

Abstract
The endocannabinoid system (ECS) is involved in a wide range of biological functions and is comprised of cannabinoid receptors and enzymes responsible for endocannabinoid synthesis and degradation. Over the past two decades, significant advances towards developing drugs and positron emission tomography (PET) tracers targeting different components of the ECS have been made. Herein, we summarized the recent development of PET tracers for imaging cannabinoid receptors 1 (CB1R) and 2 (CB2R) as well as the key enzymes monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH), particularly focusing on PET neuroimaging applications. State-of-the-art PET tracers for the ECS will be reviewed including their chemical design, pharmacological properties, radiolabeling, as well as preclinical and human PET imaging. In addition, this review addresses the current challenges for ECS PET biomarker development and highlights the important role of PET ligands to study disease pathophysiology as well as to facilitate drug discovery.
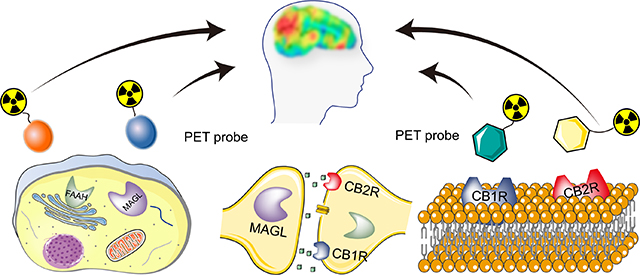
Graphical Abstract
1. INTRODUCTION
The endocannabinoid system (ECS) constitutes a lipid-based signaling system in the human central nervous system (CNS), which includes primarily two cannabinoid receptors (CB1R and CB2R), their endogenous ligands, and enzymes responsible for ligand synthesis and degradation. In the brain, 2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamine (AEA), the most well-known endogenous ligands for cannabinoid receptors, are produced by the postsynaptic membrane as “on-demand” to stimulate adjacent CB1 receptors. Scheme 1 depicts a general model for the ECS signaling in the CNS. This model, whereby post-synaptic endocannabinoid synthesis in response to a presynaptic signal acts presynaptically to depress the release of the presynaptic neurotransmitter, has been suggested to provide a protective negative feedback mechanism against over-activity of the presynaptic nerves.1 Peripheral roles of the endocannabinoid system in the regulation of processes as diverse as gut function and bone turnover are also well-described.2 These endogenous cannabinoids are mainly synthesized by membrane phospholipids on the postsynaptic membrane.3 Several catabolic pathways responsible for the termination of endocannabinoid signaling have been described.4 Among all hydrolytic enzymes, fatty acid amide hydrolase (FAAH) is the main metabolic enzyme of AEA, while in the brain monoacylglycerol lipase (MAGL) is primary metabolic enzyme of 2-AG.4–6 Nonetheless, other serine hydrolases α/β hydrolase domain-6 (ABHD6) and α/β hydrolase domain-12 (ABHD12) may be more important for endocannabinoid metabolism in microglial cells.7 Additional catabolic pathways, such as oxidation catalyzed by cyclooxygenase-2 and by some members of the cytochrome P450 (CYP) family of enzymes have also been reported and yield biologically active products,4, 8 but are outside the scope of the present article.
Scheme 1.

A general model for the ECS signaling in the nervous system
The ECS regulates important physiological processes such as pain, emotion, stress, exercise, and cognition functions.2, 9, 10 Accordingly, cannabinoid receptors have become promising targets for the management of numerous pathologies including, but not limited to, chronic pain, epilepsy, anxiety, depression, Alzheimer disease (AD), Parkinson’s syndrome (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), drug dependence, osteoporosis and liver, kidney, intestine, cardiovascular dysfunction (CVD).11–15,16 Cannabinoids, particularly phytocannabinoid preparations, also have a wide off-label use in the treatment of chronic pain, despite modest evidence of efficacy in rigorous randomized clinical trials and a high incidence of unwanted effects.17
Positron emission tomography (PET) is a non-invasive and highly sensitive molecular imaging technique that allows in vivo quantification of biochemical and pharmacological processes under healthy and diseased conditions.18 Using radioactive PET tracers, in vivo imaging can be achieved at the molecular level to obtain quantitative information on target engagement and the degree of receptor occupancy at a given drug dose. PET scanning equipment and PET probes are just like the relationship between pistol and bullet. The bullet can accurately target in vivo biomarkers and provide real-time information on tracer accumulation in the area of interest, which plays a crucial role in precision medicine and quantitative pharmacokinetic research.19, 20 Such information is invaluable and harbours enormous potential to accelerate drug development,21, 22 especially in Phase 0 clinical trials.23–26 Over the past decades, while many PET probes targeting endogenous cannabinoid receptors (CB1R and CB2R) and hydrolases (FAAH and MAGL) have substantially advanced our understanding of the ECS,27 the vast majority of reported reviews are confined to one specific ECS component such as CB1R28 or CB2R.29, 30 The last review of CNS PET did not cover the radiotracers targeting cannabinoid receptors and MAGL.31 In contrast, this review provides a comprehensive summary of PET-based ECS imaging, thereby covering CB1R-, CB2R-, FAAH- and MAGL-targeted probes, as well as presenting challenges in drug discovery, radioligand development, and medical imaging applications. For each ECS component, representative PET probes will be introduced with their pharmacological properties, radiolabeling techniques, and PET imaging in rodents/nonhuman primates (NHPs)/human subjects.
2. RECEPTOR TARGETING PET TRACERS
Both CB1R and CB2R are members of the G-protein-coupled receptor (GPCR) superfamily containing seven transmembrane structures.32 CB1R density is high in the human brain, especially in the substantia nigra, globus pallidus, putamen, hippocampus and cerebellum.33–35 CB1R is also abundantly expressed in peripheral nerves and tissues, though, in a region-specific manner.36 For example, in the gastrointestinal system, CB1R can regulate intestinal peristalsis and the secretion of gastric acid and neurotransmitter, which in turn affect appetite and digestion.37 In the cardiovascular system, CB1R is significantly upregulated under pathological conditions and promotes disease progression.38, 39 CB1R expression has also been detected in adipose tissue, bone, skin, as well as several cancer cells.36, 40
Although CB2R and CB1R share 44% structural homology, their distribution patterns in the mammalian system are fundamentally different.41 Indeed, CB2R is predominantly found in peripheral organs harboring large numbers of immune cells such as the spleen and tonsils. Further, moderate expression can be found in the cardiovascular system, bone marrow and reproductive organs.41, 42 43 Recently, the crystal structures of CB1R and CB2R have been revealed.44, 45 Hua et al.46 uncovered the structures of the inactivated, intermediate and activated states of CB1R and CB2R, respectively, which provides more accurate molecule models and theoretical supports for drug and ligand discovery.
Due to the diverse regulatory effects by chronic therapeutic exposure, and the presence of genetic splice variants, cannabinoid receptors have unique pharmacological and physiological effects and have been considered as an attractive drug target for over 20 years. CB1R plays an important role in normal physiology (cognition, appetite, memory),47 and is closely related to several neuropsychiatric48 and metabolic diseases.49, 50 CB1R agonists and inverse agonists have been extensively developed, but are accompanied by side effects impairing cognitive function and the ECS reward system.51, 52 A well-known example is rimonabant (SR141716A), a potent and selective CB1R inverse agonist developed by Sanofi-Synthelabo.53 It was found to improve lipid and glucose metabolism and became one of the most promising weight-loss drugs. However, rimonabant was later linked to rare but severe cases of depression and suicide, resulting in its withdrawal from the market in 2008.54 Currently approved drugs by the FDA include tetrahydrocannabinol, cannabidiol, and their analogs or compositions, such as Marinol® (dronabinol),55, 56 Cesamet® (nabilone)57, Sativex® (nabiximols)58, and Epidiolex® (cannabidiol) 59. CB1 negative allosteric modulators, dronabinol, and nabilone are mainly used to treat nausea, vomiting, and weight loss when traditional treatments are ineffective, and for the treatment of spasticity and pain in MS as combination therapy (nabiximols). Cannabidiol is used to treat epilepsy associated with Lennox-Gastaut Syndrome (LGS) and Dravet Syndrome (DS). In order to avoid CNS side effects, drugs that act on peripheral CB1R alone have been developed and demonstrated promising results. NEO1940 (ART27.13), a peripheral CB1R and CB2R agonist for the treatment of cancer-related anorexia and weight loss, has entered a phase II clinical trial.60 CRB-4001(JD-5037) is a peripheral CB1R inverse agonist in a phase I clinical trial, which was intended for the treatment of non-alcoholic steatohepatitis.61, 62 CB2R regulation is disrupted in peripheral inflammation, neurological diseases, cancer, and atherosclerosis.63, 64 Of note, CB2R activation is associated with anti‐inflammatory and protective effects in the CNS and periphery.65 In several neurodegenerative disease models such as AD or PD, treatment with selective CB2R agonists are effective in attenuating pro-inflammatory cytokine release and migratory activity of immune system cells.66, 67 Thus the effects of CB2R agonists may offer a new therapeutic approach for the treatment of neurodegenerative disorders, taking advantage of the notion that CB2R activation does not elicit the psychoactive side effects frequently observed with brain penetrating CB1R agonists.68, 69
CB2 agonist JBT-101 (Lenabasum) is currently in a phase III clinical trial for treating diffuse skin systemic sclerosis and dermatomyositis, and phase II clinical trial for treating cystic fibrosis, systemic sclerosis and systemic lupus erythematosus.61 CB2 agonist APD371 (Olorinab) entered phase II trials for the treatment of gastrointestinal pain.70 As mentioned above, the development of PET radiotracers for cannabinoid receptors facilitates our understanding of the ECS under normal physiological and disease states and has played an important role in the design and evaluation promising drugs that target the receptors. Several research groups have made substantial efforts towards the development of CB1R or CB2R PET radioligands, highlighted applications in CNS diseases.
Given the diversity of ECS-targeted drug discovery, the clinical availability of suitable PET probes is crucial to guide drug development as well as to identify and validate novel disease areas, where the ECS can be exploited as a pharmacological target. As such, several research groups have made substantial efforts towards the development of CB1R or CB2R PET radioligands.
2.1. PET Tracers for Imaging CB1R
Endogenous CB1R ligands have moderate affinity and are highly lipophilic, so they are not suitable for PET or SPECT (Single Photon Emission Computed Tomography) tracer development. By reducing their lipophilicity and retaining their high affinity, researchers synthesized a series of compounds for imaging CB1R in the CNS. Based on selective CB1 antagonists, PET tracers have been widely developed and selected structures are shown in Figure 1. A summary of the relevant parameters for selected CB1R specific PET tracers is presented in Table 1.
Figure 1.

Representative chemical structures for CB1R-specific PET tracers
Table 1.
Radiolabeling and PET imaging application of CB1R specific tracers
| Radioligand (RCYs) | PET imaging study |
|
|---|---|---|
| subject | brain uptake | |
| [11c]5 (16±5%a) |
Rodents:obesity (myocardial imaging)
NHP Human: SZ; obesity (myocardial imaging); alcohol dependence |
Mouse: 6.6 %ID/g (striatum, 15 min) Baboon: BP: 1.3 (putamen) Human: VT: 1.35±0.35 Human: SUVmax = 1.75±0.35 (putamen) Human: VT: 0.72±0.07 (putamen) |
| [11C]6 (2.5%±1.1 %b) | Rodents, NHP Human: SZ | Monkey: VT: 24.3 (mL/m3, striatum) Human: VT: 29.1±17.4 (putamen) Human: SUVmax= 3.5±0.5 (putamen) |
| [11C]7 (16.5%b) | Rodents, NHP | Rat: high BBB permeability, high specificity to CB1R Monkey: SUVmax = 3.3 (30 min) |
| [18F]8 (7.92±2.16%b) | Rodents, NHP | Rat: high BBB permeability, high specificly to CB1R Monkey: SUVmax = 2.75±0.75 (15 min) |
| [18F]9 (5.92%±1.34%b) | Rodents, NHP | Rat: high BBB permeability, high specificity to CB1IR Monkey: SUVmax = 5.75±0.75 (20 min) |
| [18F]10 (7.93±2.48%b) | Rodents: AD; BAT NHP Human: chronic cannabis smokers; alcohol-dependent; SZ; Psychosis |
Mouse: 1.5 %ID/mL (BAT) Monkey: SUVmax = 5.5±1.0 (20 min) Monkey: VT 35.4 (mL/m3, striatum) Human: SUVmax = 3.5±0.5 (putamen) Human: VT 24.3±7.2 (putamen) |
| [18F]11 (-) | Rodents: HD; PD NHP Human: anorexia and bulimia nervosa; episodic migraine; epilepsy; HD |
Rat, Monkey: high BBB permeability, high specificity to CB1R Human: SUVmax = 15±0.2 (putamen) Human: VT 25.1±13.3 (putamen) |
| [11C]12 (50%a) | Rodents, NHP, Human | Monkey: VT: 2.36 (mL/m3, striatum) Human: SUVmax = 2.35±0.65 (putamen) Human: VT: 3.15±0.70 (putamen) |
| [11C]15 (7.35±4.25%b) | NHP | Monkey: SUVmax = 2.2±0.5 (120 min) |
| [18F]16 (3.55±2.05%b) | NHP | Monkey: SUVmax = 1.8 (240 min) |
| [11C]7 (17±8%) | Rodents: BAT | Mouse: 2.8 %ID/mL (BAT) |
non-decay corrected RCYs
decay corrected RCYs
The data of PET imaging are listed only in the control group.
Initially, a series of PET tracers ([18F]1-[11C]4) based on rimonabant were developed for CB1R imaging and preliminary biological evaluations were carried out. However, these tracers had either high lipophilicity (logD > 4), resulting in nonspecific binding, or poor brain permeability that is not suitable for CNS imaging.71–73 To improve the binding affinity of rimonabant (Ki hCB1R = 46.2 nM, cLogD = 6.0), Fan et al. synthesized OMAR/JHU5528 (Ki hCB1R = 11 ± 7 nM, cLogD = 4.3) and its analogs by introducing cyano groups. OMAR is a CB1R antagonist, demonstrating higher specificity than earlier CB1R antagonists with less lipophilic and significantly increased Bmax/Ki ratio to 91, indicating great potential for successful in vivo imaging.74, 75 Further PET evaluations in mouse and baboons demonstrated that [11C]OMAR ([11C]5) readily entered the brain, specifically and selectively visualizing cerebral CB1 receptors. In the biodistribution experiment, the radioactive accumulation reached its maximum value (6.6 %ID/g in the striatum) at 15 min; radioactivity in the baboon brain reached its peak at about 10 min and the ratio of CB1-rich regions to CB1-poor regions was up to 2.5 and the highest binding potential (BP) value was 1.3 in the putamen.76 Wong et al.77, 78 presented the first human studies by using [11C]5, which successfully displayed the distribution of CB1R in the healthy controls and SZ patients. Maximum standard uptake value (SUVmax) in controls reached 1.4–2.1 at about 20 min post-injection and average volume of distributions (VT) ranged from 1.0–1.7. Compared with control subjects, regional total VTs of [11C]5 were higher in SZ patients, especially in the pons (0.93 vs 0.72, P < 0.05). Another study comparing the availability of CB1R between male SZ patients and healthy controls highlighted that CB1R availability of male SZ patients was moderately and globally reduced, and the use of antipsychotics and tobacco increased CB1R availability in patients79. In vitro studies have shown that the ECS plays a vital role in alcohol dependence.80–82 Neumeister et al. used [11C]5 to carry out the first in vivo experiment to demonstrate this correlation. VT in alcohol-dependent patients administered [11C]5 was approximately 20% (P = 0.023) higher than that of healthy controls.83
Compound [11C]5 was not only used to image CB1R in the CNS, but was also successfully employed to assess peripheral cardiovascular CB1R expression. Valenta et al. demonstrated that the retention rate of [11C]5 in the myocardium of obese mice was significantly higher than that of normal-weight mice.84, 85 These results may provide a theoretical basis for further clinical testing of CB1R targeted molecular imaging in cardiac metabolic diseases. However, due to the moderate lipophilicity of [11C]5, it tends to accumulate in the liver, which may hamper accurate quantitative assessment.86
Similarly, based on the structure of SR141716, Yasuno et al. reported the synthesis of [11C]MePPEP ([11C]6) (Ki hCB1R = 0.11 nM, Kb = 0.57 nM, LogD7.4 = 4.8, Kb represents an in vitor efficacy constant that is calculated using the following equation: Kb = IC50/[1 + [Agonist]/EC50], which is derived from Cheng-Prusoff relationship.87) by the reaction of the O-desmethyl precursor with [11C]CH3I. Compared to PET imaging of [11C]5 in baboon brain, the uptake kinetics of the two tracers were similar, but the peak uptake of [11C]6 was higher and SUVmax almost reached 6.0 within 10–20 minutes post injection.88 Subsequently, Donohue et al. synthesized [11C]6 and its four analogs [11F]FMePPEP ([11C]7, Kb = 0.22 nM, Log D7.4 = 5.7), [18F]FEPEP ([18F]8, Kb = 0.42 nM, Log D7.4 = 5.8), [18F]FMPEP ([18F]9, Kb = 0.19 nM, Log D7.4 = 5.7) and [18F]FMPEP-d2 ([18F]10, Ki hCB1R = 0.11 nM, LogD = 4.24),89 and demonstrated that [11C]6-7 and [18F]8-9 crossed the blood-brain barrier adequately and exhibited high specificity to CB1R in rat brain tissues. Terry et al. further evaluated these four imaging agents ([11C]7, [18F]8–10) in monkeys and SUVmax in the brain were 3.3 by 30 min, 2–3.5 at 15 min, 5–6.5 at 20 min, 4.5–6.5 by 20 min, respectively. And then they advanced the most promising agent [18F]10 to humans.90 [18F]10 uptake in the human brain was similar to [11C]6, and the peak SUV in the putamen ranged from 3.0 to 4.0, however, the retest variability of [11C]6 in plasma was relatively large (58%), as compared to [18F]10 (16%), and [18F]10 had a good retest variability of VT (14%), which had previously been reported by Terry et al.91 In subsequent studies, [11C]6 was used as a CB1R imaging agent to study the anxiogenic effects of cannabis in humans, which proved that this effect was closely related to the activation of CB1Rs on the amygdala.92 [18F]10 has been used to demonstrate a reversible downregulation of CB1Rs in chronic cannabis smokers,93, 94 and in alcohol-dependent patients,95 [18F]10 demonstrated reduced CB1R binding that did not recover after 2–4 weeks of abstinence. It is worth noting that in CB1R imaging of alcohol-dependent patients with [18F]10, the results are opposite to the [11C]5 imaging study aforementioned. Differences in experimental design or patient selection may have contributed to these divergent findings. In addition, [18F]10 has been used to study mental illnesses. Borgan et al. designed a cross-sectional, case-controlled study in patients with first-episode psychosis, demonstrating that CB1 receptors are viable therapeutic targets in psychosis.96 Besides CNS-targeted CB1R imaging, [18F]10 has been used to quantitatively monitor the density of CB1R in brown adipose tissue (BAT). Eriksson et al. and Lahesmaa et al. confirmed that [18F]10 showed specicific binding to BAT in rodents and humans, respectively.97, 98
Using taranabant as a lead compound, Merck research laboratories synthesized [18F]MK-9470 ([18F]11, IC50(human) CB1R = 0.7 nM, IC50(human) CB2R = 44 nM), a new potent CB1R inverse agonist, with a high affinity for CB1R.99 In vivo brain imaging and autoradiography distribution studies in rhesus monkeys have demonstrated reversible binding to CB1R with reasonable binding specificity. The occupancy of CB1R in the human brain has low Test-retest (TRT) variability (ca. 7%), as determined by the area under curve (AUC) of the respective time-activity curve. However, in vivo radioactive distribution of CB1R is inconsistent with ex vivo findings in the postmortem research, an inconsistency which was suggested to be related to the different properties (agonists and inverse agonists) of radioligands.100 Subsequently, Van et al. analyzed the distribution of [18F]11 in 50 healthy individuals.101 In women, as the age increased, radiotracer-receptor binding increased in the basal ganglia, the lateral iliac crest, and the limbic system, especially in the hippocampus. Men do not have these age-related characteristic changes, but men show higher [18F]11 binding in the limbic system and cortical-thalamic-cortical loop cluster.
Compound [18F]11 has been used in imaging studies of various neurological diseases: Anorexia nervosa patients demonstrated increased [18F]11 uptake (24.5%, P = 0.0003) in the brain compared to healthy controls; 102 a study investigating [18F]11 in female with episodic migraine showed that there was an increased interictal binding (average gray matter difference+16%, P = 0.009), indicating a low level of competing endogenous cannabinoids in this area;103 in patients with epilepsy of temporal lobe due to hippocampal sclerosis, [18F]11 uptake increased in the ipsilateral temporal lobe.104 In the premanifest phase of the HD, the PBA-HD (Problem Behavior Assessment for HD) scores inversely correlated with CB1R binding in prefrontal regions and cingulate cortex.105 Imaging studies of [18F]11 performed in the brains of PD patients showed that the reduced availability of CB1R is associated with various cognitive impairments106. Although the CB1R inverse agonist PET tracers highlighted above have been used in clinical research to study receptor availability in several neurological disorders, rimonabant and taranabant development was discontinued in 2008 due to neurological side effects.107
Donohue et al. developed a new selective CB1 receptor antagonist, [11C]SD5024 ([11C]12, Ki hCB1R = 0.47 nM, LogD7.4 = 3.79), which demonstrated high specific binding in the cynomolgus monkey brain.108 The brain uptake reached a peak SUV of 2.0 at 24 min and declined to 1.5 at 90 min. Tsujikawa et al. evaluated this radioligand in vitro and in vivo and compared it with the following ligands: [11C]6 (Ki hCB1R = 0.11 nM, LogD7.4 = 4.77), [11C]5 (Ki hCB1R = 2.05 nM), [18F]11 (Ki hCB1R = 0.10 nM) and [18F]10 (Ki hCB1R = 0.11 nM, LogD7.4 = 4.24).109 [11C]12 demonstrated optimal lipophilicity and precise measurement accuracy in terms of intersubject variability (22%) for VT. It is worth noting that Hjorth et al.110 employed [11C]12 in NHP imaging to obtain the pharmacokinetic profile, calculate the occupancy rate and explore the optimal dose of three novel selective CB1R antagonists developed by AstraZeneca (AZ).111 Compound [11C]13, a similar structure to [11C]12, was reported by Donohue et al. in an abstract, and it showed high specificity in the monkey brain,112 however, no further studies have been reported.
WIN 55212–2 is a cannabinoid receptor agonist that belongs to the aminoalkylindoles (AAI) family. This type of compounds has been radiolabeled for PET or SPECT imaging, but exhibited poor brain permeability and specificity.113 An important example of this compound class is [18F]14.114 Following these studies, Allen et al. synthesized a new array of aryl sulfonyl-substituted indoles as CB1R inverse agonists.115 Finnema et al. and Donohue et al. radiolabeled PipISB (Kb = 1.5 nM, cLogD = 5.1) with 18F and 11C, to afford [11C]15 and [18F]16, respectively, and further evaluated them in rhesus and cynomolgus monkeys. [11C]15 and [18F]16 crossed the blood–brain barrier (BBB) and exhibited CB1R specific and displaceable binding in monkeys.116, 117
Based on the previous research118, Yamasaki et al.119 modified 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1) to synthesize compound [11C]17 (KD = 15.3 μM, LogD = 3.76) and conducted PET imaging where this radiotracer demonstrated high specificity in the mouse BAT (2.8 %ID/mL). Zanato et al.120 labeled a fluorinated cannabis analog HU-210F with 18F ([18F]18 (Ki hCB1R = 0.98 nM, Ki hCB2R = 3.83 nM) as a potential imaging agent, but further biological evaluation has not been carried out. The latest CB1R imaging probe [18F]19, was reported by Chang et al.121 by modifying DBPR211; however, the uptake of this tracer in the mouse brain was low (< 1%).
2.2. PET Tracers for Imaging CB2R
Although CB2R expression is limited in the healthy CNS, there is a consensus that CB2R is upregulated on activated microglial cells upon neuroinflammation.63, 122 Thus, the CB2 receptor is regarded as an exceptionally promising target to exploit for neuroinflammation manifested in AD, PD and ALS.123 Despite strenuous efforts by several research groups worldwide, CB2R-targeted PET radioligand development has only moderately progressed in recent years. Reported probes have mainly been hampered by nonspecific binding due to high lipophilicity, metabolic instability, inappropriate pharmacokinetics and lack of selectivity over CB1R or other CNS targets. The search for suitable CB2R PET radioligands is further hampered by the lack of reliable information on CB2R Bmax values in the CNS as well as the lack of a highly selective CB2R antibody to confirm autoradiographic findings by histological assessment. Nonetheless, some promising examples with potential for clinical translation have emerged and will be discussed in this chapter. The structural scaffolds for CB2R PET ligands include oxoquinoline, pyridine, thiazole, triazine and oxadiazole derivatives (Figure 2). A summary of the relevant parameters for selected CB2R specific PET tracers is presented in Table 2.
Figure 2.

Representative chemical structures for CB2R-specific PET tracers
Table 2.
Radiolabeling and PET imaging application of CB2R specific tracers
| Radioligand (RCYs) | PET imaging study |
|
|---|---|---|
| subject | brain uptake | |
| [11C]20 (32±11%, 40±12%) | Rodents, NHP Human: AD | Mouse: SUVmax =1.2 (2
min) Rat: SUVmax =1.4 (1.5 min) Monkey: SUVmax = 1.5 (1.5 min) Human:low CB2R availability in AD patients |
| [11C]21 (33%a) | Rodents: neuroinflammation,
atherosclerosis Human: ALS (spinal card tissues), atherosclerosis (specimen ) |
Rat: high BBB permeability specificly to CB2R |
| [18F]22 ( ~6%a 12.0±2.7%a) | Rodents: HD Human: ALS (spinal cord tissues) |
Rat: not sensitive to the CB2R overexpression Human: no specificly to CB2R |
| [11C]23 (41.7±8.7%a) | Human: ALS (spinal cord tissues) | Human: specificly to CB2R |
| [11C]26 (-) | Rodents: neuroinflammation | Rat: specificly to CB2R |
| [18F]27 (11±4%a) | Rodents Human: ALS (spinal cord tissues) |
Rat: SUVmax = 2.7±0.3 (2 min. Spleen) Human: specificly to CB2R |
| [11C]30 (26±2%b) | Rodents: neuroinflammation, AD, cerebral ischemia | Mice:sensitive to the CB2R overexpression in the AD mice |
| [18F]31 (0.95±0.65%) | NHP: neuroinflammation | Monkey: SUVmax =0.85±0.15 (brain) |
non-decay corrected RCYs
decay corrected RCYs
Oxoquinoline derivatives have been exploited for their suitability as CB2R PET radioligands. As such, the most extensively studied candidate in this class of compounds, [11C]NE40 ([11C]20), was successfully synthesized via O-methylation of the respective phenolic precursor using either [11C]CH3I or [11C]methyl triflate.124,125 With a binding affinity (Ki CB2R) of 9.6 nM and a selectivity factor of ~ 100 over CB1R, [11C]20 proved to be specific for CB2R in the rodent spleen, while concurrently exhibiting a fast washout from the CB2R-deficient healthy brain.125 Moreover, preclinical evaluation of [11C]20 revealed specific and reversible binding to human CB2R (hCB2R) in a rat model with local hCB2R overexpression in the right striatum.126 In the same study, the authors reported a high brain uptake (SUVmax = 1.5 at 90 s) in rhesus monkeys, confirming that the tracer is able to penetrate the brain in non-human primates. In a subsequent clinical study, [11C]20 did not detect the anticipated CB2R upregulation in the brain of AD patients, thus raising concerns about the suitability of the tracer for CB2R imaging in AD.127 Particularly, given the high abundancy of CB1R in the human brain, a selectivity factor of ~ 100 might have been insufficient to avoid off-target binding of [11C]20 to CB1R. Along this line, no relationship was found between regional or global amyloid load and [11C]20 uptakes.127
Slavilk et al.128 used anisidine and diethyl 2-(ethoxymethylene)malonate to synthesize a series of novel 4-oxoquinoline derivatives based on the structure of KD2 (Ki CB2R = 1.7 nM, logD7.4 = 3.3)129. RS-016 (21) emerged as most suitable candidate, exhibiting an improved affinity (Ki = 0.7 nM) and lipophilicity (LogD7.4 = 2. 8).130 Dynamic brain PET scans of [11C]RS-016 ([11C]21) in a murine neuroinflammation model induced by lipopolysaccharide (LPS) application showed higher CB2R expression level in different brain regions including the cortex, hippocampus, and cerebellum. Further, [11C]21 was used to visualize the CB2R in human and mouse atherosclerotic lesions.130 To overcome the limitation of the short half-life of carbon-11, Slavik et al.131 developed a radiofluorinated analog of [11C]21, codenamed [18F]RS-126 ([18F]22). Although the in vitro results with [18F]22 were encouraging, no CB2R expression was detected in the brain of LPS-treated mice. The latter was attributed to the low specific activity of [18F]22. In addition, [18F]22 was rapidly metabolized in mice. Haider et al.132 improved the synthesis strategy to obtain higher radiochemical yields (RCYs) and molar activities of [18F]22. In the transgenic R6 /2 mouse model for HD, [18F]22 was not sensitive to the CB2R overexpression, although CB2R mRNA upregulation was detected by qPCR in the same tissue. Further, due to high nonspecific neuronal tissue binding, [18F]22 did not show specific CB2R binding in human ALS spinal cord sections. In sharp contrast, the newly developed hydroxylated derivative, [11C]RS-028 ([11C]23), exhibited a significantly reduced lipophilicity, thus leading to less nonspecific binding and allowing the detection of CB2R upregulation in postmortem human ALS spinal cord tissue. Notably, the spleen uptake of [11C]23 was relatively low compared to its predecessors and was accompanied by a rapid washout in vivo, potentially owing to enzymatic degradation. To investigate the influence of the O-alkyl chain length and oxygen position on metabolic stability, [18F]AH-040 and [18F]AH-043 ([18F]24 and [18F]25) were synthesized. Although these probes showed improved stability to liver enzymes, their selectivity over CB1R was substantially reduced, impeding their further development.132
Among the reported structural scaffolds of CB2R PET radioligands, 2,5,6-trisubstituted pyridines (Figure 2) have been thoroughly assessed and currently constitute the most promising candidates for clinical translation. Indeed, initial studies with [11C]RSR-056 ([11C]26, Ki hCB2R = 2.5 nM, logD7.4 = 1.94) revealed significantly increased brain uptake following LPS-induced neuroinflammation in mice. Nonetheless, it should be noted that only part of the increased [11C]26 accumulation in the brain was attributed to CB2R-specificity, as evidenced by the weak signal reduction under blockade conditions.133 The latter can be, at least in part, explained by inflammation-induced damage to the blood-brain barrier, which would ultimately result in an increased tracer delivery to the brain, independent of CB2R.134 Due to the short physical half-life of carbon-11, which confines the use of [11C]26 to facilities with an on-site cyclotron, the authors synthesized a series of fluorinated derivatives via structural modifications at positions 2 and 5 of the pyridine core. These efforts led to the development of the radiofluorinated analog [18F]27,135 with a prolonged half-life of 109.8 min, a binding affinity of 6 nM and a selectivity factor of ~ 600 over CB1R. [18F]27 proved to be CB2R-specific by in vitro autoradiography experiments of the rodent spleen. Further, CB2R receptor upregulation in postmortem spinal cord tissues of ALS patients was confirmed with [18F]27. Notwithstanding the promising in vitro results obtained with [18F]27, in vivo specificity for the CB2R-rich rodent spleen was only modest. To overcome this limitation, the authors studied the influence of structural modifications at position 6 of the pyridine core, which ultimately led to the discovery of [18F]RoSMA-18 ([18F]28), a ligand with subnanomolar affinity (Ki hCB2R = 0.7 nM) and a remarkable selectivity factor of > 12,000.136 [18F]28 revealed exceptional specificities for the CB2R-positive rodent spleen by in vitro autoradiography (71 ± 2%) and ex vivo biodistribution (86 ± 2%), which was superior to any previously reported CB2R-targeted PET probe. Indeed, the outstanding specificity was further elegantly corroborated in the CB2R knockout mouse spleen. [18F]28 was sensitive in detecting CB2R upregulation in postmortem human ALS spinal cord tissue by in vitro autoradiography. Further, PET experiments confirmed the specific and reversible CB2R binding of [18F]28 in the CB2-positive rat spleen. The authors found only intact [18F]28 in the rat brain following intravenous tracer injection. Nonetheless, in vivo defluorination was noted by substantial skull uptake, which was circumvented by replacing the hydrogen atoms in the fluoropropyl side chain with deuterium atoms to afford [18F]RoSMA-18-d6 ([18F]29), in which case no radioactivity accumulation in the skull was observed. [18F]29 is currently considered a promising candidate for clinical translation.136
A-836339 is a highly potent and selective CB2R agonist (Ki CB2R = 0.7 nM, Ki CB1R = 270 nM), which is based on a thiazole core structure137. Horti et al. radiolabeled [11C]A-836339 ([11C]30) via its desmethyl precursor and performed PET imaging in LPS-treated CD1 mice, APP/PS1 transgenic mice (mouse model for AD) as well as wild-type control animals.137 Whole body time-activity curves (TACs) for the control mice revealed a fast tracer washout from the brain, while CB2R specific binding was observed in the spleen. In LPS-treated mice, [11C]30 uptake was significantly increased in the brain (SUVmax = 2.3), as compared to the control group (SUVmax = 0.5), after five days of LPS treatment. Similarly, higher brain uptake was found in transgenic APP/PS1 strains compared to wild-type littermates, and the regional distribution was consistent with the distribution of Aβ amyloid plaques. AM630 blocking showed 29%−36% specific binding in the cerebellum, brainstem, and cortex. Attempts to develop a radiofluorinated analog of [11C]30 led to an improved CB2R affinity with Ki of 0.4 nM for CB2R and 380 nM for CB1R. Nonetheless, the rapid metabolism of this 18F analog following intravenous injection as well as the presence of radiometabolites in the brain hampered further evaluation.138 Savonenko et al. further investigated the utility of [11C]30 as a PET biomarker for neuroinflammation in AD mice.139 The authors found an increased tracer uptake in AD mice, which was blocked by the commercially available CB2R inverse agonist AM630. In sharp contrast, Pottier et al. did not detect CB2R upregulation with [11C]30 PET in two distinct rat models of cerebral ischemia.140
Triazine scaffolds have been evaluated for CB2R-targeted PET imaging by Hortala et al.141. The authors reported on the development of [18F]31, demonstrating brain penetration in rhesus macaques and baboons. Baboon pretreatment with LPS led to VT increase of about 30%, confirming previous observations with other CB2R PET probes in mice. In another study by Yrjola et al., the best performing triazine derivative, [18F]32 (EC50 = 0.60 nM), was found to suffer from rapid metabolism and elimination, rendering its pharmacokinetic properties unsuitable for in vivo PET.142
MA2 and MA3 are the analogs of highly potent CB2 agonist N-arylamide oxadiazole.143 Tissue distribution of [11C]33 and [18F]34 demonstrated high brain uptake at 2 min post injection. Notably, [18F]34 was found to specifically accumulate in brain regions with overexpression of the human CB2R in rats; however, no specific binding of [18F] 34 to the CB2 receptor was observed in PET scans in nonhuman primates.144
3. HYDROLASE TARGETING PET TRACERS
It is known that drugs that act as direct agonists or antagonists on the endocannabinoid receptors can cause drug dependence or addiction.145 Hence, as an alternative approach to achieve therapeutic effects, drugs/tool compounds that inhibit the hydrolases that metabolize AEA and 2-AG have been developed. The two main hydrolases involved in the hydrolytic metabolism of these endocannabinoids are FAAH and MAGL. FAAH is a 60kDa integral membrane hydrolytic enzyme that is highly expressed in the mammalian brain along with liver and kidney, which is responsible for terminating the endogenous cannabinoid signaling of AEA by hydrolysis of the amide bond.146, 147 MAGL, is a 33kDa serine hydrolase with high expression in the brain, BAT, adrenal glands, liver and kidneys, and acts as a critical degradative enzyme responsible for ca. 85% of 2-AG metabolism in the brain, generating glycerol and arachidonic acid (AA).148–151 As shown in Scheme 2, systemic or local inhibition of FAAH or MAGL increase levels of AEA or 2-AG, while concurrently decreasing arachidonic acid level, respectively. These changes also affect endocannabinoid signaling and suppresses eicosanoid production. Increasing levels of AEA exert effects of anti-anxiety, anti-depression, and treating addiction and withdrawal symptoms.152, 153 Similarly, increasing the level of 2-AG in the brain has effects of anti-anxiolytic, anti-depression, anti-emetic, anti-inflammation and neuroprotection.154, 155 Moreover, inhibition of MAGL (genetically/pharmacologically) has therapeutic implications for some metabolic diseases.156, 157 Numerous studies have demonstrated that FAAH and MAGL are potential therapeutic targets for a variety of human pathologies including pain, depression, (neuro)inflammatory diseases and cancer.149, 155, 158–160 For several of these indications proof-of-concept studies have been performed in several animal models with the respective knockout animals.8, 157, 161 It is worthy of note that Scheme 2 only captures main features of FAAH and MAGL in the ECS since both FAAH and MAGL can metabolize other related lipids: in the case of FAAH, blockade of its hydrolysis activity for N-palmitoylethanolamine is an interesting pharmacological approach, given the anti-inflammatory effects of this endogenous lipid.162 In the case of MAGL, it has shown that that it promotes cancer cell growth by providing long chain fatty acids from their corresponding monoacylglycerols,163, 164 and to provide arachidonic acid for prostaglandin synthesis in neuroinflammation.155
Scheme 2.

Pharmacological properties of FAAH and MALG inhibitors
Currently, a variety of FAAH and MAGL inhibitors have been developed, characterized in animal models of disease and assessed at various stages of clinical trials.165–167 Initial clinical studies with FAAH inhibitors PF-04457845,168 V158866169 and JNJ-42165279170 indicated that the compounds were well-tolerated (vide infra for a discussion with respect to FAAH inhibitor BIA10–2474171), although PF-04457845 was not efficacious in a study of osteoarthritic pain. Recently, Fazio et al. reviewed newly developed single- and dual-target FAAH inhibitors in the literature, and proposed that multi-target inhibition may be a promising strategy for the development of new effective inhibitors. Future drug design and medicinal chemistry could also take advantage of the recently disclosed human FAAH crystal structure.172 Among MAGL inhibitors, ABX-1431 has entered clinical phase II studies for neurological disorders. In this context, PET ligand development towards FAAH and MAGL has also made considerable progress for target engagement studies in drug discovery and studying pathophysiological changes during disease states.
3.1. FAAH Inhibitor based PET Tracers
A major clinical trial incident occurred in January 2016,173 which led to the death of one of the participants during the study with a FAAH inhibitor codenamed BIA 10–2474. Following the incident, there has been a solid body of evidence that the drug exhibited off-target effect.174–177 The tragic results of this trial emphasized that it is especially important to fully understand the pharmacological effects, metabolism and pharmacokinetic-pharmacodynamic (PK-PD) relationship of drugs in the body before conducting clinical trials. Using PET probes are uniquely well suited for in vivo drug evaluation to assess target engagement and correlate therapeutic dose-target occupancy relationship accurately in humans via a phase 0 study. Currently, several PET tracers for imaging FAAH have been disclosed and can be classified into two categories, namely irreversible and reversible tracers, based on different binding kinetics, as shown in Figure 3. We also summarize the pharmacological and imaging properties of FAAH PET tracers in Table 3.
Figure 3.

Representative chemical structures for FAAH-specific PET tracers
Table 3.
Radiolabeling and PET imaging application of FAAH specific tracers
| Radioligand (RCYs) | PET imaging study |
|
|---|---|---|
| subject | brain uptake | |
| [11C]36 (4%a) | Rodents, Human: cannabis addiction, alcohol use disorders, psychiatric, borderline personality disorder | Rat: SUVmax = 2±0.4 Human: SUVmax = 4.3±0.8 (putamen) |
| [11C]43 (20±4.6%b) | Rodents, NHP | Rat: SUVmax ~ 1.5 (cerebellar nucleus) Monkey: SUVmax = 2.0 (occipital cortex) |
| [11C]44 (19.5±2.5%b) | Rodents, NHP | Rat: SUVmax = 4.55±0.57 (cortex) Baboon: SUVmax=2.1 (whole brain,55 min) |
| [11C]45 (15±5%b) | Rodents | Rat: SUVmax = 7.8±0.1 (cortex,40 min) |
| [11C]46 (4.5±1.3%) | Rodents | Rat: SUVmax = 2.8±1.6 |
| [11C]47 (10.7±4.4%b) | Rodents | SUV ~ 0.8 (cortex, 90 min) |
| [11C]50 (-) | NHP, Human | Monkey: BP: 0.7±0.3, VT: 3.5±0.5 Monkey: SUVmax >1 Human: VT: 17±3 |
| [18F]51 (13%a) | Rodents | Mice: SUVmax =0.87 (cerebellar nuclei, 1.5 min) |
| [18F]52 (17±6%b) | Rodents | 9.2 %ID/g |
non-decay corrected RCYs
decay corrected RCYs
The data of PET imaging are listed only in the control group.
3.1.1. Irreversible FAAH Radiotracers
URB597 (IC50 = 4.6 nM, cLogD = 3.6) is a classic FAAH inhibitor based on O-aryl carbamate scaffold with potentially useful activity in an animal model of anxiety.178–181 A 11C-labeled URB597 analogue, biphenyl-3-yl [11C]-4-methoxyphenylcarbamate ([11C]35) was the first radiolabeled FAAH inhibitor for PET imaging studies in rodents.182 Although the LogD value (2.27) was suitable for brain penetration, a continuous washout was observed after rapid peak brain uptake, which indicated no covalent binding to FAAH.182 However, it represented a reasonable starting point for the design of radiolabeled FAAH inhibitors.
CURB (IC50 = 30 nM, LogD7.4 = 2.8), also named URB694, is an active and selective irreversible inhibitor of FAAH with improved brain penetration and plasma stability, as compared to URB597.183 As such, [11C]CURB ([11C]36) was the first reported successful irreversible PET tracer to image FAAH.184, 185 [11C]36 displayed similar ex vivo biodistribution to FAAH in the rat brain, with higher brain uptake in the cortex and lower signal in the hypothalamus. The radioactivity peaked at 5 min (SUVmax = 1.6–2.4) and no significant washout was observed, consistent with the suggested irreversible binding mechanism. High dose pretreatment using URB597 showed high specificity of binding. A polar metabolite was detected in blood, which has little effect on the imaging results as it is not blood-brain barrier permeable.185 Based on these promising preclinical studies, [11C]36 became the first PET tracer for mapping FAAH activity in the human brain.186 By performing 90-minute PET scans with arterial blood sampling on six healthy volunteers, λk3 was identified as the optimal parameter for quantifying FAAH binding and SUV reached its peak (4.3±0.8) 30 min post-injection. For the kinetic modeling of irreversible inhibitors, λk3 can be calculated by the equation K1 / k2 * k3 and constitutes a key parameter of kinetic modeling for two-tissue compartment models. Of note, λ is the ratio between the two rate constants, K1 and k2, and represents the equilibrium distribution volume of the ligand in the plasma and the organ of interest – in this case the brain. Moreover, k3 describes the influx of [11C]36 from the free and nonspecific compartment to the specifically bound compartment.187 Recently, [11C]36 has been applied in neuroimaging application of several neurological disorders, particularly in addictive diseases, such as cannabis addiction,188, 189 alcohol use disorders,190 psychiatric191 and borderline personality disorder,192 which further proves that FAAH could be a promising therapeutic target for a variety of CNS diseases.
Moreover, a series of [11C-carbonyl]-radiolabeled O-aryl carbamates were synthesized ([11C]37-[11C]42) and evaluated as radiotracers for imaging FAAH with PET by Wilson et al.193 All six radiotracers showed moderate to excellent brain uptake, good regional distribution (cortex > hypothalamus) and excellent specificity (80–95% blocking via pretreatments with URB597) for FAAH in the rat brain. Brain kinetics showed that [11C]dihydrooxazole carbamates ([11C]40–42) had much higher uptake (3.0–4.5 SUV in the cortex at 2 min) than [11C]biphenyl carbamates ([11C]36–39, 0.8–2.7 SUV in the cortex at 2 min).
2-Methylpyridin-3-yl-4-(5-(2-fluorophenyl)-4H-1,2,4-triazol-3-yl)piperidine-1-carboxylate (MFTC) is a high-affinity, selective FAAH ligand (IC50 = 0.34 nM for human FAAH)194 with moderate lipophilicity (cLogD = 3.6). In vitro studies demonstrated a similar binding affinity for FAAH as URB597 and CURB.185 [11C]MFTC ([11C])43) was successfully synthesized using bis(2-methylpyridin-3-yl)-[11C]carbonate as a new radioactive intermediate.195 PET studies of this ligand in rat brain demonstrated high uptake (SUVmax ~1.5 in the cerebellar nucleeus), and the distribution of radioactivity corresponded to FAAH expression. The accumulation of [11C]43 in the brain was successfully blocked by pretreatment with nonradioactive MFTC and URB597. Furthermore, PET imaging in monkeys brain demonstrated heterogeneous distribution and radioactivity accumulation was highest in the occipital cortex (SUVmax= 2.0); however, [11C]43 did not show high specific binding in the striatum, which was in contrast to [11C]36 and [11C]MK-3168 (a reversible inhibitor of FAAH, vide infra). In addition, NHP PET imaging showed that the tracer was gradually washed out in all regions and no plateau was reached during the 90 min scan period, suggesting that total binding in the monkey brain might be affected by nonspecific binding to some extent.
18F-Labeled PET tracers based on the dihydrooxazole scaffold have also been developed. [18F]DOPP ([18F]44) was synthesized via a multi-step automated synthesis, as a potent, selective FAAH ligand with an IC50 value of 0.82 nM for rat FAAH and a logD value of 2.33. In vitro assays showed that DOPP inhibited FAAH in a time-dependent manner. Ex vivo biodistribution studies demonstrated excellent brain uptake (> 4 SUV) and a distribution pattern consistent with FAAH patterns in the rat brain. Autoradiography of [18F]44 in the rat brain also showed high bound activity in FAAH rich regions. In addition, heterogeneous binding of [18F]44 was displaced by pretreatment with the FAAH inhibitors CURB or PF-04457845, indicating that the radiotracer bound specifically to FAAH in vitro.196 PET/magnetic resonance imaging (MRI) studies in baboons demonstrated that [18F]44 rapidly crossed the blood-brain barrier with SUVpeak of 2.1 and that the kinetic modeling parameter λk3 was appropirate for FAAH quantification.197 As a proof-of-concept, [18F]44 was employed to evaluate the potency of BIA 10–2472 in vivo in the brain, which may be useful to provide a rational approach to dose selection in clinical trials and avoid severe adverse events.198
Another 18F-labeled FAAH radiotracer, [18F]FCHC ([18F]45, IC50 = 1.2 nM), was also evaluated in rodents.199 Similar to previous results,193 ([18F]45 demonstrated high brain penetration (SUVpeak = 4.6), excellent specificity and appropriate biodistribution in rodents, warranting further evaluation in higher species.
Two urea-based irreversible tracers [11C]PF-04457845 ([11C]46, IC50 = 3.2 nM, LogD7.4 = 3.48) and [18F]PF-9811 ([18F]47, IC50 = 16 nM, LogP = 3.14) have been developed. The compounds interacted with FAAH in an analogous irreversible mechanism like other carbamate-based inhibitors.200Due to its high brain penetration, high selectivity and safety profile in humans,201 PF-04457845 was chosen as a promising candidate to be labeled with 11C via the [11C]CO2 fixation strategy.200, 202 Ex vivo biodistribution of [11C]PF-04457845 ([11C]46) in the rat brain was consistent with the known expression of FAAH and the highest uptake (4.4 SUV) was observed in the cortex. In addition, pretreatment with URB597 resulted in a 71–81% reduction of bound activity in all areas of the brain, demonstrating high specificity for FAAH.203 [18F]PF-9811 ([18F]47) was designed based on the scaffold of PF-04457845, in which the trifluoromethyl group was replaced by a fluoroethoxy functionality to facilitate the incorporation of a fluorine-18 radiolabel.204 Biodistribution experiments in rats showed good heterogeneous brain uptake, with high SUVs in the cortex, hippocampus and cerebellum (ca. 0.8 SUV at 90 min), and specific to nonspecific binding ratios of 2.3 to 2.6. Pretreatment experiments proved irreversible binding in the brain. After 90 minutes, the radioactivity of all brain regions decreased to 37%−73%, but the signal in the blood increased by a factor of 12. The authors claimed that this may have been related to irreversible binding in the brain and peripheral organs.
Dahl et al.205, 206 introduced a new and efficient fully automated [11C]CO2-fixation apparatus, and used this method to synthesize two FAAH radiotracers [11C]JNJ1661010 ([11C]48, IC50 = 12 nM) and [11C]49, with significant increased non-isolated RCY. This method can help to produce various 11C-carbonyl compounds in a highly efficient manner to facilitate rapid screening of potential FAAH PET ligands in vivo.
3.1.2. Reversible FAAH Radiotracers
Based on the binding mechanism, tissue uptake of irreversible PET tracers is frequently governed by perfusion, which is why they are described as flow limited tracers. As such, a flow limited PET tracer with irreversible binding mechanism may reveal information that is substantially confounded by organ perfusion, rather than reflecting the density/activity of the biological target. Indeed, kinetic parameter from in vivo PET quantifications, including K1, k2, k3, VT and BPND can be affected by the binding mechanism of a given tracer.207, 208 Examples of irreversible PET tracers that were shown to be spared from significant flow limiting effects include [11C]CURB184 and [18F]DOPP.197 Thus, recent research has focused on the development of reversible binding PET tracers for FAAH in order to obtain a comprehensive quantitative assessment of FAAH distribution, concentration, and availability in the brain. Currently, a limited number of reversible FAAH inhibitor PET probes have been disclosed based on pyrazole or α-ketoheterocyclic scaffolds.
A systematical evaluation of a library of FAAH inhibitors based on a pyrazole scaffold was conducted at Merck. Attributed to excellent potency (IC50(human) = 1.0 ± 0.6 nM) and selectivity, reasonable lipophilicity (LogD = 3.3), and amenability for radiolabeling, [11C]MK-3168 ([11C]50) was prepared as the first reported reversible PET tracer for imaging FAAH. In vivo PET imaging studies were performed under baseline and blocking conditions in rhesus monkeys, demonstrating consistent regional heterogeneous distribution (accumulation in the frontal cortex / striatum / hippocampus regions), good brain uptake (SUV > 1) and high specific binding (total/ nonspecific signal ~ 2:1).209 According to an abstract, the two tissue compartment model well simulated the [11C]50 TAC in monkey and human, and values of binding potential (BPND) and VT were 0.4–1 and 3–4 in the monkey respectively and the VT value in the human between 14 and 20. 210 [11C]50 has been applied to determine the optimal dose for FAAH inhibitior JNJ-42165279 in the human brain.211
FAAH inhibitors based on an α-ketoheterocyclic scaffold were comprehensively studied by Boger and co-workers.212, 213 These inhibitors exhibited reversible binding towards FAAH and other serine hydrolases. Based on the structure of OL-135 (a potent and selective FAAH inhibitor, Ki = 4.7 nM),214 Wang et al. designed two positron-emitting analogs, 18F- and 11C-labeled MPPO ([11C]51 represents the 11C-analog), with communal pyridinyl oxazole ring.215 PET imaging of 18F-analog showed rapid defluorination and high bone uptake in vivo, and failed to show sufficient brain permeability. MPPO, which exhibited an excellent potency (IC50 = 10 nM, Ki = 5.8 nM) to FAAH and selectivity over other enzymes,214 was selected as a candidate as it was amenable for 11C-labeling. The ex vivo biodistribution studies in mice indicated that the distribution of [11C]51 was consistent with the distribution of FAAH. However, only moderate brain uptake (0.8 SUV) was observed. PET imaging studies showed low-to-moderate specific binding to FAAH. Subsequent PET imaging in P-glycoprotein (PgP) / breast cancer resistance protein (Bcrp) knockout mice indicated that this tracer was not likely a substrate for Pgp/Bcrp. It should be noted here that [11C]51 showed a steady washout in the brain, indicating α-ketoheterocyclic scaffold indeed provided a roadmap for further investigation of reversible FAAH radiotracers.
Recently, Chen et al.216 discovered an improved FAAH inhibitor with a novel heterocyclic scaffold and the corresponding PET tracer [11C]FAAH-1906 ([11C]52, IC50(mice) = 11 nM, LogD = 2.9) was further studied. Ex vivo biodistribution studies demonstrated that [11C]52 possessed high brain penetration (9.2 %ID/g), reversible binding kinetics and moderate specificity. Further development of this tracer and close analogs is underway to validate this class of compounds in higher species.
3.2. MAGL Inhibitor based PET Tracers
MAGL is responsible for about 85% of 2-AG hydrolysis in the brain,5, 217, 218 and significant efforts have been contributed to drug discovery for MAGL inhibitors. So far, most MAGL inhibitors are irreversible based on carbamate and urea-bearing scaffolds, involving the formation of a covalent bond between the Ser122 of MAGL and the carbonyl group of the inhibitor. Recently, a limited number of reversible MAGL inhibitors were disclosed with high binding affinity, and advanced to potential PET tracers for imaging quantification ([11C]61-64, Figure 4). We also summarize the pharmacological and imaging properties of MAGL PET tracers in Table 4.
Figure 4.

Representative chemical structures of MAGL-specific PET tracers
Table 4.
Radiolabeling and PET imaging application of MAGL specific tracers
| Radioligand (RCYs) | PET imaging study |
|
|---|---|---|
| subject | brain uptake | |
| [11C]53 (4.5±1.5%a, 20%b) | Rodents, NHP | Rat: SUVmax= 1.75 (cerebral
cortex and striatum) Monkey: SUVmax= 0.93 (occipital cortex) |
| [11C]54 (19%b) | Rodents | Rat: SUVmax = 0.43 (cerebellum) |
| [11C]55 (40%) | Rodents, NHP | Mice, Rat: high BBB permeability, specificity to MAGL Monkey: SUVmax ~4.2, 2.5 min |
| [11C]56 (11%) | Rodents, NHP | Rat: SUVmax
>1.5 Monkey: SUVmax =1.7 (occipital cortex) |
| [11C]57 (2.5±0.4%b) | Rodents | Rat: SUVmax = 2.26 (3.5 min) |
| [11C]58 (15.0±6.8%b) | Rodents | Rat: SUVmax >1.5 |
| [11C]59 (25%) | Rodents, NHP | Rat: high BBB permeability, specificity to MAGL Monkey: SUVmax ~3.5 (40 min) |
| [18F]60 (8%a) | Rodents | Mice: SUVmax = 1.75 (2.5 min) |
| [18F]63(13.0%) | Rodents, BAT | BAT: 21.4 %ID/g |
| [18F]64(21,7±3.0%b) | Rodents, NHP | Mice: 1.0±0.1% ID/g (biod,1 min) Monkey: SUVmax >1.5 |
non-decay corrected RCYs
decay corrected RCYs
3.2.1. Irreversible MAGL Radiotracers
The first reported MAGL PET probe was described by Hicks et al. in 2014219, where carbamate based inhibitors ([11C]JW642, [11C]KML29) and two urea based inhibitor ([11C]ML30, [11C]JJKK-0048) were synthesized. However, all these inhibitors showed poor brain penetration and were not suitable to advance further PET imaging studies.
SAR127303 is a highly potent and selective carbamate-based inhibitor of MAGL developed by Sanofi, and the values of IC50 in mouse and human MAGL are 3.8 and 29 nM, respectively. SAR127303 has shown therapeutic effect on relieving inflammatory pain and epilepsy symptoms in mice.220 In 2016, Wang et al.221 labeled [11C]SAR127303 ([11C]53) with [11C]CO2 as a novel MAGL imaging agent in rodents. PET imaging of [11C]53 in rats showed 48% reduction during blocking conditions. Independently, Wang et al. 222 screened MAGL inhibitors from a series of sulfonamido-based carbamates and urea-based compounds. Among them, the most potent SAR127303 (IC50 = 39.3 nM) and TZPU (IC50 = 35.9 nM) were labeled with [11C]COCl2 to afford [11C]53 and [11C]TZPU ([11C]54). [11C]53 showed a high level of brain permeability (1.5 SUV for rats and ca.1 SUV for NHP) and heterogeneous regional brain distribution which was consistent with the distribution of MAGL in the brain. The uptake of radioactivity was almost four times higher than [11C]54 in the rat brain.222 To further assess PET quantification of [11C]53 in the rat brain, Yamasaki et al.187 successfully used an irreversible two-tissue compartment model (2-TCMi, k4 = 0) to quantify the concentration distribution of MAGL in different brain regions. Among the kinetic parameters (k3, Ki, and λk3) they have obtained, the correlation between Ki value and in vitro binding of [11C]53 was the highest.187
In 2017, Ahamed et al.223 reported on a novel MAGL PET tracer, [11C]MA-PB-1 ([11C]55), which was modified from JW642 by replacing the phenoxy group with a methoxy group. The initial uptake of [11C]55 in mouse and rat brain was comparable with that of [11C]53. However, significant washout was observed after reaching peak uptake in the rat brain, and the radioactivity remained stable after 1h. One explanation could be species differences postulated by the authors. Self-blocking experiments and chasing studies with MJN110 (a structurally distinct MAGL inhibitor) confirmed [11C]55 as a specific irreversible MAGL tracer. In rhesus monkey PET imaging of [11C]55, initial high brain uptake reached (~4.2 SUV) at 2.5 min and a steady state (2.5 SUV) reached after 20 min. This result further underlined the irreversible binding characteristics of [11C]55.
In the same year, Butler et al. disclosed a series of MAGL inhibitors bearing an azetidine scaffold to reduce the lipophilicity and increase the binding specificity.224 Subsequently, Cheng et al. further developed an array of azetidinyl carbamate-based MAGL inhibitors. Among them, the most promising MAGL-0519 was labeled with [11C]COCl2 or [11C]CH3I at two different positions and evaluated in rodents and rhesus monkeys.225 In rodent PET imaging, [11C]MAGL-0519 ([11C]56) had a high binding specificity and selectivity as well as a heterogeneous distribution with high brain uptake (SUVmax is up to 1.5), which was consistent with the distribution of MAGL. An unexpected clearance of [11C]56 in rat brain may be caused by the metabolism of the linkage between carbonyl group of the tracer and the serine residue of MAGL in vivo. In PET imaging studies with rhesus monkey, [11C]56 also had a heterogeneous distribution with peak brain uptake greater than 1.5 SUV as well as an unexpected clearance.
In 2019, Chen et al226 developed a novel array of reversible and irreversible MAGL inhibitors with a “tail switching” strategy on a piperazinyl azetidine scaffold.227–229 Among them, three promising MAGL inhibitors were labeled and evaluated in rodents, including the irreversible inhibitor [11C]MAGL-2–11 ([11C]57, IC50 = 0.88 nM, LogD = 1.9) and two reversible inhibitors [18F]MAGL-4–11 ([18F]61, IC50 = 11.7 nM, LogD = 2.7) and [11C]PAD ([11C]62, IC50 = 2.7 nM, LogD = 3.4). [11C]57 showed good brain permeability with a maximum brain SUV of 2.26 at 3.5 min post injection and no significant washout was observed during PET scan. The distribution of [11C]57 in the rat brain was heterogeneous and consistent with MAGL distribution. However, BBB permeability of two reversible tracers ([11C]PAD ([11C]62) and [18F]MAGL-4–11 ([18F]61)) was poor (0.3–0.4 SUVmax), which may be related to Pgp and Bcrp efflux. This hypothesis was validated in an experiment with Pgp/Bcrp knockout mice. Further studied in higher species are underway to verify if species difference exists for brain penetration.
In the same year, Mori et al.230 reported a novel irreversible MAGL inhibitor [11C]58 based on azetidine scaffold with a hexafluoroisopropyl (HFIP) alcohol leaving group. [11C]58 has a high MAGL potency (IC50 = 0.4 nM) and a suitable lipophilicity (cLogD=3.29). [11C]58 was labeled with [11C]COCl2 and evaluated in rodents. [11C]58 exhibited a good brain permeability with a maximum brain uptake of 1.5 SUV and specific and irreversible binding to MAGL.
Recently, an azabicyclo[3.1.0]hexane scaffold-based irreversible MAGL inhibitor, PF-06809247 with a unique trifluoromethyl glycol leaving group was labeled with carbon-11 to assess its potential as a MAGL radioligand both in vitro and in vivo.231 [11C]PF-06809247 ([11C]59) exhibited good MAGL potency (IC50 = 13 nM, cLogD = 2.7), high permeability and high specific binding in the frontal cortex, hippocampus, and cerebellum in the brain. In NHP PET imaging, [11C]59 showed high brain uptake with SUVmax ca. 3.5. Although its target occupancy and pharmacokinetics in NHP have not yet been published, [11C]59 has entered clinical trials.232 This will provide the basis for the development of MAGL targeting therapeutics and further understanding of MAGL pathophysiological changes in vivo.
In parallel, Chen et al.233 reported another azabicyclo[3.1.0]hexane scaffold-based MAGL PET tracer [18F]PF06795071 ([18F]60). PF06795071 is a promising MAGL inhibitor developed by McAllister et al. with excellent MAGL potency (IC50 = 3 nM) and excellent serine hydrolase selectivity.234 [18F]60 was 18F-labeled via spirocyclic iodonium ylide (SCIDY) strategy235–238 and further evaluated in rodents. By in vitro autoradiography, [18F]60 exhibited excellent binding specificity and heterogeneous regional brain uptake in rodent brain, consistent with MAGL distribution. In addition, [18F]60 readily crossed the BBB (SUVmax = 1.8 at 2.5 min) and exhibited heterogeneous regional brain uptake in rat PET imaging. PET evaluation in NHP is underway to verify its translation potential for human use.
3.2.2. Reversible MAGL Radiotracers
A limited number of reversible MAGL radiotracers have been reported as shown in Figure 4. [18F]61 and [11C]62 with limited brain penetration have already been mentioned above, which could be attributed to species difference.226 Subsequently, Chen et al.239 developed a novel reversible MAGL PET probe, [18F]FEPAD ([18F]63), based on the structure of [11C]62. [18F]63 showed excellent MAGL potency (IC50 = 23.8 nM) and target selectivity (> 100 fold) among any other major serine hydrolases. As a proof-of-concept, [18F]63 was used to differentiate brown and white adipose tissue in the lipid network. Indeed, [18F]63 had much higher uptake in BAT (peak value 21.4 %ID/g) than white adipose tissue (WAT, 3.5 %ID/g). Recently Hattori et al.240 developed a novel MAGL PET tracer [18F]T-401 ([18F]64, IC50(human) = 4.0 nM, LogD = 1.86) based on the piperazinyl pyrrolidine-2-one scaffold reported by Takeda pharmaceuticals. [18F]64 exhibited the highest uptake in the frontal cortex, followed by the striatum, and these results were consistent with the distribution of MAGL. The rhesus monkey PET imaging with [18F]64 exhibited reversible binding characteristics, and brain uptake of [18F]64 peaked immediately (SUVmax > 1.5) post injection, and subsequently decreased to 30–40% of the maximum at the end of 120 min scan.
4. PERSPECTIVES AND CONCLUSIONS
4.1. Opportunities in developing ECS PET tracers based on allosteric modulation
CB1R ligand development has proven challenging due to severe psychoactive adverse events observed with the majority of orthosteric CB1R ligands. The latter has prompted the focus towards allosteric CB1R modulators, providing an enhanced subtype-selectivity and reducing receptor downregulation as well as inter-receptor promiscuity – two detrimental effects observed with classical orthosteric CB1R ligands.241 Both CB1R and CB2R contain several allosteric binding sites.242, 243 Over the past few years, a number of synthetic or natural compounds were characterized as CB1R allosteric modulators.244–247 A detailed review on the pharmacological properties and therapeutic potential of these allosteric modulators has recently been reported by Khurana et al.248 Similarly, by computational modeling and molecular dynamics simulation, Dainese et al.249demonstrated that FAAH function can be fine-tuned by allosteric modulation. This discovery provided new concepts for the regulation of FAAH activity and the synthesis of allosteric FAAH inhibitors. Notably, the contemporary focus on allosteric modulators of the ECS has implications not only for therapeutic strategies but also for the development of ECS-targeted PET radioligands. Given that PET probes are crucial to facilitate drug development via target validation experiments, receptor occupancy and proper dose-finding studies, it is not surprising that there is a growing interest in radioligands that bind to allosteric sites of CB1R and CB2R. Indeed, allosteric binding sites are yet to be exploited in future tracer development programs and harbor a huge potential to improve our understanding of the ECS and to facilitate drug discovery on allosteric modulation.
4.2. Opportunities in developing PET tracers for important yet understudied biological targets within ECS
Other serine hydrolases, in addition to FAAH and MAGL, are involved in the biosynthesis and metabolism of endocannabinoids and other lipids. For some of these serine hydrolases, covalent and/or reversible inhibitors have been reported, which could potentially provide a starting point for developing PET tracers. For example, ABHD12 and ABHD6 have been shown to contribute to total brain 2-AG hydrolysis activity and may make more specialized contributions to this process, especially in cell types with low MAGL expression. In this regard, ABHD6 and ABHD12 both show distinct cellular and subcellular expression patterns compared to MAGL and may regulate different 2-AG pools.7, 250 Development of PET tracers for ABHD6 and ABHD12 may facilitate an understanding of the potentially distinct roles played by these enzymes in 2-AG metabolism and signaling. There are several highly selective inhibitors available for ABHD6 and ABHD12 such as KT182, an orally active irreversible ABHD6 inhibitor251, and DO264 – an orally active reversible ABHD12 inhibitor.252
The integral membrane serine hydrolases diacylglycerol lipase α (DAGLα) and DAGLβ have been identified as DAG lipases involved in the biosynthesis of 2-AG.253 Different isoform-selective and non-selective DAGL inhibitors have been reported, including KT109 (DAGLβ isoform-selective, non-CNS active),254 and DO34 and DH376 (DAGLα/β dual, CNS active).255 Beyond serine hydrolases, other lipases of interest include NAPE-PLD, which converts NAPEs into NAEs and is involved in the biosynthesis of AEA. Recently, a CNS-active NAPE-PLD inhibitor, LEI-401, has been reported and characterized in mice.256
Development of PET-tracers based on inhibitors of key enzymes involved in eCB metabolism is an important step in the development and characterization of new drug candidates for ECS-related diseases.
5. Summary
During the past decades, a variety of CB1R radioligands have been synthesized and evaluated. Several tracers have been transitioned into clinical research studies, of which [11C]5 and [18F]10 have gained promising results in mental disorders, such as schizophrenia and addiction disorder. PET radioligands for CB2R have shown promise in neuroinflammatory animal models and postmortem human tissues of ALS patients. It should be noted that CB2R tracer development has been plagued by the lack of a suitable antibody/ligand, which would allow the validation of autoradiographic findings and determination of CB2R Bmax in the CNS. Now that the crystal structure of CB2R has been resolved, it is envisioned that computational modeling may aid the development of a selective antibody/ligand, thereby facilitating appropriate validation of PET probes in the pipeline.45 Since a major clinical trial tragedy in 2016, few attempts have been made to develop new FAAH radiotracers, which represents a current unmet clinical need. Conversely, the development of PET probes for MAGL has recently received growing attention with a plethora of structurally distinct radiolabeled MAGL inhibitors evaluated in vivo. Reversible inhibitor PET probes for MAGL, albeit in the nascent stage, will continue to be of interest in the future.
Overall, the development of PET radioligands for ECS targets is a very energetic research field, which will lay the foundation for the research of diseases and the drugs related to the ECS. Nonetheless, only a few PET tracers displayed suitable lipophilicity and specificity from in vivo studies. Apart from improving the physical properties of the target compound, there are two primary areas of improvement needed. First of all, 18F-labeled tracers should be more actively developed because of its longer half-life than 11C, which potentially allows for longer scan times, diverse radiolabeling procedures, and remote regional distribution. The second important improvement is to develop and apply radiotracers with diverse binding characters (reversible and irreversible binding) to obtain comprehensive biological knowledge for quantification of the receptor in human subjects.257 Moreover, tracer validation requires continuous improvement of animal models, particularly regarding neuroinflammatory models that would accurately reflect alterations in the human ECS. Finally, advances in novel radiochemical technology,238 including [11C]CO2 fixation202 and SCIDY235, 237, 258 for radiofluorination, will enable the discovery and access to novel, innovative PET tracers and may ultimately improve the current standard of diagnostic and therapeutic options for patients.
Acknowledgments
L.W. and H.X. thank the support of K. C. Wong Education Foundation, the National Natural Science Foundation of China (81701751, 81871383), the Guangdong Basic and Applied Basic Research Foundation (2020A1515011192), the Fundamental Research Funds for the Central Universities (21619104) and the Project of Innovative Team for the Guangdong University (2018KCXTD001). The following funding support from the NIH (DA038000 and DA043507 to S.H.L., AG054473 and AG052414 to N.V., and DA033760 to B.F.C.), the Azrieli Foundation, Canada Foundation for Innovation, Ontario Research Fund and the Canada Research Chairs Program (to N.V.), and Swiss National Science Foundation (P2EZP3_175137 to M.A.S) are also gratefully acknowledged.
ABBREVIATION USED
- AA
arachidonic acid
- AAI
aminoalkylindoles
- ABHD6
α/β hydrolase domain-6
- ABHD12
α/β hydrolase domain-12
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- AEA
N-arachidonoylethanolamine
- BAT
brown adipose tissue
- BBB
blood-brain barrier
- BP
binding potential
- CB1R
cannabinoid receptors 1
- CB2R
cannabinoid receptors 2
- CNS
central nervous system
- CVD
cardiovascular dysfunction
- CYP
cytochrome P450
- DS
Dravet Syndrome
- ECS
endocannabinoid system
- FAAH
fatty acid amide hydrolase
- GPCR
G-protein coupled receptor
- HD
Huntington’s disease
- LGS
Lennox-Gastaut Syndrome
- MAGL
monoacylglycerol lipase
- MRI
magnetic resonance imaging
- NHP
nonhuman primate
- PD
Parkinson’s syndrome
- PBA-HD
Problem Behavior Assessment for Huntington’s disease
- PET
positron emission tomography
- PgP/Bcrp
P-glycoprotein/breast cancer resistance protein
- PK-PD
pharmacokinetic-pharmacodynamic
- RCYs
radiochemical yields
- SCIDY
spirocyclic iodonium ylide
- SPECT
Single Photon Emission Computed Tomography
- SUV
standard uptake value
- TRT
Test–retest
- V T
volume of distribution
- 2-AG
2-arachidonoylglycerol
Biography
Lu Hou is a third-year graduate student in Jinan University (JNU), majoring in imaging medicine and nuclear medicine under the guidance of Dr. Hao Xu and Dr. Lu Wang. Her main research interests are the imaging of the central nervous system and the clinical evaluation and transformation of new PET probes.
Jian Rong obtained his BSc degrees in Chemistry and Biology with Professor Wen-Jing Xiao in Central China Normal University (CCNU) in 2012, followed by his Ph.D. in Organic Chemistry with Professor Jinbo Hu in Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS) in 2017. After that, he continued his postdoctoral research with Professor Steven H. Liang in Division of Nuclear Medicine and Molecular Imaging, Department of Radiology, Massachusetts General Hospital (MGH) and Harvard Medical School (HMS). His research interests include 18F-labeling methodologies and development of PET tracers for the central and peripheral nervous system.
Achi Haider obtained his M.S. in Pharmaceutical Sciences at ETH Zurich, Switzerland, where he subsequently earned his Ph.D. in radiochemistry and molecular imaging with Professor Simon M. Ametamey in 2018. In the same year, he moved to the Department of Nuclear Medicine at the University Hospital Zurich, working with Prof. Catherine Gebhard to advance cardiovascular gender medicine with particular focus on preclinical and clinical cardiovascular PET imaging. Recently Dr. Haider joined Professor Steven H. Liang’s research group in the Division of Nuclear Medicine and Molecular Imaging at Harvard Medical School and Massachusetts General Hospital in Boston, USA.
Daisuke Ogasawara received his B.S. in 2011 and M.S. in 2013 in Pharmaceutical Sciences from Nagoya City University. He then joined the research group of Prof. Benjamin F. Cravatt at The Scripps Research Institute, where he conducted his doctoral study focusing on development of chemical tools for serine hydrolases involved in metabolism of signaling lipids. After receiving his Ph.D. in Chemistry in 2019, he joined the research group of Prof. Stuart L. Schreiber as a JSPS Overseas Research Fellow.
Cassis Varlow received her BSc in Chemical Biology from McMaster University in 2019. She continued her education at the University of Toronto in Dr. Neil Vasdev’s lab at the Centre for Addiction and Mental Health (CAMH) Brain Health Imaging Centre as an MSc candidate, and a prospective PhD student. Her research interests include in vitro radioligand characterization to de-risk the development of successful PET radiotracers for pre-clinical and human translation, for the study of neurodegenerative diseases.
Michael A. Schafroth received his B.Sc. in Chemistry in 2010 and M.Sc. in Interdisciplinary Sciences in 2012 from ETH Zürich. He then joined the research group of Prof. Erick M. Carreira, where he worked on the design and development of asymmetric iridium-catalyzed allylic substitution reactions, applications of stereodivergent methods to the synthesis of cannabinoids, and development of novel tools to study interactions of small molecules with the cell surface proteome. After receiving his Ph.D. in Chemistry in 2017, he joined the research group of Prof. Ben Cravatt at Scripps Research as a SNSF sponsored postdoctoral research associate. He currently works on the synthesis of small molecule inhibitors for serine hydrolases and is developing novel chemoprotoemic probes to modulate protein function in different biological arenas.
Lingjing Mu received her PhD in Chemistry from Nankai University, China in 1996. She worked as a postdoctoral fellow at the University of Basel and Center for Radiopharmaceutical Sciences (CRS) in Switzerland. In 2004, she was appointed as the team leader for PET chemistry at CRS. She is the Research Head of Radiopharmacy since 2010 and has working places at both University Hospital and ETH Zurich. Her research interests focus on PET tracer development and the development of new 18F-radiolabelling methods as well as the translation of new PET tracers into the clinic.
Jiefeng Gan is a third-year graduate student in JNU with the major of nuclear medicine under the supervisor of Dr. Lu Wang. Her research focus on preliminary evaluation of novel PET tracers in rodents and non-human primates.
Hao Xu received his PhD in nuclear medicine from Humboldt University of Berlin in 1996. Dr. Xu is currently the director of the Nuclear Medicine Department and PET/CT-MRI Center in the First Affiliated Hospital of Jinan University (JNUH), and the executive vice director of the Institute of Molecular and Functional Imaging in JNU. Dr. Xu’s research interests are clinical nuclear medicine and molecular imaging of cancer and neurological diseases.
Christopher J. Fowler obtained his B.A. at Cambridge University in 1975, followed by his Ph.D. in Pharmacology at the same University in 1978. Following post-doctoral posts in Sweden, Ireland and France, he was a researcher at Astra Läkemedel (now part of AstraZeneca) from 1982 to 1994. He was thereafter appointed Professor of Pharmaology at Umeå University in Sweden and took up his appointment there in 1995. Dr. Fowler’s current research interests concern the pharmacology of the endocannabinoid system. Dr. Fowler was recognized as a highly-cited researcher 2019 by the Web of Science group.
Ming-Rong Zhang received his B.S. at China Pharmaceutical University in 1985, followed by his Ph.D. in Medicinal Chemistry with Professor Masatoshi Yamato in Okayama University in 1993. Then he joined the Molecular Probe Program in National Institute of Radiological Sciences (NIRS) of Japan in 1998 and was promoted to the leader of Radiochemistry Team in 2006 and the director of Molecular Probe Program of NIRS in 2012. Dr. Zhang is currently the director of Department of Advanced Nuclear Medicine Sciences, NIRS, QST of Japan. Dr. Zhang’s research interests include radiochemistry, radiotracer and radiotherapy development, as well as clinical translation.
Neil Vasdev received his PhD in Chemistry from McMaster University, followed by a NSERC postdoc at Berkeley National Labs in 2003. He is the Director of the CAMH Brain Health Imaging Centre and the Azrieli Centre for Neuro-Radiochemistry. He is an endowed Chaired Professor at the University of Toronto & Canada Research Chair in Radiochemistry and Nuclear Medicine. He began his faculty career in Toronto in 2004 and was the Director of Radiochemistry at MGH and an Associate Professor at HMS (2011–2017). His research interests include applying novel radiochemistry methods with 11C and 18F to human PET imaging.
Simon M. Ametamey obtained his M.S. in Chemistry from the Technical University of Merseburg, Germany, in 1985. He earned doctorate degree in Organic Chemistry from the University of Zurich, Switzerland, under the supervision of with Prof. H.H. Heimgartner. Following, a postdoctoral fellowship with Prof. K. Bernhard at Hoffmann La Roche (Basel, Switzerland), he joined the Center for Radiopharmaceutical Sciences of ETH, PSI and USZ in Villigen, Switzerland, in 1991. In 1995, he became the group leader and was appointed Titularprofessor in 2006. Since then, he has co-headed the Animal Imaging Center-PET at the Institute of Pharmaceutical Sciences at ETH Zurich.
Benjamin F. Cravatt obtained his undergraduate education at Stanford University, receiving a B.S. in the Biological Sciences and a B.A. in History. He then received a Ph.D. from The Scripps Research Institute (TSRI) in 1996. Dr. Cravatt is a Professor and the Norton B. Gilula Chair of Chemical Biology in the Department of Chemistry at The Scripps Research Institute. His research group is interested in developing chemical proteomic technologies that enable protein and drug discovery on a global scale and applying these methods to characterize biochemical pathways that play important roles in human physiology and disease, especially as pertains to the nervous system and cancer.
Lu Wang received his B.S. in pharmacy from the Ocean University of China in 2010, followed by his Ph.D. in Chemical Genomics from Peking University in 2016. Dr. Wang was a senior visiting scholar at HMS/MGH (2014–2016) and has been a visiting research fellow at NIRS (2017-now). Dr. Wang is currently the Vice Director of the Center of Cyclotron and PET Radiopharmaceuticals (CCPR) in JNUH. Dr. Wang’s research interests include radiochemistry, PET tracer development and clinical translational studies.
Steven H. Liang obtained his B.S. at Tianjin University in 2003, followed by his Ph.D. in Chemistry with Professor Marco Ciufolini in the University of British Columbia in 2010. Then he started as a NSERC fellow with Professor E. J. Corey at Harvard University. In 2012, Dr. Liang accepted a junior faculty position at HMS and MGH. Dr. Liang is currently the Director of Radiochemistry and Biomarker Development, Nuclear Medicine and Molecular Imaging at MGH, and Associate Professor of Radiology at HMS. Dr. Liang’s research interests include radiochemistry, imaging biomarker and radiotherapy development, and clinical translation.
Footnotes
The authors declare no competing financial interest.
REFERENCE
- 1.Katona I; Freund TF Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med 2008, 14, 923–930. [DOI] [PubMed] [Google Scholar]
- 2.Ligresti A; De Petrocellis L; Di Marzo V From phytocannabinoids to cannabinoid receptors and endocannabinoids: pleiotropic physiological and pathological roles through complex pharmacology. Physiol. Rev 2016, 96, 1593–1659. [DOI] [PubMed] [Google Scholar]
- 3.Ohno-Shosaku T; Tanimura A; Hashimotodani Y; Kano M Endocannabinoids and retrograde modulation of synaptic transmission. Neuroscientist 2012, 18, 119–132. [DOI] [PubMed] [Google Scholar]
- 4.Ueda N; Tsuboi K; Uyama T Metabolism of endocannabinoids and related N-acylethanolamines: canonical and alternative pathways. FEBS J 2013, 280, 1874–1894. [DOI] [PubMed] [Google Scholar]
- 5.Dinh TP; Carpenter D; Leslie FM; Freund TF; Katona I; Sensi SL; Kathuria S; Piomelli D Brain monoglyceride lipase participating in endocannabinoid enactivation. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Marzo V New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov 2018, 17, 623–639. [DOI] [PubMed] [Google Scholar]
- 7.Savinainen J; Saario S; Laitinen J The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2‐arachidonoylglycerol signalling through cannabinoid receptors. Acta. physiologica 2012, 204, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martínez-Torres S; Cutando L; Pastor A; Kato A; Sakimura K; de la Torre R; Valjent E; Maldonado R; Kano M; Ozaita A Monoacylglycerol lipase blockade impairs fine motor coordination and triggers cerebellar neuroinflammation through cyclooxygenase-2. Brain behav. Immun 2019, 81, 399–409. [DOI] [PubMed] [Google Scholar]
- 9.Berardi A; Schelling G; Campolongo P The endocannabinoid system and post traumatic stress disorder (PTSD): from preclinical findings to innovative therapeutic approaches in clinical settings. Pharmacol. Res 2016, 111, 668–678. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y; Anderson HD Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol 2017, 95, 311–327. [DOI] [PubMed] [Google Scholar]
- 11.Ulugol A The endocannabinoid system as a potential therapeutic target for pain modulation. Balkan. Med. J 2014, 31, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bedse G; Romano A; Lavecchia AM; Cassano T; Gaetani S The role of endocannabinoid signaling in the molecular mechanisms of neurodegeneration in alzheimer’s disease. J. Alzheimers Dis 2015, 43, 1115–1136. [DOI] [PubMed] [Google Scholar]
- 13.Woodhams SG; Chapman V; Finn DP; Hohmann AG; Neugebauer V The cannabinoid system and pain. Neuropharmacology 2017, 124, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toczek M; Malinowska B Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci 2018, 204, 20–45. [DOI] [PubMed] [Google Scholar]
- 15.Tambaro S; Bortolato M Cannabinoid-related agents in the treatment of anxiety disorders: current knowledge and future perspectives. Recent Pat. CNS Drug Discov 2012, 7, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pertwee RG Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc. Lond. B Biol. Sci 2012, 367, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stockings E; Campbell G; Hall WD; Nielsen S; Zagic D; Rahman R; Murnion B; Farrell M; Weier M; Degenhardt L Cannabis and cannabinoids for the treatment of people with chronic noncancer pain conditions: a systematic review and meta-analysis of controlled and observational studies. Pain 2018, 159, 1932–1954. [DOI] [PubMed] [Google Scholar]
- 18.Hargreaves RJ; Rabiner EA Translational PET imaging research. Neurobiol. Dis 2014, 61, 32–38. [DOI] [PubMed] [Google Scholar]
- 19.Suridjan I; Comley RA; Rabiner EA The application of positron emission tomography (PET) imaging in CNS drug development. Brain Imaging Behav 2019, 13, 354–365. [DOI] [PubMed] [Google Scholar]
- 20.Waaijer SJH; Kok IC; Eisses B; Schröder CP; Jalving M; Brouwers AH; Lub-de Hooge MN; de Vries EGE Molecular Imaging in cancer drug development. J. Nucl. Med 2018, 59, 726–732. [DOI] [PubMed] [Google Scholar]
- 21.Bernard-Gauthier V; Collier TL; Liang SH; Vasdev N Discovery of PET radiopharmaceuticals at the academia-industry interface. Drug Discov. Today Technol 2017, 25, 19–26. [DOI] [PubMed] [Google Scholar]
- 22.Hargreaves RJ; Hoppin J; Sevigny J; Patel S; Chiao P; Klimas M; Verma A Optimizing central nervous system drug development using molecular imaging. Clin. Pharmacol. Ther 2015, 98, 47–60. [DOI] [PubMed] [Google Scholar]
- 23.Lu H; Qijun C; Lu W; Jinghao W; Weijian Y; Xiaosong W; Zhihua Z; Zhidong Z; Hao X Positron emission tomography (PET) application in the phase 0 clinical trials in drug development. Pharmacy Today (Chinese) 2020, 30, 99–105. [Google Scholar]
- 24.Guangdong Province Pharmaceutical Association. Consensus on phase 0 clinical trial application for positron radiopharmaceuticals. Pharmacy Today (Chinese). 2020, 30, 793–798. [Google Scholar]
- 25.Burt T; Yoshida K; Lappin G; Vuong L; John C; de Wildt SN; Sugiyama Y; Rowland M Microdosing and other phase 0 clinical trials: facilitating translation in drug development. Clin. Transl. Sci 2016, 9, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burt T; Vuong LT; Baker E; Young GC; McCartt AD; Bergstrom M; Sugiyama Y; Combes R Phase 0, including microdosing approaches: applying the three rs and increasing the efficiency of human drug development. Altern. Lab. Anim 2018, 46, 335–346. [DOI] [PubMed] [Google Scholar]
- 27.Varlow C; Boileau I; Wey HY; Liang SH; Vasdev N Classics in neuroimaging: imaging the endocannabinoid pathway with PET. ACS Chem. Neurosci 2020, 11, 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirvonen J In vivo imaging of the cannabinoid CB1 receptor with positron emission tomography. Clin. Pharmacol. Ther 2015, 97, 565–567. [DOI] [PubMed] [Google Scholar]
- 29.Ni R; Mu L; Ametamey S Positron emission tomography of type 2 cannabinoid receptors for detecting inflammation in the central nervous system. Acta pharmacologica Sinica 2019, 40, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spinelli F; Mu L; Ametamey SM Radioligands for positron emission tomography imaging of cannabinoid type 2 receptor. J. Labelled Comp. Radiopharm 2018, 61, 299–308. [DOI] [PubMed] [Google Scholar]
- 31.McCluskey SP; Plisson C; Rabiner EA; Howes O Advances in CNS PET: the state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 451–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shahbazi F; Grandi V; Banerjee A; Trant JF Cannabinoids and cannabinoid receptors: the story so far. iScience 2020, 23, 101301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glass M; Faull R; Dragunow M Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [DOI] [PubMed] [Google Scholar]
- 34.Herkenham M; Lynn AB; Little MD; Johnson MR; Melvin LS; De Costa BR; Rice KC Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U. S. A 1990, 87, 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackie K Cannabinoid receptors: where they are and what they do. J. Neuroendocrinol 2008, 20, 10–14. [DOI] [PubMed] [Google Scholar]
- 36.Maccarrone M; Bab I; Bíró T; Cabral GA; Dey SK; Di Marzo V; Konje JC; Kunos G; Mechoulam R; Pacher P; Sharkey KA; Zimmer A Endocannabinoid signaling at the periphery: 50 years after THC. Trends. Pharmacol. Sci 2015, 36, 277–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izzo AA; Sharkey KA Cannabinoids and the gut: new developments and emerging concepts. Pharmacol. Ther 2010, 126, 21–38. [DOI] [PubMed] [Google Scholar]
- 38.Montecucco F; Di Marzo V At the heart of the matter: the endocannabinoid system in cardiovascular function and dysfunction. Trends. Pharmacol. Sci 2012, 33, 331–340. [DOI] [PubMed] [Google Scholar]
- 39.Pacher P; Steffens S; Haskó G; Schindler TH; Kunos G Cardiovascular effects of marijuana and synthetic cannabinoids: the good, the bad, and the ugly. Nat. Rev. Cardiol 2018, 15, 151–166. [DOI] [PubMed] [Google Scholar]
- 40.Tam J; Trembovler V; Di Marzo V; Petrosino S; Leo G; Alexandrovich A; Regev E; Casap N; Shteyer A; Ledent C; Karsak M; Zimmer A; Mechoulam R; Yirmiya R; Shohami E; Bab I The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J 2008, 22, 285–294. [DOI] [PubMed] [Google Scholar]
- 41.Munro S; Thomas KL; Abu-Shaar M Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- 42.Howlett AC; Barth F; Bonner TI; Cabral G; Casellas P; Devane WA; Felder CC; Herkenham M; Mackie K; Martin BR; Mechoulam R; Pertwee RG International union of pharmacology. XXVII. classification of cannabinoid receptors. Pharmacol. Rev 2002, 54, 161–202. [DOI] [PubMed] [Google Scholar]
- 43.Van Sickle MD; Duncan M; Kingsley PJ; Mouihate A; Urbani P; Mackie K; Stella N; Makriyannis A; Piomelli D; Davison JS Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [DOI] [PubMed] [Google Scholar]
- 44.Hua T; Vemuri K; Pu M; Qu L; Han GW; Wu Y; Zhao S; Shui W; Li S; Korde A; Laprairie RB; Stahl EL; Ho JH; Zvonok N; Zhou H; Kufareva I; Wu B; Zhao Q; Hanson MA; Bohn LM; Makriyannis A; Stevens RC; Liu ZJ Crystal structure of the human cannabinoid receptor CB1. Cell 2016, 167, 750–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X; Hua T; Vemuri K; Ho JH; Wu Y; Wu L; Popov P; Benchama O; Zvonok N; Locke K; Qu L; Han GW; Iyer MR; Cinar R; Coffey NJ; Wang J; Wu M; Katritch V; Zhao S; Kunos G; Bohn LM; Makriyannis A; Stevens RC; Liu ZJ Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hua T; Li X; Wu L; Iliopoulos-Tsoutsouvas C; Wang Y; Wu M; Shen L; Johnston CA; Nikas SP; Song F; Song X; Yuan S; Sun Q; Wu Y; Jiang S; Grim TW; Benchama O; Stahl EL; Zvonok N; Zhao S; Bohn LM; Makriyannis A; Liu ZJ Activation and signaling mechanism revealed by cannabinoid receptor-G(i) complex structures. Cell 2020, 180, 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piomelli D The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci 2003, 4, 873–884. [DOI] [PubMed] [Google Scholar]
- 48.Eggan SM; Hashimoto T; Lewis DA Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch. Gen. Psychiatry 2008, 65, 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gazzerro P; Caruso M; Notarnicola M; Misciagna G; Guerra V; Laezza C; Bifulco M Association between cannabinoid type-1 receptor polymorphism and body mass index in a southern Italian population. In. J. Obesity 2007, 31, 908–912. [DOI] [PubMed] [Google Scholar]
- 50.Cota D; Steiner M-A; Marsicano G; Cervino C; Herman JP; Grubler Y; Stalla J; Pasquali R; Lutz B; Stalla GK Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 2007, 148, 1574–1581. [DOI] [PubMed] [Google Scholar]
- 51.Puighermanal E; Busquets-Garcia A; Maldonado R; Ozaita A Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Philos. Trans. R. Soc. Lond. B. Biol. Sci 2012, 367, 3254–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parsons LH; Hurd YL Endocannabinoid signalling in reward and addiction. Nat. Rev. Neurosci 2015, 16, 579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rinaldi-Carmona M; Barth F; Héaulme M; Shire D; Calandra B; Congy C; Martinez S; Maruani J; Néliat G; Caput D SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS lett 1994, 350, 240–244. [DOI] [PubMed] [Google Scholar]
- 54.Sam AH; Salem V; Ghatei MA Rimonabant: from RIO to ban. J. Obes 2011, 2011, 432607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beal JE; Olson R; Laubenstein L; Morales JO; Bellman P; Yangco B; Lefkowitz L; Plasse TF; Shepard KV Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS. J. Pain Symptom Manage. 1995, 10, 89–97. [DOI] [PubMed] [Google Scholar]
- 56.Badowski ME; Yanful PK Dronabinol oral solution in the management of anorexia and weight loss in AIDS and cancer. Ther. Clin. Risk. Manag 2018, 14, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pergolizzi JV Jr.; Taylor R; LeQuang JA; Zampogna G; Raffa RB Concise review of the management of iatrogenic emesis using cannabinoids: emphasis on nabilone for chemotherapy-induced nausea and vomiting. Cancer Chemother. Pharmacol 2017, 79, 467–477. [DOI] [PubMed] [Google Scholar]
- 58.Novotna A; Mares J; Ratcliffe S; Novakova I; Vachova M; Zapletalova O; Gasperini C; Pozzilli C; Cefaro L; Comi G; Rossi P; Ambler Z; Stelmasiak Z; Erdmann A; Montalban X; Klimek A; Davies P A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®) ), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur. J. Neurol 2011, 18, 1122–1131. [DOI] [PubMed] [Google Scholar]
- 59.Devinsky O; Verducci C; Thiele EA; Laux LC; Patel AD; Filloux F; Szaflarski JP; Wilfong A; Clark GD; Park YD Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav 2018, 86, 131–137. [DOI] [PubMed] [Google Scholar]
- 60.Artelo BIOSCIENCES. http://artelobio.com/pipeline (accessed Oct 8, 2020).
- 61.CORBUS Pharmaceuticals. https://www.corbuspharma.com/our-science/crb-4001 (accessed Oct 8, 2020).
- 62.Kale VP; Gibbs S; Taylor JA; Zmarowski A; Novak J; Patton K; Sparrow B; Gorospe J; Anand S; Cinar R; Kunos G; Chorvat RJ; Terse PS Preclinical toxicity evaluation of JD5037, a peripherally restricted CB1 receptor inverse agonist, in rats and dogs for treatment of nonalcoholic steatohepatitis. Regul. Toxicol. Pharmacol 2019, 109, 104483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maresz K; Carrier EJ; Ponomarev ED; Hillard CJ; Dittel BN Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem 2005, 95, 437–445. [DOI] [PubMed] [Google Scholar]
- 64.Cabral G; Marciano‐Cabral F Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J. Leukoc. Biol 2005, 78, 1192–1197. [DOI] [PubMed] [Google Scholar]
- 65.Benito C; Tolon RM; Pazos MR; Nunez E; Castillo AI; Romero J Cannabinoid CB2 receptors in human brain inflammation. Br. J. Pharmacol 2008, 153, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turcotte C; Blanchet MR; Laviolette M; Flamand N The CB2 receptor and its role as a regulator of inflammation. Cell Mol. Life. Sci 2016, 73, 4449–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spinelli F; Capparelli E; Abate C; Colabufo NA; Contino M Perspectives of cannabinoid type 2 receptor (CB2R) ligands in neurodegenerative disorders: structure-affinity relationship (SAfiR) and structure-activity relationship (SAR) studies. J. Med. Chem 2017, 60, 9913–9931. [DOI] [PubMed] [Google Scholar]
- 68.Nevalainen T Recent development of CB2 selective and peripheral CB1/CB2 cannabinoid receptor ligands. Curr. Med. Chem 2014, 21, 187–203. [DOI] [PubMed] [Google Scholar]
- 69.Xing C; Zhuang Y; Xu TH; Feng Z; Zhou XE; Chen M; Wang L; Meng X; Xue Y; Wang J; Liu H; McGuire TF; Zhao G; Melcher K; Zhang C; Xu HE; Xie XQ Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 2020, 180, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.ARENA Pharmaceuticals. https://www.arenapharm.com/pipeline/apd371/# (accessed Oct 8, 2010).
- 71.Mathews WB; Ravert HT; Musachio JL; Frank RA; Rinaldi-Carmona M; Barth F; Dannals RF Synthesis of [18F]SR144385: A selective radioligand for positron emission tomographic studies of brain cannabinoid receptors. J. Label. Compd. Radiopharm 1999, 42, 589–596. [Google Scholar]
- 72.Mathews WB; Scheffel U; Finley P; Ravert HT; Frank RA; Rinaldi-Carmona M; Barth F; Dannals RF Biodistribution of [18F] SR144385 and [18F] SR147963: selective radioligands for positron emission tomographic studies of brain cannabinoid receptors. Nucl. Med. Biol 2000, 27, 757–762. [DOI] [PubMed] [Google Scholar]
- 73.Mathews WB; Scheffel U; Rauseo PA; Ravert HT; Frank RA; Ellames GJ; Herbert JM; Barth F; Rinaldi-Carmona M; Dannals RF Carbon-11 labeled radioligands for imaging brain cannabinoid receptors. Nucl. Med. Biol 2002, 29, 671–677. [DOI] [PubMed] [Google Scholar]
- 74.Fan H; Ravert HT; Holt DP; Dannals RF; Horti AG Synthesis of 1-(2,4-dichlorophenyl)-4-cyano-5-(4-11C-methoxyphenyl)-N-(piperidin-1-y l)-1H-pyrazole-3-carboxamide ( 11C-JHU75528) and 1-(2bromophenyl)-4-cyano-5-(4- 11C methoxyphenyl)-N-(piperidin-1-yl)-1H -pyrazole-3-carboxamide ( 11C JHU75575) as potential radioligands for PET imaging of cerebral cannabinoid receptor. J. Label. Compd. Radiopharm 2006, 49, 1021–1036. [Google Scholar]
- 75.Fan H; Kotsikorou E; Hoffman AF; Ravert HT; Holt D; Hurst DP; Lupica CR; Reggio PH; Dannals RF; Horti AG Analogs of JHU75528, a PET ligand for imaging of cerebral cannabinoid receptors (CBI): Development of ligands with optimized lipophilicity and binding affinity. Eur. J. Med. Chem 2009, 44, 593–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horti AG; Fan H; Kuwabara H; Hilton J; Ravert HT; Holt DP; Alexander M; Kumar A; Rahmim A; Scheffel U; Wong DF; Dannals RF 11C-JHU75528: a radiotracer for PET imaging of CB1 cannabinoid receptors. J. Nucl. Med 2006, 47, 1689–1696. [PubMed] [Google Scholar]
- 77.Wong DF; Kuwabara H; Horti A; Kumar A; Brasic J; Ye W; Alexander M; Raymont V; Galecki J; Charlotte M; Cascella N PET Imaging of cannabinoid CB1 type receptors in healthy humans and patients with schizophrenia using [11C]OMAR. NeuroImage 2008, 41, T51. [Google Scholar]
- 78.Wong DF; Kuwabara H; Horti AG; Raymont V; Brasic J; Guevara M; Ye W; Dannals RF; Ravert HT; Nandi A; Rahmim A; Ming JE; Grachev I; Roy C; Cascella N Quantification of cerebral cannabinoid receptors subtype 1 (CB1) in healthy subjects and schizophrenia by the novel PET radioligand [11C]OMAR. Neuroimage 2010, 52, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ranganathan M; Cortes-Briones J; Radhakrishnan R; Thurnauer H; Planeta B; Skosnik P; Gao H; Labaree D; Neumeister A; Pittman B; Surti T; Huang Y; Carson RE; D’Souza DC Reduced brain cannabinoid receptor availability in schizophrenia. Biol. Psychiatry. 2016, 79, 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hungund BL; Szakall I; Adam A; Basavarajappa BS; Vadasz C Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J. Neurochem 2003, 84, 698–704. [DOI] [PubMed] [Google Scholar]
- 81.Basavarajappa BS Endocannabinoid system and alcohol abuse disorders. Adv. Exp. Med. Biol 2019, 1162, 89–127. [DOI] [PubMed] [Google Scholar]
- 82.Femenia T; Garcia-Gutierrez MS; Manzanares J CB1 receptor blockade decreases ethanol intake and associated neurochemical changes in fawn-hooded rats. Alcohol. Clin. Exp. Res 2010, 34, 131–141. [DOI] [PubMed] [Google Scholar]
- 83.Neumeister A; Normandin MD; Murrough JW; Henry S; Bailey CR; Luckenbaugh DA; Tuit K; Zheng MQ; Galatzer‐Levy IR; Sinha R Positron emission tomography shows elevated cannabinoid CB 1 receptor binding in men with alcohol dependence. Alcohol. Clin. Exp. Res 2012, 36, 2104–2109. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 84.Valenta I; Varga ZV; Valentine H; Cinar R; Horti A; Mathews WB; Dannals RF; Steele K; Kunos G; Wahl RL; Pomper MG; Wong DF; Pacher P; Schindler TH Feasibility evaluation of myocardial cannabinoid type 1 receptor imaging in obesity: a translational approach. JACC Cardiovasc. Imaging 2018, 11, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Valenta I; Pacher P; Dilsizian V; Schindler TH Novel myocardial PET/CT receptor imaging and potential therapeutic targets. Curr. Cardiol. Rep 2019, 21, 55. [DOI] [PubMed] [Google Scholar]
- 86.Fish KM; Hajjar RJ Myocardial cannabinoid receptor imaging in obesity. JACC Cardiovasc. imaging 2018, 11, 333–335. [DOI] [PubMed] [Google Scholar]
- 87.Cheng HC The power issue: determination of KB or Ki from IC50. A closer look at the Cheng-Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods 2001, 46, 61–71. [DOI] [PubMed] [Google Scholar]
- 88.Yasuno F; Brown AK; Zoghbi SS; Krushinski JH; Chernet E; Tauscher J; Schaus JM; Phebus LA; Chesterfield AK; Felder CC; Gladding RL; Hong J; Halldin C; Pike VW; Innis RB The PET radioligand [11C]MePPEP binds reversibly and with high specific signal to cannabinoid CB1 receptors in nonhuman primate brain. Neuropsychopharmacology 2008, 33, 259–269. [DOI] [PubMed] [Google Scholar]
- 89.Donohue SR; Krushinski JH; Pike VW; Chernet E; Phebus L; Chesterfield AK; Felder CC; Halldin C; Schaus JM Synthesis, ex vivo evaluation, and radiolabeling of potent 1,5-diphenylpyrrolidin-2-one cannabinoid subtype-1 receptor ligands as candidates for in vivo imaging. J. Med. Chem. 2008, 51, 5833–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Terry GE; Hirvonen J; Liow JS; Zoghbi SS; Gladding R; Tauscher JT; Schaus JM; Phebus L; Felder CC; Morse CL; Donohue SR; Pike VW; Halldin C; Innis RB Imaging and quantitation of cannabinoid CB1 receptors in human and monkey brains using 18F-labeled inverse agonist radioligands. J. Nucl. Med 2010, 51, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Terry GE; Liow JS; Zoghbi SS; Hirvonen J; Farris AG; Lerner A; Tauscher JT; Schaus JM; Phebus L; Felder CC; Morse CL; Hong JS; Pike VW; Halldin C; Innis RB Quantitation of cannabinoid CB1 receptors in healthy human brain using positron emission tomography and an inverse agonist radioligand. NeuroImage 2009, 48, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhattacharyya S; Egerton A; Kim E; Rosso L; Riano Barros D; Hammers A; Brammer M; Turkheimer FE; Howes OD; McGuire P Acute induction of anxiety in humans by delta-9-tetrahydrocannabinol related to amygdalar cannabinoid-1 (CB1) receptors. Sci. Rep 2017, 7, 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirvonen J; Terry GE; Halldin C; Pike VW; Innis RB Approaches to quantify radioligands that wash out slowly from target organs. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 917–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hirvonen J; Goodwin R; Li C-T; Terry G; Zoghbi S; Morse C; Pike V; Volkow N; Huestis M; Innis R Reversible and regionally selective downregulation of brain cannabinoid CB 1 receptors in chronic daily cannabis smokers. Mol. Psychiatry 2012, 17, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hirvonen J; Zanotti-Fregonara P; Umhau JC; George DT; Rallis-Frutos D; Lyoo CH; Li C-T; Hines CS; Sun H; Terry GE Reduced cannabinoid CB 1 receptor binding in alcohol dependence measured with positron emission tomography. Mol. Psychiatry 2013, 18, 916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Borgan F; Laurikainen H; Veronese M; Marques TR; Haaparanta-Solin M; Solin O; Dahoun T; Rogdaki M; Salokangas RK; Karukivi M; Di Forti M; Turkheimer F; Hietala J; Howes O; Group M In vivo availability of cannabinoid 1 receptor levels in patients with first-episode psychosis. JAMA Psychiatry 2019, 76, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eriksson O; Mikkola K; Espes D; Tuominen L; Virtanen K; Forsback S; Haaparanta-Solin M; Hietala J; Solin O; Nuutila P The cannabinoid receptor-1 is an imaging biomarker of brown adipose tissue. J. Nucl. Med 2015, 56, 1937–1941. [DOI] [PubMed] [Google Scholar]
- 98.Lahesmaa M; Eriksson O; Gnad T; Oikonen V; Bucci M; Hirvonen J; Koskensalo K; Teuho J; Niemi T; Taittonen M; Lahdenpohja S; M UD; Haaparanta-Solin M; Pfeifer A; Virtanen KA; Nuutila P Cannabinoid type 1 receptors are upregulated during acute activation of brown adipose tissue. Diabetes 2018, 67, 1226–1236. [DOI] [PubMed] [Google Scholar]
- 99.Burns HD; Van Laere K; Sanabria-Bohorquez S; Hamill TG; Bormans G; Eng WS; Gibson R; Ryan C; Connolly B; Patel S; Krause S; Vanko A; Van Hecken A; Dupont P; De Lepeleire I; Rothenberg P; Stoch SA; Cote J; Hagmann WK; Jewell JP; Lin LS; Liu P; Goulet MT; Gottesdiener K; Wagner JA; de Hoon J; Mortelmans L; Fong TM; Hargreaves RJ [18F]MK-9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid-1 receptor. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 9800–9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kent RS; De Lean A; Lefkowitz RJ A quantitative analysis of beta-adrenergic receptor interactions: resolution of high and low affinity states of the receptor by computer modeling of ligand binding data. Mol. Pharmacol 1980, 17, 14–23. [PubMed] [Google Scholar]
- 101.Van Laere K; Goffin K; Casteels C; Dupont P; Mortelmans L; de Hoon J; Bormans G Gender-dependent increases with healthy aging of the human cerebral cannabinoid-type 1 receptor binding using [18F]MK-9470 PET. NeuroImage 2008, 39, 1533–1541. [DOI] [PubMed] [Google Scholar]
- 102.Gérard N; Pieters G; Goffin K; Bormans G; Van Laere K Brain type 1 cannabinoid receptor availability in patients with anorexia and bulimia nervosa. Biol. Psychiatry. 2011, 70, 777–784. [DOI] [PubMed] [Google Scholar]
- 103.Van der Schueren BJ; Van Laere K; Gérard N; Bormans G; De Hoon JN Interictal type 1 cannabinoid receptor binding is increased in female migraine patients. Headache 2012, 52, 433–440. [DOI] [PubMed] [Google Scholar]
- 104.Goffin K; Van Paesschen W; Van Laere K In vivo activation of endocannabinoid system in temporal lobe epilepsy with hippocampal sclerosis. Brain 2011, 134, 1033–1040. [DOI] [PubMed] [Google Scholar]
- 105.Ceccarini J; Ahmad R; Van de Vliet L; Casteels C; Vandenbulcke M; Vandenberghe W; Van Laere K Behavioral symptoms in premanifest huntington disease correlate with reduced frontal CB1R levels. J. Nucl. Med 2019, 60, 115–121. [DOI] [PubMed] [Google Scholar]
- 106.Ceccarini J; Casteels C; Ahmad R; Crabbé M; Van de Vliet L; Vanhaute H; Vandenbulcke M; Vandenberghe W; Van Laere K Regional changes in the type 1 cannabinoid receptor are associated with cognitive dysfunction in Parkinson’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2348–2357. [DOI] [PubMed] [Google Scholar]
- 107.Jones D End of the line for cannabinoid receptor 1 as an anti-obesity target? Nat. Rev. Drug Discov 2008, 7, 961–962. [DOI] [PubMed] [Google Scholar]
- 108.Donohue S; Halldin C; Finnema S; Gulyas B; Pike V Synthesis and in vivo evaluation of a new PET radioligand for imaging the cannabinoid type-1 (CB1) receptors. NeuroImage, 2006, 31, T50. [Google Scholar]
- 109.Tsujikawa T; Zoghbi SS; Hong J; Donohue SR; Jenko KJ; Gladding RL; Halldin C; Pike VW; Innis RB; Fujita M In vitro and in vivo evaluation of 11C-SD5024, a novel PET radioligand for human brain imaging of cannabinoid CB1 receptors. Neuroimage 2014, 84, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hjorth S; Karlsson C; Jucaite A; Varnas K; Wahlby Hamren U; Johnstrom P; Gulyas B; Donohue SR; Pike VW; Halldin C; Farde L A PET study comparing receptor occupancy by five selective cannabinoid 1 receptor antagonists in non-human primates. Neuropharmacology 2016, 101, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wennerberg M; Cheng L; Hjorth S; Clapham JC; Balendran A; Vauquelin G Binding properties of antagonists to cannabinoid receptors in intact cells. Fundam. Clin. Pharmacol 2011, 25, 200–210. [DOI] [PubMed] [Google Scholar]
- 112.Donohue SR; Finnema SJ; Truong P; Andersson J; Gulyás B; Pike VW; Halldin C Discovery and labeling of a homochiral high affinity 3,4-diarylpyrazoline as a candidate radioligand for in vivo imaging of cannabinoid type-1 receptors. NeuroImage 2008, 41, T90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Willis PG; Pavlova OA; Chefer SI; Vaupel DB; Mukhin AG; Horti AG Synthesis and structure-activity relationship of a novel series of aminoalkylindoles with potential for imaging the neuronal cannabinoid receptor by positron emission tomography. J. Med. Chem 2005, 48, 5813–5822. [DOI] [PubMed] [Google Scholar]
- 114.Dhawan J; Deng H; Gatley SJ; Makriyannis A; Akinfeleye T; Bruneus M; Dimaio AA; Gifford AN Evaluation of the in vivo receptor occupancy for the behavioral effects of cannabinoids using a radiolabeled cannabinoid receptor agonist, R‐[125/131I] AM2233. Synapse. c [DOI] [PubMed] [Google Scholar]
- 115.Allen JB; Amegadzie KM; Gardiner GS; Hitchcock A; Hoogestraat WJ CB1 Modulator Compounds. Patent WO 2005/066126 A1, Jul 21, 2005.
- 116.Donohue SR; Halldin C; Schou M; Hong J; Phebus L; Chernet E; Hitchcock SA; Gardinier KM; Ruley KM; Krushinski JH; Schaus J; Pike VW Radiolabeling of a high potency cannabinoid subtype-1 receptor ligand, N-(4-fluoro-benzyl)-4-(3-(piperidin-1-yl)-indole-1-sulfonyl)benzamide (PipISB), with carbon-11 or fluorine-18. J. Labelled. Compd. Rad 2008, 51, 146–152. [Google Scholar]
- 117.Finnema SJ; Donohue SR; Zoghbi SS; Brown AK; Gulyas B; Innis RB; Halldin C; Pike VW Evaluation of [11C]PipISB and [18F]PipISB in monkey as candidate radioligands for imaging brain cannabinoid type-1 receptors in vivo. Synapse. 2009, 63, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.German N; Decker AM; Gilmour BP; Gay EA; Wiley JL; Thomas BF; Zhang Y Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure–activity relationship studies on 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl) pyridin-2-yl] phenyl} urea (PSNCBAM-1). J. Med. Chem 2014, 57, 7758–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamasaki T; Fujinaga M; Shimoda Y; Mori W; Zhang Y; Wakizaka H; Ogawa M; Zhang MR Radiosynthesis and evaluation of new PET ligands for peripheral cannabinoid receptor type 1 imaging. Bioorg. Med. Chem. Lett 2017, 27, 4114–4117. [DOI] [PubMed] [Google Scholar]
- 120.Zanato C; Pelagalli A; Marwick KF; Piras M; Dall’Angelo S; Spinaci A; Pertwee RG; Wyllie DJ; Hardingham GE; Zanda M Synthesis, radio-synthesis and in vitro evaluation of terminally fluorinated derivatives of HU-210 and HU-211 as novel candidate PET tracers. Org. Biomol. Chem 2017, 15, 2086–2096. [DOI] [PubMed] [Google Scholar]
- 121.Chang CP; Huang HL; Huang JK; Hung MS; Wu CH; Song JS; Lee CJ; Yu CS; Shia KS Fluorine-18 isotope labeling for positron emission tomography imaging. Direct evidence for DBPR211 as a peripherally restricted CB1 inverse agonist. Bioorg. Med. Chem 2019, 27, 216–223. [DOI] [PubMed] [Google Scholar]
- 122.Navarro G; Morales P; Rodriguez-Cueto C; Fernandez-Ruiz J; Jagerovic N; Franco R Targeting cannabinoid CB2 receptors in the central nervous system. medicinal chemistry approaches with focus on neurodegenerative disorders. Front. Neurosci 2016, 10, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen WW; Zhang X; Huang WJ Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep 2016, 13, 3391–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Raitio KH; Savinainen JR; Vepsäläinen J; Laitinen JT; Poso A; Järvinen T; Nevalainen T Synthesis and SAR studies of 2-oxoquinoline derivatives as CB2 receptor inverse agonists. J. Med. Chem 2006, 49, 2022–2027. [DOI] [PubMed] [Google Scholar]
- 125.Evens N; Muccioli GG; Houbrechts N; Lambert DM; Verbruggen AM; Van Laere K; Bormans GM Synthesis and biological evaluation of carbon-11- and fluorine-18-labeled 2-oxoquinoline derivatives for type 2 cannabinoid receptor positron emission tomography imaging. Nucl. Med. Biol 2009, 36, 455–465. [DOI] [PubMed] [Google Scholar]
- 126.Evens N; Vandeputte C; Coolen C; Janssen P; Sciot R; Baekelandt V; Verbruggen AM; Debyser Z; Van Laere K; Bormans GM Preclinical evaluation of [11C]NE40, a type 2 cannabinoid receptor PET tracer. Nucl. Med. Biol 2012, 39, 389–399. [DOI] [PubMed] [Google Scholar]
- 127.Ahmad R; Postnov A; Bormans G; Versijpt J; Vandenbulcke M; Van Laere K Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2219–2227. [DOI] [PubMed] [Google Scholar]
- 128.Slavik R; Herde AM; Bieri D; Weber M; Schibli R; Kramer SD; Ametamey SM; Mu L Synthesis, radiolabeling and evaluation of novel 4-oxo-quinoline derivatives as PET tracers for imaging cannabinoid type 2 receptor. Eur. J. Med. Chem 2015, 92, 554–564. [DOI] [PubMed] [Google Scholar]
- 129.Mu L; Bieri D; Slavik R; Drandarov K; Muller A; Cermak S; Weber M; Schibli R; Kramer SD; Ametamey SM Radiolabeling and in vitro /in vivo evaluation of N-(1-adamantyl)-8-methoxy-4-oxo-1-phenyl-1,4-dihydroquinoline-3-carboxamide as a PET probe for imaging cannabinoid type 2 receptor. J. Neurochem 2013, 126, 616–624. [DOI] [PubMed] [Google Scholar]
- 130.Meletta R; Slavik R; Mu L; Rancic Z; Borel N; Schibli R; Ametamey SM; Kramer SD; Muller Herde A Cannabinoid receptor type 2 (CB2) as one of the candidate genes in human carotid plaque imaging: Evaluation of the novel radiotracer [11C]RS-016 targeting CB2 in atherosclerosis. Nucl. Med. Biol 2017, 47, 31–43. [DOI] [PubMed] [Google Scholar]
- 131.Slavik R; Muller Herde A; Haider A; Kramer SD; Weber M; Schibli R; Ametamey SM; Mu L Discovery of a fluorinated 4-oxo-quinoline derivative as a potential positron emission tomography radiotracer for imaging cannabinoid receptor type 2. J. Neurochem 2016, 138, 874–886. [DOI] [PubMed] [Google Scholar]
- 132.Haider A; Spinelli F; Herde AM; Mu B; Keller C; Margelisch M; Weber M; Schibli R; Mu L; Ametamey SM Evaluation of 4-oxo-quinoline-based CB2 PET radioligands in R6/2 chorea huntington mouse model and human ALS spinal cord tissue. Eur. J. Med. Chem 2018, 145, 746–759. [DOI] [PubMed] [Google Scholar]
- 133.Slavik R; Grether U; Muller Herde A; Gobbi L; Fingerle J; Ullmer C; Kramer SD; Schibli R; Mu L; Ametamey SM Discovery of a high affinity and selective pyridine analog as a potential positron emission tomography imaging agent for cannabinoid type 2 receptor. J. Med. Chem 2015, 58, 4266–4277. [DOI] [PubMed] [Google Scholar]
- 134.Xaio H; Banks WA; Niehoff ML; Morley JE Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res 2001, 896, 36–42. [DOI] [PubMed] [Google Scholar]
- 135.Haider A; Kretz J; Gobbi L; Ahmed H; Atz K; Burkler M; Bartelmus C; Fingerle J; Guba W; Ullmer C; Honer M; Knuesel I; Weber M; Brink A; Herde AM; Keller C; Schibli R; Mu L; Grether U; Ametamey SM Structure-activity relationship studies of pyridine-based ligands and identification of a fluorinated derivative for positron emission tomography imaging of cannabinoid type 2 receptors. J. Med. Chem 2019, 62, 11165–11181. [DOI] [PubMed] [Google Scholar]
- 136.Haider A; Gobbi LC; Kretz J; Ullmer C; Brink A; Honer M; Woltering TJ; Muri D; Iding H; Bürkler M; Binder M; Bartelmus C; Knuesel I; Pacher P; Herde AM; Spinelli F; Ahmed H; Atz K; Keller C; Weber M; Roger S; Mu L; Grether U; Ametamey SM Identification and preclinical development of a 2,5,6-trisubstituted fluorinated pyridine derivative as a radioligand for the positron emission tomography imaging of cannabinoid type 2 receptors. J. Med. Chem 2020, 63, 10287–10306. [DOI] [PubMed] [Google Scholar]
- 137.Horti AG; Gao Y; Ravert HT; Finley P; Valentine H; Wong DF; Endres CJ; Savonenko AV; Dannals RF Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorg. Med. Chem 2010, 18, 5202–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moldovan RP; Teodoro R; Gao Y; Deuther-Conrad W; Kranz M; Wang Y; Kuwabara H; Nakano M; Valentine H; Fischer S; Pomper MG; Wong DF; Dannals RF; Brust P; Horti AG Development of a high-affinity PET radioligand for imaging cannabinoid subtype 2 receptor. J. Med. Chem 2016, 59, 7840–7855. [DOI] [PubMed] [Google Scholar]
- 139.Savonenko AV; Melnikova T; Wang Y; Ravert H; Gao Y; Koppel J; Lee D; Pletnikova O; Cho E; Sayyida N; Hiatt A; Troncoso J; Davies P; Dannals RF; Pomper MG; Horti AG Cannabinoid CB2 receptors in a mouse model of abeta amyloidosis: immunohistochemical analysis and suitability as a PET biomarker of neuroinflammation. PloS one 2015, 10, e0129618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pottier G; Gomez-Vallejo V; Padro D; Boisgard R; Dolle F; Llop J; Winkeler A; Martin A PET imaging of cannabinoid type 2 receptors with [11C]A-836339 did not evidence changes following neuroinflammation in rats. J. Cereb. Blood. Flow. Metab 2017, 37, 1163–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hortala L; Arnaud J; Roux P; Oustric D; Boulu L; Oury-Donat F; Avenet P; Rooney T; Alagille D; Barret O; Tamagnan G; Barth F Synthesis and preliminary evaluation of a new fluorine-18 labelled triazine derivative for PET imaging of cannabinoid CB2 receptor. Bioorg. Med. Chem. Lett 2014, 24, 283–287. [DOI] [PubMed] [Google Scholar]
- 142.Yrjola S; Sarparanta M; Airaksinen AJ; Hytti M; Kauppinen A; Pasonen-Seppanen S; Adinolfi B; Nieri P; Manera C; Keinanen O; Poso A; Nevalainen TJ; Parkkari T Synthesis, in vitro and in vivo evaluation of 1,3,5-triazines as cannabinoid CB2 receptor agonists. Eur. J. Pharm. Sci 2015, 67, 85–96. [DOI] [PubMed] [Google Scholar]
- 143.Ahamed M; van Veghel D; Ullmer C; Van Laere K; Verbruggen A; Bormans GM Synthesis, biodistribution and in vitro evaluation of brain permeable high affinity type 2 cannabinoid receptor agonists [11C]MA2 and [18F]MA3. Front. Neurosci 2016, 10, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Attili B; Celen S; Ahamed M; Koole M; Haute CVD; Vanduffel W; Bormans G Preclinical evaluation of [18F]MA3: a CB2 receptor agonist radiotracer for PET. Br. J. Pharmacol 2019, 176, 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stasiulewicz A; Znajdek K; Grudzień M; Pawiński T; Sulkowska AJI A guide to targeting the endocannabinoid system in drug design. Int. J. Mol. Sci 2020, 21, 2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gulyas AI; Cravatt BF; Bracey MH; Dinh TP; Piomelli D; Boscia F; Freund TF Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur. J. Neurosci 2004, 20, 441–458. [DOI] [PubMed] [Google Scholar]
- 147.McKinney MK; Cravatt BF Structure and function of fatty acid amide hydrolase. Annu. Rev. Biochem. 2005, 74, 411–432. [DOI] [PubMed] [Google Scholar]
- 148.Mulvihill MM; Nomura DK Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci 2013, 92, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tuo W; Leleu-Chavain N; Spencer J; Sansook S; Millet R; Chavatte P Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J. Med. Chem 2017, 60, 4–46. [DOI] [PubMed] [Google Scholar]
- 150.Blankman JL; Simon GM; Cravatt BF A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol 2007, 14, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Long JZ; Cravatt BF The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev 2011, 111, 6022–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lichtman AH; Shelton CC; Advani T; Cravatt BF Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain 2004, 109, 319–327. [DOI] [PubMed] [Google Scholar]
- 153.Gobbi G; Bambico FR; Mangieri R; Bortolato M; Campolongo P; Solinas M; Cassano T; Morgese MG; Debonnel G; Duranti A; Tontini A; Tarzia G; Mor M; Trezza V; Goldberg SR; Cuomo V; Piomelli D Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 18620–18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Scalvini L; Piomelli D; Mor M Monoglyceride lipase: Structure and inhibitors. Chem. Phys. Lipids. 2016, 197, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nomura DK; Morrison BE; Blankman JL; Long JZ; Kinsey SG; Marcondes MCG; Ward AM; Hahn YK; Lichtman AH; Conti B; Cravatt BF Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 2011, 334, 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gil-Ordonez A; Martin-Fontecha M; Ortega-Gutierrez S; Lopez-Rodriguez ML Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol 2018, 157, 18–32. [DOI] [PubMed] [Google Scholar]
- 157.Habib A; Chokr D; Wan J; Hegde P; Mabire M; Siebert M; Ribeiro-Parenti L; Le Gall M; Lettéron P; Pilard N; Mansouri A; Brouillet A; Tardelli M; Weiss E; Le Faouder P; Guillou H; Cravatt BF; Moreau R; Trauner M; Lotersztajn S Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut. 2019, 68, 522–532. [DOI] [PubMed] [Google Scholar]
- 158.Gaetani S; Dipasquale P; Romano A; Righetti L; Cassano T; Piomelli D; Cuomo V The endocannabinoid system as a target for novel anxiolytic and antidepressant drugs. Int. Rev. Neurobiol 2009, 85, 57–72. [DOI] [PubMed] [Google Scholar]
- 159.Piro JR; Benjamin DI; Duerr JM; Pi Y; Gonzales C; Wood KM; Schwartz JW; Nomura DK; Samad TA A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep 2012, 1, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen R; Zhang J; Wu Y; Wang D; Feng G; Tang YP; Teng Z; Chen C Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep 2012, 2, 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Habib AM; Okorokov AL; Hill MN; Bras JT; Lee MC; Li S; Gossage SJ; van Drimmelen M; Morena M; Houlden H; Ramirez JD; Bennett DLH; Srivastava D; Cox JJ Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br. J. Anaesth 2019, 123, e249–e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lo Verme J; Fu J; Astarita G; La Rana G; Russo R; Calignano A; Piomelli D The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol 2005, 67, 15–19. [DOI] [PubMed] [Google Scholar]
- 163.Nomura DK; Long JZ; Niessen S; Hoover HS; Ng S-W; Cravatt BF Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pagano E; Borrelli F; Orlando P; Romano B; Monti M; Morbidelli L; Aviello G; Imperatore R; Capasso R; Piscitelli F Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res 2017, 119, 227–236. [DOI] [PubMed] [Google Scholar]
- 165.Bisogno T; Maccarrone M Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin. Drug Discov 2013, 8, 509–522. [DOI] [PubMed] [Google Scholar]
- 166.Long JZ; Li W; Booker L; Burston JJ; Kinsey SG; Schlosburg JE; Pavon FJ; Serrano AM; Selley DE; Parsons LH; Lichtman AH; Cravatt BF Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol 2009, 5, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kohnz RA; Nomura DK Chemical approaches to therapeutically target the metabolism and signaling of the endocannabinoid 2-AG and eicosanoids. Chem. Soc. Rev 2014, 43, 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Huggins JP; Smart TS; Langman S; Taylor L; Young T An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 2012, 153, 1837–1846. [DOI] [PubMed] [Google Scholar]
- 169.Pawsey S; Wood M; Browne H; Donaldson K; Christie M; Warrington S Safety, tolerability and pharmacokinetics of FAAH inhibitor V158866: a double-blind, randomised, placebo-controlled phase I study in healthy volunteers. Drugs R. D 2016, 16, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Postnov A; Schmidt ME; Pemberton DJ; de Hoon J; van Hecken A; van den Boer M; Zannikos P; van der Ark P; Palmer JA; Rassnick S; Celen S; Bormans G; van Laere K Fatty acid amide hydrolase inhibition by JNJ-42165279: a multiple-ascending dose and a positron emission tomography study in healthy volunteers. Clin. Transl. Sci 2018, 11, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kerbrat A; Ferre JC; Fillatre P; Ronziere T; Vannier S; Carsin-Nicol B; Lavoue S; Verin M; Gauvrit JY; Le Tulzo Y; Edan G Acute neurologic disorder from an inhibitor of fatty acid amide hydrolase. N. Engl. J. Med 2016, 375, 1717–1725. [DOI] [PubMed] [Google Scholar]
- 172.Fazio D; Criscuolo E; Piccoli A; Barboni B; Fezza F; Maccarrone M Advances in the discovery of fatty acid amide hydrolase inhibitors: what does the future hold? Expert Opin. Drug Discov 2020, 1–14. [DOI] [PubMed] [Google Scholar]
- 173.Butler D; Callaway E Scientists in the dark after fatal french clinical trial. Nature 2016, 529, 263–264. [DOI] [PubMed] [Google Scholar]
- 174.Kiss LE; Beliaev A; Ferreira HS; Rosa CP; Bonifacio MJ; Loureiro AI; Pires NM; Palma PN; Soares-da-Silva P Discovery of a potent, long-acting, and CNS-active inhibitor (BIA 10–2474) of fatty acid amide hydrolase. ChemMedChem 2018, 13, 2177–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chaikin P The Bial 10–2474 Phase 1 Study-a drug development perspective and recommendations for future first-in-human trials. J. Clin. Pharmacol 2017, 57, 690–703. [DOI] [PubMed] [Google Scholar]
- 176.Farrell A Enzymology: Tracking off-targets. Nat. Chem. Biol 2017, 13, 817. [DOI] [PubMed] [Google Scholar]
- 177.Huang Z; Ogasawara D; Seneviratne UI; Cognetta III AB; am Ende CW; Nason DM; Lapham K; Litchfield J; Johnson DS; Cravatt BF Global portrait of protein targets of metabolites of the neurotoxic compound BIA 10–2474. ACS Chem. Biol 2019, 14, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ahn K; Johnson DS; Mileni M; Beidler D; Long JZ; McKinney MK; Weerapana E; Sadagopan N; Liimatta M; Smith SE; Lazerwith S; Stiff C; Kamtekar S; Bhattacharya K; Zhang Y; Swaney S; Van Becelaere K; Stevens RC; Cravatt BF Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol 2009, 16, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Piomelli D; Tarzia G; Duranti A; Tontini A; Mor M; Compton TR; Dasse O; Monaghan EP; Parrott JA; Putman D Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS. Drug. Rev 2006, 12, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ahn K; Johnson DS; Fitzgerald LR; Liimatta M; Arendse A; Stevenson T; Lund ET; Nugent RA; Nomanbhoy TK; Alexander JP; Cravatt BF Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 2007, 46, 13019–13030. [DOI] [PubMed] [Google Scholar]
- 181.Kathuria S; Gaetani S; Fegley D; Valino F; Duranti A; Tontini A; Mor M; Tarzia G; La Rana G; Calignano A; Giustino A; Tattoli M; Palmery M; Cuomo V; Piomelli D Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med 2003, 9, 76–81. [DOI] [PubMed] [Google Scholar]
- 182.Wyffels L; Muccioli GG; Kapanda CN; Labar G; De Bruyne S; De Vos F; Lambert DM PET imaging of fatty acid amide hydrolase in the brain: synthesis and biological evaluation of an 11C-labelled URB597 analogue. Nucl. Med. Biol 2010, 37, 665–675. [DOI] [PubMed] [Google Scholar]
- 183.Clapper JR; Vacondio F; King AR; Duranti A; Tontini A; Silva C; Sanchini S; Tarzia G; Mor M; Piomelli D A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem 2009, 4, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wilson AA; Garcia A; Houle S; Vasdev N [11C]-URB694 for FAAH PET imaging: A novel radiotracer for a new target. NRM 2010 Abstract 2010, 52, S24. [Google Scholar]
- 185.Wilson AA; Garcia A; Parkes J; Houle S; Tong J; Vasdev N [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl. Med. Biol 2011, 38, 247–253. [DOI] [PubMed] [Google Scholar]
- 186.Rusjan PM; Wilson AA; Mizrahi R; Boileau I; Chavez SE; Lobaugh NJ; Kish SJ; Houle S; Tong J Mapping human brain fatty acid amide hydrolase activity with PET. J. Cereb. Blood. Flow. Metab 2013, 33, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yamasaki T; Mori W; Zhang Y; Hatori A; Fujinaga M; Wakizaka H; Kurihara Y; Wang L; Nengaki N; Ohya T; Liang SH; Zhang MR First demonstration of in vivo mapping for regional brain monoacylglycerol lipase using PET with [11C]SAR127303. NeuroImage 2018, 176, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jacobson MR; Watts JJ; Da Silva T; Tyndale RF; Rusjan PM; Houle S; Wilson AA; Ross RA; Boileau I; Mizrahi R Fatty acid amide hydrolase is lower in young cannabis users. Addict. Biol 2020, e12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Boileau I; Mansouri E; Williams B; Le Foll B; Rusjan P; Mizrahi R; Tyndale RF; Huestis MA; Payer DE; Wilson AA; Houle S; Kish SJ; Tong J Fatty acid amide hydrolase binding in brain of cannabis users: imaging with the novel radiotracer [11C]CURB. Biol. Psychiatry. 2016, 80, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Best LM; Williams B; Le Foll B; Mansouri E; Bazinet RP; Lin L; De Luca V; Lagzdins D; Rusjan P; Tyndale RF Lower brain fatty acid amide hydrolase in treatment-seeking patients with alcohol use disorder: a positron emission tomography study with [11C] CURB. Neuropsychopharmacology 2020, 45, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Watts JJ; Jacobson MR; Lalang N; Boileau I; Tyndale RF; Kiang M; Ross RA; Houle S; Wilson AA; Rusjan P; Mizrahi R Imaging brain fatty acid amide hydrolase in untreated patients with psychosis. Biol. Psychiatry. 2020, 88, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kolla NJ; Mizrahi R; Karas K; Wang C; Bagby RM; McMain S; Simpson AI; Rusjan PM; Tyndale R; Houle S; Boileau I Elevated fatty acid amide hydrolase in the prefrontal cortex of borderline personality disorder: a [11C]CURB positron emission tomography study. Neuropsychopharmacology 2020, 45, 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wilson AA; Hicks JW; Sadovski O; Parkes J; Tong J; Houle S; Fowler CJ; Vasdev N Radiosynthesis and evaluation of [11C-carbonyl]-labeled carbamates as fatty acid amide hydrolase radiotracers for positron emission tomography. J. Med. Chem 2013, 56, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Aoki A; Munagata R; Kawano N; Samizu K; Oka H; Ishii T Azole Compound. U. S. Patent, 8,207,199 B2, Jun 26, 2012.
- 195.Kumata K; Yui J; Hatori A; Maeda J; Xie L; Ogawa M; Yamasaki T; Nagai Y; Shimoda Y; Fujinaga M; Kawamura K; Zhang MR Development of [11C]MFTC for PET imaging of fatty acid amide hydrolase in rat and monkey brains. ACS Chem. Neurosci 2015, 6, 339–346. [DOI] [PubMed] [Google Scholar]
- 196.Sadovski O; Hicks JW; Parkes J; Raymond R; Nobrega J; Houle S; Cipriano M; Fowler CJ; Vasdev N; Wilson AA Development and characterization of a promising fluorine-18 labelled radiopharmaceutical for in vivo imaging of fatty acid amide hydrolase. Bioorg. Med. Chem 2013, 21, 4351–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rotstein BH; Wey HY; Shoup TM; Wilson AA; Liang SH; Hooker JM; Vasdev N PET imaging of fatty acid amide hydrolase with [18F]DOPP in nonhuman primates. Mol. Pharm 2014, 11, 3832–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tong J; Mizrahi R; Houle S; Kish SJ; Boileau I; Nobrega J; Rusjan PM; Wilson AA Inhibition of fatty acid amide hydrolase by BIA 10–2474 in rat brain. J. Cereb. Blood. Flow. Metab 2017, 37, 3635–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Shoup TM; Bonab AA; Wilson AA; Vasdev N Synthesis and preclinical evaluation of [18F]FCHC for neuroimaging of fatty acid amide hydrolase. Mol. Imaging. Biol 2015, 17, 257–263. [DOI] [PubMed] [Google Scholar]
- 200.Johnson DS; Stiff C; Lazerwith SE; Kesten SR; Fay LK; Morris M; Beidler D; Liimatta MB; Smith SE; Dudley DT; Sadagopan N; Bhattachar SN; Kesten SJ; Nomanbhoy TK; Cravatt BF; Ahn K Discovery of PF-04457845: a highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med. Chem. Lett 2011, 2, 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ahn K; Smith SE; Liimatta MB; Beidler D; Sadagopan N; Dudley DT; Young T; Wren P; Zhang Y; Swaney S; Van Becelaere K; Blankman JL; Nomura DK; Bhattachar SN; Stiff C; Nomanbhoy TK; Weerapana E; Johnson DS; Cravatt BF Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther 2011, 338, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rotstein BH; Liang SH; Holland JP; Collier TL; Hooker JM; Wilson AA; Vasdev N 11CO2 fixation: a renaissance in PET radiochemistry. Chem. Commun 2013, 49, 5621–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hicks JW; Parkes J; Sadovski O; Tong J; Houle S; Vasdev N; Wilson AA Synthesis and preclinical evaluation of [11C-carbonyl]PF-04457845 for neuroimaging of fatty acid amide hydrolase. Nucl. Med. Biol 2013, 40, 740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Skaddan MB; Zhang L; Johnson DS; Zhu A; Zasadny KR; Coelho RV; Kuszpit K; Currier G; Fan KH; Beck EM; Chen L; Drozda SE; Balan G; Niphakis M; Cravatt BF; Ahn K; Bocan T; Villalobos A The synthesis and in vivo evaluation of [18F]PF-9811: a novel PET ligand for imaging brain fatty acid amide hydrolase (FAAH). Nucl. Med. Biol 2012, 39, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dahl K; Collier T; Zhang X; Liang S; Vasdev N Design and Prototype of an Automated” In-loop”[11C] CO2-Fixation Apparatus. J. Nucl. Med 2017, 58, 330–330. [Google Scholar]
- 206.Dahl K; Collier TL; Cheng R; Zhang X; Sadovski O; Liang SH; Vasdev N “In-loop” [11C]CO2 fixation: Prototype and proof of concept. J. Labelled. Comp. Radiopharm 2018, 61, 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cumming P; Vasdev N The assay of enzyme activity by positron emission tomography Neuromethods. 2012, 71, 111–135. [Google Scholar]
- 208.Pike VW PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends. Pharmacol. Sci 2009, 30, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Liu P; Hamill TG; Chioda M; Chobanian H; Fung S; Guo Y; Chang L; Bakshi R; Hong Q; Dellureficio J; Lin LS; Abbadie C; Alexander J; Jin H; Mandala S; Shiao LL; Li W; Sanabria S; Williams D; Zeng Z; Hajdu R; Jochnowitz N; Rosenbach M; Karanam B; Madeira M; Salituro G; Powell J; Xu L; Terebetski JL; Leone JF; Miller P; Cook J; Holahan M; Joshi A; O’Malley S; Purcell M; Posavec D; Chen TB; Riffel K; Williams M; Hargreaves R; Sullivan KA; Nargund RP; DeVita RJ Discovery of MK-3168: a PET tracer for imaging brain fatty acid amide hydrolase. ACS Med. Chem. Lett 2013, 4, 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Joshi A; Li W; Sanabria S; Holahan M; Purcell M; Declercq R; Depre M; Bormans G; Laere KV; Hamill T Translational studies with [11C]MK-−3168, a PET tracer for fatty acid amide hydrolase (FAAH). J. Nucl. Med 2012, 53(S1), 397. [Google Scholar]
- 211.Postnov A; Schmidt M; Penson J; Hecken AV; Zannikos P; Pemberton D; Palmer J; Hoon J. d.; Bormans G; Laere KV Kinetic modeling of fatty acid amide hydrolase (FAAH) enzyme occupancy after JNJ-42165279 inhibition based on 11C—MK-3168 PET imaging of human brain. J. Nucl. Med 2015, 56(S3), 362. [Google Scholar]
- 212.Mileni M; Garfunkle J; DeMartino JK; Cravatt BF; Boger DL; Stevens RC Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by alpha-ketoheterocycle inhibitors revealed from cocrystal structures. J. Am. Chem. Soc 2009, 131, 10497–10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Otrubova K; Boger DL α-Ketoheterocycle-based inhibitors of fatty acid amide hydrolase (FAAH). ACS Chem. Neurosci 2012, 3, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hardouin C; Kelso MJ; Romero FA; Rayl TJ; Leung D; Hwang I; Cravatt BF; Boger DL Structure-activity relationships of alpha-ketooxazole inhibitors of fatty acid amide hydrolase. J. Med. Chem 2007, 50, 3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wang L; Yui J; Wang Q; Zhang Y; Mori W; Shimoda Y; Fujinaga M; Kumata K; Yamasaki T; Hatori A; Rotstein BH; Collier TL; Ran C; Vasdev N; Zhang MR; Liang SH Synthesis and preliminary PET imaging studies of a FAAH radiotracer ([11C]MPPO) based on alpha-ketoheterocyclic scaffold. ACS Chem. Neurosci 2016, 7, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chen Z; Hou L; Gan J; Cai Q; Ye W; Chen J; Tan Z; Zheng C; Li G; Xu H; Fowler CJ; Liang SH; Wang L Synthesis and preliminary evaluation of a novel positron emission tomography (PET) ligand for imaging fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett 2020, 30, 127513. [DOI] [PubMed] [Google Scholar]
- 217.Saario SM; Savinainen JR; Laitinen JT; Jarvinen T; Niemi R Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem. Pharmacol 2004, 67, 1381–1387. [DOI] [PubMed] [Google Scholar]
- 218.Blankman JL; Simon GM; Cravatt BF A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol 2007, 14, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hicks JW; Parkes J; Tong J; Houle S; Vasdev N; Wilson AA Radiosynthesis and ex vivo evaluation of [11C-carbonyl]carbamate- and urea-based monoacylglycerol lipase inhibitors. Nucl. Med. Biol 2014, 41, 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Griebel G; Pichat P; Beeske S; Leroy T; Redon N; Jacquet A; Francon D; Bert L; Even L; Lopez-Grancha M; Tolstykh T; Sun F; Yu Q; Brittain S; Arlt H; He T; Zhang B; Wiederschain D; Bertrand T; Houtmann J; Rak A; Vallee F; Michot N; Auge F; Menet V; Bergis OE; George P; Avenet P; Mikol V; Didier M; Escoubet J Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep 2015, 5, 7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang C; Placzek MS; Van de Bittner GC; Schroeder FA; Hooker JM A novel radiotracer for imaging monoacylglycerol lipase in the brain using positron emission tomography. ACS Chem. Neurosci 2016, 7, 484–489. [DOI] [PubMed] [Google Scholar]
- 222.Wang L; Mori W; Cheng R; Yui JJ; Hatori A; Ma LL; Zhang YD; Rotstein BH; Fujinaga M; Shimoda Y; Yamasaki T; Xie L; Nagai Y; Minamimoto T; Higuchi M; Vasdev N; Zhang MR; Liang SH Synthesis and preclinical evaluation of sulfonamido-based [11C-Carbonyl]-carbamates and ureas for imaging monoacylglycerol lipase. Theranostics 2016, 6, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ahamed M; Attili B; van Veghel D; Ooms M; Berben P; Celen S; Koole M; Declercq L; Savinainen JR; Laitinen JT; Verbruggen A; Bormans G Synthesis and preclinical evaluation of [11C]MA-PB-1 for in vivo imaging of brain monoacylglycerol lipase (MAGL). Eur. J. Med. Chem 2017, 136, 104–113. [DOI] [PubMed] [Google Scholar]
- 224.Butler CR; Beck EM; Harris A; Huang Z; McAllister LA; Am Ende CW; Fennell K; Foley TL; Fonseca K; Hawrylik SJ; Johnson DS; Knafels JD; Mente S; Noell GS; Pandit J; Phillips TB; Piro JR; Rogers BN; Samad TA; Wang J; Wan S; Brodney MA Azetidine and piperidine carbamates as efficient, covalent inhibitors of monoacylglycerol lipase. J. Med. Chem 2017, 60, 9860–9873. [DOI] [PubMed] [Google Scholar]
- 225.Cheng R; Mori W; Ma L; Alhouayek M; Hatori A; Zhang Y; Ogasawara D; Yuan G; Chen Z; Zhang X; Shi H; Yamasaki T; Xie L; Kumata K; Fujinaga M; Nagai Y; Minamimoto T; Svensson M; Wang L; Du Y; Ondrechen MJ; Vasdev N; Cravatt BF; Fowler C; Zhang MR; Liang SH In vitro and in vivo evaluation of 11C-Labeled azetidinecarboxylates for imaging monoacylglycerol lipase by PET imaging studies. J. Med. Chem 2018, 61, 2278–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chen Z; Mori W; Deng X; Cheng R; Ogasawara D; Zhang G; Schafroth MA; Dahl K; Fu H; Hatori A; Shao T; Zhang Y; Yamasaki T; Zhang X; Rong J; Yu Q; Hu K; Fujinaga M; Xie L; Kumata K; Gou Y; Chen J; Gu S; Bao L; Wang L; Collier TL; Vasdev N; Shao Y; Ma JA; Cravatt BF; Fowler C; Josephson L; Zhang MR; Liang SH Design, synthesis and evaluation of reversible and irreversible monoacylglycerol lipase positron emission tomography (PET) tracers using a ‘tail switching’ strategy on a piperazinyl azetidine skeleton. J. Med. Chem 2019, 62, 3336–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Schneider G; Neidhart W; Giller T; Schmid G “Scaffold-Hopping” by topological pharmacophore search: a contribution to virtual screening. Angew. Chem. Int. Ed. Engl 1999, 38, 2894–2896. [PubMed] [Google Scholar]
- 228.Hu Y; Stumpfe D; Bajorath J Recent advances in scaffold hopping. J. Med. Chem 2017, 60, 1238–1246. [DOI] [PubMed] [Google Scholar]
- 229.Wang L; Fujinaga M; Cheng R; Yui J; Shimoda Y; Rotstein B; Zhang Y; Vasdev N; Zhang MR; Liang S Synthesis and preliminary evaluation of a 11C-labeled piperidin-4-yl azetidine diamide for imaging monoacylglycerol lipase. J. Nucl. Med 2016, 57, 1004.26966158 [Google Scholar]
- 230.Mori W; Hatori A; Zhang Y; Kurihara Y; Yamasaki T; Xie L; Kumata K; Hu K; Fujinaga M; Zhang MR Radiosynthesis and evaluation of a novel monoacylglycerol lipase radiotracer: 1,1,1,3,3,3-hexafluoropropan-2-yl-3-(1-benzyl-1H-pyrazol-3-yl)azetidine-1-[11C] carboxylate. Bioorg. Med. Chem 2019, 27, 3568–3573. [DOI] [PubMed] [Google Scholar]
- 231.Zhang L; Butler CR; Maresca KP; Takano A; Nag S; Jia Z; Arakawa R; Piro JR; Samad T; Smith DL; Nason DM; O’Neil S; McAllister L; Schildknegt K; Trapa P; McCarthy TJ; Villalobos A; Halldin C Identification and development of an irreversible monoacylglycerol lipase (MAGL) positron emission tomography (PET) radioligand with high specificity. J. Med. Chem 2019, 62, 8532–8543. [DOI] [PubMed] [Google Scholar]
- 232.ClinicalTrials.gov. Test re-test reliability of 11CPF-06809247 as a novel PET tacer. https://clinicaltrials.gov/ct2/show/NCT03100136 (accessed Oct 8, 2010).
- 233.Chen Z; Mori W; Fu H; Schafroth MA; Hatori A; Shao T; Zhang G; Van RS; Zhang Y; Hu K; Fujinaga M; Wang L; Belov V; Ogasawara D; Giffenig P; Deng X; Rong J; Yu Q; Zhang X; Papisov MI; Shao Y; Collier TL; Ma JA; Cravatt BF; Josephson L; Zhang MR; Liang SH Design, synthesis, and evaluation of 18F-Labeled monoacylglycerol lipase inhibitors as novel positron emission tomography probes. J. Med. Chem 2019, 62, 8866–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.McAllister LA; Butler CR; Mente S; O’Neil SV; Fonseca KR; Piro JR; Cianfrogna JA; Foley TL; Gilbert AM; Harris AR; Helal CJ; Johnson DS; Montgomery JI; Nason DM; Noell S; Pandit J; Rogers BN; Samad TA; Shaffer CL; da Silva RG; Uccello DP; Webb D; Brodney MA Discovery of trifluoromethyl glycol carbamates as potent and selective covalent monoacylglycerol lipase (MAGL) inhibitors for treatment of neuroinflammation. J. Med. Chem 2018, 61, 3008–3026. [DOI] [PubMed] [Google Scholar]
- 235.Rotstein BH; Stephenson NA; Vasdev N; Liang SH Spirocyclic hypervalent iodine (III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun 2014, 5, 4365. [DOI] [PubMed] [Google Scholar]
- 236.Wang L; Jacobson O; Avdic D; Rotstein BH; Weiss ID; Collier L; Chen X; Vasdev N; Liang SH Ortho‐stabilized 18F‐azido click agents and their application in PET imaging with single‐stranded DNA aptamers. Angew. Chem. Int. Ed. Engl 2015, 54, 12777–12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Rotstein BH; Wang L; Liu RY; Patteson J; Kwan EE; Vasdev N; Liang SH Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides. Chem. Sci 2016, 7, 4407–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for positron emission tomography: recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions. Angew. Chem. Int. Ed. Engl 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chen Z; Cheng R; Yang J; Shao T; Vasdev N; Ran C; Zhang M-R; Liang S A novel 18F-labeled MAG lipase biomarker for differentiating brown and white adipose tissue in the lipid network. J. Nucl. Med 2018, 59, 262–262. [Google Scholar]
- 240.Hattori Y; Aoyama K; Maeda J; Arimura N; Takahashi Y; Sasaki M; Fujinaga M; Seki C; Nagai Y; Kawamura K; Yamasaki T; Zhang MR; Higuchi M; Koike T Design, synthesis, and evaluation of (4 R)-1-{3-[2-(18F)Fluoro-4-methylpyridin-3-yl]phenyl}−4-[4-(1,3-thiazol-2-ylcarbo nyl)piperazin-1-yl]pyrrolidin-2-one ([18F]T-401) as a novel positron-emission tomography imaging agent for monoacylglycerol lipase. J. Med. Chem 2019, 2362–2375. [DOI] [PubMed] [Google Scholar]
- 241.Shekhar A; Thakur GA Cannabinoid receptor 1 positive allosteric modulators for posttraumatic stress disorder. Neuropsychopharmacology 2018, 43, 226–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Shao Z; Yan W; Chapman K; Ramesh K; Ferrell AJ; Yin J; Wang X; Xu Q; Rosenbaum DM Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol 2019, 15, 1199–1205. [DOI] [PubMed] [Google Scholar]
- 243.Pandey P; Roy KK; Doerksen RJ Negative allosteric modulators of cannabinoid receptor 2: protein modeling, binding site identification and molecular dynamics simulations in the presence of an orthosteric agonist. J. Biomol. Struct. Dyn 2020, 38, 32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Laprairie RB; Bagher AM; Kelly ME; Denovan-Wright EM Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol 2015, 172, 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bauer M; Chicca A; Tamborrini M; Eisen D; Lerner R; Lutz B; Poetz O; Pluschke G; Gertsch J Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J. Biol. Chem 2012, 287, 36944–36967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Pamplona FA; Ferreira J; Menezes de Lima O Jr.; Duarte FS; Bento AF; Forner S; Villarinho JG; Bellocchio L; Wotjak CT; Lerner R; Monory K; Lutz B; Canetti C; Matias I; Calixto JB; Marsicano G; Guimarães MZ; Takahashi RN Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 21134–21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ignatowska-Jankowska BM; Baillie GL; Kinsey S; Crowe M; Ghosh S; Owens RA; Damaj IM; Poklis J; Wiley JL; Zanda M; Zanato C; Greig IR; Lichtman AH; Ross RA A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology 2015, 40, 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Khurana L; Mackie K; Piomelli D; Kendall DA Modulation of CB1 cannabinoid receptor by allosteric ligands: pharmacology and therapeutic opportunities. Neuropharmacology 2017, 124, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Dainese E; Oddi S; Simonetti M; Sabatucci A; Angelucci CB; Ballone A; Dufrusine B; Fezza F; De Fabritiis G; Maccarrone M The endocannabinoid hydrolase FAAH is an allosteric enzyme. Sci. Rep 2020, 10, 2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Cao JK; Kaplan J; Stella N ABHD6: its place in endocannabinoid signaling and beyond. Trends. Pharmacol. Sci 2019, 40, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Manterola A; Bernal-Chico A; Cipriani R; Ruiz A; Pérez-Samartín A; Moreno-Rodríguez M; Hsu K-L; Cravatt BF; Brown JM; Rodríguez-Puertas R Re-examining the potential of targeting ABHD6 in multiple sclerosis: efficacy of systemic and peripherally restricted inhibitors in experimental autoimmune encephalomyelitis. Neuropharmacology 2018, 141, 181–191. [DOI] [PubMed] [Google Scholar]
- 252.Ogasawara D; Ichu T-A; Vartabedian VF; Benthuysen J; Jing H; Reed A; Ulanovskaya OA; Hulce JJ; Roberts A; Brown S Selective blockade of the lyso-PS lipase ABHD12 stimulates immune responses in vivo. Nat. Chem. Biol 2018, 14, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Jain T; Wager-Miller J; Mackie K; Straiker A Diacylglycerol lipaseα (DAGLα) and DAGLβ cooperatively regulate the production of 2-arachidonoyl glycerol in autaptic hippocampal neurons. Mol. Pharmacol 2013, 84, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hsu KL; Tsuboi K; Adibekian A; Pugh H; Masuda K; Cravatt BF DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat. Chem. Biol 2012, 8, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ogasawara D; Deng H; Viader A; Baggelaar MP; Breman A; den Dulk H; van den Nieuwendijk AM; Soethoudt M; van der Wel T; Zhou J Rapid and profound rewiring of brain lipid signaling networks by acute diacylglycerol lipase inhibition. Proc. Nati. Acad. Sci 2016, 113, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Mock ED; Mustafa M; Gunduz-Cinar O; Cinar R; Petrie GN; Kantae V; Di X; Ogasawara D; Varga ZV; Paloczi J; Miliano C; Donvito G; van Esbroeck ACM; van der Gracht AMF; Kotsogianni I; Park JK; Martella A; van der Wel T; Soethoudt M; Jiang M; Wendel TJ; Janssen APA; Bakker AT; Donovan CM; Castillo LI; Florea BI; Wat J; van den Hurk H; Wittwer M; Grether U; Holmes A; van Boeckel CAA; Hankemeier T; Cravatt BF; Buczynski MW; Hill MN; Pacher P; Lichtman AH; van der Stelt M Discovery of a NAPE-PLD inhibitor that modulates emotional behavior in mice. Nat. Chem. Biol 2020, 16, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kinsey SG; Wise LE; Ramesh D; Abdullah R; Selley DE; Cravatt BF; Lichtman AH Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J. Pharmacol. Exp. Ther 2013, 345, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Liang SH; Wang L; Stephenson NA; Rotstein BH; Vasdev N Facile 18F labeling of non-activated arenes via a spirocyclic iodonium(III) ylide method and its application in the synthesis of the mGluR5 PET radiopharmaceutical [18F]FPEB. Nat. Protoc 2019, 14, 1530–1545. [DOI] [PubMed] [Google Scholar]