

Abstract
To develop a novel pharmacogenetic genotyping panel, a multidisciplinary team evaluated available evidence and selected 29 genes implicated in interindividual drug response variability, including 130 sequence variants and additional copy number variants (CNVs). Of the 29 genes, 11 had guidelines published by the Clinical Pharmacogenetics Implementation Consortium. Targeted genotyping and CNV interrogation were accomplished by multiplex single‐base extension using the MassARRAY platform (Agena Biosciences) and multiplex ligation‐dependent probe amplification (MRC Holland), respectively. Analytical validation of the panel was accomplished by a strategic combination of > 500 independent tests performed on 170 unique reference material DNA samples, which included sequence variant and CNV accuracy, reproducibility, and specimen (blood, saliva, and buccal swab) controls. Among the accuracy controls were 32 samples from the 1000 Genomes Project that were selected based on their enrichment of sequence variants included in the pharmacogenetic panel (VarCover.org). Coupled with publicly available samples from the Genetic Testing Reference Materials Coordination Program (GeT‐RM), accuracy validation material was available for the majority (77%) of interrogated sequence variants (100% with average allele frequencies > 0.1%), as well as additional structural alleles with unique copy number signatures (e.g., CYP2D6*5, *13, *36, *68; CYP2B6*29; and CYP2C19*36). Accuracy and reproducibility for both genotyping and copy number were > 99.9%, indicating that the optimized panel platforms were precise and robust. Importantly, multi‐ethnic allele frequencies of the interrogated variants indicate that the vast majority of the general population carries at least one of these clinically relevant pharmacogenetic variants, supporting the implementation of this panel for pharmacogenetic research and/or clinical implementation programs.
Since the term “pharmacogenetics” was first published in the late 1950s by Friedrich Vogel, 1 the field has evolved to include a more robust understanding of human drug metabolism and interindividual response variability 2 ; candidate gene 3 and genomewide 4 association studies identifying variants implicated in drug response phenotypes; professional societies, and international consortia dedicated to pharmacogenetics research 5 , 6 and clinical implementation 6 , 7 , 8 ; drug labels incorporating information on clinically significant metabolizer phenotypes 9 ; clinical pharmacogenetic practice guidelines 10 , 11 , 12 , 13 ; and the increasing availability of clinical pharmacogenetic tests for healthcare providers. However, despite growing interest in translating pharmacogenetic discoveries into clinical practice, a number of challenges remain, 14 , 15 including evolving regulatory oversight, 16 clinical testing harmonization, 17 provider education, 18 and ongoing evaluation of clinical utility 19 and cost‐effectiveness. 20
In the United States, Clinical Laboratory Improvement Act (CLIA)‐certified laboratories evaluate clinical genetic tests using the ACCE framework: Analytic validity; Clinical validity; Clinical utility; and Ethical, legal, and social implications. 21 Support for analytical validation of pharmacogenetic testing is available from several venues, including extensive literature curation by the Pharmacogenomics Knowledgebase (PharmGKB) 22 ; star (*) allele haplotype definitions catalogued by the Pharmacogene Variation (PharmVar) Consortium 23 ; genotyping recommendations by the Association for Molecular Pathology (AMP) 11 , 12 ; reference materials from the Coriell Cell Repository/Centers for Disease Control and Prevention (CDC) Genetic Testing Reference Materials Coordination Program (GeT‐RM) 24 ; and pharmacogenetic quality assurance proficiency testing programs offered by the College of American Pathologists (CAP). 25 These resources directly support pharmacogenetic test development by clinical laboratories, which prompted our evolution from single gene tests 26 , 27 , 28 , 29 , 30 to a 29 gene pharmacogenetic panel that interrogates multi‐ethnic sequence and copy number variants (CNVs) implicated in interindividual drug response variability.
METHODS
Pharmacogenetic panel design and development
In 2016, a multidisciplinary team of pharmacogeneticists (PharmDs and PhDs), physicians (MDs), American Board of Medical Genetics and Genomics (ABMGG)‐certified clinical laboratorians, genetic counselors, bioinformaticians, software developers, laboratory supervisors, project managers, and laboratory leadership convened with the charge of designing and developing a pharmacogenetic testing solution that was centered on being clinically responsible, but scientifically progressive in order to incorporate emerging content with potential utility. The testing solution had to be technically flexible to allow for all possible testing volumes/throughput (sporadic vs. scale), as well as having a low operational cost to allow for accessible pricing in the likely event of inconsistent reimbursement. To identify genes and variants implicated in interindividual drug response variability, this team met regularly to evaluate the available sources of evidence and adjudicate the strength of association and/or validity of candidate genes and variants. The work product of that effort subsequently was put forth to the laboratory team to implement an analytical validation plan, which determined accuracy, precision, and other testing metrics related to the genes and variants included in the comprehensive pharmacogenetic panel. The multidisciplinary team continued to evaluate the appropriateness of the panel content throughout the analytical validation phase and regularly reviews literature/evidence for new candidate genes and variants, including statements by the US Food and Drug Administration (FDA) and guidelines from the American College of Medical Genetics and Genomics (ACMG), AMP, Clinical Pharmacogenetics Implementation Consortium (CPIC), Dutch Pharmacogenomics Working Group (DPWG), and other resources. Given that clinical interpretation evidence and therapeutic recommendations based on pharmacogenetic test results are continually evolving, these postanalytical aspects of the testing workflow were determined to be out of scope for this panel development/validation report.
Multiplexed targeted genotyping
The comprehensive pharmacogenetic panel uses multiplex polymerase chain reaction (PCR) and single base extension (SBE) using the Agena SpectroCHIP II and MassARRAY Analyzer 4 platform, as per manufacturer instructions (Agena Biosciences, San Diego, CA). In brief, for each sample 10–20 ng of genomic DNA was amplified in six independent 5 µl multiplex PCR reactions, which consisted of an initial denaturation step at 95°C for 2 minutes followed by 45 cycles (95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 2 minutes). Amplicons were inactivated by shrimp alkaline phosphatase (Agena Biosciences, San Diego, CA) and subjected to six corresponding multiplex SBE reactions using 2 µl of SBE reagent (Agena Biosciences), which consisted of an initial denaturation step at 95°C for 30 seconds followed by 40 cycles (95°C for 5 seconds (52°C for 5 seconds and 80°C for 5 seconds) × 5). SBE products were conditioned with resin to remove salts, spotted on a SpectroCHIP II array, and read on the MassARRAY Analyzer 4 system. Genotypes at all targeted loci were determined by SBE peak intensity and Typer software version 4.1 (Agena Biosciences, San Diego, CA), and diplotypes for selected genes were inferred by a haplotype translation table and Typer software version 4.1.
UGT1A1*28 dinucleotide repeat genotyping
Independent interrogation of the UGT1A1 promoter thymine‐adenine (TA) dinucleotide repeat variant (rs8175347; c.‐53‐52TA[6]>TA[7]) was accomplished by capillary gel electrophoresis‐based fragment sizing. Fluorescently labeled PCR (forward: 5′‐[6‐FAM]TCCCTGCTACCTTTGTGGAC‐3′; reverse: 5′‐CCTGGGACTCCACAGCCAT‐3′) consisted of an initial denaturation step at 94°C for 3 minutes followed by 30 cycles (94°C for 30 seconds, 62°C for 30 seconds, and 72°C for 40 seconds). Labeled amplicons were separated by capillary gel electrophoresis using a 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) and allele sizing was accomplished using internal positive controls and GeneMapper Software 5 (Applied Biosystems, Foster City, CA). Star (*) allele calling was based on identified repeat length: 5 repeats: *36; 6 repeats (normal): *1; 7 repeats: *28; 8 repeats: *37. 31
Multiplex ligation‐dependent probe amplification CNV detection
Multiplex ligation‐dependent probe amplification (MLPA) was performed using the Cytochrome P‐450 MLPA kit (P128‐C1; MRC‐Holland, Amsterdam, The Netherlands), as per manufacturer instructions and as previously described. 32 , 33 This commercial probe mix intersects 7 genes in the comprehensive pharmacogenetic panel: CYP1A2 (3 probes), CYP2B6 (3 probes), CYP2C19 (3 probes), CYP2C9 (5 probes), CYP2D6 (4 probes), CYP3A4 (3 probes), and CYP3A5 (3 probes). Amplified products were separated by capillary gel electrophoresis and analyzed using GeneMarker version 2.6.7 software (SoftGenetics, State College, PA). After quality control and data normalization, copy number was determined according to the following peak ratio ranges: zero copies: 0 to < 0.25, one copy: > 0.30 and < 0.70; two copies: > 0.80 and < 1.20; three copies > 1.30 and < 1.7; and four or more copies: > 1.80.
Data integration
As detailed above, diplotypes were determined for a subset of relevant genes using Typer version 4.1 software (Agena Biosciences); however, haplotypes with structural variation were also inferred by incorporating MLPA‐detected copy number using GeneMarker version 2.6.7 software (SoftGenetics, State College, PA). This data integration was most commonly performed for CYP2D6 as previously described, 33 which enabled the identification of CYP2D6 deletion (e.g., *5), duplication (e.g., x2), tandem (e.g., *36 + *10), and partial‐gene conversion (e.g., *13 and *68) haplotypes (www.pharmvar.org/gene/CYP2D6). 23 The UGT1A1*28 dinucleotide genotyping results are also manually incorporated into the Agena multiplexed genotyping results.
Validation specimens
Reference material DNA samples were identified and acquired from the Coriell Institute for Medical Research (Camden, NJ). Additional de‐identified DNA controls were isolated from peripheral blood that had previously undergone clinical genetic testing at Sema4 (formerly the Mount Sinai Genetic Testing Laboratory). Peripheral blood was collected in EDTA vacutainer tubes using standard practices and DNA was isolated using the QiaSymphony (Qiagen, Valencia, CA) or Chemagic (Perkin Elmer, Baesweiler, Germany) according to the manufacturer’s instructions. Saliva samples were collected using the Oragene Dx kit (OGD‐500; DNA Genotek, Ottawa, ON, Canada) and DNA was isolated using the QiaSymphony (Qiagen, Valencia, CA). Buccal samples were collected using the ORAcollect kit (OC‐175) and DNA was isolated using the prepIT•L2P protocol (DNA Genotek, Ottawa, ON, Canada), as per the manufacturer instructions.
Pharmacogenetic panel analytical validation: Accuracy and reproducibility
The analytical validation plan followed the Laboratory Developed Test guidelines as defined by both the CAP 34 and the Clinical Laboratory Evaluation Program at the Wadsworth Center, New York State Department of Health (NYS DOH). 35 The plan was centered on determining performance characteristics across the different technologies used in the pharmacogenetic panel, as well as defining standard operating procedures, quality control/quality assurance procedures, and validating variant detection and specimen type. Key elements of the plan included positive control reference materials (when available) to measure accuracy (sensitivity and specificity) and demonstrate intra‐run and inter‐run reproducibility. 35
Sanger sequencing
Selected variants and samples were confirmed by Sanger sequencing, which included region‐specific amplification with M13‐tagged primers, amplicon purification with shrimp alkaline phosphatase and exonuclease I (both from USB, Cleveland, OH), and bidirectional sequencing. Sequencing was performed on an ABI 3700 Sequencer (Applied Biosystems) and chromatograms were analyzed using Sequencher 5.3 (Gene Codes, Ann Arbor, MI).
Pharmacogenetic panel sequence variant carrier frequencies
The expected number of pharmacogenetic panel minor alleles per individual was estimated using 100 bootstrap resamples (with replacement) for five 1KG major populations (African, n = 661; Admixed American, n = 347; East Asian, n = 504; non‐Finnish European, n = 404; and South Asian, n = 489) and the 98 panel sequence variants present in the 1000 Genomes Project (1KG) Phase III version 5a data. The number of minor alleles observed per individual was averaged for each ancestry‐specific bootstrap and violin plots were created using the Seaborn Python Package, version 0.9.0. The minor allele was assigned as the nucleotide with the lowest mean allele frequency taken from an average of four population‐specific gnomAD (version 2.1.1) frequencies: African, Admixed American, East Asian, and non‐Finnish European (South Asian allele frequencies were not available in the gnomAD version 2.1.1 genome sites file). Of note, ‐the minor allele of the interrogated variants is not always the pharmacogenetic risk allele at all loci included in this analysis.
RESULTS
Pharmacogenetic panel design and development
All CPIC level A and B genes, 8 and those genes with CPIC guidelines in development, were considered for inclusion in the panel; however, CFTR and IFNL3 were excluded based on a perceived lack of utility, HLA‐A and HLA‐B were excluded based on technical considerations, and CACNA1S and RYR1 were excluded based on their variant density and diagnostic implication as Mendelian disease genes. In addition, all genes with a level 1A or 1B variant, as defined by PharmGKB, were included; however, ANKK1, EGFR, and XPC were excluded based on perceived limited evidence, somatic variants, and Mendelian disease association, respectively. Literature and knowledgebase review identified additional genes with evidence for inclusion in the comprehensive panel as emerging informative content (ABCG2, ADRA2A, COMT, CYP1A2, CYP2C8, DRD2, GRIK4, HTR2A, HTR2C, OPRM1, UGT2B15, and SLC6A4) 36 ; however, SLC6A4 was excluded based on technical considerations.
Specific variants for each identified gene were further evaluated for inclusion, which assessed their functional effect on protein production/activity and/or the strength of their association with a drug response phenotype, and the technical feasibility of their genotyping. This variant evaluation was performed by the multidisciplinary team noted above and led by board‐certified clinical molecular geneticists. Although assessing variant function and significance of association can be a subjective process, external resources of curated (e.g., PharmGKB 22 ) and expert committee adjudicated (e.g., PharmVar 37 ) knowledge were leveraged to inform variant selection. Rare functional variants (< 1% minor allele frequency (MAF)) were considered for inclusion in an effort to improve allele coverage across diverse multi‐ethnic populations. In addition, F2 (rs1799963) and F5 (rs6025) were included based on thrombophilia risk and their reference in the FDA warning labels of thrombocytopenia medications. The final gene and variant content of the comprehensive pharmacogenetic genotyping panel is detailed in Table 1 and Table S1 .
Table 1.
Genes and variant alleles interrogated by the pharmacogenetic panel
| Gene | Variant allelesa |
|---|---|
| ABCB1 | c.3435T>C (rs1045642), c.2677T>A/G (rs2032582) |
| ABCG2 | c.421C>A (rs2231142) |
| ADRA2A | c.‐1252G>C (rs1800544) |
| COMT | c.472G>A (rs4680) |
| CYP1A2 | *1A, *1C, *1D, *1F, *1K, *1L, *1V |
| CYP2B6 | *6 (*9), *29, *30 |
| CYP2C cluster | rs12777823 |
| CYP2C19 | *2, *3, *4, *5, *6, *7, *8, *9, *10, *16, *17, *19, *22, *24, *25, *26, *35, *36, *37 |
| CYP2C8 | *2, *3, *4 |
| CYP2C9 | *2, *3 (*18), *4, *5, *6, *8, *11, *12, *13, *15, *25, *27, *31 |
| CYP2D6 | *2, *3, *4, *5, *6, *7, *8, *9, *10, *11, *12, *13, *14, *15, *17, *18, *19, *20, *29, *30 (*40), *31, *36, *38, *41, *42, *44, *47, *49, *50, *51, *54, *55, *56, *57, *62, *64, *68, *69, *72, *100, *101, *107, *114 |
| CYP3A4 | *1B, *2, *3, *12, *17, *22 |
| CYP3A5 | *3, *6, *7 |
| CYP4F2 | *3 |
| DPYD | *2, *13, c.2846A>T (rs67376798) |
| DRD2 | c.811‐83G>T (rs1076560), c.‐585A>G (rs1799978) |
| F2 | c.*97G>A (rs1799963) |
| F5 | c.1601G>A (rs6025) |
| G6PD | Mediterranean, A+, A‐202, A‐968, A‐680, Chatam, Canton, Cosenza, Kerala‐Kalyan, Orissa |
| GRIK4 | c.83‐10039T>C (rs1954787) |
| HTR2A | c.614‐2211T>C (rs7997012), c.102C>T (rs6313), c.102C>T (rs6311) |
| HTR2C | c.‐759C>T (rs3813929), c.551‐3008C>G (rs1414334) |
| NUDT15 | *2 (*3), *4, *5 |
| OPRM1 | c.118A>G (rs1799971) |
| SLCO1B1 | c.521T>C (rs4149056; *5, *15, *17) |
| TPMT | *2, *3A, *3B, *3C, *4 |
| UGT1A1 | *6, *27, *80,b rs8175347 (*28, *36, *37) b |
| UGT2B15 | c.253T>G (rs1902023; *2) |
| VKORC1 | c.‐1639G>A (rs9923231), c.106G>T (rs61742245), c.196G>A (rs72547529) |
Brackets indicate star (*) allele haplotypes with shared variants that cannot be distinguished by genotyping.
The UGT1A1*80 variant (rs887829) is in linkage disequilibrium with the dinucleotide repeat *28 allele (rs8175347) and is used as an internal control for the independent UGT1A1*28 capillary electrophoresis test (see Methods and Results).
Pharmacogenetic panel genotyping: accuracy and reproducibility
Multiplexed targeted genotyping accuracy
To assess the accuracy of the multiplexed pharmacogenetic genotyping panel, three reference cohorts and sample sets were utilized (total n = 73; Table S2 ). Concordance was initially measured against DNA from peripheral blood specimens that were previously subjected to clinical testing at the Mount Sinai Genetic Testing Laboratory (n = 12; Table S2 ). Five genes in the current panel (CYP2C9, CYP2C19, CYP2D6, SLCO1B1, and VKORC1) were previously validated and approved by the NYS DOH using single gene targeted genotyping assays (Luminex, Austin, TX). 26 , 27 , 29 Genotype concordance between the multiplexed Agena panel and the clinical Luminex results for the 28 alleles interrogated by both platforms was 100% (280/280 alleles), and diplotype concordance was 100% (120/120 haplotypes; Table S3 ).
Of the 29 genes included in the pharmacogenetic genotyping panel, 16 (53%) were included in the previously reported pharmacogenetic GeT‐RM program. 24 As such, concordance was also measured against positive control commercial DNA from the GeT‐RM program (n = 29; Table S2 ). Differences in the specific variants included in our genotyping panel compared with the platforms used across the participating GeT‐RM laboratories resulted in intermittent discrepancies in diplotype assignment; however, these were considered concordant if the inferred star (*) alleles were appropriate based on platform. The initial genotype concordance among the 82 alleles interrogated by both our multiplexed platform and the consensus results in the GeT‐RM (i.e., confirmed by two independent laboratories/platforms) was 99.96% (2377/2378 alleles), and diplotype concordance was 99.88% (843/844 haplotypes; Table S4 ). The one inconsistent genotype was due to a single discordant heterozygous variant (NA19226: rs7900194 (CYP2C9*8)), which was confirmed by Sanger to be an Agena error and subsequently corrected with redesigned assay primers. The redesigned Agena assay and primer well were re‐tested on all validation specimens to confirm the quality and accuracy of the new oligo pool.
The third cohort used for assessing genotyping accuracy was a subset of 1KG samples (n = 32; Table S2 ), which were computationally selected using VarCover 38 based on their enrichment of variant alleles among the targeted loci of the pharmacogenetic panel. Notably, of the 130 variants genotyped by the pharmacogenetic panel, only 98 were called in the 1KG dataset and subsequently utilized for accuracy analyses. Genotyping accuracy against this dataset identified 7 discordant genotypes (including rs7900194 noted above) in 7 independent samples, which resulted in an initial sensitivity of 0.999 (741/742 minor alleles) and specificity of 0.999 (5407/5413 major alleles; Figure 1 ; Table S5 ). The discordant samples and variants were subjected to orthogonal Sanger and/or Luminex testing, which confirmed the Agena genotyping result in five of seven cases (i.e., 1KG dataset errors). The two outstanding Agena genotyping errors were rs7900194 in sample NA19226 noted above, and rs118203757 (CYP2C19*24) in sample NA20356 (Figure 1 ; Table S5 ). The rs118203757 assay was also corrected with redesigned assay primers, which was validated by retesting across all reference material specimens. After incorporating corrections (Agena and 1KG results), the optimized multiplexed panel had a sensitivity and specificity of 1.0 against the 1KG results (Figure 1 ; Table S6 ). Of note, positive control reference materials were not available for 29 sequence variants included in the comprehensive pharmacogenetic panel (Table S7 ); however, the wild‐type genotype results for these variants were all concordant with the reference material specimens. Future detection of these variants as heterozygous or homozygous for minor allele by genotyping would be confirmed by Sanger sequencing prior to reporting. 35
Figure 1.

Genotyping concordance with 1000 Genomes Project (1KG) sequence variant accuracy controls. (a) Heatmap concordance of 98 variants (y‐axis) with available genotype results in 1KG phase III version 5a across 32 selected reference DNA samples (x‐axis). Please note that the rs72549356 variant is not included in the figure due to this insertion/deletion variant not being called in the 1KG dataset; however, the variant was detected by genotyping in HG03166 and confirmed by Sanger sequencing (see Results). (b) Heatmap concordance of 98 variants (y‐axis) with available genotype results in 1KG phase III version 5a across 32 selected reference DNA samples (x‐axis) after 1KG/genotyping assay error corrections. Light blue: homozygous reference (concordant); medium blue: heterozygous (concordant); dark blue: homozygous alternate (concordant); pink: initially discordant genotype results between 1KG and the Agena genotyping (1KG error); and red: initially discordant genotype results between 1KG and the Agena genotyping (Agena error). See Results for discussion related to discordant genotyping correction among the 1KG accuracy controls.
Multiplexed targeted genotyping reproducibility
Reproducibility of the multiplexed pharmacogenetic genotyping panel was measured by subjecting reference material samples (n = 10; Table S2 ) to intra‐run and inter‐run triplicate testing (i.e., 3:1:1 validation). In summary, the intra‐run and inter‐run genotype and diplotype concordances for the 10 control samples were both 100% (1300/1300 genotypes; 340/340 haplotypes; Table S8 ).
UGT1A1 rs8175347 genotyping: Accuracy and reproducibility
To assess the accuracy of the UGT1A1 capillary electrophoresis test, genotype results were compared to GeT‐RM reference material controls 24 and additional Coriell samples that were subjected to orthogonal Sanger sequencing (total n = 32; Table S2 ). All possible rs8175347 TA dinucleotide repeat variant alleles were represented in the reference samples (i.e., *1 (6), *28 (7), *36 (5), and *37 (8)), and the results are summarized in Table S9 . TA repeat genotype concordance between the UGT1A1 capillary electrophoresis test and the reference material controls was 100% (64/64 alleles; Table S9 ). Reproducibility was tested using 23 positive control reference samples, which resulted in intra‐run and inter‐run genotype concordances of 100% (23/23 genotypes; Table S10 ). In addition, the multiplexed genotyping Agena assay includes the UGT1A1*80 defining variant (rs887829), which is in complete linkage disequilibrium with UGT1A1*28 and is used as an internal confirmatory control for the UGT1A1 capillary electrophoresis test. All reference samples genotyped for UGT1A1 by both the Agena assay and capillary electrophoresis were concordant between UGT1A1*80 and UGT1A1*28 (data not shown). Moreover, the UGT1A1*80 genotyping assay had a sensitivity (25/25 alleles) and specificity (39/39 alleles) of 1.0 with the 1KG accuracy cohort detailed above.
Pharmacogenetic panel MLPA testing: Accuracy and reproducibility
To assess the accuracy of the pharmacogenetic MLPA assay, most notably for the CYP2D6 gene (MLPA probes: exons 1, 5, 6, and 3′ downstream; Table 2 ), multiple reference cohorts, and sample sets were utilized (total n = 95; Table S2 ). Concordance was initially measured against DNA from peripheral blood specimens that were previously subjected to clinical CYP2D6 testing at the Mount Sinai Genetic Testing Laboratory (n = 20; Table S2 ). These samples were selected based on their reported CYP2D6 copy number results using the previously validated and NYS DOH‐approved xTAG CYP2D6 version 3 IVD kit (Luminex). 27 , 33 Ten of these samples had the normal 2 copies and 10 samples had full gene CYP2D6 copy number variation (3 with 1 copy, 7 with 3 copies, and 2 with 4 copies). Concordance between MLPA and the clinically reported CYP2D6 copy number results was 100% (40/40 alleles; Table S11 ).
Table 2.
Representative copy number alleles interrogated by the pharmacogenetic panel
| Sample | CNV Control | Reference | CYP2D6 | CYP2C19 | CYP2C9 | CYP2B6 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon 1 | Exon 5 | Exon 6 | 3′a | Exon 2 | Exon 6 | Exon 9 | Exon 1 | Exon 7 | Exon 8a | Exon 8b | Exon 9 | Exon 2 | Exon 3 | Exon 4 | |||
| HG00276 | CYP2D6 *4/*5 | GeT‐RM | 0.581 | 0.600 | 0.537 | 0.596 | 1.046 | 1.042 | 1.081 | 1.016 | 1.091 | 0.921 | 0.975 | 0.986 | 1.071 | 0.963 | 1.043 |
| NA02016 | CYP2D6 *2xN/*17 | GeT‐RM | 1.514 | 1.529 | 1.449 | 1.450 | 0.988 | 0.971 | 1.081 | 1.029 | 0.957 | 1.037 | 1.063 | 0.995 | 1.043 | 1.099 | 0.965 |
| NA17244 | CYP2D6 *2x2/*4x2 | GeT‐RM | 1.853 | 1.886 | 1.982 | 1.810 | 0.980 | 0.888 | 0.971 | 0.907 | 1.011 | 0.949 | 1.049 | 0.973 | 0.996 | 1.036 | 0.983 |
| NA19785 | CYP2D6 *1/*13+*2A | GeT‐RM | 0.975 | 1.438 | 1.333 | 1.394 | 0.982 | 0.966 | 0.969 | 1.025 | 1.013 | 1.023 | 0.994 | 1.032 | 0.997 | 0.971 | 1.002 |
| NA16688 | CYP2D6 *2/*36 + 10 | GeT‐RM | 1.455 | 1.456 | 1.388 | 0.928 | 1.043 | 0.862 | 1.023 | 0.925 | 1.000 | 0.895 | 0.917 | 1.021 | 1.000 | 1.040 | 1.019 |
| NA21781 | CYP2D6 *2Ax2/*68+*2 | GeT‐RM | 1.914 | 1.490 | 1.389 | 1.430 | 0.999 | 0.955 | 0.999 | 0.998 | 0.944 | 1.030 | 1.073 | 0.942 | 0.915 | 0.977 | 0.988 |
| ISMMS/Sema4_4b | CYP2C19 *36 | CMA | 1.021 | 0.982 | 0.965 | 0.952 | 0.499 | 0.458 | 0.517 | 0.928 | 0.962 | 0.953 | 0.989 | 0.961 | 0.996 | 1.015 | 1.001 |
| ISMMS/Sema4_7b | CYP2C19 *37 | CMA | 0.988 | 0.980 | 0.957 | 0.959 | 0.481 | 0.854 | 0.992 | 1.004 | 1.010 | 1.027 | 1.064 | 0.984 | 0.977 | 0.952 | 0.997 |
| ISMMS/Sema4_9b | CYP2C duplication | CMA | 0.821 | 0.829 | 0.957 | 0.854 | 1.440 | 1.665 | 1.297 | 1.209 | 1.561 | 1.368 | 1.487 | 1.577 | 0.888 | 0.886 | 0.767 |
| NA19178 | CYP2B6 *29 | CMA, TaqManc | 1.025 | 1.031 | 0.945 | 1.023 | 0.986 | 1.007 | 1.023 | 1.004 | 0.925 | 1.048 | 1.060 | 0.975 | 0.528 | 0.548 | 0.513 |
CMA, chromosomal microarray; CNV, copy number variant; GeT‐RM, Genetic Testing Reference Materials Coordination Program. 24
Multiplex ligation‐dependent probe amplification copy number results: pink shading: 1 copy; no shading: 2 copies; light blue shading: 3 copies; and dark blue shading: 4 copies.
The CYP2D6 downstream 3′ probe hybridizes to chr22:42521985_42522042 (GRCh37/hg19), which is ~ 450 bp downstream from CYP2D6 exon 9.
As reported in Botton et al. 2019. 40
As reported in Martis et al. 2013. 32
MLPA copy number concordance was also measured against reference DNA selected from the GeT‐RM program, 24 a previously reported CYP2D6 reference material project, 39 and additional Coriell samples tested internally using the xTAG CYP2D6 version 3 IVD kit (total n = 69; Table S2 ). These samples were selected based on their reported CYP2D6 structural variation, which included deletion, duplication, tandem, and conversion alleles (e.g., *5, *36, *36+*10, *68, and *13). Importantly, the MLPA CYP2D6 copy number signature coupled with genotype results allows for the detection of some tandem and CYP2D7 conversion alleles (Table 2 ). 33 Concordance between MLPA and previously reported CYP2D6 copy number results among this cohort was 100% (138/138 alleles; Table S11 ).
In addition to CYP2D6, the MLPA probe mix interrogates six additional genes in the pharmacogenetic genotyping panel (CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5); however, copy number variation has only been reported in CYP2B6 32 and more recently at CYP2C9 and CYP2C19. 40 Five positive controls with CYP2B6, CYP2C9, and/or CYP2C19 deletions or duplications were all confirmed and accurately detected by MLPA (Table 2 ; Table S11 ). Reproducibility of MLPA testing was assessed using 14 control samples, which included CYP2D6 CNVs (*5, *36, duplication, and triplication), CYP2B6*29 and CYP2C19*36. Triplicate testing resulted in intra‐run and inter‐run concordance of 100% (1,008/1,008 probes) and 99.4% (1,002/1,008 probes), respectively (Table S12 ). The six discordant probe measurements from the inter‐run testing were considered acceptable, as they were all from a single clinical specimen with compromised DNA.
Specimen validation
In addition to commercially available reference material and peripheral blood specimens, all three pharmacogenetic testing platforms were validated with saliva DNA using paired specimens from eight deidentified healthy adults. Genotype, diplotype, UGT1A1 capillary electrophoresis, and MLPA copy number results were 100% concordant across paired blood and saliva specimens (data not shown). Buccal swab DNA was also separately validated across all 3 platforms using paired saliva and buccal swab specimens from 18 de‐identified adults. Concordance was again 100% between the paired specimens (data not shown).
Multi‐ethnic allele frequencies
The average MAFs of the variants with available data in gnomAD version 2.1.1 are listed in Table S1 , and the MAFs by ethnicity of each variant are illustrated in Figure 2 . Based on the average MAF and assuming conditional independence of pharmacogenetic variants, it is estimated that > 99% of the general population carries at least one variant allele in the pharmacogenetic panel. In addition, Figure S1 displays the expected number of pharmacogenetic minor alleles (per person) as violin plots across five 1KG major populations, which suggests that individuals in the general population carry ~ 11–15 variant alleles in the pharmacogenetic panel. Differences in number of variants per individual can be observed among ancestral populations, which is evident across all pharmacogenetic panel variants as well as when restricting the analysis to only variants in genes with available CPIC guidelines (Figure S1 ).
Figure 2.
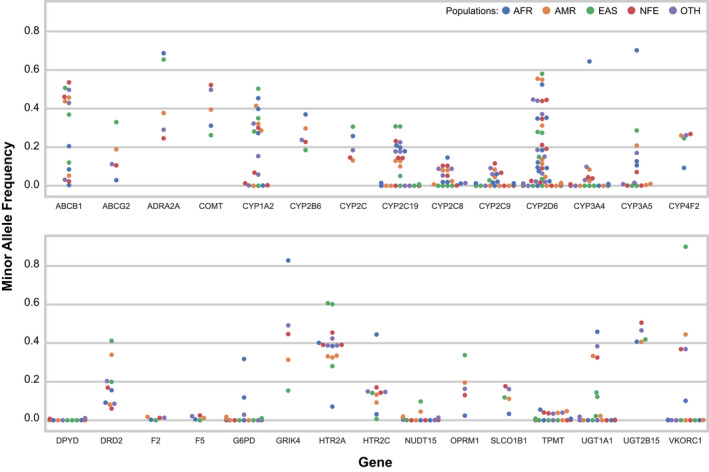
Multi‐ethnic minor allele frequencies of the pharmacogenetic panel sequence variants. Illustrated are the minor allele frequencies of the 106 (114 minor alleles) sequence variants in the pharmacogenetic panel with available data in the gnomAD 2.1.1 genomes dataset. Each dot represents the frequency of the pharmacogenetic minor allele in the corresponding gnomAD subpopulation (see legend for color coding). Minor alleles were designated as the nucleotide with the lowest mean allele frequency across four gnomAD (version 2.1.1) populations: AFR, AMR, EAS, and NFE. AFR: African; AMR: Admixed American; EAS: East Asian; NFE: non‐Finnish European; OTH: other.
DISCUSSION
The increasing enthusiasm for implementing pharmacogenetics into clinical practice, coupled with ongoing international research and clinical programs dedicated to pharmacogenetic discovery and implementation, 41 prompted our development and analytical validation of a 29 gene pharmacogenetic testing panel. Our panel is centered on multiplexed targeted genotyping, but also incorporates independent UGT1A1*28 dinucleotide repeat genotyping and copy number interrogation of specific genes and loci. The content and design were defined by a multidisciplinary team, which evaluated scientific literature and available knowledgebases to identify genes and variants implicated in interindividual drug response variability. Selected genes and variants subsequently were assessed for technical genotyping feasibility, which ultimately defined the comprehensive pharmacogenetic panel that was subjected to the analytical validation plan. The integrated platforms utilized by the panel, result integration and reporting, are typically completed in 7 days, but results can be reported in < 7 days by request.
Clinical test validation typically includes measuring assay accuracy and precision, and based on the results of > 500 tests performed on 170 independent samples, our novel pharmacogenetic genotyping panel was determined to be both accurate (sensitivity and specificity > 99%) and robust (reproducibility > 99%). Genotyping single nucleotide variants is generally considered technically feasible; however, homologous gene families and other nonunique genomic regions can be challenging to interrogate by genotyping and/or short‐read sequencing platforms. 42 The amplification chemistry and Agena assay design service enabled the accurate detection and validation of variants across homologous CYP450 genes in the pharmacogenetic panel; however, some of the originally selected variants were not amenable to the genotyping chemistry of the multiplexed panel (e.g., CYP2D6*21 (rs72549352), *59 (rs79292917), SLC6A4 rs25531, and rs4795541), including the direct interrogation of HLA‐A*31:01, HLA‐B*15:02, HLA‐B*57:01, and HLA‐B*58:01. Future iterations of the panel will likely incorporate long‐read sequencing approaches to facilitate full‐gene variant detection and haplotype phasing. 43
Our laboratory has previously reported on the value of integrating MLPA copy number with pharmacogenetic genotyping results, particularly for CYP2D6. 33 This strategy also led to our previous discoveries of loss‐of‐function structurally variant alleles in CYP2B6, 32 CYP2C19, and CYP2C9, 40 which are included in the pharmacogenetic panel. Notably, the MLPA platform includes 3–5 probes per targeted gene, and the signature of copy number results is critical for distinguishing CYP2D6 tandem and/or partial‐gene conversion alleles (e.g., *13, *36, and *68) from full‐gene deletions/duplications, which ultimately can influence subsequent phenotype prediction. 33 Our analytical MLPA validation specifically included CNV reference material with unique diplotypes, which were completely concordant after integrating the genotyping results with MLPA‐based copy number signatures.
Importantly, the frequencies of pharmacogenetic alleles can significantly differ between racial and ethnic groups, which can influence drug response variability between individuals and between populations. 44 , 45 Our pharmacogenetic panel was designed to detect both common and rare variants, with the goal of having utility for multi‐ethnic population testing. As has been previously reported by other implementation programs, 46 , 47 the frequency spectrum of our pharmacogenetic panel variants indicates that almost all individuals in the general population carry a clinically significant pharmacogenetic variant (Figure 2 and Figure S1).
Despite the ongoing advances in high‐throughput short‐read sequencing, including pharmacogenetic sequencing panels, 47 targeted genotyping is still a highly utilized and cost‐effective technology for pharmacogenetic testing by both research and clinical programs. 48 Limitations of genotyping and short‐read sequencing include the inability to directly phase variants and unambiguously detect haplotypes; however, star (*) allele haplotypes of relevant genes are inferred in our panel by leveraging the haplotype translation tables defined by PharmVar and PharmGKB. In the future, it is likely that pharmacogenetic testing will be embedded into larger genomic sequencing tests, but the inference of phased haplotypes will still be necessary. Long‐read sequencing platforms are inherently well‐positioned to directly address pharmacogenetic haplotype phasing 43 , 49 ; however, platform differences in cost, throughput, and informatics expertise suggest that long‐read pharmacogenetic sequencing may initially be most suitable for amplicon gene targets and/or smaller gene panels.
As noted above, the HLA‐A and HLA‐B genes have very high pharmacogenetic evidence, including available CPIC guidelines and FDA testing recommendations; however, they were not incorporated into our panel based on the technical challenges with interrogating the HLA region on chromosome 6p by short‐read genotyping chemistry. Other potential limitations of the panel include the omission of CACNA1S and RYR1 based on their variant density, as these genes are more suited to diagnostic full‐gene sequencing. However, the strengths of our panel include the robust validation of the sequence and CNVs incorporated into the test, the inclusion of low frequency variants and other alleles more common in under‐represented minority populations (including all AMP‐recommended tier 1 and 2 alleles 11 , 12 , 50 ), and the flexibility to efficiently run the panel for both low‐throughput and high‐throughput testing scenarios. Gene content of pharmacogenetic testing panels is a constantly evolving area, which is typically informed by the ongoing publications and communications released by professional practice agencies (e.g., ACMG, AMP, CPIC, DPWG, and the FDA). Our panel did not prioritize one of these resources over another, as some of the pharmacogenetic gene/drug pairs have inconsistent clinical practice recommendations, which ultimately is a decision for the clinical providers that pursue testing. However, the gene content of our pharmacogenetic panel covers the vast majority of genes previously reported in both conservative and progressive clinical implementation programs, 6 , 51 , 52 as well as other commercial pharmacogenetic panels. 53 Specific comparisons of commercial pharmacogenetic panel content is summarized in Table S13 ; however, given that these details can change over time it is recommended to verify these data from their direct sources if utilized in the future.
In conclusion, our validation results indicate that the 29 gene pharmacogenetic genotyping panel is accurate and reproducible. The content of the panel was driven by a multidisciplinary team, which is adjudicated on an ongoing basis. The intent of the gene content was to identify genes and variants that could provide clinical value, which includes the majority of CPIC level A and B genes. Notably, the flexibility of the comprehensive panel allows for the data to be parsed into user‐defined subpanels, including specific genes and/or variants (e.g., CYP2D6 and CYP2C19, TPMT and NUDT15), as well as clinical specialty subpanels (e.g., cardiovascular, pain, and psychiatry). Importantly, multi‐ethnic allele frequencies of the interrogated pharmacogenetic variants indicate that the vast majority of the general population carries at least one of these clinically relevant variants, supporting the implementation of this panel for pharmacogenetic research and/or clinical implementation programs.
Funding
This study was supported by Sema4, Stamford, CT.
Conflict of Interest
S.A.S., Y.S., A.J.C., R.W., N.C., H.S., G.Z., P.B., P.N., M.D., L.S., R.K., E.E.S., and L.E. are paid employees of Sema4. All other authors declared no competing interests for this work.
Author Contributions
S.A.S., E.R.S., Y.S., A.J.C., R.W., A.O.O., M.R.B., N.C., H.S., G.Z., P.B., P.N., Y.Y., M.D., L.S., R.K., E.E.S., and L.E. wrote the manuscript. S.A.S., E.R.S., Y.S., A.J.C., R.W., A.O.O., M.R.B., Y.Y., M.D., L.S., R.K., E.E.S., and L.E. designed the research. S.A.S., E.R.S., Y.S., A.J.C., R.W., A.O.O., M.R.B., N.C., H.S., G.Z., and L.S. performed the research. S.A.S., E.R.S., Y.S., M.R.B., N.C., H.S., G.Z., P.B., P.N., Y.Y., L.S., R.K., and L.E. analyzed data. E.R.S. and H.S. contributed new reagents/analytical tools.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The growing interest in implementing clinical pharmacogenetics is supported by a number of genetic testing resources, including the Pharmacogenomics Knowledgebase (PharmGKB), Pharmacogene Variation (PharmVar) Consortium, Clinical Pharmacogenetics Implementation Consortium (CPIC), Dutch Pharmacogenomics Working Group (DPWG), Association for Molecular Pathology (AMP), American College of Medical Genetics and Genomics (ACMG), Genetic Testing Reference Materials Coordination Program (GeT‐RM), and College of American Pathologists (CAP).
WHAT QUESTION DID THIS STUDY ADDRESS?
This study described and addressed the design, development, and analytical validation of a novel 29 gene pharmacogenetic genotyping panel.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study adds to current knowledge by detailing the integration of 3 molecular genetic testing platforms to interrogate 130 sequence variants and additional copy number variants implicated in interindividual drug response variability.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Allele frequency data indicate that the majority of the general population carries at least one of the variant alleles interrogated in this panel, supporting the implementation of this assay for pharmacogenetic research and/or clinical implementation programs.
Supporting information
Table S1–S13
Figure S1
Acknowledgments
The authors would like to thank Dr. Robin Everts at Agena Bioscience (San Diego, CA), Raymon Vijzelaar at MRC‐Holland (Amsterdam, The Netherlands), and Dr. Houda Hachad at Translational Software (Bellevue, WA) for technical and scientific consultation regarding the panel development.
References
- 1. Vogel, F. Moderne problem der humangenetik. Ergeb. Inn. Med. U. Kinderheilk. 12, 52–125 (1959). [Google Scholar]
- 2. Tornio, A. & Backman, J.T. Cytochrome P450 in pharmacogenetics: an update. Adv. Pharmacol. 83, 3–32 (2018). [DOI] [PubMed] [Google Scholar]
- 3. Alan R. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel‐treated patients. European Heart Journal ‐ Cardiovascular Pharmacotherapy. 2020; 6(4): 203–210. PMID: 31504375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicoletti, P. et al Shared genetic risk factors across carbamazepine‐induced hypersensitivity reactions. Clin. Pharmacol. Ther. 106, 1028–1036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Relling, M.V. et al New pharmacogenomics research network: an open community catalyzing research and translation in precision medicine. Clin. Pharmacol. Ther. 102, 897–902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rasmussen‐Torvik, L.J. et al Design and anticipated outcomes of the eMERGE‐PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin. Pharmacol. Ther. 96, 482–489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luzum, J.A. et al The pharmacogenomics research network translational pharmacogenetics program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin. Pharmacol. Ther. 102, 502–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Relling, M.V. et al The Clinical Pharmacogenetics Implementation Consortium: 10 years later. Clin. Pharmacol. Ther. 107, 171–175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Imatoh, T. , Sai, K. & Saito, Y. Pharmacogenomic information in the warning section of drug labels: a comparison between labels in the United States and those in five other countries/regions. J. Clin. Pharm. Ther. 43, 493–499 (2018). [DOI] [PubMed] [Google Scholar]
- 10. Lyon, E. et al Laboratory testing of CYP2D6 alleles in relation to tamoxifen therapy. Genetics Med. 14, 990–1000 (2012). [DOI] [PubMed] [Google Scholar]
- 11. Pratt, V.M. et al Recommendations for clinical CYP2C19 genotyping allele selection: a report of the association for molecular pathology. J. Mol. Diagn. 20, 269–276 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Pratt, V.M. et al Recommendations for clinical CYP2C9 genotyping allele selection: a joint recommendation of the association for molecular pathology and College of American Pathologists. J. Mol. Diagn. 21, 746–755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swen, J.J. et al Pharmacogenetics: from bench to byte‐ an update of guidelines. Clin. Pharmacol. Ther. 89, 662–673 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Scott, S.A. Personalizing medicine with clinical pharmacogenetics. Genetics Med. 13, 987–995 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hachad, H. , Ramsey, L.B. & Scott, S.A. Interpreting and implementing clinical pharmacogenetic tests: perspectives from service providers. Clin. Pharmacol. Ther. 106, 298–301 (2019). [DOI] [PubMed] [Google Scholar]
- 16. Hicks, J.K. et al A call for clear and consistent communications regarding the role of pharmacogenetics in antidepressant pharmacotherapy. Clin. Pharmacol. Ther. 107, 50–52 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalman, L.V. et al Pharmacogenetic allele nomenclature: International workgroup recommendations for test result reporting. Clin. Pharmacol. Ther. 99, 172–185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Owusu Obeng, A. et al Physician‐reported benefits and barriers to clinical implementation of genomic medicine: a multi‐site IGNITE‐network survey. J. Pers. Med. 8, 24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Wouden, C.H. , Bank, P.C.D. , Ozokcu, K. , Swen, J.J. & Guchelaar, H.J. Pharmacist‐initiated pre‐emptive pharmacogenetic panel testing with clinical decision support in primary care: record of PGx results and real‐world impact. Genes (Basel) 10, 416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verbelen, M. , Weale, M.E. & Lewis, C.M. Cost‐effectiveness of pharmacogenetic‐guided treatment: are we there yet? Pharmacogenomics J. 17, 395–402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanderson, S. et al How can the evaluation of genetic tests be enhanced? Lessons learned from the ACCE framework and evaluating genetic tests in the United Kingdom. Genetics Med. 7, 495–500 (2005). [DOI] [PubMed] [Google Scholar]
- 22. Barbarino, J.M. , Whirl‐Carrillo, M. , Altman, R.B. & Klein, T.E. PharmGKB: A worldwide resource for pharmacogenomic information. Wiley Interdiscip. Rev. Syst. Biol. Med. 10, e1417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nofziger, C. et al PharmVar GeneFocus: CYP2D6. Clin Pharmacol Ther. 107(1):154–170 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pratt, V.M. et al Characterization of 137 genomic DNA reference materials for 28 pharmacogenetic genes: a GeT‐RM collaborative project. J. Mol. Diagn. 18, 109–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu, A.H. Genotype and phenotype concordance for pharmacogenetic tests through proficiency survey testing. Arch. Pathol. Lab. Med. 137, 1232–1236 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Scott, S.A. , Edelmann, L. , Kornreich, R. & Desnick, R.J. Warfarin pharmacogenetics: CYP2C9 and VKORC1 genotypes predict different sensitivity and resistance frequencies in the Ashkenazi and Sephardi Jewish populations. Am. J. Hum. Genet. 82, 495–500 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scott, S.A. , Edelmann, L. , Kornreich, R. , Erazo, M. & Desnick, R.J. CYP2C9, CYP2C19 and CYP2D6 allele frequencies in the Ashkenazi Jewish population. Pharmacogenomics 8, 721–730 (2007). [DOI] [PubMed] [Google Scholar]
- 28. Scott, S.A. et al CYP2C9*8 is prevalent among African‐Americans: implications for pharmacogenetic dosing. Pharmacogenomics 10, 1243–1255 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scott, S.A. , Khasawneh, R. , Peter, I. , Kornreich, R. & Desnick, R.J. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics 11, 781–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scott, S.A. et al An allele‐specific PCR system for rapid detection and discrimination of the CYP2C19 *4A, *4B, and *17 alleles: implications for clopidogrel response testing. J. Mol. Diagn. 15, 783–789 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Barbarino, J.M. , Haidar, C.E. , Klein, T.E. & Altman, R.B. PharmGKB summary: very important pharmacogene information for UGT1A1. Pharmacogenet. Genomics 24, 177–183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martis, S. et al Multi‐ethnic cytochrome‐P450 copy number profiling: novel pharmacogenetic alleles and mechanism of copy number variation formation. Pharmacogenomics J. 13, 558–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qiao, W. et al Integrated CYP2D6 interrogation for multiethnic copy number and tandem allele detection. Pharmacogenomics 20, 9–20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jennings, L. , Van Deerlin, V.M. , Gulley, M.L. & College of American Pathologists Molecular Pathology Resource Committee . Recommended principles and practices for validating clinical molecular pathology tests. Arch. Pathol. Lab. Med. 133, 743–755 (2009). [DOI] [PubMed] [Google Scholar]
- 35. New York State Department of Health (NYSDOH) . Laboratory Standards: Clinical Laboratory Evaluation Program <https://www.wadsworth.org/regulatory/clep/clinical‐labs/laboratory‐standards>. Accessed May 15, 2020.
- 36. Whirl‐Carrillo, M. et al Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaedigk, A. et al The evolution of PharmVar. Clin. Pharmacol. Ther. 105, 29–32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scott, E.R. , Bansal, V. , Meacham, C. & Scott, S.A. VarCover: Allele Min‐Set Cover software. J. Mol. Diagn. 22, 123–131 (2020). [DOI] [PubMed] [Google Scholar]
- 39. Fang, H. et al Establishment of CYP2D6 reference samples by multiple validated genotyping platforms. Pharmacogenomics J. 14, 564–572 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Botton, M.R. et al Structural variation at the CYP2C locus: characterization of deletion and duplication alleles. Hum. Mutat. 40, e37–e51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scott, S.A. et al Institutional profile: translational pharmacogenomics at the Icahn School of Medicine at Mount Sinai. Pharmacogenomics 18, 1381–1386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mandelker, D. et al Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next‐generation sequencing. Genetics Med. 18, 1282–1289 (2016). [DOI] [PubMed] [Google Scholar]
- 43. Qiao, W. et al Long‐read single molecule real‐time full gene sequencing of cytochrome P450–2D6. Hum. Mutat. 37, 315–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalow, W. Ethnic differences in drug metabolism. Clin. Pharmacokinet. 7, 373–400 (1982). [DOI] [PubMed] [Google Scholar]
- 45. Kalow, W. & Bertilsson, L. Interethnic factors affecting drug response. Adv. Drug Res. 23, 1–53 (1994). [Google Scholar]
- 46. Chanfreau‐Coffinier, C. et al Projected prevalence of actionable pharmacogenetic variants and level A drugs prescribed among US Veterans health administration pharmacy users. JAMA Netw. Open 2, e195345 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bush, W.S. et al Genetic variation among 82 pharmacogenes: the PGRNseq data from the eMERGE network. Clin. Pharmacol. Ther. 100, 160–169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rasmussen‐Torvik, L.J. et al Concordance between Research Sequencing and Clinical Pharmacogenetic Genotyping in the eMERGE‐PGx Study. J. Mol. Diagn. 19, 561–566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liau, Y. et al Nanopore sequencing of the pharmacogene CYP2D6 allows simultaneous haplotyping and detection of duplications. Pharmacogenomics 20, 1033–1047 (2019). [DOI] [PubMed] [Google Scholar]
- 50. Pratt, V.M. et al Recommendations for clinical warfarin sensitivity genotyping allele selection: a report of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn. 22, 847–859 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dunnenberger, H.M. et al Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annu. Rev. Pharmacol. Toxicol. 55, 89–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van der Wouden, C.H. et al Development of the PGx‐Passport: a panel of actionable germline genetic variants for pre‐emptive pharmacogenetic testing. Clin. Pharmacol. Ther. 106, 866–873 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Precision Medicine Advisors .Pharmacogenomic Resources <https://www.precisionmedicineadvisors.com/pgx‐resources>. Accessed May 15, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1–S13
Figure S1