

Abstract
Antibody drug conjugates (ADCs) provide targeted delivery of cytotoxic agents directly inside tumor cells. However, many ADCs targeting solid tumors have exhibited limited clinical efficacy, in part, due to insufficient penetration within tumors. To better understand the relationship between ADC tumor penetration and efficacy, previously applied Krogh cylinder models that explore tumor growth dynamics following ADC administration in preclinical species were expanded to a clinical framework by integrating clinical pharmacokinetics, tumor penetration, and tumor growth inhibition. The objective of this framework is to link ADC tumor penetration and distribution to clinical efficacy. The model was validated by comparing virtual patient population simulations to observed overall response rates from trastuzumab‐DM1 treated patients with metastatic breast cancer. To capture clinical outcomes, we expanded upon previous Krogh cylinder models to include the additional mechanism of heterogeneous tumor growth inhibition spatially across the tumor. This expansion mechanistically captures clinical response rates by describing heterogeneous ADC binding and tumor cell killing; high binding and tumor cell death close to capillaries vs. low binding, and high tumor cell proliferation far from capillaries. Sensitivity analyses suggest that clinical efficacy could be optimized through dose fractionation, and that clinical efficacy is primarily dependent on the ADC‐target affinity, payload potency, and tumor growth rate. This work offers a mechanistic basis to predict and optimize ADC clinical efficacy for solid tumors, allowing dosing strategy optimization to improve patient outcomes.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Antibody drug conjugates (ADCs) provide targeted delivery of potent cytotoxic agents to tumor cells via membrane antigen binding and internalization. Although it is known that ADC efficacy is diminished by heterogeneous disposition and binding within solid tumors, optimal dosing strategies to homogenize disposition across solid tumors remains unclear.
WHAT QUESTION DID THIS STUDY ADDRESS?
What is the impact of various dosing regimens on the intra‐tumoral disposition of ADCs in solid tumors and subsequent impact on tumor volume reduction?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study provides a mechanistic modeling framework that describes ADC pharmacokinetics and tumor penetration by incorporating tumor growth inhibition via ADC binding radially across solid tumors, mechanistically linked to clinical response rates and relapse or resistance to ADC therapies.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This work provides a mechanistic basis of ADC efficacy in solid tumors, allowing improvements in dosing strategy to improve patient outcomes.
Antibody drug conjugates (ADCs) provide targeted delivery of potent cytotoxic agents to tumor cells by binding to antigens that are specifically expressed and/or upregulated on tumor cells. The eight ADCs that are currently approved and commercialized as therapeutic agents for treatment of multiple solid and hematological malignancies have heralded in a new wave of clinical development of ADCs, 1 with an additional upcoming approval in relapsed/refractory myeloma. There are currently > 50 ADCs in clinical development for various hematological malignancies and solid tumors, and > 200 clinical trials involving ADCs. 2 Despite the clinical successes with ADCs, designing a successful ADC therapy is complex and primarily involves balancing efficacy and adverse effects associated with the potent cytotoxic agent conjugated to the antibody.
Recent research has identified that increasing ADC penetration of solid tumors increases both treatment efficacy and survival. 3 Mechanistic Krogh cylinder models have been developed previously to capture the relationship between ADC tumor penetration and efficacy, 4 , 5 , 6 , 7 , 8 , 9 , 10 suggesting that fractionating dosing 11 or co‐administering ADCs with naked antibodies improve solid tumor penetration 10 in preclinical species. Although these models provide a basis to explore ADC mechanisms preclinically, analogous mechanistic models for optimizing ADC clinical efficacy in solid tumors have not been reported. Additionally, incorporating mechanisms and virtual clinical populations to capture key clinical outcomes, such as resistance/relapse and overall response rate, may provide insight into properties driving clinical efficacy while providing a platform to model ADC dosing for solid tumors clinically.
In the current work, we have developed a mechanistic model framework, composed of three integrated submodels for ADCs, that is parameterized with: (i) human pharmacokinetics (PKs) using a minimal physiologically‐based pharmacokinetic (PBPK) model, (ii) spatial distribution of ADCs across the tumor using a Krogh cylinder model, and (iii) a tumor growth inhibition model. The model was extended from previously published models by incorporating heterogeneous ADC binding and tumor growth inhibition spatially across the tumor, mechanistically capturing clinical response rate and relapse/resistance. The model was calibrated and verified using reported clinical data for Ado‐trastuzumab Emtansine (T‐DM1) and was used to identify the key ADC parameters that impact the spatial distribution and tumor growth inhibition properties of T‐DM1 as a case study for ADCs. In addition, the model was used to characterize the impact of dose and dosing schedule on the intra‐tumor spatial distribution of ADCs and the resulting impact on tumor volume reduction.
METHODS
Model development
A mechanistic ADC PK/clinical response model was developed using the Simbiology application in MATLAB version 2017a (Mathworks, Natick, MA). Representative Model Code in Systems Biology Markup Language (SBML) format is provide in the supplementary information. Figure 1 provides an overview of the strategy used for the model development and application. Three submodels were sequentially built and linked to create an integrated model that captures ADC PK (Figure 2a ), distribution across the tumor (Figure 2b ), and clinical response (Figure 2c ). Model parameters were derived from literature or optimized to clinical data, as summarized in Table S1 .
Figure 1.
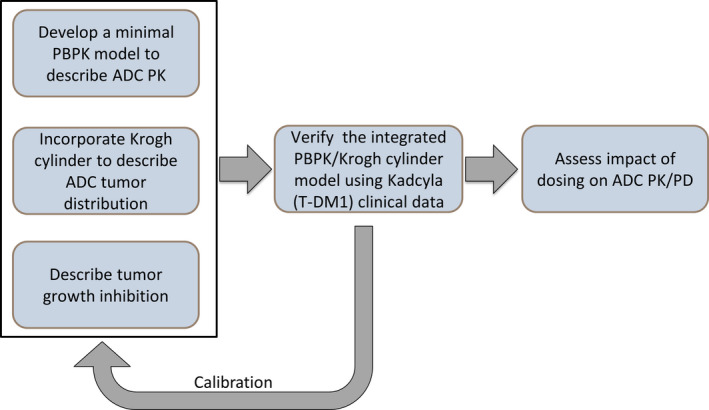
Model development workflow. First, the ADC model was developed by integrating three submodels: (1) a minimal PBPK submodel to capture ADC PK, (2) a Krogh cylinder submodel to capture ADC distribution within the tumor, and (3) a tumor growth inhibition submodel to capture ADC PD. The integrated ADC model was next verified with T‐DM1 clinical PK/PD in metastatic breast cancer as a test case. The verified model was then applied to examine the impact of dosing on PK/PD of a hypothetical ADC. ADC, antibody‐drug conjugate; PBPK, physiologically‐based pharmacokinetic; PD, pharmacodynamic; PK, pharmacokinetic; T‐DM1, Ado‐trastuzumab emtansine.
Figure 2.
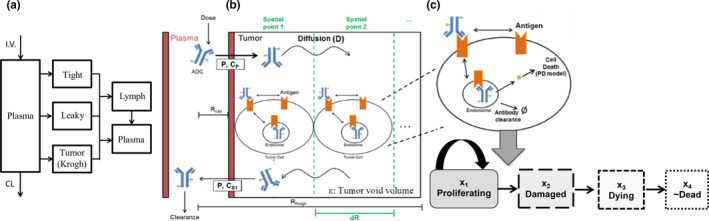
Schematic of the mechanistic ADC model. (a) A minimal PBPK model captures ADC PK and disposition to plasma, lymph, tight, leaky, and tumor vasculature compartments. Tight and leaky tissue compartments represent lumped organs as described in ref. 13 ADC penetration from the tumor vasculature into and across the tumor is modeled by (b) the Krogh cylinder. The Krogh radius (RKrogh) defines the maximal distance the ADC diffuses into the tumor (i.e., RKrogh is half the intercapillary distance within the tumor). ADC diffusion (D) across the tumor is modeled at discrete spatial points at distances dR apart. ADC penetration across the capillary wall is driven by the capillary radius (Rcap), capillary permeability (Cp), and the ADC concentration in the tumor vasculature (CP), and at the spatial point next to the capillary wall in the tumor (CS1). The ADC concentration within the tumor is modified by the tumor void volume (ɛ). (c) At each spatial point, the ADC binds to the antigen present on the cell surface and is internalized, where the toxin is released via antibody degradation and/or linker cleavage. The released toxin causes cell death which is captured by the Simeoni tumor growth inhibition model. 20 The tumor growth inhibition is modeled by four transit compartments, labeled here as different cell populations for conceptual ease: proliferating, damaged, dying, or nearly dead cells. ADC, antibody‐drug conjugate; CL, clearance; PBPK, physiologically‐based pharmacokinetic; PD, pharmacodynamics.
Minimal PBPK model of T‐DM1
A minimal PBPK submodel, based on published PBPK models for antibodies, 12 , 13 was built to capture T‐DM1 PK (Figure 2a ). The minimal PBPK model consists of plasma, lymph, leaky tissue, tight tissue, and tumor vasculature compartments. The tumor vasculature compartment estimates ADC available to the tumor as input to the Krogh cylinder submodel, detailed in the next section. All ADC species are assumed to have the same drug antibody ratio (DAR), given by the observed average DAR (3.5 for T‐DM1). All toxin molecules are assumed to deconjugate from the antibody simultaneously when the antibody is degraded or nonspecific linker deconjugation occurs. The plasma ADC concentration ([ADCP]) is determined by:
where V p is the plasma volume, tt refers to tight tissue, lt refers to leaky tissue, tm refers to tumor vasculature, lm refers to lymph, and σ are the tissue reflection coefficients. Brackets indicate ADC concentration within the specific tissue. refers to the plasma flow rate to each tissue, and refers to the lymph flow rate from a tissue, where is the lymph flow rate to plasma. is linear clearance via nonspecific linker deconjugation, whereas V max and describe nonlinear systemic clearance, chosen due to T‐DM1 exhibiting nonlinear elimination. 14 Dose defines the i.v. dose amount/rate of T‐DM1.
The ADC concentration in non‐tumorous tissue j (tight and leaky) is:
where is the tissue volume.
The unconjugated antibody is formed by ADC clearance via nonspecific linker deconjugation (). Furthermore, total unconjugated trastuzumab systemic clearance in plasma is assumed linear (), taken from the trastuzumab population PKs model. 15 This assumption allows tumor volume reduction (TVR) to be calibrated by the T‐DM1 effect alone, as the relative contributions of T‐DM1 and unconjugated trastuzumab to TVR cannot be determined due to lacking individual patient data. This assumption is supported by clinical and preclinical data showing that T‐DM1 produces cell death and TVR, whereas single‐agent trastuzumab induces cytostasis. 16 , 17
Tumor biodistribution model
A Krogh cylinder submodel was built to capture the ADC spatial distribution across the tumor (Figure 2b ), as described in the Supplementary Methods . The Krogh submodel was expanded from previously published models 8 , 18 , 19 to incorporate the radial tumor mass at each individual spatial point. The radial tumor mass dynamically changes based on tumor cell growth and death at each spatial point, as described in the next section. The radial antigen concentration changes dynamically with the radial tumor mass by altering the antigen synthesis rate at each spatial point i () as follows:
where wi is the radial tumor mass for each spatial point, and t 0 indicates baseline (time = 0). As the ADC spatial distribution across the tumor cannot be verified with clinical data, model predicted treatment efficacy was calibrated to clinical overall response rate (ORR) for T‐DM1 in metastatic breast cancer, as described in the next section.
Tumor volume model
Radial tumor volume is captured based on the tumor growth inhibition model by Simeoni et al. (Figure 2c ), 20 as described in the Supplementary Methods . The Simeoni model was modified to account for radial tumor growth and inhibition as a function of the proliferating cells at each spatial point as follows:
where L 0 is the tumor exponential growth rate, L 1 is the tumor linear growth rate, Ψ is a constant that allows the tumor growth to switch from exponential to linear growth, is the maximal tumor cell killing by the toxin, is the toxin potency, and is the radial intracellular toxin concentration. Total tumor volume is determined by the sum of radial tumor volumes.
Model calibration and verification
This model was designed to explore ADC clinical efficacy in solid tumors using T‐DM1 as a case study. The model was calibrated to T‐DM1 PK by fitting the clearance parameters CL_deconj (nonspecific linker deconjugation) and maximal rate of metabolism (Vmax) and Km (ADC systemic clearance; Table S1 ) to observed clinical PK (peak plasma concentration (Cmax) and area under the curve (AUC)) of T‐DM1 in metastatic breast cancer. 21 T‐DM1 clinical efficacy was calibrated to the ORRs from the EMILIA study 22 and was verified using the ORRs from the MARIANNE study. 23 The lack of publicly available longitudinal tumor volume data led us to use the ORR end point as a measure of verification of the model. Clinical efficacy was calibrated by fitting the Emax parameter (Table S1 ) to the reported clinical ORRs from monotherapy T‐DM1 dosed at 3.6 mg/kg in metastatic breast cancer. 23 The observed ORR was calculated as the percent of responsive patients, those achieving complete response (CR; tumor undetectable) or partial response (PR; sum of longest diameters reduced > 30% from baseline), defined by the RECIST criteria. 24 The model was verified by predicting the ORR from MARIANNE. 23 Predicted ORR was determined by simulating 500 patients across 100 trials by varying parameters shown in Table S1 .
Sensitivity analyses
A generalized ADC and tumor were modeled to assess the impact of parameters and dosing schedule on TVR. The impact of dosing schedule on TVR was simulated while accounting for dose fractionation, such that the total dose was maintained constant across dosing scenarios of every week (QW), every 2 weeks (Q2W), every 3 weeks (Q3W), and every 4 weeks (Q4W). Confidence intervals for ORR simulations were obtained by simulating 500 patients across 10 trials. Local sensitivity analysis was performed to understand how individual ADC or tumor parameters can be manipulated to direct TVR. The impact of parameters on TVR was determined by ranging each parameter individually across orders of magnitude relative to the baseline parameter value (Table S1 ). Four parameters (K D, IC50, antigen expression, and tumor doubling time) were chosen for sensitivity analyses as they are the parameters most readily tuned by the choice of ADC and tumor type.
RESULTS
Evaluation of minimal PBPK and spatial TVR model of ADC using T‐DM1 PK and ORR in patients with metastatic breast cancer
The minimal PBPK model accurately captures T‐DM1 clinical PK data (Figure 3a ). The model simulated PK parameters were compared with observed PK parameters of T‐DM1 in patients with metastatic breast cancer at doses ranging from 0.1 mg/kg to 3.6 mg/kg (Table S2 ). The model predicted AUC and Cmax across all the dose levels were within 50% prediction error of observed data, resulting in acceptance that the PBPK model construct predicts T‐DM1 PK. In particular, the clinical dose (3.6 mg/kg) was well captured by the model with < 25% prediction error for AUC and Cmax (Table S2 ), and exposures in this cohort (N = 15) were representative of exposures in studies conducted in larger patient populations (N = 272 total). Overall, the PBPK simulated profiles were in agreement with the observed PK profile of total trastuzumab and T‐DM1 (Figure 3a ).
Figure 3.
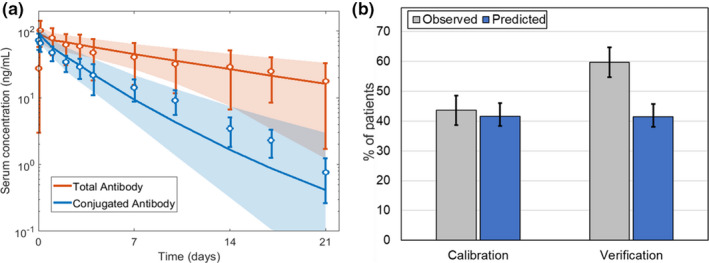
ADC model verification with T‐DM1 clinical pharmacokinetics (PKs)/pharmacodynamics as a test case. (a) Observed (n = 15) vs. predicted plasma PK for total trastuzumab (T‐DM1 conjugated and unconjugated antibody) and T‐DM1 (conjugated antibody only) following a single T‐DM1 clinical dose at 3.6 mg/kg. 21 (b) The observed clinical objective response rates (ORRs) for monotherapy T‐DM1 dosed at 3.6 mg/kg is compared to model predicted. 22 , 23 All observed data are given as median ± 95% confidence interval, and simulated data is represented as the median ± 95% confidence interval represented by the a shaded region or b error bars. Predicted confidence intervals were obtained by simulating 100 virtual patients across 10 groups. AUC, area under the curve; Cmax, maximum concentration.
The model construct was also able to predict the clinical ORR (Figure 3b ) with a prediction error of 30%. The prediction error was mostly attributed to trial‐to‐trial variability that was observed across the clinical trials. In six clinical studies with T‐DM1, the reported ORR ranged from 26% to 64% with a mean of 43 ± 13%. 25 These ORRs are highly dependent on patient characteristics in the specific study and can influence the end points. In the absence of trial specific patient characteristics like human epidermal growth factor receptor 2 (HER2) expression, the virtual patient populations for the calibration and verification simulations were varied randomly. Although the model was optimized by calibrating Emax to overall % ORR, the model accurately predicted the relative contribution of CRs (observed: 1.0%; predicted: 2.0%), PRs (observed: 42.6%; predicted: 40.6%), and stable disease or progressive disease (observed: 56.4%; predicted: 57.4%), as shown in Figure S1 . 22 Importantly, the addition of modeling radial tumor mass was required to accurately predict the relative contribution of complete and partial responses (Figure S1 a). When Emax was optimized to overall % ORR and radial tumor mass was not modeled, the model overpredicted complete responses and underpredicted partial response rates (Figure S1 a). Radial tumor mass modeling captured response rates as it mechanistically describes tumor resistance and relapse (Figure S1 b), resulting from high tumor cell growth far from capillaries relative to high tumor cell death close to capillaries (Figure S1 c). Thus, the model mechanistically represents both PK and variability in post‐treatment TVR as a clinical outcome for ADC therapy.
Influence of ADC properties (target affinity, payload potency, and choice of dosing schedule) on TVR and clinical outcomes
ADC efficacy, measured by TVR, is highly dependent on both the ADC and tumor properties. The impact of key parameters dictating ADC (K D and IC50) and tumor (antigen expression and tumor doubling time) properties on total TVR (Figure 4 top row) and spatial TVR (Figure 4 bottom row) were examined via sensitivity analysis and with respect to dose fractionation (QW, Q2W, Q3W, or Q4W dosing; Figure 4 ), whereas keeping the total dose administered over the 4 week period constant. These four parameters were chosen as they are sensitive to driving TVR (Figure S2 ) and are parameters that are either tunable based on the manufactured properties of the ADC (K D and IC50) or are clinically measurable properties of the tumor (antigen expression and tumor doubling time). Sensitivity analysis revealed an optimal K D range to achieve maximal TVR, when all other parameters held constant; a K D too low or too high is ineffective at inhibiting tumor growth (Figure 4a ). Conversely, IC50 (Figure 4b ) values exhibit linear TVR responses that follow an anticipated distribution. As toxin potency increases (lower IC50), the ability to kill tumor cells increases (Figure 4b ). Similar to the scenario with K D, an optimal range for antigen expression is observed (Figure 4c ) due to the close relationship antigen expression and K D have in determining target binding. The tumor doubling time expressed a linear response with TVR (i.e., as the tumor doubling time increased, the ability to kill the tumor decreased (Figure 4d )), indicating the tumor cell death rate by the toxin cannot overcome the tumor cell proliferation rate.
Figure 4.
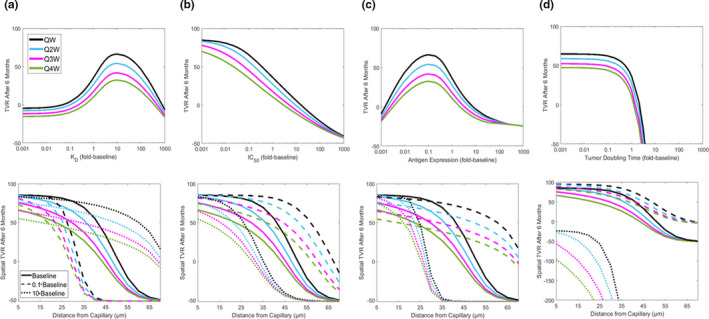
ADC model sensitivity analysis. Sensitivity analyses were conducted on two key parameters tunable by the ADC properties (a) dissociation constant (K D) between the ADC and antigen and (b) toxin potency (IC50), and two parameters based on the tumor type (c) antigen expression and (d) tumor doubling time defined by the linear tumor growth parameter. (Top row) The total TVR after 6 months of treatment was determined by altering each parameter across a range of three orders of magnitude lower and three orders of magnitude higher than the model baseline value. (Bottom row) Spatial TVR, the reduction in tumor volume at each spatial point across the tumor, was determined after 6 months of treatment for each parameter at baseline and 1 order of magnitude higher and lower than baseline. For each sensitivity analysis, the ADC was fractionated to QW, Q2W, Q3W, and Q4W dosing. ADC, antibody‐drug conjugate; IC50, half maximal inhibitory concentration; K D, dissociation constant; TVR, tumor volume reduction.
This sensitivity analysis also showed that ADC and tumor properties impact the ability of the ADC to inhibit growth across the tumor (Figure 4 bottom row). Although high tumor cell reduction was achieved close to capillaries (< 20 µm) for nearly all parameter sets and dosing frequencies, the ADC and tumor parameters both drastically alter tumor cell reduction far from capillaries (> 55 µm). For example, although it may be counterintuitive that increasing K D leads to higher efficacy, sensitivity analysis shows that maximal TVR is observed with K D = 5 nM (10‐fold the baseline value) as it allows the ADC to distribute and bind across a tumor with antigen expression of 106 receptors/cell (Figure 4a ). Thus, tumor cells farthest from capillaries (70 µm) are growth inhibited at K D = 5 nM (spatial TVR ≈ 0%), whereas with lower K D = 0.05 nM (0.1‐fold the baseline value) the ADC binds so strongly to tumor cells near the capillary that it does not distribute across the entire tumor (spatial TVR far from capillary ≈ −50%; Figure 4a ). Overall, this sensitivity analysis reveals that the success of an ADC therapy is driven by the ability of the ADC to target tumor cells farthest from capillaries.
For all parameter sets, increased TVR resulted from dose fractionation with more frequent dosing, with QW dosing achieving up to ~ 30% greater TVR than Q4W dosing (Figure 4 top). Although dose fractionation results in lower maximal receptor occupancy (RO), ~ 40% average ROmax across the tumor for QW compared with ~ 65% for Q4W, fractionated dosing maintains higher average RO across time, ~ 35% ROmean for QW, and ~ 20% for Q4W over 1 month (Figure 5a,b ). This more frequent dosing provides near constant RO across the tumor over a 28‐day cycle for QW dosing (Figure 5c ), whereas Q4W dosing loses target engagement by the end of the cycle (Figure 5d ). Additionally, fractionated dosing leads to lower off‐target maximum and average RO (Figure S3 ), which indicates fractionated dosing is also favorable in terms of safety. Overall, model predictions indicate that sustained RO across the tumor achieved by fractionated ADC dosing is safer and more effective than less frequent higher doses.
Figure 5.
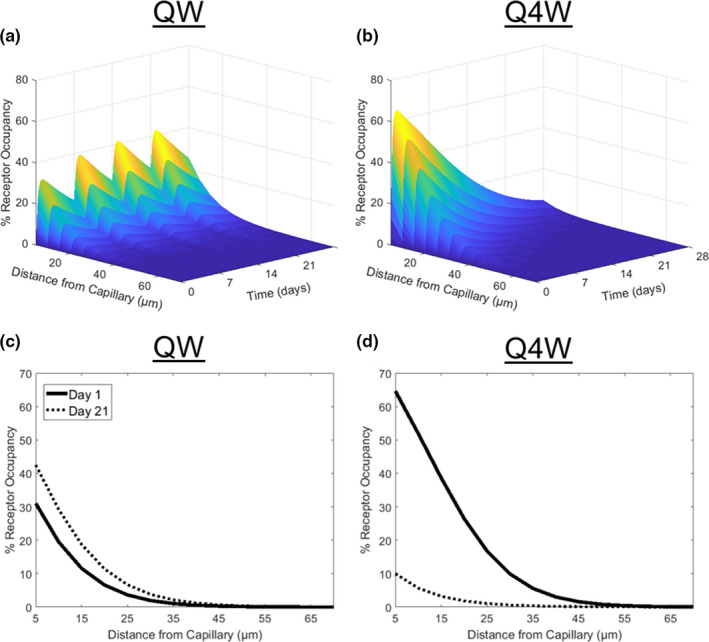
Impact of fractionated ADC dosing on receptor occupancy. (a, b) Simulated percent receptor occupancy across time and a theoretical tumor over 1 month is given for ADC dosing fractionated at (a) 10 mg QW or (b) 40 mg Q4W. RO radially across the tumor at day 1 (solid line) and day 21 (dashed line) are given for the (c) QW and (d) Q4W dose schedules.
Interactions across multiple parameters can also be elucidated with this model construct. Take the interaction between payload potency and target expression as a case study (Figure 6 ). Tumor cells with low target expression will be virtually unaffected by an ADC with low payload potency (Figure 6 bottom left). Although partial tumor reduction can be achieved with the same low potency payload in tumors with high target expression, the treatment would be ineffective overall as deep tumor cells far from capillaries are not affected (Figure 6 bottom right). Conversely, high payload potency can be effective in tumors with both low and high target expression (Figure 6 top). This case study highlights the ability for the model to predict efficacy given specific ADC and tumor properties of interest.
Figure 6.
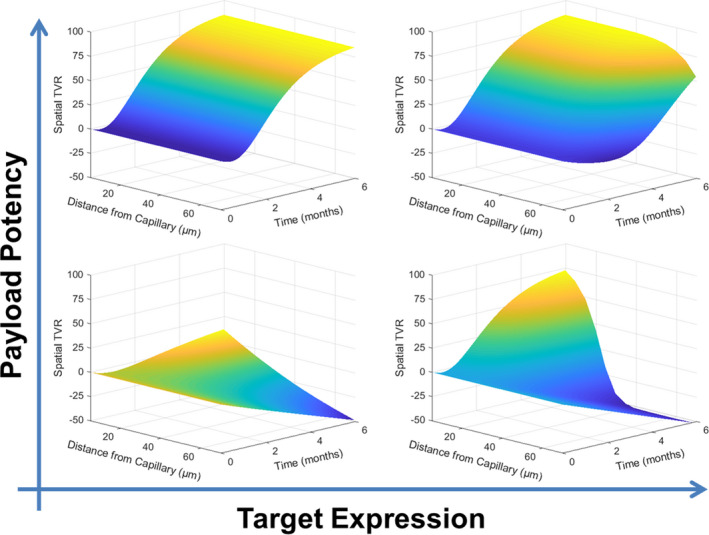
Impact of payload potency and target expression on percent tumor kill. A hypothetical ADC with either high (IC50 = 10 pM) or low (IC50 = 0.10 µM) potency targeted a generalized tumor with either high (107 receptors/cell) or low (105 receptors/cell) antigen expression. Spatial TVR, the reduction in tumor volume at each spatial point across the tumor, is given as a function of time in each scenario. ADC, antibody‐drug conjugate; IC50, half maximal inhibitory concentration; RO, receptor occupancy; TVR, tumor volume reduction.
Extending sensitivity analysis into virtual patient populations quantifies the relationship between TVR and clinical response rates (Figure 7 ). These population simulations provide insight into when dose fractionation may or may not be beneficial for efficacy end points. For example, given an ADC dosed Q4W, a compromise could be reached to allow the ADC to have weaker affinity if the dose is fractionated to maintain or increase efficacy (Figure 7a ). Such a compromise may not be feasible for the toxin as dose fractionation does not increase efficacy if a less potent toxin is used (Figure 7b ). The model allows a similar argument to be made around tumor selection; lower antigen expression is permissible with dose fractionation to increase efficacy (Figure 7c ), but increased efficacy cannot be achieved without a higher dose for fast growing tumors (Figure 7d ). This case study highlights the potential for the model construct to predict probability of success for ADCs and tumor properties.
Figure 7.
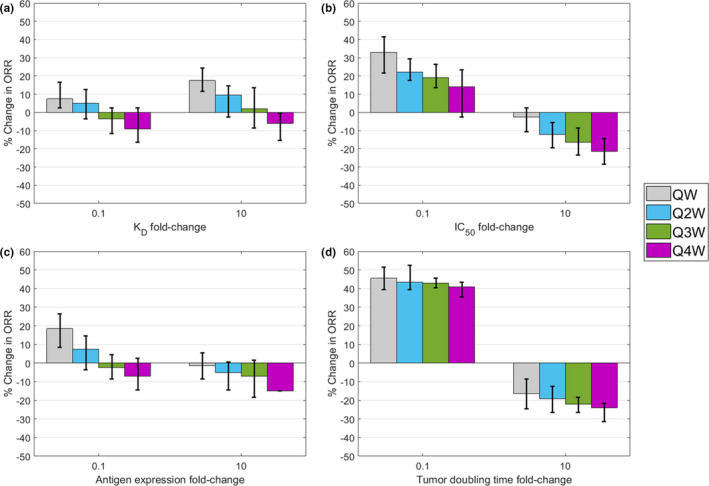
Relationship between dose fractionation and parameters on % ORR. The % ORR after 6 months of treatment was determined for patients receiving doses fractionated to QW, Q2W, Q3W, or Q4W using baseline parameter values (Table S1 ). The effects of four key parameters (a) K D, (b) IC50, (c) antigen expression, and (d) tumor doubling time for each dose schedule were determined by altering the parameter 10‐fold higher and lower than the baseline value. The % ORR given parameter alteration and dose schedule are represented as the difference from the % ORR given by Q4W dose scheduling with baseline parameters. A positive % ORR indicates higher efficacy, and a negative % ORR indicates lower efficacy, relative to Q4W dose scheduling with baseline parameters. Virtual patients were defined by calibrating parameter variability to the clinical response rate observed for T‐DM1. 22 Variability was estimated by simulating 100 random patients 10 times for each treatment condition, and error bars represent the 95% confidence interval. ORR, Overall response rate; IC50, half maximal inhibitory concentration; K D, dissociation constant.
DISCUSSION
In this work, a quantitative ADC modeling framework that mechanistically describes PK, pharmacodynamics, and clinical efficacy was developed and verified. The model utility, as described by the example of T‐DM1 in metastatic breast cancer, provides a mechanistic translational approach to enable decision making during drug development with respect to ADC property design, clinical population selection, and dose optimization. The model predictions of tumor volume change and clinical efficacy, along with recent experimental advances, highlight the importance of understanding ADC distribution within tumors and suggests greater utility of the modeling and simulation framework as described herein.
Extensive studies in preclinical species have shown that antibodies and ADCs suffer from poor penetration in solid tumors. 10 These studies have used antibodies and ADCs conjugated with near‐infrared fluorophores and confocal microscopy of histology slices to show the heterogenous distribution of these biologics in tumor xenograft mouse models. These findings were compared with predictions from a Krogh cylinder model, both showing that these biologics tend to have a perivascular distribution in vivo, thereby validating the use of the Krogh cylinder model. Other studies included dosing antibodies along with ADCs to improve the spatial distribution and efficacy of these ADCs. 3 The Krogh cylinder model was able to predict these results, increasing confidence in the ability of the model to make predictions about the spatial distribution of antibodies and ADCs in solid tumors.
This model provides a mechanistic basis supporting that dose fractionation may be beneficial for clinical efficacy while reducing toxicity concerns, consistent with the recent clinical learning from gemtuzumab ozogamicin 26 , 27 and additional preclinical studies, 28 , 29 along with clinical benefits observed from other therapies. 30 , 31 It should be noted that these model predictions of potential benefit from dose fractionation stems from predicted RO by the ADC in leaky tissue and does not account for toxicity concerns by the unconjugated toxin, 32 , 33 which is outside the scope of this study. Hinrichs et al. have shown that, for ADCs with pyrrolobenzodiazepine payloads, dose fractionation results in similar antitumor activity in mouse xenograft models, as efficacy closely related to ADC exposure. 28 Additionally, dose fractionation gave a better safety profile in rats and monkeys, as toxicity was associated with Cmax. 28 Clinical evidence also showed that dose fractionation resulted in better efficacy and safety of gemtuzumab ozogamicin in hematological malignancies. 34 Whereas studies such as these provide preclinical and clinical evidence in favor of ADC dose fractionation, no clinical evidence of ADC dose fractionation efficacy/safety in solid tumors exist to our knowledge. Testing the hypothesis that ADC dose fractionation improves clinical efficacy and safety profiles in solid tumors may address the question of its benefit over the traditional dosing strategies and will have the potential to greatly improve patient outcomes. An important consideration for dose fractionation is the potential target mediated drug disposition (TMDD) of ADCs. TMDD is highly dependent on the target and the specific ADC but can cause significant nonlinearity in the PKs of ADCs. For ADCs that undergo TMDD, low doses have an increased clearance that will lead to reduced efficacy. Therefore, the dose selection of fractionated doses will be critical in achieving high efficacy.
Another utility of the modeling framework is in translational simulation scenarios to evaluate dose fractionation of ADC therapies, which may have not been adopted by clinicians due to toxicity concerns seen with other therapies. 35
The model sensitivity analyses provided mechanistic insight into how ADCs and tumor properties can be stratified to predict population responses. For example, sensitivity analysis led to a counterintuitive hypothesis that lower ADC affinity to the target increases tumor targeting and killing. Mechanistically, this appears due to a lower affinity allowing the ADC to penetrate deeper into the tumor and bind to the target more homogenously across the tumor, a prediction verified by recent in vivo findings. 36 Another hypothesis derived from sensitivity analysis is that the tumor growth rate is a critical parameter affecting the success of the ADC therapy. Clinical evidence supports this hypothesis that higher tumor growth rates result in decreased efficacy; the anti‐PD1 antibody toripalimab showed a 83.9% response rate (partial response + stable disease) in patients with low pretreatment tumor growth rates compared with a 26.5% a response rate in patients with high pretreatment tumor growth rates. 37 Tumor growth rates have also been identified as a biomarker of clinical survival (progression free and overall) across multiple cancers. 38 , 39 Overall, this model allows for relationships between ADC or tumor properties with clinical outcome to be explored and understood mechanistically.
Although this ADC construct provides a mechanistic basis to model dosing based on both ADC and tumor properties, there are current limitations outside the scope of this study that can be addressed in future model extensions to provide greater pharmacological relevancy. The major limitation is that there is limited clinical data (T‐DM1) to verify this model framework. An additional limitation is the model does not describe payload PKs and disposition dynamics. Free DM1 disposition dynamics was not included in this model because free DM1 is assumed to not contribute to TVR 21 and DM1 has been shown to have minimal bystander killing effect in vitro. 40 , 41 However, applicability to other ADCs may necessitate incorporating additional payload considerations, particularly applying a DAR range rather than average DAR used here, or including payload disposition if there is significant bystander effect. Payload release is complicated by multiple contribution routes, including release by on‐target antigen binding, off‐target binding to antigen expressed in healthy tissues or Fc receptors, and nonspecific deconjugation. 42 , 43 Understanding payload deconjugation mechanistically can help optimize dosing or ADC properties, such as linker type.
Modeling drug‐drug interaction potential would also be greatly beneficial to clinical trial design. Payloads particularly can be victims or perpetrators of enzymes and transporters, such as brentuximab vedotin, which uses the monomethyl auristatin E payload that is a substrate and inhibitor of CYP3A4. 44 Currently, the tumor is assumed spatially homogenous with regard to cell type, antigen expression, and perfusion, whereas tumors are known to be highly heterogeneous. 45 , 46 Incorporating tumor heterogeneity may improve tumor growth and efficacy predictions by allowing the model to capture the variability that is seen across trials. It can also be useful in extending this framework to other tumor types that have varying levels of heterogeneity. This information is typically qualitative in nature and is based on tumor biopsies from patients but implementation in the model would require detailed and robust quantitative data. Model updates to incorporate additional biology will provide greater insights into ADC pharmacology and extend model applications to additional ADC design considerations and clinical trial optimization.
This work provides a framework for mechanistic understanding of the interplay between ADCs and tissue heterogeneity to identify optimal dosing strategies and potentially improve patient outcomes.
Funding
This work was sponsored by AbbVie Inc. AbbVie contributed to the study design, research, and interpretation of data, and the writing, review, and approval of the publication.
Conflict of Interest
S.B. and M.S. are employees and may hold AbbVie stock or stock options. J.W., M.S.C., and J.P.G. are former employees of AbbVie and may hold AbbVie stock or stock options.
Author Contributions
All authors wrote the manuscript, designed the research, performed the research, and analyzed the data.
Data Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Table S2
Supplementary Material
Supplementary Material
Acknowledgment
The authors acknowledge the medical writing support that was provided by Wesley Wayman, an employee of AbbVie.
References
- 1. Goldenberg, D.M. & Sharkey, R.M. Sacituzumab govitecan, a novel, third‐generation, antibody‐drug conjugate (ADC) for cancer therapy. Expert Opin. Biol. Ther. 20, 871–885 (2020). [DOI] [PubMed] [Google Scholar]
- 2. Dott, J. , Abila, B. & Wuerthner, J.U. Current trends in the clinical development of antibody‐drug conjugates in oncology. Pharm. Med. 32, 259–273 (2018). [Google Scholar]
- 3. Cilliers, C. , Menezes, B. , Nessler, I. , Linderman, J. & Thurber, G.M. Improved tumor penetration and single‐cell targeting of antibody‐drug conjugates increases anticancer efficacy and host survival. Cancer Res. 78, 758–768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah, D.K. , Haddish‐Berhane, N. & Betts, A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab‐vedotin. J. Pharmacokinet. Pharmacodyn. 39, 643–659 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Shah, D.K. et al A priori prediction of tumor payload concentrations: preclinical case study with an auristatin‐based anti‐5T4 antibody‐drug conjugate. AAPS J. 16, 452–463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh, A.P. et al Evolution of antibody‐drug conjugate tumor disposition model to predict preclinical tumor pharmacokinetics of trastuzumab‐emtansine (T‐DM1). AAPS J. 18, 861–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khot, A. , Tibbitts, J. , Rock, D. & Shah, D.K. Development of a translational physiologically based pharmacokinetic model for antibody‐drug conjugates: a case study with T‐DM1. AAPS J. 19, 1715–1734 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vasalou, C. , Helmlinger, G. & Gomes, B. A mechanistic tumor penetration model to guide antibody drug conjugate design. PLoS One 10, e0118977 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Singh, A.P. & Shah, D.K. Measurement and mathematical characterization of cell‐level pharmacokinetics of antibody‐drug conjugates: a case study with trastuzumab‐vc‐MMAE. Drug Metab. Dispos. 45, 1120–1132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cilliers, C. , Guo, H. , Liao, J. , Christodolu, N. & Thurber, G.M. Multiscale modeling of antibody‐drug conjugates: connecting tissue and cellular distribution to whole animal pharmacokinetics and potential implications for efficacy. AAPS J. 18, 1117–1130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Betts, A.M. et al Preclinical to clinical translation of antibody‐drug conjugates using PK/PD modeling: a retrospective analysis of inotuzumab ozogamicin. AAPS J. 18, 1101–1116 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Shah, D.K. & Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 39, 67–86 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Cao, Y. , Balthasar, J.P. & Jusko, W.J. Second‐generation minimal physiologically‐based pharmacokinetic model for monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 40, 597–607 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chudasama, V.L. et al Semi‐mechanistic population pharmacokinetic model of multivalent trastuzumab emtansine in patients with metastatic breast cancer. Clin. Pharmacol. Ther. 92, 520–527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruno, R. et al Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother. Pharmacol. 56, 361–369 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Barok, M. , Tanner, M. , Koninki, K. & Isola, J. Trastuzumab‐DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab‐resistant breast cancer cells in vivo. Breast Cancer Res. 13, R46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Teicher, B.A. & Doroshow, J.H. The promise of antibody‐drug conjugates. N. Engl. J. Med. 367, 1847–1848 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Thurber, G.M. & Weissleder, R. A systems approach for tumor pharmacokinetics. PLoS One 6, e24696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khera, E. , Cilliers, C. , Bhatnagar, S. & Thurber, G.M. Computational transport analysis of antibody‐drug conjugate bystander effects and payload tumoral distribution: implications for therapy. Mol. Sys. Design Eng. 3, 73–88 (2018). [Google Scholar]
- 20. Simeoni, M. et al Predictive pharmacokinetic‐pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 64, 1094–1101 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Girish, S. et al Clinical pharmacology of trastuzumab emtansine (T‐DM1): an antibody‐drug conjugate in development for the treatment of HER2‐positive cancer. Cancer Chemother. Pharmacol. 69, 1229–1240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verma, S. et al Trastuzumab emtansine for HER2‐positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perez, E.A. et al Trastuzumab emtansine with or without pertuzumab versus trastuzumab plus taxane for human epidermal growth factor receptor 2‐positive, advanced breast cancer: primary results from the phase III MARIANNE study. J. Clin. Oncol. 35, 141–148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Therasse, P. et al New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 92, 205–216 (2000). [DOI] [PubMed] [Google Scholar]
- 25. Peddi, P.F. & Hurvitz, S.A. Trastuzumab emtansine: the first targeted chemotherapy for treatment of breast cancer. Future Oncol. 9, 319–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Norsworthy, K.J. et al FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33‐positive acute myeloid leukemia. Oncologist. 23, 1103–1108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taksin, A.L. et al High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 21, 66–71 (2007). [DOI] [PubMed] [Google Scholar]
- 28. Hinrichs, M.J.M. et al Fractionated dosing improves preclinical therapeutic index of pyrrolobenzodiazepine‐containing antibody drug conjugates. Clin. Cancer Res. 23, 5858–5868 (2017). [DOI] [PubMed] [Google Scholar]
- 29. Hedrich, W.D. , Fandy, T.E. , Ashour, H.M. , Wang, H. & Hassan, H.E. Antibody‐drug conjugates: pharmacokinetic/pharmacodynamic modeling, preclinical characterization, clinical studies, and lessons learned. Clin. Pharmacokinet. 57, 687–703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ho, K.F. et al IMRT dose fractionation for head and neck cancer: variation in current approaches will make standardisation difficult. Acta Oncol. 48, 431–439 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Manabe, Y. et al Toxicity and efficacy of three dose‐fractionation regimens of intensity‐modulated radiation therapy for localized prostate cancer. J. Radiat. Res. 55, 494–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lucas, A.T. et al Factors affecting the pharmacology of antibody‐drug conjugates. Antibodies 7, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinrichs, M.J. & Dixit, R. Antibody drug conjugates: nonclinical safety considerations. AAPS J. 17, 1055–1064 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pilorge, S. et al Fractionated gemtuzumab ozogamicin and standard dose cytarabine produced prolonged second remissions in patients over the age of 55 years with acute myeloid leukemia in late first relapse. Am. J. Hematol. 89, 399–403 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Farrell, M.J. et al Radiation dose and fractionation for limited‐stage small‐cell lung cancer: survey of US radiation oncologists on practice patterns. Clin. Lung Cancer 20, 13–19 (2019). [DOI] [PubMed] [Google Scholar]
- 36. Tsumura, R. et al Influence of the dissociation rate constant on the intra‐tumor distribution of antibody‐drug conjugate against tissue factor. J. Control Release 284, 49–56 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Tang, B. et al Tumor growth rate as an early indicator of the efficacy of anti‐PD‐1 immunotherapy in advanced melanoma. J. Clin. Oncol. 37, e21050 (2019). [Google Scholar]
- 38. Stein, W.D. et al Tumor regression and growth rates determined in five intramural NCI prostate cancer trials: the growth rate constant as an indicator of therapeutic efficacy. Clin. Cancer Res. 17, 907–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dromain, C. et al Tumor growth rate as a metric of progression, response, and prognosis in pancreatic and intestinal neuroendocrine tumors. BMC Cancer 19, 66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ogitani, Y. , Hagihara, K. , Oitate, M. , Naito, H. & Agatsuma, T. Bystander killing effect of DS‐8201a, a novel anti‐human epidermal growth factor receptor 2 antibody‐drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 107, 1039–1046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van der Lee, M.M. et al The preclinical profile of the duocarmycin‐based HER2‐targeting ADC SYD985 predicts for clinical benefit in low HER2‐expressing breast cancers. Mol. Cancer Ther. 14, 692–703 (2015). [DOI] [PubMed] [Google Scholar]
- 42. Han, T.H. & Zhao, B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody‐drug conjugates. Drug Metab. Dispos. 42, 1914–1920 (2014). [DOI] [PubMed] [Google Scholar]
- 43. Hoffmann, R.M. et al Antibody structure and engineering considerations for the design and function of antibody drug conjugates (ADCs). Oncoimmunology 7, e1395127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen, Y. et al Physiologically based pharmacokinetic modeling as a tool to predict drug interactions for antibody‐drug conjugates. Clin. Pharmacokinet. 54, 81–93 (2015). [DOI] [PubMed] [Google Scholar]
- 45. Thurber, G.M. , Schmidt, M.M. & Wittrup, K.D. Antibody tumor penetration: transport opposed by systemic and antigen‐mediated clearance. Adv. Drug Deliv. Rev. 60, 1421–1434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gillies, R.J. , Schornack, P.A. , Secomb, T.W. & Raghunand, N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia 1, 197–207 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Table S2
Supplementary Material
Supplementary Material