

Abstract
Advances in cancer treatment have significantly improved the survival of patients with cancer, but, unfortunately, many of these treatments also have long‐term complications. Cancer treatment‐related cardiotoxicities are becoming a significant clinical problem that a new discipline, Cardio‐Oncology, was established to advance the cardiovascular care of patients with growing cancer populations. Anthracyclines are a class of chemotherapeutic agents used to treat many cancers in adults and children. Their clinical use is limited by anthracycline‐induced cardiotoxicity (AIC), which can lead to heart failure. Early‐onset cardiotoxicity appears within a year of treatment, whereas late‐onset cardiotoxicity occurs > 1 year and even up to decades after treatment completion. The pathophysiology of AIC was hypothesized to be caused by generation of reactive oxygen species that lead to lipid peroxidation, defective mitochondrial biogenesis, and DNA damage of the cardiomyocytes. The accumulation of anthracycline metabolites was also proposed to cause mitochondrial damage and the induction of cardiac cell apoptosis, which induces arrhythmias, contractile dysfunction, and cardiomyocyte death. This paper will provide a general overview of cardiotoxicity focusing on the effect of anthracyclines and their epigenetic molecular mechanisms on cardiotoxicity.
CLINICAL BACKGROUND OF ANTHRACYCLINES
Newer early cancer detection technologies and novel cancer therapies have significantly improved the prognosis of cancer in the last few decades. 1 , 2 However, one class of seminal anticancer agents, anthracyclines, continues to play a significant role in cancer treatment worldwide since their discovery in the1960s. The World Health Organization (WHO) names anthracyclines on its list of essential medicines 3 and researchers all over the world still display substantial interest in the drugs’ anticancer and cardiotoxic mechanisms through numerous clinical trials and research. 4 The American Cancer Society estimates that there are around 1,806,590 new cancer cases in 2020 in the United States, and ~ 16,850 of those cases will be for pediatric patients ≤ 19 years old. 5 The continual rise of cancer cases in both adult and pediatric populations supports the anthracyclines’ role as a significant player in antitumor treatment, but their cardiotoxicity remains an obstacle in cancer pharmacotherapy.
There are currently four anthracyclines that are commonly used in clinical practice: doxorubicin, daunorubicin, epirubicin, and idarubicin, with doxorubicin being the most common. Doxorubicin is used to treat numerous cancers for both adult and pediatric populations, such as breast cancer, Hodgkin’s lymphoma in adults, and acute lymphoblastic leukemia, and acute myeloid leukemia in pediatrics. 6 , 7 Daunorubicin is usually utilized in the treatment of acute leukemias in both adults and pediatrics, whereas epirubicin is mostly used for breast cancer in adults and relapsed sarcoma in children. 8 Idarubicin use in adult and pediatric cancers is still minimal compared with the other anthracyclines. 8 Doxorubicin and its precursor, daunorubicin, were the first anthracyclines to be discovered and the first to be put into clinical practice. Epirubicin has a larger volume of distribution and longer terminal half‐life than doxorubicin (doxorubicin terminal half‐life = 1–3 hours, epirubicin 31–35 hours). Idarubicin has a higher cellular uptake than daunorubicin, of which it is a derivative, and is the most lipophilic anthracycline. 4 Table 1 summarized the cardiotoxicity risk of each anthracycline compared with doxorubicin. 9 The succeeding anthracyclines, such as idarubicin, epirubicin, and mitoxantrone, are far less cardiotoxic than doxorubicin, but cardiotoxicity remains problematic with all anthracyclines. 10
Table 1.
The cardiotoxicity risk of each anthracycline compared to doxorubicin
| Anthracycline |
Clinical cardiotoxicity P value |
Cardiotoxicity risk comparison |
|---|---|---|
| Doxorubicin | Reference | — |
| Daunorubicin | N/A | Although a trend suggested that daunorubicin was less cardiotoxic than doxorubicin, a definitive conclusion could not be drawn because of limited statistical power |
| Epirubicin | 0.008 | 61% lower risk than doxorubicin |
| Idarubicin | N/A | Little difference in risk of any cardiotoxic event (clinical and subclinical) with comparable therapeutic efficacy |
| Liposomal doxorubicin | < 0.0001 | 22% lower risk than doxorubicin |
| Dexrazoxane with doxorubicin or epirubicin | < 0.0001 | 79% lower risk than doxorubicin |
ANTHRACYCLINES AND CARDIOTOXICITY
Anthracyclines are highly effective anticancer drugs that have contributed to a 5‐year survival of 80% among different cancer types. 4 The mechanism of action was hypothesized to be targeting tumor DNA via topoisomerase II enzyme inhibition, protein, and cell membrane dysfunction of rapidly dividing cancer cells. This disruption of DNA replication and transcription prevents the replication of cancer cells. 11
Despite their contribution to improved survival, the use of anthracyclines has been plagued by the development of early and late‐onset cardiotoxicity in patients with cancer contributing to increased cardiac morbidity and mortality. Anthracyline‐induced cardiotoxicity (AIC) is observed in about 57% of the patients treated with anthracyclines and appears as myocardial cell injury, progresses to subclinical left ventricular dysfunction, then, if not addressed in a timely manner, leads to symptomatic heart failure (HF). 12 The investigation into the cardiotoxic effects of anthracyclines has been of important focus in the area of Cardio‐Oncology research. Studies have shown that higher cumulative doses of chemotherapy increase the probability of developing myocardial dysfunction and HF. 13 The risk of cardiotoxicity associated with doxorubicin rises dramatically when the cumulative dose of doxorubicin is higher than 250–300 mg/m2. Other factors, such as female sex, age above 60 years, pediatric population, and previous radiotherapy have also been associated with a higher risk of cardiotoxicity. 14 Comorbid conditions, such as cardiac diseases, arterial hypertension, diabetes, dyslipidemia, and renal failure, are also significant risk factors. 15 , 16
Patients treated with anthracyclines are five times more likely to have reduced left ventricular ejection fraction or develop HF compared with those treated with other non‐anthracycline chemotherapy. 4 AIC can be acute or chronic, with acute cardiotoxicity occurring during the treatment or immediately afterward; this entails pericarditis‐myocarditis and arrhythmias, which are both possibly reversible. The chronic form could occur decades after the end of treatment and hold severe and clinically significant consequences, like morbidity and mortality, in which case long‐term therapy will be required. 10 Some patients can tolerate high doses without cardiotoxicity, whereas others developed cardiotoxicity at low doses. This variability in susceptibility to AIC suggests a genetic component. 17
MEDICINAL CHEMISTRY OF ANTHRACYCLINES AND ITS IMPACT ON CARDIOTOXICITY
A basic anthracycline structure consists of two main portions, a sugar portion (L‐daunosamine) and a nonsugar organic portion commonly referred to as an aglycone (Figure 1 ). 18 The daunosamine sugar and the flat, aromatic part of the aglycone ring system bind to DNA, whereas the aglycone’s ring is thought to be a bridge between the DNA and topoisomerase II. 19 DNA intercalation commences the anthracycline action against tumor growth. The aglycone rings insert themselves in between DNA’s two strands, positioning the aglycone ring along the DNA’s long axis to stabilize the complex. The daunosamine sugar then binds to the DNA’s minor groove at the DNA‐topoisomerase interface to initiate the DNA poisoning process. 19
Figure 1.
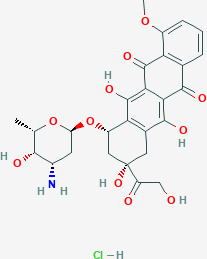
Doxorubicin structure.
The cardiotoxicity associated with the anthracycline drug class is thought to be derived from cytotoxic free radicals, which are formed during anthracycline breakdown. They are formed by a one‐electron reduction of the anthracycline quinone to hydroquinone by NAPDH/CYP450 reductase. Free radicals of importance are reactive oxygen species (ROS), such as the superoxide radical ion and the hydroxyl radical. 20 Superoxide radical ions produce hydrogen peroxide when they react, a product that is instrumental to the cardiotoxic side effects. Catalase is an enzyme that breaks down hydrogen peroxide into water and oxygen, which are ultimately harmless on their own. However, hydrogen peroxide is converted to the toxic hydroxyl radical while in the presence of ferrous ion through the process known as the Fenton reaction. 21 Anthracyclines are known to interfere with normal ferritin‐iron mobilization, which results in iron accumulation in the body; this, along with the frequent use of anthracyclines during treatment, almost guarantees the steady production of cytotoxic hydroxyl radicals in a patient undergoing anthracycline therapy. Cytotoxic hydroxyl radicals are produced throughout the body and especially within the heart, leading ultimately to acute cardiotoxicity. 20 Cardiac tissue is especially susceptible to free radical damage caused by anthracyclines due to its low amounts of catalase, forcing hydrogen peroxide formed in the myocardium to proceed down the Fenton pathway, producing more cytotoxic hydroxyl radicals, and setting the patient down a path of steady cardiotoxicity sustained by anthracycline therapy.
MECHANISM OF ACTION OF ANTHRACYCLINE‐INDUCED CARDIOTOXICITY
The mechanism of AIC is very complicated. Although the exact mechanism remains unclear, multiple processes have been revealed to be involved in the development of AIC. Most of these processes focus on cardiomyocyte injury and death. The most common and widely accepted mechanism of action of AIC is the anthracycline‐induced generation of ROS. 20 The ROS are formed through at least two pathways, a nonenzymatic and an enzymatic pathway. In the nonenzymatic pathway, anthracyclines react with ferric ion and produce free radicals. In the enzymatic pathway, anthracyclines react with the mitochondrial respiratory chain and other cytochrome‐containing enzymes to produce free radicals. 22 These free radicals can then circulate to the heart, damaging numerous cell components, such as proteins, nucleic acids, and cell membrane lipids, eventually leading to cell death and cardiotoxicity. 22 Anthracyclines have different modes of action for efficacy and toxicity (Figure 2 ): (i) to interfere with replication of proliferating cancer cells by interacting with Topoisomerase 2α, which increases DNA breaks and prevents DNA and RNA synthesis by inducing the apoptosis for cancer cells (efficacy), and (ii) to induce cardiotoxicity by interacting with Topoisomerase 2β that leads to increasing iron release and accumulating in mitochondria causing mitochondrial dysfunction, which is associated with ROS formation and apoptosis to cardiac cells (toxicity). 23
Figure 2.

Proposed mechanisms of anthracycline‐induced cardiotoxicity and cancer cell death. ROS, reactive oxygen species.
Doxorubicin, the most common anthracycline used in cancer treatment, is particularly harmful to the heart muscle because it has direct effects on the mitochondria and is associated with doxorubicin‐induced cardiotoxicity (DIC). 4 The pumping function of the heart to circulate blood flow throughout the entire body requires a significant amount of energy production from the mitochondria. 10 Mitochondria is an active site where most of the ROS are produced as a result of electrons escaping from the electron transport chain and captured by oxygen; this makes it the home of superoxide production and primarily leads to lipid peroxidation and DNA damage in cardiomyocytes, 10 , 24 these studies focused particularly on doxorubicin, making this mechanism of cardiotoxicity specific to doxorubicin only. It is currently not clear if other anthracyclines confer cardiotoxicity through the same mechanism. Other proposed mechanisms of DIC include the accumulation of cardiotoxic metabolites in the heart, disruption of calcium homeostasis, and induction of apoptosis. 25
A recent study has demonstrated the role of ferroptosis in DIC. 26 Ferroptosis is a new form of regulated cell death that is characterized by iron release from heme degradation to be accumulated to the fetal level. The authors reported that doxorubicin increases the accumulation of the nuclear factor erythroid 2 (Nrf2) gene, which translocates to the nucleus and binds to the antioxidant response element. This binding activates the heme oxygenase (decycling) 1 (Hmox1) and promotes the expression, facilitating the release of free iron, which leads to ferroptosis and HF. 26 The heme oxygenase enzymes, Hmox1 and Hmox2, have a role in catalyzing hydroxylation of heme to carbon monoxide, ferrous iron, and biliverdin. 27 In addition, they found that inhibiting ferroptosis significantly reduced DIC. 26
Another process attributing to AIC is the poisoning of topoisomerase, an enzyme that is crucial for DNA synthesis and replication in all cell types, this helps to cease the growth of proliferating cancer cells, but it also leads to the damage and apoptosis of cardiac myocytes. 20 Other factors that contribute to cardiotoxicity are the anthracyclines’ planar structure and its interaction with DNA and the downregulation of transcription factors during anthracycline exposure, these transcription factors are necessary for cardiac sarcomere synthesis and maintenance. 28
EPIGENETICS AND AIC
Along with researching anthracyclines’ clinical data, scientists are currently using pharmacogenomic approaches to determine which patients are at higher inherent risk of developing AIC. The field of epigenomics is the study of variations in gene expression that are inheritable but without changes in the genomic DNA sequence. 29 These variations include DNA methylation, post‐translational histone modification, and microRNAs. Current advances in epigenetic studies show that DNA methylation can play an important role in the pharmacodynamics of different drugs by regulating the expression of specific drug‐metabolizing enzymes. 30 DNA methylation is associated with transcriptional repression, and it has been implicated in different cardiovascular diseases, such as atherosclerosis, coronary heart disease, and abdominal aortic aneurysm. 30 Epigenetic therapies have not yet been widely tested in pediatric patients, but studies currently underway are providing some rationale for their use. 31
In addition to the several hypotheses for the association between genetic variants and susceptibility to cardiotoxicity, 32 , 33 , 34 , 35 recent studies showed that the epigenetic mechanism might play a role in promoting long‐term doxorubicin toxicity. 36 , 37 Previous study showed that long‐term exposure to doxorubicin causes a persistent and irreversible alteration of metabolism and gene expression in mitochondria. 38 Mitochondrial metabolism changes by the environment, and has an effect on the epigenomic landscape of nuclear DNA. When the energy is produced, the enzymes use mitochondrial‐derived adenosine triphosphate and acetyl coenzyme A to phosphorylate and acetylate chromatin, to help in increasing gene expression. When the metabolism decreased the gene expression also reduced. 39
miRNAS and anthracycline‐induced cardiotoxicity
MiRNAs are small, non‐coding RNA molecules (∼ 20–25 nucleotides) that play important roles in eukaryotic gene regulation. Currently, around 2,800–3,000 human miRNAs have been identified. 40 A miRNA can regulate multiple target mRNAs, and multiple miRNAs together can work as a functional cluster to coordinate the expression of a particular gene. It is involved in cell proliferation, differentiation, apoptosis, angiogenesis, etc. 41 MiRNAs also play vital roles in the pathophysiology of cardiovascular diseases, such as myocardial infarction, hypertrophy, fibrosis, HF, etc. 42 MiRNAs have been considered as critical regulators during heart development. Genetic studies demonstrated that miRNA changes during the process of cardiac differentiation of stem cells, such as miR‐1, miR‐208, miR‐133, and miR‐499, are strongly associated with cardiac maturation. 43
Detecting early biomarkers for toxicity is essential to manage anticancer treatment. Several clinical studies reported that some cardiac biomarkers, such as troponin I and troponin T, could be used to confirm cardiotoxicity, and high levels of troponin indicate there is irreversible myocardial cell injury in patients with cancer who received chemotherapy. B‐type natriuretic peptides and N‐terminal fragment of the prohormone brain natriuretic peptide are also considered routine biomarkers for cardiac dysfunction and HF diagnosis during chemotherapy. 44 , 45 , 46 The main disadvantage of these biomarkers is that they cannot be used as an early indicator of cardiotoxicity because their expressions occur after the irreversible cardiac damage. So there is an urgent need to identify new potential early detection biomarkers to provide early warning for chemotherapy‐induced cardiotoxicity.
Circulating miRNAs have been the focus of research as biomarkers for diagnosis and prevention, given their ease of detection in blood using noninvasive procedures. The origin of circulating miRNAs can be identified because of miRNA specificity in tissue. However, several circulating miRNAs in plasma still have a completely unknown origin and functionality. 47 The stability of circulating miRNAs makes them promising candidates for biomarkers for different diseases.
The miRNAs currently have an essential role as potential biomarkers in toxicological studies and are used as biomarkers for drug‐induced tissue injury. Several studies reported the association between AIC and the dysregulation of circulating miRNA. For example, miR‐34a‐5p, miR‐199a‐3p, miR‐423‐5p, and miR‐126‐3p were upregulated after epirubicin treatment. 48 In another study, miRNAs, such as miR‐143, miR‐361, and miR21, were downregulated in patients with breast cancer treated with doxorubicin. 49 MiR‐1 was upregulated in patients with breast cancer after doxorubicin treatment and showed strong ability to distinguish between patients with and without DIC more than troponin I, which increases its potential to be a promising new biomarker. 50 MiR‐133b, miR‐146a, and miR‐423‐5p were also upregulated with doxorubicin treatment in patients with breast cancer. 50 A pilot study included a different type of cancers in children (acute myeloid leukemia, acute lymphoid leukemia, hepatoblastoma, Hodgkin’s lymphoma, etc.) demonstrated higher expression of circulating miR‐29b and miR‐499 after anthracycline treatment (mainly doxorubicin) in patients who developed cardiotoxicity confirming by high‐sensitivity troponin T increase ≥ 5 ng/L from baseline. 51
Circulating miRNAs were also used as biomarkers in drug safety assessment because of their tissue specificity and early release in plasma after tissue injury. Several studies reported the association of specific circulating miRNAs and cardiotoxicity, 47 hepatotoxicity, 52 and, nephrotoxicity. 53 , 54 In this review, we have summarized the studies reporting miRNAs that are associated with AIC using in vivo and in vitro approaches (Table 2 , Figure 3 ).
Table 2.
List of miRNAs associated with AIC from previous studies in different models
| miRNA | Model | Sample | Anthracyclines type | Regulation | Putative target pathway | References |
|---|---|---|---|---|---|---|
| miR‐17‐5p | Human | Blood | EC‐D | Down | Not reported | 74 |
| miR‐210 | Human | Blood | EC‐D | Down | Not reported | 74 |
| let‐7a | Human | Plasma | DOX | Up | Not reported | 49 |
| let‐7f | Human | Blood | EC‐D | Down | Not reported | 74 |
| let‐7f‐2‐3p | Cell lines | Cardiomyocytes | DOX | Up | Long noncoding RNA NEAT1 inhibits XPO1‐mediated HAX‐1 nuclear export | 75 |
| Let‐7g | Rat | Heart tissue | DOX, L‐DOX | Down | Not reported | 76, 77 |
| miR‐1 | Human/rat/mice | Plasma/heart tissue | DOX | Up | Not reported | 50, 78, 79 |
| miR‐1 | Cell lines | Cardiomyocytes from treated animals | Epirubicin | Down | Suppress the PI3K/AKT/mTOR and NF‐κB signalling pathways | 80 |
| miR‐122‐5P | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐125 | Human | Plasma | DOX | Up | Not reported | 49 |
| miR‐126‐3p | Human | Plasma | Epirubicin | Up | Not reported | 48 |
| miR‐127‐3p | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐1303 | Cell lines | Cardiomyocytes | DOX | Down | Not reported | 82 |
| miR‐130a | Cell lines | Cardiac cells | DOX | Up | Apoptosis pathway | 83 |
| miR‐133a | Rat, mice | Plasma | DOX | Up | Not reported | 78, 84 |
| miR‐133a‐3p | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐133a‐3p | Mice | Plasma | DOX | Up | Not reported | 85 |
| miR‐133b | Mice/human | Plasma | DOX | Up | Not reported | 50, 84 |
| miR‐1‐3p | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐140‐3p | Mice | Plasma | DOX | Down | Not reported | 85 |
| miR‐140‐5p | Rat/mice | Heart tissue | DOX | Up | Mitochondrial apoptosis, oxidative stress | 86, 87 |
| miR‐143 | Human | Plasma | DOX | Down | Not reported | 49 |
| miR‐145 | Human | Plasma | DOX | Up | Not reported | 49 |
| miR‐146a | Human | Plasma | DOX | Up | Not reported | 50 |
| miR‐146a | Rat | Neonatal rat cardiac myocytes | DOX | Up | ErbB4 | 88 |
| miR‐146a | Cell lines | (hPSC‐CM) | DOX | Down | MMPs via the Fos/AP‐1 pathway | 89 |
| miR‐15b | Cell lines | (hPSC‐CM) | DOX | Down | TGFβ‐pathway | 89 |
| miR‐15b‐5p | Rat | Cardiomyocytes from treated animals | DOX | Up | Apoptosis, oxidative stress, and mitochondria damage | 90 |
| miR‐182‐5p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐187‐3p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐191 | Human | Plasma | DOX | Up | Not reported | 49 |
| miR‐199a‐3p | Human | Plasma | Epirubicin | Up | Not reported | 48 |
| miR‐1a | Mice | Plasma | DOX | Up | Not reported | 84 |
| miR‐200c | Mice | Heart tissue | DOX | Up | Zinc finger E‐box | 91 |
| miR‐206 | Rat | Plasma | DOX | Up | Not reported | 78 |
| miR‐208a | Rat | Heart tissue | DOX, L‐DOX | Down | Myosin heavy chain expression | 76 |
| miR‐208a | Mice | Heart tissue | DOX | Up | GATA4 | 92 |
| miR‐208a | Human | Plasma | DOX | Not released | Not reported | 93 |
| miR‐208b | Rat | Rat heart | DOX | Up | Not reported | 94 |
| miR‐20a | Human | Blood | EC‐D | Down | Not reported | 74 |
| miR‐21 | Mice | Heart tissue | DOX | Up | Not reported | 79, 95 |
| miR‐21 | Rat/mice | Mouse heart tissues and rat H9C2 cardiomyocytes | DOX | Up | B cell translocation gene 2 | 96 |
| miR‐21 | Human | Plasma | DOX | Down | Not reported | 49 |
| miR‐215 | Rat | Rat heart | DOX | Up | Not reported | 94 |
| miR‐215‐5p | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐216b | Rat | Rat heart | DOX | Up | Not reported | 94 |
| miR‐23a | Rat | Primary neonatal rat ventricular myocytes | DOX | Up | PGC‐1α/p‐Drp1, thereby inhibiting mitochondria‐dependent apoptosis | 97 |
| miR‐29b | Rat | Heart tissue | DOX | Down | Mitochondria‐dependent pathway by directly targeting Bax | 98 |
| miR‐29b | Human | Plasma | Anthracycline | Up | Not reported | 51 |
| miR‐30 | Rat | Cardiomyocytes from treated animals | DOX | Down | Pro‐apoptotic gene BNIP3L/NIX | 99 |
| miR‐30a | Rat | Cardiomyocytes from treated animals | DOX | Up | Autophagy in a miR‐30e/beclin‐1 signal pathway | 100 |
| miR‐30e | Rat | Cardiomyocytes from treated animals | DOX | Up | Autophagy in a miR‐30e/beclin‐1 signal pathway | 100 |
| miR‐301b‐3p | Mice | Plasma | DOX | Up | Not reported | 85 |
| miR‐30c | Rat | Cardiomyocytes from treated animals | DOX | Up | Autophagy in a miR‐30e/beclin‐1 signal pathway | 100 |
| miR‐320a | Mice | Heart tissue | DOX | Up | Not reported | 101 |
| miR‐32‐5p | Mice | Plasma | DOX | Up | Not reported | 85 |
| miR‐339 | Mice | Plasma | DOX | Down | Not reported | 84 |
| miR‐34a | Mice, rat/cell lines | Plasma, heart tissue/(hPSC‐CM) | DOX | Up | Not reported | 84, 89, 102, 103 |
| miR‐34a‐3p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐34a‐5p | Mice/rat | Cardiomyocytes/plasma | DOX | Up | Not reported | 81, 85, 104 |
| miR‐34a‐5p | Human | Plasma | Epirubicin | Up | Not reported | 48, 104 |
| miR‐34b‐3p | Mice | Plasma | DOX | Up | Not reported | 85 |
| mir‐34c | Rat | Rat heart | DOX | Up | Not reported | 94 |
| miR‐34c‐3p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐34c‐5p | Mice/cell lines | Plasma/cardiomyocytes | DOX | Up | Not reported | 82, 85 |
| miR‐361 | Human | Plasma | DOX | Down | Not reported | 49 |
| miR‐367 | Rat | Rat heart | DOX | Up | Not reported | 94 |
| miR‐378 | Human | Blood | EC‐D | Down | Not reported | 74 |
| miR‐423‐5p | Human | Plasma | DOX/epirubicin | Up | Not reported | 48, 50 |
| miR‐431‐5p | Mice | Plasma | DOX | Down | Not reported | 85 |
| miR‐4423‐3p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐451a | Mice | Plasma | DOX | Down | Not reported | 85 |
| miR‐455‐3‐p | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐486‐3p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐486‐5p | Cell lines | Cardiomyocytes | DOX | Up | Not reported | 82 |
| miR‐499 | Human | Plasma | Anthracycline | Up | Not reported | 51 |
| mir‐499‐5p | Mice | Plasma | DOX | Up | Not reported | 85 |
| miR‐499a‐5p | Mice | Plasma | DOX | Down | Not reported | 81 |
| miR‐500a‐3p | NA | Cardiomyocytes | DOX | Down | Not reported | 105 |
| miR‐532‐3p | NA | Cardiomyocytes | DOX | Up | Not reported | 105 |
| miR‐532‐3p | Mice | Mice heart and cardiomyocytes | DOX | Up | Mitochondrial fission and apoptosis | 106 |
| miR‐6236 | Mice | Plasma | DOX | Down | Not reported | 84 |
| miR‐6240 | Mice | Plasma | DOX | Down | Not reported | 84 |
| miR‐6946 | Mice | Plasma | DOX | Down | Not reported | 84 |
| miR‐7058 | Mice | Plasma | DOX | Up | Not reported | 84 |
| miR‐93‐5p | Mice | Plasma | DOX | Down | Not reported | 85 |
AIC, anthracycline‐induced cardiotoxicity; Akt, protein kinase B; AP‐1, Activator protein 1; BAX , Bcl‐2 Associated X‐protein; BNIP3L, BCL2 Interacting Protein 3 Like; DOX, doxorubicin; EC‐D, epirubicin/cyclophosphamide followed by docetaxel; Erb‐B2, Receptor Tyrosine Kinase 4; GATA4, GATA Binding Protein 4; HAX1, HCLS1 Associated Protein X‐1; hPSC‐CM, human induced pluripotent stem cell‐derived cardiomyocytes; MHC, major histocompatibility complex; miRNA, microRNA; MMPs, Matrix metallopeptidases; mTOR, The mammalian target of rapamycin; NEAT1, Nuclear Enriched Abundant Transcript 1; NF‐κB, Nuclear factor kappa B; PI3K, Phosphoinositide 3‐kinase; PGC‐1α, coactivator 1‐alpha; TGFβ, Transforming growth factor beta; XPO1, Exportin 1.
Figure 3.
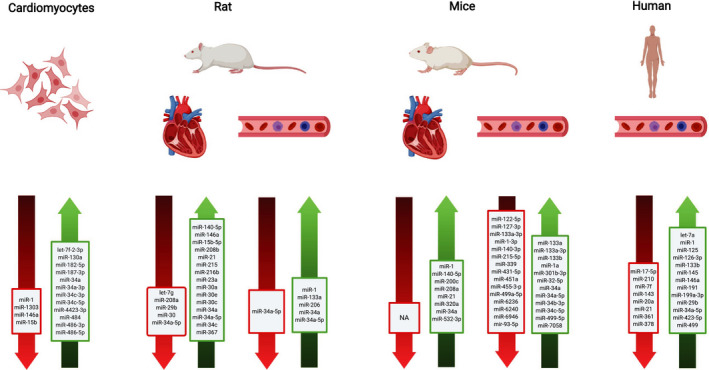
Summary of most of microRNAs regulation detected in different models treated with doxorubicin.
Methylation and AIC
A previous study showed that treating H9c2 (rat heart‐derived cell line) with doxorubicin decreased the DNA methyltransferase I mRNA expression, which might reduce the mitochondrial DNA (mtDNA) methylation level. mtDNA plays an important role in the upregulation of some mitochondrial genes and maintains the function of mitochondria. Downregulation of DNA methyltransferase I was associated with mtDNA hypomethylation under oxidative stress. This finding supports the presence of dynamic mitochondrial epigenetic mechanisms associated with doxorubicin treatment, 55 as illustrated in Figure 4 . Another study found that rats treated with doxorubicin had decreased global DNA methylation in the heart, these changes were associated with alteration in mRNA expression of different functional gene groups, and disruption in cardiac mitochondrial biogenesis, which was demonstrated by decreasing of mtDNA levels and alteration in transcript levels of mitochondrial genes encoded by nuclear and mitochondrial genomes. 56 This study also showed that doxorubicin disturbs the mitochondrial‐dependent production of the main acetyl and methyl donors (acetyl coenzyme A and S‐adenosylmethionine, respectively) and consequently imprints a long‐lasting toxic epigenetic effect that can be detected in metabolic transcriptome and metabolome. 56
Figure 4.
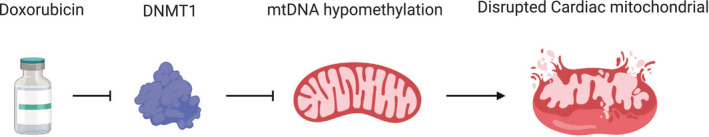
The effect of doxorubicin on DNA methyltransferase I (DNMT1), which leads to mitochondrial DNA (mtDNA) hypomethylation and disruption of the cardiac mitochondrial cell.
Another study reported that variability in gene expression of cardiac aldo‐keto reductases (AKR7A2), the most abundant anthracyclines reductase in the heart, might have an impact on the metabolism of anthracycline substrates and subsequently increasing the risk of AIC. The authors also found there was a relative linear correlation between DNA methylation status in specific CpG sites of the AKR7A2 locus and intracardiac synthesis of cardiotoxic alcohol metabolites, and they concluded this correlation might be potentially associated with cardiac AKR7A2 gene expression and maximal anthracyclines reductase activity. 57 Moreover, in a different study, DIC in Wistar rats was decreased by co‐administrating the major biological methyl donor S‐adenosylmethionine. 58
MANAGEMENT OF CARDIOTOXICITY
There are many strategies to manage AIC without altering the efficacy of antitumor treatment. The most utilized strategies are (i) dose reduction, (ii) changing the treatment frequency from classical dosing of once every 2–3 weeks to weekly or continuous infusion dosing, 59 (iii) managing cardiovascular risk factors of patients with cancer, 60 and (iv) using cardioprotective agents. 61 , 62 , 63
Dexrazoxane (Zinecard®) is currently the only cardioprotective agent that is approved for AIC by the US Food and Drug Administration (FDA). 64 Dexrazoxane is an iron chelator that binds to free irons and prevent anthracyclines‐iron complex formation. By preventing oxygen free radical formation, dexrazoxane protects cardiac cells from cardiotoxicity. 65 The clinical efficacy of dexrazoxane was previously attributed to its ability to chelate iron, which prevents the formation of ROS that would ultimately damage the myocardium. 66 This initial hypothesis of dexrazoxane’s cardioprotective mechanism of action gradually became inadequate, because it could not explain why other iron chelators have not been shown to protect against AIC as well. 66 New research indicated that dexrazoxane’s clinical efficacy against cardiotoxicity comes from its role as a catalytic inhibitor of topoisomerase II. Although both anthracyclines and dexrazoxane target topoisomerase, the distinction in their mechanisms is their effects on DNA breakage. 66 Doxorubicin blocks the reformation of double‐stranded DNA after the DNA has been cleaved by topoisomerase II. This prevents the replication of cancer cells, but it also results in DNA double‐stranded breaks that remain in the body and eventually lead to cell death. 10 In contrast, dexrazoxane changes the configuration of topoisomerase, preventing anthracyclines from binding to it altogether and thereby prevents cardiomyocyte death, mitochondrial dysfunction, and the suppression of antioxidant gene expression. 67 Dexrazoxane also binds iron before it enters cardiomyocytes, which prevents the formation of the iron‐anthracycline complex; which prevents the free radical formation and, ultimately, cardiac damage. 67 Dexrazoxane’s contrasting mechanism to the anthracyclines helps protect the heart from the anthracyclines’ cardiotoxicity by preventing the creation of DNA breaks and their eventual damage to the myocardium. Although dexrazoxane has been proven to help protect against AIC, its use in pediatrics has been limited because of its adverse effects.
There are debatable concerns in adult and pediatric patients that are receiving dexrazoxane that might reduce the response of anthracyclines on tumor cells and increase the risk of secondary malignancies. 65 At least two randomized clinical trials have shown a three‐fold increase in the incidence of primary, secondary malignancies in acute myeloid leukemia and pediatric patients with Hodgkin’s disease compared with controls after dexrazoxane prescription. 68 A retrospective cohort performed in US hospitals for secondary acute myeloid leukemia demonstrated no association with increased risk of secondary malignancies in pediatric patients with cancer. 69 Conflicting results in pediatric studies emphasize the necessity for more research in this population. Today, there are limited options for cardiac protection in cancer therapy, and the need continues to rise as anthracycline use increases in both pediatric and adult patients with cancer. Additional studies on dexrazoxane and its side effects are warranted to move toward more stable and effective cancer treatment regimens.
PHARMACOGENOMICS OF AIC
Pharmacogenomics, or the identification of genomic determinants of drug response or adverse effects, is a tool that has been used in individualized medication therapy. 70 The Clinical Pharmacogenomics Implementation Consortium (CPIC) develops and publishes peer‐reviewed gene‐drug guidelines to facilitate clinical implementation of pharmacogenomic test. There have been a few pharmacogenomic studies of AIC in adults and pediatric patients with cancer, 34 , 71 , 72 but none have reached CPIC level strength of evidence. However, there was a recommendation published by the Canadian Clinical Practice Recommendations Group that aims to encourage cancer institutes to perform germline DNA pharmacogenomic testing in all pediatric patients with cancer who will receive anthracyclines, such as doxorubicin or daunorubicin to reduce the incidence of cardiotoxicity. 73 The genetic variants recommended to be tested include retinoic acid receptor gamma (RARG) rs2229774, solute carrier transporters (SLC28A3) rs7853758 and UDP‐glucuronosyltransferase family 1A, isoform 6 (UGT1A6*4) rs17863783 variants. These tests were not recommended for adults and other pediatric patients with cancer receiving different types of anthracyclines. 73 In our opinion, these germline variants need to be validated in independent cohort studies or randomized trials before they can be implemented in the clinical setting.
CONCLUSION
In summary, AIC is a severe problem associated with the administration of anthracyclines in many patients with cancer in adults and pediatrics. Current literature supports the important role of epigenetic changes, including miRNA and DNA methylation in the development of AIC, mediated through disrupting mitochondrial biogenesis, increasing ROS release, inducing apoptosis, and ferroptosis of cardiac cells. The miRNAs have been reported in several studies to be affected by drugs, and the potential of miRNAs as biomarkers and diagnostic tools has been considered. In addition, the main aspect discussed for the advantage of miRNA is the alteration of miRNA expression in cardiac cells upon treatment with anthracyclines, circulating miRNAs play an important role as a promising early biomarker of cardiotoxicity. Epigenetics studies showed that DNA methylation could affect the pharmacodynamics of different drugs by regulating the expression of specific drug‐metabolizing enzymes. Further investigation of different epigenetic mechanisms associated with cardiotoxicity might have the potential to decrease the risk of AIC.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work. As an Editor‐in‐Training for Clinical and Translational Science, Sonal Singh was not involved in the review or decision process for this paper.
References
- 1. Falzone, L. , Salomone, S. & Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 9, 1300 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seebacher, N.A. , Stacy, A.E. , Porter, G.M. & Merlot, A.M. Clinical development of targeted and immune based anti‐cancer therapies. J. Exp. Clin. Cancer Res. 38, 156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization . WHO Model List of Essential Medicines. 1–43 (World Health Organization, Austria, 2013). <https://www.who.int/medicines/publications/essentialmedicines/18th_EML.pdf>. [Google Scholar]
- 4. McGowan, J.V. , Chung, R. , Maulik, A. , Piotrowska, I. , Walker, J.M. & Yellon, D.M. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc. Drugs Ther. 31, 63–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siegel, R.L. , Miller, K.D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 6. Asselin, B.L. et al Cardioprotection and safety of dexrazoxane in patients treated for newly diagnosed T‐cell acute lymphoblastic leukemia or advanced‐stage lymphoblastic non‐Hodgkin lymphoma: a report of the Children’s Oncology Group randomized trial Pediatric Oncology Group. J. Clin. Oncol. 34, 854–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jarfelt, M. , Andersen, N.H. & Hasle, H. Is it possible to cure childhood acute myeloid leukaemia without significant cardiotoxicity? Br. J. Haematol. 175, 577–587 (2016). [DOI] [PubMed] [Google Scholar]
- 8. Feijen, E.A.M. et al Derivation of anthracycline and anthraquinone equivalence ratios to doxorubicin for late‐onset cardiotoxicity. JAMA Oncol. 5, 864–871 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith, L.A. et al Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta‐analysis of randomised controlled trials. BMC Cancer 10, 337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Angsutararux, P. , Luanpitpong, S. & Issaragrisil, S. Chemotherapy‐induced cardiotoxicity: overview of the roles of oxidative stress. Oxid. Med. Cell. Longev. 2015, 13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scully, R.E. & Lipshultz, S.E. Anthracycline cardiotoxicity in long‐term survivors of childhood cancer. Cardiovasc. Toxicol. 7, 122–128 (2007). [DOI] [PubMed] [Google Scholar]
- 12. Wojtukiewicz, M.Z. , Omyła, J. , Kozłowski, L. & Szynaka, B. Cardiotoxicity of anthracycline. Postpy Hig i Med doświadczalnej. 54, 467–485 (2020). [PubMed] [Google Scholar]
- 13. Mulrooney, D.A. et al Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the childhood cancer survivor study cohort. BMJ 339, b4606 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mele, D. et al Current views on anthracycline cardiotoxicity. Heart Fail. Rev. 21, 621–634 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Vaitiekus, D. et al Impact of arterial hypertension on doxorubicin‐based chemotherapy‐induced subclinical cardiac damage in breast cancer patients. Cardiovasc. Toxicol. 20, 321–327 (2020). [DOI] [PubMed] [Google Scholar]
- 16. Neuendorff, N.R. et al Anthracycline‐related cardiotoxicity in older patients with acute myeloid leukemia : a Young SIOG review paper Pathophysiology of ARLVD. Blood Adv. 4, 762–775 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lipshultz, S.E. , Colan, S.D. , Gelber, R.D. , Perez‐Atayde, A.R. , Sallan, S.E. & Sanders, S.P. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N. Engl. J. Med. 324, 808–815 (1991). [DOI] [PubMed] [Google Scholar]
- 18. National Center for Biotechnology Information . PubChem database. Doxorubicin Hydrochloride, CID=443939. <https://pubchem.ncbi.nlm.nih.gov/compound/Doxorubicin‐Hydrochloride>.
- 19. Shaul, P. et al The structure of anthracycline derivatives determines their subcellular localization and cytotoxic activity. ACS Med. Chem. Lett. 4, 323–328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marinello, J. , Delcuratolo, M. & Capranico, G. Anthracyclines as topoisomerasE II poisons: from early studies to new perspectives. Int. J. Mol. Sci. 19, 3480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koutsoukis, A. , Ntalianis, A. , Repasos, E. , Kastritis, E. , Dimopoulos, M.A. & Paraskevaidis, I. Cardio‐oncology: a focus on cardiotoxicity. Eur. Cardiol. Rev. 13, 64–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gammella, E. , Maccarinelli, F. , Buratti, P. , Recalcati, S. & Cairo, G. The role of iron in anthracycline cardiotoxicity. Front. Pharmacol. 5, 1–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Henriksen, P.A. Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart 104, 971–977 (2018). [DOI] [PubMed] [Google Scholar]
- 24. Lipshultz, S.E. , Alvarez, J.A. & Scully, R.E. Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart 94, 525–533 (2007). [DOI] [PubMed] [Google Scholar]
- 25. Minotti, G. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229 (2004). [DOI] [PubMed] [Google Scholar]
- 26. Fang, X. et al Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA. 116, 2672–2680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maines, M.D. The Heme Oxygenase System: a regulator of second messenger gases. Ann. Rev. Pharmacol. Toxicol. 37, 517–554 (1997). [DOI] [PubMed] [Google Scholar]
- 28. Volkova, M. & Russell, R. III Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 7, 214 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weinhold, B. Epigenetics: the science of change. Environ. Health Perspect. 114, A160 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Metzinger, L. , De Franciscis, S. & Serra, R. The management of cardiovascular risk through epigenetic biomarkers. Biomed. Res. Int. 2017, 1–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lawlor, E.R. & Thiele, C.J. Epigenetic changes in pediatric solid tumors: promising new targets. Clin. Cancer Res. 18, 2768–2779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Linschoten, M. , Teske, A.J. , Cramer, M.J. , van der Wall, E. & Asselbergs, F.W. Chemotherapy‐related cardiac dysfunction: a systematic review of genetic variants modulating individual risk. Circ. Genomic. Precis. Med. 11, e001753 (2018). [DOI] [PubMed] [Google Scholar]
- 33. Aminkeng, F. et al Recommendations for genetic testing to reduce the incidence of anthracycline‐induced cardiotoxicity. Br. J. Clin. Pharmacol. 82, 683–695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Visscher, H. et al Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline‐induced cardiotoxicity in children. Pharmacogenomics 16, 1065–1076 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Visscher, H. et al Pharmacogenomic prediction of anthracycline‐induced cardiotoxicity in children. J. Clin. Oncol. 30, 1422–1428 (2016). [DOI] [PubMed] [Google Scholar]
- 36. Pang, B. et al Drug‐induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 4, 1908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montaigne, D. et al Stabilization of mitochondrial membrane potential prevents doxorubicin‐induced cardiotoxicity in isolated rat heart. Toxicol. Appl. Pharmacol. 244, 300–307 (2010). [DOI] [PubMed] [Google Scholar]
- 38. Richard, C. et al Oxidative stress and myocardial gene alterations associated with doxorubicin‐induced cardiotoxicity in rats persist for 2 months after treatment cessation. J. Pharmacol. Exp. Ther. 339, 807–814 (2011). [DOI] [PubMed] [Google Scholar]
- 39. Wallace, D.C. & Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10, 12–31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Londina, E. et al Analysis of 13 cell types reveals evidence for the expression of numerous novel primate‐ and tissue‐specific microRNAs. Proc. Natl. Acad. Sci USA 112, E1106–E1115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang, S.‐J. et al The miR‐30 family: versatile players in breast cancer. Tumor Biol. 39, 1010428317692204 (2017). [DOI] [PubMed] [Google Scholar]
- 42. Wojciechowska, A. , Braniewska, A. & Kozar‐Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 26, 865–874 (2017). [DOI] [PubMed] [Google Scholar]
- 43. Small, E.M. & Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 469, 336–342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liao, D. et al Involvement of neurotrophic signaling in doxorubicin‐induced cardiotoxicity. Exp. Ther. Med. 19, 1129–1135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Słowik, A. et al Anthracycline‐induced cardiotoxicity prevention with angiotensin‐converting enzyme inhibitor ramipril in low‐risk breast cancer women: results of a prospective randomized study. Kardiol. Pol. 78, 131–137 (2020). [DOI] [PubMed] [Google Scholar]
- 46. Ebrahimi, A. et al Drug‐induced myocardial dysfunction – recommendations for assessment in clinical and pre‐clinical studies. Expert Opin. Drug Safety 19, 281–294 (2020). [DOI] [PubMed] [Google Scholar]
- 47. Skála, M. , Hanousková, B. , Skálová, L. & Matoušková, P. MicroRNAs in the diagnosis and prevention of drug‐induced cardiotoxicity. Arch. Toxicol. 93, 1–9 (2019). [DOI] [PubMed] [Google Scholar]
- 48. Frères, P. et al Variations of circulating cardiac biomarkers during and after anthracycline‐containing chemotherapy in breast cancer patients. BMC Cancer 18, 102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Todorova, V.K. , Makhoul, I. , Wei, J. & Klimberg, V.S. Circulating miRNA profiles of doxorubicin‐induced cardiotoxicity in breast cancer patients. Ann. Clin. Lab. Sci. 47, 115–119 (2017). [PubMed] [Google Scholar]
- 50. Rigaud, V.O.‐C. et al Circulating miR‐1 as a potential biomarker of doxorubicin‐induced cardiotoxicity in breast cancer patients. Oncotarget. 8, 6994–7002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leger, K.J. , Leonard, D. , Nielson, D. , de Lemos, J.A. , Mammen, P.P.A. & Winick, N.J. Circulating microRNAs: potential markers of cardiotoxicity in children and young adults treated with anthracycline chemotherapy. J. Am. Heart Assoc. 6, e004653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin, H. , Ewing, L.E. , Koturbash, I. , Gurley, B.J. & Miousse, I.R. MicroRNAs as biomarkers for liver injury: Current knowledge, challenges and future prospects. Food Chem. Toxicol. 110, 229–239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alexander, H. , Gert, M. Circulating miRNAs as biomarkers of kidney disease. Clin. Kidney J. 10, 27–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koturbash, I. et al microRNAs as pharmacogenomic biomarkers for drug efficacy and drug safety assessment. Biomark. Med. 9, 1153–1176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferreira, L.L. , Cunha‐Oliveira, T. , Veloso, C.D. , Costa, C.F. , Wallace, K.B. & Oliveira, P.J. Single nanomolar doxorubicin exposure triggers compensatory mitochondrial responses in H9c2 cardiomyoblasts. Food Chem. Toxicol. 124, 450–461 (2019). [DOI] [PubMed] [Google Scholar]
- 56. Ferreira, A. et al Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology 390, 63–73 (2017). [DOI] [PubMed] [Google Scholar]
- 57. Hoefer, C.C. , Quiñones‐Lombraña, A. , Blair, R.H. & Blanco, J.G. Role of DNA methylation on the expression of the anthracycline metabolizing enzyme AKR7A2 in human heart. Cardiovasc. Toxicol. 16, 182–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Russo, S. , Filippelli, W. , Ferraraccio, F. , Berrino, L. , Guarino, V. & Rossi, F. Effects of S‐adenosylmethionine (SAMe) on doxorubicin‐induced cardiotoxicity in the rat. J. Med. 25, 65–89 (1994). [PubMed] [Google Scholar]
- 59. van Dalen, E. , van der Pal, H. & Kremer, L. Different anthracycline derivates for reducing cardiotoxicity in cancer patients. Cochrane Database Syst. Rev. 3, CD005006 (2004). [DOI] [PubMed] [Google Scholar]
- 60. Zamorano, J.L. et al 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: the Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 19, 9–42 (2017). [DOI] [PubMed] [Google Scholar]
- 61. Olivieri, J. et al Modern management of anthracycline‐induced cardiotoxicity in lymphoma patients: low occurrence of cardiotoxicity with comprehensive assessment and tailored substitution by nonpegylated liposomal doxorubicin. Oncologist. 22, 422–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Armenian, S.H. et al Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 35, 893–911 (2017). [DOI] [PubMed] [Google Scholar]
- 63. Henry, M.L. , Niu, J. , Zhang, N. , Giordano, S.H. & Chavez‐MacGregor, M. Cardiotoxicity and cardiac monitoring among chemotherapy‐treated breast cancer patients. JACC Cardiovasc. Imaging 11, 1084–1093 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang, J. et al Research progress of cardioprotective agents for prevention of anthracycline cardiotoxicity. Am. J. Transl Res. 8, 2862–2875 (2016). [PMC free article] [PubMed] [Google Scholar]
- 65. Ganatra, S. et al Upfront dexrazoxane for the reduction of anthracycline‐induced cardiotoxicity in adults with preexisting cardiomyopathy and cancer: a consecutive case series. CardioOncology. 5, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yeh, E.T.H. et al Mechanisms and clinical course of cardiovascular toxicity of cancer treatment I. Oncology. Semin. Oncol. 46, 397–402 (2019). [DOI] [PubMed] [Google Scholar]
- 67. Vejpongsa, P. & Yeh, E.T.H. Prevention of anthracycline‐induced cardiotoxicity: challenges and opportunities. J. Am. Coll. Cardiol. 64, 938–945 (2014). [DOI] [PubMed] [Google Scholar]
- 68. Tebbi, C.K. et al Dexrazoxane‐associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric hodgkin's disease. J Clin Oncol 25, 493–500 (2007). 10.1200/jco.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 69. Seif, A.E. et al Dexrazoxane exposure and risk of secondary acute myeloid leukemia in pediatric oncology patients. Pediatr. Blood Cancer. 62, 704–709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Collins, F.S. & Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aminkeng, F. et al A coding variant in RARG confers susceptibility to anthracycline‐induced cardiotoxicity in childhood cancer. Nat. Genet. 47, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Dalen, E.C. , van der Pal, H.J.H. , Kok, W.E.M. , Caron, H.N. & Kremer, L.C.M. Clinical heart failure in a cohort of children treated with anthracyclines: a long‐term follow‐up study. Eur. J. Cancer 42, 3191–3198 (2006). [DOI] [PubMed] [Google Scholar]
- 73. Aminkeng, F. et al Recommendations for genetic testing to reduce the incidence of anthracycline‐induced cardiotoxicity. Br. J. Clin. Pharmacol. 82, 683–695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Qin, X. , Chang, F. , Wang, Z. & Jiang, W. Correlation of circulating pro‐angiogenic miRNAs with cardiotoxicity induced by epirubicin/cyclophosphamide followed by docetaxel in patients with breast cancer. Cancer Biomark. 23, 473–484 (2018). [DOI] [PubMed] [Google Scholar]
- 75. Liu, Y. et al Upregulation of let‐7f‐2‐3p by long noncoding RNA NEAT1 inhibits XPO1‐mediated HAX‐1 nuclear export in both in vitro and in vivo rodent models of doxorubicin‐induced cardiotoxicity. Arch. Toxicol. 93, 3261–3276 (2019). [DOI] [PubMed] [Google Scholar]
- 76. Novak, J. , Sana, J. , Stracina, T. , Novakova, M. & Slaby, O. Doxorubicin and liposomal doxorubicin differentially affect expression of miR‐208a and let‐7g in rat ventricles and atria. Cardiovasc. Toxicol. 17, 355–359 (2017). [DOI] [PubMed] [Google Scholar]
- 77. Fu, J. , Peng, C. , Wang, W. , Jin, H. , Tang, Q. & Wei, X. Let‐7 g is involved in doxorubicin induced myocardial injury. Environ. Toxicol. Pharmacol. 33, 312–317 (2012). [DOI] [PubMed] [Google Scholar]
- 78. Nishimura, Y. et al Plasma miR‐208 as a useful biomarker for drug‐induced cardiotoxicity in rats. J. Appl. Toxicol. 35, 173–180 (2015). [DOI] [PubMed] [Google Scholar]
- 79. Razavi‐Azarkhiavi, K. et al The cardiotoxic mechanism of doxorubicin (DOX) and pegylated liposomal DOX in mice bearing C‐26 colon carcinoma: a study focused on microRNA role for toxicity assessment of new formulations. Pharm Res. 34, 1849–1856 (2017). [DOI] [PubMed] [Google Scholar]
- 80. Wu, J. et al Cardioprotective effect of paeonol against epirubicin‐induced heart injury via regulating miR‐1 and PI3K/AKT pathway. Chem. Biol. Interact. 286, 17–25 (2018). [DOI] [PubMed] [Google Scholar]
- 81. Ruggeri, C. et al A specific circulating MicroRNA cluster is associated to late differential cardiac response to doxorubicin‐induced cardiotoxicity in vivo. Dis. Markers. 2018, 8395651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chaudhari, U. , Nemade, H. , Gaspar, J.A. , Hescheler, J. , Hengstler, J.G. & Sachinidis, A. MicroRNAs as early toxicity signatures of doxorubicin in human‐induced pluripotent stem cell‐derived cardiomyocytes. Arch. Toxicol. 90, 3087–3098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pakravan, G. et al Downregulation of miR‐130a, antagonized doxorubicin‐induced cardiotoxicity via increasing the PPARγ expression in mESCs‐derived cardiac cells. Cell Death Dis. 9, 758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hanousková, B. et al Imatinib‐induced changes in the expression profile of microRNA in the plasma and heart of mice‐a comparison with doxorubicin. Biomed. Pharmacother. 115, 108883 (2019). [DOI] [PubMed] [Google Scholar]
- 85. Gioffré, S. et al Plasmatic and chamber‐specific modulation of cardiac microRNAs in an acute model of DOX‐induced cardiotoxicity. Biomed. Pharmacother. 110, 1–8 (2019). [DOI] [PubMed] [Google Scholar]
- 86. Zhao, L. , Tao, X. , Qi, Y. , Xu, L. , Yin, L. & Peng, J. Protective effect of dioscin against doxorubicin‐induced cardiotoxicity via adjusting microRNA‐140‐5p‐mediated myocardial oxidative stress. Redox Biol. 16, 189–198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhao, L. et al MicroRNA‐140‐5p aggravates doxorubicin‐induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 15, 284–296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Horie, T. et al Acute doxorubicin cardiotoxicity is associated with miR‐146a‐induced inhibition of the neuregulin‐ErbB pathway. Cardiovasc. Res. 87, 656–664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Holmgren, G. , Synnergren, J. , Andersson, C.X. , Lindahl, A. & Sartipy, P. MicroRNAs as potential biomarkers for doxorubicin‐induced cardiotoxicity. Toxicol. In Vitro 34, 26–34 (2016). [DOI] [PubMed] [Google Scholar]
- 90. Wan, G.‐X. , Cheng, L. , Qin, H.‐L. , Zhang, Y.‐Z. , Wang, L.‐Y. & Zhang, Y.‐G. MiR‐15b‐5p is involved in doxorubicin‐induced cardiotoxicity via inhibiting Bmpr1a signal in H9c2 cardiomyocyte. Cardiovasc. Toxicol. 19, 264–275 (2019). [DOI] [PubMed] [Google Scholar]
- 91. Beji, S. et al Doxorubicin upregulates CXCR4 via miR‐200c/ZEB1‐dependent mechanism in human cardiac mesenchymal progenitor cells. Cell Death Dis. 8, e3020 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tony, H. , Yu, K. & Qiutang, Z. MicroRNA‐208a silencing attenuates doxorubicin induced myocyte apoptosis and cardiac dysfunction. Oxid. Med. Cell. Longev. 2015, 597032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Oliveira‐Carvalho, V. , Ferreira, L.R.P. & Bocchi, E.A. Circulating mir‐208a fails as a biomarker of doxorubicin‐induced cardiotoxicity in breast cancer patients. J. Appl. Toxicol. 35, 1071–1072 (2015). [DOI] [PubMed] [Google Scholar]
- 94. Vacchi‐Suzzi, C. et al Perturbation of microRNAs in rat heart during chronic doxorubicin treatment. PLoS One 7, e40395 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sun, H. et al Nucleolin protects against doxorubicin‐induced cardiotoxicity via upregulating microRNA‐21. J. Cell. Physiol. 233, 9516–9525 (2018). [DOI] [PubMed] [Google Scholar]
- 96. Tong, Z. et al MiR‐21 Protected cardiomyocytes against doxorubicin‐induced apoptosis by targeting BTG2. Int. J. Mol. Sci. 16, 14511–14525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Du, J. et al Inhibition of miR‐23a attenuates doxorubicin‐induced mitochondria‐dependent cardiomyocyte apoptosis by targeting the PGC‐1α/Drp1 pathway. Toxicol. Appl. Pharmacol. 369, 73–81 (2019). [DOI] [PubMed] [Google Scholar]
- 98. Jing, X. , Yang, J. , Jiang, L. , Chen, J. & Wang, H. MicroRNA‐29b regulates the mitochondria‐dependent apoptotic pathway by targeting bax in doxorubicin cardiotoxicity. Cell. Physiol. Biochem. 48, 692–704 (2018). [DOI] [PubMed] [Google Scholar]
- 99. Roca‐Alonso, L. et al Myocardial MiR‐30 downregulation triggered by doxorubicin drives alterations in β‐adrenergic signaling and enhances apoptosis. Cell Death Dis. 6, e1754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lai, L. , Chen, J. , Wang, N. , Zhu, G. , Duan, X. & Ling, F. MiRNA‐30e mediated cardioprotection of ACE2 in rats with doxorubicin‐induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci. 169, 69–75 (2017). [DOI] [PubMed] [Google Scholar]
- 101. Yin, Z. et al miR‐320a mediates doxorubicin‐induced cardiotoxicity by targeting VEGF signal pathway. Aging (Albany, NY) 8, 192–207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Piegari, E. et al MicroRNA‐34a regulates doxorubicin‐induced cardiotoxicity in rat. Oncotarget 7, 62312–62326 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Desai, V.G. et al Early biomarkers of doxorubicin‐induced heart injury in a mouse model. Toxicol. Appl. Pharmacol. 281, 221–229 (2014). [DOI] [PubMed] [Google Scholar]
- 104. Zhu, J.N. et al Activation of miR‐34a‐5p/Sirt1/p66shc pathway contributes to doxorubicin‐induced cardiotoxicity. Sci. Rep. 7, 11879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Revathidevi, S. et al Screening for the 3’UTR polymorphism of the PXR gene in South Indian breast cancer patients and its potential role in pharmacogenomics. Asian Pac. J. Cancer Prev. 17, 3971–3977 (2016). [PubMed] [Google Scholar]
- 106. Wang, J.X. et al MicroRNA‐532‐3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. 6, e1677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]