

Abstract
Fibrosis is a common pathological response to inflammation in many peripheral tissues and can prevent tissue regeneration and repair. Here, we identified persistent fibrotic scarring in the central nervous system (CNS) following immune cell infiltration in the experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis. Using lineage tracing and single-cell sequencing in EAE, we determined that the majority of the fibrotic scar is derived from proliferative CNS fibroblasts, not pericytes or infiltrating bone marrow-derived cells. Ablating proliferating fibrotic cells using cell-specific expression of herpes thymidine kinase led to an increase in oligodendrocyte lineage cells within the inflammatory lesions and a reduction in motor disability. We further identified that interferon gamma pathway genes are enriched in CNS fibrotic cells, and the fibrotic cell-specific deletion of Ifngr1 resulted in reduced fibrotic scarring in EAE. These data delineate a framework for understanding the CNS fibrotic response.
Introduction
Fibrosis, defined as the pathological deposition of collagen I (Col1)-rich extracellular matrix, is a common response to injury and inflammation in peripheral organs such as the heart, kidney, liver and lung 1–5. Following tissue damage, extracellular matrix deposits provide essential structure to areas of injury. Fibrotic scarring occurs if the secretion of these proteins is left unchecked and the matrix overcomes the tissue, which can cause organ damage and in severe cases organ failure6,7. Only a handful of reports have evaluated fibrosis in the CNS, and have largely done so in the context of spinal cord injury (SCI)8–10. Little is known about the presence or extent of fibrosis in the CNS in response to neuroinflammation and its role in disease recovery.
Following SCI, a scar forms that has two components: an outer glial scar made up of reactive astrocytes that surrounds the site almost immediately following injury and an inner fibrotic scar made up of extracellular matrix (ECM) proteins, predominately Col1, that appears a few days post injury9,11. While the origin and role of the glial scar have been studied12–14, much less is known about the fibrotic component. The fibrotic scar is important for recovery as it seals the injury site, which has been hypothesized to limit the influx of blood cells and toxins into the brain parenchyma, but also prevents repair in the long term as axons are not able to grow past ECM proteins10,15. Interestingly, if fibrotic scar formation is completely ablated following SCI, the lesion does not seal, leaving a cavity that is not compatible with regeneration16. Meanwhile reducing, but not completely inhibiting, fibrotic scar formation following SCI promotes functional recovery by enabling axon regeneration17–21. This suggests that modulating specific aspects of fibrotic scarring may be an important therapeutic strategy to improve CNS repair.
Much less is known about the presence of fibrotic scarring in response to neuroinflammation and how this may affect tissue repair. In multiple sclerosis (MS), a neuroinflammatory disease characterized by CNS lesions with immune cell infiltration and demyelination, a number of studies have reported Col1 deposition surrounding blood vessels and an increase in fibrotic gene expression in lesions22–25. In EAE, a mouse model for MS, an increase in collagen-producing cells has been reported in demyelinating lesions22. Following neuroinflammation, such as that which occurs in MS, this fibrotic scar may act to lessen the severity of disease by limiting immune cell infiltration or it may inhibit repair by limiting neuronal function and/or inhibiting the entry of progenitor cells into the lesions. As there are no approved treatments for MS that repair damaged tissue, understanding what factors prevent tissue repair in neuroinflammatory conditions such as MS could lead to new therapeutics.
Determining the origin and mechanisms of fibrotic scar formation could influence treatment options for any CNS disorder with fibrotic scarring. In SCI, the origin of Col1-producing fibrotic cells has been controversial. Some reports propose that a subset of pericytes are activated to give rise to Col1-expressing cells in the scar, whereas other groups have suggested the fibrotic cells derive from the proliferation and migration of resident CNS fibroblasts which produce Col1 at rest 16,26–29. Recent single-cell sequencing studies showed that indeed these are distinct populations in health: pericytes are embedded in the basement membrane of capillaries, and fibroblasts are associated with large vessels in the parenchyma, meninges and choroid plexus30,31. However, these cell types can often be confused with each other as both express similar surface markers such as PDGFRβ, and thus it is unclear which cells drive the fibrotic response in the CNS. Furthermore, very little is known about the molecular mechanisms that lead to CNS fibrotic scar formation and how these mechanisms can be targeted to modulate fibrotic scar formation and enhance recovery.
Here, we show that there is extensive fibrotic scar formation following immune cell infiltration which contributes to disease severity. This fibrotic scar is found in every neuroinflammatory lesion and lasts for months following lesion formation. Using lineage tracing and single-cell sequencing we demonstrate that the scar forms largely from the proliferation and migration of CNS fibroblasts, and that inhibiting CNS fibroblast proliferation leads to decreased fibrotic scarring and reduced disability in the chronic stages of disease. We further show that interferon gamma (IFNγ) signaling regulates the amplitude of fibrotic scar formation, identifying a potential therapeutic target to modulate levels of scar formation.
Results
Immune cell infiltration drives CNS fibrosis
To determine if a fibrotic scar forms following neuroinflammation, we induced EAE in Col1a1-GFP mice and examined both collagen 1 (Col1) protein expression and the number and localization of Col1a1-GFP+ cells in the spinal cord. In health, Col1 protein and Col1a1-GFP+ cells were found in the meninges and associated with large parenchymal vessels (Fig 1a, Extended Data Fig 1a). In EAE, a robust Col1+ fibrotic scar was found in every lesion examined starting at 5 days post symptom onset (5 d PSO) and remained for the duration of the experiment (60 days PSO) (Fig 1a, Extended Data Fig 1b–d). Col1 deposition coincided with a massive increase in the number of Col1a1-GFP+ cells throughout the parenchymal lesions that peaked at 10 days PSO, and maintained their numbers throughout the course of the experiment (Fig 1a,b, Extended Data Fig 1e,f). Fibrotic scar formation and expansion of Col1a1-GFP+ cells were observed secondary to the initial influx of immune cells and the onset of motor symptoms (Fig 1b–d, Extended Data Fig 1b,d), suggesting that fibrosis is likely a response to, and not causal of, immune cell infiltration and symptom initiation.
Figure 1: Fibrotic scar formation occurs in EAE lesions.
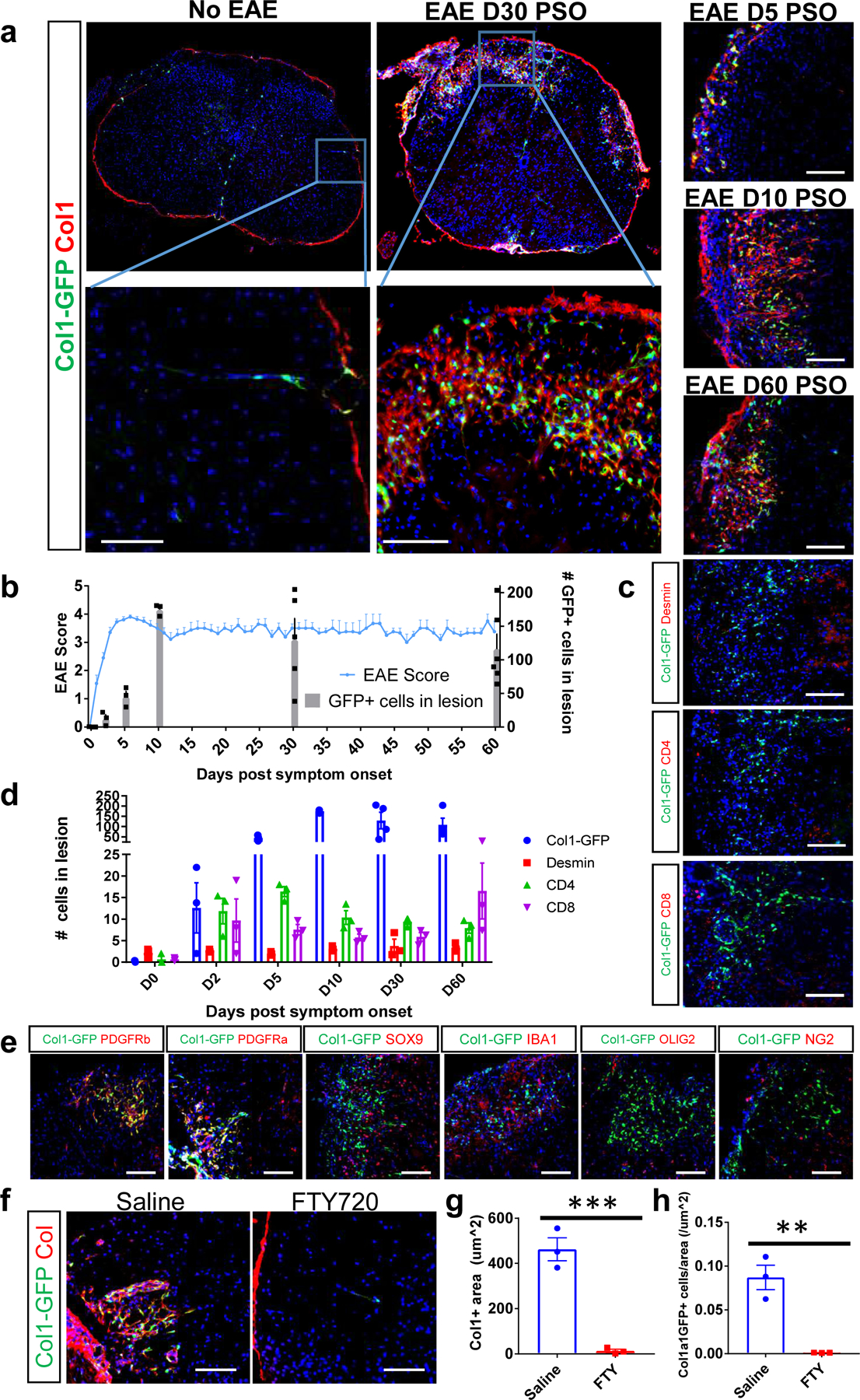
a. Spinal cord sections from Col1a1-GFP mice in health or with EAE were stained for collagen I (red) to label the fibrotic scar, DAPI to label nuclei (blue) and visualized for the GFP reporter (green). Left: whole spinal cord cross sections (top) with specific area magnified (bottom) for Col1a1-GFP mice in health or 30 days EAE post symptom onset (PSO). Right: Magnified spinal cord cross section areas from Col1a1-GFP mice 5, 10 or 60 days PSO. b. Quantification of the number of Col1a1-GFP+ cells per lesion (bar graph) was compared with EAE motor symptoms (line graph) over the course of EAE, (± s.e.m.; n=3–6 per time point for cell quantification, 4–11 for EAE score). c. Spinal cord sections from Col1a1-GFP mice were stained with desmin, CD4 or CD8 (red) over the course of EAE, and the number of positive cells for each marker was quantified in (d), ± s.e.m., n= 3–4 mice per marker per time point. e. Spinal cords from Col1a1-GFP mice with EAE at 10 or 30 days PSO were stained with DAPI (blue) and molecular markers for various cell types (red). GFP+ cells are immune-reactive for both PDGFRα and PDGFRβ, but not for markers of astrocytes (SOX9), microglia/macrophages (IBA1), oligodendrocyte lineage cells (OLIG2) or mural cells (NG2). f. Spinal cord sections from Col1a1-GFP mice with EAE treated daily with either saline or 2 mg/kg FTY720 and collected when the mice administered saline reached 8–10 d PSO were stained with Col1 in red. The collagen area and number of Col1a1-GFP+ cells per area is quantified in g, h, ± s.e.m, n = 3 per condition, ***p = 0.0009, **p = 0.0035 by an unpaired, two-tailed t-test. Scale bars = 100 µm.
We further immunostained fibrotic spinal cords with a series of cellular markers to determine which co-localize with the Col1a1-GFP+ fibrotic cells. Col1a1-GFP+ cells were immune-reactive for both PDGFRα and PDGFRβ which often mark fibroblasts in peripheral organs. The Col1a1-GFP+ cells did not stain positive for markers of astrocytes (SOX9, GFAP), microglia/macrophages (IBA1, CD11b) or mural cells (NG2, Desmin) (Fig 1e). Additionally, while Col1a1-GFP+ cells increased substantially during disease, there was not a comparable increase in Desmin+ cells in the lesions over time (Fig 1c,d). Staining for OLIG2, a marker for oligodendrocyte lineage cells, demonstrated that these cells were outside the Col1-stained region, suggesting that this scar may be blocking oligodendrocyte lineage cells from entering the lesion site and repairing demyelinated axons (Fig 1e). In contrast to the clear lamination of the fibrotic and glial scars that occurs following SCI9, we observed intermixing of GFAP+ reactive astrocyte processes and Col1a1-GFP+ fibrotic cells (Extended Data Fig 1c). These results demonstrate that a robust Col1+ fibrotic scar forms in response to immune cell infiltration, and the Col1-secreting cells increase in the lesion over time and are immunoreactive for both PDGFRα and PDGFRβ.
To determine the relative contributions of inflammation and demyelination to fibrotic scarring, we inhibited inflammation in EAE using fingolimod (FTY720) which prevents immune cell exit from lymph nodes and is used as a treatment for MS in human patients32,33. Mice administered saline following EAE had robust demyelination, motor symptoms, fibrotic scarring and Col1a1-GFP+ cell expansion, whereas mice administered FTY720 did not experience motor symptoms or demyelination and had no discernable fibrotic scar or increase in scar-forming cells (Fig 1f–h). Thus, inhibition of immune cell infiltration in the EAE model reduced both demyelination and fibrotic scarring. To further delineate the roles of inflammation and demyelination we looked for fibrotic scarring in mice with the cuprizone and lysolecithin (LPC) models of demyelination. Through immunostaining for collagen 1 protein and infiltrating immune cells, we found that mice administered cuprizone, which does not lead to widespread CNS immmune cell infiltration, did not have fibrotic scarring in the areas of demyelination, while mice administered LPC, which does lead to immune cell entry into demyelinated areas, did have a fibrotic scar (Extended Data Fig 2). Together, these experiments suggest that fibrotic scarring is associated with immune cell infiltration-driven demyelination.
CNS fibroblasts overwhelmingly form the fibrotic scar in neuroinflammation
Conflicting studies have reported that CNS fibrotic scar formation is derived from either the expansion of Col1-expressing CNS fibroblasts or pericytes turning on the expression of collagen. Single-cell sequencing of vascular cells in the CNS has demonstrated that there are indeed distinct populations of pericytes (along capillaries and post-capillary venules) and fibroblasts (associated primarily with large vessels), however, it is likely that these cells have been confused for each other in the literature as both cell types express PDGFRβ30.
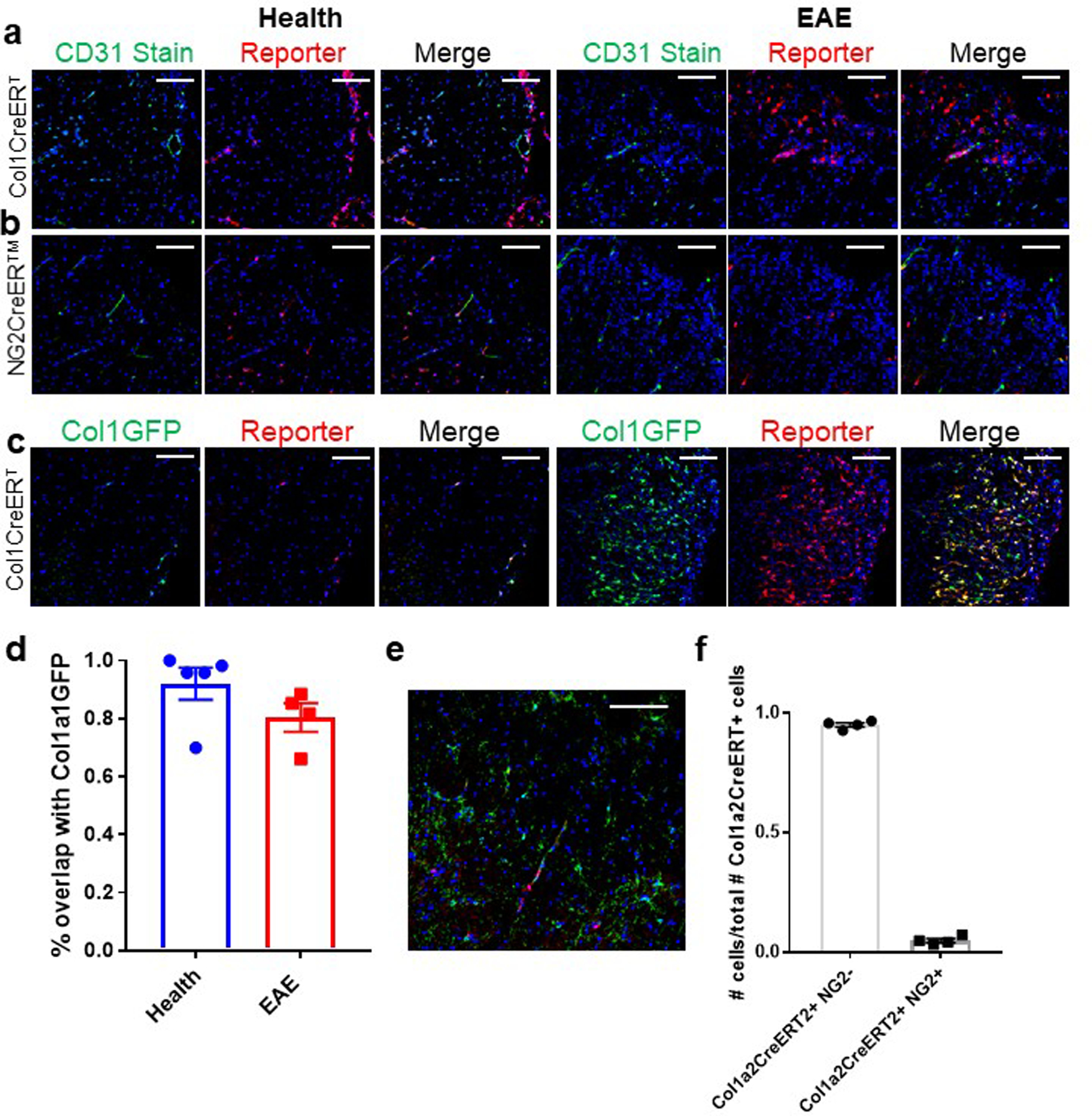
To determine the identity of the scar forming cells we performed lineage tracing experiments with the Rosa-lsl-tdTomato reporter mouse line mated to tamoxifen-inducible cre transgenic lines: NG2CreER™ to label mural cells (pericytes and vascular smooth muscle cells [vSMCs]) and oligodendrocyte precursor cells, aSMACreERT2 to label vSMCs and Col1a2CreERT to label cells that express Col1 at rest. We injected mice from each strain with tamoxifen at 6 weeks of age to induce expression of tdTomato within the specific cell populations and induced EAE at 12 weeks of age. We collected spinal cords at 10 days PSO to assess the presence of any cells that were downstream of the genetically labeled NG2, aSMA, or Col1a2 cells within the Col1+ fibrotic scar. We observed a 70-fold increase in the number of Col1a2CreERT labeled cells in the injury site that co-localized with the Col1+ fibrotic scar, without a similar increase in the NG2CreER™ or aSMACreERT2 traced cells (Fig 2a–d). We then bred the Col1a2CreERT reporter mice with the Col1a1-GFP mice and found that over 90% of GFP+ cells were also tdTomato+ in health and over 80% in EAE (Extended Data Fig 3c,d). Additionally, less than 5% of Col1a2CreERT labeled cells were NG2+ (Extended Data Fig 3e,f). This demonstrates that the vast majority of fibrotic scar-forming cells arose from the expansion of cells that expressed Col1 at rest, but not the proliferation of mural cells (vSMCs/pericytes) or other cell populations turning on the expression of Col1.
Figure 2: Scar-forming cells arise from cells that produce collagen at rest, and not mural cells.
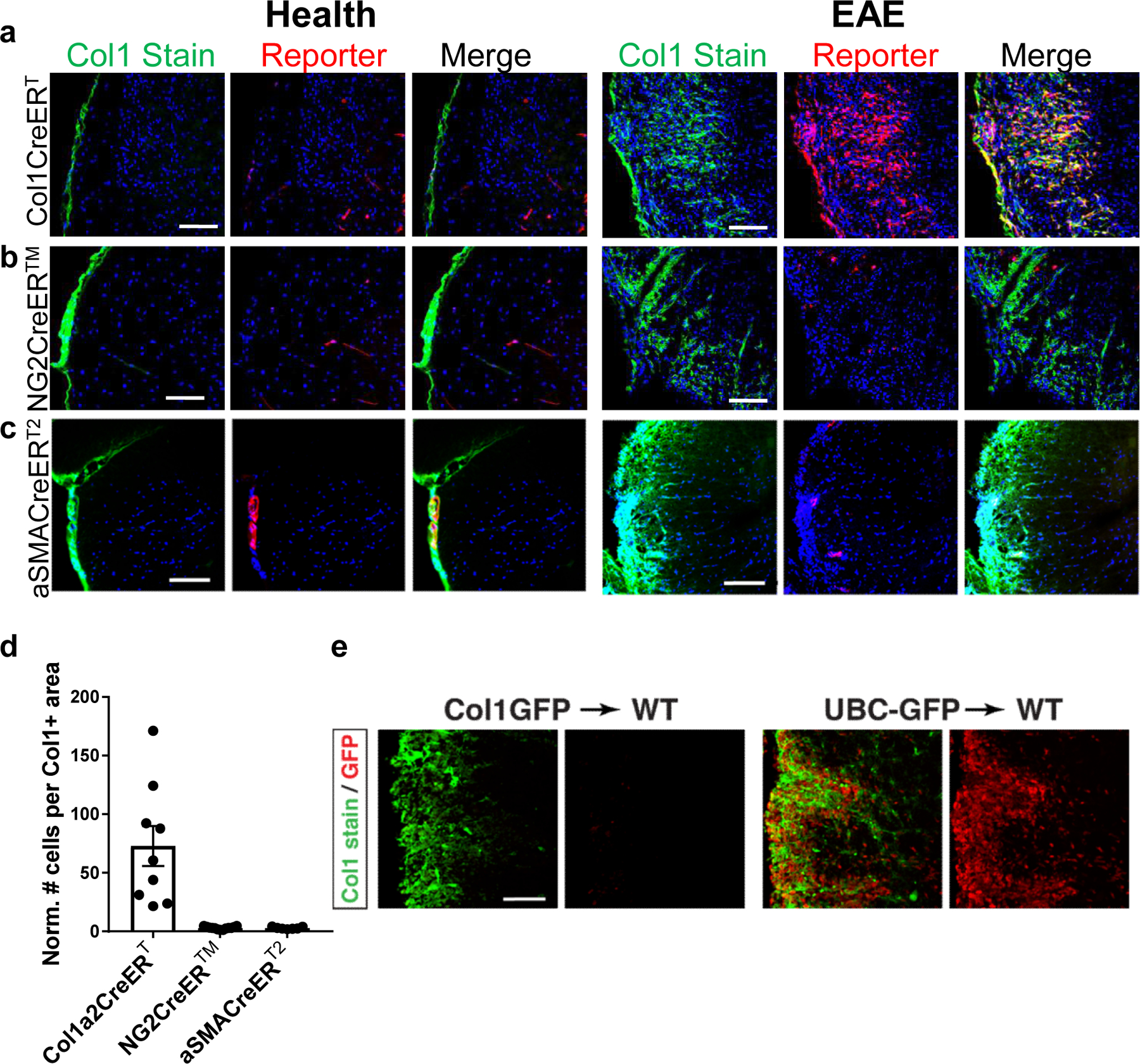
a–d. Sections from spinal cords of Col1a2CreERT;Rosa-tdTomato (a), NG2CreER™;Rosa-tdTomato (b), or aSMACreERT2;Rosa-tdTomato (c) mice with EAE were stained for Col1 (green) and DAPI (blue) and visualized for tdTomato reporter (red). d. The number of reporter cells within the Col1+ area was normalized to the number of reporter cells in white matter in health (± s.e.m., n=7 health, 9 EAE for Col1a2CreERT, n=7 health, 8 EAE for NG2CreER™, n=4 health, 7 EAE for aSmaCreERT2, p<0.0001 using a one way ANOVA with multiple comparisons, p<0.0001 for Col1a2CreERT vs. NG2CreER™, p=0.0001 for Col1a2CreERT vs. aSMACreERT2 and p>0.9999 for NG2CreER™ vs aSMACreERT2). e. Spinal cord sections from mice with Col1a1-GFP donor bone marrow (left) or UBC-GFP donor bone marrow (right) with EAE at 10 days PSO were stained with Col1 (green), and the GFP was visualized in red (representative images for experiment with n=8 for Col1a1-GFP, n=5 for UBC-GFP). All scale bars = 100 µm.
To determine whether the CNS fibrotic cells derive from the bone marrow, we transplanted wild type mice with bone marrow from either Col1GFP or UBC-GFP (pan-cellular ubiquitous expression of GFP) mice. Recipients of UBC-GFP bone marrow displayed massive infiltration of GFP+ cells into EAE lesions, confirming that donor bone marrow derived immune cells can infiltrate the CNS in this transplantation model. Col1GFP bone marrow recipients displayed a robust Col1+ fibrotic scar, but had no Col1GFP+ cells in lesions. These results indicate that Col1-expressing fibrotic cells were derived from the host, and not bone marrow derived fibrocytes or immune cells that migrated into EAE lesions (Fig 2e).
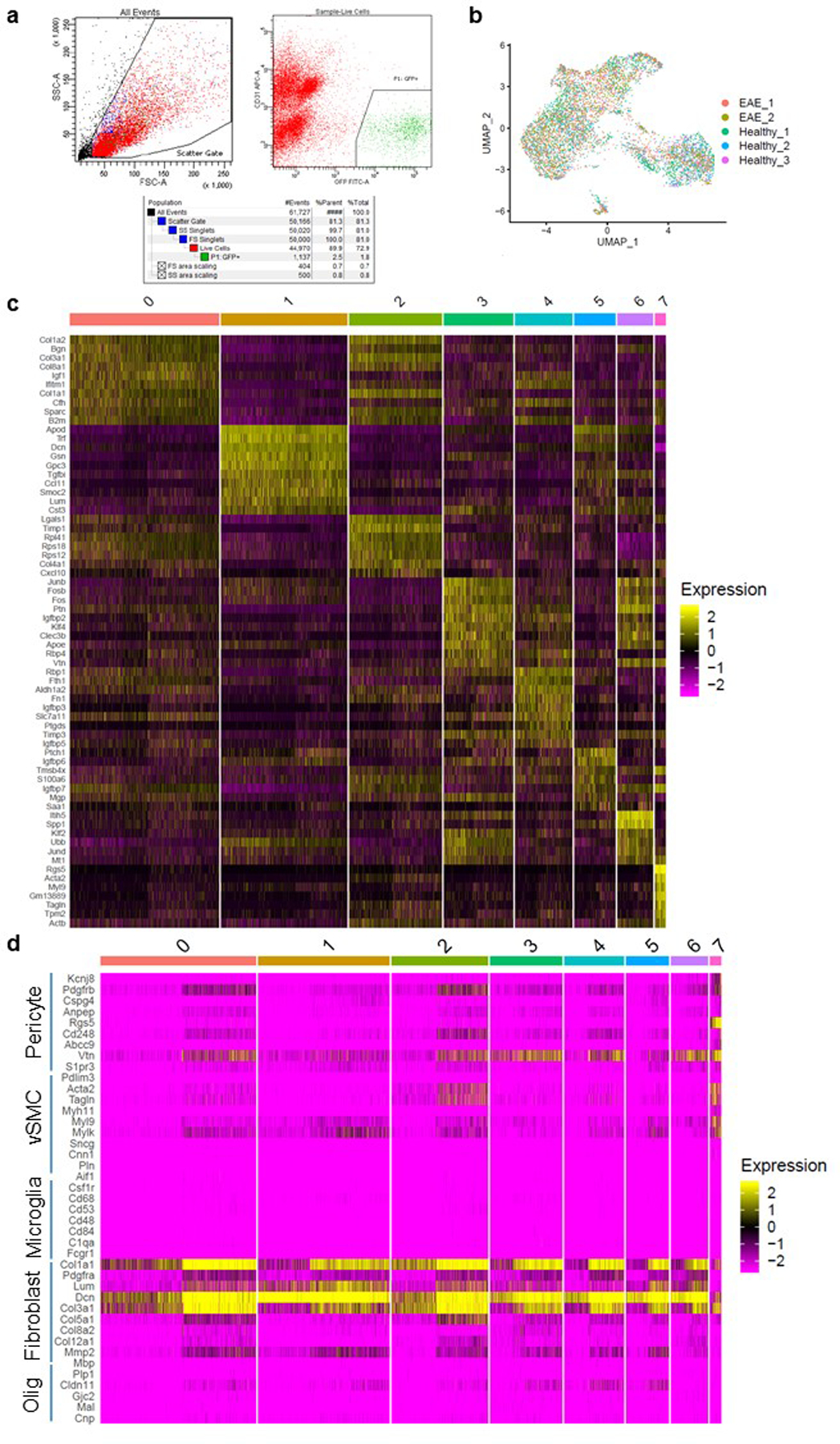
To further evaluate the cellular identities of Col1-secreting cells in health and EAE, we performed single-cell RNA sequencing of GFP+ cells from Col1a1-GFP mice in health and EAE at 5–7 days PSO. The cells clustered into 8 clusters (0–7), which could be subdivided into three major classes that were clearly distinct on the UMAP plot : Class 1 (clusters 0,2,3,4,6), Class 2 (clusters 1,5) and Class 3 (cluster 7) (Fig 3b). Cells from both health and EAE were found in each class of cells, and each cluster within the class, suggesting that there is not a unique cell population that turns on the expression of Col1a1 in EAE. This is consistent with the lineage tracing results which demonstrate that the fibrotic cells result from cells that express Col1 in health. Class 1 and Class 2, representing 98% of cells, were characterized as fibroblasts and Class 3 as stromal cells using SingleR, a computational method for unbiased cell type recognition of single-cell data sets using thousands of bulk RNA-seq reference datasets from all organs (Fig 3b,d)34. Indeed, the expression of pericyte and vSMC-specific genes were low in clusters 0–6 (classes 1 and 2), whereas canonical fibroblast-specific genes were highly expressed in these clusters that make up 98% of the Col1a1-GFP+ cells. Cluster 7 expressed a combination of pericyte and vSMC-specific genes (Fig 3e, Extended Data Fig 4d)30. Although there were Col1GFP+ mural cells identified in cluster 7, these represented a small minority (<2%) of the Col1-producing cells, and this cluster displayed the lowest Col1 expression of any of the clusters. This data, combined with NG2CreER™ and aSMACreERT2 lineage tracing studies, demonstrate that mural cells are not major contributors to fibrotic scar formation.
Figure 3: Scar-forming cells have the transcriptional profile of fibroblasts at the single cell level.
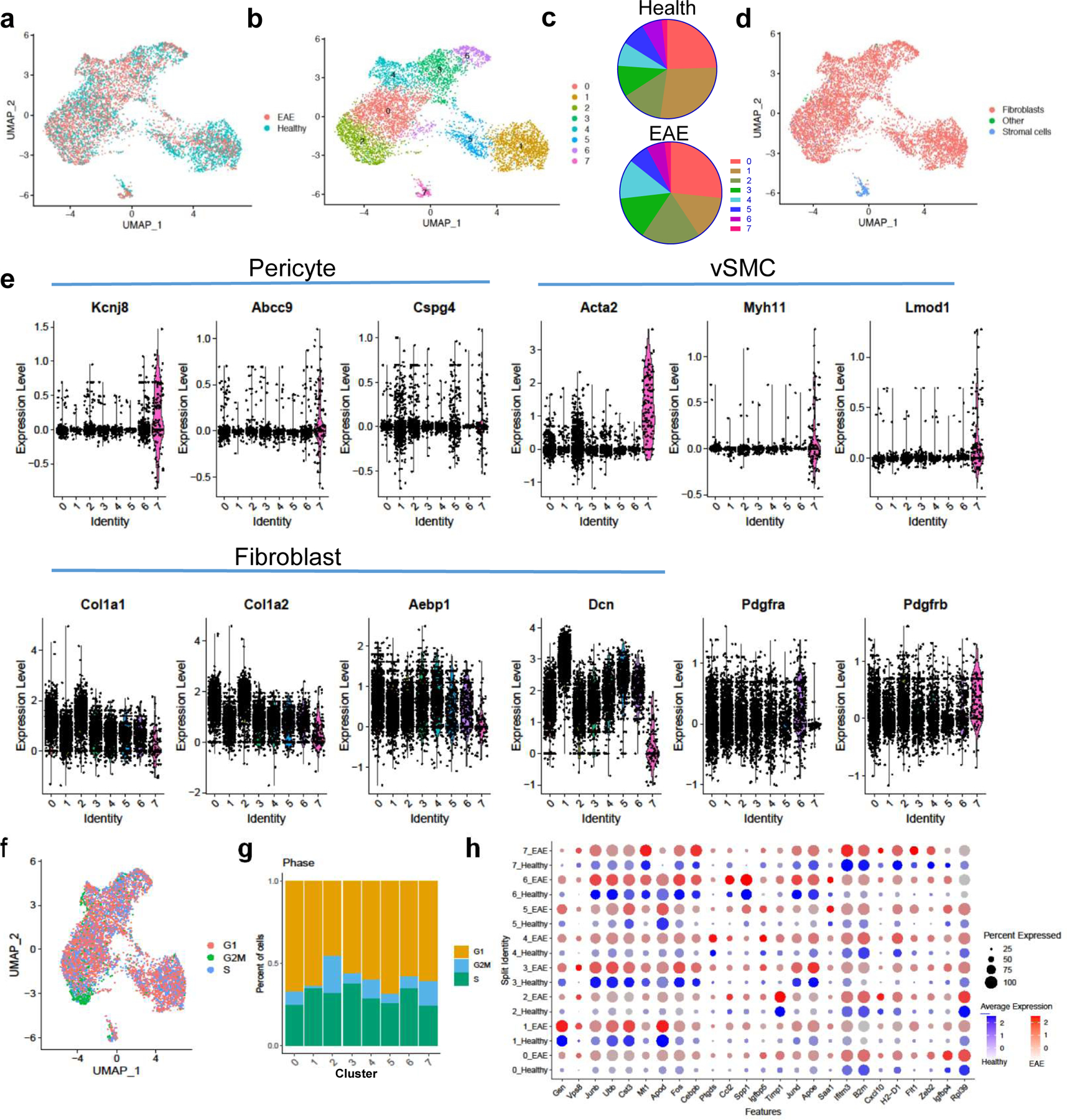
a–d. Col1a1-GFP+ cells from spinal cords of healthy mice (n=3 samples with 2–3 spinal cords each) and mice with EAE 5–7 days PSO (n=2 samples, 2 spinal cords each) were transcriptionally profiled at the single cell level and clustered using Seurat v3. a. UMAP plot with the sample identity (Health vs. EAE) labeled for each cell. b. UMAP plot of the clustering analysis reveals 8 clusters that could be subdivided into three classes: Class 1 (clusters 0,2,3,4,6), Class 2 (clusters 1,5), and Class 3 (cluster 7). c. Pie charts showing the percentage of each cluster relative to the total number of cells in health or EAE. d. UMAP plot with each cell labeled with its cellular identity determined using SingleR and the Immgen reference dataset. e. Violin plots of the expression levels of pericyte, vSMC and fibroblast-specific genes per cluster. f. UMAP plot showing the transcriptional cell cycle identity per cell. g. Bar graph showing the percentage of transcriptional signatures of the different steps of the cell cycle in each cluster. h. Dot plot of selected genes that are expressed at greater levels in EAE than in health in at least one cluster.
The proportions of each cell population were very similar in health versus EAE, with small increases in Class 1 clusters (0, 2, 3, 4) and decreases in Class 2 clusters (1,5) in EAE (Fig 3a–c). However, within clusters, genes indicative of activated fibroblasts were expressed more highly in EAE cells, indicating that there was a shift towards a more activated fibroblast in EAE (Fig 3h). Proliferation analysis of the cells revealed that each cluster has some proportion of cells with a transcriptional profile indicative of the S and G2/M phases of the cell cycle, with cells in cluster 2 having the largest proportion of cells in the G2/M stage (Fig 3f,g). Cluster 0 has the most enriched expression of extracellular matrix proteins such as collagens, and cluster 3 is enriched for the transcription factors Fos, Fosb and Junb, which are expressed in activated fibroblasts and required for stretch-induced ECM production (Extended Data Fig 4c)35,36. These transcription factors are most highly expressed in cells in this cluster from EAE (Fig 3h). Therefore, these data indicate that there are two classes of Col1+ fibroblasts that produce the fibrotic scar which can be divided into clusters that define specific states of these cells, such as dividing and actively producing high levels of ECM.
Taken together these lineage tracing and single-cell sequencing studies indicate that fibrotic cells arise overwhelmingly from the expansion of CNS fibroblasts expressing Col1 in health, and not pericytes/vSMCs that turn on Col1 production or infiltrating bone marrow derived cells. These studies further identify potential fibroblast cell states specifically associated with neuroinflammatory fibrotic scar formation.
Ablation of proliferating fibrotic cells reduces disease severity in EAE
To determine how fibrotic scar formation affects the progression of neuroinflammatory disease, we generated transgenic mice expressing the herpes simplex virus thymidine kinase (HTK) in Col1-expressing cells, where the administration of ganciclovir (GCV) would kill these cells that are dividing following EAE induction37,38. Test (Col1a2CreERT; lox-stop-lox-HTK [fHTK]) and littermate control (lox-stop-lox-HTK) mice both were injected with tamoxifen at 6 weeks to induce HTK expression specifically in the fibrotic cells of the fHTK mice, and EAE was induced at 12 weeks. GCV (25 mg/kg) was administered to all mice daily starting at day 8 post EAE induction to continually prevent fibrotic cell proliferation in the fHTK mice, and tissue was collected 30 days post EAE induction. This resulted in a significant ablation of the Col1+ fibrotic scar in fHTK mice with most residual Col1 expression observed around blood vessels (Fig 4a,b). Extracellular matrix proteins such as periostin and collagen 3 and the fibroblast antigen ER-TR7 were also reduced by this paradigm (Extended Data Fig 5a). There was no apparent effect on immune cell infiltration or reactive gliosis (Fig 4c, Extended Data Fig 5a–c). Therefore, a reduction in fibrotic cell proliferation led to a major reduction in parenchymal fibrotic scar formation but still left some perivascular Col1.
Figure 4: Reducing fibrotic scar formation reduces disease severity in EAE.
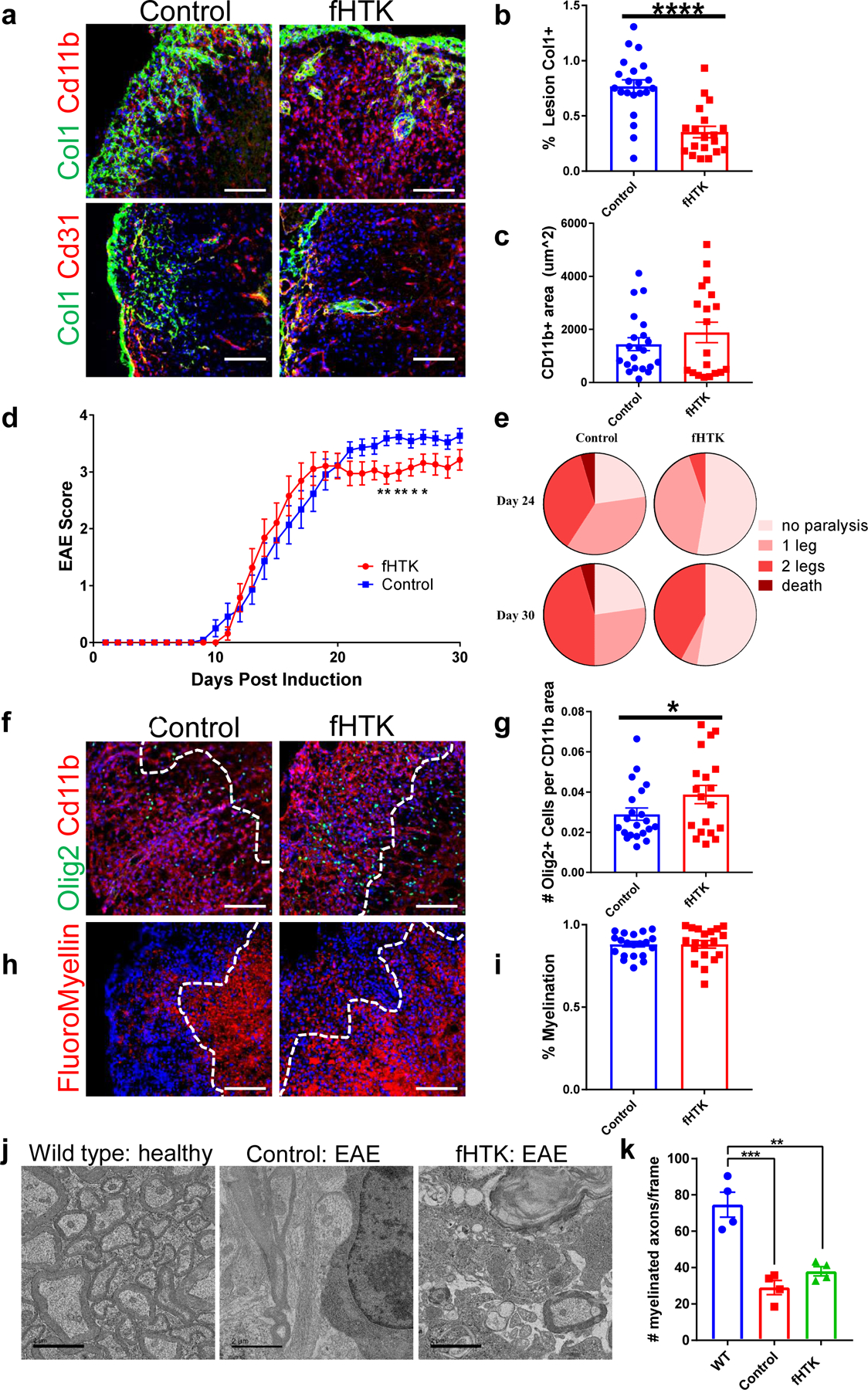
a. Spinal cord sections from fibrotic cell-specific herpes thymidine kinase (fHTK) mice and littermate controls 30 days after EAE immunization were stained for Col1 (green), DAPI (blue) and CD11b (top, red) to label immune cells or CD31 (bottom, red) to label endothelial cells. b. Quantification of the percentage of the area of immune infiltration (denoted by CD11b) that is Col1+, ± s.e.m., **p <0.0001 by Student’s two-tailed t-test, n=21 control, 19 fHTK. c. Quantification of the total lesion size, denoted by Cd11b staining, ± s.e.m., n = 21 control, 19 fHTK, p = 0.33 by Student’s two-tailed t-test. d. EAE score for the fHTK mice and controls up to 30 days post EAE induction, ± s.e.m., **p<0.01, *p <0.05 by two-tailed Mann-Whitney test, n=22 control, 19 fHTK. e. Pie charts depicting the percentages of control and fHTK mice that were paralyzed at day 24 and day 30 post EAE induction. f. Spinal cords from control and fHTK mice were stained for OLIG2 (green), CD11b (red) and DAPI (blue) with the area of the CD11b+ lesion traced with a dotted white line. g. The number of OLIG2+ cells per CD11b+ lesion was quantified comparing the fHTK and control mice, ± s.e.m., n = 21 control, 19 fHTK, *p = 0.038 by Student’s one-tailed t-test. h. Spinal cord sections from fHTK mice and controls at 30 days post EAE induction were stained for FluoroMyelin (red) and DAPI (blue) with the area of the CD11b+ lesion traced with a dotted white line. i. Quantification of the percentage of the total white matter area that is FluoroMyelin positive, ± s.e.m., n = 21 control, 19 fHTK, p = 0.96 by Student’s two-tailed t-test. j. Electron microscopy images of spinal cord sections from healthy wild type mice, and fHTK mice and control mice both at 30 days post EAE induction. k. Quantification of the # of myelinated axons from the 3 groups per 3000x picture frame, n = 4 per group, ***p = 0.0002, **p = 0.0012, using a one-way ANOVA with multiple comparisons. Scale bars for immunofluorescence images = 100 µm, scale bars for EM images = 2 µm.
While there was no difference in the onset of EAE motor symptoms, fHTK mice had EAE scores around 0.5 points lower than littermate controls at the chronic stages of disease (Fig 4d). While numerically small, a scoring difference of 0.5 at this point in the EAE curve represents the difference between mice with the use of both back paws and mice that have at least one hind limb paralyzed, thus a reduction in the fibrotic scar led to a significant reduction in the deterioration of motor ability (Fig 4e). Additionally, fHTK mice had more OLIG2+ cells per lesion area than controls although there were no differences in total myelin areas between groups (Fig 4f–i). To further characterize the identity of the OLIG2+ cells, we co-labeled these cells for the mature oligodendrocyte marker CC1. We found no difference between the percentages of OLIG2+ CC1+ and OLIG2+CC1− cells between groups, suggesting that the increase in OLIG2+ cells in lesions of fHTK mice were not specific to a particular stage in the oligodendrocyte lineage (Extended Data Fig 5d).
To understand how fibrosis, and specifically depositions of Col1, would decrease OLIG2+ cell entry into lesions, we performed in vitro experiments with primary, cultured OPCs to determine how Col1 affects OPC proliferation, differentiation, and migration. For these experiments we examined primary OPCs seeded in cell culture wells treated with poly-L-lysine (PLL) alone, PLL + Col1, PLL + fibronectin, and PLL + laminin in order to compare the effects of Col1 to those of extracellular matrix proteins of the basement membrane. We found that Col1 did not have an effect on cell proliferation or differentiation but significantly decreased OPC migration across a transwell insert (Extended Data Fig 6). In contrast, the basement membrane matrix proteins fibronectin and laminin both significantly increased migration, which corresponds to the fact that OPCs migrate along CNS vessels during development39.
These results suggest that fibrosis impairs the ability of reparative myelin-forming oligodendrocyte linage cells to migrate into the lesions. However, ablation of proliferative fibrotic cells alone does not promote axonal remyelination or full symptomatic recovery in EAE. We found a profound loss of neurofilament positive axons within lesions in both control in fHTK mice, suggesting that although more oligodendrocyte lineage cells were able to enter the lesion, there was a lack of healthy axons to myelinate (Extended Data Fig 5e). This was further demonstrated by electron microscopy images where both control and fHTK mice had significantly less myelinated axons than healthy, wild type mice (Fig 4j,k). Therefore, combining anti-fibrotic approaches with therapeutics to maintain axon integrity might prove synergistic in the treatment of diseases such as MS.
Interferon gamma signaling regulates the amplitude of fibrotic scarring
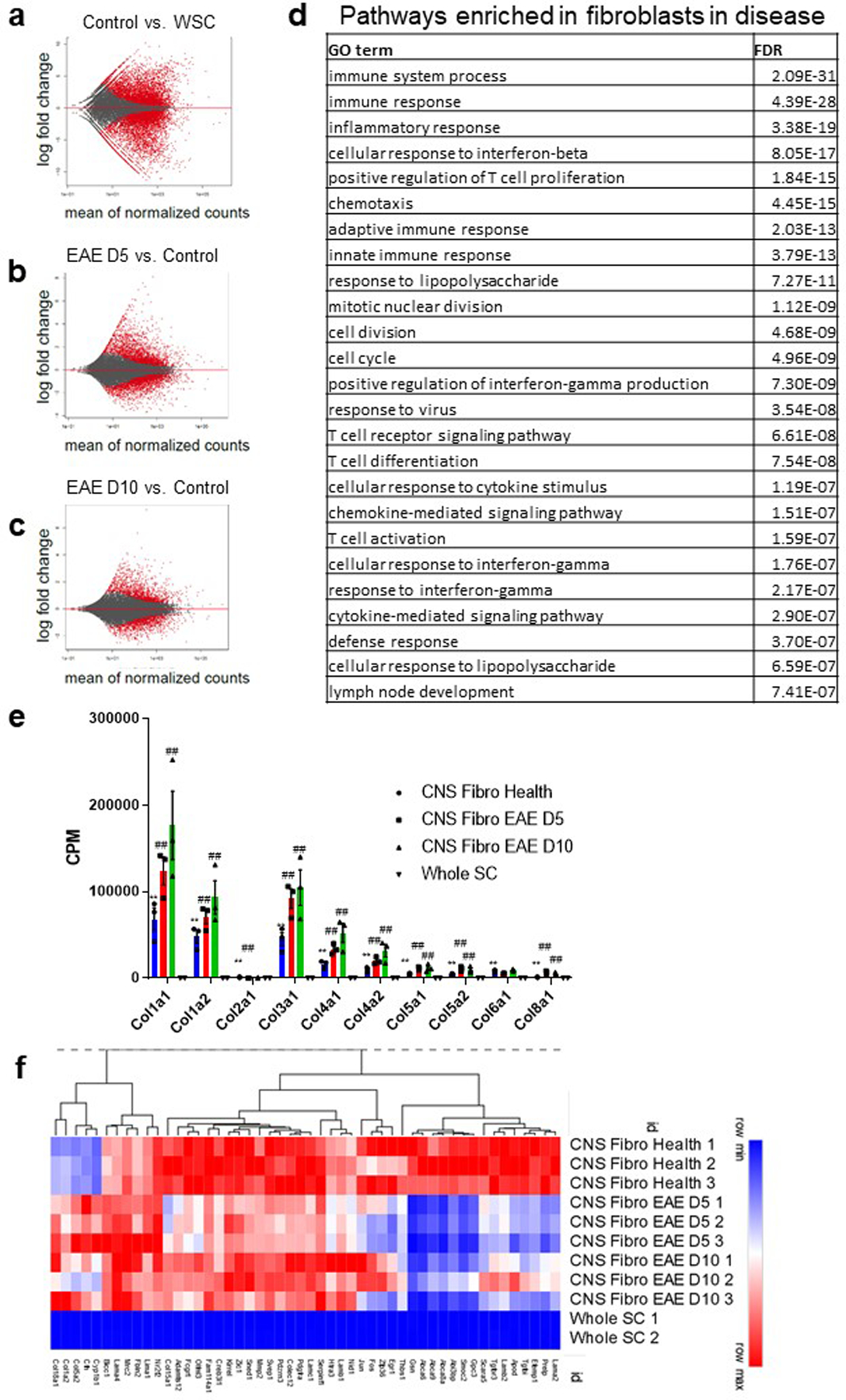
To gain deeper insight into the molecular mechanisms of neuroinflammatory fibrosis, we performed bulk RNA sequencing on Col1a1-GFP+ cells sorted from spinal cords of healthy mice, mice with EAE 5 and 10 days PSO, and whole spinal cord homogenate from healthy mice. Col1a1, Col1a2 and Col3a1 were some of the most highly enriched genes in both the healthy and EAE GFP+ cells compared to the whole spinal cord and their expression continued to increase throughout the course of EAE (Extended Data Fig 7). When comparing Col1a1-GFP+ cells in health and EAE, there were more significant differentially expressed genes 5 days PSO (2516 upregulated, 2278 downregulated) than 10 days PSO (1414 upregulated, 1122 downregulated). Many of the genes highly upregulated at 5 days PSO are involved in inflammatory signaling, and their expression largely peaked at 5 days PSO and moderately decreased by 10 days PSO (Fig 5a, Extended Data Fig 7). A smaller subset of genes was increased at 5 days PSO and continued to increase at 10 days PSO, and included many genes encoding collagen subunits (Extended Data Fig 7). These data suggest that Col1+ cells turn on inflammatory signaling pathways early in disease progression when there is the most cell proliferation and continue depositing extracellular matrix once their expansion is complete.
Figure 5: Fibrotic cells upregulate interferon gamma pathway genes in EAE.
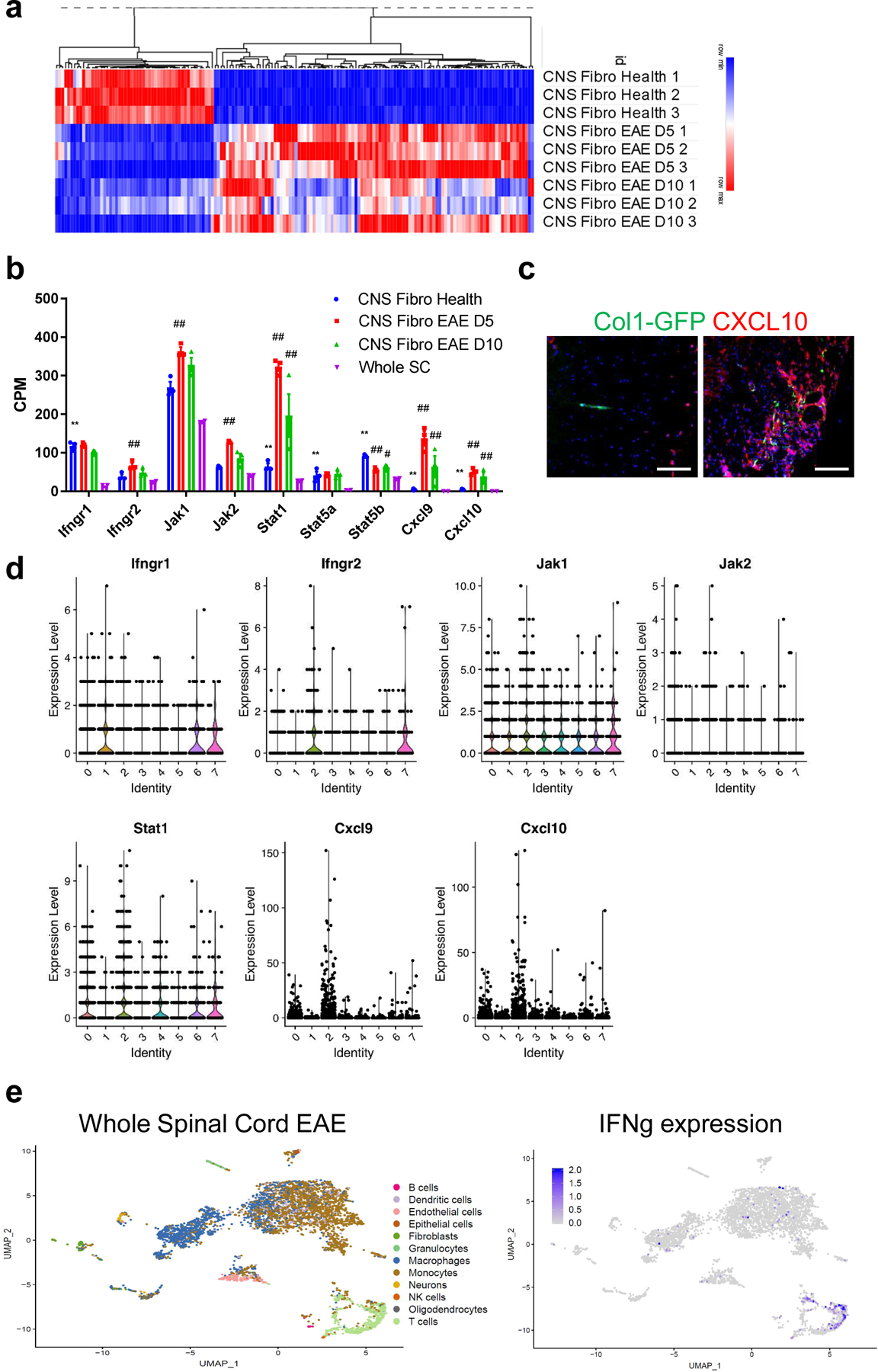
a. Heat map of the expression levels of the top 100 differentially expressed genes by FDR in Col1a1-GFP+ cells from EAE 5 days PSO and 10 days PSO (CNS Fibro EAE D5, D10) compared to Col1a1-GFP+ cells from health (CNS Fibro Health). b. mRNA levels in counts per million (CPM) of interferon gamma pathway and target genes from the sequencing of whole spinal cord tissue (Whole SC, n = 2) and CNS Fibro Health, CNS Fibro EAE D5 and CNS Fibro EAE D10 (n = 3 each), ± s.e.m., *FDR < 0.05, **FDR < 0.01 to Whole SC, #FDR < 0.05, ##FDR < 0.01 to CNS Fibro Health. c. Spinal cord sections from Col1a1-GFP mice in health (left) and EAE (right) were stained with CXCL10 (red). d. Violin plots from the single-cell sequencing dataset of Col1a1-GFP+ cells described in Figure 3 depicting the total RNA counts for genes in the interferon gamma pathway. e. UMAP plot of individual cells sequenced from a whole spinal cord of a wild type mouse with EAE 4 days PSO with their assigned cell type identity using SingleR and the Immgen reference dataset. e. Interferon gamma expression is indicated in blue, and is found predominately in T cells in EAE. All scale bars = 100 µm.
Pathway analysis identified IFNγ signaling is enriched in EAE fibrotic cells compared to the whole spinal cord (Extended Data Fig 7d). We found that fibrotic cells express IFNγ receptors (IFNGR1, IFNGR2) and downstream signaling molecules (JAK1, JAK2, STAT1, STAT5a, STAT5b) in both health and EAE, and upregulate IFNγ target genes CXCL9 and CXCL10 following EAE (Fig 5b,c)40–42. In the single-cell sequencing dataset, IFNγ pathway genes were expressed throughout each of the different clusters of both Class 1 and Class 2 fibroblasts (Fig 5d). IFNγ target genes CXCL9 and CXCL10 were expressed mainly in Class 1 cluster 2, which is the cluster with the highest proportion of actively dividing cells, suggesting that this IFNγ signaling may regulate fibroblast proliferation. Taken together these data reveal that CNS fibroblasts express the machinery to respond to IFNγ signaling, but this signaling cascade is only induced following EAE. To determine which cells in the EAE lesion are secreting IFNγ, we performed single-cell sequencing on whole spinal cords from EAE mice. IFNγ was highly expressed in a cluster of cells that were identified as T cells (Fig 5e). These data suggest that during EAE, T cells secrete IFNγ signals to CNS fibroblasts.
The role of IFNγ signaling in fibrosis in peripheral tissues has been debated43–47. To determine whether IFNγ signaling in fibrotic cells is necessary for CNS fibrotic scar formation, we selectively deleted Ifngr1 from fibrotic cells prior to EAE induction. Mice with fibrotic cell-specific Ifngr1 deletion (Ifngr1f/f;Col1a2CreERT [fIFNγ]) and littermate controls (Ifngr1f/f) were injected with tamoxifen at 6 weeks of age, induced with EAE at 12 weeks of age, collected 30 days post EAE induction, and analyzed for the extent of fibrotic scar formation. fIFNγ mice had a significant reduction in fibrotic scar formation (Fig 6a,b), although not to the extent of the reduction in the cell ablation paradigm. There were no differences in motor disability, lesion size, or myelination between groups (Fig 6c–f). To determine if IFNγ is sufficient for scar formation, we analyzed tissue from mice administered cuprizone for 5 weeks with astrocyte-specific overexpression of IFNγ48. We found no fibrotic scar in the areas of demyelination, suggesting that an induction of IFNγ is not sufficient for scar formation (Extended Data Fig 2b,c). Together, these experiments confirm that IFNγ signaling in CNS fibrotic cells regulates the amplitude of fibrotic scarring, but on its own is not sufficient to initiate fibrotic scarring.
Figure 6: Interferon gamma signaling regulates scar formation following neuroinflammation.
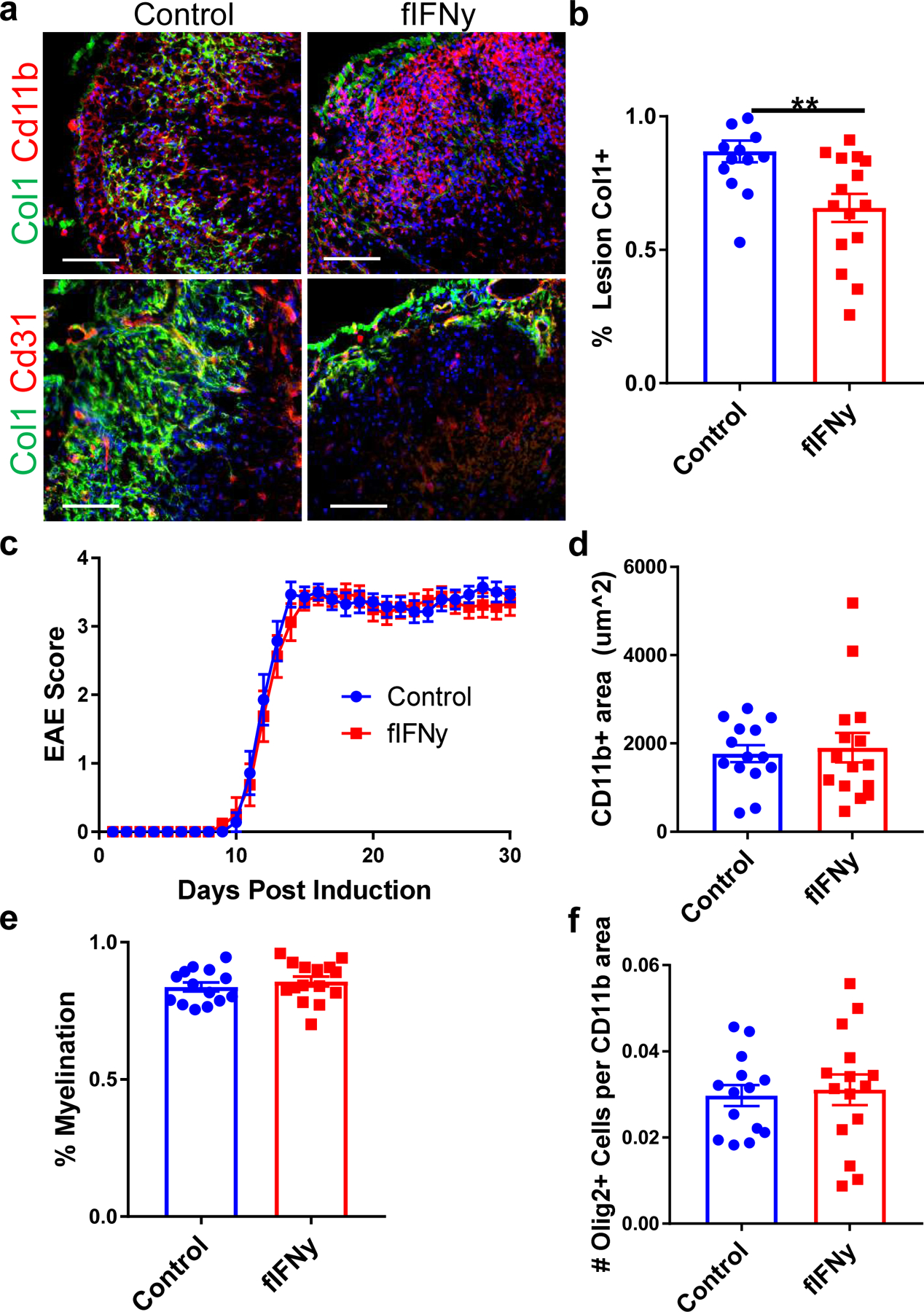
a. Spinal cord sections from fibrotic cell-specific IFNγ knockout mice (fIFNγ, IFNγfl/fl; Col1a2CreERT) and littermate controls (IFNγfl/fl) stained for Col1 (green), CD11b (red) or CD31 (red), and DAPI (blue). The amount of fibrotic scar covering the lesion was quantified in (b), ± s.e.m., **p = 0.0039 by Student’s two-tailed t-test, n=14 control, 15 fIFNγ. c. EAE score for the fIFNγ mice and controls up to 30 days post EAE induction (± s.e.m., n= 14 control, 15 fIFNγ). d. Quantification of the CD11b+ immune cell area in the control and fIFNγ groups 30 days post EAE immunization, ± s.e.m., p = 0.61 by Student’s two-tailed t-test, n= 14 control, 15 fIFNγ. e. Quantification of the percentage of the total white matter area that is FluoroMyelin positive for the control and fIFNγ groups, ± s.e.m., p = 0.44 by Student’s two-tailed t-test, n= 14 control, 15 fIFNγ. f. Quantification of OLIG2+ cells per CD11b+ lesion area between the control and fIFNγ groups, ± s.e.m., p = 0.76 by Student’s two-tailed t-test, n= 14 control, 15 fIFNγ.
Discussion
In this study we demonstrate that a dense, Col1+ fibrotic scar forms in the spinal cord following immune infiltration in mouse models of demyelination and plays a role in regulating disease severity. Combined with previous characterizations of fibrotic scarring following SCI our data suggest that there is a robust fibrotic response to both injury and inflammation in the CNS, and that activation and proliferation of CNS fibroblasts may be a common response across many different neurological disorders. This has wide implications for our understanding of the pathophysiology and repair of a wide array of neurological diseases, and should stimulate future research into the fibrotic response in various human neurological and neurodegenerative diseases.
Using lineage tracing and single-cell sequencing, we illustrate that this fibrotic scar derives overwhelmingly from the proliferation and migration of CNS fibroblasts found in the meninges and surrounding large blood vessels in health. This is in contrast to previous studies reporting that a subtype of pericytes forms the fibrotic scar following SCI16,17,21. These studies used a GlastCreER reporter to label scar-forming cells, and denoted these cells as Type A pericytes due to their expression of PDGFRβ and perivascular localization. As these qualities are also true of fibroblasts, it is possible that some Type A pericytes are fibroblasts. Indeed, Glast (Slc1a3) is strongly expressed in fibroblasts in addition to astrocytes and pericytes30. Our lineage tracing experiments suggest that the scar originates from cells that express Col1 in health, but not NG2+ or aSMA+ cells, and our single-cell sequencing data classify the vast majority of these cells as fibroblasts. We did find that a small portion (2%) of collagen-expressing cells in health and EAE expressed typical markers of mural cells (vSMCs/pericytes). While these cells had lower collagen transcript expression than fibroblasts, we cannot formally rule out their contribution to fibrosis. The proportion of this scar that comes from the CNS fibroblasts in the meninges vs. those surrounding large blood vessels remains unknown, but we hypothesize that fibroblasts from both regions may play a role in scar formation. Techniques such as in vivo, two-photon imaging could be used to further delineate the roles of CNS fibroblasts from different regions.
Interestingly, when we ablated proliferating fibrotic cells, although the fibrotic scar was greatly diminished, there was still Col1 deposition largely restricted to the area around blood vessels. This remaining Col1 deposition may derive from the incomplete recombination of the Col1a2CreERT used in the ablation paradigm, scar tissue deposition by CNS fibroblasts that aren’t actively proliferating, and/or scar deposition by vascular cells such as endothelial cells, pericytes or vSMCs. A recent study found that in EAE, endothelial cells take up myelin debris which leads to an endothelial-mesenchymal transition and an upregulation in the expression of extracellular matrix proteins49. While we did not find robust Col1a1-GFP reporter expression in CNS endothelial cells (Extended Data Fig 4a), it is possible that CNS endothelial cells express Col1 protein without turning on the Col1a1-GFP promoter, thus leading to perivascular Col1 accumulation. Interestingly, we found a small proportion of mural cells secrete Col1, suggesting that these cells may regulate the perivascular Col1 deposition.
Fibrotic scarring could potentially influence the course of neuroinflammatory disease by restricting immune cell trafficking into inflammatory lesions, or by preventing tissue repair and regeneration. We found that reducing fibrotic scar formation by preventing fibrotic cell proliferation decreases motor disability and increases the number of oligodendrocyte lineage cells in the lesion in the chronic stages of disease without affecting immune cell entry or myelination. Additionally, collagen proteins inhibit OPC migration in vitro. This suggests that fibrotic scarring limits the ability of cells with repair potential (oligodendrocyte lineage cells) from entering the demyelinating lesion, however, these cells are still not capable of remyelination. This lack of remyelination may stem from either the presence of oligodendrocyte precursor differentiation inhibition cues, or from axon degeneration that occurs in the EAE model. Indeed, we found robust axon degeneration in this EAE model (Fig 4j,k, Extended Data Fig 5e). Therefore, combining therapeutics that inhibit scar formation with those that preserve axon integrity may synergize to enhance repair following neuroinflammation. As there are no changes in myelination after reducing fibrotic scarring, it is not completely clear how reducing the fibrotic scar decreases the EAE score in the chronic stages of disease.
This study also implicates interferon gamma signaling in CNS fibrotic scar formation, unveiling a new molecular mechanism that could be of interest for CNS disorders with scar tissue deposition. As Ifngr1 deletion from fibrotic cells did not completely ablate the fibrotic scar in neuroinflammatory lesions, other pathways are also likely involved in fibrotic scar formation. Additionally, interferon gamma is mainly secreted by adaptive immune cells, and thus this signal may be utilized to amplify fibrotic scar formation when adaptive immunity is involved in neuroinflammatory lesion formation. Pathways involved in fibrosis in peripheral tissues such as the TGFβ pathway and Wnt pathway could also be playing a role in CNS fibrosis following a variety of triggers6,50. Our RNA sequencing dataset of Col1+ cells in health and EAE will be widely useful to understand other drivers of CNS scar formation, and could influence treatment options for SCI, stroke and other neurological injuries and inflammatory diseases with fibrotic scarring.
Methods
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Col1a1-GFP mice from David Brenner (C57BL/6 background) were used to label Col1 producing cells for imaging and FACS sorting analysis. Rosa-lsl-tdTomato mice, Charles River Laboratories 007909 (C57BL/6 background), were crossed to Col1a2CreERT mice, Jackson Labs 029567 (C57BL/6 background), Ng2CreER™ mice, Jackson Labs 008538 (C57BL/6 background), or aSMACreERT2 mice (previously described)51 for lineage tracing analysis. B6;129S7-Hprt1tm2(Pgk1-Pac/Tk)Brd/Mmucd, MMRRC 010860-UCD (C57BL/6Try c-Brd mixed background), (lox-stop-lox-HTK) mice were crossed to Col1a2CreERT for the fibrosis ablation experiments. Ifngr1fl/fl mice, Jackson Labs 025394 (C57BL/6 background), were crossed to Col1a2CreERT mice for the IFN mechanistic studies. UBC-GFP (004353) reporter mice and CD45.1 (002014) mice used in bone marrow transplant (BMT) experiments were purchased from Jackson Labs. GFAP/tTA mice on the C57BL/6 background were mated with TRE/IFN-γ mice on the C57BL/6 background to produce GFAP/tTA;TRE/IFN-γ double transgenic mice and were maintained in the Popko lab for cuprizone studies. Animal protocols were approved by IACUC at UCSD, UCSF and Northwestern and we have followed all ethical regulations in the use of mice for this study. Male mice were used for fHTK experiments as the gene is X linked and for the Col1a1-GFP regional quantifications in health. Female mice were used for bone marrow transplant studies and the Col1a1-GFP cell number in EAE quantifications. Male and female mice were used for all other experiments. Mice were used between 2 and 6 months of age unless otherwise noted, and were housed in a temperature controlled environment (68 to 72 degrees Fahrenheit for UCSD and 40–70% humidity) with a 12 hour light/dark cycle.
METHOD DETAILS
EAE:
EAE was induced through 2 subcutaneous injections of myelin oligodendrocyte glycoprotein (MOG) in Freud’s adjuvant and an IP injection of pertussis toxin (PTX) (Hooke Laboratories EK-2110). 24 hours after the initial injections a second IP injection of PTX was administered. 120–160 ng PTX was used for each dose based on manufacturer’s instructions for the PTX lot. Mice were induced at 10–12 weeks of age unless notified otherwise. The following EAE score system was used to assess motor outcomes:
0.5 – tip of tail limp
1 – whole tail limp
2 – mouse does not instantly turn over when flipped
2.5 – wobbly gait
3 – mouse dragging backside
3.5 – paralysis of one hind limb
4 – paralysis of both hind limbs
5 – moribund
Mice were scored every day following the induction of EAE and until tissue was collected.
FTY720 injections:
Col1a1-GFP mice were injected with either saline or 2 mg/kg FTY720 I.P. starting on the day of EAE induction and continuing daily until tissue was collected on day 8–10 post symptom onset for the saline mice.
Cuprizone model of demyelination:
GFAP/tTA;TRE/IFN-γ double transgenic mice were maintained on 0.05 mg/ml doxycycline (Sigma, #D9891) in drinking water from conception. At six weeks of age doxycycline was discontinued in half the mice to induce CNS expression of IFN-γ, and all mice were administered a 0.2% cuprizone diet (Envigo, #TD.160049). Cuprizone feeding lasted for 5 weeks and then mice were placed back to a normal diet for up to 3 weeks to allow remyelination to occur. Mice were then perfused and brains were removed, post-fixed with 4% PFA and cryopreserved in 30% sucrose. The tissue were then embedded in OCT for sectioning.
LPC model of demyelination:
Demyelinated lesions were produced in the ventrolateral or dorsal spinal cord white matter of 8- to 10-week-old female C57bl6 WT mice as previously described52. Anesthesia was induced and maintained with inhalational isoflurane and oxygen supplemented with 0.05 ml of buprenorphine (Vetergesic; 0.05 mg/ml) given subcutaneously. Having exposed the spinal vertebrae at the level of T12/T13, tissue was cleared overlying the intervertebral space, and the dura was pierced with a dental needle just lateral to midline. A Hamilton needle was advanced through the pierced dura at an angle of 45°, and 0.5 μl 1% lysolecithin (L-α- lysophosphatidylcholine; Sigma L4129) was injected into the ventrolateral white matter. Mice were perfused transcardially at 5 days post lesioning (5dpl), 7dpl, or 14dpl with 4% paraformaldehyde, postfixed for 4 hours, and cryoprotected overnight in 30% sucrose. Spinal cords were frozen in OCT for storage and cryostat sections cut at 15 micrometer thickness.
Tissue slice collection and staining:
Mice were anesthetized through an intraperitoneal injection of a ketamine/xylene cocktail and then perfused transcardially with D-PBS followed by 4% paraformaldehyde (Electron Microscopy Sciences 15714-S) using a Dynamax peristaltic pump. Spinal cords were dissected and placed in 30% sucrose until frozen in 1:2 30% sucrose:OCT and sectioned into 10 uM slices. Sections were blocked with 5% Normal Goat Serum and permeabilized in 0.2% Triton X-100 in D-PBS followed by an overnight incubation at 4ºC with the following primary antibodies in antibody buffer (NaCl 150 mM, Tris Base 50 mM, 1% BSA, L-Lysine 100 mM, 0.02% Sodium azide in water). Primary antibodies used were: Col1 abcam ab21286 1:750, CD11b Bio-Rad MCA711 1:1000, CD45 Bio-Rad MCA1031 1:1000, GFAP abcam ab7260 1:500, CD31 BD Biosciences 553370 1:1000, PDGFRb eBioscience 14–1402-82 clone APB5 1:500, PDGFRa BD Biosciences 558774 clone APA5 1:500, SOX9 abcam ab185966 1:500, IBA1 Wako 019–19741 1:500, OLIG2 EMD Millipore AB9610 1:500, NG2 EMD Millipore MAB5384 1:500, CD4 eBioscience 16–0041081 1:1000, Col3 abcam ab7778 1:500, Periostin R&D Systems MAB3548 clone 345613 1:1000, ER-TR7 Novus NB100–64932 1:1000, CD8 clone 53–6.7 eBioscience 14–0081-82 1:1000, Actin, α-smooth muscle Sigma-Aldrich A2547 1:500., Neurofilament heavy polypeptide abcam ab8135 1:1000, Desmin abcam ab8592 1:1000, CC1 Calbiochem OP80 1:500, CXCL10 abcam ab9938 1:1000, MBP Abcam ab40390 1:1000. Slides were then washed with D-PBS and incubated at room temperature for 1.5 hours with the following secondary antibodies, all 1:1000 in D-PBS: Goat-anti-Rabbit-Alexa 488 (ThermoFisher A11034), Goat-anti-Rat-Alexa 488 (ThermoFisher A11006), Goat-anti-Rabbit-Alexa 594 (ThermoFisher R37117), Goat-anti-Rat-Alexa 594 (ThermoFisher A11007), Goat-anti-Mouse-Alexa 647 (ThermoFisher A-21235). Following secondary incubation slides were washed and DAPI Fluoromount-G (SouthernBiotech, 0100–20) was added. Images were taken with an Axio Imager D2 (Carl Zeiss) and digital camera (Axiocam HRm, Carl Zeiss) using the AxioVision software (AxioVis4O V 4.8.2.0) and contrasted using Adobe Photoshop. For all fluorescence experiments, tissue from multiple mice was stained and representative images were chosen for publication. For light sheet microscopy images, mice were perfused with 100 µL of a tomato lectin (Vector Laboratories DL-1177) in dPBS prior to paraformaldehyde and spinal cord tissue was dissected and incubated overnight with 4% PFA followed by a PBS wash. Cords were then cleared in a solution of 8% SDS, 10% N-butyldiethanolamine, 3% 1-Thioglycerol in PBS for 72 hours and then imaged on a Ziess Z.1 light sheet microscope at the UCSD School of Medicine Microscopy Core. For confocal images, Col1a1-GFP+ mouse spinal cords stained with CD31 were imaged on a Leica SP8 Confocal microscope at the UCSD School of Medicine Microscopy Core.
Lineage Tracing:
All mice received intraperitoneal (IP) injections of 2 mg tamoxifen in sterile corn oil for 3 consecutive days at 6 weeks of age to induce tdTomato expression. EAE was induced at 12 weeks of age as described above. Mice were scored based on the EAE scoring system described above and tissue was collected 10 days post symptom onset and stained for collagen I. The total collagen area per section was traced using Image J 1.52a and the number of tdTomato+ reporter cells within this area was quantified and compared between groups. This number was normalized to the average of the number of reporter cells per white matter area for each of the reporters in age-matched healthy mice. For Col1a2CreERT analysis n=7 health and n=9 EAE. For the NG2CreER™ analysis n=10 health and n=13 EAE. For aSmaCreERT2 analysis n=4 health and n=7 EAE. Males and females were used in all groups.
Bone Marrow Transplantation:
Female 12-week-old Col1a1-GFP or UBC-GFP mice were used as bone marrow donors. UBC-GFP mice express enhanced GFP under the direction of the human ubiqutin C promoter. GFP is expressed in all tissues examined, is uniform within cell lineages and remains constant throughout development and in injury paradigms (JAX: 004353). Female CD45.1 mice were used as recipients for cell transplantation. This strain carries the CD45.1 pan-leukocyte marker to distinguish donor-derived cells from recipient CD45.2 cells. Recipient mice were irradiated with 900 rads, split dose, 3 hours apart using a cesium source. Purified donor cells (4 × 106) from bone marrow were injected intravenously with 200,000 spleen helper cells, and hematopoietic reconstitution was monitored in the peripheral blood based on either GFP or CD45.1 expression. Recipients with ≥99% donor chimerism were considered reconstituted. Note that no GFP+ cells were observed in the blood from Col1GFP recipients. Transplanted mice were kept on antibiotic-containing food for 2 weeks. All mice were maintained at UCSF in accordance with IACUC approved protocols.
Tissue Dissociation and FACS Sorting:
To obtain single cell suspensions of CNS fibroblasts, spinal cords from Col1a1-GFP mice were dissected, chopped with a #10 blade, and enzymatically dissociated with papain (Worthington Biochemical, LK003176, 1 vial per sample) containing DNase (125 U/ml, Worthington LS002007) for 1.5 hours at 35°C. This was followed by mechanical trituration in a solution containing ovomucoid (2 mg/ml, Roche 109878) and DNase (125 U/ml) and a second enzymatic digestion with 1.0 mg/ml Collagenase Type 2 (Worthington Biochemical, LS004176) and 0.4 mg/ml Neutral Protease (Worthington Biochemical, LS02104) at 35°C for 30 minutes. Next, myelin was removed with myelin removal beads (MACS Miltenyi Biotec 130–096-433) and LS columns (MACS Miltenyi Biotec 130–042-401) on a MidiMACS separator (MACS Miltenyi Biotec 130–042-302). Samples were then blocked with Rat IgG 1:100 (Sigma Aldrich I8015) for 25 minutes on ice. Cell suspensions were re-suspended in buffer containing DAPI 1:50 and Rat-anti-CD31-Alexa 647 (Molecular Probes A14716) 1:100 and incubated in the fridge for 30 minutes. Suspensions were washed twice with buffer and live, CNS fibroblasts were FACS sorted into Trizol (Invitrogen 15596026) based on GFP fluorescence using an ARIA II sorter at the Flow Cytometry Core at the VA Hospital in La Jolla, CA. Forward scatter and side scatter analysis were also used as gates to limit the sorting to single, live cells using FACSDiva v8 software.
Single-cell sequencing:
Tissue was dissociated and FACS sorted as described above into PBS + 0.05% BSA. 3 samples were collected of the Col1a1-GFP+ cells in health group, each containing 2–3 spinal cords of Col1a1-GFP females. For the Col1a1-GFP+ cells in EAE group 2 samples each containing 2 spinal cords from Col1a1-GFP mice 5 and 7 days after EAE symptom onset were combined. For the whole spinal cord in EAE 1 spinal cord from a wild type mouse with EAE 4 days after symptom onset was used. Following FACS sorting the single cell suspension was brought to the UCSD IGM core, run through the 10X Genomics pipeline (v2) and sequenced on an Illumnia HiSeq4000 or NovaSeq 6000. The sequencing files were run through the 10X CellRanger 2.1.1 pipeline to generate gene counts data, and then analyzed using Seurat v3. We used the sctransform pipeline to analyze individual datasets, and the integration pipeline to integrate the healthy and EAE samples53,54. In all analyses a 200 non-zero genes cut-off was used. We next filtered out Col1a1 negative cell types by identifying clusters where the majority of cells do not express Col1a1 or Col1a2. This strategy removed 463 total cells. Following stringent criteria, 6,509 cells (93.4% of total cells) were analyzed using unsupervised clustering. Following the filtering, the counts were reanalyzed. Resolution parameter of clustering was set to 0.5. Wilcox method was used for differential expression analysis, with min.pct = 0.25 and logfc.threshold = 0.25. SingleR v1.4 was used to annotate cells using the Immgen reference dataset55.
CNS fibrotic scar prevention and interferon gamma receptor deletion:
All mice received intraperitoneal (IP) injections of 2 mg tamoxifen in sterile corn oil for 3 consecutive days at 6 weeks of age to induce cre expression. EAE was induced at 12 weeks of age as described above. Administration of GCV (Sigma Y0001129) began on day 8 post EAE induction through a subcutaneous injection at a concentration of 25 mg/kg in dPBS and occurred each following day until tissue was collected. For the HTK experiment males were used as the HTK gene is X-linked, n = 22 control and 19 fHTK. One of the control mice died during the course of EAE so it was not included in the histology quantifications. For the IFNγ deletion experiments, tamoxifen and EAE were administered in males and females. Mice were collected 30 days after EAE induction. n= 14 control, 15 fIFNγ.
Electron Microscopy:
Control and fHTK mice at day 30 post EAE induction and healthy, C57/bl6 mice were anesthetized through an intraperitoneal injection of a ketamine/xylene cocktail and then perfused transcardially with D-PBS followed by 2% paraformaldehyde and 2.5% glutaraldehyde in a 0.15M sodium cacodylate buffer. 1mm^3 spinal cord sections were dissected and placed in the fixing solution until processed at the CMM UCSD Electron Microscopy Core. Samples were then placed in 1% osmium in 0.15M sodium cacodylate for 1–2 hours on ice, washed 5×10 min in 0.15M sodium cacodylate buffer and rinsed in ddH2O on ice. Samples were incubated in 2% uranyl acetate for 1 to 2 hrs at 4C, dehydrated at increasing concentrations of ethanol on ice and then dry acetone for 15 min at room temperature, placed in 50:50 ethanol: Durcupan for 1hr at room temperature and then incubated in 100% Durcupan overnight. Tissue was embedded in Durcupan in 60C oven for 36 to 48hrs. Ultrathin sections (60nm) were cut on a Leica microtome with a Diamond knife followed by post staining with both uranyl acetate and lead. Images were captured on FEI Spirit Tecnai TEM at 80KV with Eagle 4kx4k camera. Images at 3000x were used for analysis in which all myelinated axons per frame were counted for each of the different conditions.
Oligodendrocyte precursor cell (OPC) cultures:
Primary rat OPCs were isolated from cortical hemispheres of postnatal day 7 rat brains as previously described56. Briefly, rat cortices were minced and dissociated in papain (Worthington) for 75 min at 37°C with periodic shaking. Following trituration, the suspension was immersed in 0.2% BSA at room temperature and subjected to sequential immunopanning consisting of two 30 min incubations in negative selection plates (Ran-2 and Gal-C) and one 45 min incubation for positive selection (O4). Selection plates were prepared by incubating dishes overnight at room temperature with goat IgG and IgM secondary antibodies (Jackson ImmunoResearch) in 50 mM Tris-HCl. Antibodies Ran-2, Gal-C, or O4 were added after washing with DPBS (Invitrogen). OPCs were dissociated from the positive selection dish with 0.05% Trypsin-EDTA (Invitrogen) and purified OPCs subsequently seeded onto 12mm coverslips coated with poly-L-lysine (PLL, Sigma-Aldrich), or PLL with 10 ug/mL of: laminin (EMD Millipore CC095), fibronectin (Sigma-Aldrich F0635), or collagen I (Aviva Systems Biology OPED00033). OPCs were seeded at a density of 15,000 cells per coverslip in DMEM (Invitrogen) supplemented with B27 (Invitrogen), N2 (Invitrogen), N-acetylcysteine (Sigma-Aldrich), forskolin (Sigma-Aldrich), penicillin-streptomycin (Invitrogen), and PDGF-AA (Peprotech) and cultured overnight at 37°C, 5% CO2. The following day, OPCs were either incubated in 10 uM EdU with PDGF-AA for 2 hours, or cultured for 3 days following the removal of PDGF-AA from media with or without the addition of 50 nM T3 (Sigma-Aldrich). Immunohistochemistry: Cultures were fixed in 4% (w/v) paraformaldehyde (PFA) in DPBS for 15 minutes and dehydrated. Cultures were blocked and permeabilized in 10% goat serum in DPBS containing 0.1% (v/v) Triton X-100 for 1 h at room temperature. Primary antibodies were diluted in 10% goat serum and incubated overnight at 4°C. Secondary antibodies were diluted in 10% goat serum with DAPI and incubated for 1 h at room temperature. The following primary antibodies were used: rat monoclonal anti-PDGFRa (BD Biosciences, 558774, 1:200); rabbit polyclonal anti-Olig2 (EMD Millipore AB9610, 1:1000); rat monoclonal anti-MBP (Bio-Rad/Serotec, MCA409S, 1:500). Alexa Fluor 488 and Alexa Fluor 594 IgG secondary antibodies (rat, rabbit, 1:1000) were used to detect fluorescence. The incorporation of EdU by proliferating cells was detected via the Click-iT EdU Cell Proliferation Kit (Invitrogen C10340) after incubation in primary and secondary antibodies. Images were obtained on a Zeiss Axio Imager Z1 microscope. Cells were quantified from randomly selected fields of view per coverslip under 10x magnification. For migration experiments, transwell membranes (Corning, 8.0 μm pore polycarbonate membrane insert) were coated on both sides with PLL and 10ug/ml laminin, fibronectin, or collagen I was added to the top of the membrane. OPCs were seeded onto the membranes in DMEM + B27, N2, N-acetylcysteine, forskolin, and pen/strep. DMEM + B27, N2, N-acetylcysteine, forskolin, and pen/strep + 10ng/ml PDGF-AA was added to the bottom of the well. Cells were scraped off the top of the membrane 24 hours after initial seeding and fixed with 4% PFA in DPBS for 15 minutes. Cells were then blocked and permeabilized in 10% goat serum with 0.1% Triton, incubated in rabbit monoclonal anti-PDGFRa antibody (1:500, gift from WB Stallcup) overnight at 4°C and then incubated in secondary antibody & DAPI for 1 hour at room temperature (goat anti-rabbit Alexa Fluor 488 1:1000). The transwell membranes were then cut out and mounted for imaging, and the number of cells on the bottom of the transwell were counted. For all in vitro experiments, 3 cortical cultures, each containing cells from 3 pooled postnatal day 7 rats, were split into 3 replicates. The 9 total replicates were analyzed for each experiment. Statistics were calculated using Prism with a one-way ANOVA with Sidak’s.
Bulk RNA sequencing:
For bulk RNA sequencing of CNS fibroblasts we collected and analyzed 3 samples each for the control, EAE 5 days PSO and EAE 10 days PSO groups. 2–3 spinal cords from Col1a1-GFP females of 3–4 months of age were combined for each sample. 2 samples were collected for the whole spinal cord homogenate, each consisting of 1 spinal cord of a Col1a1-GFP female at 3 months of age. RNA was purified from the FACS-sorted fibroblasts using the Qiagen RNA Isolation Microkit. The RNA was then tested for quality and concentration at the UCSD IGM Core using an Agilent Bioanalyzer. Stranded, cDNA libraries were made using the SMARTer Stranded Total RNA-Seq Kit - Pico Input Mammalian (Clontech) and then the samples were sequenced on an Illumnia HiSeq4000, 100 cycles, paired ends. Sequence reads were aligned to Ensembl mm9 v67 mouse whole genome using Tophat v 2.0.11 and Bowtie 2 v 2.2.1 using parameters –m 2 –a 5 –p 7. The resulting files were then sorted using SAMtools v.0.1.19 and count tables generated using HTSeq-0.6.1. Differential expression analysis and statistical analysis including p values and FDR was performed using DESeq2. Heat maps were prepared using the Morpheus software (https://software.broadinstitute.org/morpheus) and clustered using hierarchical clustering with a metric of one minus the pearson correlation.
QUANTIFICATION AND STATISTICAL ANALYSIS
EAE time course quantifications:
To calculate the number of Col1a1-GFP+ cells per lesion over time, tissue sections from Col1a1-GFP mice with EAE were stained for CD45. CD45 areas were traced using Image J and the number of Col1a1-GFP+ cells within this area were counted at different time points throughout EAE. To calculate the number of pericytes and T cells in EAE lesions over time, tissue sections from Co1la1GFP or c57/Bl6 mice induced with EAE and collected at different time points post symptom onset were stained with Desmin, CD4, or CD8. DAPI was then administered to all slides and used to determine the lesion location due to increased cell density. The number of Desmin, CD4 or CD8+ cells within the lesion were counted for each mouse using Image J. 3–6 mice were quantified per group with at least 4 spinal cord sections (two thoracic and two lumbar) analyzed from each mouse.
LPC and cuprizone scar tissue quantifications:
To quantify the area of demyelination that is Col1+ in the LPC mouse model, spinal cord cross sections from 4 mice 7 days post LPC administration were stained for Col1 and FlouroMyelin. The total Col1+ area and Flouromyelin- area were quantified using Image J and the Col1+ area/FlouroMyelin- area was reported. To quantify the area of demyelination that is Col1+ in the cuprizone mouse model with and without induced CNS interferon gamma expression, spinal cord cross sections from 3 mice with doxycycline and 3 mice without doxycycline that were 3 weeks after the start of cuprizone administration were stained for Col1 and DAPI. The total Col1+ area and area of the corpus callosum were quantified using Image J and the Col1+ area/corpus callosum area was reported.
fHTK quantifications:
To determine the extent of fibrotic scar tissue reduction, control and fHTK spinal cord tissue was collected as described above and stained for collagen I and CD11b. The total area of both stains was traced using Image J and the Col1+ area/CD11b+ area was reported. To quantify the % myelination per group, tissue sections were stained with FluoroMyelin (Thermo Fisher F34652) 1:300 in dPBS at room temperature. The total white matter area was traced using Image J and compared to the total area of the myelin stain. To calculate the number of OLIG2+ cells per lesion tissue sections were stained for OLIG2 and CD11b. CD11b areas were traced using Image J and the number of OLIG2+ cells within this area were counted and compared between groups. To calculate the percentage of OLIG2+ cells that were CC1+, sections were stained with OLIG2 and CC1 using the Vector Labs Mouse on Mouse Basic Kit (BMK-2202). DAPI was then administered to all slides and used to determine the lesion location due to increased cell density. The percentage of OLIG2+ cells also positive for CC1 within the lesion were counted for each mouse using Image J. To calculate the number of CD4+ and CD8+ cells per lesion, tissue sections were stained for CD4 or CD8. Lesion areas were traced using DAPI in Image J and the number of CD4+ or CD8+ cells within this area were counted and compared between groups. To calculate the number of axons in the lesions of control and fHTK mice, tissue sections were stained for CD11b and neurofilament heavy polypeptide. Quantifications were done using Cell Profiler. For all quantifications at least 6 spinal cord sections (3 thoracic and 3 lumbar) were analyzed from each mouse.
Col1a1-GFP regional analysis:
Brains and spinal cords of 4 adult, male, Col1a1-GFP mice were collected, sectioned and stained for CD31 in red and smooth muscle actin in far red. The length of the total vasculature and smooth muscle actin positive vasculature and the number of GFP+ cells associated with the vasculature for the different brain regions were quantified using Image J.
Col1a2CreERT analysis:
All mice were injected with tamoxifen, induced with EAE and tissue was collected and sectioned in the same way as the lineage training mice. For the Col1a1-GFP; Col1a2CreERT overlap analysis, 6 spinal cord cross sections per mouse were imaged and the percentage of Col1a1-GFP+ cells that were also positive for the tdTomato reporter was calculated using Image J. n=5 health and n=4 EAE. For the Col1a2CreERT; NG2 overlap analysis, spinal cord sections from Col1a2CreERT mice were stained for NG2. At least 6 spinal cross sections per mouse were imaged and the number of Col1a2CreERT labeled cells positive for NG2 and the number of Col1a2CreERT labeled cells negative for NG2 were counted using Image J. n = 4 health.
Statistics:
The statistics and n values used for each experiment are described in the figure legends. All error bars presented are ± standard error of the mean and n refers to the number of animals used in the experiment in every case. Animals were assigned to groups based on genotype when appropriate or randomly when mice of the same genotype were split into groups. Investigators were blinded for data collection and experimental analysis. Sample sizes were selected based on variability of the measurement and values of difference between conditions. Data distribution was assumed to be normal. Statistics were calculated using Prism 7 and Microsoft Excel 2013.
Extended Data
Extended Data Figure 1. Col1a1-GFP+ cell localization in health and following EAE.
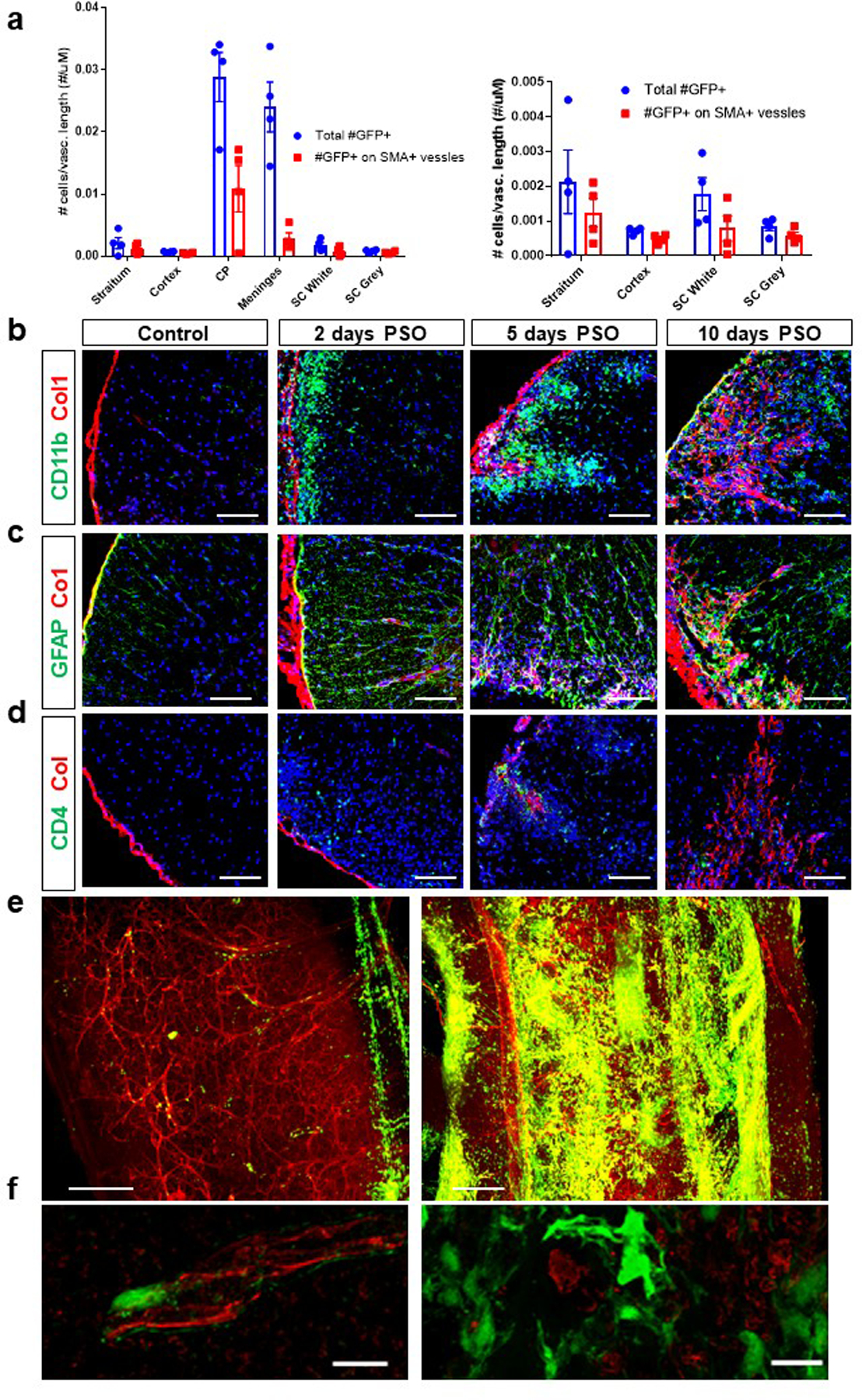
a. Analysis of the number of Col1a1-GFP+ cells per total vascular length and smooth muscle actin (SMA)+ vascular length in different CNS regions in healthy adult mice. CP= choroid plexus, SC= spinal cord white or grey matter, ± s.e.m., n=4, Col1a1-GFP mice. b-d. Spinal cord sections from wild type mice in health or with EAE at 2, 5 or 10 days PSO were stained with Col1 (red), DAPI (blue) and CD11b (b, green), GFAP (c, green), or CD4 (d, green) Scale bars = 100 µm. e. Light sheet microscopy image of a Col1a1-GFP mouse in health (left) and one with EAE (right) 10 days PSO perfused with tomato lectin and optically cleared, scale bars = 200 µm. f. Confocal microscopy images of Col1a1-GFP spinal cords from health (left) and EAE (right) stained for CD31 in red, scale bars = 10 µm.
Extended Data Figure 2. Fibrotic scarring is present in the LPC, but not cuprizone, model of demyelination.
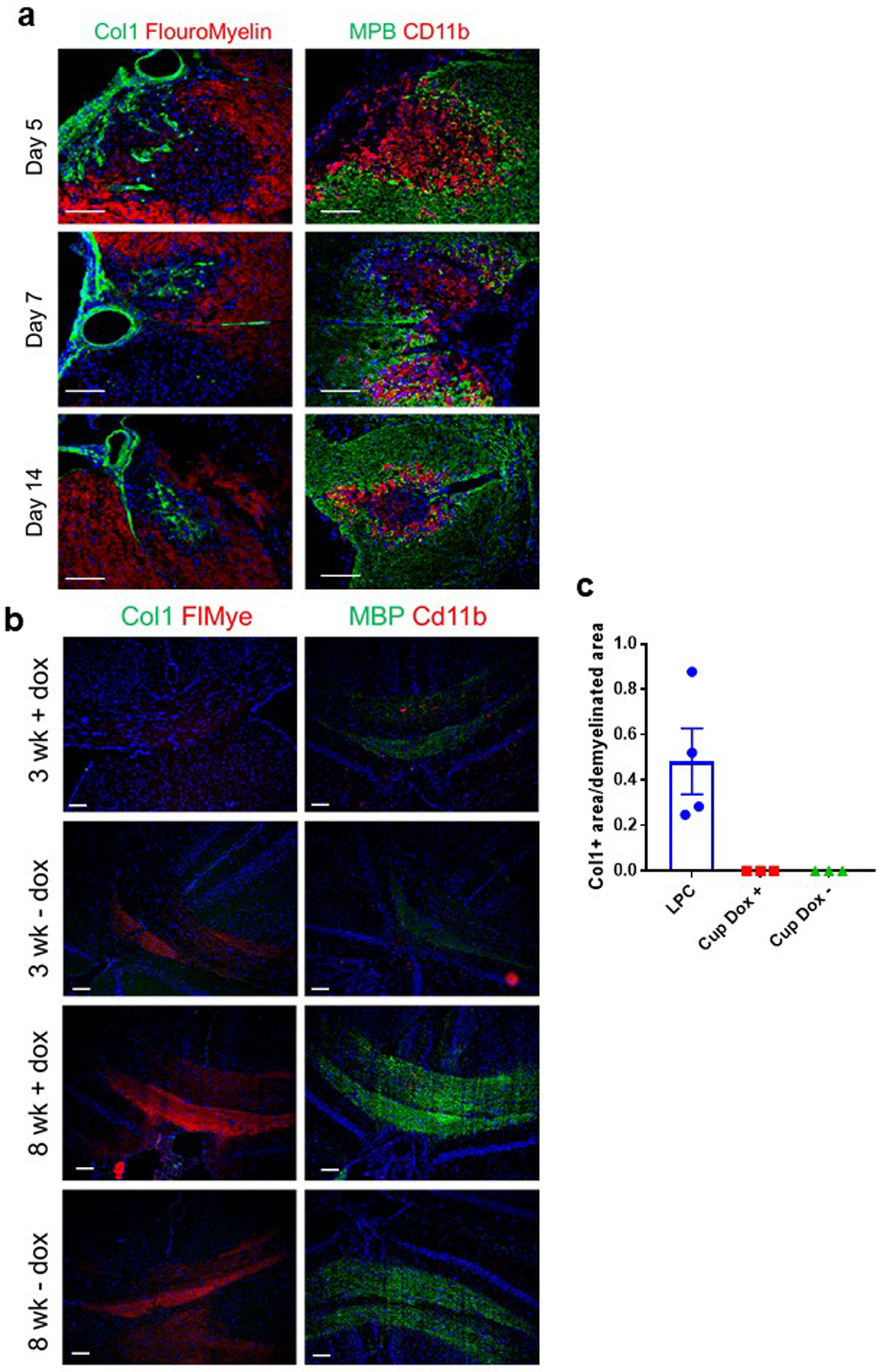
a. Spinal cord sections from mice either 5, 7, or 14 days post LPC injection into the spinal cord stained for either Col1 (green) and FluoroMyelin (red) or myelin basic protein (MBP) (green) and Cd11b (red). b. Brain sections in the area of the corpus callosum from GFAP/tTA;TRE/IFN-γ mice following cuprizone administration. GFAP/tTA;TRE/IFN-γ mice received doxycycline starting from birth, and half of the mice were taken off of doxycycline at 6 weeks of age (- dox) to induce the expression of interferon gamma in the CNS. Both the +dox and -dox groups were divided into two groups. The first group (3 wk) was given cuprizone for 3 weeks and then analyzed at the end of the 3 week period. The second group (8 wk) was given cuprizone for 5 weeks and then analyzed 3 weeks after completion of the cuprizone administration. All brains were stained for either Col1 (green) and FluoroMyelin (red) or myelin basic protein (MBP) (red) and Cd11b (green).c. Quantification of the proportion of the demyelinated area that is Col1+ from mice 7 days post LPC injection (n=4) or mice 3 weeks post cuprizone administration with and without doxycycline (n=3 each), ± s.e.m.
Extended Data Figure 3. Col1a2CreERT and NG2CreERTM reporter expression.
Spinal cords of Col1a2CreERT;Rosa-tdTomato (a) or NG2CreER™;Rosa-tdTomato (b) mice in health or EAE 10 d PSO were stained with CD31 in green and DAPI in blue. Scale bars = 100 µm. c. Spinal cords of Col1a2CreERT;Rosa-tdTomato;Col1a1-GFP mice in health or 10 days EAE PSO were imaged for both reporters. The percentage of Col1a1-GFP+ cells that were also positive for the tomato reporter in health and EAE is quantified in (d), ± s.e.m., n = 5 health, 4 EAE. e. Col1a2CreERT;Rosa-tdTomato mice were stained with NG2 in red, and the proportion of Col1a2CreERT+ cells that were NG2+ and NG2− were quantified in (f), ± s.e.m., n = 4. Scale bars = 100 µm
Extended Data Figure 4. Single-cell sequencing: cell purification and cluster expression.
a. Sample FACS plots of the purification of Col1a1-GFP+ cells used for the single-cell sequencing analysis of Col1a1-GFP+ cells in health and EAE. b. UMAP plot of the single-cell RNA-seq dataset of Col1a1-GFP+ cells in health and EAE with the individual sample identity labeled for each cell. c. Heat map depicting the expression of the 10 most differentially expressed genes in each cluster based on the logFC of the dataset. d. Heat map depicting the expression levels of genes specific to the labeled cell types, Olig = oligodendrocyte.
Extended Data Figure 5. Effects of reducing fibrotic scar formation on immune cell infiltration, oligodendrocyte lineage cell populations and axon numbers.
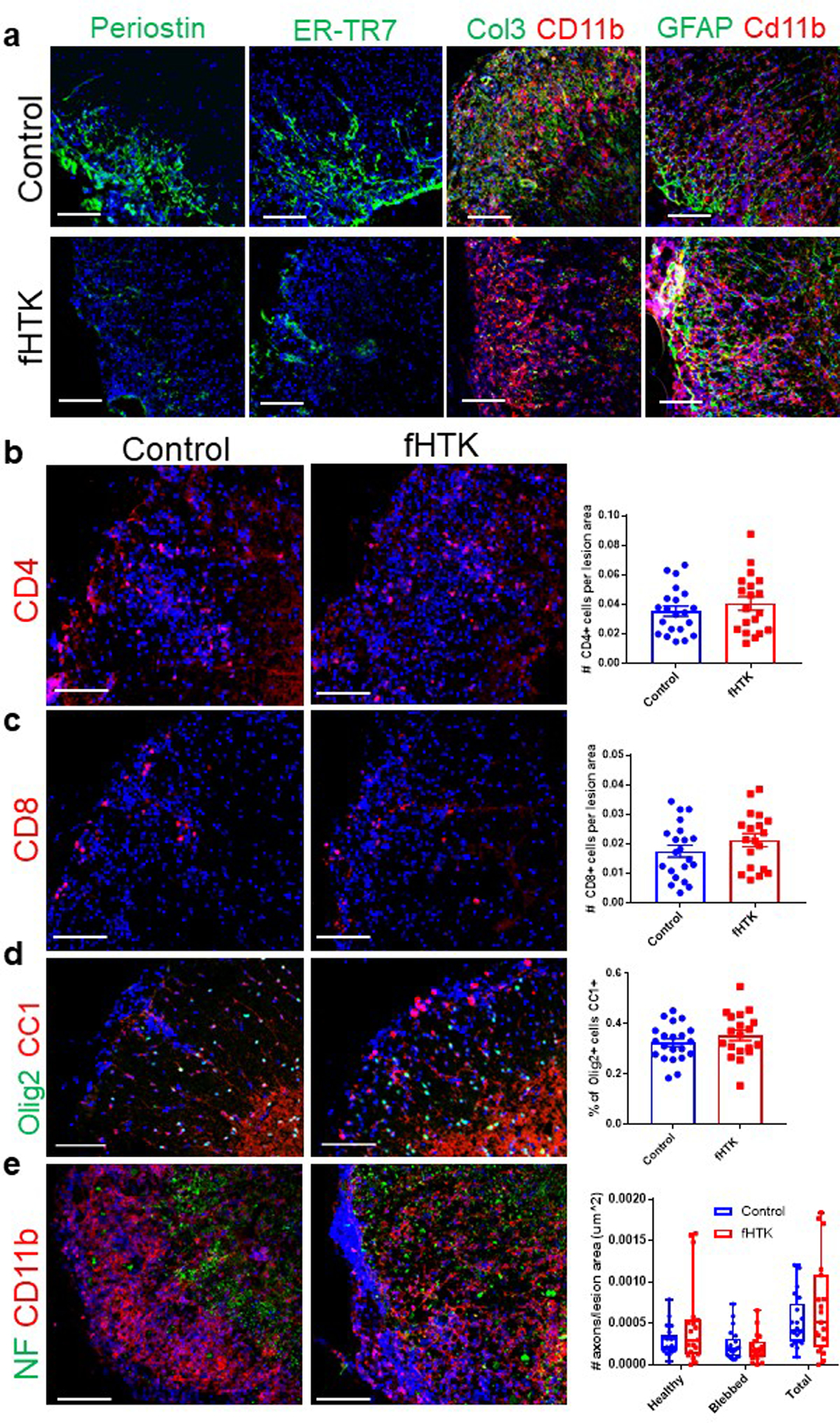
a. Spinal cord sections from fHTK mice and controls were stained for DAPI (blue) and periostin (green), ER-TR7 (green), Col3 (green) and CD11b (red), or GFAP (green) and Cd11b (red). b. Spinal cord sections from fHTK mice and controls were stained for CD4 (red, left) and DAPI (blue) and the number of CD4+ cells per lesion area was compared between groups (right), p = 0.38 by Student’s two-tailed t-test, ± s.e.m., n=21 control and 19 fHTK. c. Spinal cord sections from fHTK mice and controls were stained for CD8 (red, left) and DAPI (blue) and the number of CD8+ cells per lesion area was compared between groups (right), ± s.e.m., p = 0.22 by Student’s two-tailed t-test, n=21 control and 19 fHTK. d. Spinal cord sections from fHTK mice and controls were stained for OLIG2 in green and CC1 in red, and the percent of OLIG2+ cells that were also CC1+ was quantified, ± s.e.m., p = 0.26 by Student’s two-tailed t-test e. Spinal cord sections from fHTK mice and controls were stained for neurofilament heavy polypeptide (NF) in green and CD11b in red, and the number of healthy, blebbed and total axons was quantified, ± s.e.m.. Scale bars = 100 µm
Extended Data Figure 6. Col1 reduces OPC migration, but not proliferation or differentiation, in vitro.
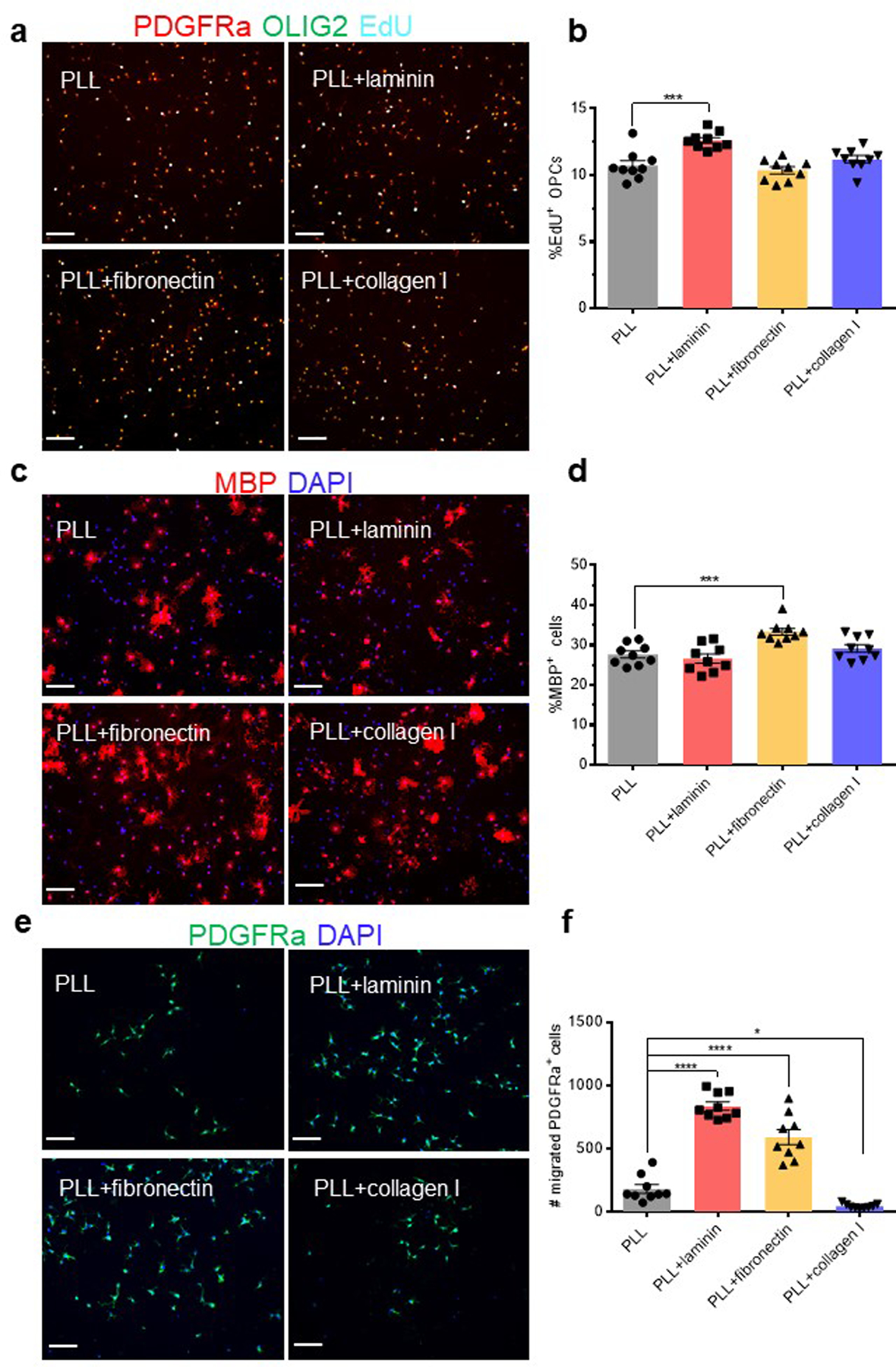
a. Representative images of OLIG2 (green) and PDGFRa (red) staining with EdU labeling (cyan) in rat OPC cultures on PLL, laminin, fibronectin, or collagen I after a 2 hour incubation in 10 uM EdU. b, Quantification of the percentage of EdU+ OPCs (OLIG2+PDGFRa+) for cultures represented in a., ± s.e.m., n = 9 replicates. c. Representative images of MBP (red) staining in rat OPC cultures on PLL, laminin, fibronectin, or collagen I three days after removal of PDGF-AA. Cell nuclei detected with DAPI (blue). d. Quantification of MBP+ cells over total cells (DAPI+) for cultures represented in c., ± s.e.m., n = 9 replicates. e. Representative images of PDGFRa (green) staining with DAPI in rat OPC cultures that had migrated through transwells coated with PLL, laminin, fibronectin, or collagen I following a 24 hour incubation. f, Quantification of PDGFRa+ cells on the underside of each transwell for the cultures represented in e., ± s.e.m., n = 9 replicates. Data displayed represent 3 replicates of 3 samples, each containing cells from three pooled postnatal day 7 rats. Comparisons were performed using one-way ANOVA with Sidak’s post hoc tests, ****p<0.0001, ***p<0.001, *p<0.05. Scale bars = 100 µm
Extended Data Figure 7. RNA sequencing analysis of Col1a1-GFP+ cells from the spinal cord in health and EAE.
a–c. MA plots comparing the transcriptome of CNS fibroblasts in health with whole spinal cord tissue (a), CNS fibroblasts in health with CNS fibroblasts EAE D5 (b) or D10 (c) PSO with red dots signifying genes with FDR < 0.1. d. Pathway analysis using DAVID Bioinformatics Resources 6.8, NIAID/NIH, GOTERM_BP_DIRECT for genes with a log2 fold change greater than 2 for CNS fibroblasts EAE D5 PSO compared to CNS fibroblasts in health. e. CPM of collagen genes from the bulk sequencing of whole spinal cord tissue (Whole SC, n = 2), CNS fibroblasts from health (CNS Fibro Health, n = 3) CNS fibroblasts 5 days PSO (CNS Fibro EAE D5, n = 3) and 10 days PSO (CNS Fibro EAE D10, n = 3), ± s.e.m., *FDR < 0.05, **FDR < 0.01 to Whole SC, #FDR < 0.05, ##FDR < 0.01 to CNS Fibro Health. f. Heat map of the expression levels of the top genes differentially expressed in CNS Fibro Health (each expressed at least 1 CPM in each control sample) compared to the whole spinal cord by FDR.
Acknowledgments:
We would like to thank Kristen Jepsen and the UCSD Institute of Genomic Medicine Genomics center, Tara Lambardo and the UCSD Veterans Hospital Flow Cytometry Core, Ying Jones and the CMM UCSD Electron Microscopy Core and Jennifer Santini and Marcy Erb at the UCSD Microscopy Core. We would like to thank David Brenner, UCSD Health Sciences, for kindly supplying the Col1GFP mice.
Funding:
RD and TA are funded by NIH/NINDS R01 NS119615. RD is funded by NIH/NINDS R01 NS091281–01A1, R01 NS103844, National Multiple Sclerosis Society Grant and UCSF Program for Breakthrough Biomedical Science. CED is funded by the UCSD Graduate Training Program in Cellular and Molecular Pharmacology through an institutional training grant from the National Institute of General Medical Sciences, T32 GM007752 and NIH/NINDS F31 NS108651. TA is funded by NIH/NINDS 5K08NS096192. BP is funded by NIH/NINDS, R01 NS034939. The UCSD Microscopy Core is funded by NINDS NS047101.
Footnotes
Competing interests: The authors declare no competing interests.
DATA AVAILABILTY:
All RNA sequencing and single-cell sequencing raw and processed data files have been uploaded to GEO and can be accessed using the following accession codes: GSE135186 for Single Cell Transcriptional Profile of Col1a1-GFP+ cells in health and EAE, GSE135044 for Transcriptional Profile of Col1a1-GFP+ CNS cells in health and EAE, and GSE135185 for Single cell transcriptional profile of mouse spinal cord in EAE.
REFERENCES
- 1.Bataller R & Brenner DA Liver fibrosis. The Journal of clinical investigation 115, 209–218, doi: 10.1172/JCI24282 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SB & Kalluri R Mechanistic connection between inflammation and fibrosis. Kidney international. Supplement, S22–26, doi: 10.1038/ki.2010.418 (2010). [DOI] [PMC free article] [PubMed]
- 3.Travers JG, Kamal FA, Robbins J, Yutzey KE & Blaxall BC Cardiac Fibrosis: The Fibroblast Awakens. Circulation research 118, 1021–1040, doi: 10.1161/CIRCRESAHA.115.306565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lederer DJ & Martinez FJ Idiopathic Pulmonary Fibrosis. New England Journal of Medicine 378, 1811–1823, doi: 10.1056/NEJMra1705751 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Mack M Inflammation and fibrosis. Matrix Biology 68–69, 106–121, doi: 10.1016/J.MATBIO.2017.11.010 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Wynn TA Cellular and molecular mechanisms of fibrosis. The Journal of pathology 214, 199–210, doi: 10.1002/path.2277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rockey DC, Bell PD & Hill JA Fibrosis — A Common Pathway to Organ Injury and Failure. New England Journal of Medicine 372, 1138–1149, doi: 10.1056/NEJMra1300575 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Fernández-Klett F & Priller J The Fibrotic Scar in Neurological Disorders. Brain Pathology 24, 404–413, doi: 10.1111/bpa.12162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Shea TM, Burda JE & Sofroniew MV Cell biology of spinal cord injury and repair. The Journal of Clinical Investigation 127, 3259–3270, doi: 10.1172/JCI90608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawano H et al. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res 349, 169–180, doi: 10.1007/s00441-012-1336-5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cregg JM et al. Functional regeneration beyond the glial scar. Experimental neurology 253, 197–207, doi: 10.1016/j.expneurol.2013.12.024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faulkner JR et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 2143–2155, doi: 10.1523/JNEUROSCI.3547-03.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yiu G & He Z Glial inhibition of CNS axon regeneration. Nature reviews. Neuroscience 7, 617–627, doi: 10.1038/nrn1956 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson MA et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200, doi: 10.1038/nature17623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brazda N & Müller HW Pharmacological modification of the extracellular matrix to promote regeneration of the injured brain and spinal cord. Progress in Brain Research 175, 269–281, doi: 10.1016/S0079-6123(09)17518-0 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Göritz C et al. A Pericyte Origin of Spinal Cord Scar Tissue. Science 333 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Dias DO & Göritz C Fibrotic scarring following lesions to the central nervous system. Matrix Biology, doi: 10.1016/j.matbio.2018.02.009 (2018). [DOI] [PubMed]
- 18.Hellal F et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science (New York, N.Y.) 331, 928–931, doi: 10.1126/science.1201148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshioka N, Hisanaga S-I & Kawano H Suppression of fibrotic scar formation promotes axonal regeneration without disturbing blood-brain barrier repair and withdrawal of leukocytes after traumatic brain injury. The Journal of Comparative Neurology 518, 3867–3881, doi: 10.1002/cne.22431 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Zhu Y et al. Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol Dis 74, 114–125, doi: 10.1016/j.nbd.2014.10.024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dias DO et al. Reducing Pericyte-Derived Scarring Promotes Recovery after Spinal Cord Injury. Cell 173, 153–165.e122, doi: 10.1016/j.cell.2018.02.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yahn SL et al. Fibrotic scar after experimental autoimmune encephalomyelitis inhibits oligodendrocyte differentiation. Neurobiology of Disease 134, 104674–104674, doi: 10.1016/J.NBD.2019.104674 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Horssen J, Bö L, Dijkstra CD & de Vries HE Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiology of Disease 24, 484–491, doi: 10.1016/j.nbd.2006.08.005 (2006). [DOI] [PubMed] [Google Scholar]
- 24.van Horssen J, Dijkstra CD & de Vries HE The extracellular matrix in multiple sclerosis pathology. Journal of Neurochemistry 103, 1293–1301, doi: 10.1111/j.1471-4159.2007.04897.x (2007). [DOI] [PubMed] [Google Scholar]
- 25.Mohan H et al. Extracellular Matrix in Multiple Sclerosis Lesions: Fibrillar Collagens, Biglycan and Decorin are Upregulated and Associated with Infiltrating Immune Cells. Brain Pathology 20, no–no, doi: 10.1111/j.1750-3639.2010.00399.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soderblom C et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 13882–13887, doi: 10.1523/JNEUROSCI.2524-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly KK et al. Col1a1+ perivascular cells in the brain are a source of retinoic acid following stroke. BMC neuroscience 17, 49–49, doi: 10.1186/s12868-016-0284-5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birbrair A et al. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem cell research & therapy 5, 122–122, doi: 10.1186/scrt512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guimarães-Camboa N et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell stem cell 0, 968–973, doi: 10.1016/j.stem.2016.12.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanlandewijck M et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature, doi: 10.1038/nature25739 (2018). [DOI] [PubMed]
- 31.Muhl L et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nature Communications 11, 3953, doi: 10.1038/s41467-020-17740-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aktas O, Küry P, Kieseier B & Hartung HP Fingolimod is a potential novel therapy for multiple sclerosis. Nat Rev Neurol 6, 373–382, doi: 10.1038/nrneurol.2010.76 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Chun J & Hartung H-P Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clinical neuropharmacology 33, 91–101, doi: 10.1097/WNF.0b013e3181cbf825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology 20, 163–172, doi: 10.1038/s41590-018-0276-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie T et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 22, 3625–3640, doi: 10.1016/j.celrep.2018.03.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramachandran A et al. FosB regulates stretch-induced expression of extracellular matrix proteins in smooth muscle. The American journal of pathology 179, 2977–2989, doi: 10.1016/j.ajpath.2011.08.034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y-T, Levasseur R, Vaishnav S, Karsenty G & Bradley A Bigenic Cre/loxP, puDeltatk conditional genetic ablation. Nucleic acids research 32, e161–e161, doi: 10.1093/nar/gnh158 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voskuhl RR et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 11511–11522, doi: 10.1523/JNEUROSCI.1514-09.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsai HH et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 351, 379–384, doi: 10.1126/science.aad3839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroder K, Hertzog PJ, Ravasi T & Hume DA Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75, 163–189, doi: 10.1189/jlb.0603252 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Saha B, Jyothi Prasanna S, Chandrasekar B & Nandi D Gene modulation and immunoregulatory roles of Interferonγ. Cytokine 50, 1–14, doi: 10.1016/J.CYTO.2009.11.021 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Rawlings JS, Rosler KM & Harrison DA The JAK/STAT signaling pathway. Journal of cell science 117, 1281–1283, doi: 10.1242/jcs.00963 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Oldroyd SD, Thomas GL, Gabbiani G & El Nahas AM Interferon-γ inhibits experimental renal fibrosis. Kidney International 56, 2116–2127, doi: 10.1046/j.1523-1755.1999.00775.x (1999). [DOI] [PubMed] [Google Scholar]
- 44.Chen ES, Greenlee BM, Wills-Karp M & Moller DR Attenuation of lung inflammation and fibrosis in interferon-gamma-deficient mice after intratracheal bleomycin. Am J Respir Cell Mol Biol 24, 545–555, doi: 10.1165/ajrcmb.24.5.4064 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Kim JH et al. Natural killer T (NKT) cells attenuate bleomycin-induced pulmonary fibrosis by producing interferon-gamma. The American journal of pathology 167, 1231–1241, doi: 10.1016/s0002-9440(10)61211-4 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King TE et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. The Lancet 374, 222–228, doi: 10.1016/S0140-6736(09)60551-1 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Li J & Li D Losartan reduces myocardial interstitial fibrosis in diabetic cardiomyopathy rats by inhibiting JAK/STAT signaling pathway. Int J Clin Exp Pathol 8, 466–473 (2015). [PMC free article] [PubMed] [Google Scholar]
- 48.Lin W et al. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain 129, 1306–1318, doi: 10.1093/brain/awl044 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Zhou T et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nature Neuroscience, 1–1, doi: 10.1038/s41593-018-0324-9 (2019). [DOI] [PMC free article] [PubMed]
- 50.Burgy O & Königshoff M The WNT signaling pathways in wound healing and fibrosis. Matrix Biology 68–69, 67–80, doi: 10.1016/J.MATBIO.2018.03.017 (2018). [DOI] [PubMed] [Google Scholar]
Method only References
- 51.Wendling O, Bornert JM, Chambon P & Metzger D Efcient temporally controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis 47, 14–18 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Niu J et al. Aberrant oligodendroglial-vascular interactions disrupt the blood–brain barrier, triggering CNS infammation. Nat. Neurosci 22, 709–718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heng TSP et al. Te Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Mei F et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med 20, 954–960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All RNA sequencing and single-cell sequencing raw and processed data files have been uploaded to GEO and can be accessed using the following accession codes: GSE135186 for Single Cell Transcriptional Profile of Col1a1-GFP+ cells in health and EAE, GSE135044 for Transcriptional Profile of Col1a1-GFP+ CNS cells in health and EAE, and GSE135185 for Single cell transcriptional profile of mouse spinal cord in EAE.