

Abstract
Enzyme replacement with ectonucleotide pyrophosphatase phospodiesterase‐1 (ENPP1) eliminates mortality in a murine model of the lethal calcification disorder generalized arterial calcification of infancy. We used protein engineering, glycan optimization, and a novel biomanufacturing platform to enhance potency by using a three‐prong strategy. First, we added new N‐glycans to ENPP1; second, we optimized pH‐dependent cellular recycling by protein engineering of the Fc neonatal receptor; finally, we used a two‐step process to improve sialylation by first producing ENPP1‐Fc in cells stably transfected with human α‐2,6‐sialyltransferase (ST6) and further enhanced terminal sialylation by supplementing production with 1,3,4‐O‐Bu3ManNAc. These steps sequentially increased the half‐life of the parent compound in rodents from 37 hours to ~ 67 hours with an added N‐glycan, to ~ 96 hours with optimized pH‐dependent Fc recycling, to ~ 204 hours when the therapeutic was produced in ST6‐overexpressing cells with 1,3,4‐O‐Bu3ManNAc supplementation. The alterations were demonstrated to increase drug potency by maintaining efficacious levels of plasma phosphoanhydride pyrophosphate in ENPP1‐deficient mice when the optimized biologic was administered at a 10‐fold lower mass dose less frequently than the parent compound—once every 10 days vs. 3 times a week. We believe these improvements represent a general strategy to rationally optimize protein therapeutics.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The accepted standard for frequency of blood based enzyme therapy is once a day. For example, hemophilia is treated with intravenous doses up to four times a day to control bleeding, whereas asfotase alpha is dosed six times a week subcutaneously.
WHAT QUESTION DID THIS STUDY ADDRESS?
We sought to determine the effect of a rational combinatorial strategy of optimization on drug potency, pharmacokinetics (PKs), and pharmacodynamics. We utilized a three‐prong approach, in which we rationally searched for beneficial N‐linked glycosylation, and combined these with enhancement of pH dependent recycling of the Fc domain and glyco‐polishing techniques implemented during bioproduction.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Our findings demonstrate that the PKs and potency of blood based enzyme therapeutics can be rationally optimized to enable of sub‐milligram/kg bimonthly dosing regimens delivered via subcutaneous injections. Such therapies are routinely used in enzyme replacement therapy (ERT) for metabolic disorders.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
By vastly improving PK and potency of ERT with the methods described, the cost of goods, patient compliance, and therapeutic efficacy of ERT can be greatly improved. We posit that if our results translate into humans, the methods described will shift the impetus for drug development in appropriate metabolic disorders away from gene therapy and toward protein replacement therapy.
The ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) enzyme family catalyzes phosphoryl‐transfer reactions on extracellular phosphoanhydrides. The products of these reactions, such as ATP, lysophosphatidic acid, and pyrophosphate, are essential extracellular signaling molecules that govern whole organismal fate through the regulation of essential biological functions, such as angiogenesis, 1 , 2 , 3 cell motility, 4 , 5 tumor metastasis, 6 , 7 , 8 , 9 , 10 bone mineralization, 11 , 12 , 13 , 14 vascular calcification, 15 , 16 , 17 and hemostasis. 18 Nonenzymatic phosphoryl‐transfer reactions that produce these critical signaling molecules have some of the slowest reaction rates known; as a result, living systems depend on tremendous acceleration from ENPPs that serves as a critical biologic catalyst. Quantitatively, ENPPs accelerate reaction rates by as much as 1027‐fold 19 and, as outlined below, a devasting impact on normal biological function occurs with ENPP deficits.
ENPP1, the first enzyme identified in this family, generates nucleoside 5′‐monophosphates and phosphoanhydride pyrophosphate (PPi) from extracellular nucleotide triphosphates. ENPP1 is the only human enzyme that synthesizes extracellular PPi with deficiencies resulting in potentially lethal disorders of ectopic calcification in infants to skeletal disorders of rickets and osteoporosis in children and adults. Levels of PPi in healthy individuals are ~ 2.0–2.2 μM, 20 compared with patients with heterozygous ENPP1 deficiency who exhibit half‐normal plasma PPi, 21 whereas homozygous ENPP1‐deficient patients exhibit nearly absent levels of plasma PPi typically below 250 nM. 15 , 16
Although ignored as clinical analytes, the physiologic effects of low plasma PPi and nucleoside 5′‐monophosphates are severe. PPi is a strong endogenous mineralization inhibitor, 22 and nucleoside 5′‐monophosphates have potent effects on developing vasculature. In a condition called generalized calcification of infancy (GACI), 16 , 23 homozygous ENPP1 deficiency induces vascular calcifications in the large arteries and heart, as well as significant arterial stenosis, due to deficiencies in plasma PPi 24 and nucleoside 5′‐monophosphates, 25 respectively. Arterial calcifications in GACI may be detected as early as the third trimester of pregnancy, and infants with GACI usually present in the first week of life “in crisis” whereupon they are transferred to the neonatal intensive care unit to receive supportive care. In some cases, patients are treated with bisphosphonate‐based drugs that ameliorate certain symptoms; 26 nevertheless, by 6 months of age, ~ 40–60% of affected patients die.
Patients surviving past 6 months undergo what appears to be an adaptive physiologic response by elevating plasma FGF23 and wasting phosphate, which results in disease stabilization. 26 Remarkably, in some patients, vascular calcification regresses or disappears 27 but this improvement occurs at the expense of bone formation. Survivors, however, are afflicted with a phosphate wasting rickets termed autosomal recessive hypophosphatemic rickets type‐2 (ARHR2), 13 , 14 a severe form of nonvitamin D dependent rickets that may induce skeletal deformities and growth retardation in children with and without a history of GACI. Patients with ARHR2 continue to calcify as they mature and frequently develop conductive hearing loss 28 , 29 and painful tendon and joint calcifications that limit mobility and adversely affect quality of life. 30 In some instances, potentially fatal recurrent cardiovascular calcifications occur in adolescents and young adults. 31
The clinical consequences of ENPP deficiency are severe and therapeutic options are limited because the bioactive products of this enzyme (PPi and nucleoside 5′‐monophosphates) are not bioavailable. We therefore developed an enzyme replacement strategy consisting of the extracellular domain of ENPP1 fused to the Fc domain of IgG1. 24 The murine isoform of ENPP1 fused to the murine Fc domain exhibited a biologic half‐life of ~ 6 hours and required daily subcutaneous doses of 8–1 g/kg to normalize plasma PPi, and induced a complete biochemical and clinical response in murine models of GACI. 24 The human isoform of ENPP1‐Fc, possessing 80.2% homology and 93.7% identity to murine ENPP1, exhibited a half‐life of 40 hours in mice and required 3 weekly doses of 2.5 mg/kg to normalize murine plasma PPi. For comparison, current blood‐based enzyme therapeutics in humans are dosed as frequently as four times a day in the case of recombinant factor VIII for hemophilia A, six times a week in the case of Asphotase alpha for hypophosphatasia, and as infrequently as once every four days in the case of recombinant antihemophilic factor VIII‐Fc (ELOCTATE) for bleeding prophylaxis in hemophilia A.
In this study, we further enhanced the potency of human ENPP1‐Fc through a series of glycosylation and protein engineering strategies combined with novel biomanufacturing methods, increasing the plasma half‐life of ENPP1‐Fc in mice by ~ 5.5‐fold (204 vs. 37 hours), and the area under the curve (AUC) by 13‐fold (45,000 vs. 3,400 measured as the optical density (mOD)/min hour) without compromising the enzymic activity of the fusion protein. The combination of methods resulted in a therapeutic, which normalized plasma PPi in mice at concentrations ≈ 10 times lower than the starting compound when dosed less frequently—once every 10 days vs. 3 times a week. The combination of techniques provides a template for the optimization of blood based human enzyme therapeutics.
METHODS
Mice
All animal procedures complied with the US National Institutes of Health guide for the care and use of laboratory animals and were approved by the Animal Care and Use Committee of Yale University. Animal care and maintenance were provided through Yale University Animal Resource Center. Pharmacokinetic analysis was performed on 6‐week‐old C57BL/6J male mice purchased from The Jackson Laboratory. Pharmacodynamic (PD) analysis was performed on Enpp1asj/asj mice (Jackson Laboratory stock number 012810). All animals were fed a regular chow diet and given access to food and water ad libitum.
Three‐dimensional structural modeling
A model of the tertiary structure of ENPP‐1 was based on murine ENPP1, PDB file 4GTW in the Protein Data Bank to derive the RaptorX predicted protein structure. 32 Protein structure visualization and assessment were carried out using PyMOL3 version 2.0.7.
Cloning and protein expression and purification
The protein sequence is listed in Figure S1 . Details on the cloning, expression, and purification are included in the Supplementary Methods.
Glycan analysis
Glycan analysis was performed at the Complex Carbohydrate Research Center (Athens, GA). Approximately 400 µg of each enzyme sample was dissolved in 50 µL digestion buffer (50 mM aq. NH4CO3), subjected to reduction (25 µL, 25 mM DTT, 45 minutes, 45°C), followed by alkylation (25 µL, 90 mM iodoacetamide, 45 minutes, RT) in the dark. The samples were then dialyzed against nanopure water using a 4kDa tube dialyzer overnight. Then N‐glycans were released by enzymatic cleavage with PNGase F at 37°C and purified with a C18 Sep‐Pak cartridge. The carbohydrate fraction was eluted with 5% acetic acid and dried by lyophilization. Released N‐linked oligosaccharides were permethylated by using methyl iodide in DMSO/NaOH, dried with nitrogen gas and profiled by matrix‐assisted laser desorption ionization‐time of flight analysis. After reduction/alkylation, an aliquot of each sample (20 µg) was treated with sequencing grade trypsin and analyzed by liquid chromatography tandem mass spectrometry. The glycopeptides were detected, assigned, and quantitated by search using Byonic software and manual methods.
For high‐performance anion‐exchange chromatography‐pulsed amperometric detection analysis samples were dissolved in water, and an aliquot (~ 160 µg) was allocated for neutral and amino sugars and a similar amount of aliquot was taken for sialic acid analysis. The aliquots for neutral and amino sugars were hydrolyzed with 2 M trifluoroacetic acid (4 hours at 100°C), whereas the aliquots for sialic acids were hydrolyzed with 2 M acetic acid (3 hours at 80°C). The monosaccharides were analyzed by high‐performance anion‐exchange chromatography‐pulsed amperometric detection using a Dionex ICS3000.
Enzyme kinetics
Cell media from transiently transfected Chinese hamster ovary (CHO) cells was analyzed for ENPP1 enzyme activity by diluting 10 µL of conditioned media into 90 µL of buffer containing 1 M Tris pH 8.0, 50 mM NaCl, 20 µM CaCl2, 20 µM ZnCl2, and 1 mM thymidine 5′‐monophosphate p‐nitrophenyl (TMP‐pNP) from Sigma‐Aldrich (cat #T4510). The mean velocity of the chromogenic product was measured in triplicate in the linear range using a Synergy Mx microplate reader with Gen5 software, and reported as change at absorbance units per minute (O.D./min) at 405 nm using Gen5 software.
Enzyme kinetics were measured at room temperature at 2 concentrations 50 ng and 100 ng in 100 μL of a buffer containing 250 mM Trizma pH 9.4, 500 mM NaCl, 0.05% Triton X‐100. The absorbance at 405 nM was recorded every 4 seconds immediately after the addition of 8 concentrations of the TMP‐pNP substrate (50, 100, 200, 300, 400, 500, 750, and 1,000 μM) and the mean initial velocity of change of absorbance in unit time was derived from the first 2 time points and TMP‐pNP concentration dependence of the initial velocity was used to calculate the maximal rate of metabolism (Vmax) and kinetic metabolite (Km) using the equation Vinit = Vmax*[TMP‐pNP]/(Km + [TMP‐pNP]). The data was analyzed with GraphPad Prism 8.
Pharmacokinetics and pharmacodynamics
Six‐week‐old C57BL/6J male mice were injected with a single subcutaneous dose of purified enzyme and blood was collected up to four times retro‐orbitally at various time points. The blood was centrifuged at 900 g for 10 minutes from which the plasma was transferred to a new tube and frozen. We estimated the total drug exposure, expressed AUC, as the integral of the enzyme activity vs. time curve, and not protein concentration vs. time curve, because enzyme therapeutics are conventionally quantitated and dosed as activity units and not concentrations to account for batch to batch variations in enzyme preparations. Enzyme activity units are therefore viewed as more precise measures of drug exposure than protein concentrations. Enzyme activity was quantitated as the initial enzyme velocity in murine plasma, measured as the optical density change at 405 nm per unit time (mOD min‐1) due to hydrolysis of the colorimetric substrate TMP‐pNP by the enzyme. We quantitated activity units by diluting 2 µL of murine plasma into 98 µL of TMP buffer (1 M Tris pH=8.0, 50 mm NaCl, 20 μm CaCl2, 20 μM ZnCl2, 1mM TMP‐pNP), and recording changes in optical density per minute ((O.D./min) × 10‐3, or mO.D./min) at 405 nm.
For half‐life calculations, the velocity values were converted to percent activity and plotted in GraphPad Prism 8. Half‐life of enzyme activity was calculated from its correspondant pharmacokinetic (PK) parameters elimination () and absorption () rate constants, which were determined by plotting the fractional enzymatic activity (F) of ENPP1‐Fc as function of time and then by fitting the experimental curves to Eq. 1:
| (1) |
Equation 1 Relationship between systemic fractional enzyme activity in serum and time of a drug administered to a subcutaneous depot in a single injection.
The elimination half‐life is then determined from half‐life = ln(2)/k e. The relationship between AUC and the elimination rate constant () can best be understood by Eq. 2:
| (2) |
Equation 2: Relationship between AUC, , and peak plasma concentration (Cmax).
Mice of the same age and weight were used for all PK experiments, and volume of distribution was assumed to be constant between the animals. Blood samples were collected at 4 time points that initially were between 12 and 75 hours postinjection; later, with PK improvements, time points were taken between 21 and 263 hours postinjection. The initial fractional activity from the first bleed was assigned a fractional activity of 0.9. Mice were also immunosuppressed with an i.p. injection of anti‐CD4 clone Gk1.5 24 hours before s.c. injection of enzyme to mitigate any negative immune responses mounted against the longer lasting therapeutic. Plasma PPi was measured in murine plasma as previously described. 33 The data were analyzed and visualized with GraphPad Prism 8.
RESULTS
Design criteria for addition of new N‐glycans to ENPP1‐Fc
Based on evidence that therapeutic proteins benefit from the addition of N‐glycans (e.g., insulin 34 and erythropoietin 35 ), we applied this strategy to ENPP1‐Fc. Although the general principle of adding an N‐glycan to a therapeutic protein is established, there are no established guidelines to add the glycan for maximal benefit. Nevertheless, we used a semirational approach, which first required knowledge of existing glycans. We began by noting that the crystal structure of murine Enpp1 exhibits four glycans, 36 which we reasoned were also present in (highly homologous) human ENPP1. Human ENPP1 has six N‐glycan consensus sequences (N‐GCS; Figure S2 a), which we incorporated into structural models with genetic data from patients with GACI to help predict beneficial glycovariants. Specifically, we created structural models integrating putative glycosylation sites with the locations of ENPP1 loss of function mutations present in patients with GACI (Figure S2 b). We then used the protein sequence database to identify N‐GCS in ENPP family members (ENPP2‐ENPP7) from all mammalian species with available protein sequences, and modeled these sites onto mouse Enpp1 (PDB 4GTW). Using these tools, we established a list of potential N‐GCS sites by avoiding sites near inactivating ENPP1 mutations and sites that would disrupt disulfide crosslinking.
Referencing the list, we introduced additional N‐GCS (consisting of Asn‐X‐Ser/Thr where X is any amino acid except proline) into the parent isoform (hENPP1‐Fc, construct #770). We specifically targeted disordered regions on the exterior surface of ENPP1‐Fc that permitted the introduction of an N‐GCS through a single amino acid substitution. If the location of a proposed N‐GCS were near inactivating mutations in patiens with GACI or interfered with the disulfide crosslinking, the modifications were not pursued. By using these criteria, we selected 31 possible sites to add N‐glycans, attempting to individually sample the entire surface area of ENPP1 (Figure 1a and Table S1 ). Each of these 31 N‐GCS were introduced individually, or in combination, into the parent ENPP1‐Fc (construct #770) via site directed mutagenesis, resulting in a final pool of 53 ENPP1‐Fc glycoforms (Table S1 ). Protein production and hydrolytic velocity of the pNP‐TMP colorimetric substrate (Figure S3 , green bars) was measured in each isoform after transient expression in CHO cells. The nine most promising N‐GCS isoforms were then established as stably selected CHO K1 cell clones, and the glycoforms were individually expressed and purified to homogeneity.
Figure 1.
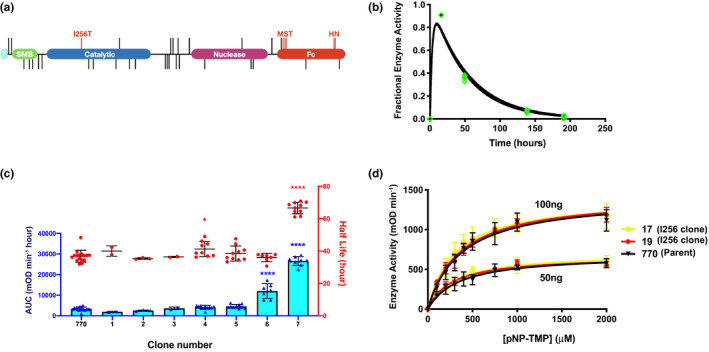
Pharmacokinetic effects of additional N‐Glycans. (a) Domain structure of the parent clone drawn to scale with the position of all mutations indicated by vertical lines. The two tandem somatomedin B domains (green), catalytic domain (blue) and endonuclease domain (purple), of human ENPP1 was fused N‐terminally with the signal sequence of human ENPP7 (cyan) and c‐terminally with the Fc domain of human IgG1 (red). The Fc domain contains two sets of mutations; M242Y/S254T/T246E (MST) and H433K/N434F (HN), which were introduced to enhance Fc neonatal receptor (FcRn) mediated endosomal recycling. All other lines indicate attempts to introduce new N‐glycosylation sequons (N‐GCS) into the protein. Sequons that reduced enzymatic activity in vitro are positioned below the domain structure whereas those that maintained activity are positioned above. Only the I256T mutation, which creates a new glycan at position Asn254, enhanced pharmacokinetic (PK) properties of the protein in vivo. (b) PK analysis of the parent clone 770 tested in 5 animals fitted to curve described in Eq. 1. The fractional enzyme activity, corresponding to the fractional activity compared with peak plasma concentration (Cmax), was sampled in each mouse at 4 separate times over 200 hours (individual data points overlaid in green). (c) PK effects of additional N‐GCS engineered into the parent clone (770). Area under the curve (AUC, left y axis) in blue and half‐life (right y‐axis) overlayed in red. Error bars represent SD of the mean. Blue and red asterisks represent significance of AUC and half‐life measurement, respectively, compared with clone 770. *P < 0.05, ****P < 0.0001, analysis of variance (ANOVA) comparison of means. (d) Steady‐state Michaelis–Menten kinetic assays performed at two enzyme concentrations comparing 770 (in black) with clones possessing the I256T mutation (clones 17 in yellow and 19 in red). Each point represents the average of six measurements of each enzyme at each concentration. Error bars denote SDs of the mean. mOD, measured as the optical density; TMP‐pNP, thymidine 5′‐monophosphate p‐nitrophenyl.
PK effects of adding N‐glycans to ENPP1‐Fc
The half‐life and AUC of stably expressed ENPP1‐Fc glycoforms were tested in C57BL6 mice using a single subcutaneous injection, with doses varying from 5 mg/kg for the early constructs to 0.3 mg/kg for the latter more potent constructs. The parent ENPP1‐Fc (clone 770) yielding a half‐life of 37 hours and an AUC of 3,382 mOD/min hour (Table 1 , Figure 1b ); the effect of adding an additional N‐GCS was modest with the notable exception of the I256T mutation, designed to add an N‐glycan at asparagine 254 in the catalytic domain near the active site. This N‐glycan, introduced because it is present in human ENPP3, increased the AUC of ENPP1‐Fc by ~ 8‐fold and half‐life by a factor of 1.8 (construct 7, Table 1 and Figure 1c ).
Table 1.
Pharmacokinetic effects of added N‐GCS
| Construct | Signal sequence | Catalytic domain | Nuclease domain | Linker 2 | Half‐life, hours | AUC, mOD/min hour |
|---|---|---|---|---|---|---|
| 770 | 37 | 3,382 | ||||
| 1 | E592N | 40 | 1,935 | |||
| 2 | K369N/I371T | 35 | 2,561 | |||
| 3 | C25N K27T | 36 | 3,636 | |||
| 4 | V29N | E864N L866T | 41 | 4,134 | ||
| 5 | C25N K27T | S766N | 38 | 4,537 | ||
| 6 | C25N K27T |
E864N L866T |
36 | 11,997 | ||
| 7 | I256T | 66 | 26,596 |
The color shades correspond to the domain coloring of the ENPP1 cartoon presented in Figure 1a, i.e., the listed mutations in the table are colored according to their domain location in the cartoon in figure 1a.
AUC, area under the curve; mOD, measured as the optical density; N‐GCS, N‐glycan consensus sequences.
To determine whether the ~ 8‐fold increase in AUC resulted from an increased half‐life or a gain in catalytic activity, we compared the Michaelis–Menten kinetic constants of the parent ENPP1‐Fc with two I256T containing constructs (clone 17 and clone 19) at two different concentrations, and found no significant differences in the Km or Kcat (Figure 1d ). We then confirmed that the mutation resulted in the predicted glycan at Asn254 using mass spectrometry, by identifying that the I256T clone exhibited increased sialyl glycopeptide peaks in the digested peptide fragment 241SGTFFWPGSDVEINGTFPDIYK262 compared with the parent ENPP1‐Fc (Figure S4 ). As demonstrated in Figure 2b , the increase in AUC in the I256T clone appears to primarily result from an increased Cmax due to the presence of an additional glycosylation.
Figure 2.
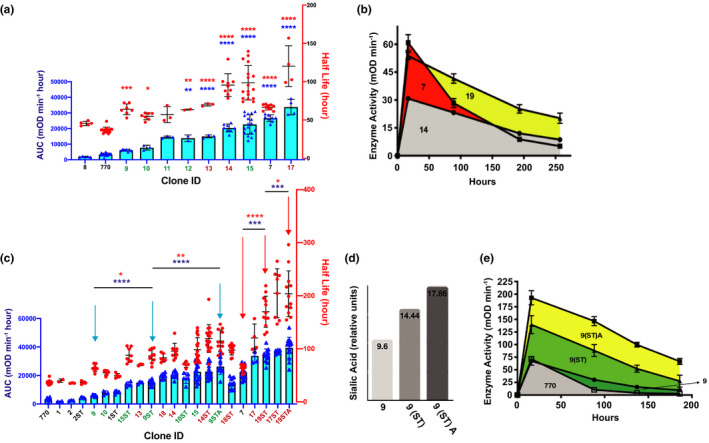
Pharmacokinetic (PK) effects of combining N‐glycans, Fc mutations, and glyco‐polishing. (a) PK effects of Fc mutations—overlays of area under the curve (AUC; left y axis, blue symbols) and half‐life (right y‐axis, red symbols). Clones containing the FcH433K/N434F (HN) mutation are in green font and clones containing the Fc‐M242Y/S254T/T246E (MST) mutation are in ox‐blood font. 13 , 14 , 17 , 18 , 19 Blue and red asterisks represent significance of AUC and half‐life measurements, respectively. (b) Activity vs. time plots of clones 14, 7, and 19 used to derive AUC data. Animals were dosed subcutaneously with 1 mg/kg of each construct. Data points represent means and SDs of individual data displayed in a and c. Increased drug absorption from the subcutaneous depot is reflected by the increased maximal enzyme activity (peak plasma concentration (Cmax)) in construct 7 and 19 compared with construct 14. Construct 7 and 19 possess the I256T glycosylation, unlike construct 14. (c) PK effects of glycopolishing represented by overlays of AUC (left y axis, blue symbols) and half‐life (right y‐axis, red symbols). PK constants of glycoforms expressed in unmodified Chinese hamster ovary (CHO) cells—clone numbers only—are compared with those expressed in CHO cells stably transfected with α‐2,6‐sialytransferase (α‐2,6‐ST) with or without 1,3,4‐O‐Bu3ManNAc supplementation—clone numbers followed by ST or STA, respectively. To avoid clutter, statistical significance is only denoted in cohorts marked by arrows to highlight two examples of progressive improvements in PK via glycopolishing discussed in text. (d) Anion‐exchange chromatography with pulsed amperometric detection (HPAE‐PAD) of clone 9 grown in CHO K1 cells alone or stably transfected with human α‐2,6‐ST or a combination of α‐2,6‐ST and the sialic acid precursor 1,3,4‐O‐Bu3ManNAc shows a progressive increase in the percentage of sialic acid content with each treatment. (e) Activity vs. time plots comparing clone 770 with glycopolished forms of clone 9 to demonstrate the progressive effects of glycopolishing on Cmax and AUC. Animals were dosed subcutaneously with 5 mg/kg of each construct. Data points represent the means and SD of individual data displayed in c. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001, ANOVA comparison of means. mOD, measured as the optical density.
PK effects of optimizing pH‐dependent FcRn recycling
Mutations in the Fc domain of therapeutic antibodies are known that enhance the pH dependent interactions of Fc with Fc neonatal receptor (FcRn) and thereby extend circulatory half‐life. 37 We examined the effect of two sets of such Fc mutations—H433K/N434F (i.e., “HN” mutations) and M242Y/S254T/T246E (“MST” mutations, Figure S5 ). The MST‐induced changes in the pH dependent affinity of the Fc domain for murine, monkey, and human FcRns are well‐documented in the literature. 37 , 38 , 39 Both Fc variants were combined with N‐GCS isoforms to test the combined effect of hyperglycosylation and enhanced FcRn recycling. In general, the Fc MST mutations increased AUC to a greater degree than the Fc HN mutations (Table 2 , Figure 2a ). For example, adding the MST mutations to parent ENPP1‐Fc (construct 14) increased AUC by ~ 6‐fold and half‐life by ~ 2.6‐fold, in comparison to the HN mutations (constructs 9–12), which increased AUC ≤ 4.1‐fold in the presence of additional glycans (Table 2 and Figure 2a ). However, in some cases, specific N‐GCS decreased AUC when combined with the Fc mutations (i.e., the combination of an N‐glycan at residue 766 with either Fc mutation was detrimental; MST in construct 8 vs. construct 14 and HN in construct 9 vs. construct 11; Table 2 and Figure 2a ). Reproducibility was assessed by establishing two independent CHO cell clones producing the same ENPP1‐Fc glycoform (clones 11 and 12), and yielded no statistical difference in AUC or half‐life (Table 2 ). Finally, the improvement in AUC achieved by optimizing FcRn recycling was less than that achieved by adding an N‐glycan at residue 254 (clone 7 vs. clone 14; Figure 2a ). Drug clearance, demonstrated in the activity vs. time plots, shows that Fc optimization enhanced PK by increasing half‐life (Figure 2b ), whereas the N254 N‐glycan enhanced AUC by increasing plasma drug absorption (i.e., increasing Cmax; greater maximal activity of clone 7 in Figure 2b ). Finally, combining the MST Fc mutation with the N254 N‐glycan increased both half‐life and Cmax, as demonstrated by clone 19 in Figure 2b .
Table 2.
Pharmacokinetic effects combining N‐GCS and Fc mutations
| Construct | Signal sequence | Catalytic domain | Nuclease domain | Fc domain | Half‐life, hours | AUC, mOD/min hour |
|---|---|---|---|---|---|---|
| 8 | S766N | M883Y S885T T887E | 45 | 1,912 | ||
| 770 | 37 | 3,382 | ||||
| 9 | V29N | S766N | H1064K N1065F | 65 | 6,047 | |
| 10 | C25N K27T | S766N | H1064K N1065F | 55 | 7,735 | |
| 11 | V29N | H1064K N1065F | 57 | 14,506 | ||
| 12 | V29N | H1064K N1065F | 63 | 13,812 | ||
| 13 | V29N | E592N | M883Y S885T T887E | 70 | 14,978 | |
| 14 | M883Y S885T T887E | 95 | 20,360 | |||
| 15 | V29N | E592N | H1064K N1065F | 99 | 22,690 | |
| 7 | I256T | 66 | 26,596 | |||
| 17 | V29N | I256T | P534N V536T | M883Y S885T T887E | 120 | 33,751 |
The color shades correspond to the domain coloring of the ENPP1 cartoon presented in Figure 1a, i.e., the listed mutations in the table are colored according to their domain location in the cartoon in figure 1a.
AUC, area under the curve; mOD, measured as the optical density; N‐GCS, N‐glycan consensus sequences.
PK effects of “humanized” α‐2,6‐sialyation
The CHO cells are used for biomanufacturing protein therapeutics due to glycosylation patterns, which are similar to human and therefore avoid safety concerns (for example, CHO cells do not produce the immunogenic alpha‐Gal epitope 40 , 41 ). Differences between human and hamster glycosylation do exist, however, which may compromise efficacy. Specifically, CHO cells used in biomanufacturing lack α‐2,6‐linked sialic acids, which may increase protein stability and half‐life. 41 , 42 To test this hypothesis, we established a CHO cell line stably expressing human ß‐galactoside α‐2,6‐sialytransferase (α‐2,6‐ST; Figure S6 ) and evaluated ENPP1‐Fc glycoforms produced in these cells. We found that production of ENPP1‐Fc in these cells improved half‐life and AUC of glycoforms; for example, expressing clones 9 and 10 in α‐2,6‐ST cells increased AUC 1.3‐fold and 1.6‐fold, respectively (Table 3 and Figure 2c ).
Table 3.
Pharmacokinetic effects of increased and α‐2,6‐siaylation
| Construct | Signal sequence | Catalytic domain | Nuclease domain | Fc domain | Half‐life, hours | AUC, mOD/min hour |
|---|---|---|---|---|---|---|
| 770 | 37 | 3,381 | ||||
| 1 | E592N | 40 | 1,935 | |||
| 2 | K369N/I371T | 35 | 2,561 | |||
| 2(ST) | K369N/I371T | 36 | 4,426 | |||
| 9 | V29N | S766N | H1064K N1065F | 65 | 6,047 | |
| 10 | C25N K27T | S766N | H1064K N1065F | 55 | 7,735 | |
| 1(ST) | E592N | 49 | 8,379 | |||
| 15(ST) | V29N | E592N | H1064K N1065F | 88 | 13,871 | |
| 13 | V29N | E592N | M883Y S885T T887E | 70 | 14,978 | |
| 9(ST) | V29N | S766N | H1064K N1065F | 86 | 15,099 | |
| 18 | E592N | M883Y S885T T887E | 83 | 19,638 | ||
| 14 | M883Y S885T T887E | 96 | 20,360 | |||
| 10(ST) | C25N K27T | S766N | H1064K N1065F | 70.1 | 18,207 | |
| 15 | V29N | E592N | H1064K N1065F | 99 | 22,620 | |
| 14(ST) | M883Y S885T T887E | 119 | 22,793 | |||
| 9(ST) A | V29N | S766N | H1064K N1065F | 115 | 26,312 | |
| 18(ST) | E592N | M883Y S885T T887E | 97.5 | 14,263 | ||
| 7 | I256T | 67 | 26,598 | |||
| 17 | V29N | I256T | P534N, V536T | M883Y S885T T887E | 120 | 33,752 |
| 19(ST) | I256T | M883Y S885T T887E | 170 | 35,252 | ||
| 17(ST) | V29N | I256T | P534N, V536T | M883Y S885T T887E | 205 | 36,595 |
| 19(ST) A | I256T | M883Y S885T T887E | 204 | 44,742 |
The color shades correspond to the domain coloring of the ENPP1 cartoon presented in Figure 1a, i.e., the listed mutations in the table are colored according to their domain location in the cartoon in figure 1a.
AUC, area under the curve; mOD, measured as the optical density.
PK effects of increased flux‐based sialylation from 1,3,4‐O‐Bu3ManNAc
Benefits derived from producing ENPP1 in α‐2,6‐ST overexpressing CHO cells include a gain in human‐like α‐2,6‐linked sialic acids and an overall increase in sialylation due to the combined effect of a‐2,3‐sialic and a‐2,6‐sialic acids, which improves serum half‐life by concealing circulating proteins from asialoglycoprotein receptors. 41 , 43 To exploit the benefits of increased sialylation, we supplemented the production cells with 1,3,4‐O‐Bu3ManNAc, a “high‐flux” metabolic precursor that supplies flux into the sialic acid biosynthetic pathway and increases glycoconjugate sialylation. 44 , 45 This metabolite provided added benefit when incorporated into our biomanufacturing platform, as illustrated by clone 9 (Figure 2c ). This ENPP1‐Fc glycoform exhibited modestly increased biologic effects when produced in standard CHO cells; AUC increased by 2.5‐fold when expressed in CHO cells overexpressing α‐2,6‐ST, but expression of the clone in cell culture media with 1,3,4‐O‐Bu3ManNAc increased AUC an additional ~ 1.7‐fold for a cumulative benefit of 4.3‐fold in AUC (clones 9, 9(ST), and 9(ST)A, respectively; Table 3 and Figure 2e ). Finally, expressing clone 19(ST) in media containing 1,3,4‐O‐Bu3ManNAc yielded the best performing ENPP1‐Fc glycoform, increasing AUC by ~ 13‐fold above baseline (clones 770 vs. 19(ST)A, Figure 3 a,b). Mass spectrometry analysis confirmed that sialic acid content was increased in 19(ST)A (Figure 3c ).
Figure 3.
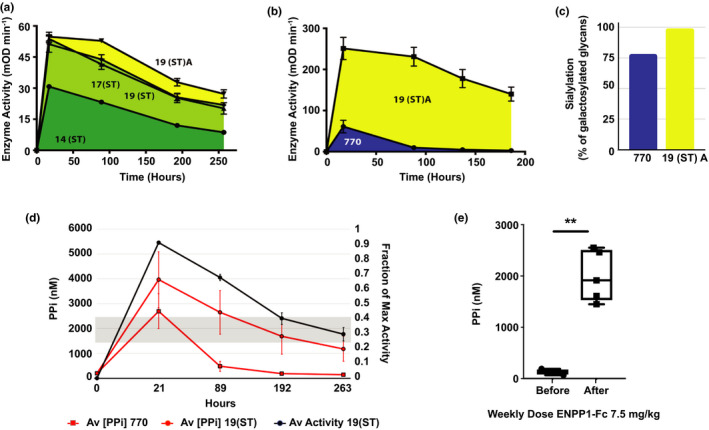
Pharmacokinetic and pharmacodynamic effects of optimized ectonucleotide pyrophosphatase phospodiesterase‐1 (ENPP1‐Fc). (a) Activity vs. time plots comparing Fc mutated clones without (clone 14(ST)) and with the I256T mutation and further glycopolishing (clones 17(ST), 19(ST), and 19(ST)A). Animals were dosed subcutaneously with 1 mg/kg of each construct. Data points represent the means and SDs of data individually displayed in Figure 2c . The area under the curve (AUC) for Fc‐M242Y/S254T/T246E (MST) containing clones (shaded area) was enhanced by the I256T mutation and further increased by 1,3,4‐O‐Bu3ManNAc supplementation. (b) Activity vs. time plots to highlight the AUC of the parent clone (770) with the final optimized glycoform (clone 19(ST)A), demonstrating a nearly 18‐fold increase in AUC in the final product. Animals were dosed subcutaneously with 5 mg/kg of each construct. Data points represent the means and SDs of data individually shown in Figure 2c . (c) Matrix‐assisted laser desorption ionization‐tandem time of flight (MALDI‐TOF/TOF) analysis for N‐glycan profiling revealed that the percentage of glycans containing sialic acid is higher in clone 19(ST)A (99.2%) compared with parent clone 770 (78.4%) when calculated based on the structures that contains at least one galactose for transfer of sialic acid. (d) The pharmacodynamic effect after a single dose of 0.3 mg/kg of either the parent clone 770 (red squares) or the optimized ENPP1‐Fc clone 19(ST) (red circles), as measured by plasma (phosphoanhydride pyrophosphate (PPi)) (left y‐axis) in Enpp1asj/asj mice. Physiological levels of PPi in normal mice (shaded grey) is between 1.5 and 2.5 μM PPi while Enpp1asj/asj mice have nearly undetectable amounts. A single dose of clone 770 restores physiological levels of PPi that return to baseline in < 89 hours whereas clone 19(ST) maintained or exceeded physiological levels for 263 hours. Data points represent the mean and SD of five animals. (e) Plasma [PPi] in Enpp1asj/asj mice before and after dosing animals with 7.5 mg/kg per week with clone 770 in 3 equally divided doses (on Monday, Wednesday, and Friday). Box and violin plots represent individual data points with minimum to maximum distribution. **P < 0.01, Student’s paired t‐test.
In vivo confirmation of the disease‐reversing ability of bio molecularly engineered ENPP1‐Fc
The results presented above demonstrate that by evaluating a modestly sized panel of glycosylation‐engineered and protein‐engineered variants of ENPP1‐Fc and by further producing these nascent therapeutics using a novel biomanufacturing platform that ensures a high level of “humanized” sialylation, we were able to identify specific candidates with substantially improved PK properties. To close this study, we confirmed that the newly engineered forms of ENPP1‐Fc retained disease‐reversing activity in vivo by monitoring PPi in plasma, which is a biomarker for clinical efficacy of ENPP1 enzyme replacement therapy. 16 To compare the PD effects of the above alterations, we dosed Enpp1asj/asj mice with a single subcutaneous dose of 770 and 19(ST) and measured plasma PPi and enzyme activity in plasma for 11 days (Figure 3d ). Plasma PPi in mice dosed with parent ENPP1‐Fc (clone 770) required a weekly dose of 7.5 mg/kg to maintain plasma PPi in the normal range (Figure 3e ), whereas a single dose of 0.3 mg/kg of 19(ST) elevated plasma PPi at or above the normal range for ~ 250 hours (Figure 3d ), representing a PK gain of ~ 37‐fold. Figure 3d also demonstrates that plasma PPi was more variable than plasma enzyme concentration, suggesting a physiological tug‐of‐war between the PPi generating ENPP1‐Fc and the PPi degrading enzyme ALPL, which may elevate when plasma PPi levels are too high.
DISCUSSION
In this study, we sequentially applied strategies to optimize the pharmacologic and PD properties of a therapeutic enzyme designed to treat ENPP1 deficiency while maintaining full catalytic activity. Our approach differed from previous biologic optimization studies in two ways. First, whereas the various strategies (e.g., Fc fusion proteins and mutations, building in N‐glycosylation sites, and using downstream biomanufacturing strategies to increase sialylation) have been used individually in the past, they have not been combined to aggregate benefit, and second, glycan engineering and glycopolishing have never been used to optimize enzyme therapeutics. Our studies demonstrate the significant benefit resulting from sequentially combining these complementary strategies in enzyme biologics.
In the current work, our first attempts to improve ENPP1‐Fc were inspired by a growing number of reports where addition of N‐glycans to therapeutic proteins (so‐called “hyper‐glycosylation”) improved their effectiveness. Erythropoietin (EPO) provides a groundbreaking example; specifically, Aranesp (darbepoetin alfa) is a glycoengineered form of human recombinant EPO where two N‐glycans were added to EPO’s three endogenous N‐glycans, resulting in an approximately threefold enhancement of serum half‐life. 35 , 46 In examples that have not yet reached the clinic, a non‐canonical N‐glycan improves the neutralizing action of an HIV‐targeting antibody 47 and N‐glycans added to insulin (a normally unglycosylated therapeutic protein) improve its function. 34
Our efforts to similarly glycoengineer ENPP1‐Fc by adding one or more N‐GCS yielded negligible benefit with one notable exception. Specifically, we identified a single beneficial N‐GCS that provided substantially greater improvement than any of other techniques we used singly—the I256T mutation, which introduced a glycosylation in residue 254. This N‐GCS was found by referencing other ENPP family members and avoiding areas of ENPP1 inactivating mutations present in GACI patients. These strategies allowed us to prioritize several dozen N‐GCS sites for high throughput screening, which identified N‐GCS sites in every ENPP1 protein domain designed to shield the ENPP1 protein surface. We found that the introduction of N‐GCS reduced either the protein expression or the catalytic activity of most of these ENPP1 glycoforms with the substantial benefit only observed in one instance, the I256T mutation in clone 7. Interestingly, this N‐GCS was an outlier not designed to cover the surface of the ENPP1 protein, instead the glycan is present in the ENPP3 insertion loop near the catalytic residue responsible for the nucleophilic attack of the catalyst on the substrate. The introduction of this site into ENPP1 increased AUC by approximately eightfold, primarily by increasing Cmax after subcutaneous dosing, presumably by enhancing the absorbance of the subcutaneous bolus into the blood (Figure c 1 and Figure 2b ).
In our second strategy, based on protein engineering, we optimized FcRn recycling of the Fc domain; in contrast to the glycoengineering strategy, this method enhanced PK by increasing serum half‐life with little effect on Cmax. Quantitatively comparing the two techniques, adding a glycan at position 254 had a greater impact than Fc optimization, increasing AUC by ~ 8‐fold compared with an ~ 6‐fold increase from MST Fc mutations (clones 7 and 14 vs. 770; Figure 2a ). Using both techniques in combination increased AUC 10‐fold above the parent 770.
Glyco‐engineering and protein‐engineering represented by clone 17, usually applied separately unlike our combined approach, constitute the current limits of “upstream” efforts to improve therapeutic proteins. In this study, we reasoned that additional benefits could be obtained by implementing downstream biomanufacturing advances. We experimentally demonstrated this premise by expressing ENPP1‐Fc with the combined I256T and MST mutations in α‐2,6‐ST overexpressing CHO cells and growing the cultures in the sialic acid precursor 1,3,4‐O‐Bu3ManNAc; these 2 steps increased the AUC an additional 3‐fold, resulting in a biologic with an ~ 13‐fold increase over the parent biologic (Clone 19(ST) A vs. 770; Figure 3b ), demonstrating the importance of glyco‐polishing for improving PK properties. The PD effect of these changes was substantial; the increase in potency of the optimized therapeutic, as judged by normalization of plasma PPi, was ~ 37‐fold (i.e., whereas construct 770 required 7.5 mg/kg to normalize plasma PPi for 7 days, 0.3 mg/kg of construct 19(ST) was able to normalize plasma PPi for 10.4 days). The overall strategy with a step‐by‐step illustration in PK improvements is provided in Figure 4 .
Figure 4.
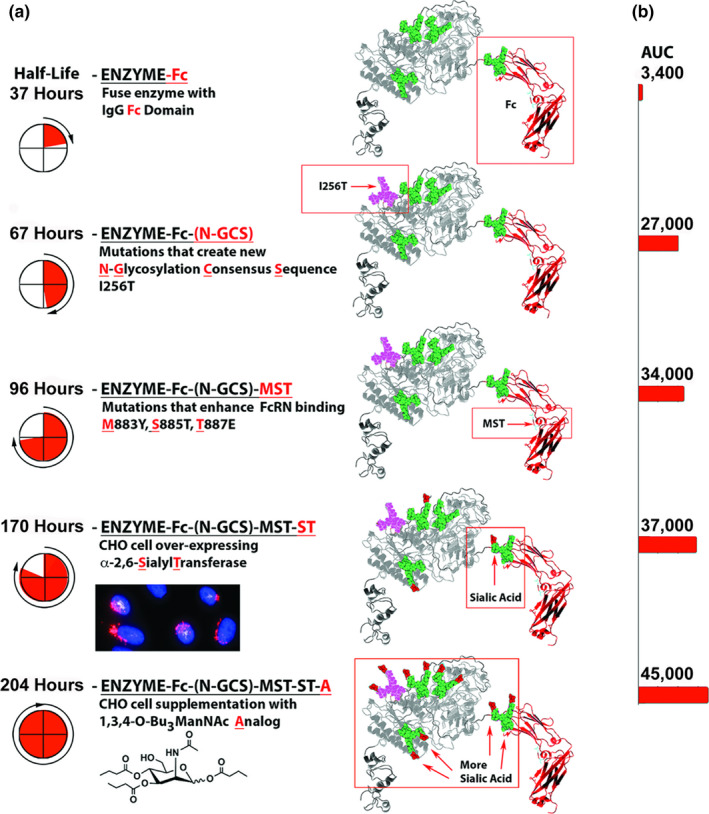
Summary of the protein engineering steps. (a) Half‐life and (b) area under the curve (AUC).
Cost of goods, dosing frequency, and complications associated with immune reactions present barriers to the development of enzyme replacement therapy, and reducing dose levels and frequencies address critical commercial and clinical barriers to the development of these products. Our studies demonstrate that integrating protein design with techniques that enhance biologic PK enables the rational optimization of biologic enzymes to overcome these problems. Although the results must be reproduced in primates to determine their applicability to humans, we note that the murine half‐life of our best construct is comparable to the primate half‐life of a biologic, which was predictive of a bimonthly dose frequency. 48 We also note that interspecies scaling and prediction of drug clearance in humans is well predicted by allometric scaling alone. 49 , 50 Moreover, the clearance rates of macro‐molecules with MW> 69 kDa tend to be significantly lower in humans than in lower order mammals, such as rodents, 51 suggesting that bimonthly human dosing is a conservative estimate for the dose frequency of the optimized constructs, based on our murine data. Less frequent dosing would be clinically preferable for the chronic therapy required in GACI and ARHR2, as well as better tolerated in other diseases of vascular and soft tissue calcification induced by low PPi such as pseudoxanthoma elasticum and chronic kidney disease bone mineralization disorder, which may also respond to ENPP1‐Fc therapy.
Funding
Funding from Inozyme Pharma and the National Institutes of Health through R01 DK121326 to D.T.B., and from the National Institutes of Health through R01 CA112314 and R21 CA249381 (awarded to K.J.Y.), and R35GM136656 (awarded to E.M.D.L.C.). Glycan analysis was supported in part by NIH grants 4P41GM103490‐14 and P41GM10349010 to Parastoo Azadi.
Conflicts of Interest
P.R.S. and D.T.B. are inventors of patents owned by Yale University, which describe therapeutics for ENPP1 deficiency. D.T.B and E.D.L.C. are equity holders and receive research and/or consulting support from Inozyme Pharma, Inc. All other authors declared no competing interests for this work.
Author Contributions
P.R.S., S.G., W.C., E.D.L.C., K.J.Y., and D.T.B. wrote the manuscript. P.R.S., K.J.Y., and D.T.B. designed the research. P.R.S., K.Z., A.A., D.K., C.T.S., C.A., S.G., and D.T.B. performed the research. P.R.S., K.Z., S.G., W.C., E.D.L.C., K.J.Y., and D.T.B. analyzed the data.
Supporting information
Supplementary Material
Acknowledgment
D.T.B. acknowledges helpful discussions with Steven Jungles (Inozyme), Denis Schrier (Inozyme), and Christopher Crean (xyzagen).
References
- 1. van Meeteren, L.A. et al Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1‐phosphate. J. Biol. Chem. 280, 21155–21161 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Tanaka, M. et al Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 281, 25822–25830 (2006). [DOI] [PubMed] [Google Scholar]
- 3. Nam, S.W. et al Autotaxin (NPP‐2), a metastasis‐enhancing motogen, is an angiogenic factor. Cancer Res. 61, 6938–6944 (2001). [PubMed] [Google Scholar]
- 4. Manning, T.J. Jr , Rosenfeld, S.S. & Sontheimer, H. Lysophosphatidic acid stimulates actomyosin contraction in astrocytes. J. Neurosci. Res. 53, 343–352 (1998). [DOI] [PubMed] [Google Scholar]
- 5. Umezu‐Goto, M. et al Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 158, 227–233 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nam, S.W. , Clair, T. , Campo, C.K. , Lee, H.Y. , Liotta, L.A. & Stracke, M.L. Autotaxin (ATX), a potent tumor motogen, augments invasive and metastatic potential of ras‐transformed cells. Oncogene 19, 241–247 (2000). [DOI] [PubMed] [Google Scholar]
- 7. Hoelzinger, D.B. , Nakada, M. , Demuth, T. , Rosensteel, T. , Reavie, L.B. & Berens, M.E. Autotaxin: a secreted autocrine/paracrine factor that promotes glioma invasion. J. Neurooncol. 86, 297–309 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Saunders, L.P. et al Identification of small‐molecule inhibitors of autotaxin that inhibit melanoma cell migration and invasion. Mol. Cancer Ther. 7, 3352–3362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu, S. et al Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 15, 539–550 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kishi, Y. et al Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J. Biol. Chem. 281, 17492–17500 (2006). [DOI] [PubMed] [Google Scholar]
- 11. Okawa, A. , Nakamura, I. , Goto, S. , Moriya, H. , Nakamura, Y. & Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Gen. 19, 271–273 (1998). [DOI] [PubMed] [Google Scholar]
- 12. Nakamura, I. et al Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL). Hum. Gen. 104, 492–497 (1999). [DOI] [PubMed] [Google Scholar]
- 13. Levy‐Litan, V. et al Autosomal‐recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am. J. Hum. Gen. 86, 273–278 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lorenz‐Depiereux, B. , Schnabel, D. , Tiosano, D. , Hausler, G. & Strom, T.M. Loss‐of‐function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal‐recessive hypophosphatemic rickets. Am. J. Hum. Genet. 86, 267–272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rutsch, F. et al Mutations in ENPP1 are associated with 'idiopathic' infantile arterial calcification. Nat. Genetics 34, 379–381 (2003). [DOI] [PubMed] [Google Scholar]
- 16. Rutsch, F. et al PC‐1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am. J. Pathol. 158, 543–554 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li, Q. , Guo, H. , Chou, D.W. , Berndt, A. , Sundberg, J.P. & Uitto, J. Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy. Dis. Mod. Mech. 6, 1227–1235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Albright, R.A. et al NPP4 is a procoagulant enzyme on the surface of vascular endothelium. Blood 120, 4432–4440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zalatan, J.G. , Fenn, T.D. , Brunger, A.T. & Herschlag, D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: implications for mechanism and evolution. Biochemistry 45, 9788–9803 (2006). [DOI] [PubMed] [Google Scholar]
- 20. O'Neill, W.C. , Sigrist, M.K. & McIntyre, C.W. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol. Dial. Transplant. 25, 187–191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oheim, R. et al Human heterozygous ENPP1 deficiency is associated with early onset osteoporosis, a phenotype recapitulated in a mouse model of Enpp1 deficiency. J. Bone Mineral Res. 35, 528–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meyer, J.L. Can biological calcification occur in the presence of pyrophosphate? Arch. Biochem. Biophys. 231, 1–8 (1984). [DOI] [PubMed] [Google Scholar]
- 23. Bryant, J. & White, W. A case of calcification of the arteries and obliterative endarteritis, associated with hydronephrosis, in a child aged six months. Guy's Hosp. Rep. 55, 17–28 (1901). [Google Scholar]
- 24. Albright, R.A. et al ENPP1‐Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat. Commun. 6, 10006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nitschke, Y. , Yan, Y. , Buers, I. , Kintziger, K. , Askew, K. & Rutsch, F. ENPP1‐Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp. Mol. Med. 50, 139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rutsch, F. et al Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Cir. Cardiovas. Genet. 1, 133–140 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sholler, G.F. , Yu, J.S. , Bale, P.M. , Hawker, R.E. , Celermajer, J.M. & Kozlowski, K. Generalized arterial calcification of infancy: three case reports, including spontaneous regression with long‐term survival. J. Pediatrics 105, 257–260 (1984). [DOI] [PubMed] [Google Scholar]
- 28. Brachet, C. , Mansbach, A.L. , Clerckx, A. , Deltenre, P. & Heinrichs, C. Hearing loss is part of the clinical picture of ENPP1 loss of function mutation. Horm. Res. Paediatr. 81, 63–66 (2014). [DOI] [PubMed] [Google Scholar]
- 29. Steichen‐Gersdorf, E. , Lorenz‐Depiereux, B. , Strom, T.M. & Shaw, N.J. Early onset hearing loss in autosomal recessive hypophosphatemic rickets caused by loss of function mutation in ENPP1. J. Pediatr. Endocrinol. Metab. 28, 967–970 (2015). [DOI] [PubMed] [Google Scholar]
- 30. Kotwal, A. et al Clinical and biochemical phenotypes in a family with ENPP1 mutations. J. Bone Mineral Res. 35, 662–670 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marrott, P.K. , Newcombe, K.D. , Becroft, D.M. & Friedlander, D.H. Idiopathic infantile arterial calcification with survival to adult life. Pediatr. Cardiol. 5, 119–122 (1984). [DOI] [PubMed] [Google Scholar]
- 32. Kallberg, M. et al Template‐based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oheim, R. et al Human heterozygous ENPP1 deficiency is associated with early onset osteoporosis, a phenotype recapitulated in a mouse model of Enpp1 deficiency. J. Bone Miner. Res. 35, 528–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guan, X. et al Chemically precise glycoengineering improves human insulin. ACS Chem. Biol. 13, 73–81 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Egrie, J.C. , Dwyer, E. , Browne, J.K. , Hitz, A. & Lykos, M.A. Darbepoetin alfa has a longer circulating half‐life and greater in vivo potency than recombinant human erythropoietin. Exp. Hematol. 31, 290–299 (2003). [DOI] [PubMed] [Google Scholar]
- 36. Kato, K. et al Structural insights into cGAMP degradation by Ecto‐nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 9, 4424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dall'Acqua, W.F. et al Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J. Immunol. 169, 5171–5180 (2002). [DOI] [PubMed] [Google Scholar]
- 38. Dall'Acqua, W.F. , Kiener, P.A. & Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 281, 23514–23524 (2006). [DOI] [PubMed] [Google Scholar]
- 39. Robbie, G.J. et al A novel investigational Fc‐modified humanized monoclonal antibody, motavizumab‐YTE, has an extended half‐life in healthy adults. Antimicrob. Agents Chemother. 57, 6147–6153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung, C.H. et al Cetuximab‐induced anaphylaxis and IgE specific for galactose‐alpha‐1,3‐galactose. N. Engl. J. Med. 358, 1109–1117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buettner, M.J. , Shah, S.R. , Saeui, C.T. , Ariss, R. & Yarema, K.J. Improving immunotherapy through glycodesign. Front. Immunol. 9, 2485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yin, B. et al Glycoengineering of Chinese hamster ovary cells for enhanced erythropoietin N‐glycan branching and sialylation. Biotechnol. Bioengineering 112, 2343–2351 (2015). [DOI] [PubMed] [Google Scholar]
- 43. Ellies, L.G. et al Sialyltransferase ST3Gal‐IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc. Natl. Acad. Sci. USA 99, 10042–10047 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aich, U. et al Regioisomeric SCFA attachment to hexosamines separates metabolic flux from cytotoxicity and MUC1 suppression. ACS Chem. Biol. 3, 230–240 (2008). [DOI] [PubMed] [Google Scholar]
- 45. Almaraz, R.T. et al Metabolic oligosaccharide engineering with N‐Acyl functionalized ManNAc analogs: cytotoxicity, metabolic flux, and glycan‐display considerations. Biotechnol. Bioeng. 109, 992–1006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sinha, S.D. et al Efficacy, tolerability and safety of darbepoetin alfa injection for the treatment of anemia associated with chronic kidney disease (CKD) undergoing dialysis: a randomized, phase‐III trial. BMC Nephrol. 20, 90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song, R. , Oren, D.A. , Franco, D. , Seaman, M.S. & Ho, D.D. Strategic addition of an N‐linked glycan to a monoclonal antibody improves its HIV‐1‐neutralizing activity. Nat. Biotechnol. 31, 1047–1052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weng, Y. et al Glyco‐engineered long acting FGF21 variant with optimal pharmaceutical and pharmacokinetic properties to enable weekly to twice monthly subcutaneous dosing. Sci. Rep. 8, 4241 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mahmood, I. Pharmacokinetic allometric scaling of antibodies: application to the first‐in‐human dose estimation. J. Pharm. Sci. 98, 3850–3861 (2009). [DOI] [PubMed] [Google Scholar]
- 50. Mahmood, I. Designing first‐in‐human dose of coagulation factors: application of pharmacokinetic allometric scaling. Haemophilia 20, 32–38 (2014). [DOI] [PubMed] [Google Scholar]
- 51. Huh, Y. , Smith, D.E. & Feng, M.R. Interspecies scaling and prediction of human clearance: comparison of small‐ and macro‐molecule drugs. Xenobiotica 41, 972–987 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material