

Abstract
Recent functional magnetic resonance imaging (fMRI) studies have demonstrated that moment-to-moment variability in the blood oxygen level-dependent (BOLD) signal is related to age differences, cognition, and symptomatic Alzheimer disease (AD). However, no studies have examined BOLD variability in the context of preclinical AD. We tested relationships between resting-state BOLD variability and biomarkers of amyloidosis, tauopathy, and neurodegeneration in a large (N=321), well-characterized sample of cognitively normal adults (age=39–93), using multivariate machine learning techniques. Further, we controlled for cardiovascular health factors, which may contaminate resting-state BOLD variability estimates. BOLD variability, particularly in the default mode network, was related to CSF amyloid-β42, but was not related to CSF phosphorylated tau-181. Further, BOLD variability estimates were also related to markers of neurodegeneration, including CSF neurofilament light protein, hippocampal volume, and a cortical thickness composite. Notably, relationships with hippocampal volume and cortical thickness survived correction for cardiovascular health and also contributed to age-related differences in BOLD variability. Thus, BOLD variability may be sensitive to preclinical pathology, including amyloidosis and neurodegeneration in AD-sensitive areas.
Keywords: BOLD variability, Alzheimer disease, amyloid, neurodegeneration, resting-state fMRI
1. INTRODUCTION
Accumulating evidence suggests that Alzheimer disease (AD) is marked by a preclinical period of amyloid (A) deposition, hyperphosphorylated tau (T) aggregation, and neurodegeneration (N) in the years leading up to cognitive impairment and clinical diagnosis of dementia (Bateman et al., 2012; Hardy and Higgins, 1992; Jack et al., 2016, 2013). Since it is hypothesized that there is little reversible neuronal damage during this period, the identification of early AD biomarkers in the absence of clinical diagnosis is a critical step for improving early detection and prediction of AD. Functional magnetic resonance imaging (fMRI) has been used to non-invasively probe differences in task-related neural activity and functional connectivity of brain networks in both the preclinical and clinical stages of AD (for review, see Sheline and Raichle, 2013; Sperling et al., 2010). In addition to these methods, which are respectively based on mean levels of task-related change or spontaneous correlation in the blood oxygen-level dependent (BOLD) signal, more recent fMRI methods have considered moment-to-moment BOLD variability as a potential signal that might be distinct from task activation and functional connectivity approaches (for review, see Garrett et al., 2013; Grady and Garrett, 2014).
Initial studies of BOLD variability focused on relationships with healthy age differences and cognition. These studies have reported that BOLD variability throughout a wide range of grey matter areas is negatively related to age (e.g., Garrett et al., 2010, 2011; Hu et al., 2014; Nomi et al., 2017) and positively related to task performance (e.g., Burzynska et al., 2015; Garrett et al., 2011; Grady and Garrett, 2018). However, there are some mixed reports of region-specific positive relationships with age and negative relationships with some task measures, even within the same studies reporting broad patterns in the opposite direction (e.g., Burzynska et al., 2015; Garrett et al., 2011, 2010; Nomi et al., 2017). A variety of theoretical accounts for these relationships have been proposed, although the specific mechanism is still unclear. For instance, greater BOLD variability might optimize flexible responses in situations of environmental uncertainty (Deco et al., 2011; Grady and Garrett, 2018) or might afford efficient communication within and between networks (Burzynska et al., 2015). In the present report, we explore the clinical utility of BOLD variability as a potential signal sensitive to preclinical AD pathology.
Previous studies have examined BOLD variability in the context of early cognitive decline by comparing cognitively normal controls to patient populations with mild cognitive impairment (MCI) or very mild to moderate dementia (Clinical Dementia Rating, CDR 0.5–2). Most of these studies have characterized frequency-specific BOLD fluctuations using the absolute or fractional amplitude of low-frequency fluctuation (ALFF or fALFF). Although not a direct measure of variability per se, both ALFF and standard deviation (SD) approaches capture the degree of fluctuation in BOLD signal over time. Interestingly, there have been consistent reports that fluctuation power in the precuneus and posterior cingulate is lower in cognitively impaired individuals (Han et al., 2011; Liu et al., 2014; Zhao et al., 2015). Notably, these regions are associated with early amyloid deposition (Mintun et al., 2006) and dysfunction in early AD (Lustig et al., 2003; Sperling et al., 2009). However, there have been inconsistent findings in other regions, including mixed reports of AD-related increases and decreases in BOLD variability within medial temporal, prefrontal, parietal, and temporal regions (Han et al., 2012, 2011; Liu et al., 2014; Xi et al., 2012; Zhao et al., 2015). A recent study reported AD-related increases in BOLD SD also in superior frontal, precentral, and putamen regions (Scarapicchia et al., 2018). These studies suggest that BOLD fluctuations might be sensitive to early clinical stages of AD; however, the direction and anatomical specificity of these relationships remains unclear, perhaps in part due to small samples (average N = 23 per group, range = 16 to 34).
Although there is growing interest in BOLD variability in early clinical stages of AD, it has been understudied in the context of preclinical AD. This area might yield important findings for assessing the clinical utility of BOLD variability, as well as potential mechanisms. Specifically, if BOLD variability is indeed sensitive to clinically-relevant biomarkers, it might serve as a non-invasive, reliable signal of dysfunction in the preclinical stage. Moreover, although previous age relationships with BOLD variability have been interpreted to reflect healthy aging processes, it is possible that preclinical AD processes contribute to these “healthy” age relationships, as has been shown in functional connectivity (Brier et al., 2014) and neuropsychological measures (Sliwinski et al., 1996). By first characterizing relationships between BOLD variability and preclinical AD biomarkers, we can begin to disentangle the influences of healthy aging and early pathological processes. Two recent studies (Good et al., 2020; Zhang et al., 2020) have demonstrated relationships between BOLD variability and structural estimates in the medial temporal lobe (possibly reflecting AD-related neurodegeneration), but no studies have examined relationships with amyloid or tau pathology.
The present study aims to examine relationships between BOLD variability and established biomarkers of preclinical AD pathology in a large, well-characterized sample of cognitively normal (CDR 0) older adults. Specifically, we will evaluate relationships with three categories of AD biomarkers: amyloidosis (A: cerebrospinal fluid [CSF] amyloid β 42 and amyloid PET), tauopathy (T: CSF phosphorylated tau-181), and neurodegeneration (N: CSF total tau, CSF neurofilament light protein, hippocampal volume, and a cortical thickness signature) (AT(N); Jack et al., 2016). Importantly, we will maximally control for head motion and global signal artifacts, since there is evidence that individual differences in these properties contribute to BOLD variability (Millar et al., 2020). In addition, we will examine any observed relationships after controlling for a cardiovascular health composite (CVH) and white matter hyperintensity burden (WMH), which have recently been shown to contribute to age relationships with BOLD variability (Millar et al., 2020; Tsvetanov et al., 2015, 2019). Hence, we will evaluate the sensitivity of BOLD variability to biomarkers above and beyond potential contaminating factors. Further, we will apply a network-based ROI approach to assess anatomical specificity of relationships with BOLD variability. Specifically, if BOLD variability indeed reflects a meaningful signal, one should expect that relationships with biomarkers should follow an anatomical structure at the level of networks, particularly those that are sensitive to preclinical AD pathology (cf., Sheline and Raichle, 2013). Finally, we will apply multivariate machine learning techniques to generate continuous predictions for untrained cases. These methods can evaluate the extent to which imaging measures offer predictive sensitivity to clinically-relevant processes (for review, see Cole and Franke, 2017; Nielsen et al., 2020). In sum, this study is designed to evaluate the potential clinical utility of BOLD variability as a sensitive signal related to biomarkers of preclinical AD and begin to disentangle the influences of healthy age differences and early pathological processes on BOLD variability.
2. METHODS AND MATERIALS
2.1. Participants
As described previously (Millar et al., 2020), 323 older adult participants were selected from a larger longitudinal cohort at the Charles and Joanne Knight Alzheimer Disease Research Center (ADRC) at Washington University in St. Louis. The sample was selected on the basis of being cognitively normal as assessed by the clinical dementia rating scale (CDR 0; Morris, 1993), absence of severe psychiatric conditions, availability of at least one biomarker measure (see below), and a usable resting-state fMRI scan as defined by low mean head motion (FD < .20, see Millar et al., 2020). Table 1 provides the demographics of the sample. All procedures were approved by the Human Research Protection Office at Washington University in St. Louis. All participants provided informed consent prior to all procedures.
Table 1.
Demographic and summary measures of the sample.
| Measure (units) | N | Mean (SD) | Range | |
|---|---|---|---|---|
| Demographic | Age (years) | 321 | 66.76 (9.28) | 39 – 93 |
| Sex (N female / N male) | 321 | 194 / 127 | NA | |
| Education (years) | 321 | 16.01 (2.56) | 10 – 20 | |
| Race (N white / N black / N Asian) | 321 | 299 / 20 / 2 | NA | |
| Mean Head Motion (mm FD) | 321 | 0.14 (0.04) | 0.05 – 0.2 | |
| MMSE (score) | 265 | 29.16 (1.1) | 26 – 30 | |
| CSF Biomarkers | Aβ42 (pg/mL) | 250 | 1389.54 (604.38) | 307.6 – 3385 |
| pTau (pg/mL) | 250 | 19.75 (9.93) | 8 – 70.08 | |
| tTau (pg/mL) | 250 | 216.94 (92.77) | 80 – 609.3 | |
| NfL (pg/mL) | 102 | 1518.82 (781.47) | 391.75 – 4124.7 | |
| Imaging Biomarkers | PIB PET (mean cortical SUVR) | 181 | 1.23 (0.53) | 0.86 – 3.77 |
| Adjusted hippocampal volume (mm3) | 318 | 7815.49 (951.86) | 4861.98 – 10123.83 | |
| Cardiovascular | Pulse (BPM) | 314 | 67.96 (9.74) | 41 – 108 |
| Systolic Blood Pressure (mmHg) | 319 | 125.44 (15.87) | 88 – 168 | |
| BMI (kg/m2) | 319 | 26.51 (4.74) | 14.09 – 47.37 | |
| WMH volume (mm3) | 231 | 13117.19 (16300.84) | 48.45 – 86112.11 |
FD = framewise displacement, MMSE = Mini Mental State Examination, Aβ42 = CSF Amyloid β peptide 42, pTau = CSF phosphorylated tau-181, tTau = CSF total tau, NfL = CSF neurofilament light, PIB PET = [11C]-Pittsburgh Compound B Positron Emission Tomography, SUVR = standard uptake ratio, BPM = beats per minute, BMI = body mass index, WMH = white matter hyperintensities.
2.2. Cardiovascular Health Measures
Following previous methods (Millar et al., 2020; Tsvetanov et al., 2019), we calculated a composite of cardiovascular health (CVH). CVH measures included resting pulse, systolic blood pressure, and body mass index (BMI), and white matter hyperintensity (WMH) lesion volume. WMH volumes were assessed with a fluid-attenuated inversion recovery (FLAIR) sequence, after segmentation using the Lesion Segmentation Tool (LST; Schmidt et al., 2012) for SPM 8. However, as reported in a mostly overlapping sample (Millar et al., 2020), correlations among the four measures were fairly small (rs = −.08 to .16). Moreover, WMH volume alone was strongly related to both age (r = .53, p < .001) and BOLD variability (r = −.21, p < .001), in contrast to other CVH measures. Hence, we used a CVH composite (including WMH) and the singular estimate of WMH. Importantly, the low correlations with CVH measures suggest that WMH might capture specific variance that is not broadly related to CVH.
2.3. CSF Biomarkers
Cerebrospinal fluid (CSF) was collected via lumbar puncture using methods described previously (Fagan et al., 2007). After overnight fasting, 20- to 30-mL samples of CSF were collected, then aliquoted (500 μL) in polypropylene tubes, and stored at −80°C. CSF amyloid β peptide 42 (Aβ42), phosphorylated tau-181 (pTau), and total tau (tTau) were measured with Elecsys immunoassays (Roche Diagnostics, Basel, Switzerland) (Schindler et al., 2018). A single lot of assays for each analyte was used to avoid lot-to-lot variability. Aβ42, pTau, and tTau are well established biomarkers in the AT(N) framework (Jack et al., 2016). Using previously reported cutoffs (Schindler et al., 2018), 33.6% of the sample was identified with positive amyloid pathology, 39.6% with positive pTau, and 26.8% with positive tTau, suggesting that these individuals are in a preclinical stage of AD and are at increased risk for cognitive decline (Jack et al., 2018). We tested whether BOLD SD was sensitive to continuous variance in these biomarkers within the cognitively normal sample (CDR = 0, MMSE > 25, see Table 1).
CSF neurofilament light (NfL) was measured with an ELISA immunoassay (Uman Diagnostics, Umeå, Sweden). NfL is an emerging biomarker of axonal damage, which has been shown to be modestly elevated in clinical-stage AD and predictive of cognitive decline in AD samples, but is also elevated in a wide range of neurodegenerative diseases (for review, see Gordon, 2020). Thus, NfL may reflect a non-specific marker of neurodegeneration.
2.4. PIB-PET Imaging
Amyloid burden was imaged with positron emission tomography (PET) using a [11C]-Pittsburgh Compound B (PIB) tracer (Mintun et al., 2006). Regions of interest were segmented automatically using FreeSurfer 5.3 (Fischl, 2012). Regional standard uptake ratios (SUVRs) were modeled from the 30- to 60-minute post-injection window, using the cerebellum as a reference region. Global amyloid burden was defined as the mean of SUVRs from regions associated with elevated PIB retention in early-stages of AD (Mintun et al., 2006), including bilateral precuneus, prefrontal cortex, gyrus rectus, and lateral temporal regions (Su et al., 2013).
2.5. Scanning Protocol and Preprocessing
MRI data were obtained using two separate Siemens Trio 3T scanners equipped with a standard 12-channel head coil. One-way analyses of variance (ANOVAs) revealed that there were significant differences between participants across scanners in measures of CSF Aβ42 (F = 4.090, p = .007) and NfL (F = 5.142, p = .026), but not in pTau (F = 1.333, p = .264), PIB SUVR (F = 0.145, p = .865), hippocampal volume (F = 0.047, p = .986), or AD cortical signature (F = 1.300, p = .275). In order to examine the possibility that biomarker relationships with BOLD variability might be confounded by differences in the scanners, we replicated the results with each biomarker after controlling for scanner as a factor of non-interest. Overall, the relationships were consistent after controlling for scanner differences (see Supplementary Results).
Structural and functional scans were acquired using methods described previously (Brier et al., 2012; Millar et al., 2020; Wisch et al., 2020). Structural scans were acquired with a sagittal T1-weighted magnetization-prepared rapid gradient echo sequence (MPRAGE; TR = 2400 ms, TE = 3.16 ms, flip angle = 8°, field of view = 256 mm, 1-mm isotropic voxels), as well as an oblique T2-weighted fast spin echo sequence (FSE; TR = 3200 ms, TE = 455 ms, 256 × 256 acquisition matrix, 1-mm isotropic voxels). Functional scans were acquired with an interleaved whole-brain echo planar imaging sequence (EPI; TR = 2200 ms, TE = 27 ms, flip angle = 90°, field of view = 256 mm, 4-mm isotropic voxels). Participants completed two consecutive 6-minute runs (164 volumes each) of functional imaging, during which they were instructed to stay awake and fixate on a visual crosshair.
Both resting state runs were processed together, using conventional methods (Brier et al., 2012; Shulman et al., 2010), as well as conservative nuisance rejection including global signal regression and frame-wise motion censoring (Fox et al., 2009). See the Supplementary Methods for detailed description of these procedures. Motion-related differences in the number of censored frames might confound BOLD variability estimates. Thus, we analyzed BOLD variability within a subset of 120 randomly-selected usable frames from either run for each participant. Two participants with fewer than 120 usable frames were excluded (final N = 321).
2.6. Structural MRI Measures
T1-weighted images were subjected to volumetric segmentation with FreeSurfer 5.3 (Fischl, 2012). Regional volumetric estimates were corrected for intra-cranial volume using regression normalization (Buckner et al., 2004). Bilateral volumetric estimates were summed across hemispheres, while thickness estimates were averaged across hemispheres. An AD cortical signature composite was defined as the mean of thickness estimates from entorhinal cortex, fusiform gyrus, inferior, middle, and superior temporal gyri, superior and inferior parietal lobules, posterior cingulate gyrus, and precuneus. Thinning in these regions has been associated with estimates of tau in CSF and PET (Wang et al., 2016, 2015). Thus, the cortical signature may reflect neurodegeneration that is characteristic of, but not necessarily specific to, AD-related pathology. To assess the specificity of structural relationships, we also examined thickness in control regions sensitive to age-related thinning and/or minimally sensitive to AD: superior frontal, precentral, and rostral anterior cingulate gyri (Bakkour et al., 2013; Fjell et al., 2009).
2.7. Calculation of BOLD Variability
As described previously (Millar et al., 2020), final BOLD data were averaged across voxels within 298 grey matter ROIs (Power et al., 2011; see Seitzman et al., 2020 for a figure), including 243 10-mm cortical spheres, 28 8-mm subcortical spheres, and 27 8-mm spheres in the cerebellum. Importantly, each ROI has been assigned to one of 14 networks, including: somatomotor (SM), lateral somatomotor (SML), cingulo-opercular (CO), auditory (AUD), default mode (DMN), parietal memory (PMN), visual (VIS), fronto-parietal (FPN), salience (SAL), subcortical (SUB), ventral attention (VAN), dorsal attention (DAN), and cerebellum (CER) networks, as well as ROIs that were unassigned to a network (N/A). In each ROI, we calculated SD of BOLD signal over the 120 selected usable frames. Of course, because the BOLD variability approach is based on the SD within an ROI rather than correlations between ROIs, as in functional connectivity, we considered variability within ROIs or networks as our primary fMRI measure of interest.
2.8. Support Vector Regression
Support vector regression (SVR) is a supervised machine learning technique in which a model is trained to identify multivariate relationships. We used this approach to generate continuous predictions about label values (i.e., AT(N) biomarkers) from models trained on multivariate feature sets (i.e., BOLD SDs in the 298 ROIs). SVR analyses were conducted using the e1071 package in R (Meyer et al., 2017). We performed epsilon-insensitive SVR, as described previously (Dosenbach et al., 2010; Millar et al., 2020; Nielsen et al., 2019). Briefly, SVR fits a regression line in multivariate space between the feature set and the label values. A tube of width epsilon is defined around the regression line. Data points outside this tube are penalized, while points inside the tube are not. The penalty factor C determines the trade-off between training error and model complexity. All SVR analyses were performed with epsilon = 0.00001 and C = infinity, based on previous reports predicting age from functional connectivity (Dosenbach et al., 2010; Nielsen et al., 2019) and BOLD variability (Millar et al., 2020).
Importantly, the SVR model is trained on a subset of cases, allowing for the assessment of model prediction in an unseen testing set. Specifically, we evaluated predictive accuracy using 10-fold cross-validation. For each fold, a non-overlapping set of 10% of the sample served as the testing set. The remaining 90% served as the training set. Thus, across the 10 folds, the SVR model predicted a label value for each participant. We quantified predictive accuracy as R2 between the model-predicted and the true label values for each participant. We tested the predictive accuracy of SVR models trained on the full feature set of BOLD SD values from all 298 ROIs. Specifically, we tested the performance of these models to predict AT(N) biomarkers.
2.9. Assessment of Network Specificity of Relationships
As described previously (Millar et al., 2020), network specificity of relationships with BOLD SD was assessed in two ways. First, univariate network-level relationships were tested using a bootstrap approach. Specifically, we randomly generated 10,000 samples by resampling the dataset with replacement. In each bootstrap sample, we calculated the Pearson correlation coefficients between the biomarker and BOLD SD in each ROI, uncorrected for multiple comparisons. We then averaged the correlation coefficients across ROIs within each network. Across bootstrap samples, we then calculated the empirical 95% confidence interval for each network-level correlation.
Second, we assessed the multivariate predictive accuracy of networks using network-specific feature selection. Each network-specific SVR model predicted biomarker labels from a limited feature set of regions restricted to a single network and was evaluated using 10-fold cross validation. Since larger networks should perform better simply due to a greater number of features, which might capture a related signal by chance, we compared SVR performance for network-specific feature sets to a bootstrapped distribution of 10,000 randomly selected feature sets (i.e., random regions from any network), matched in the number of features. Hence, this distribution is an appropriate null model to test whether signals are localized to specific networks or instead broadly distributed throughout the brain (Nielsen et al., 2020).
3. RESULTS
3.1. Relationships with Amyloid Biomarkers
3.1.1. CSF Aβ42.
As shown in Fig. 1A, there were positive trends in most networks such that reduced CSF Aβ42, indicative of preclinical amyloidosis, was related to lower BOLD SD. However, these trends did not reach statistical significance in any network. Interestingly, as shown in Fig. 1B, SVR models successfully predicted CSF Aβ42 from the full feature set of BOLD SD in all 298 ROIs (R2 = .028, p = .007). Hence, there may be a multivariate pattern in BOLD SD that captures a small, but significant portion of variance in CSF Aβ42. We evaluated this multivariate relationship in specific networks using network-driven feature selection. As shown in Fig. 2A, BOLD SD within the DMN was particularly successful in predicting CSF Aβ42 (R2 = .047). This level of performance might be expected, since the DMN includes a large number of individual features and may capture amyloid-related signal by chance. Hence, we compared SVR performance from the DMN to a bootstrapped distribution of 10,000 randomly selected feature sets of equal set size from any network. SVR performance from the DMN significantly outperformed this bootstrapped distribution (empirical p = .025). Hence, although the multivariate relationship between BOLD SD and CSF Aβ42 is relatively small, it may be specific to networks associated with early amyloid deposition (Mintun et al., 2006).
Figure 1.
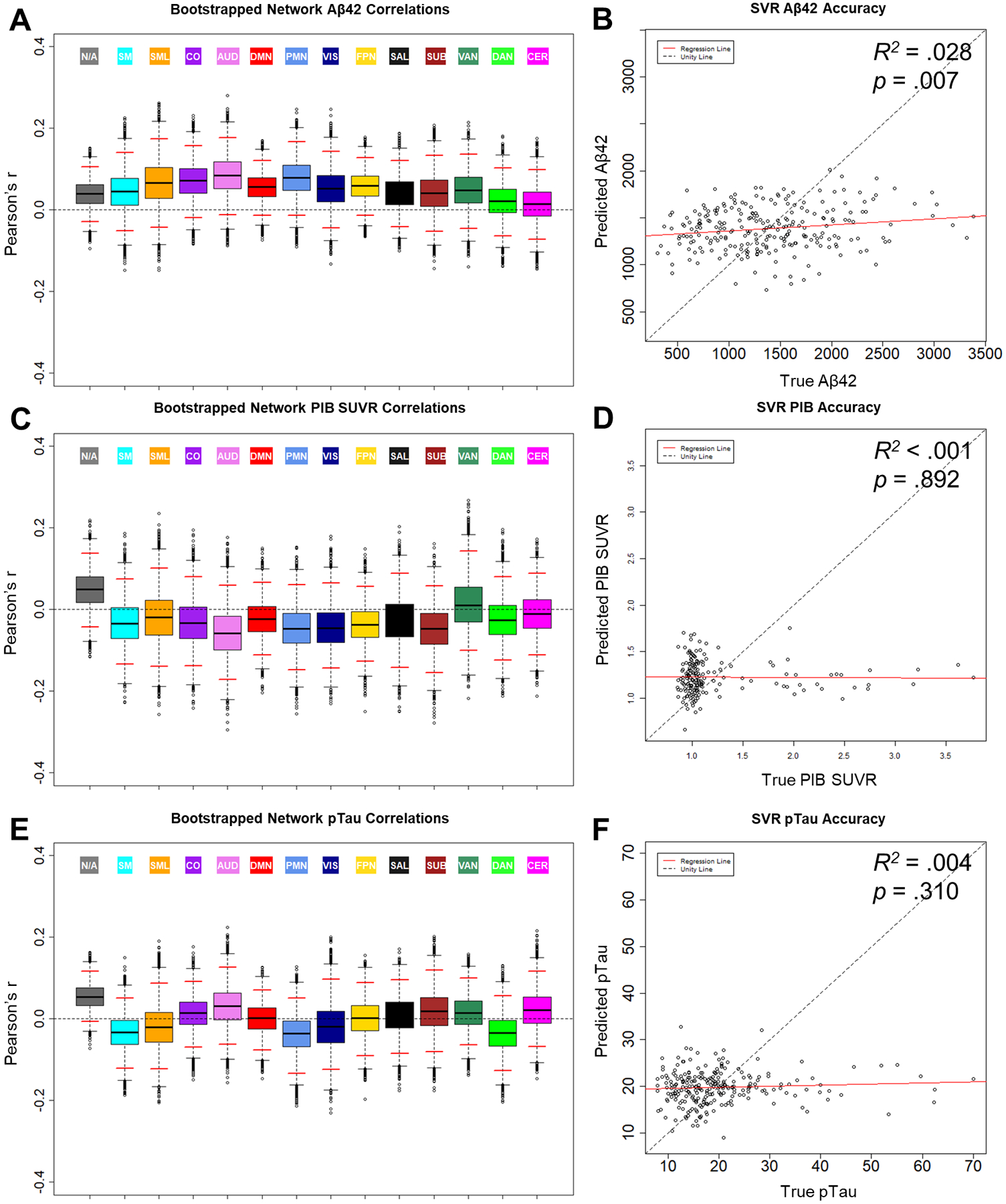
Relationships between BOLD SD and biomarkers of amyloid and tau: CSF Aβ42 (A & B), PIB SUVR (C & D), and CSF pTau (E & F). Boxplots (A, C, & E) display the bootstrapped distribution of network average Pearson correlation values between BOLD SD and the biomarkers. Solid black lines denote the median of the distribution. Dotted whisker lines denote the spread of datapoints up to 1.5 times beyond the width of the IQR. Datapoints beyond the whiskers denote possible outliers. Solid red lines denote the empirical 95% confidence interval of the bootstrapped samples. Scatterplots (B, D, & F) display SVR prediction results for the biomarkers. Label values predicted by the model are plotted as a function of the true value. Aβ42 = amyloid β peptide 42, PIB = [11C]-Pittsburgh Compound B, SUVR = standard uptake ratio, pTau = phosphorylated tau-181, SVR = support vector regression
Figure 2.
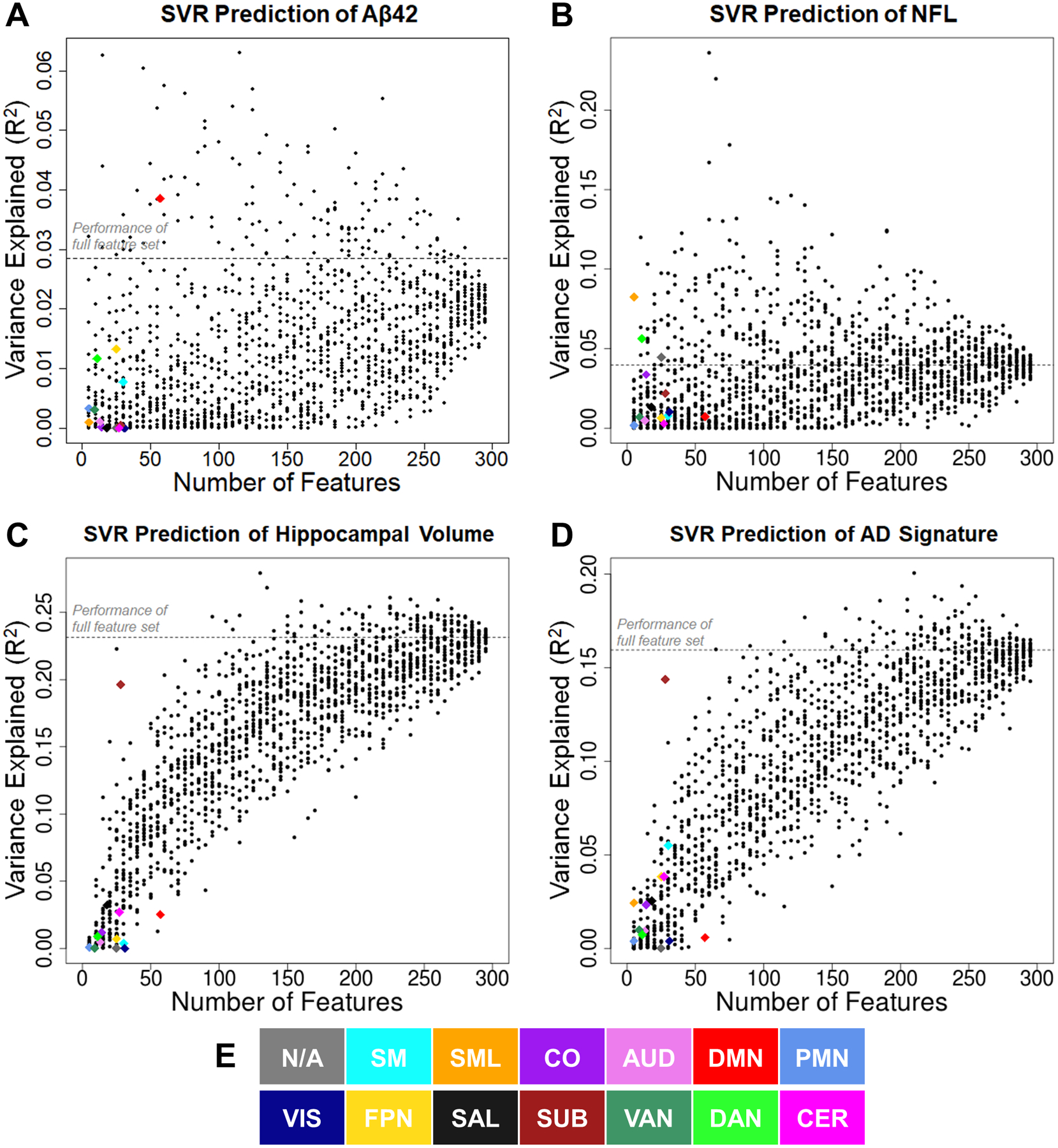
Performance of SVR models predicting Aβ42 (A), NfL (B), hippocampal volume (C), and AD signature (D) across a range of feature sets (from 5 to 295). Colored diamonds denote anatomical feature selection schemes, in which features included only ROIs from a specific network. Each network-specific model was compared to 10,000 simulated models using randomly selected feature sets from any functional network. For simplicity, only 25 of the simulated models are plotted for each feature set size (black dots). E. Color key for network identities. Aβ42 = amyloid β peptide 42, NfL = neurofilament light, AD signature = Alzheimer disease signature region cortical thickness composite
3.1.2. PIB-PET.
Next, we examined relationships between BOLD SD and PIB-PET estimates of amyloid deposition. As shown in Fig. 1C, PIB SUVR was not related to BOLD SD in any network. Moreover, as shown in Fig. 1D, SVR models were not able to predict PIB SUVR from BOLD SD (R2 < .001, p = .892). Thus, the relationship between amyloid and BOLD SD observed in the CSF does not replicate using a PET imaging marker of amyloid deposition.
3.2. Relationships with Tau Biomarkers
3.2.1. pTau.
As shown in Fig. 1E, pTau was not related to BOLD SD in any network. Moreover, as shown in Fig. 1F, SVR models were not able to predict pTau (R2 = .004, p = .310) (tTau SVR R2 = .006, p = .217). Although CSF pTau and tTau are hypothesized to reflect distinct processes of tauopathy and neurodegeneration (Jack et al., 2016), they are highly correlated in AD samples (r = .98, p < .001 in the present sample). Unsurprisingly, relationships with BOLD SD were consistent for both pTau and tTau. Hence, we only report findings for pTau in Fig. 1.
Since we evaluated biomarker relationships continuously within a cognitively normal sample, it is possible that disease heterogeneity might contribute to the null relationships with tau pathology. Specifically, we did not exclude Aβ42-negative participants with elevated pTau, which might reflect suspected non-Alzheimer pathophysiology (SNAP; Vos et al., 2013). Thus, we examined relationships with pTau within specific subsamples, including Aβ42-positive (likely preclinical AD), Aβ42-negative (likely SNAP), and apolipoprotein (APOE) ε4-positive (increased genetic risk of AD) participants. However, the lack of relation between BOLD SD and CSF pTau was consistent within each subsample (see Supplementary Results).
3.3. Relationships with Biomarkers of Neurodegeneration
3.3.1. NfL.
As shown in Fig. 3A, there were marginal negative relationships between NfL and BOLD SD in somatomotor, auditory, visual, frontoparietal, and dorsal attention networks. Further, as shown in Fig. 3B, SVR models marginally predicted NfL from BOLD SD (R2 = .040, p = .044). Again, we evaluated the network specificity of this multivariate relationship using network-driven feature selection. As shown in Fig. 2B, BOLD SD in the lateral somatomotor network was particularly successful in predicting NfL (R2 = .082). SVR performance from these features significantly outperformed randomly selected regions (p = .01).
Figure 3.
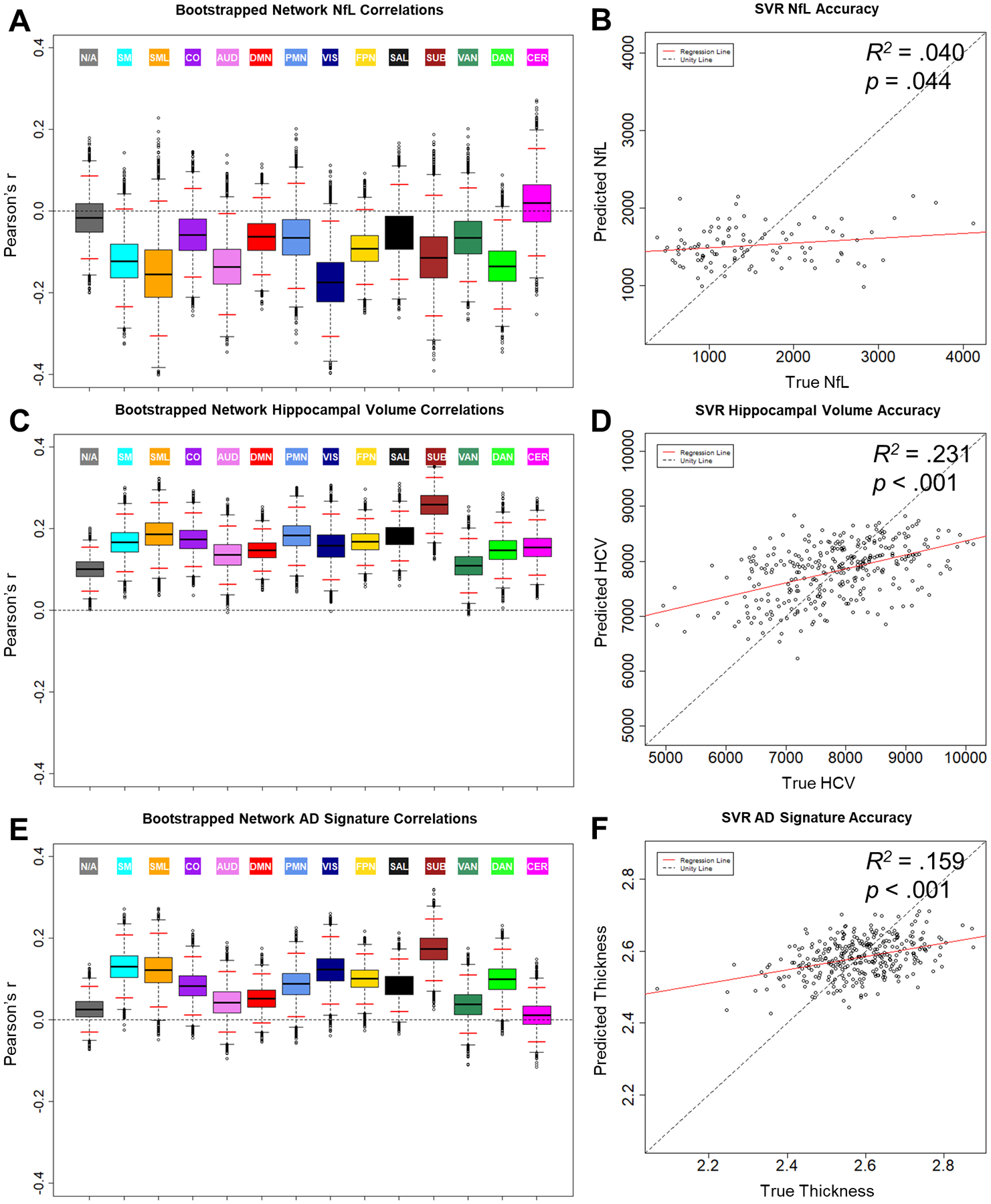
Relationships between BOLD SD and biomarkers of neurodegeneration: CSF NfL (A & B), hippocampal volume (C & D), and AD signature (E & F). Boxplots (A, C, & E) display the distribution and empirical 95% confidence interval (red lines) of bootstrapped distributions of network average Pearson correlations. Scatterplots (B, D, & F) display biomarker measures predicted by SVR model as a function of true score. NfL = neurofilament light, AD signature = Alzheimer disease signature region cortical thickness composite
3.3.2. Hippocampal volume.
As shown in Fig. 3C, there were significant positive relationships between hippocampal volume (HCV) and BOLD SD in all networks. This relationship was strongest in the subcortical network. Further, as shown in Fig. 3D, SVR models successfully predicted HCV (R2 = .231, p < .001). Again, we evaluated the network specificity of this relationship using network-driven feature selection. As shown in Fig. 2C, BOLD SD in the subcortical network was particularly successful in predicting HCV (R2 = .196). SVR performance from these features significantly outperformed randomly selected regions (empirical p = .003). Hence, although HCV is related to BOLD SD in a broad range of networks beyond the hippocampus, there appear to be particularly strong relationships in subcortical areas.
3.3.3. AD Signature.
As shown in Fig. 3E, there were marginal to significant positive relationships between the AD signature cortical thickness composite and BOLD SD in a range of sensory, motor, and association networks. Further, as shown in Fig. 3F, SVR models successfully predicted AD signature cortical thickness (R2 = .159, p < .001). Similar to HCV, BOLD SD in the subcortical network was particularly successful in predicting the AD signature (R2 = .144, see Fig. 2D). SVR performance from these features significantly outperformed randomly selected regions (empirical p < .001), as did lateral somatomotor features (R2 = .024, empirical p = .004). Notably, these networks were also particularly sensitive to other neurodegenerative measures included in the present report: respectively, HCV and NfL.
3.3.4. Control Regions.
In addition to the AD-sensitive regions, we also tested relationships with estimates of thickness in regions sensitive to age-related cortical thinning and/or minimally sensitive to AD. To summarize, we found that BOLD SD was somewhat sensitive to thickness in these regions, but in comparison to the HCV and AD signature relationships, the correlations were much smaller and the SVR models exhibited reduced predictive accuracy. See the Supplementary Results for full details.
3.4. CVH- and WMH-Related Influences on Biomarker Relationships
Other studies have previously demonstrated that vascular factors might contribute to age differences in BOLD SD (Tsvetanov et al., 2015, 2019). Thus, it is possible that relationships with CSF and structural biomarkers might also be sensitive to vascular mechanisms. To examine this possibility, we attempted to replicate SVR prediction of CSF Aβ42, NfL, HCV, and AD cortical thickness signature using residual values of BOLD SD after regressing out either the CVH composite or WMH burden. This method has been shown to attenuate SVR prediction of age and cognition from BOLD SD (Millar et al., 2020).
As shown in Table 2, only the relationship with CSF Aβ42 was attenuated after controlling for CVH. Multivariate relationships with NfL, HCV, and the AD signature cortical thickness estimates remained marginally to highly significant with largely comparable, but somewhat reduced, predictive accuracy. In contrast, BOLD SD relationships with all biomarkers were drastically reduced after controlling for WMH. However, even after controlling for WMH, BOLD SD still captured a significant portion of variance in both HCV and the AD cortical thickness signature, although, as noted, the predictive accuracy was much smaller.
Table 2.
Performance of SVR models (R2 statistic) predicting biomarker measures.
| Biomarker | SVR R2 (Original) | SVR R2 (Partial CVH) | SVR R2 (Partial WMH) |
|---|---|---|---|
| Aβ42 | .028** | .008 | .001 |
| NfL | .040* | .040^ | .011 |
| HCV | .231*** | .195*** | .080*** |
| AD Signature | .159*** | .089*** | .022* |
Performance is presented for original models, as well as after statistically controlling for cardiovascular health composite (CVH) and white matter hyperintensity (WMH) values in BOLD SD estimates. Aβ42 = CSF Amyloid β peptide 42, NfL = CSF neurofilament light, HCV = adjusted hippocampal volume, AD Signature = Alzheimer Disease cortical thickness signature
p < .10;
p < .05;
p < .01;
p < .001
It is somewhat surprising that controlling for CVH and WMH produced such different patterns in the relationships with biomarkers of neurodegeneration, especially considering that WMH is included in the CVH composite. One possible explanation is that WMH may capture a distinct source of variance from general cardiovascular health. Indeed, as noted, we observed only weak relationships among the CVH composite measures. Specifically, WMH may reflect progressive small vessel disease associated with cardiovascular health across the lifespan (Pantoni, 2010; Schmidt et al., 1999), whereas the other CVH composite factors (i.e., pulse, blood pressure, BMI) may be more sensitive to current fluctuations in CVH and modulated by medication and lifestyle factors. It is possible that cumulative injury, captured by WMH, may be particularly important in explaining relationships between BOLD variability and neurodegeneration. In this light, WMH may be sensitive to co-occurring neurodegeneration, as opposed to CVH per se. Moreover, it is also possible that WMH may also be sensitive to distinct pathological processes, including, for instance, traumatic brain injury (Marquez De La Plata et al., 2007) or multiple sclerosis (Filippi et al., 2011). Thus, the other estimates of CVH may add a distinct, unrelated signal to the composite. However, it is also worth noting that overall WMH levels were relatively low in this sample, due to screening of the cohort. Hence, it is possible that different patterns might be observed in a sample with greater WMH burden.
3.5. Biomarker-Related Influences on Age Relationships
Finally, after characterizing relationships between BOLD SD and biomarkers of amyloid deposition and neurodegeneration, we examined whether these biomarkers might contribute to age-related variance in BOLD SD. To that end, we attempted to replicate relationships between age and BOLD SD using residual BOLD SD values after regressing out one of the related biomarkers. Importantly, we compared these results to analyses controlling for WMH, which as discussed, has been shown to attenuate age relationships with BOLD SD.
As shown in Fig. 4A, in the original BOLD SD values, we replicated the general pattern of significant negative relationships with age. SVR models successfully predicted age from these original BOLD SD values (R2 = .239, p < .001). After controlling for WMH, we replicated non-significant negative trend-level relationships between age and BOLD SD, as shown in Fig. 4B. SVR prediction of age was not successful when BOLD SD was corrected for WMH (R2 < .001, p = .754), consistent with previous results (Millar et al., 2020).
Figure 4.
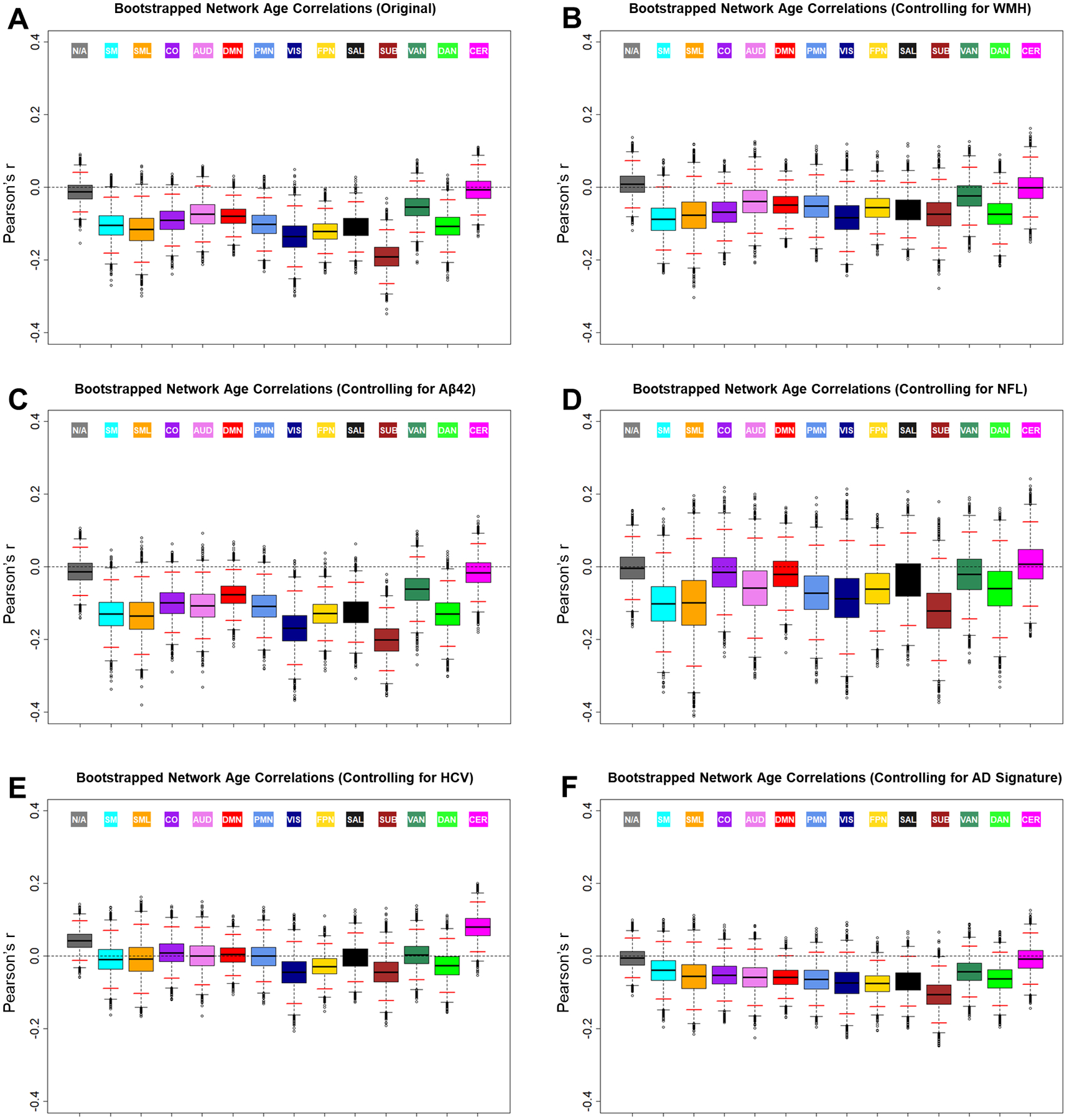
Boxplots of the distribution and 95% confidence interval (red lines) of bootstrapped distributions of network average Pearson correlations between BOLD SD and age. Relationships are shown for uncorrected BOLD SD (A), as well as after controlling for WMH (B), Aβ42 (C), NfL (D), HCV (E), and AD signature (F). WMH = white matter hyperintensity volume, Aβ42 = amyloid β peptide 42, NfL = neurofilament light, HCV = hippocampal volume, AD signature = Alzheimer disease signature region cortical thickness composite
In contrast, after controlling for CSF Aβ42, significant negative relationships with age were observed in most of the same networks as the original analyses, as shown in Fig. 4C. SVR prediction of age was highly successful after correcting for CSF Aβ42 (R2 = .172, p < .001), suggesting that age- and amyloid-related influences on BOLD SD might be distinct. After controlling for NfL, negative trend-level relationships with age were preserved in many networks, although these relationships were no longer significant, as shown in Fig. 4D. This change may be the result of the relatively small sample size of participants with NfL (N = 102). Thus, relationships between age and BOLD SD might be less stable in this subsample. However, SVR prediction of age was highly successful after correcting for NfL (R2 = .140, p < .001).
Turning to the structural biomarkers, after controlling for HCV, negative relationships with age were largely eliminated. Rather, as shown in Fig. 4E, only positive trends were observed in cerebellum and unassigned regions. After controlling for the AD cortical thickness signature, negative trend-level relationships with age were observed, but were not significant in most networks, as shown in Fig. 4F. SVR prediction of age was not successful after correcting for HCV (R2 < .001, p = .748) or the AD cortical thickness signature (R2 < .001, p = .969). Thus, age relationships with resting-state BOLD SD might be sensitive to a mechanism related to grey matter structure in AD-sensitive regions.
4. DISCUSSION
The present study aimed to evaluate relationships between resting-state BOLD variability and established biomarkers of early pathological processes associated with AD. To review the noteworthy results, we found small, but significant relationships between BOLD variability and CSF measures of Aβ42 and NfL, but no relationships with tTau or pTau. Further, we found stronger relationships with structural MRI measures of HCV and the AD signature cortical thickness, but no relationships with PIB-PET imaging of amyloid deposition. Importantly, relationships with the structural biomarkers survived correction for a CVH composite and WMH burden, although the predictive accuracy was reduced. Finally, we demonstrated that inverse age relationships with BOLD variability were reduced or eliminated after controlling for structural biomarkers, but not after controlling for CSF Aβ42. We now discuss each of these findings, focusing on their implications for the interpretation of resting-state BOLD variability and potential for application as a clinically-relevant biomarker.
4.1. Relationships with Amyloid Deposition
The most novel aspect of this study was the evaluation of relationships between resting-state BOLD variability and biomarkers of AD-related pathology in cognitively normal individuals. As preclinical AD pathology begins in the brain, some of the earliest observable changes include a reduction of CSF Aβ42 and increased uptake on PET amyloid imaging (Bateman et al., 2012). We observed trend-level relationships between CSF Aβ42 and BOLD variability across a wide range of networks, suggesting that early pathological reductions in CSF Aβ42 may be associated with reduced BOLD variability. Importantly, multivariate SVR models successfully predicted CSF Aβ42 from BOLD variability, particularly within the DMN, which includes regions associated with early amyloid deposition (Buckner et al., 2005). Hence, network-specific estimates of BOLD variability in the DMN might capture significant, but small, portions of variance in early amyloid pathology. This finding is consistent with previous reports that BOLD variability in specific DMN regions (i.e., precuneus and posterior cingulate) is reduced in MCI and early symptomatic AD samples (Han et al., 2011; Liu et al., 2014; Zhao et al., 2015). Here we extend upon these results to include novel relationships with preclinical amyloidosis. Moreover, this finding is also in line with previous reports that both task-related over-activation (Sperling et al., 2009) and reduced functional connectivity (Hedden et al., 2009; Wang et al., 2013) within the DMN are also associated with preclinical amyloidosis.
Interestingly, relationships between BOLD variability and CSF Aβ42 did not survive correction for a CVH composite or WMH burden. Similar corrections have been shown to reduce or eliminate age relationships with BOLD variability (Millar et al., 2020; Tsvetanov et al., 2015, 2019). Hence, the influence of amyloid on BOLD variability may be sensitive to mechanisms related to CVH or WMH burden. For instance, amyloid pathology might lead to microhemorrhages as observed in cerebral amyloid angiopathy (Pantoni, 2010; Vinters, 1987).
Although the CSF Aβ42 results suggest that resting-state BOLD variability may be sensitive to early amyloid pathology, we did not observe relationships with PIB-PET imaging of amyloid deposition. One limitation of these analyses is that the continuous estimates of PIB SUVR are highly skewed, potentially hindering statistical tests. Hence, we supplemented the reported analyses with additional approaches, including a log-transform of the SUVR values, as well as dichotomous SVR prediction of individuals above or below a threshold of PIB positivity. However, these methods did not increase sensitivity. Another potential explanation for the inconsistency is relative differences in statistical power for the two measures, due to differences in sample size. Indeed, there were fewer participants with PIB imaging than there were with CSF Aβ42 (Ns = 181 vs. 250). Alternatively, it is has been noted that changes in CSF amyloid might be observed earlier in the disease progression than changes in PET binding (Vlassenko et al., 2016). Thus, CSF Aβ42 might capture distinct aspects of amyloid pathology that are more strongly related to BOLD variability than PET measures. Further work is necessary to evaluate the consistency of BOLD variability relationships with CSF and PET measures of amyloid.
4.2. Relationships with Tauopathy
In contrast to CSF Aβ42, we did not observe relationships between resting-state BOLD variability and CSF pTau. This pattern was consistent within Aβ42-positive, Aβ42-negative, and APOE ε4-positive subsamples in which pTau variance might be driven by distinct disease processes (see Supplementary Results). Moreover, this finding is consistent with recent demonstrations that measures of amyloidosis, but not tauopathy, are related to reduced global intra-network functional connectivity using fMRI (Wisch et al., 2020), as well as disruption of hubs within multiplex functional networks using MEG (Yu et al., 2017). Thus, functional estimates of variability and connectivity might be sensitive to early (amyloid), but not intermediate (tau), stages of preclinical AD pathology (Jack et al., 2016).
Although we failed to detect relationships with CSF pTau, we did not examine relationships between BOLD variability and regional tau accumulation as imaged using tau PET. These measures were available in the cohort, but there was excessive temporal delay between the tau PET scans and the resting-state fMRI sessions evaluated in the current study. Tau PET might be more sensitive to BOLD variability and should be investigated in future studies.
4.3. Relationships with Neurodegeneration
The strongest relationships with resting-state BOLD variability were observed in measures of grey matter volume and cortical thickness. Specifically, BOLD variability in a wide range of networks was sensitive to HCV and an AD signature cortical thickness composite. Although HCV and AD signature areas are associated with neurodegeneration in early AD, it is unclear whether BOLD variability captures AD-specific pathology in these relationships. In the absence of large, consistent relationships with amyloid deposition, BOLD SD might instead capture non-specific neurodegeneration that may arise through distinct pathological mechanisms. However, relationships with AD-related structural measures were relatively strong compared to control regions that are not typically as sensitive to AD (see Supplementary Results), suggesting that BOLD SD may be particularly sensitive to neurodegeneration in areas that are characteristic of, but not specific to, AD-related pathology. Thus, BOLD variability might be sensitive to two stages of pathological progression – early amyloidosis and later neurodegeneration – which resemble recent demonstrations in functional connectivity (Wisch et al., 2020).
The present results are consistent with a recent demonstration that greater low-frequency BOLD SD in the precuneus was associated with greater HCV in a sample of 96 AD, 96 aMCI, and 48 cognitively normal participants (Zhang et al., 2020). However, another study in a smaller sample of 20 cognitively normal and 20 “at-risk” participants (defined as low performance on the Montreal cognitive assessment) demonstrated relationships in the opposite direction, such that BOLD SD in medial temporal areas was associated with smaller medial temporal volumes (Good et al., 2020). The present results instead suggest that HCV might be positively related to BOLD SD throughout a wide range of networks in a large (N = 318) cognitively normal sample. Inconsistencies between these findings may be the consequence of differences in clinical staging of the samples, sample size, or processing of the functional and structural estimates.
The AD-sensitive structural measures demonstrated particularly strong relationships with BOLD variability in the subcortical network. Although subcortical areas are not typically implicated in early sporadic AD pathology, other studies have reported unique findings regarding BOLD variability in these areas. For instance, Garrett and colleagues (2018) found that BOLD variability in the thalamus demonstrated a particularly strong relationship with network integration (defined as resting-state PCA dimensionality). Moreover, aging studies have reported mixed findings of age-related increases (Garrett et al., 2011, 2010; Guitart-Masip et al., 2016) and decreases (Millar et al., 2020; Nomi et al., 2017) in subcortical BOLD variability. Together, these findings suggest that subcortical areas might serve a unique role in age- and AD-related differences in BOLD SD, possibly related to characteristics of network organization and structural atrophy. These relationships should be further examined in future studies.
It is also noteworthy that BOLD variability relationships with HCV and AD signature cortical thickness were reduced, yet remained significant, after controlling for CVH and WMH burden. Hence, BOLD variability may be sensitive to a neurodegeneration component, even after controlling for influences related to CVH and/or small vessel disease pathology. However, evidence from CSF biomarkers only partly supports this interpretation. Specifically, BOLD variability was marginally sensitive to NfL, a marker of non-specific axonal neurodegeneration. However, relationships between NfL and BOLD SD did not survive correction for CVH and WMH, suggesting that they might be mediated by these factors.
4.4. Heathy Aging vs. Early Pathological Influences on BOLD Variability
One aim of the present study was to evaluate whether early pathological processes might contribute to age differences in resting-state BOLD variability. Notably, we replicated negative relationships with age after correcting for CSF Aβ42 and NfL, both of which were marginally related to BOLD variability in the current sample. Hence, age relationships with BOLD variability are not likely driven by amyloid deposition or axonal neurodegeneration mechanisms.
However, age relationships with BOLD variability were eliminated or drastically reduced after correcting for HCV or AD signature cortical thickness. Hence, age-related differences in BOLD variability may be driven in part by grey matter neurodegeneration. These findings stand in contrast to a recent report from Tsvetanov and colleauges (2019), in which age relationships with BOLD variability were totally eliminated after controlling for cardiovascular health and cerebral blood flow, but not after controlling for grey matter volume. Notably, Tsvetanov and colleauges (2019) corrected estimates of BOLD variability for grey matter volume within each overlapping voxel or independent component. In contrast, in the present report, we corrected estimates of BOLD variability in all ROIs for specific structural estimates of only HCV or AD signature cortical thickness, and thus the interpretation of structural-functional relationships is slightly different. Hence, the present results suggest that age-related differences in BOLD variability throughout the brain may be sensitive to neurodegenerative processes in these AD-sensitive regions, which were not examined by Tsvetanov and colleauges (2019).
4.5. Limitations & Future Directions
Although we present novel relationships between BOLD variability and CSF biomarkers of Aβ42 and NfL, our interpretations of these relationships are limited by their relatively small effect sizes. This may be in part due to our conservative control of motion and global signal artifacts in our analytic approach. The relatively small magnitude of these effects is also likely a consequence of testing these relationships in cognitively normal individuals, which of course is critical to the aim of testing relationships with preclinical pathology. Hence, these relationships should be replicated in an independent sample.
Additionally, as noted, MRI data were collected on two separate Siemens Trio 3T scanners and thus, there may be scanner-related differences in both functional and structural measures. Although we replicated the main biomarker results in residualized estimates after regressing out scanner differences (see Supplementary Results), future studies should replicate these effects with large samples collected on the same scanner or applying more advanced methods to harmonize data across scanners (Fortin et al., 2018; Yu et al., 2018).
Also, as noted, our findings regarding the influence of CVH and WMH may be limited based on selection of the sample. Overall levels of WMH were relatively low in comparison to the general aging population, due to screening of the cohort. Thus, these results should be replicated in a sample with greater vascular risk.
Finally, our analyses were limited to cross-sectional relationships among BOLD SD, the AT(N) biomarkers, cardiovascular measures, and age. Thus, although we observed reductions in sensitivity of BOLD SD after correcting for cardiovascular or structural measures, we cannot assume that they reflect causal contributions to the biomarker and age relationships, as they may be driven by accounting for shared variance among the measures.
4.6. Conclusions
Resting-state BOLD variability is potentially a clinically-informative signal, but it is currently understudied in the area of preclinical AD. The present study provides novel insights into the sensitivity of resting-state BOLD variability to preclinical pathological processes, as well as healthy age differences. Specifically, reductions in BOLD variability within the DMN may reflect subtle preclinical dysfunction as a consequence of amyloid deposition in these areas, in line with previously noted differences in task activation and functional connectivity. Additionally, reduced BOLD variability, particularly in subcortical regions, may also be sensitive to non-specific neurodegenerative processes in AD-sensitive regions, including hippocampus and the AD signature regions. This neurodegeneration-related change may largely contribute to observed age relationships with BOLD variability, in addition to possible cardiovascular and WMH-related factors. Hence, differences in resting-state BOLD variability may reflect a combined influence of cardiovascular and neuronal factors.
Supplementary Material
Acknowledgements
This research was part of P.M.’s dissertation and was supported by the National Institutes of Health (P01-AG026276, P01-AG03991, P-50 AG05681, R01-MH118031, R01-AG052550, R01-AG057680, T32-AG000030-41, 1S10RR022984-01A1, 1S10OD018091-01, K01 AG053474), with generous support from the Paula and Rodger O. Riney Fund and the Daniel J. Brennan MD Fund. We thank the participants for their dedication to this project, Jan Duchek, Doug Garrett, Denise Head, and Steve Petersen for helpful comments, Jo Etzel for consultation on support vector regression analyses, Dimitre Tomov for technical and programming support, and Julie Wisch for data curation support.
Footnotes
Disclosure Statement
The authors declare no competing interests.
References
- Bakkour A, Morris JC, Wolk DA, Dickerson BC, 2013. The effects of aging and Alzheimer’s disease on cerebral cortical anatomy: Specificity and differential relationships with cognition. Neuroimage 76, 332–344. 10.1016/j.neuroimage.2013.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC, 2012. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med 367, 795–804. 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Thomas JB, Snyder AZ, Benzinger TLS, Zhang D, Raichle ME, Holtzman DM, Morris JC, Ances BM, 2012. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J. Neurosci 32, 8890–9. 10.1523/JNEUROSCI.5698-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Thomas JB, Snyder AZ, Wang L, Fagan AM, Benzinger TLS, Morris JC, Ances BM, 2014. Unrecognized preclinical Alzheimer disease confounds rs-fcMRI studies of normal aging. Neurology 83, 1613–1619. 10.1212/WNL.0000000000000939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Head D, Parker J, Fotenos AF, Marcus D, Morris JC, Snyder AZ, 2004. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: Reliability and validation against manual measurement of total intracranial volume. Neuroimage 23, 724–738. 10.1016/j.neuroimage.2004.06.018 [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis g, Morris JC, Mintun MA, 2005. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci 25, 7709–17. 10.1523/JNEUROSCI.2177-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzynska AZ, Wong CN, Voss MW, Cooke GE, McAuley E, Kramer AF, 2015. White matter integrity supports BOLD signal variability and cognitive performance in the aging human brain. PLoS One 10, 1–17. 10.1371/journal.pone.0120315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JH, Franke K, 2017. Predicting Age Using Neuroimaging: Innovative Brain Ageing Biomarkers. Trends Neurosci 40, 681–690. 10.1016/j.tins.2017.10.001 [DOI] [PubMed] [Google Scholar]
- Corder EH, Sauners AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA, 1993. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science (80-.) 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Deco G, Jirsa VK, McIntosh AR, 2011. Emerging concepts for the dynamical organization of resting-state activity in the brain. Nat. Rev. Neurosci 12, 43–56. 10.1038/nrn2961 [DOI] [PubMed] [Google Scholar]
- Dosenbach NUF, Nardos B, Cohen AL, Fair DA, Power JD, Church J. a, Nelson SM, Wig GS, Vogel AC, Lessov-schlaggar CN, Barnes KA, Dubis JW, Feczko E, Coalson RS Jr, J.R.P., Barch DM, Petersen SE, Schlaggar BL, 2010. Prediction of Individual Brain Maturity Using fMRI. Science (80-.) 329, 1358–1361. 10.1126/science.1194144.Prediction [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM, 2007. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch. Neurol 64, 343–349. 10.1001/archneur.64.3.noc60123 [DOI] [PubMed] [Google Scholar]
- Filippi M, Rocca MA, De Stefano N, Enzinger C, Fischer E, Horsfield MA, Inglese M, Pelletier D, Comi G, 2011. Magnetic Resonance Techniques in Multiple Sclerosis: The Present and the Future. Neurol. Rev 68, 1514–1520. 10.1093/brain/123.5.1066 [DOI] [PubMed] [Google Scholar]
- Fischl B, 2012. FreeSurfer. Neuroimage2 62, 774–781. 10.1016/j.neuroimage.2012.01.021.FreeSurfer [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, Dale AM, Walhovd KB, 2009. High consistency of regional cortical thinning in aging across multiple samples. Cereb. Cortex 19, 2001–2012. 10.1093/cercor/bhn232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin JP, Cullen N, Sheline YI, Taylor WD, Aselcioglu I, Cook PA, Adams P, Cooper C, Fava M, McGrath PJ, McInnis M, Phillips ML, Trivedi MH, Weissman MM, Shinohara RT, 2018. Harmonization of cortical thickness measurements across scanners and sites. Neuroimage 167, 104–120. 10.1016/j.neuroimage.2017.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Zhang D, Snyder AZ, Raichle ME, 2009. The global signal and observed anticorrelated resting state brain networks. J. Neurophysiol 101, 3270–83. 10.1152/jn.90777.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett DD, Epp SM, Perry A, Lindenberger U, 2018. Local temporal variability reflects functional integration in the human brain. Neuroimage 183, 776–787. 10.1016/j.neuroimage.2018.08.019 [DOI] [PubMed] [Google Scholar]
- Garrett DD, Kovacevic N, McIntosh AR, Grady CL, 2011. The importance of being variable. J. Neurosci 31, 4496–4503. 10.1523/JNEUROSCI.5641-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett DD, Kovacevic N, McIntosh AR, Grady CL, 2010. Blood oxygen level-dependent signal variability is more than just noise. J. Neurosci 30, 4914–4921. 10.1523/JNEUROSCI.5166-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett DD, Lindenberger U, Hoge RD, Gauthier CJ, 2017. Age differences in brain signal variability are robust to multiple vascular controls. Sci. Rep 7, 1–13. 10.1038/s41598-017-09752-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett DD, Samanez-Larkin GR, MacDonald SWS, Lindenberger U, McIntosh AR, Grady CL, 2013. Moment-to-moment brain signal variability: A next frontier in human brain mapping? Neurosci. Biobehav. Rev 37, 610–624. 10.1016/j.neubiorev.2013.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good TJ, Villafuerte J, Ryan JD, Grady CL, Barense MD, 2020. Resting state bold variability of the posterior medial temporal lobe correlates with cognitive performance in older adults with and without risk for cognitive decline. eNeuro 7 10.1523/ENEURO.0290-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, 2020. Neurofilaments in disease: what do we know? Curr. Opin. Neurobiol 61, 105–115. 10.1016/j.conb.2020.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL, Garrett DD, 2018. Accepted Manuscript: Brain signal variability is modulated as a function of internal and external demand in younger and older adults. Neuroimage 169, 510–523. 10.1016/j.neuroimage.2017.12.031 [DOI] [PubMed] [Google Scholar]
- Grady CL, Garrett DD, 2014. Understanding variability in the BOLD signal and why it matters for aging. Brain Imaging Behav. 8, 274–283. 10.1007/s11682-013-9253-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart-Masip M, Salami A, Garrett DD, Rieckmann A, Lindenberger U, Bäckman L, 2016. BOLD Variability is Related to Dopaminergic Neurotransmission and Cognitive Aging. Cereb. Cortex 26, 2074–2083. 10.1093/cercor/bhv029 [DOI] [PubMed] [Google Scholar]
- Han Y, Lui S, Kuang W, Lang Q, Zou L, Jia J, 2012. Anatomical and functional deficits in patients with amnestic mild cognitive impairment. PLoS One 7 10.1371/journal.pone.0028664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wang J, Zhao Z, Min B, Lu J, Li K, He Y, Jia J, 2011. Frequency-dependent changes in the amplitude of low-frequency fluctuations in amnestic mild cognitive impairment: A resting-state fMRI study. Neuroimage 55, 287–295. 10.1016/j.neuroimage.2010.11.059 [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA, 1992. Alzheimer’s Disease : The Amyloid Cascade Hypothesis. Science (80-.) 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Hedden T, Van Dijk KRA, Becker JA, Mehta A, Sperling RA, Johnson KA, Buckner RL, 2009. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J. Neurosci 29, 12686–12694. 10.1523/JNEUROSCI.3189-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Chao HHA, Zhang S, Ide JS, Li CSR, 2014. Changes in cerebral morphometry and amplitude of low-frequency fluctuations of BOLD signals during healthy aging: Correlation with inhibitory control. Brain Struct. Funct 219, 983–994. 10.1007/s00429-013-0548-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust WJ, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling RA, Elliott C, Masliah E, Ryan L, Silverberg N, 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 14, 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B, 2016. A new classification system for AD, independent of cognition A / T / N : An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 0, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ, 2013. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang S, Zhang X, Wang Z, Tian X, He Y, 2014. Abnormal amplitude of low-frequency fluctuations of intrinsic brain activity in Alzheimer’s disease. J. Alzheimer’s Dis 40, 387–397. 10.3233/JAD-131322 [DOI] [PubMed] [Google Scholar]
- Lustig C, Snyder AZ, Bhakta M, O’Brien KC, McAvoy M, Raichle ME, Morris JC, Buckner RL, 2003. Functional deactivations: change with age and dementia of the Alzheimer type. Proc Natl Acad Sci U S A 100, 14504–14509. 10.1073/pnas.2235925100\r2235925100 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez De La Plata C, Ardelean A, Koovakkattu D, Srinivasan P, Miller A, Phuong V, Harper C, Moore C, Whittemore A, Madden C, Diaz-Arrastia R, Devous M, 2007. Magnetic resonance imaging of diffuse axonal injury: Quantitative assessment of white matter lesion volume. J. Neurotrauma 24, 591–598. 10.1089/neu.2006.0214 [DOI] [PubMed] [Google Scholar]
- Meyer D, Dimitriadou E, Hornik K, Weingessel A, Leisch F, Chang C-C, Lin C-C, 2017. Misc Functions of the Department of Statistics, Probability Theory Group (E1071), TUWien. Compr. R Arch. Netw [Google Scholar]
- Millar PR, Petersen SE, Ances BM, Gordon BA, Benzinger TLS, Morris JC, Balota DA, 2020. Evaluating the sensitivity of resting-state BOLD variability to age and cognition after controlling for motion and cardiovascular influences: A network-based approach. Cereb. Cortex 1–16. 10.1093/cercor/bhaa138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, MacH RH, Klunk WE, Mathis CA, Dekosky ST, Morris JC, 2006. [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology 67, 446–452. 10.1212/01.wnl.0000228230.26044.a4 [DOI] [PubMed] [Google Scholar]
- Morris JC, 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–4. [DOI] [PubMed] [Google Scholar]
- Nielsen AN, Barch DM, Petersen SE, Schlaggar BL, Greene DJ, 2020. Machine Learning With Neuroimaging: Evaluating Its Applications in Psychiatry. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 1–8. 10.1016/j.bpsc.2019.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AN, Greene DJ, Gratton C, Dosenbach NUF, Petersen SE, Schlaggar BL, 2019. Evaluating the Prediction of Brain Maturity From Functional Connectivity After Motion Artifact Denoising. Cereb. Cortex 29, 2455–2469. 10.1093/cercor/bhy117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomi JS, Bolt TS, Ezie CEC, Uddin LQ, Heller AS, 2017. Moment-to-Moment BOLD Signal Variability Reflects Regional Changes in Neural Flexibility across the Lifespan. J. Neurosci 37, 5539–5548. 10.1523/JNEUROSCI.3408-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojemann JG, Akbudak E, Snyder AZ, McKinstry RC, Raichle ME, Conturo TE, 1997. Anatomic localization and quantitative analysis of gradient refocused echo-planar fMRI susceptibility artifacts. Neuroimage 6, 156–167. 10.1006/nimg.1997.0289 [DOI] [PubMed] [Google Scholar]
- Pantoni L, 2010. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. 10.1016/S1474-4422(10)70104-6 [DOI] [PubMed] [Google Scholar]
- Power JD, Barnes K. a., Snyder AZ, Schlaggar BL, Petersen SE, 2012. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 59, 2142–2154. 10.1016/j.neuroimage.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Cohen AL, Nelson SM, Wig GS, Barnes KA, Church J. a., Vogel AC, Laumann TO, Miezin FM, Schlaggar BL, Petersen SE, 2011. Functional Network Organization of the Human Brain. Neuron 72, 665–678. 10.1016/j.neuron.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarapicchia V, Mazerolle EL, Fisk JD, Ritchie LJ, Gawryluk JR, 2018. Resting State BOLD Variability in Alzheimer’s Disease: A Marker of Cognitive Decline or Cerebrovascular Status? Front. Aging Neurosci 10, 1–13. 10.3389/fnagi.2018.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler SE, Gray JD, Gordon BA, Xiong C, Batrla-Utermann R, Quan M, Wahl S, Benzinger TLS, Holtzman DM, Morris JC, Fagan AM, 2018. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimer’s Dement. 14, 1460–1469. 10.1016/j.jalz.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt P, Gaser C, Arsic M, Buck D, Förschler A, Berthele A, Hoshi M, Ilg R, Schmid VJ, Zimmer C, Hemmer B, Mühlau M, 2012. An automated tool for detection of FLAIR-hyperintense white-matter lesions in Multiple Sclerosis. Neuroimage 59, 3774–3783. 10.1016/j.neuroimage.2011.11.032 [DOI] [PubMed] [Google Scholar]
- Schmidt R, Fazekas F, Kapeller P, Schmidt H, Hartung HP, 1999. MRI white matter hyperintensities: Three-year follow-up of the Austrian Stroke Prevention Study. Neurology 53, 132–139. 10.1212/wnl.53.1.132 [DOI] [PubMed] [Google Scholar]
- Seitzman BA, Gratton C, Marek S, Raut RV, Dosenbach NUF, Schlaggar BL, Petersen SE, Greene DJ, 2020. A set of functionally-defined brain regions with improved representation of the subcortex and cerebellum. Neuroimage 206, 1–17. 10.1016/j.neuroimage.2019.116290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Raichle ME, 2013. Resting state functional connectivity in preclinical Alzheimer’s disease. Biol. Psychiatry 74, 340–347. 10.1016/j.biopsych.2012.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GL, Pope DLW, Astafiev SV, McAvoy MP, Snyder AZ, Corbetta M, 2010. Right hemisphere dominance during spatial selective attention and target detection occurs outside the dorsal frontoparietal network. J. Neurosci 30, 3640–3651. 10.1523/JNEUROSCI.4085-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwinski MJ, Lipton RB, Buschke H, Stewart W, 1996. The effects of preclinical dementia on estimates of normal cognitive functioning in aging. Journals Gerontol. - Ser. B Psychol. Sci. Soc. Sci 51, 217–225. 10.1093/geronb/51B.4.P217 [DOI] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA, 2010. Functional alterations in memory networks in early alzheimer’s disease. NeuroMolecular Med. 12, 27–43. 10.1007/s12017-009-8109-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, LaViolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA, 2009. Amyloid Deposition Is Associated with Impaired Default Network Function in Older Persons without Dementia. Neuron 63, 178–188. 10.1016/j.neuron.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, Mintun MA, Benzinger TLS, 2013. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One 8 10.1371/journal.pone.0073377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetanov KA, Henson RN, Jones PS, Mutsaerts H-J, Fuhrmann D, Tyler LK, Cam-CAN, Rowe JB, 2019. The effects of age on resting-state BOLD signal variability is explained by cardiovascular and neurovascular factors. bioRxiv 836619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetanov KA, Henson RNA, Tyler LK, Davis SW, Shafto MA, Taylor JR, Williams N, Rowe JB, 2015. The effect of ageing on fMRI: Correction for the confounding effects of vascular reactivity evaluated by joint fMRI and MEG in 335 adults. Hum. Brain Mapp 36, 2248–2269. 10.1002/hbm.22768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinters HV, 1987. Cerebral amyloid angiopathy a critical review. Stroke 18, 311–324. 10.1161/01.STR.18.2.311 [DOI] [PubMed] [Google Scholar]
- Vlassenko AG, McCue L, Jasielec MS, Su Y, Gordon BA, Xiong C, Holtzman DM, Benzinger TLS, Morris JC, Fagan AM, 2016. Imaging and cerebrospinal fluid biomarkers in early preclinical alzheimer disease. Ann. Neurol 80, 379–387. 10.1002/ana.24719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJB, Xiong C, Visser PJ, Jasielec MS, Hassenstab JJ, Grant EA, Cairns NJ, Morris JC, Holtzman DM, Fagan AM, 2013. Preclinical Alzheimer’s disease and its outcome: A longitudinal cohort study. Lancet Neurol. 12, 957–965. 10.1016/S1474-4422(13)70194-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Benzinger TLS, Hassenstab JJ, Blazey TM, Owen C, Liu J, Fagan AM, Morris JC, 2015. Spatially distinct atrophy is linked to beta-amyloid and tau in preclinical Alzheimer disease. Neurology 84, 1254–1260. 10.1212/WNL.0000000000001401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Benzinger TLS, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, Ances BM, 2016. Evaluation of Tau imaging in staging Alzheimer disease and revealing interactions between β-Amyloid and tauopathy. JAMA Neurol. 73, 1070–1077. 10.1001/jamaneurol.2016.2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brier MR, Snyder AZ, Thomas JB, Fagan AM, Xiong C, Benzinger TLS, Holtzman DM, Morris JC, Ances BM, 2013. Cerebrospinal fluid Aβ42, phosphorylated tau181, and resting-state functional connectivity. JAMA Neurol. 70, 1242–1248. 10.1001/jamaneurol.2013.3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisch JK, Roe CM, Babulal GM, Schindler SE, Fagan AM, Benzinger TLS, Morris JC, Ances BM, 2020. Resting State Functional Connectivity Signature Differentiates Cognitively Normal from Individuals Who Convert to Symptomatic Alzheimer’s Disease. J. Alzheimer’s Dis 1–11. 10.3233/JAD-191039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, Zhao X, Wang P, Guo Q, Jiang H, Cao X, He Y, Yan C, 2012. Spontaneous brain activity in mild cognitive impairment revealed by amplitude of low-frequency fluctuation analysis: A resting-state fMRI study. Radiol. Medica 117, 865–871. 10.1007/s11547-011-0780-8 [DOI] [PubMed] [Google Scholar]
- Yu M, Engels MMA, Hillebrand A, Van Straaten ECW, Gouw AA, Teunissen C, Van Der Flier WM, Scheltens P, Stam CJ, 2017. Selective impairment of hippocampus and posterior hub areas in Alzheimer’s disease: An MEG-based multiplex network study. Brain 140, 1466–1485. 10.1093/brain/awx050 [DOI] [PubMed] [Google Scholar]
- Yu M, Linn KA, Cook PA, Phillips ML, McInnis M, Fava M, Trivedi MH, Weissman MM, Shinohara RT, Sheline YI, 2018. Statistical harmonization corrects site effects in functional connectivity measurements from multi-site fMRI data. Hum. Brain Mapp 39, 4213–4227. 10.1002/hbm.24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zuo X, Ng KK, Su J, Chong X, Shim HY, Qin M, Ong W, Loke YM, Choo BL, Jun E, Chong Y, W, Z.X., Hilal S, Venketasubramanian N, Tan BY, Chen CL, Zhou JH, 2020. Distinct BOLD variability changes in the default mode and salience networks in Alzheimer ‘ s disease spectrum and associations with cognitive decline. Sci. Rep 10, 1–12. 10.1038/s41598-020-63540-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZL, Fan FM, Lu J, Li HJ, Jia LF, Han Y, Li KC, 2015. Changes of gray matter volume and amplitude of low-frequency oscillations in amnestic MCI: An integrative multi-modal MRI study. Acta radiol. 56, 614–621. 10.1177/0284185114533329 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.