharmacokinetic profile, specifically relating to the half-life and the distribution in CNS tissues/brain permeability of the final compound. For the first two cases, several examples reporting binding features retrieved from PDB data allowed us to highlight the key role of the morpholine chemotype in establishing key interactions. For the latter, we compared morpholine-containing compounds with analogues lacking this ring, in order to evaluate not only the potency but also the PK/PD properties relevant for the CNS drug discovery. The morpholine ring has attracted the attention of a large area of the medicinal chemistry community for the development of compounds with a plethora of different biological activities, as demonstrated by the increasing number of research articles and review papers. We do expect in the future that the morpholine chemotype will be taken into account increasingly for drug development in the field of CNS drug discovery.
Acknowledgments
Airalzh Onlus-COOP Italia (fellowship to LC), MIUR (“Progetto Dipartimenti di Eccellenza 2018-2022” allocated to Department of Chemistry “Ugo Schiff”) and Fondazione Cassa di Risparmio di Pistoia e Pescia (Bando Giovani@Ricerca scientifica 2018, fellowship to EL) are acknowledged for financial support.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Kesselheim A. S.; Hwang T. J.; Franklin J. M. (2015) Two decades of new drug development for central nervous system disorders. Nat. Rev. Drug Discovery 14, 815–816. 10.1038/nrd4793. [DOI] [PubMed] [Google Scholar]
- Gribkoff V. K.; Kaczmarek L. K. (2017) The need for new approaches in CNS drug discovery. Neuropharmacology 120, 11–19. 10.1016/j.neuropharm.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose A. K.; Herbertz T.; Hudkins R. L.; Dorsey B. D.; Mallamo J. P. (2012) Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 3, 50–68. 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalatsa A.; Schatzlein A. G.; Uchegbu I. F. (2014) Strategies to deliver peptide drugs to the brain. Mol. Pharmaceutics 11, 1081–1093. 10.1021/mp400680d. [DOI] [PubMed] [Google Scholar]
- Lau J. L.; Dunn M. K. (2018) Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 26, 2700–2707. 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- Neves V.; Aires-Da-Silva F.; Corte-Real S.; Castanho M. A. R. B. (2016) Antibody approaches to treat brain diseases. Trends Biotechnol. 34, 36–48. 10.1016/j.tibtech.2015.10.005. [DOI] [Google Scholar]
- Ryan P.; Patel B.; Makwana V.; Jadhav H. R.; Kiefel M.; Davey A.; Reekie T. A.; Rudrawar S.; Kassiou M. (2018) Peptides, peptidomimetics, and carbohydrate-peptide conjugates as amyloidogenic aggregation inhibitors for Alzheimer’s disease. ACS Chem. Neurosci. 9, 1530–1551. 10.1021/acschemneuro.8b00185. [DOI] [PubMed] [Google Scholar]
- Sweeney M. D.; Sagare A. P.; Zlokovic B. V. (2018) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Chandrasekaran R. Y.; Hou X.; Troutman M. D.; Verhoest P. R.; Villalobos A.; Will Y. (2010) Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 1, 420–434. 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. (2016) Central nervous system multiparameter optimization desirability: application in drug discovery. ACS Chem. Neurosci. 7, 767–775. 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- Rankovic Z. (2015) CNS drug design: balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 58, 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Danon J. J.; Reekie T. A.; Kassiou M. (2019) Challenges and opportunities in central nervous system drug discovery. Trends Chem. 1, 612–624. 10.1016/j.trechm.2019.04.009. [DOI] [Google Scholar]
- Wijtmans R.; Vink M. K. S.; Schoemaker H. E.; van Delft F. L.; Blaauw R. H.; Rutjes F. P. J. T. (2004) Biological relevance and synthesis of C-substituted morpholine derivatives. Synthesis 2004, 641–662. 10.1055/s-2004-816003. [DOI] [Google Scholar]
- Ilaš J.; Anderluh P. Š.; Dolenc M. S.; Kikelj D. (2005) Recent advances in the synthesis of 2H-1,4-benzoxazin-3-(4H)-ones and 3,4-dihydro-2H-1,4-benzoxazines. Tetrahedron 61, 7325–7348. 10.1016/j.tet.2005.05.037. [DOI] [Google Scholar]
- Pal’chikov V. A. (2013) Morpholines. Synthesis and biological activity. Russ. J. Org. Chem. 49, 787–814. 10.1134/S1070428013060018. [DOI] [Google Scholar]
- Tzara A.; Xanthopoulos D.; Kourounakis A. P. (2020) Morpholine as a scaffold in medicinal vhemistry: an update on synthetic strategies. ChemMedChem 15, 392–403. 10.1002/cmdc.201900682. [DOI] [PubMed] [Google Scholar]
- Kourounakis A. P.; Xanthopoulos D.; Tzara A. (2020) Morpholine as a privileged structure: a review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev. 40, 709–752. 10.1002/med.21634. [DOI] [PubMed] [Google Scholar]
- Al-Ghorbani M.; Begum B.; Zabiulla A.; Mamatha S. V.; Khanum S. A. (2015) Piperazine and morpholine: synthetic preview and pharmaceutical applications. J. Chem. Pharm. Res. 7, 281–301. [Google Scholar]
- Naim M. J.; Alam O.; Alam M. J.; Alam P.; Shrivastava N. (2015) A review on pharmacological profile of morpholine derivatives. Int. J. Pharmacol. Pharm. Sci. 3, 40–51. [Google Scholar]
- Rupak K.; Vulichi S. R.; Suman K. (2016) Emphasizing morpholine and its derivatives (maid): typical candidate of pharmaceutical importance. Int. J. Chem. Sci. 14, 1777–1788. [Google Scholar]
- Arshad F.; Khan M. F.; Akhtar W.; Alam M. M.; Nainwal L. M.; Kaushik S. K.; Akhter M.; Parvez S.; Hasan S. M.; Shaquiquzzaman M. (2019) Revealing quinquennial journey of morpholine: a SAR based review. Eur. J. Med. Chem. 167, 324–356. 10.1016/j.ejmech.2019.02.015. [DOI] [PubMed] [Google Scholar]
- Kumari A.; Singh R. K. (2020) Morpholine as ubiquitous pharmacophore in medicinal chemistry: deep insight into the structure-activity relationship (SAR). Bioorg. Chem. 96, 103578. 10.1016/j.bioorg.2020.103578. [DOI] [PubMed] [Google Scholar]
- Rekka E. A.; Kourounakis P. N. (2010) Medicinal chemistry of 2,2,4-substituted morpholines. Curr. Med. Chem. 17, 3422–3430. 10.2174/092986710793176276. [DOI] [PubMed] [Google Scholar]
- Hocine S.; Berger G.; Hanessian S. (2020) Design and synthesis of backbone-fused, conformationally constrained morpholine-proline chimeras. J. Org. Chem. 85, 4237–4247. 10.1021/acs.joc.9b03413. [DOI] [PubMed] [Google Scholar]
- Baskar R.; Baby C.; Moni M. S.; Subramanian K. (2013) Synthesis, characterization and dynamic NMR studies of a novel chalcone based N-substituted morpholine derivative. J. Mol. Struct. 1040, 90–97. 10.1016/j.molstruc.2013.02.029. [DOI] [Google Scholar]
- Cotten J. F.; Keshavaprasad B.; Laster M. J.; Eger E. I.; Yost C S. (2006) The ventilator stimulant doxapram inhibits TASK tandem pore (K2P) potassium channel function but does not affect minimum alveolar anesthetic concentration. Anesth. Analg. 102, 779–785. 10.1213/01.ane.0000194289.34345.63. [DOI] [PubMed] [Google Scholar]
- Stoops W. W.; Strickland J. C.; Alcorn J. L.; Hays L. R.; Rayapati A. O.; Lile J. A.; Rush C. R. (2019) Influence of phendimetrazine maintenance on the reinforcing, subjective, performance, and physiological effects of intranasal cocaine. Psychopharmacology 236, 2569–2577. 10.1007/s00213-019-05227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet U. (2003) Moclobemide: therapeutic use and clinical studies. CNS Drug Rev. 9, 97–140. 10.1111/j.1527-3458.2003.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu C.; Kulkarni J. (2019) Psychosis induced by antidepressant treatment with reboxetine. Aust. N. Z. J. Psychiatry 53, 1227–1228. 10.1177/0004867419865612. [DOI] [PubMed] [Google Scholar]
- Walsh S. L.; Heilig M.; Nuzzo P. A.; Henderson P.; Lofwall M. R. (2013) Effects of the NK1 antagonist, aprepitant, on response to oral and intranasal oxycodone in prescription opioid abusers. Addict. Biol. 18, 332–343. 10.1111/j.1369-1600.2011.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale J. J.; Mills S. G.; MacCoss M.; Finke P. E.; Cascieri M. A.; Sadowski S.; Ber E.; Chicchi G. G.; Kurtz M.; Metzger J.; et al. (1998) Structural optimization affording 2-(R)-(1-(R)-3, 5-bis(trifluoromethyl) phenylethoxy)-3-(S)-(4-fluoro)phenyl-4- (3-oxo-1,2,4-triazol-5-yl)methylmorpholine, a potent, orally active, long acting morpholine acetal human NK-1 receptor antagonist. J. Med. Chem. 41, 4607–4614. 10.1021/jm980299k. [DOI] [PubMed] [Google Scholar]
- Hargreaves R.; Arjona Ferreira J. C.; Hughes D.; Brands J.; Hale J.; Mattson B.; Mills S. (2011) Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Ann. N. Y. Acad. Sci. 1222, 40–48. 10.1111/j.1749-6632.2011.05961.x. [DOI] [PubMed] [Google Scholar]
- Hobbs H.; Bravi G.; Campbell I.; Convery M.; Davies H.; Inglis G.; Pal S.; Peace S.; Redmond J. (2019) and Summers, D. Discovery of 3-oxabicyclo[4.1.0]heptane, a non-nitrogen containing morpholine isostere, and its application in novel inhibitors of the PI3K-AKT-mTOR pathway. J. Med. Chem. 62, 6972–6984. 10.1021/acs.jmedchem.9b00348. [DOI] [PubMed] [Google Scholar]
- Li J.; Ji J.; Xu R.; Li R. (2019) Indole compounds with N-ethyl morpholine moieties as CB2 receptor agonists for anti-inflammatory management of pain: synthesis and biological evaluation. MedChemComm 10, 1935–1947. 10.1039/C9MD00173E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T.; Waku K. (2002) Cannabinoid receptors and their endogenous ligands. J. Biochem. 132, 7–12. 10.1093/oxfordjournals.jbchem.a003200. [DOI] [PubMed] [Google Scholar]
- Janero D. R.; Vadivel S. K.; Makriyannis A. (2009) Pharmacotherapeutic modulation of the endocannabinoid signalling system in psychiatric disorders: drug-discovery strategies. Int. Rev. Psychiatry 21, 122–133. 10.1080/09540260902782778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman M.; Nagarathnam D.; Gopal D.; He H. M.; Lin C. M.; Hamel E. (1992) Synthesis and evaluation of analogs of (Z)-1-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethene as potential cytotoxic and antimitotic agents. J. Med. Chem. 35, 2293. 10.1021/jm00090a021. [DOI] [PubMed] [Google Scholar]
- Haubrich D. R.; Ward S. J.; Baizman E.; Bell M. R.; Bradford J.; Ferrari R.; Miller M.; Perrone M.; Pierson A. K.; Saelens J. K.; et al. (1990) Pharmacology of pravadoline: a new analgesic agent. J. Pharmacol Exp Ther. 255, 511–522. [PubMed] [Google Scholar]
- Shi Y.; Duan Y. H.; Ji Y. Y.; Wang Z. L.; Wu Y. R.; Gunosewoyo H.; Xie X. Y.; Chen J. Z.; Yang F.; Li J.; et al. (2017) Amidoalkylindoles as potent and selective cannabinoid type 2 receptor agonists with in vivo efficacy in a mouse model of multiple sclerosis. J. Med. Chem. 60, 7067–7083. 10.1021/acs.jmedchem.7b00724. [DOI] [PubMed] [Google Scholar]
- Reggio P. H. (2010) Endocannabinoid binding to the cannabinoid receptors: what is known and what remains unknown. Curr. Med. Chem. 17, 1468–1486. 10.2174/092986710790980005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Pu M.; Pu M.; Qu L.; Han G. W.; Wu Y.; Zhao S.; Shui W.; Li S.; et al. (2016) Crystal structure of the human cannabinoid receptor CB1. Cell 167, 750–762. 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Hua T.; Vemuri K.; Ho J.-H.; Wu X.; Wu L.; Popov P.; Benchama O.; Zvonok N.; et al. (2019) Crystal structure of the human cannabinoid receptor CB2. Cell 176, 459–467. 10.1016/j.cell.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett A. C. (2002) The cannabinoid receptors. Prostaglandins Other Lipid Mediators 68, 619–631. 10.1016/S0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Rajesh M.; Mukhopadhyay P.; Haskó G.; Huffman J. W.; Mackie K.; Pacher P. (2008) CB2 cannabinoid receptor agonists attenuate TNF-alpha-induced human vascular smooth muscle cell proliferation and migration. Br. J. Pharmacol. 153, 347–357. 10.1038/sj.bjp.0707569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone R. P.; Kendall D. A. (2015) Minireview: from the bench, toward the clinic: therapeutic opportunities for cannabinoid receptor modulation. Mol. Endocrinol. 29, 801–813. 10.1210/me.2015-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M.; Grether U.; Fingerle J.; Grim T. W.; Fezza F.; de Petrocellis L.; Ullmer C.; Rothenhäusler B.; Perret C.; van Gils N.; et al. (2017) Cannabinoid CB 2 receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 8, 13958. 10.1038/ncomms13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee M. H.; Nevo I.; Bayewitch M. L.; Zagoory O.; Vogel Z. (2000) Functional role of tryptophan residues in the fourth transmembrane domain of the CB(2) cannabinoid receptor. J. Neurochem. 75, 2485–2491. 10.1046/j.1471-4159.2000.0752485.x. [DOI] [PubMed] [Google Scholar]
- Su T. P.; Su T. C.; Nakamura Y.; Tsai S. Y. (2016) The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol. Sci. 37, 262–278. 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy J. D.; Roth B. L. (2015) Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 150, 129–142. 10.1016/j.pharmthera.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz J. L.; Cuberes R.; Berrocal J.; Contijoch M.; Christmann U.; Fernández A.; Port A.; Holenz J.; Buschmann H.; Laggner C.; et al. (2012) Synthesis and biological evaluation of the 1-arylpyrazole class of σ(1) receptor antagonists: identification of 4-{2-[5-methyl-1-(naphthalen-2-yl)-1H-pyrazol-3-yloxy]ethyl}morpholine (S1RA, E-52862). J. Med. Chem. 55, 8211–8224. 10.1021/jm3007323. [DOI] [PubMed] [Google Scholar]
- Castany S.; Codony X.; Zamanillo D.; Merlos M.; Verdú E.; Boadas-Vaello P. (2019) Repeated sigma-1 receptor antagonist MR309 administration modulates central neuropathic pain development after spinal cord injury in mice. Front. Pharmacol. 10, 222. 10.3389/fphar.2019.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Blázquez P.; Pozo-Rodrigálvarez A.; Merlos M.; Garzón J. (2018) The sigma-1 receptor antagonist, S1RA, reduces stroke damage, ameliorates post-stroke neurological deficits and suppresses the overexpression of MMP-9. Mol. Neurobiol. 55, 4940–4951. 10.1007/s12035-017-0697-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Teegarden B. R.; Choi J. S.; Strah-Pleynet S.; Decaire M.; Jayakumar H.; Dosa P. I.; Casper M. D.; Pham L.; Feichtinger K.; et al. (2010) Discovery and structure-activity relationship of 3-methoxy N-(3-(1-methyl-1Hpyrazol- 5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide (APD791): a highly selective 5-hydroxytryptamine2A receptor inverse agonist for the treatment of arterial thrombosis. J. Med. Chem. 53, 4412–4421. 10.1021/jm100044a. [DOI] [PubMed] [Google Scholar]
- Przyklenk K.; Frelinger A. L.; Linden M. D.; Whittaker P.; Li Y.; Barnard M. R.; Adams J.; Morgan M.; Al-Shamma H.; Michelson A. D. (2010) Targeted inhibition of the serotonin 5HT2A receptor improves coronary patency in an in vivo model of recurrent thrombosis. J. Thromb. Haemostasis 8, 331–340. 10.1111/j.1538-7836.2009.03693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H. J.; Park E.-J.; Kim M. J.; Kang S. Y.; Lee S. H.; Kim H. J.; Lee K. N.; Jung M. E.; Lee M.; Kim M. S.; et al. (2011) Design and synthesis of novel arylpiperazine derivatives containing the imidazole core targeting 5-HT(2A) receptor and 5-HT transporter. J. Med. Chem. 54, 6305–6318. 10.1021/jm200682b. [DOI] [PubMed] [Google Scholar]
- Quartara L.; Maggi C. A. (1997) The tachykinin NK1 receptor. Part I: Ligands and mechanisms of cellular activation. Neuropeptides 31, 537–563. 10.1016/S0143-4179(97)90001-9. [DOI] [PubMed] [Google Scholar]
- Steinhoff M. S.; von Mentzer B.; Geppetti P.; Pothoulakis C.; Bunnett N. W. (2014) Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol. Rev. 94, 265–301. 10.1152/physrev.00031.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöppe J.; Ehrenmann J.; Klenk C.; Rucktooa P.; Schütz M.; Doré A. S.; Plückthun A. (2019) Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 10, 17. 10.1038/s41467-018-07939-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edited with PyMOL 0.99rc6, DeLano Scientific LLC; Palo Alto, CA, USA, 2006. [Google Scholar]
- Celanire S.; Wijtmans M.; Talaga P.; Leurs R.; de Esch I. J. P. (2005) Histamine H3 receptor antagonists reach out for the clinic. Drug Discovery Today 10, 1613–1627. 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- Gemkow M. J.; Davenport A. J.; Harich S.; Ellenbroek B. A.; Cesura A.; Hallett D. (2009) The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discovery Today 14, 509–515. 10.1016/j.drudis.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Dvorak C. A.; Apodaca R.; Xiao W.; Jablonowski J. A.; Bonaventure P.; Dugovic C.; Shelton J.; Lord B.; Miller K.; Dvorak L. K.; et al. (2009) Diamine-based human histamine H3 receptor antagonists: (4-aminobutyn-1-yl) benzylamines. Eur. J. Med. Chem. 44, 4098–4106. 10.1016/j.ejmech.2009.04.049. [DOI] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crupi R.; Impellizzeri D.; Cuzzocrea S. (2019) Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 12, 20. 10.3389/fnmol.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki S.; Ago Y.; Palucha-Paniewiera A.; Matrisciano F.; Pilc A. (2013) mGlu2/3 and mGlu5 receptors: potential targets for novel antidepressants. Neuropharmacology 66, 40–52. 10.1016/j.neuropharm.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Childress E. S.; Wieting J. M.; Felts A. S.; Breiner M. M.; Long M. F.; Luscombe V. B.; Rodriguez A. L.; Cho H. P.; Blobaum A. L.; Niswender C. M.; et al. (2019) Discovery of novel central nervous system penetrant metabotropic glutamate receptor subtype 2 (mGlu2) negative allosteric modulators (NAMs) based on functionalized pyrazolo[1,5-a]pyrimidine-5-carboxamide and thieno[3,2-b]pyridine-5-carboxamide cores. J. Med. Chem. 62, 378–384. 10.1021/acs.jmedchem.8b01266. [DOI] [PubMed] [Google Scholar]
- Gatta E.; Cupello A.; Di Braccio M.; Grossi G.; Ferruzzi R.; Roma G.; Robello M. (2010) New 1,5-benzodiazepine compounds: activity at native GABA(A) receptors. Neuroscience 166, 917–923. 10.1016/j.neuroscience.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Colestock T.; Wallach J.; Mansi M.; Filemban N.; Morris H.; Elliott S. P.; Westphal F.; Brandt S. D.; Adejare A. (2018) Syntheses, analytical and pharmacological characterizations of the ‘legal high’ 4-[1-(3-methoxyphenyl)cyclohexyl]morpholine (3-MeO-PCMo) and analogues. Drug Test. Anal. 10, 272–283. 10.1002/dta.2213. [DOI] [PubMed] [Google Scholar]
- Szeto J. Y. Y.; Walton C. C.; Rizos A.; Martinez-Martin P.; Halliday G. M.; Naismith S. L.; Chaudhuri K. R.; Lewis S. J. G. (2020) Dementia in long-term Parkinson’s disease patients: a multicentre retrospective study. NPJ. Parkinsons Dis. 6, 20. 10.1038/s41531-019-0106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkouzi A.; Vedam-Mai V.; Eisinger R. S.; Okun M. S. (2019) Emerging therapies in Parkinson disease — repurposed drugs and new approaches. Nat. Rev. Neurol. 15, 204–223. 10.1038/s41582-019-0155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I.; Kim J. W.; Dawson V. L.; Dawson T. M. (2014) LRRK2 pathobiology in Parkinson’s disease. J. Neurochem. 131, 554–565. 10.1111/jnc.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell M. J.; Mirescu C.; Basu K.; Cheewatrakoolpong B.; et al. (2015) MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharmacol. Exp. Ther. 355, 397–409. 10.1124/jpet.115.227587. [DOI] [PubMed] [Google Scholar]
- Mori W.; Yamasaki T.; Hattori Y.; Zhang Y.; Kumata K.; Fujinaga M.; Hanyu M.; Nengaki N.; Zhang H.; Zhang M.-R. (2020) Radiosynthesis and evaluation of 4-(6-[18F]Fluoro-4-(5-isopropoxy-1H-indazol-3-yl)pyridin-2-yl)-morpholine as a novel radiotracer candidate targeting leucine-rich repeat kinase 2. RSC Med. Chem. 11, 676–684. 10.1039/C9MD00590K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. W.; Hopkins C. R. (2017) Return of D4 dopamine receptor antagonists in drug discovery. J. Med. Chem. 60, 7233–7243. 10.1021/acs.jmedchem.7b00151. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Singh S.; Shukla S. (2018) Physiological and functional basis of dopamine receptors and their role in neurogenesis: possible implication for Parkinson’s disease. J. Exp. Neurosci. 12, 1179069518779829. 10.1177/1179069518779829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach R. H.; Luedtke R. R. (2018) Challenges in the development of dopamine D2- and D3-selective radiotracers for PET imaging studies. J. Labelled Compd. Radiopharm. 61, 291–298. 10.1002/jlcr.3558. [DOI] [PubMed] [Google Scholar]
- Blagg J.; Allerton C. M.; Batchelor D. V.; Baxter A. D.; Burring D. J.; Carr C. L.; Cook A. S.; Nichols C. L.; Phipps J.; Sanderson V. G.; et al. (2007) Design and synthesis of a functionally selective D3 agonist and its in vivo delivery via the intranasal route. Bioorg. Med. Chem. Lett. 17, 6691–6696. 10.1016/j.bmcl.2007.10.059. [DOI] [PubMed] [Google Scholar]
- Todorovic M.; Wood S. A.; Mellick G. D. (2016) Nrf2: a modulator of Parkinson’s disease?. J. Neural Transm. 123, 611–619. 10.1007/s00702-016-1563-0. [DOI] [PubMed] [Google Scholar]
- Choi J. W.; Kim S.; Park J.-H.; Kim H. J.; Shin S. J.; Kim J. W.; Woo S. Y.; Lee C.; Han S. M.; Lee J.; et al. (2019) Optimization of vinyl sulfone derivatives as potent nuclear factor erythroid 2-related factor 2 (Nrf2) activators for Parkinson’s disease therapy. J. Med. Chem. 62, 811–830. 10.1021/acs.jmedchem.8b01527. [DOI] [PubMed] [Google Scholar]
- Woo S. Y.; Kim J. H.; Moon M. K.; Han S. H.; Yeon S. K.; Choi J. W.; Jang B. K.; Song H. J.; Kang Y. G.; Kim J. W.; et al. (2014) Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson’s disease therapy. J. Med. Chem. 57, 1473–1487. 10.1021/jm401788m. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Disease International (2019) World Alzheimer Report 2019: Attitudes to dementia, Alzheimer’s Disease International, London.
- Beyreuther K.; Masters C. L. (1991) Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1, 241–251. 10.1111/j.1750-3639.1991.tb00667.x. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J.; Hardy J. (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–560. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia S. S.; St George-Hyslop P. H. (2002) Gamma-secretase, notch, abeta and Alzheimer’s disease: where do the presenilins fit in?. Nat. Rev. Neurosci. 3, 281–290. 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Hu X.; Das B.; Hou H.; He W.; Yan R. (2018) BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 215, 927–940. 10.1084/jem.20171831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno A. B.; Agejas J.; Broughton H.; Dally R.; Durham T. B.; Espinosa J. F.; González R.; Hahn P. J.; Marcos A.; Rodríguez R.; et al. (2017) Optimization of hydroxyethylamine transition state isosteres as aspartic protease inhibitors by exploiting conformational preferences. J. Med. Chem. 60, 9807–9820. 10.1021/acs.jmedchem.7b01304. [DOI] [PubMed] [Google Scholar]
- Rombouts F. J. R.; Tresadern G.; Delgado O.; Martínez-Lamenca C.; Van Gool M.; García-Molina A.; Alonso de Diego S. A.; Oehlrich D.; Prokopcova H.; Alonso J. M.; et al. (2015) 1,4-Oxazine β-secretase 1 (BACE1) inhibitors: from hit generation to orally bioavailable brain penetrant leads. J. Med. Chem. 58, 8216–8235. 10.1021/acs.jmedchem.5b01101. [DOI] [PubMed] [Google Scholar]
- Gijsen H. J. M.; Alonso de Diego S. A.; De Cleyn M.; García-Molina A.; Macdonald G. J.; Martínez-Lamenca C.; Oehlrich D.; Prokopcova H.; Rombouts F. J. R.; Surkyn M.; et al. (2018) Optimization of 1,4-oxazine β-secretase 1 (BACE1) inhibitors toward a clinical candidate. J. Med. Chem. 61, 5292–5303. 10.1021/acs.jmedchem.8b00304. [DOI] [PubMed] [Google Scholar]
- Brodney M. A.; Beck E. M.; Butler C. R.; Barreiro G.; Johnson E. F.; Riddell D.; Parris K.; Nolan E. C.; Fan Y.; Atchison K.; et al. (2015) Utilizing structures of CYP2D6 and BACE1 complexes to reduce risk of drug-drug interactions with a novel series of centrally efficacious BACE1 inhibitors. J. Med. Chem. 58, 3223–3252. 10.1021/acs.jmedchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti R.; Lenci E.; Menchi G.; Pupi A.; Trabocchi A. (2017) Design and synthesis of bicyclic acetals as Beta Secretase (BACE1) inhibitors. Bioorg. Med. Chem. 25, 5077–5083. 10.1016/j.bmc.2017.03.030. [DOI] [PubMed] [Google Scholar]
- Mundlapati V. R.; Gautam S.; Sahoo D. K.; Ghosh A.; Biswal H. S. (2017) Thioamide, a hydrogen bond acceptor in proteins and nucleic Acids. J. Phys. Chem. Lett. 8, 4573–4579. 10.1021/acs.jpclett.7b01810. [DOI] [PubMed] [Google Scholar]
- van Bergen L. A. H.; Alonso M.; Palló A.; Nilsson L.; De Proft F.; Messens J. (2016) Revisiting sulfur H-bonds in proteins: the example of peroxiredoxin AhpE. Sci. Rep. 6, 30369. 10.1038/srep30369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calugi L.; Lenci E.; Innocenti R.; Trabocchi A. (2020) Synthesis of morpholine derivatives using the Castagnoli-Cushman reaction as BACE1 inhibitors: unexpected binding activity of cyclic thioamides. Bioorg. Med. Chem. Lett. 30, 127211. 10.1016/j.bmcl.2020.127211. [DOI] [PubMed] [Google Scholar]
- Wiley J. C.; Hudson M.; Kanning K. C.; Schecterson L. C.; Bothwell M. (2005) Familial Alzheimer’s disease mutations inhibit gamma-secretase-mediated liberation of beta-amyloid precursor protein carboxy-terminal fragment. J. Neurochem. 94, 1189–1201. 10.1111/j.1471-4159.2005.03266.x. [DOI] [PubMed] [Google Scholar]
- Josien H.; Bara T.; Rajagopalan M.; Clader J. W.; Greenlee W. J.; Favreau L.; Hyde L. A.; Nomeir A. A.; Parker E. M.; Song L.; et al. (2009) Novel orally active morpholine N-arylsulfonamides γ-secretase inhibitors with low CYP 3A4 liability. Bioorg. Med. Chem. Lett. 19, 6032–6037. 10.1016/j.bmcl.2009.09.055. [DOI] [PubMed] [Google Scholar]
- Josien H.; Bara T.; Rajagopalan M.; Asberom T.; Clader J. W.; Favreau l.; Greenlee W. J.; Hyde L. A.; Nomeir A. A.; Parker E. M.; et al. (2007) Small conformationally restricted piperidine N-arylsulfonamides as orally active γ-secretase inhibitors. Bioorg. Med. Chem. Lett. 17, 5330–5335. 10.1016/j.bmcl.2007.08.013. [DOI] [PubMed] [Google Scholar]
- de Matos A. M.; Martins A.; Man T.; Evans D.; Walter M.; Oliveira M. C.; López Ó.; Fernandez-Bolaños J. G.; Dätwyler P.; Ernst B.; et al. (2019) Design and synthesis of CNS-targeted flavones and analogues with neuroprotective potential against H2O2- and Aβ1–42-induced toxicity in SH-SY5Y human neuroblastoma cells. Pharmaceuticals 12, 98. 10.3390/ph12020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Song M.; Liu X.; Kang S.; Duong D. M.; Seyfried N. T.; Cao X.; Cheng L.; Sun Y. E.; Ping Yu S.; et al. (2015) Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 6, 8762. 10.1038/ncomms9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Obianyo O.; Dall E.; Du Y.; Fu H.; Liu X.; Kang S. S.; Song M.; Yu S. P.; Cabrele C.; et al. (2017) Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nat. Commun. 8, 14740. 10.1038/ncomms14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis P. T. (2005) The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. 10, 6–9. 10.1017/S1092852900014164. [DOI] [PubMed] [Google Scholar]
- Cai Z. (2014) Monoamine oxidase inhibitors: promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 9, 1533–1541. 10.3892/mmr.2014.2040. [DOI] [PubMed] [Google Scholar]
- Manera C.; Martinelli A.; Nencetti S.; Romagnoli F.; Rossello A.; Giannaccini G.; Scatizzi R.; Cozzini P.; Domiano P. (1994) X-ray analysis, theoretical studies and aadrenergic biopharmacological properties of 1-(2,5 dimethoxyphenyl)-2- aminoethanol and its morpholine analogue. Eur. J. Med. Chem. 29, 519–525. 10.1016/0223-5234(94)90144-9. [DOI] [Google Scholar]
- Repsold B. P.; Malan S. F.; Joubert J.; Oliver D. W. (2018) Multi-targeted directed ligands for Alzheimer’s disease: design of novel lead coumarin conjugates. SAR QSAR Environ. Res. 29, 231–255. 10.1080/1062936X.2018.1423641. [DOI] [PubMed] [Google Scholar]
- Bertron J. L.; Cho H. P.; Garcia-Barrantes P. M.; Panarese J. D.; Salovich J. M.; Nance K. D.; Engers D. W.; Rook J. M.; Blobaum A. L.; Niswender C. M.; et al. (2018) The discovery of VU0486846: steep SAR from a series of M1 PAMs based on a novel benzomorpholine core. Bioorg. Med. Chem. Lett. 28, 2175–2179. 10.1016/j.bmcl.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook J. M.; Bertron J. L.; Cho H. P.; Garcia-Barrantes P. M.; Moran S. P.; Maksymetz J. T.; Nance K. D.; Dickerson J. W.; Remke D. H.; Chang S.; et al. (2018) A novel M1 PAM VU0486846 exerts efficacy in cognition models without displaying agonist activity or cholinergic toxicity. ACS Chem. Neurosci. 9, 2274–2285. 10.1021/acschemneuro.8b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Nie J.; Ma X.; Wei Y.; Peng Y.; Wei X. (2019) Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 18, 26. 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Cheng H.; Roberts T. M.; Zhao J. J. (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discovery 8, 627–644. 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton R. A.; Sabatini D. M. (2017) mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pópulo H.; Lopes J. M.; Soares P. (2012) The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 13, 1886–1918. 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heras-Sandoval D.; Avila-Muñoz E.; Arias C. (2011) The phosphatidylinositol 3-kinase/mTor pathway as a therapeutic target for brain aging and neurodegeneration. Pharmaceuticals 4, 1070–1087. 10.3390/ph4081070. [DOI] [Google Scholar]
- Heras-Sandoval D.; Pérez-Rojas J. M.; Hernández-Damián J.; Pedraza-Chaverri J. (2014) The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signalling 26, 2694–2701. 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Sun C.; Xu S.; Wu C.; Wu J.; Xu M.; Zhao H.; Chen L.; Zeng W.; Zheng P. (2014) Design, synthesis, anticancer activity and docking studies of novel 4-morpholino-7,8-dihydro-5H-thiopyrano-[4,3-d]pyrimidine derivatives as mTOR inhibitors. Bioorg. Med. Chem. 22, 6746–6754. 10.1016/j.bmc.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Liu K. K.-C.; Bailey S.; Dinh D. M.; Lam H.; Li C.; Wells P. A.; Yin M. J.; Zou A. (2012) Conformationally-restricted cyclic sulfones as potent and selective mTOR kinase inhibitors. Bioorg. Med. Chem. Lett. 22, 5114–5117. 10.1016/j.bmcl.2012.05.104. [DOI] [PubMed] [Google Scholar]
- Helwa A. A.; Gedawy E. M.; Taher A. T.; El-Ansary A. K.; Abou-Seri S. M. (2020) Synthesis and biological evaluation of novel pyrimidine-5-carbonitriles featuring morpholine moiety as antitumor agents. Future Med. Chem. 12, 403–421. 10.4155/fmc-2019-0146. [DOI] [PubMed] [Google Scholar]
- Zask A.; Kaplan J.; Verheijen J. C.; Richard D. J.; Curran K.; Brooijmans N.; Bennett E. M.; Toral-Barza L.; Hollander I.; Ayral-Kaloustian S.; et al. (2009) Morpholine derivatives greatly enhance the selectivity of mammalian target of rapamycin (mTOR) inhibitors. J. Med. Chem. 52, 7942–7945. 10.1021/jm901415x. [DOI] [PubMed] [Google Scholar]
- Maira S.-M.; Pecchi S.; Huang A.; et al. (2012) Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 11, 317–328. 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- Beaufils F.; Cmiljanovic N.; Cmiljanovic V.; et al. (2017) 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a potent, brain-penetrant, orally bioavailable, pan-class I PI3K/mTOR inhibitor as clinical candidate in oncology. J. Med. Chem. 60, 7524–7538. 10.1021/acs.jmedchem.7b00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M.; Hall M. N. (2014) Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 15, 155–162. 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Saxton R. A.; Sabatini D. M. (2017) mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M.; Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293. 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino P. B.; Nathanson K. L.; Henske E. P. (2006) The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356. 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Talboom J. S.; Velazquez R.; Oddo S. (2015) The mammalian target of rapamycin at the crossroad between cognitive aging and Alzheimer’s disease. NPJ. Aging Mech. Dis. 1, 15008. 10.1038/npjamd.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Rudge D. G.; Koos J. D.; Vaidialingam B.; Yang H. J.; Pavletich N. P. (2013) mTOR kinase structure, mechanism and regulation. Nature 497, 217–223. 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupak K.; Vulichi S. R.; Suman K. (2016) Emphasizing morpholine and its derivatives (MAID): A typical candidates of pharmaceutical importance. Int. J. Chem. Sci. 14, 1777–1788. [Google Scholar]
- Degorce S. L.; Bodnarchuk M. S.; Cumming I. A.; Scott J. S. (2018) Lowering lipophilicity by adding carbon: one-carbon bridges of morpholines and piperazines. J. Med. Chem. 61, 8934–8943. 10.1021/acs.jmedchem.8b01148. [DOI] [PubMed] [Google Scholar]
- Rageot D.; Bohnacker T.; Melone A.; et al. (2018) Discovery and preclinical characterization of 5 - [4,6-Bis({3-oxa-8-azabicyclo[3.2.1]octan-8-yl})-1,3,5-triazin-2-yl]-4-(difluoromethyl)pyridin-2-amine (PQR620), a highly potent and selective mTORC1/2 inhibitor for cancer and neurological disorders. J. Med. Chem. 61, 10084–10105. 10.1021/acs.jmedchem.8b01262. [DOI] [PubMed] [Google Scholar]
- Bonazzi S.; Goold C. P.; Gray A.; Thomsen N. M.; Nunez J.; Karki R. G.; Gorde A.; Biag J. D.; Malik H. A.; Sun Y.; et al. (2020) Discovery of a brain-penetrant ATP-competitive inhibitor of the mechanistic target of rapamycin (mTOR) for CNS disorders. J. Med. Chem. 63, 1068–1083. 10.1021/acs.jmedchem.9b01398. [DOI] [PubMed] [Google Scholar]