

Abstract
Covalent drugs have been intensively studied in some very important fields such as anti-tumor and anti-virus, including the currently global-spread SARS-CoV-2. However, these drugs may interact with a variety of biological macromolecules and cause serious toxicology, so how to reactivate the inhibited targets seems to be imperative in the near future. Organophosphate was an extreme example, which could form a covalent bound easily with acetylcholinesterase and irreversibly inhibited the enzyme, causing high toxicology. Some nucleophilic oxime reactivators for organophosphate poisoned acetylcholinesterase had been developed, but the reactivation process was still less understanding. Herein, we proposed there should be a pre-reactivated pose during the reactivating process and compounds whose binding pose was easy to transfer to the pre-reactivated pose might be efficient reactivators. Then we refined the previous reactivators based on the molecular dynamic simulation results, the resulting compounds L7R3 and L7R5 were proven as much more efficient reactivators for organophosphate inhibited acetylcholinesterase than currently used oximes. This work might provide some insights for constructing reactivators of covalently inhibited targets by using computational methods.
Keywords: Covalent drugs, Organophosphate, Acetylcholinesterase, Oximes, Pre-reactivated pose
Graphical abstract

1. Introduction
In recent years, aspirin, the penicillins, omeprazole and clopidogrel were found to interact with targets in the form of covalent bonds [1]. It has been recognized that covalent drugs possessed several advantages compared to their non-covalent counterparts, such as longer curative effect, lower treatment dose and less resistance. Consequently, covalent drugs were intensively researched in fields such as anti-cancer and anti-virus [2]. Recently, two promising antiviral drug candidates were reported exhibiting excellent inhibitory activity towards SARS-CoV-2 through a covalent interaction [3]. However, for a long time, it was suggested to avoid introducing electrophilic groups (such as epoxy, acridine, Michael receptor, etc.) into the molecular structure of drugs, because they may interact with a variety of biological macromolecules and cause serious toxicology. For an extreme example, organophosphates (OP) could form a covalent bound easily with the catalytic serine residue of acetylcholinesterase (AChE), and exhibit acute toxicity [4]. As a result, OPs can be used as pesticides (e.g., paraoxon, parathion, and dichlorvos, Fig. 1 ) and nerve agents in armed conflicts (e.g., sarin, VX, tabun and soman, Fig. 1) [5]. The broad use of the pesticides leads to a serious public health issue with about 3,000,000 acute intoxications and over 200,000 fatalities annually worldwide [6,7]. Nerve agents could be used as weapons of mass destruction (Iran-Iraq War) or used for terrorist attacks (e.g., subway attack in Tokyo in 1995) and murder (e.g., Kim Jong-nam’s Killing in Kuala Lumpur in 2017) [8]. Thus it is worth studying reactivation of covalently inhibited targets. In this paper, design, synthesis and evaluation of reactivators for OP inhibited hAChE was used as a prologue, which could provide a good reference for further research about reactivation for covalently inhibited targets.
Fig. 1.

Chemical structures of some organophosphates and currently used pyridinium oxime reactivators.
OPs inhibit AChE irreversibly though phosphorylation of the catalytic serine residue (Ser203) [9], causing accumulation of neurotransmitter acetylcholine (ACh) at both peripheral and central cholinergic synapses, leading to cholinergic crisis, respiratory distress, convulsive seizures, and ultimately death [10,11]. Previously, pyridinium aldoximes (e.g., pralidoxime (2-PAM), obidoxime, HI-6, Fig. 1) were developed as AChE reactivators [[12], [13], [14], [15]]. However, due to their permanent positive charge, these quaternary reactivators are unable to cross the blood-brain barrier (BBB) and show poor central reactivation efficiency [[16], [17], [18]].
To overcome this obstacle, various non-quaternary reactivators were studied, such as monoisonitrosoacetone (MINA) [[19], [20], [21]] (Fig. 1) and amidine-oximes (Fig. 1) [22,23]. These nucleophilic aldoximes were thought to remove the phosphyl moiety from the active site (A site) serine in phosphyl conjugates and restore the enzyme’s activity [24]. But these nonquaternary oximes were much less efficient reactivators than 2-PAM in vitro due to their low affinities to the inhibited enzyme. Noteworthily, it was found that a peripheral site (P-site) was located at the entrance of the active gorge in AChE, which could be served as the binding site for distinctive substrates [25]. In an earlier study, we had firstly reported a series of salicylic aldoxime conjugates, the biological evaluation results confirmed that connection of proper peripheral ligand (PSL) to single aldoximes would dramatically improve reactivating ability for OP inhibited AChE [26]. Enlightened by the isonicotinamide ligand in structure of HI-6, its bioisostere aminobenzamides were introduced as simple PSLs, then the efficient uncharged reactivators L6M1R3 and L6M1R5 (Fig. 3) with ortho-methyl or chloro substituted salicylaldoximes were constructed [27]. However, understanding of the reactivation process was important for rational design of new efficient reactivators [28].
Fig. 3.

Design of novel hybrid nonquaternary salicylaldoxime reactivators.
It was noteworthy that Allgardsson et al. and Driant et al. had used the density functional theory (DFT) and QM/MM approaches to analysis the reactivation mechanism recently. They figured a plausible near-attack conformation of HI-6 and Sarin, in which the oxime oxygen of HI-6 is within van der Waals contact distance (3.3 Å) of the sarin phosphorus atom [29,30]. Based on these important findings, we proposed there should be a pre-reactivated pose following the binding of reactivator with AChE at the binding pose, in which the oxime group located near to the P atom of the modified Ser203. For example, in the crystal structure of VX inhibited hAChE in complex with HI-6 (PDB code: 6CQW) [31], the isonicotinamide was located at the P site, the aldoximes was located at the A site and the distance of the O atom of nucleophilic aldoximes and P atom of VX was 9.5 Å (Fig. 2 ). The aldoximes should overcome the steric hindrance and rotate to reach the pre-reactivated pose before reactivating the inhibited hAChE. Since other OPs would modify the Ser with bigger substrates, the steric hindrance for rotation might be different and thus reduce the scope of application. Hence we investigated the binding interaction of L6M1R3 with inhibited AChE by molecular dynamic (MD) simulations, and proposed that compounds whose binding pose was easy to transfer to the pre-reactivated pose might be more efficient and broad-spectrum non-quaternary reactivators (Fig. 3).
Fig. 2.

Binding interaction of HI-6/inhibited hAChE complex (PDB code: 6CQW). (A) The structure of HI-6/inhibited hAChE complex. The distance of the O atom of nucleophilic aldoximes and the P atom of VX was marked in red. (B) HI-6 located in the P site in surface model. (C) There is a space between HI-6 and VX modified Ser203.
As a proof of concept, various drug induced simple aromatic or aliphatic moieties were screened for proper PSLs, and the efficient methyl or chloro substituted salicylaldoximes were reserved to construct novel reactivators. Encouragingly, the in vitro evaluation demonstrated that the resulting conjugates L7R3 and L7R5 (Fig. 3) exhibited much higher reactivating efficiency than currently used oximes 2-PAM, obidoxime and HI-6 for both nerve agents (VX, sarin and tabun) and pesticides (paraoxon, parathion and dichlorvos) poisoning in most cases. Interestingly, the following MD simulation study found that L7R3 and L7R5 could form stable pre-reactivated poses. This work may provide some insights for constructing efficient reactivators of covalently inhibited targets.
2. Results and discussion
2.1. Design of new reactivator to research pre-reactivated pose
To investigate into the binding interaction of reactivates, molecular docking was carried out to construct the conformation of our previously reported L6M1R3 in the VX-inhibited hAChE by using the “SYBYL-X 2.0” software [32]. Specially, the binding pocket was explored according to the ligand from the crystal structure of VX inhibited hAChE in complex with HI-6 (PDB code: 6CQW, resolution 2.28 Å) [31].
Firstly, we conducted 50 ns molecular dynamics simulations for the complex of hAChE-L6M1R3 to get a further insight into the binding poses and pre-reactivated poses. The root mean square deviation (RMSD) was monitored during the simulation time to investigate the stability of the binding pose of L6M1R3 in hAChE. We found that most of the residues during the molecular dynamics showed slight fluctuation with stable RMSD value lower than 1.8 Å and only a few residues at the terminus had greater variation with big RMSD value (Fig. 4 A). From RMSD analysis, hAChE reached equilibrium state at approximately 5 ns, with the RMSD value of 1.4 Å (Fig. 4A). However, the conformation of L6M1R3 had slight changes and then kept stable after 7 ns with the RMSD value of 1.7 Å (Fig. 4A). The part of aldoximes of L6M1R3 located at A site kept similar conformation. The distance of the O atom of nucleophilic aldoximes and P atom of VX was kept at 11.4 Å during first 20 ns and changed to 10.4 Å for the last 30 ns (Fig. 4A). On the other side, the aminobenzamide part of L6M1R3 had great changes and was moved out of the P site (Fig. 4B and C).
Fig. 4.

Molecular dynamics study to investigate the Binding Interaction of L6M1R3/inhibited hAChE complex. (A) The RMSD of inhibited AChE and L6M1R3, the distance between the P atom of VX and the O atom of aldoximes, and the RMSD of inhibited AChE during MD simulation. (B) The structure of the docking molecular L6M1R3 in AChE (grays). (C) The docking pose (gray) and the structure after MD simulation (deep salmon) of L6M1R3/inhibited hAChE complex in surface model, suggesting the aminobenzamide was moved out of the P site. (D) Binding free energy decomposition of L6M1R3/inhibited hAChE system. (E) The binding interaction of L6M1R3/inhibited hAChE in 3D view, 2D view and in surface model.
To explore the interactions in the two systems, MM/GBSA free energy calculation was performed [33]. The total binding free energy was −26.9 ± 4.7 kcal/mol in L6M1R3/inhibited hAChE system (Supplementary Table S1). According to the results, the ΔEvdw term contributed greatly to the binding (−45.7 ± 2.8 kcal/mol), indicating that the interactions were primarily mediated by VDW interactions. The energy decomposition in this system was calculated. The π-π interaction between Tyr341 and benzaldoxime, and the hydrophobic interactions between Trp286 or Tyr72 and the linker contributed more favorable energies with value lower than −2.0 kcal/mol (Fig. 4D and E). As the aldoximes was located far away from P atom of VX and there was enough space between them, rotating the aldoximes to reach pre-reactivated pose was possible (Fig. 4E) However, the π-π interaction between Tyr341 and benzaldoxime, as well the hydrogen bond between Tyr124 and aldoxime would prohibit the rotation (Fig. 4E). Actually, we tried but failed to generate the pre-reactivated pose in L6M1R3/inhibited hAChE system with different starting poses of ligand, suggesting that the pre-reactivated pose might be unstable. Although L6M1R3 was well docked with the inactive hAChE, the rotation was blocked to generate pre-reactivated pose during the reactivation process, we guessed that a smaller linker and PSL would be better for the rotation of ligand to reach pre-reactivated pose.
On the other side, we also calculated the volume of L6M1R3 and HI-6 by DFT calculation, and found the volume of HI-6 (with value of 335 Å) was much smaller than L6M1R3 (with value of 507 Å) (Fig. 5 ). Moreover, the length of L6M1R3 was 2.9 Å longer than HI-6 (Fig. 5). These results also suggested the L6M1R3 might not be easy to research the pre-reactivated pose. Compounds employing small volumes and lengths should be designed as better reactivators.
Fig. 5.

The volumes and lengths of HI-6, L6M1R3 and L7R3.
2.2. Proof of concept by synthesis and biological evaluation
2.2.1. Design and synthesis
A representative synthesis route highlighted in Scheme 1 was used to prepare the conjugate template L7R3 as shown in Fig. 2. R3 was synthesized in a similar way described in our previously paper [26,27]. Condensation of L7 and R3 to afford the intermediates L7R3-d1, then it was readily converted to the final oxime conjugates L7R3 by treating with hydroxylammonium chloride. The rest conjugates (Table 1 ) described in this paper were all synthesized in a similar way to that of L7R3.
Scheme 1.

Preparation of conjugates L7R3. Conditions and reagents: a) DCM, NEt3, r.t. 81%; b) HONH2HCl, NaOAc, EtOH/DCM, 76%.
Table 1.
a. Reactivation of VX, sarin and tabun inhibited hAChE by newly synthesized reactivators and the reference oximes (0.1 mM, the reactivation time was 30 min for VX and sarin, was 120 min for tabun). b. IC50 of the reference and new synthesized reactivators.
| Compound | R1 | R2 | Reactivation (%) |
IC50 (μM) | ||
|---|---|---|---|---|---|---|
| VX | sarin | tabun | ||||
| 2-PAM | – | – | 50.6 ± 4.0 | 26.8 ± 3.4 | 13.4 ± 0.3 | 996 ± 107 |
| HI-6 | – | – | 66.4 ± 1.2 | 67.9 ± 4.6 | 6.9 ± 0.6 | 636 ± 148 |
| obidoxime | – | – | 78.5 ± 3.4 | 22.4 ± 2.5 | 36.7 ± 1.7 | 2169 ± 234 |
| L7R3 | 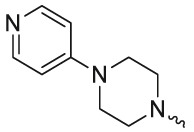 |
-Me | 80.2 ± 0.7 | 90.6 ± 4.1 | 28.3 ± 0.5 | 484 ± 93.3 |
| L7R5 | -Cl | 61.3 ± 3.4 | 69.6 ± 1.4 | 26.3 ± 0.4 | 262 ± 30.8 | |
| L8R3 |  |
-Me | 1.9 ± 0.1 | 1.7 ± 0.3 | 1.1 ± 0.1 | 5149 ± 1917 |
| L8R5 | -Cl | 1.1 ± 0.8 | 1.5 ± 0.2 | 1.2 ± 0.2 | 4275 ± 1064 | |
| L9R3 | 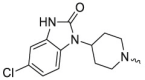 |
-Me | 2.0 ± 0.3 | 1.9 ± 0.1 | 2.3 ± 0.4 | 1905 ± 229 |
| L9R5 | -Cl | 0.4 ± 0.1 | 0.6 ± 0.3 | 1.3 ± 0.2 | 4148 ± 492 | |
| L10R3 |  |
-Me | 1.0 ± 0.2 | 0.5 ± 0.1 | 1.4 ± 0.1 | 3183 ± 801 |
| L10R5 | -Cl | 0.4 ± 0.2 | 0.8 ± 0.1 | 1.8 ± 0.1 | 3035 ± 689 | |
| L11R3 |  |
-Me | 26.6 ± 1.2 | 0.4 ± 0.1 | 0.4 ± 0.3 | 96.7 ± 14.4 |
| L11R5 | -Cl | 1.2 ± 0.1 | 1.1 ± 0.2 | 1.2 ± 0.2 | 826 ± 407 | |
| L12R3 |  |
-Me | 1.0 ± 0.1 | 0.2 ± 0.1 | 0.6 ± 0.2 | 5305 ± 1180 |
| L12R5 | -Cl | 1.4 ± 0.2 | 1.1 ± 0.1 | 1.5 ± 0.1 | 4177 ± 770 | |
| L13R3 |  |
-Me | 1.4 ± 0.5 | 1.4 ± 0.4 | 1.5 ± 0.2 | 3269 ± 601 |
| L13R5 | -Cl | 0.5 ± 0.2 | 0.2 ± 0.1 | 0.6 ± 0.1 | 5917 ± 1459 | |
| L14R3 |  |
-Me | 1.8 ± 0.1 | 1.3 ± 0.3 | 0.9 ± 0.2 | 3255 ± 406 |
| L15R3 | 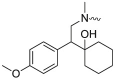 |
-Me | 2.3 ± 0.8 | 0.9 ± 0.1 | 1.2 ± 0.5 | 1256 ± 344 |
| L15R5 | -Cl | 1.6 ± 0.5 | 2.8 ± 0.5 | 0.4 ± 0.3 | 2505 ± 411 | |

2.2.2. hAChE reactivation and inhibition experiments
Guided by the computation results described above, the simple and small ligand pyridine linked to the piperidine were used as PSL along with the linker, the resulting salicylic aldoxime conjugates L7R3 and L7R5 were synthesized and tested firstly. The in vitro experiments were conducted with human acetylcholinesterase serving as enzyme source. 2-PAM, HI-6 and obidoxime were used as reference reactivators. Three most common nerve agents (VX, sarin and tabun) were used for the in vitro reactivation experiment. It was encouraging that both L7R3 and L7R5 emerged as quite efficient reactivators for both VX and sarin inhibited hAChE in a primary reactivation evaluation experiment. L7R3 displayed even higher reactivation potency than 2-PAM, HI-6 and obidoxime for VX- and sarin-hAChE conjugates at concentration of 100 μM. The PSL of them was simple basic aromatic pyridine and the linker was relatively long and flexible piperazine. Moreover, it was exciting that L7R3 and L7R5 also showed reactivating efficiency for the notorious stubborn tabun poisoning. Meanwhile, the inhibition experiment is necessary for these oximes because strong inhibition of hAChE would result in heavy toxicity. Recently, a series of nonquaternary pyridine aldoximes linked to various PSLs were reported as equal or more efficient reactivators for OP inhibited hAChE in comparison to the pyridinium oximes HI-6 and obidoxime [[34], [35], [36], [37], [38], [39]], nevertheless, these new-generation aldoxime reactivators were relatively heavy inhibitors of hAChE (IC50 < 20 μM). It was encouraging that both conjugates L7R3 and L7R5 (IC50 values were 484 ± 93.3 and 262 ± 30.8 μM respectively, Table 1) were much slighter inhibitors of hAChE in contrast to these reported pyridine aldoxime reactivators.
The primary success encouraged us to proceed for a broader attempt, various drug induced simple aromatic or aliphatic moieties (such as pyrazole, triazole, sulfathiazole, benzimidazolinone, venlafaxine, morpholine and pyridine) were screened for proper PSLs, and both relative long flexible and inflexible linkers were used to construct novel hybrid nonquaternary salicylaldoximes, such as piperidine, piperazine and benzene or pyridine ring (Table 1). The reactivation results showed that most of the newly synthesized compounds were inefficient reactivators for VX, sarin and tabun inhibited hAChE. Both aromatic and aliphatic moieties containing PSLs connected to the inflexible aromatic linkers could not reactivate the hAChE inhibited by any of the tested nerve agents, such as the L8, L10, L12, L13 and L14 series. Meanwhile, the aromatic benzimidazolinone and venlafaxine derivative PSLs (L9 and L15 series) connected to flexible linkers such as piperidine also hardly reactivated the inhibited enzyme. It was noteworthy that all these inefficient reactivators also exhibited quite slight inhibition potency towards hAChE with IC50 higher than 500 μM L11R3 was an exception among those containing inflexible aromatic linkers; it showed moderate reactivating capacity for hAChE poisoned by VX. Interestingly enough, L11R3 was also moderate inhibitor of hAChE, while the choloro substituted weak inhibitor L11R5 could not reactivate VX-hAChE at all; nevertheless, both of them were inefficient reactivators for sarin or tabun inhibited hAChE.
Experiments were performed in duplicate at 37 °C in phosphate buffer (0.10 M, pH 7.4), data shows the average and standard deviation.
Due to their efficiency for sarin, VX and tabun inhibited hAChE, L7R3 and L7R5 were tested for reactivation of hAChE inhibited by three commonly used pesticides (paraoxon, parathion and dichlorvos) and the most stubborn nerve agent soman [40], the results were presented in Table 2 .
Table 2.
Reactivation of paraoxon, parathion, dichlorvos and soman inhibited hAChE by selected reactivators and the reference oximes (0.1 mM, the reactivation time was 30 min).
| Compound | Reactivation (%) |
|||
|---|---|---|---|---|
| paraoxon | parathion | dichlorvos | soman | |
| 2-PAM | 54.0 ± 0.7 | 44.0 ± 0.8 | 38.0 ± 0.9 | 1.0 ± 0.1 |
| HI-6 | 31.5 ± 7.6 | 29.2 ± 0.7 | 7.1 ± 0.3 | 37.5 ± 1.9 |
| obidoxime | 90.7 ± 1.6 | 84.1 ± 1.6 | 38.5 ± 1.0 | 6.8 ± 0.2 |
| L7R3 | 86.1 ± 1.4 | 79.1 ± 1.9 | 38.5 ± 0.3 | 3.9 ± 0.2 |
| L7R5 | 64.1 ± 0.4 | 53.4 ± 1.1 | 25.9 ± 0.4 | 9.5 ± 0.1 |
Experiments were performed in duplicate at 37 °C in phosphate buffer (0.10 M, pH 7.4), data shows the average and standard deviation.
For pesticides inhibited hAChE, obidoxime showed the most promising result among the quaternary oximes tested, while HI-6 seemed to be even weaker reactivator than 2-PAM. Both L7R3 and L7R5 demonstrated reactivating efficiency for all three used pesticides, L7R3 was superior to L7R5 at the concentration of 100 μM and was almost as equal efficient as the best quaternary oxime obidoxime. In terms of soman poisoning, L7R5 demonstrated a little reactivation potency, but was still much lower than that of HI-6, while the other tested compounds were inefficient. Yet in any case, structural optimization of L7R5 holds promise for discovery of better nonquaternary reactivators for soman inhibited hAChE.
2.2.3. Determination of reactivation kinetics
Determination of maximal reactivation rate constant k r, dissociation constant K D and second order reactivation rate constant k r2 (k r2 = k r/K D) would help get a deep and total comprehension of the reactivating ability. Results of the reactivation kinetics constants are reported in Table 3 . For VX-hAChE conjugates, the k r of L7R3 (27.5 ± 1.8∗10−3min−1) was almost equivalent to that of HI-6 and obidoxime (25.7 ± 3.9 and 30.4 ± 1.4∗10−3min−1 respectively); the K D of L7R3 (17.3 ± 2.5 μM) was half or less lower than that of HI-6 and obidoxime (60.3 ± 13.6 and 47.9 ± 3.6 μM respectively), resulting its 2.5-fold–3.5-fold higher k r2 than the reference oximes. In terms of L7R5, although the k r was much lower (16.7 ± 1.5∗10−3min−1), its final k r2 was 2-fold–3-fold higher than HI-6 or obidoxime as a result of its lower K D value (13.3 ± 2.1 μM). It could be concluded that the elevation of the reactivating efficiency mainly due to increased affinity towards the inhibited enzyme, which indicated by lower K D values. Interestingly, L7R3 and L7R5 also exhibited higher inhibition ability towards hAChE than HI-6 and obidoxime, and the heaviest inhibitor L7R5 (IC50 = 262 ± 30.8 μM) displayed the highest affinity (K D = 13.3 ± 2.1 μM) towards VX-hAChE conjugate. For other inhibited hAChE conjugates, same phenomena were observed with the exception for obidoxime reactivated sarin-hAChE with the lowest K D value (2.1 ± 0.3 μM). Hence we further proposed that oximes showing proper inhibition ability towards the hAChE would more likely produce efficient reactivators for the poisoned enzyme conjugates. For sarin-hAChE, both L7R3 and L7R5 outperformed obidoxime and they were just slightly less efficient than HI-6. Quite remarkably, both L7R3 and L7R5 exhibited 2-fold more reactivation efficiency than the best tested quaternary oxime obidoxime for tabun poisoning, which mainly due to their greatly increased affinity towards tabun-hAChE adduct too. It was delightful that promising results were also got in the reactivation experiment for pesticides poisoning. L7R3 emerged as the best reactivator, it was 6.6-fold, 5.9-fold and 2-fold more efficient than obidoxime for paraoxon, parathion and dichlorvos poisoning respectively. L7R5 was less efficient than L7R3, but it was still 2.3-fold and 2.2-fold more efficient than obidoxime and was equally efficient for dichlorvos poisoning.
Table 3.
Reactivation rate constant (kr), dissociation constant (KD), second order reactivation rate constant (kr2). (kr2 = kr/KD).
| kr/10−3min−1 | KD/μM | kr2/mM−1min−1 | kr/10−3min−1 | KD/μM | kr2/mM−1min−1 | |
|---|---|---|---|---|---|---|
| VX | Sarin | |||||
| HI-6 | 25.7 ± 3.9 | 60.3 ± 13.6 | 0.43 | 27.1 ± 3.1 | 29.3 ± 6.2 | 0.92 |
| Obidoxime | 30.4 ± 1.4 | 47.9 ± 3.6 | 0.63 | 1.69 ± 0.1 | 2.1 ± 0.3 | 0.8 |
| L7R3 | 27.5 ± 1.8 | 17.3 ± 2.5 | 1.59 | 11.4 ± 0.6 | 13.1 ± 1.5 | 0.87 |
| L7R5 | 16.7 ± 1.5 | 13.3 ± 2.1 | 1.28 | 6.0 ± 0.5 | 6.7 ± 1.3 | 0.90 |
| Paraoxon | Parathion | |||||
| Obidoxime | 62.8 ± 7.5 | 6.8 ± 2.0 | 9.23 | 55.9 ± 1.8 | 5.3 ± 0.47 | 10.56 |
| L14R3 | 41.6 ± 4.3 | 0.68 ± 0.38 | 60.93 | 46.6 ± 3.7 | 0.75 ± 0.30 | 62.08 |
| L14R5 | 19.7 ± 2.2 | 0.66 ± 0.33 | 29.90 | 20.6 ± 2.1 | 0.90 ± 0.35 | 22.84 |
| Tabun | Dichlorvos | |||||
| Obidoxime | 5.2 ± 0.5 | 91.7 ± 13.6 | 0.06 | 11.3 ± 0.36 | 1.2 ± 0.26 | 9.35 |
| L14R3 | 0.88 ± 0.05 | 7.6 ± 1.6 | 0.12 | 18.8 ± 0.19 | 0.99 ± 0.42 | 19.02 |
| L14R5 | 0.56 ± 0.07 | 4.2 ± 1.8 | 0.13 | 7.9 ± 0.54 | 0.84 ± 0.22 | 9.46 |
Experiments were performed in duplicate at 37 °C in phosphate buffer (0.10 M, pH 7.4), data shows the nonlinear fitting results and standard deviation.
Generally, the kinetic evaluation revealed that both L7R3 and L7R5 were very promising reactivator candidates with broad-spectrum activity. They were almost as equal efficient reactivators as HI-6 and obidoxime for sarin-hAChE conjugate, and performed much better than these currently used quaternary oximes for VX and especially for the stubborn tabun inhibited hAChE. Meanwhile, they were also much more efficient reactivators for pesticide-hAChE conjugates than the best tested quaternary oxime obidoxime, while our previously reported conjugates L6M1R3 and L6M1R5 were less efficient than HI-6 and obidoxime for nerve agents poisoning and much less efficient than obidoxime for pesticides poisoning [27].
2.2.4. Molecular dynamic simulations to generate pre-reactivated pose
To test our previous theory, molecular dynamic simulations were conducted by using the new reactivator L7R3. Firstly, we compared volume and length of L7R3 with L6M1R3 and HI-6, and found L7R3 employed a small volume and shorter length than L6M1R3. Despite the volume of L7R3 was greater than HI-6, the length of L7R3 was similar to HI-6 (Fig. 5). Then we carried out the MD simulations of L7R3 (Fig. 6 ) with the inhibited hAChE. The inhibited hAChE showed slight variation with similar RMSD value as above, and reached equilibrium state after approximately 5 ns with the RMSD value of 1.4 Å (Fig. 6A). The conformation of L7R3 was stable after 13 ns with the RMSD value of 0.6 Å (Fig. 6A). Interestingly, the pyridine moiety of L7R3 moved from the P site to a pocket formed by Trp286, Val294, Tyr341, and Gly342, named as R site (Fig. 6B and C). While the aldoxime part of L7R3 located at A site with a similar conformation. The distance between the O atom of nucleophilic aldoximes and P atom of VX kept at very short distance with value of 3.8 Å during MD simulation. To further explore the interactions, MM/GBSA free energy calculation was performed. The total binding free energy was −34.1+± 3.2 kcal/mol in L7R3/inhibited hAChE system (Supplementary Table S2). Similarly, the interactions were primarily mediated by VDW interactions as the ΔEvdw term contributed greatly to the binding (−48.2 ± 2.9 kcal/mol). The energy decomposition was carried out for this system. The hydrophobic interaction of VX with L7R3 contributed the most favorable energy with a value of −2.8 kcal/mol (Fig. 6B). Beside the binding energy of pyridine with R site, the residues near the VX, such as Trp86, Gly121, and Tyr124, contributed more favorable energy via hydrophobic interaction. As distance between the O atom of aldoximes and P atom of VX (3.8 Å) was similar to that in the DFT calculated pre-reactivated pose (3.65 Å), this binding mode was thought to be the pre-reactivated pose (Fig. 6D). The lower binding free energy illustrated that the pose was stable. The short distance between the O atom of aldoximes and P atom of VX would facilitate the nucleophilic attack. In addition, the hydrogen migration process would avoid the resistance of rotation. The results suggested the L7R3 would reactivate the inhibited hAChE through an easy and efficient way.
Fig. 6.

Molecular dynamics study to investigate the binding interaction of L7R3/inhibited hAChE complex. (A) The RMSD of inhibited AChE and L7R3, the distance between the P atom of VX and the O atom of aldoximes, and the RMSD of inhibited AChE during MD simulation. (B) Binding free energy decomposition of L7R3/inhibited hAChE system. (C) The structure before (gray) and after (deep salmon) MD simulation of L7R3/inhibited hAChE complex in surface model. (D) The binding interaction of L7R3/inhibited hAChE in 3D view, 2D view and in surface model.
It was interesting that the biological results were in accordance with the MD simulation results in some extent. The binding energy of pyridine with the R site and the residues near the VX with the aldoximes of L7R3 via hydrophobic interaction might accounted for its increased affinity towards the VX-hAChE conjugates, and the readily formed pre-reactivated pose might account for its high k r value. We thus believe that MD simulations could be used as a rational and efficient method for design and construction of more efficient and broad-spectrum reactivators.
Finally, some suggestions for construction of reactivators for covalently inhibited targets were concluded based on the above work. Firstly, proper affinity seems to be important for the reactivators to anchor to the inhibited targets, while heavy inhibition potency should be avoided. Secondly, a reactivation moiety may play important role during the reactivation process, such as the nucleophilic oxime in this study, which could form a pre-reactivated pose and finish the reactivation process. In addition, formation of a pre-reactivated pose may promote the reactivation process, where the DFT and MM/GBSA free energy calculation and MD simulations could be used as efficient tools for construction of functional reactivators.
3. Conclusions
Given to the potential toxicology threat of covalent drugs, a primary study of the reactivators for highly toxic OPs inhibited hAChE was conducted in this paper. We firstly used the DFT calculation to analyze the reactivation process and proposed that formation of a pre-reactivated pose was very important for efficient nonquaternary reactivators. Then molecular dynamic simulation was conducted to analyze the binding interaction of our previously reported reactivator L6M1R3 with VX-hAChE conjugate, then MM/GBSA free energy calculation was performed and we proposed that a smaller peripheral site ligand and linker would facilitate the reactivating process. As a proof of concept, some small PSLs were used to construct novel nonquaternary salicylic aldoxime reactivators. The in vitro biological evaluation experiments were conducted for both nerve agents and pesticides inhibited hAChE. The inhibition and reactivation results demonstrated that the pyridine bearing conjugates L7R3 and L7R5 were equal or even more efficient reactivators for both nerve agents and pesticides poisoning in comparison to currently approved quaternary oximes 2-PAM, obidoxime and HI-6, while they exhibited slight inhibition potency for the enzyme. Additionally, the reactivation kinetic experiments confirmed that both L7R3 and L7R5 were almost as equal efficient reactivators as HI-6 and obidoxime for sarin-hAChE conjugates, and they were 2-fold or more efficient reactivators for VX and especially the stubborn tabun inhibited hAChE. Moreover, L7R3 exhibited the highest reactivation potency for the tested pesticides inhibited enzyme and L7R5 also performed equal or higher reactivation efficiency in comparison to the best tested quaternary reactivator obidoxime. The kinetic study also revealed that the great elevation of reactivation potency were mainly due to increased affinity for inhibited hAChE, which seems to have some relationship with enzyme inhibition ability. Interestingly, molecular dynamic simulation results could partially accounted for the experimental data, which suggested that structural based computation method could be used for rational design of more efficient nonquaternary reactivators. This work might provide some insights for reactivation of covalently inhibited targets by using computational methods, and we thought that a proper affinity, a reactivation moiety and formation of a pre-reactivated pose might be useful for the reactivation process. Currently, our group is engaging in further study for development of more widely reactivators for various covalently inhibited targets.
4. Experimental section
4.1. Chemicals
All reagents and solvents were used as received from commercial sources. All synthesized compounds were determined to possess a purity of more than 95%, as evidenced by high-performance liquid chromatography analysis. 1H NMR and 13C NMR spectra were recorded at 400 MHz and 100 MHz on a Bruker-400 instrument in CDCl3 or DMSO‑d 6, respectively. Proton and carbon chemical shifts are expressed in parts per million (ppm) relative to internal tetramethylsilane (TMS) and coupling constants (J) are expressed in Hertz (Hz).
General Method for the Preparation of 2-hydroxy-5-methyl- 3-((4-(pyridin-4-yl) piperazin-1-yl) methyl) benzaldehyde oxime L7R3: The intermediates R3 or R5 was prepared as we described previously by using 2-hydroxy-5-methyl-benzaldehyde or 5-chloro- 2-hydroxybenzaldehyde [37]. To a solution of 1-(pyridin-4-yl) piperazine (L7) (0.16 g, 0.98 mmol) in DCM (10 mL) was added triethylamine (0.21 g, 2.07 mmol) and R3 (0.19 g, 1.03 mmol), the mixture was stirred at room temperature for 2 h. After concentration under reduced pressure, the residue was purified by silica gel chromatography (DCM/MeOH = 25/1, v/v) to afford the intermediate L7R3-d1 (0.26 g, 81%) as a white solid. 1H NMR (400 MHz, DMSO) δ 10.12 (s, 1H), 8.28 (d, J = 5.7 Hz, 2H), 7.40 (s, 1H), 7.28 (s, 1H), 6.68 (d, J = 5.7 Hz, 2H), 3.71 (s, 2H), 3.50–3.35 (m, 4H), 2.80–2.63 (m, 4H), 2.33 (s, 3H).Then to a solution of L7R3-d1 (0.25 g, 0.80 mmol) in ethanol (15 mL) was added hydroxylammonium chloride (0.10 g, 1.43 mmol) and anhydrous sodium acetate (0.14 g, 1.70 mmol). The mixture was stirred at room temperature for 3 h. After filtration, the residue was purified by silica gel chromatography (DCM/MeOH = 20/1, v/v) to afford the title compound L7R3 (0.20 g, 76%) as a white solid. 1H NMR (400 MHz, DMSO) δ 11.32 (s, 1H), 11.07–10.40 (m, 1H), 8.30 (s, 1H), 8.18 (d, J = 6.1 Hz, 2H), 7.24 (s, 1H), 7.04 (s, 1H), 6.87 (d, J = 6.1 Hz, 2H), 3.65 (s, 2H), 3.45–3.26 (m, 4H), 2.68–2.43 (m, 4H), 2.22 (s, 3H). 13C NMR (101 MHz, DMSO) δ 155.24, 153.27, 148.64, 147.94, 132.16, 128.24, 127.46, 123.30, 118.15, 108.72, 57.76, 52.07, 45.75, 20.47. HRMS (ESI+, m/z): cal. for C18H22N4O2 [M+H]+ 327.1776; found, 327.1815.
5-chloro-2-hydroxy-3-((4-(pyridin-4-yl)piperazin-1-yl)methyl)benzaldehyde oxime (L7R5): The title compound was obtained in a manner similar to that used for L7R3 as a white solid (0.15 g, 67%) but using R5. 1H NMR (400 MHz, DMSO) δ 11.58 (s, 1H), 8.32 (s, 1H), 8.18 (d, J = 4.5 Hz, 2H), 7.47 (s, 1H), 7.28 (s, 1H), 6.85 (d, J = 4.5 Hz, 2H), 3.70 (s, 2H), 3.46–3.21 (m, 4H), 2.70–2.51 (m, 4H). 13C NMR (101 MHz, DMSO) δ154.55, 153.79, 148.98, 146.07, 129.86, 125.52, 122.87, 119.72, 108.33, 56.88, 51.51, 45.21. HRMS (ESI+, m/z): cal. for C11H19 35Cl N4O2 [M+H]+ 347.1275; found, 347.1269.
The other reported oxime conjugates were synthesized in a manner similar to that used for L7R3 and details of analyses were reported in the Supporting Information.
2. Computational methods.
Details and results of the computational methods were described in the Supporting Information, including: Preparation of protein crystal structures, Molecular dynamics simulation, Trajectory analysis, Calculation of binding free energies and Molecular docking study.
4.2. General in vitro AChE screening information
The in vitro experiments were conducted with human acetylcholinesterase (hAChE, 20 U/mL, dissolved in 20 mM HEPES, pH 8.0, contain 0.1% TRITON X-100, from Sigma-Aldrich) serving as enzyme source. 2-PAM and pesticides (paraoxon, parathion, phorate and dichlorvos) were from commercial sources. HI-6 and obidoxime were synthesized according to the literature protocols [41,42]. Sarin, VX, tabun and soman were from Anti chemical command and Engineering Institute of the Chinese people’s Liberation Army. A solution of oxime (10 mM) were prepared in water containing 10% acetic acid and it was further diluted by PBS (0.1 M, pH = 7.4) to the required concentrations. The final concentration of acetic acid in the incubation mixture was <1% and had no effect on the biological essay through a control experiment. Biological evaluation experiment were conducted in 96-well plate, the enzyme activity was measured by the time-dependent hydrolysis of acetylthiocholine (ATCh) in which the product (thiocholine) was detected by reaction with the Ellman’s reagent, 5, 5′-dithiodis-2-nitrobenzoic acid (DTNB) and absorbance at 412 nm [43]. No oximolysis of ATCh by the tested oximes was detected and the enzyme activity in the control remained constant during the experiment.
4.3. hAChE inhibition experiments
The oxime solutions (oxime final concentrations: 1000, 100, 10, 1, 0.1, 0.01 μM, each sample was measured duplicate in parallel in 96-well plate) were incubated with diluted hAChE solutions for 30 min at 25 °C. A positive control was run in parallel. The percentage of enzyme activity (%Activity) was calculated as the ratio of the inhibited enzyme activity and activity in the control (100% activity). IC50 values were calculated by non-linear fitting using the standard IC50 equation: %Activity = 100∗IC50/(IC50+[Ox]).The details of experimental procedures were described in the Supporting Information.
4.4. hAChE reactivation experiments
Four most common nerve agents (VX, sarin tabun and soman) and three pesticides (paraoxon, parathion and dichlorvos) were used for the in vitro reactivation experiment. Initially the concentrations of different nerve agents and pesticides were determined by a pre-experiment similar to the inhibition experiment to attain an inhibition plateau between 90% and 97%, the final concentration of the OPs in the incubation mixture were as followings: VX, 8∗10−8; sarin, 4∗ 10−7; tabun, 6∗10−7; soman, 4∗10−8, paraoxon, 7∗10−8, parathion, 3∗10−6, dichlorvos, 2∗10−6. Next, the inhibited hAChE was incubated with different oximes (0.1 mM) for reactivation during a time of 30 min at 37 °C. The percentage of reactivated enzyme (%Reactivation) was calculated as the ratio of the recovered enzyme activity and activity in the control. The details of experimental procedures were described in the Supporting Information.
4.5. Determination of reactivation kinetics
Initially the diluted hAChE was intoxicated by different nerve agents or pesticides to attain an inhibition plateau between 90% and 97% as we described above; then the inhibited hAChE was incubated with different oximes at different concentrations for reactivation during a time more than 150 min at 37 °C, and the reactivation rate at different time intervals were measured. The experimental details were described in the Supporting Information. The observed first-order rate constant K obs for each oxime concentration, the dissociation constant K D of inhibited enzyme-oxime conjugates (EP-OX) and the maximal reactivation rate constant k r were calculated by non-linear fitting using the standard oxime concentration dependent reactivation equation derived from the following scheme [44].

In this scheme, EP is the phosphylated enzyme, [EP-OX] is the reversible Michaelis-type complex between EP and the oxime [OX], E is the active enzyme and P-OX the phosphylated oxime. K D is equal to the ratio (k -1 + k r)/k 1, and it typically approximates the dissociation constant of the [EP-OX] complex, where from it follows that: k r2 = k r /K D.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The financial support from the “National Key R&D program of China” (NO. 2018YFA0507900) and the “National Natural Science Foundation of China” (NO. 81703413).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113286.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Bauer R.A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Singh J., Petter R.C., Baillie T.A., Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 3.Dai W.H., Zhang B., Jiang X.M., Su H.X., Li J., Zhao Y., Xie X., Jin Z.M., Peng J.J., Liu F.J., Li C.P., Li Y., Bai F., Wang H.F., Cheng X., Cen X.B., Hu ShL., Yang X.N., Wang J., Liu X., Xiao G.F., Jiang H.L., Rao Z.H., Zhang L.K., Xu Y.C., Yang H.T., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor P., Goodman Gilman’ s, Hardman J.G., Limbird L.E. In: The Pharmacological Basis of Therapeutics. tenth ed. Goodman A.G., editor. McGraw-Hill; New York: 2001. p. 175. [Google Scholar]
- 5.Jeyaratnam J. Acute pesticide poisoning: a major global health problem. World Health Stat. Q. 1990;43:139–144. [PubMed] [Google Scholar]
- 6.Eddleston M., Karalliedde L., Buckley N., Fernando R., Hutchinson G., Isbister G., Konradsen F., Murray D., Piola J.C., Senanayake N., Sheriff R., Singh S., Siwach S.B., Smit L. Pesticide poisoning in the developing world eaminimum pesticides list. Lancet. 2002;360:1163–1167. doi: 10.1016/s0140-6736(02)11204-9. [DOI] [PubMed] [Google Scholar]
- 7.Eddleston M., Buckley N.A., Eyer P., Dawson A.H. Management of acute organophosphorus pesticide poisoning. Lancet. 2008;371:597–607. doi: 10.1016/S0140-6736(07)61202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tu A.T. Toxicological and chemical aspects of sarin terrorism in Japan in 1994 and 1995. Toxin Rev. 2007;26:231–274. [Google Scholar]
- 9.Bajgar J. Organophosphates/nerve agent poisoning: mechanism of action, diagnosis, prophylaxis, and treatment. Adv. Clin. Chem. 2004;38:151–216. doi: 10.1016/s0065-2423(04)38006-6. [DOI] [PubMed] [Google Scholar]
- 10.Marrs T.C. Organophosphate poisoning. Pharmacol. Ther. 1993;58:51–66. doi: 10.1016/0163-7258(93)90066-m. [DOI] [PubMed] [Google Scholar]
- 11.Sidell F.R., Borak J. Chemical warfare agents: II. Nerve agents. Ann. Emerg. Med. 1992;21:865–871. doi: 10.1016/s0196-0644(05)81036-4. [DOI] [PubMed] [Google Scholar]
- 12.Jokanović M., Stojiljković M.P. Current understanding of the application of pyridinium oximes as cholinesterase reactivators in treatment of organophosphate poisoning. Eur. J. Pharmacol. 2006;553:10–17. doi: 10.1016/j.ejphar.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 13.Jokanović M., Prostran M. Pyridinium oximes as cholinesterase reactivators. Structure-activity relationship and efficacy in the treatment of poisoning with organophosphorus compounds. Curr. Med. Chem. 2009;16:2177–2188. doi: 10.2174/092986709788612729. [DOI] [PubMed] [Google Scholar]
- 14.Jokanović M. Medical treatment of acute poisoning with organophosphorus and carbamate pesticides. Toxicol. Lett. 2009;190:107–115. doi: 10.1016/j.toxlet.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Mercey G., Verdelet T., Renou J., Kliachyna M., Baati R., Nachon F., Jean L., Renard P.Y. Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. Acc. Chem. Res. 2012;45:756–766. doi: 10.1021/ar2002864. [DOI] [PubMed] [Google Scholar]
- 16.Shih T.M., Skovira J.W., O’Donnell J.C., McDonough J.H. In vivo reactivation by oximes of inhibited blood, brain and peripheral tissue cholinesterase activity following exposure to nerve agents in Guinea pigs. Chem. Biol. Interact. 2010;187:207–214. doi: 10.1016/j.cbi.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Lorke D.E., Kalasz H., Petroianu G.A., Tekes K. Entry of oximes into the brain: a review. Curr. Med. Chem. 2008;15:743–753. doi: 10.2174/092986708783955563. [DOI] [PubMed] [Google Scholar]
- 18.Little P.J., Scimeca J.A., Martin B.R. Distribution of [3H]diisopropylfluorophosphate, [3H]soman, [3H]sarin, and their metabolites in mouse brain. Drug Metab. Dispos. 1988;16:515–520. [PubMed] [Google Scholar]
- 19.Rutland J.P. The effect of some oximes in sarin poisoning. Br. J. Pharmacol. Chemother. 1958;13:399–403. doi: 10.1111/j.1476-5381.1958.tb00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shih T.M., Skovira J.W., O’Donnell J.C., McDonough J.H. Treatment with tertiary oximes prevents seizures and improves survival following sarin intoxication. J. Mol. Neurosci. 2010;40:63–69. doi: 10.1007/s12031-009-9259-7. [DOI] [PubMed] [Google Scholar]
- 21.Worek F., Thiermann H. Reactivation of organophosphate-inhibited human acetylcholinesterase by isonitrosoacetone (MINA): a kinetic analysis. Chem. Biol. Interact. 2010;194:91–96. doi: 10.1016/j.cbi.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Kalisiak J., Ralph E.C., Zhang J., Cashman J.R. Amidine-oximes: reactivators for organophosphate exposure. J. Med. Chem. 2011;54:3319–3330. doi: 10.1021/jm200054r. [DOI] [PubMed] [Google Scholar]
- 23.Kalisiak J., Ralph E.C., Cashman J.R. Nonquaternary reactivators for organophosphate-inhibited cholinesterases. J. Med. Chem. 2012;55:465–474. doi: 10.1021/jm201364d. [DOI] [PubMed] [Google Scholar]
- 24.Kassa J. Review of oximes in the antidotal treatment of poisoning by organophosphorus nerve agents. J. Toxicol. Clin. Toxicol. 2002;40:803–816. doi: 10.1081/clt-120015840. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberry T.L., Johnson J.L., Cusack B., Thomas J.L., Emani S., Venkatasubban K.S. Interactions between the peripheral site and the acylation site in acetylcholinesterase. Chem. Biol. Interact. 2005;181:157–158. doi: 10.1016/j.cbi.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 26.Wei Z., Liu Y.Q., Wang S.Z., Yao L., Nie H.F., Wang Y.A., Liu X.Y., Zheng Z.B., Li S. Conjugates of salicylaldoximes and peripheral site ligands: novel efficient nonquaternary reactivators for nerve agent-inhibited acetylcholinesterase. Bioorg. Med. Chem. 2017;25:4497–4505. doi: 10.1016/j.bmc.2017.06.041. [DOI] [PubMed] [Google Scholar]
- 27.Wei Z., Bi H., Liu Y.Q., Nie H.F., Yao L., Wang S.Z., Yang J., Wang Y.A., Liu X., Zheng Z.B. Design, synthesis and evaluation of new classes of nonquaternary reactivators for acetylcholinesterase inhibited by organophosphates. Bioorg. Chem. 2018;81:681–688. doi: 10.1016/j.bioorg.2018.09.025. [DOI] [PubMed] [Google Scholar]
- 28.Koellner G., Kryger G., Millard C.B., Silman I., Sussman J.L., Steiner T. Active-site gorge and buried water molecules in crystal structures of acetylcholinesterase from Torpedo californica. J. Mol. Biol. 2000;296:713–735. doi: 10.1006/jmbi.1999.3468. [DOI] [PubMed] [Google Scholar]
- 29.Allgardsson A., Berg L., Akfur C., Hörnberg A., Worek F., Linusson A., Ekström F.J. Structure of a prereaction complex between the nerve agent sarin, its biological target acetylcholinesterase, and the antidote HI-6. Proc. Natl. Acad. Sci. U.S.A. 2016;113:5514–5519. doi: 10.1073/pnas.1523362113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Driant T., Nachon F., Ollivier C., Renard P.Y., Derat E. On the influence of the protonation states of active site residues on AChE reactivation: a QM/MM approach. Chembiochem. 2017;18:666–675. doi: 10.1002/cbic.201600646. [DOI] [PubMed] [Google Scholar]
- 31.Bester S.M., Guelta M.A., Cheung J., Winemiller M.D., Bae S.Y., Myslinski J., Pegan S.D., Height J.J. Structural insights of stereospecific inhibition of human acetylcholinesterase by VX and subsequent reactivation by HI-6. Chem. Res. Toxicol. 2018;31:1405–1417. doi: 10.1021/acs.chemrestox.8b00294. [DOI] [PubMed] [Google Scholar]
- 32.Tripos International . Tripos International; St. Louis, MO, USA: 2012. Sybyl-X 2.0. [Google Scholar]
- 33.Wang E., Sun H., Wang J., Wang Zhe, Liu H., Zhang J.Z.H., Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- 34.Mercey G., Verdelet T., Saint-André G., Gillon E., Wagner A., Baati R., Jean L., Nachon F., Renard P.Y. First efficient uncharged reactivators for the dephosphylation of poisoned human acetylcholinesterase. Chem. Commun. 2011;47:5295–5297. doi: 10.1039/c1cc10787a. [DOI] [PubMed] [Google Scholar]
- 35.Mercey G., Renou J., Verdelet T., Kliachyna M., Baati R., Gillon E., Arboléas M., Loiodice M., Nachon F., Jean L., Renard P.Y. Phenyltetrahydro-isoquinoline-pyridinaldoxime conjugates as efficient uncharged reactivators for the dephosphylation of inhibited human acetylcholinesterase. J. Med. Chem. 2012;55:10791–10795. doi: 10.1021/jm3015519. [DOI] [PubMed] [Google Scholar]
- 36.Renou J., Mercey G., Verdelet T., Păunescu E., Gillon E., Arboléas M., Loiodice M., Kliachyna M., Baati R., Nachon F., Jean L., Renard P.Y. Syntheses and in vitro evaluations of uncharged reactivators for human acetylcholinesterase inhibited by organophosphorus nerve agents. Chem. Biol. Interact. 2013;203:81–84. doi: 10.1016/j.cbi.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 37.Kliachyna M., Santoni G., Nussbaum V., Renou J., Sanson B., Colletier J.P., Arboléas M., Loiodice M., Weik M., Jean L., Renard P.Y., Nachon F., Baati R. Design, synthesis and biological evaluation of novel tetrahydroacridine pyridine- aldoxime and -amidoxime hybrids as efficient uncharged reactivators of nerve agent-inhibited human acetylcholinesterase. Eur. J. Med. Chem. 2014;78:455–467. doi: 10.1016/j.ejmech.2014.03.044. [DOI] [PubMed] [Google Scholar]
- 38.Renou J., Loiodice M., Arboléas M., Baati R., Jean L., Nachon F., Renard P.Y. Tryptoline-3-hydroxypyridinaldoxime conjugates as efficient reactivators of phosphylated human acetyl and butyrylcholinesterases. Chem. Commun. 2014;50:3947–3950. doi: 10.1039/c4cc00561a. [DOI] [PubMed] [Google Scholar]
- 39.McHardy S.F., Bohmann J.A., Corbett M.R., Campos B., Tidwell M.W., Thompson P.M., Bemben C.J., Menchaca T.A., Reeves T.E., Cantrell W.R., Jr., Bauta W.E., Lopez A., Maxwell D.M., Brecht K.M., Sweeney R.E., McDonough J. Design, synthesis, and characterization of novel, nonquaternary reactivators of GF-inhibited human acetylcholinesterase. Bioorg. Med. Chem. Lett. 2014;24:1711–1714. doi: 10.1016/j.bmcl.2014.02.049. [DOI] [PubMed] [Google Scholar]
- 40.Clement J.G., Lockwood P.A. HI-6, an oxime which is an effective antidote of soman poisoning: a structure-activity study. Toxicol. Appl. Pharmacol. 1982;64:140–146. doi: 10.1016/0041-008x(82)90332-5. [DOI] [PubMed] [Google Scholar]
- 41.Hsiao L.Y.Y., Getzville N.Y., Musallam H.A., Damascus Md. U.S. Patent; 1992. Bis-methylene Ether Pyridinium Compound Preparation. [Google Scholar]
- 42.Eddolls J., Mccormack P., Hodgson A. H.K. Patent; 2013. Process for the Manufacture of HI-6 Dimethanesulfonate. [Google Scholar]
- 43.Ellman G.L., Courtney K.D., Andres V., Jr., Feather-stone R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 44.Worek F., Thiermann H., Szinicz L., Eyer P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem. Pharmacol. 2004;68:2237–2248. doi: 10.1016/j.bcp.2004.07.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.