

Abstract
Epidemiologically, high-density lipoprotein (HDL) cholesterol levels have been inversely associated to cardiovascular (CV) events, although a Mendelian Randomisation Study had failed to establish a clear causal role. Numerous atheroprotective mechanisms have been attributed to HDL, the main being the ability to promote cholesterol efflux from arterial walls; anti-inflammatory effects related to HDL ligands such as S1P (sphingosine-1-phosphate), resolvins and others have been recently identified. Experimental studies and early clinical investigations have indicated the potential of HDL to slow progression or induce regression of atherosclerosis. More recently, the availability of different HDL formulations, with different phospholipid moieties, has allowed to test other indications for HDL therapy. Positive reports have come from studies on coronary stent biocompatibility, where the use of HDL from different sources reduced arterial cell proliferation and thrombogenicity. The observation that low HDL-C levels may be associated with an enhanced risk of heart failure (HF) has also suggested that HDL therapy may be applied to this condition. HDL infusions or apoA-I gene transfer were able to reverse heart abnormalities, reduce diastolic resistance and improve cardiac metabolism. HDL therapy may be effective not only in atherosclerosis, but also in other conditions, of relevant impact on human health.
Key messages
High-density lipoproteins have as a major activity that of removing excess cholesterol from tissues (particularly arteries).
Knowledge on the activity of high-density lipoproteins on health have however significantly widened.
HDL-therapy may help to improve stent biocompatibility and to reduce peripheral arterial resistance in heart failure.
Keywords: HDL, HDL-cholesterol, HDL therapy, inflammation, rice milk phospholipids, resolvins
Introduction
High density lipoproteins (HDL) are a major fraction of circulating lipoproteins. Epidemiological and clinical evidence has suggested the existence of an inverse association between HDL-C levels and CHD risk, although recently a U-shaped association between HDL cholesterol concentrations and all-cause mortality was found, i.e. both extreme high and low concentrations of HDL being associated with all-cause mortality risk [1]. A large clinical experience and basic studies have supported the concept that the antiatherogenic role of high HDL-C is mediated by the removal of excess cholesterol from the extrahepatic tissues, carrying it back to the liver for metabolisation, the so-called reverse cholesterol transport [2,3]. In this review article the present status of HDL and its pro-effluxing and anti-cell proliferating properties will be discussed, potentially resulting in an effective HDL therapy for, particularly, coronary conditions.
While the postulated vascular benefits have not been supported by a Mendelian Randomisation Study [4], an extensive evaluation of genetic and secondary causes of severe HDL deficiency and CV disease pointed out that HDL deficiency may be associated with ATP-binding cassette transporter (ABCA1), lecithin-cholesterol acyltransferase (LCAT), apoliprotein (apoA)-I or lipoprotein lipase (LPL) mutations or variants, the highest prevalence of ASCVD being observed in the ABCA1 (Tangier disease) and apoA-I variant groups [5]. The Mendelian Randomisation Study only offered a limited view of HDL associated risk, in particular being just related to the single nucleotide polymorphisms (SNPs) of endothelial lipase [4].
A limitation of linking CV risk to HDL-C levels is the possible presence of a reduced functionality of HDL. “HDL dysfunction” may lead to a reduced ability to promote cholesterol removal from macrophages, but also to the maintenance of vascular endothelial function through a variety of effects on vascular tone, inflammation and endothelial cell homeostasis and integrity [6,7]. Bettering HDL function has proven challenging. Very recently Ossoli et al. [8] gave evidence that HDL dysfunction observed during an acute coronary syndrome (ACS) can be corrected by adding recombinant human LCAT. In vitro incubation of ACS patients’ plasma with recombinant human LCAT restores the ability of HDL to promote endothelial NO production, possibly related to significant modifications in HDL phospholipid (PL) classes. Lipid species in HDL are more than 200 individual molecules, PL representing approximately 50% of the total [9]. In addition to phosphatidylcholine (PC) the HDL-PL are represented by phosphatidylethanolamine (PE), phosphatidylserine (PS) and others.
A significant role of the sphingomyelin (SM) species, representing 5–10% in weight of total has been recognised in a number of recent reports. The SM derivative sphingosine-1-phosphate (S1P) in particular follows phosphorylation of cell membrane derived sphingosine through SphK1 (sphingosine kinase 1) and SphK2 (sphingosine kinase 2) [10]. S1P rapidly reaches high plasma concentrations being bound to albumin or HDL. This bioactive PL can influence the quality and quantity of HDL dependent function, particularly with the binding partner apolipoprotein M; ApoM deficient mice do not carry S1P and show a functional deficiency of HDL [11]. Cellular protection may be exerted also by way of opening of the mitochondrial channels, exerted by S1P, apo AI, clusterin and miRNA [12,13]. The mechanism appears to be that of induction of STAT3, subsequently bound by mitochondria, decreasing the permeability transition pore and preventing apoptosis [14].
S1P, although not frequently studied, is probably crucial in providing the antioxidant activity in the HDL proteome [15,16]. Among the proposed mechanisms of HDL/S1P signalling is a stimulated eNOS dependent arterial vasodilation through S1P and also S1P3 receptors in the coronary microcirculation [17]. The anti-inflammatory effects of HDL/SP1 occur partially because of the capacity of ApoM(+)HDL to act as a biased agonist of the S1P1 endothelial cell receptor, partially explaining the cardiovascular protective functions of HDL [18]. The role of inflammation in atherosclerosis development/progression has been recently reviewed [19].
Changes with inflammation are further exerted on the HDL proteome, consisting of over 85 associated proteins [20,21]. Typical changes after inflammation are substitutions of protective proteins like apo AI, apoAIV, transthyretin or RBP (Retinol Binding Protein), with proinflammatory proteins such as serum amyloid A (SAA), complement 3 and lipopolysaccharise binding protein (LBP). In the proteomic evaluation of HDL subspecies [22], 16 novel subspecies were characterised, among others, by the presence of apo AIV, apoCI, Apo CII and apoJ, being associated with differential functions, including anti-inflammatory activity. The presence of apo E in HDL has become of interest after the observation that HDL containing apo E is diet responsive being particularly raised after unsaturated fat, becoming more biologically active [23].
Stimulation of endothelial cells by HDL leads to prostacyclin production, by raising supply of arachidonate and cyclooxygenase-2 expression [24]; HDL-C levels are positively correlated with the stable metabolite of prostacyclin 6-keto PGF1a [25]. The potential effects of HDL on resolvin formation have been also described. Resolvins, specialised metabolic products of omega-3-polyunsaturated fatty acids [26], are markedly increased in the initial phases of acute inflammatory responses and can reduce atherosclerosis in the absence of cholesterol lowering [27]. There are as yet no clear findings on a potential association of HDL in reducing/inhibiting the activity of resolvins, but it is well defined that thromboxane A2 biosynthesis is reduced by large HDL particles, modulating the prostacyclin/TxA2 balance [28].
The association between HDL and arterial protection is thus linked to: HDL-C levels, HDL protein moiety, mainly characterised by apoA-I and finally by HDL function, apparently modified in different clinical conditions, i.e. ranging from patients with myocardial infarction [29], to those with heart failure with reduced ejection fraction [30]. Evaluation of HDL-C levels, still widely applied and of clinical value, thus offers a limited view of CV risk [31].
Recent approaches aiming to elevate HDL-C levels have mostly shown unsatisfactory results [32,33]. Well established drugs, such as fenofibrate and extended release nicotinic acid (ER-NA) were not associated with decreased CV risk in patients on statin treatment [34,35], even though several authors have highlighted some issues affecting the design of the studies [36]. Fenofibrate, in particular, did reduce CV risk in hypertriglyceridemic patients with low HDL-C levels [37], probably indicating the importance of a targeted selection of patients. Among cholesteryl ester transfer protein (CETP) inhibitors, markedly raising HDL-C, only anacetrapib in the large REVEAL study did reduce CV risk by approximately 10%, but certain safety concerns associated to the drug [38]. in particular a very long half-life and prolonged permanence in tissues, consequent to a direct uptake by the adipocytes [39], led to drug discontinuation. Trials with this drug class well support the notion of quality vs quantity of HDL particle levels [40]. The HDL infusion therapy has been tested in different conditions with initially very positive results, particularly with the mutant apoA-IMilano [41]. However, more recent investigations with this and other HDL formulations did not confirm the initial positive findings, for reasons needing further evaluation (see below).
Extended studies on HDL and its potential therapeutic approaches have, however, led to a number of newer findings related to CV risk and its prevention. Among these, a reduced atherogenic inflammation [42] and an improved control of hematopoietic stem cell proliferation, monocytosis and neutrophilia, all leading to an improved cholesterol effluxing capacity of HDL [43]. These findings have stimulated research into better focussed and more effective HDL based therapies.
HDL – the present day biology
HDL is traditionally viewed as a dominant factor in the process of reverse cholesterol transport (RCT). HDL, removing cholesterol from the arterial walls by way of the transporters ABCA1 and ABCG1 can effectively raise HDL-cholesterol back-transport as a mechanism of arterial protection [42]. However, this is now rated as just one of several protective mechanisms and, interestingly, drugs raising HDL-C do improve cholesterol efflux, but this is not always associated with a reduced CV risk (CETP antagonists) [44]. HDL is responsible for key effects on macrophage induced inflammation and reduced endoplasmic reticulum (ER) stress and apoptosis [45,46] and the same membrane transporters can control monocyte activation, adhesiveness and inflammation.
The ATP binding cassette transporter ABCA1 can be induced in the arterial wall macrophage by Liver X receptors (LXR)/retinoid X receptor (RXR) activation, thus promoting efflux of cholesterol into lipid poor apoA-I and HDL particles [47]. ABCA1/G1 deficient macrophages show, in addition to reduced efflux, a raised inflammatory response, partly related to increased surface expression of the toll like receptor A4 (TLRA4) increasing signalling via MYD88 and TLR dependent pathways, all linked to raised cell cholesterol content [48]. HDL linked transporters also suppress extensive proliferation of hematopoietic stem cells and in general clonal haematopoiesis (CH) [49].
Very low HDL-C levels have been found to be associated with an increased risk of autoimmune diseases in individuals from the general population [50] possibly consequent to a defective control of immune function by HDL, ranging from regulation of hemotopoietic stem cells to modulation of immune cells by surface receptors [51]. Interesting, in a neighbouring study (Copenhagen City Heart Study) the autoimmune disease distribution was U-shaped, both low and high levels of HDL-C being associated with the disease.
Activation of cholesterol efflux by the ABCA1/G1 transporters can prevent inflammasome activation and atherogenesis, as shown by conditions such as Tangier disease, carrying a loss off-functional mutation ABCA1, increased myeloid cholesterol content and a marked decrease in plasma interleukin (IL)-1β and IL-18 levels [52]. MACabcdko mice, a model of Tangier disease, show inflammasome activation in particular of the NLRP3/caspase [53,54].
The association between atherosclerosis development and inflammatory conditions has led to the investigation of factors having an impact in particular on plaque development and rupture. The presence of cholesterol crystals in the growing plaque leads to activation of the NLRP3 inflammasome [55] with a major impact in atherosclerosis development and as a potential trigger in plaque progression and rupture. Inhibition of the inflammasome can be thus a key protective mechanism of HDL, considering the potential to reduce plaques and events [56].
Another possible pathway mediating HDL-A-I atheroprotective function can be by raised expression of angiopoietin like-4 (ANGPTL-4), a known inhibitor of lipoprotein lipase [57] in endothelial cells: ANGPTL-4 reduces lipid uptake in macrophages [58]. By evaluating gene expression in whole genome microarrays, raised expression of ANGPTL-4 in EA-HY926 endothelial cells was found as one of the most up-regulated and biologically relevant molecules [59]. Gene induction was directly blocked by the presence of inhibitors of the AKT or p38 MAP kinases. A FOX01 inhibitor or a FOX01-specific siRNA enhanced ANGPTL-4 expression thus indicating that FOX01 functions as an inhibitor of ANGPTL-4, versus HDL-apoA-I blocking FOXO1 and activating ANGPTL4. These novel findings are of special interest in in view of the current work on the association of ANGPTL-4 with CV disease risk [60].
HDL therapy – the coronary atheroma target (Figure 1)
Figure 1.

Therapeutic effects of different formulations and delivery methods of HDL/APOA-I(Milano) in the major clinical manifestations of cardiovascular disease. AI(M): Apolipoprotein 1 (Milano variant); VGT: Vascular Gene Therapy; HDL: High-Density Lipoproteins; HF: heart failure; rAAV: recombinant Adeno-Associated Viral vectors; tAIM: human APOA-IMilano sequence exogenously expressed in genetically modified rice plants; NIH: neointimal hyperplasia; SMC: smooth muscle cell; cardiac met: cardiac metabolism. Some images and pictures in the figure were from Servier Medical Art by Servier (https://smart.servier.com/.. ), licenced under a Creative Commons Attribution 3.0 Unported Licence (https://creativecommons.org/licenses/by/3.0/.. ).
Early knowledge on the potential role of HDL in lipid removal from plaques led to initial studies in animal models, particularly in cholesterol-fed rabbits, infused with isolated HDL in an effort to reduce aortic lesions. In these early studies, administration of homologous HDL to atherosclerotic rabbits, not only mitigated lesion development but was able to regress established lesions [61,62]. In a number of further reports these same authors pointed out different aspects of this antiatherosclerotic effect, among others reduction of prostacyclin release in smooth muscle cells (SMC), dependent of cyclooxygenase-2 expression [63].
A significant step forward was provided by the development of the intravascular ultrasound (IVUS) technology. A clinical study comparing IVUS with the classical coronary angiographic evaluation [64] had reported that in patients with familial combined hyperlipidaemia there was an inverse correlation between HDL-C levels and plaque thickness, reported as maximal intima index. This observation generated interest in a possible study, at that time, with the mutant apo AIMilano (AIM) as a possible inducer of atheroma regression. Carriers of the mutant (Cys-Arg substitution at position 173 of apo AI) are, in fact, characterised by extreme reductions of HDL-C levels in the presence of clear cardiovascular protection [65]. Turnover studies had indicated that the dimeric form of AIMilano (AIM/AIM) is characterised by an optimal permanence in blood after a single infusion (10 days vs 5 days for the wild type protein) together with an excellent capacity to remove tissue cholesterol [66].
The optimal activity of the dimer [67] led to the final decision to evaluate a direct effect of AIM/AIM on a focal atheroma. This last was generated by an electric injury in the common carotid arteries of cholesterol-fed rabbits and characterised by a massive lipid and macrophage accumulation in a well reproducible fashion [68]. Direct assessment of local delivery of AIM was by way of the external carotid, positioning the catheter’s tip proximally from the focal plaque. AIM-dipalmitoylphosphatidylcholine (DPPC) complexes were given as single doses of 0.25–1 g of protein over 90 min and were followed by an impressive shrinking of the carotid plaques, up to –30% at the end of infusion [69]. The direct effect on lipid removal was confirmed by a histochemical evaluation and was also possible to assess the presence of immunochemically detectable AIM upon the final sacrifice of the animals 3 days later, confirming a prolonged permanence within the plaque. Interestingly, in a more recent study led by Spanish investigators, AIM in rabbits’ aortas was detected up to 6 months after two infusions of AIM dimer [70].
The successful completion of these animal studies prompted the clinical evaluation with a somewhat similar protocol in coronary patients undergoing an invasive procedure. ACS patients with at least a 20% coronary luminal narrowing were randomised to receive 5 weekly infusions of graded doses, i.e. either 15 or 45 mg/kg. The treated groups had a 4.2% decrease from baseline of the total coronary plaque volume, as compared to no changes in the placebo group, the maximal effect occurring in patients with the largest plaque volume at baseline [41].
This at the time astonishing result was followed by the rapid acquisition of the developing company (Esperion Therapeutics) by Pfizer, allowing a more extensive investigation of the protein’s effect. Unfortunately, a newer preparation of AIM tested in coronary patients led to severe allergic reactions resulting in one death. The reason, not reported in the literature, was the presence of small protein contaminants in the newer biotechnological formulation of AIM. This led to a halting of the clinical trials in spite of still ongoing basic studies with different methodologies. In 2009, development of AIM was transferred to the Medicines Company (MDCO), now responsible for development.
In more recent years, three products have been tested in human trials mainly with the IVUS method (Figure 2). They are the AIM dimer, and two preparations of normal human A-I complexed in different formulations. Different PL components have been also the object of evaluation in reconstituted (r) products. The most frequently selected PL components in rHDL in clinical/animal trials has been either dimyristoylphosphatidylcholine (DMPC) or 1-palmitoyl-2-oleylphosphocholine (POPC). More recently also addition of PS or Sph have been tested. POPC appears to be easier to handle, being of moderate fluidity, quite stable and of lower cost, thus being mainly used in clinical studies, e.g. in the case of AIMilano trials [41,71]. Recently, SM as the PL component appeared to have some benefit in terms of anti-inflammatory properties [72]. Finally, enrichment of HDL particle lipidome with PS appears to display more cardioprotective properties vs those without PS both in vitro and in mice [73].
Figure 2.
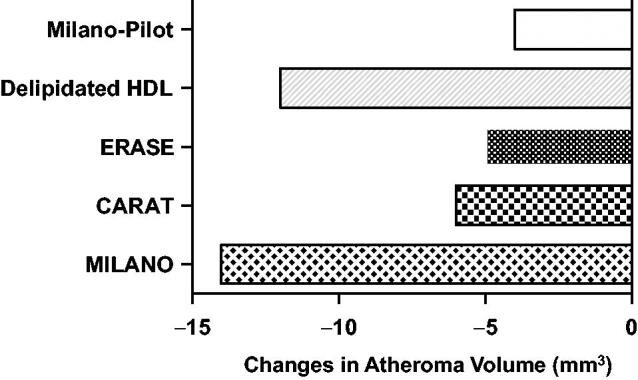
Intravascular ultrasound studies of HDL (high-density lipoprotein) mimetics. From bottom to top: change in atheroma volume infusing HDL mimetics containing apoA-I (apolipoprotein A-I) Milano in 2003 (MILANO), wild-type apoA-I and sphingomyelin (CARAT), wild-type apoA-I (ERASE), autologous delipidated HDL (Delipidated HDL), and apo A-I Milano in 2016 (MILANOPILOT) [75].
Infusions of high dose blood-derived reconstituted HDL (CSL-111) given as 40 or 80 mg/kg weekly for 4 weeks (ERASE Study) resulted in a small, not statistically significant, reduction in coronary atheroma volume compared with placebo, but a significant change in coronary score [74,75]. The 80 mg/kg proved to have liver side effects, not allowing completion of the study. Availability of this newer product provided an interesting opportunity to evaluate the direct effect of infusions in patients with femoral atherosclerotic disease. A single rHDL infusion reduced plaque lipid content, macrophage size and inflammatory mediators such as VCAM-1 [76]. A refined product (CSL-112) proved to be safe and highly efficacious in promoting ABCA1-mediated cholesterol efflux in the AEGIS-1 trial [77], not associated, however, with a promotion of atheroma regression by IVUS [78].
Since CSL-112 appears to enhance cholesterol efflux similarly in healthy individuals and stable atherosclerotic patients [79], CSL-112 is being evaluated in the AEGIS II study, a randomised placebo-controlled study evaluating a single infusion of 6 g of apo AI in 17,000 patients with myocardial infarction (MI). It has been noted that this dose may be relatively small compared with the total plasma apo AI pool and it also possible that a late enrolment of patients (7 days after MI) may not be suitable to mitigate the injury of ischemia/reperfusion [11].
The second product, a sphingosine-enriched HDL preparation from Cerenis (CER-001) was evaluated with a similar protocol in patients with ACS (CHI-SQUARE study) given as 6 weekly infusions (3 mg/kg, 6 mg/kg or 12 mg/kg of CER-001) vs placebo. There was no reduction in coronary atherosclerosis on IVUS or quantitative coronary angiography (QCA) and no difference in major CV events at each dose tested [80], but a following analysis suggested efficacy of the lowest CER-001 dose [81]. However, in a subsequent clinical trial, 10 infusions of 3 mg/kg CER-001 in addition to statins were not able to replicate this positive finding [78].
This same research group last attempted to repeat the original study on AIM (now MDCO-216) this time testing 120 patients randomised to receive either placebo (n = 60) or MDCO-216 (20 mg/kg; n = 52) for 5 weekly infusions. Differently from the original trial this study did not report any significant benefit in terms of regression or reduced progression in the examined IVUS images [82]. An accompanying Editorial [83] indicated that possibly the case of AIM might be different in acute vs stable coronary conditions, in this latter case indicating benefit (and also the object of a positive comment in the earlier study by the same author) [84]. The Author pointed out in addition that in at least one single study [85] on AIM gene-transfected mice, these animals did not efficiently mobilise macrophage cholesterol, whereas this occurred to a higher extent in wild type (WT) mice, thus concluding for a possibly smaller benefit for AIM in terms of direct effluxing capacity. Macrophages separated from normal mice vs gene transfected animals are, however, different: macrophages exposed to external AIM have a clearly higher capacity to mobilise cholesterol [86]. The interpretation by Rader is thus certainly not well founded, since an evaluation of AIM vs AI on cholesterol efflux in gene transfected mice is not comparable. As pointed out by other investigators [87] the lack of changes in lipoprotein profile in AIM treated patients appears to differ from that in the prior study by Nissen et al. [41]. In addition, hsCRP was raised by AIM in this last study [82], thus possibly indicating that some structural features may differ from the original apolipoprotein, as noted in earlier clinical reports [88].
Other approaches to the evaluation of vascular benefit of HDL therapy have relied on magnetic resonance imaging (MRI). By this method Hovingh et al. [89] tested the CER-001 product (8 mg/kg given for 12 biweekly infusions) by 3-TMRI scan of the carotids in 23 patients with genetically confirmed homozygosity or compound heterozygosity for LDL-R, apoB, PCSK9 or LDL RAP1 mutations. After CER-001 infusions apoA-I increased from a mean of 114.8 ± 20.7 mg/dL to 129.3 ± 23 mg/dL. Mean vessel wall area (primary endpoint) was reduced from 17.23 to 16.75 mm2 (p = 0.008) This study indicated that the HDL treatment regimen can reduce the vessel wall area of the carotids, indicative of a reduced plaque extent. Mean carotid wall thickness most specifically was reduced by approximately 2.5%.
Novel areas have been explored in order to take advantage of the properties of AIM in CV prevention or, better, atheroma treatment. One successful approach in an animal model has been that of vascular gene therapy. Gene therapy with viral vectors was earlier successfully achieved with the apoA-I gene in apoE-KO animals [90]. The hypothesis that gene therapy using AIM might be more efficacious than using recombinant AIM as a standard infusion in reducing atherosclerosis was initially tested by Wang et al [91] by bone marrow transplantation in female mice lacking both the apoE and apoA-I genes. Bone marrows from donor mice, transduced with a retroviral vector expressing wild-type (WT) apoA-I or AIM, were transplanted into double KO females that were fed a high-cholesterol diet and sacrificed after 24 weeks from transplantation. Wild-type A-I gene therapy reduced aortic atherosclerosis by 25%, a result which is inferior compared with that obtained with apoAIM (–65%). Interestingly there were no differences in circulating cholesterol levels between the two animal groups.
More recently, the same authors investigated a more classical gene therapy approach by using the recombinant adeno-associated virus (rAAV)8 vector for apoAIM in apoA-I/apoE double KO mice after a high cholesterol diet for 20 weeks. The animals were placed on a low-cholesterol diet and injected with empty rAAV (controls) or maintained on the same low-cholesterol diet and iv injected, once, with the rAAV8 vector expressing AIM. At the 40-week endpoint, rAAV8 AIM recipients showed significant regression of atherosclerosis compared to the mice euthanized after the 20 weeks of high cholesterol diet (i.e. before starting treatment) as well as to those animals receiving the empty vector [92]. These data show that whereas dietary mediated cholesterol lowering may halt atherosclerosis progression, it does not induce regression, elicited instead by AAV8 mediated apoAIM gene therapy.
Another approach to gene therapy may be that of transducing arterial endothelial cells with the helper dependent adenoviral (HdAd) vector expressing apoA-I [93]. High fat fed rabbits underwent bilateral carotid artery gene transfer, one artery receiving a control vector (Hd Null) and the other receiving an apoA-I expressing vector. After 24 weeks on a high fat diet, HdAd apoA-I treated arteries had 30% less intima media lesion volume (p = 0.03) with concomitant reduction in intimal macrophage and muscle cell content (−23% and −32% respectively). Treated arteries had also decreased intimal inflammatory markers such as VCAM-I, ICAM-1, MCP-1 and TNF-α. Thus, local vascular gene therapy may offer great benefit, in reducing atherosclerotic lesion growth and intima inflammation. More recently, the same Authors [94] reported that with concurrent ABCA1 overexpression a raised cholesterol efflux capacity and further reduced inflammation can be elicited.
A total new approach is that of exploiting a possible transport system in the intestine, capable of delivering AIM from the oral route. This was attempted by Romano et al. [95] by using genetically modified rice plants. Engineered plasmids were introduced into Agrobacterioum tumefaciens by electroporation, allowing transformation of Oriza sativa SSP Japonica Rosa Marchetti. Total genomic DNA, isolated from leaves of putative transgenic rice plants clearly showed successful genetic modification. Expression was found in pulps and seeds, then processed to “rice milk”, it was minimal in leaves, stems and roots of the transgenic rice. Features of the transgenic “rice milk” are indicated in a specific patent (n° PCT/IB2006/054948). The transfected AIM protein was not degraded and, interestingly, it was detected primarily in the dimeric form, the one evaluated in experimental and clinical trials for efficacy in atherosclerosis regression.
The possible anti-inflammatory activity of the “rice milk” containing AIM was tested in oxLDL-challenged THP1 macrophages in vitro. In this model AIM rice milk could prevent MCP1 production, the decrease being proportional to AIM concentrations, also reducing foam cell formation. Additionally, exposure to 0.1 or 0.5 μg/mL of AIM significantly raised cholesterol efflux from cells. Finally, “rice milk” was well tolerated, allowing to evaluate its activity on apoE–/– mice fed a Western diet for 8 weeks. In the following 3 weeks mice were maintained on the same diet and treated by gavage with 10 mL/kg “rice milk” for 5 days a week. AIM rice milk treatment caused a markedly reduced extent of atherosclerotic plaques i.e. about 50%, compared to those of mice receiving wild-type rice milk. Significant reductions occurred both at the aortic arch and at the aortic sinus levels (Figure 3).
Figure 3.

En face pictures of aortic arches from Apo E–/– mice on a Western diet, treated with AIM rice milk vs mice fed wild type rice milk for 3 weeks (10 ml/kg by gavage, 5 days a week). AIM rice milk caused a markedly reduced extent of atherosclerotic plaques, i.e. about 50%, compared to mice receiving normal rice milk [95].
These findings are certainly quite provocative. ApoA-I mimetic peptides are absorbed at the small intestinal level when orally delivered, to a very small extent [96]. The presence of HDL cell transporters such ABCA1 in the intestine may not be responsible for an improved uptake. Since HDL biogenesis receives a significant contribution from the intestine [97], these hypothetical mechanisms will need to be evaluated in appropriate animal and possibly human studies, where availability of this type of milk may lead to a very simple approach to HDL therapy.
“HDL therapy” may provide a very effective tool to reduce arterial lesions in classical models of animal atherosclerosis. Clinical results are less clear, some early very positive findings having been not fully duplicated. Reasons for the recent failures are difficult to define, but do certainly encourage further work in this area. Very recently, in addition, clear differences have been found in mouse models of early- versus late-stage atherosclerosis [98]. In apoE–/– mice on a high-fat diet for 8 or 34 weeks, i.e. with early- and late-stage disease, infusions of human apoA-I resulted in clearly different outcomes. ApoA-I infusions had minimal effects on atherosclerotic plaque sizes and composition in mice with late-stage disease, whereas early stage-atheromas were markedly reduced by treatment: besides a 30.2% reduction in plaque area, a 51.2% reduction in macrophage content and increased plaque SMC content were detected. The Authors provide as an explanation both a reduced cholesterol effluxing capacity of apo B depleted plasma from late-stage mice and lower anti-apoptotic and anti-inflammatory activities. It thus appears that both the anti-atheromatous and cellular effects of HDL infusions are reduced in late-stage vs early-stage disease and this may guide future clinical studies.
HDL and stent biocompatibility
The wide use of stenting during percutaneous coronary interventions (PCIs) has resulted in a reduced mortality rate from CVD [99]. Improvements in stent biocompatibility and reduced incidence of restenosis have been provided by novel generations of stents, incorporating antiproliferative drugs or new designs [100]. To achieve further improvements it would be desirable to dispose of an agent suppressing SMC proliferation and neointimal hyperplasia [101], reducing platelet activation and thrombus formation [102], improving endothelial repair [103] and inhibiting monocyte recruitment [104]. All these are properties of HDL and a number of reports have indicated that HDL-therapy may improve stent biocompatibility.
Epidemiological data, on the other hand, have indicated that patients with higher HDL-C have improved stent patency at one year [105] and alternatively, an LDL to HDL cholesterol ratio below 1.5 leads to a lower frequency of non-target lesion interventions after PCI [106].
Extensive studies in animal models have provided fundamental information on the potential properties of HDL-therapy in preventing stent failure. Among the most significant targeted mechanisms for improving stent patency has been the prevention of damage to the endothelial cell layer, initiating a cascade of proinflammatory events. Adenoviral overexpression of apoA-I reduces neointimal formation after carotid artery wire injury [107] and similar positive effects were found after vein grafting [108]. Relative to neointimal hyperplasia (NIH), based on in vivo studies with infusions of recombinant AIM complexed with phospholipids, in a murine model of stent, alternate day infusions of HDL similarly reduced in stent neointimal area after stent deployment [109]. A similar effect was achieved by intramural delivery of an AIM phospholipid complex in stented porcine coronary arteries [110].
Studies on the mechanisms of the inflammatory response that cause NIH after vascular injury have highlighted the involvement of chemokines in promoting SMC proliferation and migration. Incubations of SMCs have in fact shown that a range of chemokines can all increase SMC proliferation [111]. Evaluation of the influence of HDL on this mechanism found that preincubation of SMCs with rHDL (apoA-I plus phosphatidylcholine) significantly reduces SMC proliferation as well as the expression of chemokines promoting proliferation [112]. By siRNA knock down of the scavenger receptor SR-B1, this was found to be crucial in the mediation of these effects [101]. In order to achieve local inhibition of proliferation, apoA-I immobilised on a stainless steel surface similar to a stent surface has also allowed effective antagonism to the proliferation of attached SMCs [113].
The mechanism of protection of endothelial cells (EC) and of promotion of endothelial repair is attributed to increased NO endothelial synthase (eNOS) [114] and prevention of apoptosis [115]. Increased eNOS by HDL can inhibit leukocyte adhesion, modulate vascular dilatation and regulate local cell growth, reduce SMC proliferation and inhibit platelet aggregation, thus further contributing to stent biocompatibility [116]. An additional contribution to re-endothelialization by HDL is provided by raised endothelial progenitor cell (EPC) number [117] effectively regulating NIH cell growth [118]. Mechanisms leading to re-endothelialization are thus a crucial factor in HDL mediated improvement in stent biocompatibility and more prolonged patency. Increased endothelial cells can further reduce focal inflammation and in particular SMC proliferation.
While antagonism to inadequate re-endothelialization might be of benefit in preventing the risk of late and very late stent thrombosis, endothelial coverage may be also the best predictor of late stent thrombosis in patients implanted with drug eluting stents [119]. Dual anti-platelet therapy (DAPT) has resulted in improved prevention of myocardial infarction or repeated coronary interventions compared to single regimens [120]. The second antiplatelet agent added to aspirin has been initially clopidrogel, followed by prasugrel and ticagrelor. The optimal duration of DATP has been prolonged over time and now many cardiologists give treatments longer than the standard duration of 12 months [121]. Still, availability of an agent such as rHDL, reducing aggregation and improving endothelialization, is rated by many of value towards improving stent bioacompatibility, reducing thrombosis and maintaining patency. Immobilisation of apoA-I rHDL on stent surfaces can reduce thrombosis [113] and many studies have reported antithrombotic effects of HDLs and AIM [122]. A single rHDL infusion can reduce platelet activity and platelet aggregation ex-vivo by >50% in diabetics [123]. Diabetics with elevated HDL-C (≥40 mg/dL) have a 12% lower risk of developing stent thrombosis compared to similar patients with lower HDL-C (≤40 mg/dL) [124].
HDL therapy has a definite potential to reduce the risk of stent occlusion particularly due to thrombosis. Since HDL therapy may have efficacy on atherosclerotic plaque formation, applicability after stent insertion is most reasonable. In stents, neoatherosclerosis is an important contributing factor to loss of patency, particularly after drug eluting stents. These novel lesions do not differ from native atherosclerosis containing macrophage foam cells, areas of calcification and necrotic cores. They occur, however, after a relatively short time versus classical lesions. The pleiotropic effects of HDL including antagonism to LDL oxidation [125], reduction in cell adhesion molecules [126] and suppression of chemokine expression [127] are all factors potentially responsible for reducing neoatherosclerosis.
As yet only anectodal, reports have indicated benefit of HDL infusions in the course of PTCA [101]. Interest in this area is high and may lead to further technological developments allowing local provision of HDL in the course of new stent positioning.
HDL therapy for heart failure
Heart failure is probably the most significant vascular epidemics of the twenty-first century [128]. Although a number of new drug developments have brought hope in the management of this disease, e.g. combinations of angiotensin II/natriuretic peptide cleavage antagonists [129], still therapeutic approaches are mainly symptom oriented, without a clear impact on the basic disease. Among the numerous potential determinants of the increased heart failure (HF) risk, an independent association between decreased HDL-C levels and HF incidence was reported in the Framingham Heart Study [130]. Further, reduced HDL-C and apoA-I levels are independent predictors of an unfavourable evolution of HF in patients with CHD [131].
The pleiotropic properties of HDL may be certainly of interest on the potential improving effect on myocardial function [20]. HDL, in particular has been shown to down-regulate the angiotensin II type 1 (AT1) receptor [132] and by this mechanism it can inhibit AT1 induced cardiac hypertrophy [133]. In isolated cardiomyocytes, HDL reduces biochemical stress induced autophagy and hypertrophy. Cardiac hypertrophy was antagonised in vivo by the continuous infusion of HDL, possibly mediated by the down-regulation of the AT1 receptor [134].
While there are no clinical studies directly addressing the effects of apoA-I or AIM on clinical HF, two studies have examined two different aspects of HDL therapy. The first [135] employed gene therapy. The selective HDL raising AAV8 A-I gene transfer was performed at 12 weeks of age in male LDLr–/– mice. In order to obtain pressure overload, two weeks later, mice were treated by transverse aortic constriction (TAC) or sham operation. Gene therapy led to a rise of HDL-cholesterol of 1.47 fold and 1.45 fold in TAC operated and sham treated mice, respectively. A significantly lower mortality was noted in the AAV8 AI TAC mice compared to controls (HR for mortality of 0.543; 95% CI: 0.282–1.05). Heart weights, and in particular atrial weights, were significantly reduced in the AAV8 A-I TAC mice. A significant reduction of lung weights were found and, upon microscopic evaluation, there was clear indication of reduced apoptosis (–46.7%) in AAV8 AI TAC mice vs controls and marked reduction in the nitrotyrosine positive areas. Capillary density and relative vascularity were higher in gene transfected mice with a prompt decrease of interstitial and perivascular fibrosis. Morphological changes were accompanied by significantly improved diastolic function with lower end-diastolic pressure.
While these findings were related to an animal model preceding fully established HF, in a follow up study the same investigators used a more targeted approach, i.e. that of infusing recombinant AIM phospholipid particles in mice with established HF [136]. Mice underwent TAC or sham operations at 14 weeks of age, and 8 weeks later were randomised to HDL therapy (5 i.p. injections of recombinant HDLMilano 100 mg/kg or an equivalent volume of control buffer) at 48 h intervals starting at day 56. Endpoint analyses were performed at day 65. There was clear evidence of an improved clinical picture of HF with reduced lung weights in AIM treated mice (–25.3%), lower tissue fibrosis and increased relative vascularity compared to control TAC mice. The peak rate of isovolumetric relaxation in AIM treated TAC mice were 30.4% higher vs reference TAC mice. The significant improvement of diastolic function and cardiac metabolism clearly indicate the clinical potential in the treatment of HF, potentially linked to improved cardiac flow and possibly reduced TGF-β1 induced collagen deposition [137]. Improvement of diastolic function is consistent with previous observations on the effect of gene therapy with an E1E3E4-deleted human apoA-I vector in LDL-receptor deficient [138] and diabetic mice [139].
Conclusions
HDL therapy can address a potentially wide range of targets, both in clinical atherosclerosis and neighbouring fields. The use of isolated HDL per se has shown significant vascular benefit as early as in the ’90s and these findings have been repeatedly confirmed particularly in the rabbit model. Studies in rabbits with focal atheromas had an indirect confirmation in intravascular ultrasound studies in men after repeated infusions of HDL containing the mutant AIM. While these findings were not apparently confirmed in recent years, doubt remains on the clinical conditions of treated patients and, more so, on some apparent structural differences between the originally studied AIM and the newer developed preparation. A potential breakthrough in the use of HDL therapy may be the recently described oral administration of engineered rice milk enriched with recombinant AIM. This will, of course, need to be properly tested in humans also providing convincing data on absorption. Cholesterol efflux as evaluated in vitro after exposure to AI differs from the case where, e.g. AIM is injected into the circulation. Cholesterol efflux is the crucial mechanism in the HDL protective mechanism and there is evidence that HDL-mediated cholesterol efflux capacity (CEC) is impaired in STEMI patient [140]. CEC is reduced also in different HDL subspecies in coronary patients. A large study of 1,609 MI patients showed that CEC in the acute phase of the event is inversely associated with all-cause mortality evaluated after a median follow up of 1.9 years (interquartile range: 1.5–4.2 years) regardless of HDL-C levels [141].
Newer, possibly even more exciting uses of HDL therapy are the improvement of stent biocompatibility and the treatment of heart failure (HF). In the first case, a number of reports have indicated that HDL, either infused or inserted by gene transfer into the arterial wall, may reduce atherosclerosis also by inhibiting neoatheroma formation, platelet aggregation and thrombogenicity. At present only anecdotical reports have been provided on the use of HDL in the course of coronary procedures, but the field is of extreme clinical interest.
Finally, treatment of experimental HF with HDL therapy, more recently with HDL enriched with recombinant AIM, has provided data indicative of improved cardiac function associated with reduced lung weights, interstitial fibrosis and relative vascularity. The isovolumetric relaxation were over 30% higher in HDL-AIM treated mice, thus suggesting that recombinant HDL may emerge as a novel treatment modality for HF.
Overall, the availability of HDL therapy, be it with wild type apo AI or probably better with recombinant AIM, opens up an area of potential extreme value in CV therapy, by improving life quality and expectation in coronary and non coronary patients.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Madsen CM, Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J. 2017;38(32):2478–2486. [DOI] [PubMed] [Google Scholar]
- 2.Monette JS, Hutchins PM, Ronsein GE, et al. . Patients with coronary endothelial dysfunction have impaired cholesterol efflux capacity and reduced HDL particle concentration. Circ Res. 2016;119(1):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allard-Ratick MP, Kindya BR, Khambhati J, et al. . HDL: fact, fiction, or function? HDL cholesterol and cardiovascular risk. Eur J Prev Cardiolog. 2019;123(10):1736–1737. [DOI] [PubMed] [Google Scholar]
- 4.Voight BF, Peloso GM, Orho-Melander M, et al. . Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian Randomisation Study. Lancet. 2012;380(9841):572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geller AS, Polisecki EY, Diffenderfer MR, et al. . Genetic and secondary causes of severe HDL deficiency and cardiovascular disease. J Lipid Res. 2018;59(12):2421–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol. 2003;23(10):1724–1731. [DOI] [PubMed] [Google Scholar]
- 7.Manzini S, Pinna C, Busnelli M, et al. . Beta2-adrenergic activity modulates vascular tone regulation in lecithin: cholesterol acyltransferase knockout mice. Vascul Pharmacol. 2015;74:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ossoli A, Simonelli S, Varrenti M, et al. . Recombinant LCAT (Lecithin:Cholesterol Acyltransferase) rescues defective HDL (High-Density Lipoprotein)-mediated endothelial protection in acute coronary syndrome. Arterioscler Thromb Vasc Biol. 2019;39(5):915–924. [DOI] [PubMed] [Google Scholar]
- 9.Kontush A, Lhomme M, Chapman MJ. Unraveling the complexities of the HDL lipidome. J Lipid Res. 2013;54(11):2950–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sattler KJ, Elbasan S, Keul P, et al. . Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol. 2010;105(6):821–832. [DOI] [PubMed] [Google Scholar]
- 11.Sposito AC, de Lima-Junior JC, Moura FA, et al. . Reciprocal multifaceted interaction between HDL (High-Density Lipoprotein) and myocardial infarction. Arterioscler Thromb Vasc Biol. 2019;39(8):1550–1564. [DOI] [PubMed] [Google Scholar]
- 12.Boengler K, Hilfiker-Kleiner D, Heusch G, et al. . Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105(6):771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duarte FV, Palmeira CM, Rolo AP. The role of microRNAs in mitochondria: small players acting wide. Genes. 2014;5(4):865–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White CR, Giordano S, Anantharamaiah GM. High-density lipoprotein, mitochondrial dysfunction and cell survival mechanisms. Chem Phys Lipids. 2016;199:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elsoe S, Ahnstrom J, Christoffersen C, et al. . Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221(1):91–97. [DOI] [PubMed] [Google Scholar]
- 16.Brites F, Martin M, Guillas I, et al. . Antioxidative activity of high-density lipoprotein (HDL): mechanistic insights into potential clinical benefit. BBA Clin. 2017;8:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolle M, Klockl L, Wiedon A, et al. . Regulation of endothelial nitric oxide synthase activation in endothelial cells by S1P1 and S1P3. Biochem Biophys Res Commun. 2016;476(4):627–634. [DOI] [PubMed] [Google Scholar]
- 18.Galvani S, Sanson M, Blaho VA, et al. . HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. 2015;8(389):ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruscica M, Tokgözoğlu L, Corsini A, et al. . PCSK9 inhibition and inflammation: a narrative review. Atherosclerosis. 2019;288:146–155. [DOI] [PubMed] [Google Scholar]
- 20.Shah AS, Tan L, Long JL, et al. . Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. 2013;54(10):2575–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riwanto M, Landmesser U. High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J Lipid Res. 2013;54(12):3227–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furtado JD, Yamamoto R, Melchior JT, et al. . Distinct proteomic signatures in 16 HDL (High-Density Lipoprotein) subspecies. Arterioscler Thromb Vasc Biol. 2018;38(12):2827–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morton AM, Furtado JD, Mendivil CO, et al. . Dietary unsaturated fat increases HDL metabolic pathways involving apoE favorable to reverse cholesterol transport. JCI Insight. 2019;4(7):pii:124620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. 2012;111(8):1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nofer JR, Kehrel B, Fobker M, et al. . HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis. 2002;161(1):1–16. [DOI] [PubMed] [Google Scholar]
- 26.Calder PC Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans. 2017;45(5):1105–1115. [DOI] [PubMed] [Google Scholar]
- 27.Salic K, Morrison MC, Verschuren L, et al. . Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis. 2016;250:158–165. [DOI] [PubMed] [Google Scholar]
- 28.Oravec S, Demuth K, Myara I, et al. . The effect of high density lipoprotein subfractions on endothelial eicosanoid secretion. Thromb Res. 1998;92(2):65–71. [DOI] [PubMed] [Google Scholar]
- 29.Vilahur G, Gutierrez M, Casani L, et al. . Hypercholesterolemia abolishes high-density lipoprotein-related cardioprotective effects in the setting of myocardial infarction. J Am Coll Cardiol. 2015;66(21):2469–2470. [DOI] [PubMed] [Google Scholar]
- 30.Hunter WG, McGarrah RW 3rd, Kelly JP, et al. . High-density lipoprotein particle subfractions in heart failure with preserved or reduced ejection fraction. J Am Coll Cardiol. 2019;73(2):177–186. [DOI] [PubMed] [Google Scholar]
- 31.Barter P, Genest J. HDL cholesterol and ASCVD risk stratification: a debate. Atherosclerosis. 2019;283:7–12. [DOI] [PubMed] [Google Scholar]
- 32.Riaz H, Khan SU, Rahman H, et al. . Effects of high-density lipoprotein targeting treatments on cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiolog. 2019;26(5):533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pirillo A, Catapano AL. Increasing high-density lipoprotein cholesterol levels for cardiovascular benefit: the end of a dream? Eur J Prev Cardiolog. 2019;26(5):531–532. [DOI] [PubMed] [Google Scholar]
- 34.Group AS, Ginsberg HN, Elam MB, et al. . Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362(17):1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Investigators A-H, Boden WE, Probstfield JL, et al. . Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. [DOI] [PubMed] [Google Scholar]
- 36.Goldfine AB, Kaul S, Hiatt WR. Fibrates in the treatment of dyslipidemias–time for a reassessment. N Engl J Med. 2011;365(6):481–484. [DOI] [PubMed] [Google Scholar]
- 37.Elam MB, Ginsberg HN, Lovato LC, et al. . Association of fenofibrate therapy with long-term cardiovascular risk in statin-treated patients with type 2 diabetes. JAMA Cardiol. 2017;2(4):370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferri N, Corsini A, Sirtori CR, et al. . Present therapeutic role of cholesteryl ester transfer protein inhibitors. Pharmacol Res. 2018;128:29–41. [DOI] [PubMed] [Google Scholar]
- 39.Johns DG, LeVoci L, Krsmanovic M, et al. . Characterization of anacetrapib distribution into the lipid droplet of adipose tissue in mice and human cultured adipocytes. Drug Metab Dispos. 2019;47(3):227–233. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita S, Ruscica M, Macchi C, et al. . Cholesteryl ester transfer protein: an enigmatic pharmacology – antagonists and agonists. Atherosclerosis. 2018;278:286–298. [DOI] [PubMed] [Google Scholar]
- 41.Nissen SE, Tsunoda T, Tuzcu EM, et al. . Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290(17):2292–2300. [DOI] [PubMed] [Google Scholar]
- 42.Tall AR Plasma high density lipoproteins: therapeutic targeting and links to atherogenic inflammation. Atherosclerosis. 2018;276:39–43. [DOI] [PubMed] [Google Scholar]
- 43.Murphy AJ, Westerterp M, Yvan-Charvet L, et al. . Anti-atherogenic mechanisms of high density lipoprotein: effects on myeloid cells. Biochim Biophys Acta. 2012;1821(3):513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sirtori CR, Yamashita S, Francesca Greco M, et al. . Recent advances in synthetic pharmacotherapies for dyslipidaemias. Eur J Prev Cardiolog. 2019:2047487319845314. [DOI] [PubMed] [Google Scholar]
- 45.Francis GA, Heinecke JW. Advances in high density lipoprotein formation and metabolism: a tribute to John F. Oram (1945-2010). Biochim Biophys Acta. 2012;1821(3):343–344. [DOI] [PubMed] [Google Scholar]
- 46.Mollazadeh H, Atkin SL, Butler AE, et al. . The effect of statin therapy on endoplasmic reticulum stress. Pharmacol Res. 2018;137:150–158. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz K, Lawn RM, Wade DP. ABC1 gene expression and ApoA-I-mediated cholesterol efflux are regulated by LXR. Biochem Biophys Res Commun. 2000;274(3):794–802. [DOI] [PubMed] [Google Scholar]
- 48.Yvan-Charvet L, Welch C, Pagler TA, et al. . Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118(18):1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westerterp M, Murphy AJ, Wang M, et al. . Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013;112(11):1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madsen CM, Varbo A, Nordestgaard BG. Low HDL cholesterol and high risk of autoimmune disease: two population-based cohort studies including 117341 individuals. Clin Chem. 2019;65(5):644–652. [DOI] [PubMed] [Google Scholar]
- 51.Catapano AL, Pirillo A, Bonacina F, et al. . HDL in innate and adaptive immunity. Cardiovasc Res. 2014;103(3):372–383. [DOI] [PubMed] [Google Scholar]
- 52.Westerterp M, Fotakis P, Ouimet M, et al. . Cholesterol efflux pathways suppress inflammasome activation, netosis and atherogenesis. Circulation. 2018;138(9):898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy AJ, Dragoljevic D, Tall AR. Cholesterol efflux pathways regulate myelopoiesis: a potential link to altered macrophage function in atherosclerosis. Front Immunol. 2014;5:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paramel Varghese G, Folkersen L, Strawbridge RJ, et al. . NLRP3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc. 2016;5(5):pii:e003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duewell P, Latz E. Assessment and quantification of crystal-induced lysosomal damage. Methods Mol Biol. 2013;1040:19–27. [DOI] [PubMed] [Google Scholar]
- 56.Sano S, Oshima K, Wang Y, et al. . Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the il-1beta/nlrp3 inflammasome. J Am Coll Cardiol. 2018;71(8):875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dijk W, Kersten S. Regulation of lipid metabolism by angiopoietin-like proteins. Curr Opin Lipidol. 2016;27(3):249–256. [DOI] [PubMed] [Google Scholar]
- 58.Oteng AB, Ruppert PMM, Boutens L, et al. . Characterization of ANGPTL4 function in macrophages and adipocytes using Angptl4-knockout and Angptl4-hypomorphic mice. J Lipid Res. 2019;60(10):1741–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Theofilatos D, Fotakis P, Valanti E, et al. . HDL-apoA-I induces the expression of angiopoietin like 4 (ANGPTL4) in endothelial cells via a PI3K/AKT/FOXO1 signaling pathway. Metabolism. 2018;87:36–47. [DOI] [PubMed] [Google Scholar]
- 60.Dewey FE, Gusarova V, O’Dushlaine C, et al. . Inactivating variants in angptl4 and risk of coronary artery disease. N Engl J Med. 2016;374(12):1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Badimon JJ, Badimon L, Galvez A, et al. . High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab Invest. 1989;60(3):455–461. [PubMed] [Google Scholar]
- 62.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85(4):1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinals M, Martinez-Gonzalez J, Badimon JJ, et al. . HDL-induced prostacyclin release in smooth muscle cells is dependent on cyclooxygenase-2 (Cox-2). Arterioscler Thromb Vasc Biol. 1997;17(12):3481–3488. [DOI] [PubMed] [Google Scholar]
- 64.Hausmann D, Johnson JA, Sudhir K, et al. . Angiographically silent atherosclerosis detected by intravascular ultrasound in patients with familial hypercholesterolemia and familial combined hyperlipidemia: correlation with high density lipoproteins. J Am Coll Cardiol. 1996;27(7):1562–1570. [DOI] [PubMed] [Google Scholar]
- 65.Franceschini G, Sirtori CR, Capurso A 2nd, et al. . A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980;66(5):892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roma P, Gregg RE, Meng MS, et al. . In vivo metabolism of a mutant form of apolipoprotein A-I, apo A-IMilano, associated with familial hypoalphalipoproteinemia. J Clin Invest. 1993;91(4):1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soma MR, Donetti E, Parolini C, Sirtori CR, Fumagalli R, Franceschini G. Recombinant apolipoprotein A-IMilano dimer inhibits carotid intimal thickening induced by perivascular manipulation in rabbits. Circ Res. 1995;76(3):405–411. [DOI] [PubMed] [Google Scholar]
- 68.Chiesa G, Di Mario C, Colombo N, et al. . Development of a lipid-rich, soft plaque in rabbits, monitored by histology and intravascular ultrasound. Atherosclerosis. 2001;156(2):277–287. [DOI] [PubMed] [Google Scholar]
- 69.Chiesa G, Monteggia E, Marchesi M, et al. . Recombinant apolipoprotein A-I(Milano) infusion into rabbit carotid artery rapidly removes lipid from fatty streaks. Circ Res. 2002;90(9):974–980. [DOI] [PubMed] [Google Scholar]
- 70.Giannarelli C, Cimmino G, Ibanez B, et al. . Acute ApoA-I Milano administration induces plaque regression and stabilisation in the long term. Thromb Haemost. 2012;108(6):1246–1248. [DOI] [PubMed] [Google Scholar]
- 71.Kallend DG, Reijers JA, Bellibas SE, et al. . A single infusion of MDCO-216 (ApoA-1 Milano/POPC) increases ABCA1-mediated cholesterol efflux and pre-beta 1 HDL in healthy volunteers and patients with stable coronary artery disease. Eur Heart J Cardiovasc Pharmacother. 2016;2(1):23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwendeman A, Sviridov DO, Yuan W, et al. . The effect of phospholipid composition of reconstituted HDL on its cholesterol efflux and anti-inflammatory properties. J Lipid Res. 2015;56(9):1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Darabi M, Kontush A. Can phosphatidylserine enhance atheroprotective activities of high-density lipoprotein?. Biochimie. 2016;120:81–86. [DOI] [PubMed] [Google Scholar]
- 74.Tardif JC, Gregoire J, L’Allier PL, et al. . Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297(15):1675–1682. [DOI] [PubMed] [Google Scholar]
- 75.Di Bartolo BA, Psaltis PJ, Bursill CA, et al. . Translating evidence of HDL and plaque regression. Arterioscler Thromb Vasc Biol. 2018;38(9):1961–1968. [DOI] [PubMed] [Google Scholar]
- 76.Shaw JA, Bobik A, Murphy A, et al. . Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103(10):1084–1091. [DOI] [PubMed] [Google Scholar]
- 77.Michael Gibson C, Korjian S, Tricoci P, et al. . Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I, after acute myocardial infarction: the AEGIS-I Trial (ApoA-I event reducing in ischemic syndromes I). Circulation. 2016;134(24):1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nicholls SJ, Andrews J, Kastelein JJP, et al. . Effect of serial infusions of CER-001, a Pre-beta high-density lipoprotein mimetic, on coronary atherosclerosis in Patients following acute coronary syndromes in the CER-001 atherosclerosis regression acute coronary syndrome trial: a randomized clinical trial. JAMA Cardiol. 2018;3(9):815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gille A, D’Andrea D, Tortorici MA, et al. . CSL112 (Apolipoprotein A-I [Human]) enhances cholesterol efflux similarly in healthy individuals and stable atherosclerotic disease patients. Arterioscler Thromb Vasc Biol. 2018;38(4):953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tardif JC, Ballantyne CM, Barter P, et al. . Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J. 2014;35(46):3277–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kataoka Y, Andrews J, Duong M, et al. . Regression of coronary atherosclerosis with infusions of the high-density lipoprotein mimetic CER-001 in patients with more extensive plaque burden. Cardiovasc Diagn Ther. 2017;7(3):252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nicholls SJ, Puri R, Ballantyne CM, et al. . Effect of infusion of high-density lipoprotein mimetic containing recombinant apolipoprotein a-i milano on coronary disease in patients with an acute coronary syndrome in the MILANO-PILOT trial: a randomized clinical trial. JAMA Cardiol. 2018;3(9):806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rader DJ Apolipoprotein A-I infusion therapies for coronary disease: two outs in the ninth inning and swinging for the fences. JAMA Cardiol. 2018;3(9):799–801. [DOI] [PubMed] [Google Scholar]
- 84.Rader DJ High-density lipoproteins as an emerging therapeutic target for atherosclerosis. JAMA. 2003;290(17):2322–2324. [DOI] [PubMed] [Google Scholar]
- 85.Alexander ET, Weibel GL, Joshi MR, et al. . Macrophage reverse cholesterol transport in mice expressing ApoA-I milano. Arterioscler Thromb Vasc Biol. 2009;29(10):1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Franceschini G, Calabresi L, Chiesa G, et al. . Increased cholesterol efflux potential of sera from ApoA-IMilano carriers and transgenic mice. Arterioscler Thromb Vasc Biol. 1999;19(5):1257–1262. [DOI] [PubMed] [Google Scholar]
- 87.Garcia-Ropero A, Santos-Gallego CG, Badimon JJ. High-density lipoprotein-targeted therapies-not dead yet. JAMA Cardiol. 2018;3(12):1254. [DOI] [PubMed] [Google Scholar]
- 88.Reijers JAA, Kallend DG, Malone KE, et al. . MDCO-216 does not induce adverse immunostimulation, in contrast to its predecessor etc-216. Cardiovasc Drugs Ther. 2017;31(4):381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hovingh GK, Smits LP, Stefanutti C, et al. . The effect of an apolipoprotein A-I-containing high-density lipoprotein-mimetic particle (CER-001) on carotid artery wall thickness in patients with homozygous familial hypercholesterolemia: the Modifying Orphan Disease Evaluation (MODE) study. Am Heart J. 2015;169(5):736–742 e1. [DOI] [PubMed] [Google Scholar]
- 90.Cimmino G, Giannarelli C, Chen W, et al. . Adeno-associated virus serotype 8 ApoA-I gene transfer reduces progression of atherosclerosis in ApoE-KO mice: comparison of intramuscular and intravenous administration. J Cardiovasc Pharmacol. 2011;57(3):325–333. [DOI] [PubMed] [Google Scholar]
- 91.Wang L, Sharifi BG, Pan T, et al. . Bone marrow transplantation shows superior atheroprotective effects of gene therapy with apolipoprotein A-I Milano compared with wild-type apolipoprotein A-I in hyperlipidemic mice. J Am Coll Cardiol. 2006;48(7):1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang L, Tian F, Arias A, et al. . Comparative effects of diet-induced lipid lowering versus lipid lowering along with apo A-I Milano Gene therapy on regression of atherosclerosis. J Cardiovasc Pharmacol Ther. 2016;21(3):320–328. [DOI] [PubMed] [Google Scholar]
- 93.Wacker BK, Dronadula N, Bi L, et al. . Apo A-I (Apolipoprotein A-I) vascular gene therapy provides durable protection against atherosclerosis in hyperlipidemic rabbits. Arterioscler Thromb Vasc Biol. 2018;38(1):206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stamatikos A, Dronadula N, Ng P, et al. . ABCA1 overexpression in endothelial cells in vitro enhances ApoAI-mediated cholesterol efflux and decreases inflammation. Hum Gene Ther. 2019;30(2):236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Romano G, Reggi S, Kutryb-Zajac B, et al. . APOA-1Milano muteins, orally delivered via genetically modified rice, show anti-atherogenic and anti-inflammatory properties in vitro and in Apoe(–/–) atherosclerotic mice. Int J Cardiol. 2018;271:233–239. [DOI] [PubMed] [Google Scholar]
- 96.Chattopadhyay A, Navab M, Hough G, et al. . A novel approach to oral apoA-I mimetic therapy. J Lipid Res. 2013;54(4):995–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brunham LR, Kruit JK, Iqbal J, et al. . Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116(4):1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morton J, Bao S, Vanags LZ, et al. . Strikingly different atheroprotective effects of apolipoprotein A-I in early- versus late-stage atherosclerosis. JACC Basic Transl Sci. 2018;3(2):187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hunink MG, Goldman L, Tosteson AN, et al. . The recent decline in mortality from coronary heart disease, 1980–1990. The effect of secular trends in risk factors and treatment. JAMA. 1997;277(7):535–542. [PubMed] [Google Scholar]
- 100.Bonaa KH, Mannsverk J, Wiseth R, et al. . Drug-eluting or bare-metal stents for coronary artery disease. N Engl J Med. 2016;375(13):1242–1252. [DOI] [PubMed] [Google Scholar]
- 101.Vanags LZ, Tan JTM, Galougahi KK, et al. . Apolipoprotein A-I reduces in-stent restenosis and platelet activation and alters neointimal cellular phenotype. JACC Basic Transl Sci. 2018;3(2):200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lerch PG, Spycher MO, Doran JE. Reconstituted high density lipoprotein (rHDL) modulates platelet activity in vitro and ex vivo. Thromb Haemost. 1998;80(2):316–320. [PubMed] [Google Scholar]
- 103.Tso C, Martinic G, Fan WH, et al. . High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler Thromb Vasc Biol. 2006;26(5):1144–1149. [DOI] [PubMed] [Google Scholar]
- 104.Barter PJ, Nicholls S, Rye KA, et al. . Antiinflammatory properties of HDL. Circ Res. 2004;95(8):764–772. [DOI] [PubMed] [Google Scholar]
- 105.Topakian R, Sonnberger M, Nussbaumer K, et al. . Postprocedural high-density lipoprotein cholesterol predicts carotid stent patency at 1 year. Eur J Neurol. 2008;15(2):179–184. [DOI] [PubMed] [Google Scholar]
- 106.Fukuda Y, Miura S, Tsuchiya Y, et al. . Lower frequency of non-target lesion intervention in post-successful percutaneous coronary intervention patients with an LDL to HDL cholesterol ratio below 1.5. Int J Cardiol. 2011;149(1):120–122. [DOI] [PubMed] [Google Scholar]
- 107.De Geest B, Zhao Z, Collen D, et al. . Effects of adenovirus-mediated human apo A-I gene transfer on neointima formation after endothelial denudation in apo E-deficient mice. Circulation. 1997;96(12):4349–4356. [DOI] [PubMed] [Google Scholar]
- 108.Feng Y, Gordts SC, Chen F, et al. . Topical HDL administration reduces vein graft atherosclerosis in apo E deficient mice. Atherosclerosis. 2011;214(2):271–278. [DOI] [PubMed] [Google Scholar]
- 109.Ameli S, Hultgardh-Nilsson A, Cercek B, et al. . Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90(4):1935–1941. [DOI] [PubMed] [Google Scholar]
- 110.Kaul S, Rukshin V, Santos R, et al. . Intramural delivery of recombinant apolipoprotein A-IMilano/phospholipid complex (ETC-216) inhibits in-stent stenosis in porcine coronary arteries. Circulation. 2003;107(20):2551–2554. [DOI] [PubMed] [Google Scholar]
- 111.Schober A, Zernecke A, Liehn EA, et al. . Crucial role of the CCL2/CCR2 axis in neointimal hyperplasia after arterial injury in hyperlipidemic mice involves early monocyte recruitment and CCL2 presentation on platelets. Circ Res. 2004;95(11):1125–1133. [DOI] [PubMed] [Google Scholar]
- 112.van der Vorst EP, Vanags LZ, Dunn LL, et al. . High-density lipoproteins suppress chemokine expression and proliferation in human vascular smooth muscle cells. Faseb J. 2013;27(4):1413–1425. [DOI] [PubMed] [Google Scholar]
- 113.Vanags LZ, Tan JTM, Santos M, et al. . Plasma activated coating immobilizes apolipoprotein A-I to stainless steel surfaces in its bioactive form and enhances biocompatibility. Nanomedicine. 2017;13(7):2141–2150. [DOI] [PubMed] [Google Scholar]
- 114.Mineo C, Yuhanna IS, Quon MJ, et al. . High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003;278(11):9142–9149. [DOI] [PubMed] [Google Scholar]
- 115.Sugano M, Tsuchida K, Makino N. High-density lipoproteins protect endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Biochem Biophys Res Commun. 2000;272(3):872–876. [DOI] [PubMed] [Google Scholar]
- 116.Tamagaki T, Sawada S, Imamura H, et al. . Effects of high-density lipoproteins on intracellular pH and proliferation of human vascular endothelial cells. Atherosclerosis. 1996;123(1-2):73–82. [DOI] [PubMed] [Google Scholar]
- 117.van Oostrom O, Nieuwdorp M, Westerweel PE, et al. . Reconstituted HDL increases circulating endothelial progenitor cells in patients with type 2 diabetes. Arterioscler Thromb Vasc Biol. 2007;27(8):1864–1865. [DOI] [PubMed] [Google Scholar]
- 118.Werner N, Junk S, Laufs U, et al. . Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003;93(2):e17–24. [DOI] [PubMed] [Google Scholar]
- 119.Finn AV, Joner M, Nakazawa G, et al. . Pathological correlates of late drug-eluting stent thrombosis: strut coverage as a marker of endothelialization. Circulation. 2007;115(18):2435–2441. [DOI] [PubMed] [Google Scholar]
- 120.Moussa I, Oetgen M, Roubin G, et al. . Effectiveness of clopidogrel and aspirin versus ticlopidine and aspirin in preventing stent thrombosis after coronary stent implantation. Circulation. 1999;99(18):2364–2366. [DOI] [PubMed] [Google Scholar]
- 121.Park SJ, Park DW, Kim YH, et al. . Duration of dual antiplatelet therapy after implantation of drug-eluting stents. N Engl J Med. 2010;362(15):1374–1382. [DOI] [PubMed] [Google Scholar]
- 122.Li D, Weng S, Yang B, et al. . Inhibition of arterial thrombus formation by ApoA1 Milano. Arterioscler Thromb Vasc Biol. 1999;19(2):378–383. [DOI] [PubMed] [Google Scholar]
- 123.Calkin AC, Drew BG, Ono A, et al. . Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation. 2009;120(21):2095–2104. [DOI] [PubMed] [Google Scholar]
- 124.Kansara P, Qureshi W, Jurkovitz C, et al. . Definite stent thrombosis: does high density lipoprotein cholesterol level matter?. J Am Coll Cardiol. 2015;65(10):A224. [Google Scholar]
- 125.Mertens A, Holvoet P. Oxidized LDL and HDL: antagonists in atherothrombosis. Faseb J. 2001;15(12):2073–2084. [DOI] [PubMed] [Google Scholar]
- 126.Calabresi L, Franceschini G, Sirtori CR, et al. . Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem Biophys Res Commun. 1997;238(1):61–65. [DOI] [PubMed] [Google Scholar]
- 127.Bursill CA, Castro ML, Beattie DT, et al. . High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2010;30(9):1773–1778. [DOI] [PubMed] [Google Scholar]
- 128.Roger VL Heart failure epidemic: it’s complicated. Circulation. 2018;138(1):25–28. [DOI] [PubMed] [Google Scholar]
- 129.Morrow DA, Velazquez EJ, DeVore AD, et al. . Clinical outcomes in patients with acute decompensated heart failure randomly assigned to sacubitril/valsartan or enalapril in the PIONEER-HF trial. Circulation. 2019;139(19):2285–2288. [DOI] [PubMed] [Google Scholar]
- 130.Velagaleti RS, Massaro J, Vasan RS, et al. . Relations of lipid concentrations to heart failure incidence: the Framingham Heart Study. Circulation. 2009;120(23):2345–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhao Q, Li J, Yang J, et al. . Association of total cholesterol and HDL-C levels and outcome in coronary heart disease patients with heart failure. Medicine. 2017;96(9):e6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Van Linthout S, Spillmann F, Lorenz M, et al. . Vascular-protective effects of high-density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension. 2009;53(4):682–687. [DOI] [PubMed] [Google Scholar]
- 133.Lin L, Gong H, Ge J, et al. . High density lipoprotein downregulates angiotensin II type 1 receptor and inhibits angiotensin II-induced cardiac hypertrophy. Biochem Biophys Res Commun. 2011;404(1):28–33. [DOI] [PubMed] [Google Scholar]
- 134.Lin L, Liu X, Xu J, et al. . High-density lipoprotein inhibits mechanical stress-induced cardiomyocyte autophagy and cardiac hypertrophy through angiotensin II type 1 receptor-mediated PI3K/Akt pathway. J Cell Mol Med. 2015;19(8):1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Amin R, Muthuramu I, Aboumsallem JP, et al. . Selective HDL-raising human apo a-i gene therapy counteracts cardiac hypertrophy, reduces myocardial fibrosis, and improves cardiac function in mice with chronic pressure overload. Int J Mol Sci. 2017;18(9):pii:E2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aboumsallem JP, Mishra M, Amin R, et al. . Successful treatment of established heart failure in mice with recombinant HDL (Milano). Br J Pharmacol. 2018;175(21):4167–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Spillmann F, Miteva K, Pieske B, et al. . High-density lipoproteins reduce endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. 2015;35(8):1774–1777. [DOI] [PubMed] [Google Scholar]
- 138.Gordts SC, Van Craeyveld E, Muthuramu I, et al. . Lipid lowering and HDL raising gene transfer increase endothelial progenitor cells, enhance myocardial vascularity, and improve diastolic function. PLoS One. 2012;7(10):e46849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Van Linthout S, Spillmann F, Riad A, et al. . Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation. 2008;117(12):1563–1573. [DOI] [PubMed] [Google Scholar]
- 140.Annema W, Willemsen HM, de Boer JF, et al. . HDL function is impaired in acute myocardial infarction independent of plasma HDL cholesterol levels. J Clin Lipidol. 2016;10(6):1318–1328. [DOI] [PubMed] [Google Scholar]
- 141.Guerin M, Silvain J, Gall J, et al. . Association of serum cholesterol efflux capacity with mortality in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2018;72(25):3259–3269. [DOI] [PubMed] [Google Scholar]