

SUMMARY
Cancer cells, like microbes, live in complex metabolic environments. Recent evidence suggests that microbial behavior across metabolic environments is well described by simple empirical growth relationships, or growth laws. Do such empirical growth relationships also exist in cancer cells? To test this question, we develop a high-throughput approach to extract quantitative measurements of cancer cell behaviors in systematically altered metabolic environments. Using this approach, we examine relationships between growth and three frequently studied cancer phenotypes: drug-treatment survival, cell migration, and lactate overflow. Drug-treatment survival follows simple linear growth relationships, which differ quantitatively between chemotherapeutics and EGFR inhibition. Cell migration follows a weak grow-and-go growth relationship, with substantial deviation in some environments. Finally, lactate overflow is mostly decoupled from growth rate and is instead determined by the cells’ ability to maintain high sugar uptake rates. Altogether, this work provides a quantitative approach for formulating empirical growth laws of cancer.
Graphical Abstract

In Brief
Kochanowski et al. quantify cancer cell phenotypes across systematically altered in vitro metabolic environments to search for phenotype-growth relationships, similar to the growth laws found in microbes. Three case studies highlight examples in which such growth relationships are clearly operating (cancer drug survival), weakly present (cell migration), or absent (lactate overflow).
INTRODUCTION
Cancer cells share recurrent phenotypic alterations, including deregulated growth, increased cell migration, and elevated nutrient uptake (Hanahan and Weinberg, 2000, 2011; Pavlova and Thompson, 2016). Effort has gone into elucidating the impact of genetics on these phenotypes, for example, the mutations in signaling pathways that enable cancer cells to grow in the absence of growth signals or to resist apoptosis signals (Sanchez-Vega et al., 2018). In comparison, less is known about how microenvironments affect cancer cell phenotypes. This is particularly true for metabolic environments: although cancer cells live in complex metabolic environments, encompassing a diverse range of nutrients and concentrations (Hensley et al., 2016; Kamphorst et al., 2015; Reznik et al., 2018; Sullivan et al., 2019), their impact on cancer cell phenotypes is only poorly understood.
A common approach to investigate the impact of metabolic environments in vitro is to subject cancer cells to pairs of defined culture media in which the concentration of one individual component has been altered. Such efforts have been instrumental, for example, in elucidating how changes to methionine (Mentch et al., 2015; Wang et al., 2019), glucose (Birsoy et al., 2014), and glutamine (Chen et al., 2019; Timmerman et al., 2013) availability affect cancer cell growth. However, the extent to which changes in a metabolic environment affect phenotypes other than growth is often unclear. Moreover, a limited number of pairwise comparisons may not provide insight into how complex metabolic environments affect cancer cell phenotypes. In particular, such pairwise comparisons make it difficult to detect overarching relationships by which different metabolic environments affect a phenotype of interest.
Evidence from microbes suggests that such overarching relationships may exist. Recent works have shown that for many microbial phenotypes, such as antibiotic survival (Brauner et al., 2016; Fung et al., 2010), colony expansion (Cremer et al., 2019), constitutive gene expression (Berthoumieux et al., 2013; Gerosa et al., 2013; Kochanowski et al., 2017), or overflow metabolism (Basan et al., 2015; Brauer et al., 2008), the impact of metabolic environments follows simple empirical relationships that relate cell behavior to the growth rate supported by each environment. These empirical relationships, or growth laws, have been instrumental in elucidating the interplay between metabolic activity and growth and in developing predictive phenomenological models of cell behavior (Basan et al., 2015; Berthoumieux et al., 2013; Cremer et al., 2019; Kochanowski et al., 2017). However, whether cancer cell phenotypes also follow such empirical growth relationships is unclear, and current efforts are hampered by a lack of commercially available culture media with altered nutrient composition.
Here, we develop a combined experimental and computational approach to monitor cancer cell behavior across systematically altered metabolic environments and search for empirical growth relationships (Figure 1). As a proof of concept, we examine the impact of metabolic environments on three cancer cell phenotypes commonly studied in the context of cancer metabolism: survival of treatment with lethal doses of cancer drugs (Hardeman et al., 2017; Momcilovic et al., 2017; Viale and Draetta, 2016; Viswanathan et al., 2017), cell migration (Yizhak et al., 2014; Zanotelli et al., 2018), and lactate overflow (Jain et al., 2012; Ortmayr et al., 2019). We find that across diverse metabolic environments, empirical growth relationships are clearly pronounced for drug-treatment survival, weakly present for cell migration, and absent for lactate overflow. Follow-up metabolomics and perturbation experiments suggest that lactate overflow is instead determined by the cells’ ability to maintain high rates of sugar uptake. Overall, this work provides a framework to disentangle the complex interplay of metabolic environment, growth, and cancer cell behavior and takes the first step toward formulating the empirical growth laws of cancer.
Figure 1.
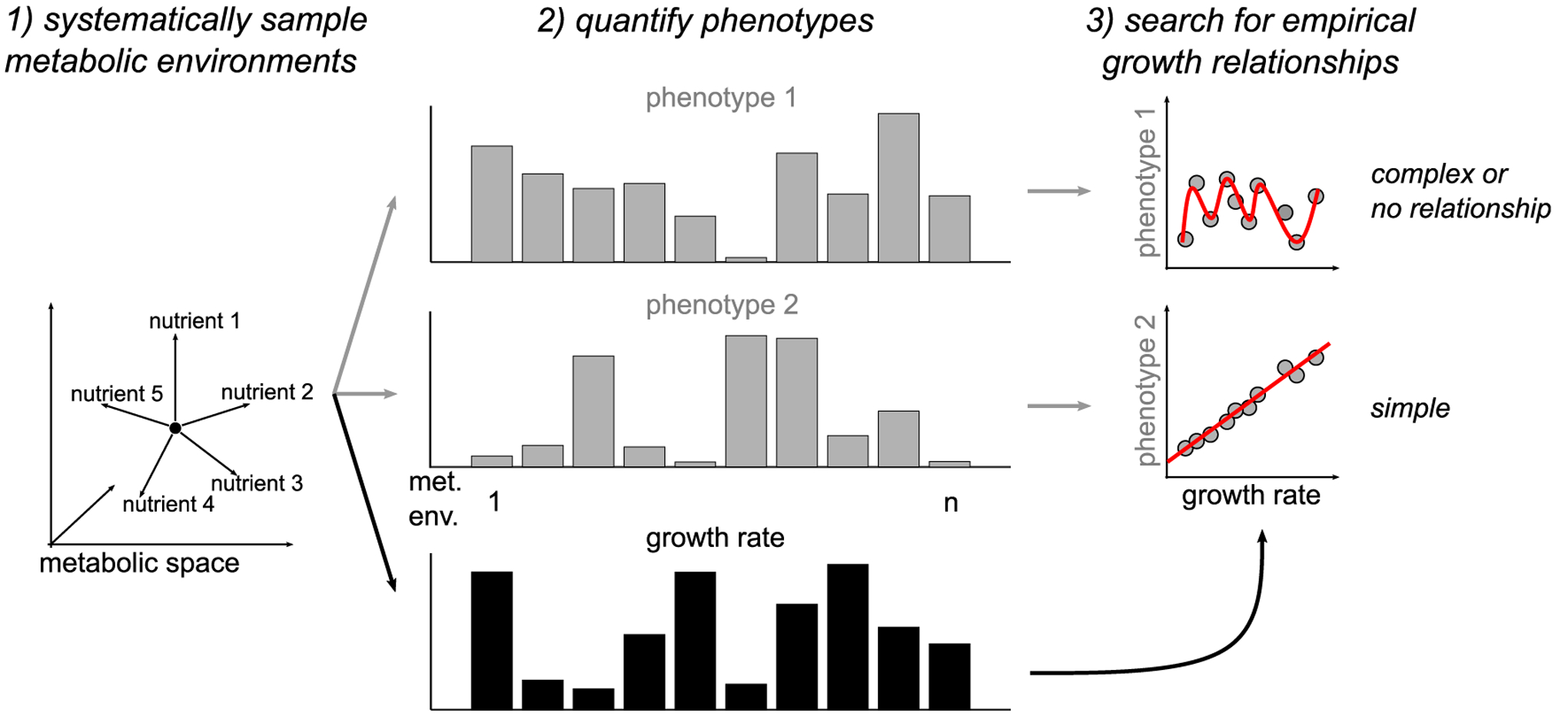
Schematic of approach to search for empirical growth relationships across metabolic environments
RESULTS
Experimental workflow to generate defined metabolic environments
To enable systematic examination of growth relationships between in vitro metabolic environment and cancer cell behavior, we adapted a synthetic media formulation (Cantor et al., 2017) and used automated liquid transfer to expose cells to diverse arrays of culture media with distinct nutrient composition in a 96- or 384-well plate format (Figure 2A; see STAR methods). We benchmarked this workflow by examining the impact of systematically altering amino acid and sugar composition on cell viability and growth in two adherent human cancer cell lines with different tissues of origin: PC9 (non-small cell lung cancer) and A375 (melanoma), established in vitro models of EGFR- and BRAF-driven cancer, respectively (Kochanowski et al., 2018; Shaffer et al., 2017; Sharma et al., 2010). Altogether, we examined more than 100 distinct nutrient compositions (Figures 2B and S1), using a standard tissue culture medium (RPMI 1640) as a reference point (see the list of used nutrients in Tables S1 and S2).
Figure 2. Experimental pipeline to cultivate cancer cells in multiple in vitro metabolic environments.

(A) Schematic of the experimental pipeline.
(B) Top row: cell number at 72 h (relative to 0 h) of PC9 cells upon amino acid depletion (diamonds) or replacement of glucose for alternative sugars (circles). Overall, >100 metabolic environments with unique nutrient composition were tested. Bottom row: corresponding maximal growth rate (relative to the reference condition). Error bars denote standard deviation (n = 2–3).
The data recapitulated many qualitative findings from previous reports, such as the well-established essentiality of 13 amino acids for mammalian cell growth in vitro (Eagle, 1955) and the ability of some nutrients to support growth (galactose, mannose, fructose, and maltose) or extend cell survival without growth (sorbitol) when glucose is absent (Figures 2B and S1A). In addition, these data enabled us to observe relationships between metabolic environments and cell growth that are difficult to detect without systematic, high-throughput metabolic perturbations. First, the impact of complete nutrient dropout on cell survival varied considerably among the essential nutrients (typically within the 10-fold range, with some more extreme deviations), as well as between the two cell lines (e.g., glucose and threonine in Figure S1A). Second, for most essential nutrients, the maximal growth rate only diminished once nutrient concentrations were reduced more than 10-fold compared with standard cell culture conditions (Figures 2B and S1B), highlighting the importance of examining nutrients over a range of concentrations. These data demonstrated our ability to systematically and quantitatively examine cellular responses across multiple metabolic environments in vitro and highlight the range of cell growth rates supported by these diverse conditions.
Identify empirical growth relationships in three common cancer phenotypes
Using this workflow, we next wanted to identify the growth relationships by which these different metabolic environments affect cancer cell phenotypes. We chose to investigate three phenotypes that are frequently studied in the context of cancer metabolism and can be readily quantified in vitro: survival of cancer drug treatment (Hardeman et al., 2017; Momcilovic et al., 2017; Viale and Draetta, 2016; Viswanathan et al., 2017), cell migration (Yizhak et al., 2014; Zanotelli et al., 2018), and lactate overflow (Jain et al., 2012; Ortmayr et al., 2019) (i.e., the rapid conversion of glucose to lactate in the presence of oxygen; DeBerardinis and Chandel, 2020). For all three phenotypes, both metabolism and cell growth have been implicated as determinants of cell behavior. For example, slow growth has been associated with increased tolerance to both chemotherapeutics (Baguley et al., 1995; Pearl Mizrahi et al., 2016) and targeted therapy (Roesch et al., 2010, 2013), and lactate overflow is considered a common feature of many fast-growing cell types (DeBerardinis and Chandel, 2020; Vander Heiden et al., 2009). However, the extent and direction of these postulated growth relationships are still largely unclear. For example, both negative (Atkins et al., 2019; Giese et al., 1996; Tiek et al., 2018) and positive (Garay et al., 2013) relationships between growth rate and cell migration have been reported in the literature.
To systematically explore the relationships among metabolic environment, growth, and cell phenotype in a constant genetic background, we quantified each phenotype in one cell line (PC9) across ~20 diverse metabolic environments that support exponential growth for the duration of the experiment across a range of growth rates. Each phenotype was quantified using established experimental approaches (Forcina et al., 2017; Jain et al., 2012; Jaqaman et al., 2008). Specifically, we examined (1) survival of treatment with lethal doses of cancer drugs (Hardeman et al., 2017; Momcilovic et al., 2017) by quantifying the fraction of dead cells in a population (termed lethal fraction) over time with fluorescent microscopy (Forcina et al., 2017) (Figures 3A and 3B; see STAR methods), (2) cell migration by automatically tracking (Jaqaman et al., 2008) the undirected movement of hundreds of individual cells with high-temporal resolution microscopy (Figures 4A and 4B; see STAR methods), and (3) lactate overflow by quantifying lactate secretion rates using a recently published experimental protocol (Jain et al., 2012) (Figures 5A and 5B; see STAR Methods). To identify empirical growth relationships for each phenotype, we developed an analytical approach based on the Akaike Information Criterion (AIC) (Burnham et al., 2011) to test whether any low-dimensional polynomial fit better explains the observed phenotype-growth relationship across metabolic environments than the null hypothesis (i.e., no relationship between phenotype and growth rate; see STAR methods and Figure S2 for illustrations of the analytical approach).
Figure 3. Divergent impacts of metabolic environments on chemotherapy and targeted therapy survival in PC9 cells.
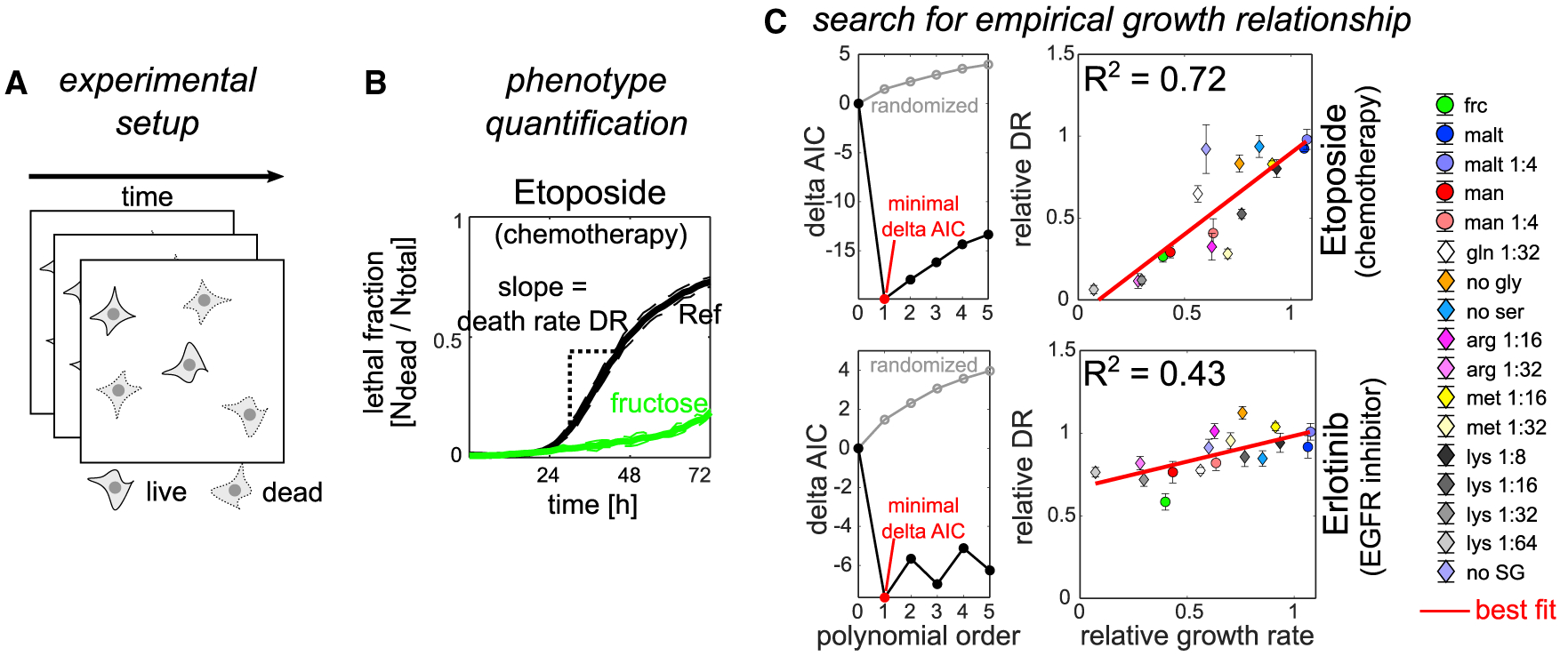
(A) Schematic of the experimental approach.
(B) Example lethal fraction time courses highlighting the analytical approach to quantify drug survival. Black, reference condition. Green, fructose. Dashed lines denote standard deviation (n = 3).
(C) Search for an empirical growth relationship for etoposide (top row) or erlotinib (bottom row). Left: delta AIC (ΔAIC) (compared with zero polynomial order) as a function of polynomial order. Black, original data. Gray, median of randomizing data labels 10,000 times. Red circle, polynomial order with minimal ΔAIC (= best fit). Right: death rate (relative to the reference condition) across metabolic environments plotted as a function of growth rate. Red line, best fit. Error bars denote standard deviation (n = 3).
Figure 4. Impact of metabolic environments on cancer cell migration in PC9 cells follows a grow-and-go scheme.

(A) Schematic of the experimental approach.
(B) Example cell tracking data highlighting the analytical approach to quantify cell migration. Left: rose plot of individual tracks captured (with track number n) over 18 h in fructose. Each track was realigned to the origin. Right: corresponding distribution of average speed across tracks. Black, reference condition (all nutrients present at a concentration corresponding to standard RPMI 1640 media). Green, fructose. Shown are distributions of three separate replicates. Red dashed line, median of distribution across all replicates.
(C) Search for an empirical growth relationship for cell migration. Left: ΔAIC (compared with zero polynomial order) as a function of polynomial order. Black, original data. Gray, median of randomizing data labels 10,000 times. Red circle, polynomial order with minimal ΔAIC (= best fit). Right: population speed (median of the average speed shown in B) plotted against the corresponding growth rate in 19 metabolic environments. Red line, best fit. Error bars denote standard deviation (n = 3).
Figure 5. Lactate overflow is largely decoupled from growth rate across metabolic environments in PC9 cells.

(A) Schematic of the experimental approach.
(B) Example data highlighting the analytical approach to quantify the lactate secretion rate. Left: cell number time courses. Black, reference condition (glucose). Green, fructose. Right: corresponding produced lactate.
(C) Search for an empirical growth relationship for the lactate secretion rate. Left: ΔAIC (compared with zero polynomial order) as a function of polynomial order. Red circle, polynomial order with minimal ΔAIC (= best fit). Right: lactate secretion rate as a function of growth rate relative to the reference condition (all nutrients present at a concentration corresponding to standard RPMI 1640 media) across metabolic environments in PC9 cells. Red line, best fit. Error bars denote standard deviation (n = 3).
First, we examined the impact of metabolic environments on the efficacy of five cancer therapeutics with distinct modes of action. With our experimental approach, we measured cell survival in high doses of three commonly used chemotherapeutics (topoisomerase inhibitor etoposide, folate antimetabolite pemetrexed, and microtubule inhibitor paclitaxel) and two EGFR inhibitors (reversible inhibitor erlotinib and covalent inhibitor osimertinib) as examples of targeted therapy (Kochanowski et al., 2018). In metabolic environments that resemble standard tissue culture conditions, all tested drugs readily killed most cells within 72 h (maximal lethal fraction of ~0.5 or higher; black lines in Figure S3A), but across metabolic environments, cell survival varied substantially (example in Figure 3B; red lines in Figure S3A). In all tested cancer therapeutics, the rate of cell death (see STAR methods) across conditions was well described by a linear empirical growth relationship (R2 of the fit between 0.43 and 0.72; see Figures 3C, S3B, and S3C), with most conditions deviating less than 2-fold from the predicted growth relationship (Figure S3D). However, this empirical growth relationship differed quantitatively among the tested drugs, yielding two distinct sets of parameters (Figure S3E). Specifically, although chemotherapeutics showed a strong growth relationship across conditions (up to an ~10-fold difference in death rate compared with the reference condition), EGFR inhibitor efficacy was only moderately affected (a maximally 2-fold difference in death rate). Other metrics of drug efficacy, such as the drug-induced-proliferation rate (Harris et al., 2016), showed similar growth-dependent patterns (Figure S3F). Altogether, these data suggested that the well-established negative relationship between growth rate and cancer drug sensitivity across genetic backgrounds (Baguley et al., 1995) holds true in cells of a common genetic background grown across multiple metabolic environments. However, the extent of this effect depends on the cancer drug being used, with chemotherapeutic efficacy being more sensitive to metabolic environment than targeted EGFR inhibition.
Second, we examined the impact of metabolic environments on cell migration. As reported previously (Takai et al., 2014), PC9 cells were highly motile in metabolic environments that resemble standard tissue culture conditions (black line in the right panel of Figure 4B; Figure S4A). Moreover, there was large variability in cell motility across metabolic environments (Figures 4C and S4A). This pattern was robust to changes in the number of cells seeded, suggesting that it is not merely a function of cell density (Figure S4B). Across metabolic environments, these data were best described by a linear empirical growth relationship (R2 of the fit = 0.56; see Figure 4C). However, some conditions deviated substantially from this growth relationship. Most notably, upon complete glutamine dropout, cells showed lower motility than predicted based on the growth rate (Figure S4C), echoing previous reports showing that glutaminase inhibition reduces breast cancer cell migration (Wang et al., 2010). Additional cell motility metrics yielded similar results (Figure S4D). Altogether, these data suggest that metabolic environment does not affect cell migration through a grow-or-go trade-off (Giese et al., 1996). Instead, these data are consistent with a grow-and-go scheme, in line with recent work reporting a positive correlation between growth rate and cell migration across a panel of cancer cell lines in a fixed environment (Garay et al., 2013).
Third, we examined the impact of metabolic environments on lactate overflow (Figures 5A and 5B). As with the cancer drug survival and cell migration phenotypes, we found substantial variability in lactate secretion rate across metabolic environments (Figure 5C). However, in contrast to the previous two phenotypes, this variability was not linked to the growth rate, and no simple empirical growth relationship could account for the observed lactate overflow pattern across metabolic environments (Figure 5C). To test whether this lack of an empirical growth relationship across metabolic environments is specific to PC9 cells, we quantified lactate secretion rates in the same metabolic environments for three additional cancer cell lines with different tissues of origin and driver mutations (A375, A549, and SKBR3) and one non-cancerous cell line (HEK293T). In all tested cell lines, no simple growth relationship could account for the observed lactate overflow pattern (Figure 6). Thus, although lactate overflow is considered a common feature of many fast-growing cell types (DeBerardinis and Chandel, 2020; Vander Heiden et al., 2009), our data suggest that this phenotype is largely decoupled from growth.
Figure 6. No simple empirical growth relationship accounts for lactate overflow across metabolic environments in diverse cell lines.
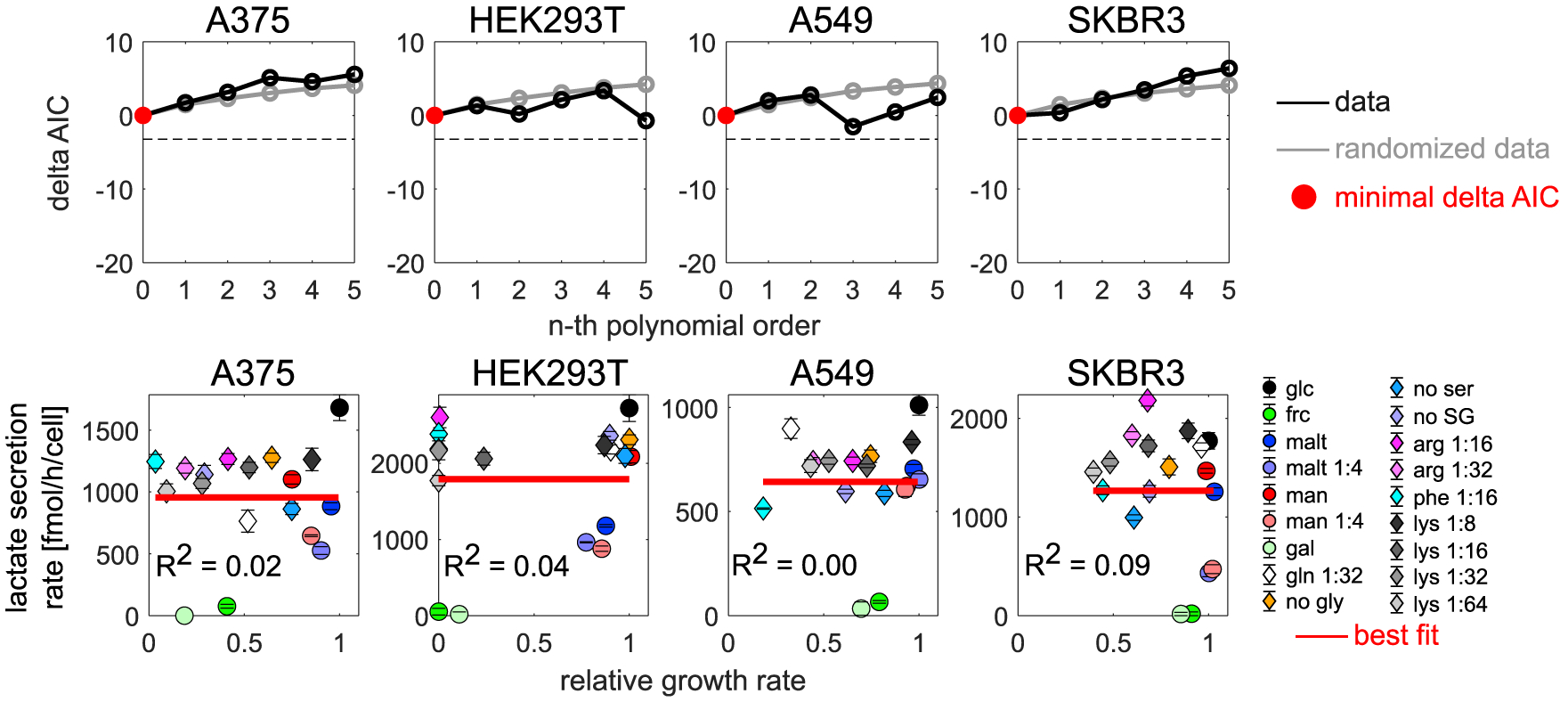
Top row: ΔAIC (compared with zero polynomial order) as a function of polynomial order. Red circle, polynomial order with minimal ΔAIC (= best fit). Horizontal dashed line, ΔAIC cutoff of −3.22 (the respective model must be at least five times more likely than the null model; see STAR methods). Bottom row: lactate secretion rate as a function of growth rate relative to the reference condition (all nutrients present at a concentration corresponding to standard RPMI 1640 media) across metabolic environments. Red line, best fit. Error bars denote standard deviation (n = 3).
Overall, by systematically quantifying cell behavior across multiple metabolic environments, we were able to identify empirical growth relationships in three cancer cell phenotypes. The data revealed three novel insights about the interplay of growth, metabolic environment, and cancer cell behavior. First, the impact of metabolic environment on drug survival followed simple linear growth relationships; however, this differed substantially between chemotherapeutics and EGFR inhibitors. Second, the impact of metabolic environment on cell migration was best explained by a linear positive growth relationship, albeit with some substantially deviating conditions. Third, lactate overflow was largely decoupled from growth not only in PC9 cells but also in other genetically diverse cell lines. Thus, our approach not only put prior intuition on the postulated relationship between growth and cancer cell phenotypes on a quantitative footing but also revealed cases (i.e., lactate overflow) in which differences in growth alone cannot account for the impact of different metabolic environments on cancer cell behavior.
Follow-up on lactate overflow: identifying the underlying mechanism
In the remainder of this study, we aimed to examine the relationship between metabolic environment and lactate overflow in more detail. Although our data showed that growth rate alone could not account for the observed lactate overflow pattern across metabolic environments, closer observation suggested a general pattern separating two types of metabolic environments. Specifically, amino acid variation had limited impact on lactate secretion rate regardless of supported growth rate, whereas there were wide ranges in lactate secretion depending on the supplied sugar (Figure S5A). Moreover, we observed matching changes in the uptake rate of the respective sugars, resulting in a strong proportional relationship between lactate secretion and sugar uptake (Figures 7A, S5B, and S5C). In most cases, sugar concentrations did not drop below 50% of the initial concentration, suggesting sufficient sugar supply over the course of the experiment (Figure S5B). Thus, our data suggested that lactate overflow was coupled to sugar uptake rate instead of growth rate. These findings extend reports showing that lactate secretion and glucose uptake correlate across genetically diverse cell lines grown in a fixed metabolic environment (Chen et al., 2019; Jain et al., 2012; Ortmayr et al., 2019). Our data show that this positive correlation is maintained across sugars in a fixed genetic background and reaffirm the notion that lactate is largely derived from glycolysis (DeBerardinis and Chandel, 2020).
Figure 7. Investigating the source of variability in lactate secretion rate across metabolic environments.

(A) Lactate overflow correlates strongly with sugar uptake rate. Lactate secretion rate plotted against sugar uptake rate across metabolic environments in PC9 cells (top) and A375 cells (bottom). Continuous black line, 1:1 molar relationship of lactate/sugar uptake. Dashed line, 2:1 molar relationship. Error bars denote standard deviation (n = 3).
(B) Using metabolomics to examine potential sources for variation in lactate overflow. Left: schematic of mammalian sugar utilization and catabolism by oxidative phosphorylation or lactate fermentation. Boxed metabolites (e.g., G6P) denote hexose-phosphates (Hexose-Ps). Right: ratio of AMP/ATP relative to the reference condition and Hexose-P plotted against the corresponding lactate secretion rate (relative to the reference condition) in PC9 and A375 cells. Error bars denote standard deviation (n = 3).
(C) Ability to increase lactate fermentation determines energetic response to complex I inhibition. ATP concentration (determined with CellTiterGlo, normalized to signal at t = 0 min) upon complex I inhibition with 1 μM rotenone in PC9 cells growing in conditions with high (glucose), intermediate (1:4 mannose [man]), or low (fructose [frc]) lactate secretion rates (indicative of the ability to increase lactate overflow if needed). Error bars denote standard deviation (n = 2).
Next, we wanted to elucidate the source of variability in lactate overflow across metabolic environments. A possible explanation for the low lactate secretion rates in metabolic environments, such as fructose and galactose, is that in these cases, ATP production through respiration (which has a higher ATP yield than glycolysis; Figure 7B) already matches the cell’s ATP demand. As a result, additional ATP production through increased glycolysis would be unnecessary. To test this hypothesis, we quantified the ratio of AMP to ATP (a measure of the energetic state of the cell) in metabolic environments with low/intermediate/ high rates of lactate secretion in two cell lines (PC9 and A375) using targeted metabolomics. Overall, both cell lines showed a similar metabolome response in the different metabolic environments (Figures S6A and S6B). Importantly, the AMP to ATP ratio showed a negative relationship with the lactate secretion rate across metabolic environments (Figure 7B), as did, e.g., the guanosine monophosphate (GMP) to guanosine triphosphate (GTP) ratio (Figure S6C). These results suggested that cells growing in environments with low lactate secretion rates are energy limited. Moreover, the observed reduction in the NADH to NAD ratio in environments with low lactate secretion rates (Figure S6C) argued that this energy limitation was not due to a bottleneck in the electron transfer chain (which would instead manifest as an increase in the NADH to NAD ratio, as shown recently; Gui et al., 2016).
Why do cells growing in these metabolic environments with alternative sugars such as fructose not simply increase glycolysis to meet their energy demands? These alternative sugars have distinct entry routes into glycolysis (Figure 7B), which rely on metabolic reactions (i.e., transport and internal conversion reactions) unique to each sugar. A parsimonious explanation is that metabolic bottlenecks somewhere along these unique entry routes render cells unable to increase glycolysis to match their energy demands. Although pinpointing metabolic bottlenecks remains challenging even in microbial systems (Liu et al., 2016), we reasoned that intracellular metabolite data may enable us to distinguish between two potential bottlenecks for each sugar: the internal conversion step (which often manifests as an accumulation of the immediate substrate) or the sugar transport (in which no such accumulation is observed). Our metabolomics data cannot distinguish the hexose-phosphate species unique to each sugar utilization pathway. Nevertheless, the dramatic increase in the total hexose-phosphate pool during growth on galactose, mannose, and fructose (Figure 7B) pointed toward internal bottlenecks, as suggested for galactose-grown HEK293T cells (Oh et al., 2020). In contrast, the lack of hex-ose-phosphate accumulation for maltose pointed toward transport bottlenecks.
Finally, to test the hypothesis that cells are unable to increase the glycolysis of alternative sugars such as fructose, we examined the impact of electron transfer chain inhibition, which forces cells to increase glycolysis to maintain energy balance. During growth on glucose, cells compensated for rotenone inhibition of complex I inhibition by increasing the rate of sugar uptake (Figure S6D), allowing cells to maintain high ATP levels (Figure 7C, left). In contrast, rotenone treatment in metabolic environments with intermediate and low lactate rates (mannose and fructose, respectively) caused a dramatic drop in ATP (Figure 7C, middle and right), in line with previous reports (Marroquin et al., 2007). We could mimic this drop in ATP with rotenone treatment during growth on glucose by inhibiting sugar uptake with the specific GLUT1 inhibitor Bay876 (Figure S6E), which by itself does not affect ATP levels (Figure S6F). Altogether, our results suggest that the low lactate secretion rates observed in slow-growth metabolic environments such as fructose or galactose are driven by the cells’ inability to increase glycolysis because of metabolic bottlenecks in the respective utilization pathways.
DISCUSSION
Here, we systematically searched for empirical growth relationships in the interplay between in vitro metabolic environments and three common cancer cell phenotypes: drug survival, cell migration, and lactate overflow. Quantifying cell behavior in different metabolic environments—with varying sugar and amino acid concentrations—revealed strong empirical growth relationships for chemotherapeutic survival and weak growth relationships for cell motility. In contrast, lactate overflow was largely decoupled from growth and instead was determined by the cells’ ability to maintain high rates of sugar uptake.
The empirical growth relationships across metabolic environments reported here are remarkably similar to the growth laws (Basan, 2018; Scott and Hwa, 2011) by which nutrient availability governs analogous phenotypes in microbes. For example, as we have shown here for chemotherapeutic survival in cancer cells, there is a positive linear relationship between growth and antibiotic survival in bacteria (Brauner et al., 2016; Fung et al., 2010). Similarly, our observed empirical growth relationship for cancer cell migration echoes the impact of growth rate on bacterial colony expansion rate (Cremer et al., 2019). This resemblance is particularly striking when comparing the patterns of overflow metabolism in cancer cells and microbes: as observed here in cancer cells, the main determinant of microbial overflow metabolism is not growth rate per se but rather the type of nutrient limitation cells face in their environment (Basan et al., 2015; Brauer et al., 2008). Thus, our findings suggest that the phenotypic response of cancer cells to changes in metabolic environment may be shaped by metabolic constraints similar to those found in bacteria and may adhere to similar empirical growth laws. Moreover, our findings have implications beyond cancer, because many phenotypes investigated here are not exclusive to cancer cells. For example, lactate overflow is found in many other cell types, such as activated T cells (Brand et al., 1988). Although we have used cancer cells as a test case, this work may guide future efforts to examine whether the observed empirical growth relationships hold for other cell types.
One notable exception to these growth relationships was the moderate impact of metabolic environments on EGFR inhibitor efficacy (Figure 3). Despite recent reports highlighting the importance of metabolism for targeted therapy survival (Hangauer et al., 2017; Momcilovic et al., 2017; Raha et al., 2014), EGFR inhibitor efficacy changed only moderately across metabolic environments for comparable drug concentrations. A possible explanation for this discrepancy is that the targeted metabolic perturbations typically used in the literature (i.e., direct inhibition of metabolic enzymes with small-molecule compounds; Hangauer et al., 2017; Momcilovic et al., 2017; Raha et al., 2014) may trigger changes in metabolic activity that are distinct from the changes in metabolic environments used here. Moreover, even though the prior literature had hinted at a negative correlation between cell growth and targeted therapy survival (Roesch et al., 2010, 2013), the data presented here suggest that, at least in the context of EGFR inhibition, metabolically induced slow growth is not sufficient to dramatically increase EGFR inhibitor survival. Future efforts may test the extent to which these findings generalize to other EGFR inhibitors or other models of targeted therapy.
There are several limitations of this study. First, the selection of metabolic environments focused on varying amino acid and sugar concentrations. Clearly, this selection is not exhaustive, and other nutrients present in vivo (Cantor et al., 2017; Vande Voorde et al., 2019) (lipids, dicarboxylic acids, and vitamins) could be explored in future studies. Similarly, we did not explore the impact of varying oxygen concentrations, which can differ substantially in vivo from standard cell culture conditions (Ast and Mootha, 2019), on the observed empirical growth relationships. Although the impact of oxygen concentration on lactate overflow is well established, changes in oxygen availability have also been found to affect cancer cell migration (e.g., Sahlgren et al., 2008) and drug sensitivity (e.g., Ahmadi et al., 2014; Frederiksen et al., 2007; Murakami et al., 2014; Wei et al., 2013). Future efforts may examine how the interplay between nutrient and oxygen availability affects the empirical growth relationships identified here. Moreover, although using an established cell culture formulation (RPMI 1640) as a reference point facilitated comparison with prior literature, the nutrient concentrations that cancer cells encounter in vivo are likely to differ substantially. Nevertheless, because more physiological media formulations (Cantor et al., 2017; Vande Voorde et al., 2019) and quantitative in vivo information about the metabolic environment of tumors are becoming available (Sullivan et al., 2019), this study may serve as a template for identifying changes in microenvironments that are critical for a phenotype of interest.
Second, we did not tackle the question of how different genetic backgrounds affect the observed empirical growth relationships. Our data suggest that metabolic environments may affect lactate secretion rates in similar ways across diverse cell lines. However, it is not clear whether the same is true for drug survival or cell migration. Future efforts may use the approaches presented here to examine the impact of genetic background (i.e., different driver mutations) on the relationship between metabolic environment and these phenotypes—and potentially on other phenotypes of interest.
Third, in this proof-of-concept study we focused on simple 2D models of cancer that can be readily studied and manipulated in vitro. However, these simple 2D models do not capture the full complexity of tumors, such as the differential nutrient requirements in vivo compared with in vitro (Davidson et al., 2016; Elia et al., 2019). Moreover, we focused on simple phenotypes that can be quantified accurately at high throughput. For example, we quantified cell death during treatment with lethal cancer drug doses as a proxy for drug efficacy across metabolic environments. However, this approach does not capture the impact of metabolic environments on cytostatic drugs or doses. Similarly, we quantified undirected cell motility as a proxy for the migratory potential of cancer cells. However, cancer cell migration (i.e., during metastasis) is a complex process in vivo and involves steps such as tissue invasion (Hanahan and Weinberg, 2011; Stuelten et al., 2018) that were not examined here. Nevertheless, the experimental platform developed here to systematically generate defined metabolic environments is compatible with emerging in vitro platforms (Welf et al., 2016) to study more complex phenotypes, including proliferation and invasiveness in complex 3D cell cultures (Kaukonen et al., 2017). Thus, future efforts may use the present work as a starting point to systematically search for empirical growth relationships in more complex cancer phenotypes.
Finally, although we focused on the phenotypic characterization of cells across metabolic environments, we did not assess how the observed empirical growth relationship between metabolic environment and phenotype is established mechanistically. For example, we did not address the mechanisms underlying the differences in cell motility observed here (Figure 4). Previous work has highlighted the importance of ATP production for cell motility (Yizhak et al., 2014; Zanotelli et al., 2018). Consistently, we find that inhibition of ATP production has a dramatic effect on PC9 cell motility (Figures S7A and S7B). Another candidate mechanism is mTOR, which acts as a nutrient sensor (Moss- mann et al., 2018; Zoncu et al., 2011) and whose inhibition also impairs cell motility in PC9 cells (Figures S7A and S7B). These preliminary data may guide future efforts to examine the impact of metabolic environments on mTOR activity, as well as other molecular markers of cell state, including cell-cycle-stage distribution, in more detail.
For lactate overflow, our data already point toward a potential mechanism, namely, that lactate secretion is determined not by growth rate but rather by the cells’ ability to maintain high rates of sugar uptake. This would be consistent with recent work showing that glucose import is a key flux-controlling step in glycolysis (Tanner et al., 2018). Moreover, the elevated AMP to ATP ratio (indicating energy limitation) that we observed in environments with low sugar uptake suggests that the additional ATP provided by elevated glycolysis constitutes a major contribution to the cell’s energy balance. The physiological role of decoupling lactate overflow from growth rate (e.g., during amino acid limitation) is currently unclear. Preliminary experiments showed that inhibition of glucose uptake (and thus lactate overflow) does not affect cell viability during amino acid limitation (Figures S7C and S7D), suggesting that lactate overflow may be dispensable for cell survival during this type of nutrient-limitation stress. Further studies may explore whether these findings generalize to other types of nutrient stresses.
In conclusion, our study provides a quantitative approach for systematically examining the impact of different metabolic environments on cancer cell behavior. This allowed us to identify empirical relationships between growth rate and different cancer cell phenotypes. The empirical growth relationships uncovered here are a key step toward formulating the phenotypic growth laws of cancer.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Lani Wu (lani.wu@ucsf.edu).
Materials availability
This study did not create new unique reagents.
Data and code availability
All data generated in the study are available at Mendeley Data: https://doi.org/10.17632/ss9wgrdk4p
Dataset 01: PC9 and A375 cell viability and growth rate across metabolic environments. Source data for Figures 2 and S1.
Dataset 02: PC9 cancer drug survival across metabolic environments. Source data for Figures 3 and S3.
Dataset 03: PC9 cell migration across in vitro metabolic environments. Source data for Figures 4 and S4.
Dataset 04: Lactate secretion / sugar uptake rates for 5 cell lines. Source data for Figures 5, 6, 7A, and S5.
Dataset 05: Relative intracellular metabolite concentrations across metabolic environments in PC9 and A375 cells. Source data for Figures 7B and S6.
Dataset 06: Lactate secretion and sugar uptake rates for PC9 and A375 cells treated with different metabolic inhibitors. Source data for Figure S6D.
Dataset 07: ATP levels of PC9 cells during acute treatment with different metabolic inhibitors. Source data for Figures S6E and S6F.
Dataset 08: ATP levels of PC9 cells utilizing different sugars upon acute treatment with Complex I inhibitor rotenone. Source data for Figure 7C.
Dataset 09: PC9 cell migration upon treatment with metabolic inhibitors. Source data for Figures S7A and S7B.
Dataset 10: PC9 and A375 relative viability upon GLUT1 inhibition with Bay876 growing in reference condition, or at 1:32 reduced lysine concentration. Source data for Figures S7C and S7D.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PC9 cells were obtained from the Minna Laboratory at UT Southwestern, and all other cell lines used in here were obtained from the UCSF cell culture facility. PC9, A375, HEK293T, and SKBR3 cell lines harboring H2B-mCherry were constructed using lentiviral gene transfer. H2B-mCherry transfer vector was a gift from the Yang lab at Columbia University. After transfection, cells harboring mCherry were FACS sorted. Cell identity was confirmed using STR profiling, and cells were examined and found negative for mycoplasma contamination.
METHOD DETAILS
Media
Cell lines were maintained in phenol-red free RPMI 1640 media (GIBCO, 11835–030) supplemented with 5% fetal bovine serum (Gemini Bioproducts, CAT 100–106, LOT A15G00I) and 1% antibiotic-antimycotic (Gemini Bioproducts, CAT 400–101, LOT F23S00J), which was sterile-filtered (0.22 μm membrane, Olympus, CAT 25–227) before usage.
Unless stated otherwise, all experiments were performed in defined synthetic cell culture media, termed reconstituted media, based on a recently published formulation (Cantor et al., 2017), with the following modifications: First, amino acids were used at the same concentration as in standard RPMI 1640 media with exception of alanine (not included in RPMI 1640 media, used at 0.1 mM) and arginine (used at 0.115 mM). Second, additional polar metabolites listed in the published formulation (Cantor et al., 2017) were omitted. Third, glucose was used at 5.55 mM concentration (half the concentration of RPMI 1640 media). Fourth, phenol red was omitted to avoid interference with fluorescence imaging. Finally, Sytox Green (Life Technologies, CAT S7020, used at 20 nM) was added to detect dead cells based on their green fluorescent signal. See Table S1 for the exact media composition. In experiments involving nutrient replacement, the respective nutrient was omitted and replaced with one alternative nutrient per condition. See Table S2 for the full list of alternative nutrients. All media and nutrients were sterile-filtered (0.22 μm membrane, Olympus, CAT 25–227) before usage.
Cultivation
Cell cultivation was performed as follows: Cultures grown in maintenance media as described above (to 60%–80% confluence) were trypsinized for 5 min at 37C (0.25% Trypsin, Gemini Bioproducts, CAT 400–151), centrifuged (RT, 300 g, 3 min), and re-suspended in pre-heated reconstituted media lacking amino acids and glucose.
For benchmarking and drug survival experiments, nutrients were transferred to 384-well plates (Corning, CAT 353962) using an ECHO liquid handler (Labcyte ECHO 525/650) to the designated concentrations, and cell suspensions were added (50 μL culture volume, seeding 1000 cells per well). Plates were sealed with BreathEasy foil (Neta Scientific, CAT RPI-248738) to minimize evaporation and incubated at 37C with 5% CO2 for 16–20h before starting the time course experiments to allow cells to adhere to the plate bottom. In the case of drug survival experiments, cancer drugs (or DMSO as solvent control) were added 1:1000 using an ECHO liquid handler immediately before starting the time lapse microscopy experiments. Final cancer drug concentrations were: Etoposide 5 μM; Pemetrexed: 2 μM; Paclitaxel 5 μM; Erlotinib: 1 μM; Osimertinib: 0.1 μM.
For cell migration experiments, nutrients were transferred to 96-well IncuCyte ImageLock plates (Essen Biosciences, CAT 4379) using an ECHO liquid handler (Labcyte ECHO 525/650) to the designated concentrations, and cell suspensions were added (100 μL culture volume, seeding 3000 cells per well). Plates were sealed with BreathEasy foil (Neta Scientific, CAT RPI-248738) to minimize evaporation and incubated at 37C with 5% CO2 for 16–20h before starting the time lapse microscopy experiments to allow cells to adhere to the plate bottom.
For lactate overflow, metabolomics, and acute rotenone treatment experiments, nutrients were transferred to 96-well plates (Corning, CAT 353219) using an ECHO liquid handler (Labcyte ECHO 525/650) to the designated concentrations, and cell suspensions were added (100 μL culture volume, seeding 5000 cells per well). Plates were sealed with BreathEasy foil (Neta Scientific, CAT RPI-248738) to minimize evaporation and incubated at 37C with 5% CO2 for 12h before starting the time lapse microscopy experiments to allow cells to adhere to the plate bottom.
Time lapse microscopy
Cell cultures were monitored over time (at 37C and 5% CO2) using the IncuCyte S3 automated imaging system (Essen Biosciences): at regular intervals (every 2h for benchmarking, drug survival, and lactate overflow experiments, every 15 min for cell migration experiments) phase/RFP/GFP images were taken of each well (one image per well, settings: 10x objective, 300/400 ms acquisition time for GFP/RFP). From these images, the total number of cells (defined as the number of objects in the RFP channel) and the number of dead cells (defined as the number of objects which overlap in RFP and GFP channels) were extracted using built-in IncuCyte Analysis Software. The number of live cells per image and time point was calculated as the total number of cells minus the number of dead cells, and normalized to the first time point to yield time courses of relative cell numbers. The lethal fraction per image and time point was calculated as the number of dead cells divided by the total number of cells (Forcina et al., 2017). For A549 cells (which did not harbor H2B-mCherry), confluence in phase images was converted to cell number using a separate calibration curve obtained with rapid-red nuclear dye (Essen Biosciences, CAT 4706, used 1:2000).
Metabolomics
Relative intracellular metabolite concentrations were quantified following previously published protocols (Guder et al., 2017; Ortmayr et al., 2019). Briefly, cells were cultivated in 96-well plate format as described above in the designated metabolic environments for at least 24h. Subsequently, media were removed, cells were washed once with 100 μL 75 mM ammonium carbonate (pH 7.4, 37°C), and 100 μL quenching/extraction solution was added (40% methanol, 40% acetonitrile, 20% water, −20°C). Plates were kept at −20°C for 2h, centrifuged (4000 rpm, 5 min), and 50 μL metabolite extracts were transferred to conical storage plates (ThermoFisher, CAT AB-1058), sealed (ThermoFisher, CAT AB-0745) and kept at −80°C. Prior to measurement, 50 μL metabolite extracts of E. coli grown on 13C glucose were added to serve as internal 13C standard (Guder et al., 2017). Metabolite concentrations in extracts were then quantified by liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS) as described before (Guder et al., 2017), and for each metabolite and sample peak intensity was normalized to the respective 13C peak intensity and the confluence (at time of extraction as a proxy for biomass). Subsequently, metabolite signals were normalized for each cell line to the reference condition to obtain relative metabolite concentrations.
Response to acute rotenone treatment
Cells were cultivated in 96-well plate format as described above in the designated metabolic environments for at least 24h. Rotenone was then added to a final concentration of 1 μM. After 15 to 60 mins, ATP levels (proxy for the cellular energetic state) were quantified with CellTiter-Glo 2.0 (Promega, CAT G9243) following the manufacturers’ protocol. ATP signals were normalized to DMSO-treated controls grown in the respective metabolic environments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of growth and death rates
Growth rates were calculated from cell number time courses following previously published approaches for cancer (Harris et al., 2016) and microbial (Balaban et al., 2019; Kochanowski et al., 2017) cell cultures. Briefly, time-dependent growth rate μ(t) was estimated by linear regression of the relative cell number measurement (in log scale) within a sliding window of 9 consecutive time points (corresponding to a 16h time window, equivalent to the approximate doubling time of PC9 and A375 cells in standard media). The maximal value of μ(t) across the time course was defined as the maximal growth rate μmax of the respective culture. Conversely, the time-dependent death rate DR(t) was estimated by linear regression of the lethal fraction measurement within a sliding window of 9 consecutive time points. The maximal value of DR(t) across the time course was defined as the death rate of the respective culture. All calculations were performed using MATLAB (MATLAB 2019a).
Quantification of cell motility from time course experiments
Motility of individual cells from time lapse microscopy experiments was quantified based on a previously published single-particle tracking algorithm (Jaqaman et al., 2008) using custom-made MATLAB scripts (MATLAB 2019a). First, stacks of RFP images (18h duration, imaging interval 15 min) were imported for each well, and Nuclei were identified by LoG detection (MATLAB command edge, LoG threshold 0.025). Starting from the first time point, x/y positions of nuclei centroids were extracted in consecutive pairs of images (MATLAB command regionprops). Next, the Euclidean distance between each centroid in image 1 and image 2 was calculated (MATLAB command pdist2) and used to solve the linear assignment problem (MATLAB command matchpairs, cost of being unmatched: 20). Single cell tracks were constructed from links between centroids in consecutive images, only considering tracks with a length > 10 consecutive time points ( = 2.5h). The average speed of each track was calculated as the mean Euclidean distance across all time points within a track, and converted into the final unit (micrometer/h) based on the objective-specific conversion factor of 1.24 μm/pixel and a frame rate of 1 image every 15 min. To obtain a population level metric of cell motility, the median of average track speeds within the well was calculated.
Quantification of lactate secretion and sugar uptake
Lactate secretion and sugar uptake rates were quantified following a previously published approach (Jain et al., 2012). Briefly, lactate secretion was quantified as follows: First, the amount of lactate, which had been produced over the course of the experiment (36–48h experiment duration), was quantified from culture supernatants using a commercial lactate quantification kit (Megazyme, CAT K-LATE) in 96-well plate format following the manufacturers’ protocol. Second, the lactate secretion rate was calculated by normalizing the molar amount of lactate produced by the area under the growth curve (Jain et al., 2012) (calculated from cell number time courses quantified as described above).
Similarly, sugar uptake was determined by first quantifying the amount of sugar, which had been consumed over the course of the experiment (i.e., initial sugar concentration minus remaining sugar concentration at sampling time), from culture supernatants using commercial enzymatic assays (Megazyme, CAT K-GLUC for glucose; CAT K-MANGL for mannose/fructose; CAT K-ARGA for galactose; CAT K-MASUG for maltose) in 96-well plate format following the manufacturers’ protocol. The sugar uptake rate was then calculated by normalizing the molar amount of sugar consumed by the area under the growth curve, as described for the lactate secretion rate quantification.
Analytical approach to identify empirical growth relationships
Empirical growth relationships were identified as follows: Polynomial fits with varying polynomial orders (from 0th to 5th order) were estimated (MATLAB command polyval) for each phenotype and corresponding growth rate across all quantified conditions. For each fit the respective Aikaike Information Criterion (AIC) (Burnham et al., 2011) was calculated using Equation 1:
where n denotes the number of conditions, RSS denotes the residual sum of squares, and k denotes the number of parameters in the model. To test whether any polynomial fit better explains the observed phenotype-growth relationship than the null hypothesis (i.e., 0th order polynomial fit, corresponding to a constant phenotype across conditions), the difference in AIC (ΔAIC) was calculated as AIC(ith order) – AIC(0th order). Negative ΔAIC values indicate that the respective model better explains the observed data than the null hypothesis. The model with the lowest ΔAIC was selected as the best empirical growth relationship estimate, using an ΔAIC cut-off of below −3.22 (indicating that the respective model is at least five times more likely than the null model (Burnham et al., 2011)). Finally, to test for biases in the data distribution, this procedure was repeated 10000 times while randomly permuting the data (MATLAB command randperm), and the median ΔAIC was calculated.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Etoposide | Sigma-Aldrich | E1383 |
| Paclitaxel | Sigma-Aldrich | T1912 |
| Pemetrexed | Cayman Chemical Company | 14269 |
| Erlotinib | Selleck Chemicals | S1023 |
| Osimertinib | Selleck Chemicals | S7297 |
| Bay876 | Cayman Chemical Company | 19961 |
| Rotenone | Sigma-Aldrich | R8875 |
| Oxamate | Sigma-Aldrich | O2751 |
| 2-Deoxyglucose | Sigma-Aldrich | D6134 |
| Oligomycin | Sigma-Aldrich | 75351 |
| FCCP | Sigma-Aldrich | C2920 |
| Bromopyruvate | Sigma-Aldrich | 16490 |
| Torin 1 | Cell Signaling Technology | 14379 |
| Critical commercial assays | ||
| CellTiter-Glo 2.0 Cell Viability Assay | Promega | G9242 |
| L-Lactic Acid Assay Kit | Megazyme | K-LATE |
| Glucose Assay Kit | Megazyme | K-GLUC |
| Mannose Assay Kit | Megazyme | K-MANGL |
| Galactose Assay Kit | Megazyme | K-ARGA |
| Maltose Assay Kit | Megazyme | K-MASUG |
| Deposited data | ||
| Raw and analyzed data | This study; Mendeley data | http://dx.doi.org/10.17632/ss9wgrdk4p.2 |
| Experimental models: cell lines | ||
| PC9 | Present from Minna laboratory at UT Southwestern | N/A |
| A375 | UCSF cell culture facility | N/A |
| SKBR3 | UCSF cell culture facility | N/A |
| HEK293T | UCSF cell culture facility | N/A |
| A549 | UCSF cell culture facility | N/A |
| Software and algorithms | ||
| MATLAB | https://www.mathworks.com | 2019A |
Highlights.
Quantitative approach to search for phenotype-growth relationships in cancer cells
Cancer drug survival shows strong growth relationship
Cell migration follows a weak grow-and-go relationship
Lactate overflow is largely decoupled from growth rate
ACKNOWLEDGMENTS
The authors thank Alain Bonny for assistance with fluorescence-activated cell sorting (FACS). K.K. received postdoctoral fellowships from the European Molecular Biology Organization (long-term fellowship ALTF 1167-2016) and the Swiss National Science Foundation (2EZP3 - 172200), S.J.A. is supported by NIH GM112690, and L.F.W. is supported by NCI-NIH R01 CA184984. The authors thank Bernadett Gaal, Mattia Zampieri, Maike Roth, Heinz Hammerlindl, Xiaoxiao Sun, Louise Heinrich, and Leanna Morinishi for their feedback on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108647.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmadi M, Ahmadihosseini Z, Allison SJ, Begum S, Rockley K, Sadiq M, Chintamaneni S, Lokwani R, Hughes N, and Phillips RM (2014). Hypoxia modulates the activity of a series of clinically approved tyrosine kinase inhibitors. Br. J. Pharmacol 171, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ast T, and Mootha VK (2019). Oxygen and mammalian cell culture: are we repeating the experiment of Dr. Ox? Nat. Metab 1, 858–860. [DOI] [PubMed] [Google Scholar]
- Atkins RJ, Stylli SS, Kurganovs N, Mangiola S, Nowell CJ, Ware TM, Corcoran NM, Brown DV, Kaye AH, Morokoff A, et al. (2019). Cell quiescence correlates with enhanced glioblastoma cell invasion and cytotoxic resistance. Exp. Cell Res 374, 353–364. [DOI] [PubMed] [Google Scholar]
- Baguley BC, Marshall ES, Whittaker JR, Dotchin MC, Nixon J, McCrystal MR, Finlay GJ, Matthews JHL, Holdaway KM, and van Zijl P (1995). Resistance mechanisms determining the in vitro sensitivity to paclitaxel of tumour cells cultured from patients with ovarian cancer. Eur. J. Cancer 31A, 230–237. [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, et al. (2019). Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol 17, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basan M (2018). Resource allocation and metabolism: the search for governing principles. Curr. Opin. Microbiol 45, 77–83. [DOI] [PubMed] [Google Scholar]
- Basan M, Hui S, Okano H, Zhang Z, Shen Y, Williamson JR, and Hwa T (2015). Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature 528, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoumieux S, de Jong H, Baptist G, Pinel C, Ranquet C, Ropers D, and Geiselmann J (2013). Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol. Syst. Biol 9, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand K, Aichinger S, Forster S, Kupper S, Neumann B, Nürnberg W, and Ohrisch G (1988). Cell-cycle-related metabolic and enzymatic events in proliferating rat thymocytes. Eur. J. Biochem 172, 695–702. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, and Botstein D (2008). Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 19, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner A, Fridman O, Gefen O, and Balaban NQ (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol 14, 320–330. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR, and Huyvaert KP (2011). AIC model selection and multimodel inference in behavioral ecology: some background, observations, and comparisons. Behav. Ecol. Sociobiol 65, 23–35. [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louis-saint A Jr., Lewis CA, and Sabatini DM (2017). Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P-H, Cai L, Huffman K, Yang C, Kim J, Faubert B, Boroughs L, Ko B, Sudderth J, McMillan EA, et al. (2019). Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Mol. Cell 76, 838–851.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer J, Honda T, Tang Y, Wong-Ng J, Vergassola M, and Hwa T (2019). Chemotaxis as a navigation strategy to boost range expansion. Nature 575, 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2020). We need to talk about the Warburg effect. Nat. Metab 2, 127–129. [DOI] [PubMed] [Google Scholar]
- Eagle H (1955). Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514. [DOI] [PubMed] [Google Scholar]
- Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, Boon R, Escalona-Noguero C, Torrekens S, Verfaillie C, et al. (2019). Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcina GC, Conlon M, Wells A, Cao JY, and Dixon SJ (2017). Systematic Quantification of Population Cell Death Kinetics in Mammalian Cells. Cell Syst 4, 600–610.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen LJ, Sullivan R, Maxwell LR, Macdonald-Goodfellow SK, Adams MA, Bennett BM, Siemens DR, and Graham CH (2007). Chemosensitization of cancer in vitro and in vivo by nitric oxide signaling. Clin. Cancer Res 13, 2199–2206. [DOI] [PubMed] [Google Scholar]
- Fung DKC, Chan EWC, Chin ML, and Chan RCY (2010). Delineation of a bacterial starvation stress response network which can mediate antibiotic tolerance development. Antimicrob. Agents Chemother 54, 1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay T, Juhász É, Molnár E, Eisenbauer M, Czirók A, Dekan B, László V, Hoda MA, Döme B, Tímár J, et al. (2013). Cell migration or cytokinesis and proliferation?—revisiting the “go or grow” hypothesis in cancer cells in vitro. Exp. Cell Res 319, 3094–3103. [DOI] [PubMed] [Google Scholar]
- Gerosa L, Kochanowski K, Heinemann M, and Sauer U (2013). Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol. Syst. Biol 9, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese A, Loo MA, Tran N, Haskett D, Coons SW, and Berens ME (1996). Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 67, 275–282. [DOI] [PubMed] [Google Scholar]
- Guder JC, Schramm T, Sander T, and Link H (2017). Time-Optimized Isotope Ratio LC-MS/MS for High-Throughput Quantification of Primary Metabolites. Anal. Chem 89, 1624–1631. [DOI] [PubMed] [Google Scholar]
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, and Vander Heiden MG (2016). Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RAA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardeman KN, Peng C, Paudel BB, Meyer CT, Luong T, Tyson DR, Young JD, Quaranta V, and Fessel JP (2017). Dependence On Glycolysis Sensitizes BRAF-mutated Melanomas For Increased Response To Targeted BRAF Inhibition. Sci. Rep 7, 42604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LA, Frick PL, Garbett SP, Hardeman KN, Paudel BB, Lopez CF, Quaranta V, and Tyson DR (2016). An unbiased metric of antiproliferative drug effect in vitro. Nat. Methods 13, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza a.L., Kafri R, Kirschner MW, Clish CB, and Mootha VK (2012). Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 336, 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukonen R, Jacquemet G, Hamidi H, and Ivaska J (2017). Cell-derived matrices for studying cell proliferation and directional migration in a complex 3D microenvironment. Nat. Protoc 12, 2376–2390. [DOI] [PubMed] [Google Scholar]
- Kochanowski K, Gerosa L, Brunner SF, Christodoulou D, Nikolaev YV, and Sauer U (2017). Few regulatory metabolites coordinate expression of central metabolic genes in Escherichia coli. Mol. Syst. Biol 13, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanowski K, Morinishi L, Altschuler S, and Wu L (2018). Drug persistence—from antibiotics to cancer therapies. Curr. Opin. Syst. Biol 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Link H, Liu L, Du G, Chen J, and Sauer U (2016). A dynamic pathway analysis approach reveals a limiting futile cycle in N-acetylglucosamine overproducing Bacillus subtilis. Nat. Commun 7, 11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, and Will Y (2007). Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci 97, 539–547. [DOI] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gómez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, et al. (2015). Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab 22, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Magyar C, Braas D, Graeber T, Jackson NJ, Czernin J, Emberley E, et al. (2017). Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep 18, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossmann D, Park S, and Hall MN (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. [DOI] [PubMed] [Google Scholar]
- Murakami A, Takahashi F, Nurwidya F, Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K, Shimada N, et al. (2014). Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS ONE 9, e86459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SL, Cheng LYJ, J Zhou JF, Henke W, and Hagen T (2020). Galactose 1-phosphate accumulates to high levels in galactose-treated cells due to low GALT activity and absence of product inhibition of GALK. J. Inherit. Metab. Dis 43, 529–539. [DOI] [PubMed] [Google Scholar]
- Ortmayr K, Dubuis S, and Zampieri M (2019). Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun 10, 1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl Mizrahi S, Gefen O, Simon I, and Balaban NQ (2016). Persistence to anti-cancer treatments in the stationary to proliferating transition. Cell Cycle 15, 3442–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, Wilson C, Merchant M, and Settleman J (2014). The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res 74, 3579–3590. [DOI] [PubMed] [Google Scholar]
- Reznik E, Luna A, Aksoy BA, Liu EM, La K, Ostrovnaya I, Creighton CJ, Hakimi AA, and Sander C (2018). A Landscape of Metabolic Variation across Tumor Types. Cell Syst 6, 301–313.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, and Herlyn M (2010). A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, et al. (2013). Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23, 811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlgren C, Gustafsson MV, Jin S, Poellinger L, and Lendahl U (2008). Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 105, 6392–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. ; Cancer Genome Atlas Research Network (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M, and Hwa T (2011). Bacterial growth laws and their applications. Curr. Opin. Biotechnol 22, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuelten CH, Parent CA, and Montell DJ (2018). Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat. Rev. Cancer 18, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai E, Tsukimoto M, Harada H, and Kojima S (2014). Autocrine signaling via release of ATP and activation of P2X7 receptor influences motile activity of human lung cancer cells. Purinergic Signal 10, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, White E, Toettcher JE, and Rabinowitz JD (2018). Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst 7, 49–62.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiek DM, Rone JD, Graham GT, Pannkuk EL, Haddad BR, and Riggins RB (2018). Alterations in Cell Motility, Proliferation, and Metabolism in Novel Models of Acquired Temozolomide Resistant Glioblastoma. Sci. Rep 8, 7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ, et al. (2013). Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, and Tardito S (2019). Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MGMG, Cantley LCLC, and Thompson CBCB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, and Draetta GF (2016). Metabolic features of cancer treatment resistance. Recent Results Cancer Res 207, 135–156. [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, and Cerione RA (2010). Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, Teo CC, Ang HYK, Peh KLE, Yuan J, et al. (2019). Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med 25, 825–837. [DOI] [PubMed] [Google Scholar]
- Wei W, Shi Q, Remacle F, Qin L, Shackelford DB, Shin YS, Mischel PS, Levine RD, and Heath JR (2013). Hypoxia induces a phase transition within a kinase signaling network in cancer cells. Proc. Natl. Acad. Sci. USA 110, E1352–E1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welf ES, Driscoll MK, Dean KM, Schäfer C, Chu J, Davidson MW, Lin MZ, Danuser G, and Fiolka R (2016). Quantitative Multiscale Cell Imaging in Controlled 3D Microenvironments. Dev. Cell 36, 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Le Dévédec SE, Rogkoti VM, Baenke F, de Boer VC, Frezza C, Schulze A, van de Water B, and Ruppin E (2014). A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol. Syst. Biol 10, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotelli MR, Goldblatt ZE, Miller JP, Bordeleau F, Li J, VanderBurgh JA, Lampi MC, King MR, and Reinhart-King CA (2018). Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol. Biol. Cell 29, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, and Sabatini DM (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol 12, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in the study are available at Mendeley Data: https://doi.org/10.17632/ss9wgrdk4p
Dataset 01: PC9 and A375 cell viability and growth rate across metabolic environments. Source data for Figures 2 and S1.
Dataset 02: PC9 cancer drug survival across metabolic environments. Source data for Figures 3 and S3.
Dataset 03: PC9 cell migration across in vitro metabolic environments. Source data for Figures 4 and S4.
Dataset 04: Lactate secretion / sugar uptake rates for 5 cell lines. Source data for Figures 5, 6, 7A, and S5.
Dataset 05: Relative intracellular metabolite concentrations across metabolic environments in PC9 and A375 cells. Source data for Figures 7B and S6.
Dataset 06: Lactate secretion and sugar uptake rates for PC9 and A375 cells treated with different metabolic inhibitors. Source data for Figure S6D.
Dataset 07: ATP levels of PC9 cells during acute treatment with different metabolic inhibitors. Source data for Figures S6E and S6F.
Dataset 08: ATP levels of PC9 cells utilizing different sugars upon acute treatment with Complex I inhibitor rotenone. Source data for Figure 7C.
Dataset 09: PC9 cell migration upon treatment with metabolic inhibitors. Source data for Figures S7A and S7B.
Dataset 10: PC9 and A375 relative viability upon GLUT1 inhibition with Bay876 growing in reference condition, or at 1:32 reduced lysine concentration. Source data for Figures S7C and S7D.