

Abstract
The Drosophila melanogaster male embryonic gonad is an advantageous model to study various aspects of developmental biology including, but not limited to, germ cell development, piRNA biology, and niche formation. Here, we present a dissection technique to live-image the gonad ex vivo during a period when in vivo live-imaging is highly ineffective. This protocol outlines how to transfer embryos to an imaging dish, choose appropriately-staged male embryos, and dissect the gonad from its surrounding tissue while still maintaining its structural integrity. Following dissection, gonads can be imaged using a confocal microscope to visualize dynamic cellular processes. The dissection procedure requires precise timing and dexterity, but we provide insight on how to prevent common mistakes and how to overcome these challenges. To our knowledge this is the first dissection protocol for the Drosophila embryonic gonad, and will permit live-imaging during an otherwise inaccessible window of time. This technique can be combined with pharmacological or cell-type specific transgenic manipulations to study any dynamic processes occurring within or between the cells in their natural gonadal environment.
Introduction
The Drosophila melanogaster testis has served as a paradigm for our understanding of many dynamic cellular processes. Studies of this model have shed light on stem cell division regulation1,2,3, germ cell development4,5, piRNA biology6,7,8, and niche-stem cell signaling events9,10,11,12,13. This model is advantageous because it is genetically tractable14,15 and is one of the few where we can live-image stem cells in their natural environment3,16,17,18. However, live-imaging of this model has been limited to adult tissue and early embryonic stages, leaving a gap in our knowledge of gonadal dynamics in the late embryo, the precise stage when the niche is first forming and beginning to function.
The late stage embryonic gonad is a sphere, consisting of somatic niche cells at the anterior, and germ cells encysted by somatic gonadal cells throughout more posterior regions19. This organ can be imaged live in vivo up until early embryonic Stage 1717,20,21. Further imaging is prevented due to initiation of large-scale muscle contractions. These contractions are so severe that they push the gonad out of the imaging frame, and such movement cannot be corrected with imaging software. Our lab is interested in unveiling the mechanisms of niche formation, which occurs during this elusive period for live-imaging. Therefore, we generated an ex vivo approach to live image the gonad starting at embryonic Stage 16, facilitating the study of the cell dynamics during this crucial period of gonad development. Previous work from our lab shows that this ex vivo imaging faithfully recapitulates in vivo gonad development17. This technique is the first and only of its kind for the Drosophila embryonic gonad.
Here, we present the dissection protocol required for ex vivo live-imaging of the gonad during late embryonic stages. This protocol can be combined with pharmacological treatments, or transgenic manipulation of specific cell lineages within the gonad. Using this technique, we have successfully imaged the steps of stem cell niche formation17. This imaging approach is thus instrumental for the field of stem cell biology, as it will enable visualization of the initial stages of niche formation in real time within its natural environment15,17. While this method is beneficial for the field of stem cell biology, it is additionally applicable for visualizing any dynamic processes occurring in the gonad during this developmental timepoint, including cellular rearrangements22, cell adhesion2,12,23, and cell migration23. This dissection protocol will thus enhance our understanding of many fundamental cell biological processes.
Protocol
1. Day-before-dissection preparation
-
Electrolytically sharpen a tungsten needle24 so that the resulting diameter is approximately 0.03 mm. Adjust the voltage supplied to approximately 14 V, and use 3.3 M NaOH. Sharpening should take no more than 1 or 2 min. CAUTION: NaOH is highly corrosive and will cause burns upon contact with skin. Wear gloves and goggles while handling, and work inside a fume hood.
NOTE: After use, store NaOH in a polypropylene Falcon tube.
Make the prepared imaging media. In a 15 mL conical tube, combine 4.25 mL of Schneider’s media with 750 μL of Fetal Bovine Serum (FBS, 15% final concentration) and 27.5 μL of Penicillin-Streptomycin (0.05 U/μL of Penicillin, 0.05 μg/μL final concentration). Store this prepared imaging media at 4 °C.
-
Make heptane-glue solution. Add about 0.5 mL heptane to a 20 mL sealable vial stuffed with about 20 cm double-sided tape. Rock on a nutator for about an hour, or stir with a pipette tip until a consistency between that of water and glycerol is achieved. This solution of dissolved glue will last for several days before the heptane evaporates. To freshen the solution, add an additional 0.5 mL heptane, and rock.
NOTE: An effective heptane-glue solution can be prepared using various ratios of heptane to tape, as well as varying durations of rocking/stirring. The above specifications are merely suggestions for preparing or freshening one such adequate solution.
2. Embryo collection—15–17 h before dissection
-
Add fresh yeast paste to an apple juice agar plate25. Set up an embryo collection cage by adding adult flies (less than 10 days old) to an empty food bottle, perforated with small holes26. Cap the collection cage using the yeasted agar plate, taped to the bottle, and place the cage with the plate side down in a 25 °C dark incubator for 1 h.
NOTE: The flies must express a transgene that will fluorescently mark the gonad, for example, six4-eGFP::Moesin20, because dissection of embryos will take place under a stereo-fluorescent microscope. See Table of Materials for a list of genotypes used to mark the gonad.
-
Remove the cage from the incubator and discard this first collection. Replace it with a freshly yeasted agar plate and place the cage back in the incubator for 2 h.
NOTE: The first embryo collection is used to clear females of fertilized, developing embryos, to achieve a tightly timed second collection of embryos.
-
Remove the agar plate from the cage and place it (with yeasted side facing upward) on a moist paper towel inside a sealable plastic container. Place this humid chamber in a 25 °C incubator to age the embryos for 14.5 h (final ages, 14.5–16.5 h after egg lay).
NOTE: Immediately before beginning step 2.4, complete steps 3.1 and 3.2.
Remove the agar plate from the incubator, and using a squirt bottle, add enough water to the agar plate to dissolve the yeast paste by brushing lightly with a paint brush. Rinse the embryos and dissolved yeast into a small mesh screen basket sitting inside a weigh boat. Rinse the basket with the water squirt bottle until most yeast paste has filtered through the mesh.
-
Remove the water from the weigh boat and place the basket back inside. Dechorionate the embryos by immersing them in a 50% bleach solution, using a squirt bottle. The bleach solution should have a depth of 3–5 mm. Keep the embryos submerged in bleach for ~2 min, with occasional swirling.
CAUTION: Bleach is corrosive and can irritate or damage eyes and the respiratory tract. Wear gloves and goggles while handling bleach.
NOTE: During this time, complete as much as possible of step 3.3. Dechorionation progress can be checked by placing the basket under a stereo microscope and checking for the absence of the dorsal appendages. Submerge the embryos in the bleach solution for additional time, if necessary.
Discard the bleach, and thoroughly rinse the embryos inside the basket with water from a squirt bottle (for ~3 s, blotting the mesh basket with paper towels; repeat 2–3 times).
Table of Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| Alfa Aesar Tungsten wire | Fisher Scientific | AA10408G6 | 0.25mm (0.01 in.) dia., 99.95% (metals basis) |
| D. melanogaster: His2Av::mRFP1 | Bloomington Drosophila Stock Center (BDSC) | FBtp0056035 | Schuh, Lehner, & Heidmann, Current Biology, 2007 |
| D. melanogaster: nos-lifeact::tdtomato | Gift from Ruth Lehmann Lab | Benjamin Lin: nos5’- Lifeact-tdtomato p2a tdkatushka2 Caax nos3’ | |
| D. melanogaster: P-Dsix4-eGFP::Moesin | FBtp0083398 | Sano et al., PLoS One, 2012 | |
| Diamond-tipped knife | |||
| Fetal Bovine Serum | GIBCO | 10082 | |
| Imaging dish | MatTek | P35GC-1.5-14-C | |
| Insulin, bovine | Sigma | l0516 | Store aliquots at 4 °C |
| Needle holder | Fisher Scientific | 08-955 | |
| Nytex basket | |||
| Penicillin/streptomycin | Corning | 30-002-Cl | |
| Ringer’s solution | 2 mM MgCl2, 2 mM CaCl2, 130 mM NaCl, 5mM KCl, 36 mM Sucrose, 5mM Hepe’s Buffer; adjusted with NaOH until pH of 7.3 is achieved | ||
| Schneider’s Insect Media | GIBCO | 21720-024 | |
3. Day-of-dissection preparation
Add 30 μL insulin (10 mg/mL) to 1,500 μL prepared imaging media (see step 1.2) in a 1.5 mL Eppendorf tube(0.2 mg/mL final concentration). Mix well and leave the tube on the benchtop to equilibrate to room temperature.
- Prepare coverslip strips coated with glue.
- Use a diamond-tipped knife to cut a 22 mm × 22 mm coverslip into four, equally sized strips (Figure 1A).
- Use forceps to pick up one strip and spread a total of approximately 30 μL of heptane-glue solution onto both sides of the strip (Figure 1B). To achieve an even layer of glue residue, tilt the strip at various angles while the heptane evaporates.
- Store the glue-covered strip in an empty slot of a coverslip box; to maintain its stickiness, place the strip in a slanted, but upright position, leaning against the edges of the box to minimize contact with box surfaces (Figure 1C). Close the box so that particulates in the air do not coat the glue and lessen its stickiness.
Place the following items on the benchtop: a 6-inch glass Pasteur pipette, a microscope slide, a P200 and a P1000 pipetteman with the appropriate pipette tips, Ringer’s solution27 (pH adjusted to 7.3 with NaOH), and an uncovered, Poly-D-Lysine-coated 35 mm imaging dish.
- Transfer embryos from the mesh screen basket to a small watch glass filled with 500–750 μL heptane.
-
Blot the sides and bottom of the basket dry with a tissue wipe. Moisten a paintbrush in the heptane, touch the paintbrush to the embryos (the hydrophobic vitelline membrane should adhere to the bristles), and dip the paintbrush back into the heptane in the watch glass (embryos should sink to bottom).NOTE: The next steps must be performed as quickly as possible to prevent the embryos from drying out. Embryos should not be exposed to air for more than 20 s.
-
Transfer the embryos onto the microscope slide using a Pasteur pipette (Figure 1D). Draw embryos into the pipette slowly and limit them to the narrow portion of the pipette. Pipette embryos onto the slide slowly, such that they aggregate just inside the tip of the pipette before flowing onto the slide. Twist the corner of a tissue wipe into a fine tip, and wick away heptane from embryos on the slide. They will aggregate and cover a smaller area, making it easier to capture them on a glue-covered strip in the next step.
With forceps, pick up a glue-covered strip, and gently touch it to the embryos (Figure 1E). Place the strip in the imaging dish, embryo side-up, and just outside of the Poly-D-Lysine-coated center. Press the strip onto the dish using forceps, to ensure it is fixed in place. Immediately flood the dish with 2–3.5 mL Ringer’s solution; submerge the embryos first to prevent them from drying out (Figure 1F).
Figure 1: Mounting and hydrating embryos prior to dissection.
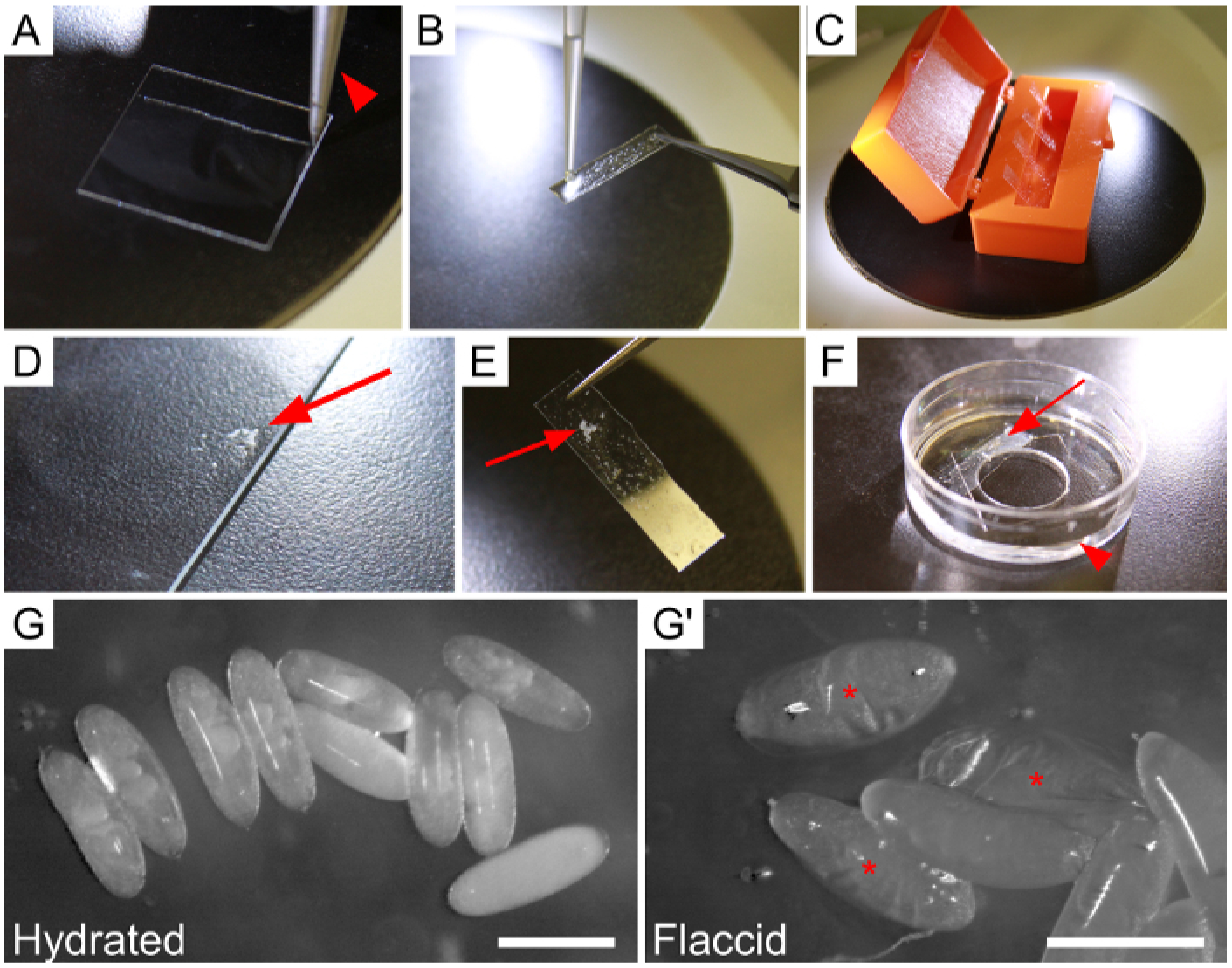
(A–F) Steps for adhering embryos to a strip of glue-coated coverslip, and securing the strip to an imaging dish. (A) Coverslip scored once by diamond-tipped knife (knife indicated by arrowhead) to create a strip. (B) Application of heptane-glue to severed coverslip strip, held with a pair of forceps. (C) Four glue-coated strips drying in a coverslip box. (D–E) Arrows point to embryos. (D) Dechorionated embryos that have been collected in heptane and expelled onto the edge of a microscope slide. Excess heptane was removed with a kimwipe. (E) Glue-coated strip with embryos attached. (F) The final setup of the dish immediately prior to dissection. Note the shallow layer of Ringer’s solution (arrowhead) and placement of the strip (arrow) above the inner dissection circle. (G–G’) Embryos adhered to the strip in the dish. (G) Properly-hydrated, turgid embryos. (G’) Embryos that have become dehydrated due to prolonged air exposure, evident by collapsed vitelline membranes. Asterisks indicate flaccid embryos. Scale bars are 0.5 mm.
4. Dissection
NOTE: These steps must be carried out under a stereo-fluorescent microscope.
-
Devitellinize 10–15 embryos.
NOTE: This may range from two embryos, for beginners, to fifteen embryos, for experts.-
Select Stage 16 embryos based on gut morphology (Figure 2). At this stage, embryos have three gut constrictions that create four stacked gut segments (Figure 2B,B’). To begin devitellinization, pierce the selected embryo at one end, preferably the anterior, with the tungsten needle. The embryo may pop out of its vitelline membrane, but if not, peel the membrane off of the embryo.NOTE: Embryos that are too young to dissect have non-regionalized guts (Figure 2C). Dissection of Stage 17 embryos is possible, but more challenging than dissection of Stage 16 embryos because the cuticle is beginning to develop at this stage. Early Stage 17 embryos present with four gut segments that are shifted relative to one another (Figure 2D,D’).
-
Hook the needle through the embryo in a region far from the gonads. Transfer the hooked embryo to the Poly-lysine-coated region of the dish (from here onwards referred to as simply “cover slip”) and drag it against the bottom until it adheres. Repeat these steps, and arrange devitellinized embryos in a row along the top of the Poly-lysine-coated cover slip (Figure 3A), leaving plenty of space below for further dissection.NOTE: The gonads appear as small spherical aggregates of cells that should fluoresce brightly, depending on the specific marker used and transgene copy number. At this stage, the gonads are located laterally in segment A5, at approximately 70%–80% of embryo length from anterior. Throughout the Dissection protocol, the tissue may stick to the needle. To rid the needle of the debris, raise it just above the surface of the Ringer’s solution. Devitellinization can be untidy as long as the gonad proper is disturbed as little as possible.
-
- Finesse the gonads out of the embryo and onto the dish (Figure 3C,D).
-
First, filet the embryo to expose its interior (Figure 3C). Slice through the embryo from its center, moving the needle posteriorly, between the gonads. Tease out some internal tissue, as this will stick to the dish much better than the external cuticle. The stickiness of the tissue will enable the next manipulations to reveal the gonad and allow it to adhere to the cover slip.NOTE: If tissue is not sticking to the dish, try coaxing it to an uncontaminated region of the coated cover slip; the outer regions of the cover slip will be particularly sticky. As stated in step 4.1.2, it does not matter if the dissection manipulations result in a mangled embryo carcass, as long as the gonads remain unscathed.
-
Use the needle to slice around a gonad until a piece of tissue, including the gonad, is separated from the remaining carcass. With the needle, draw this tissue to a fresh region of coated cover slip, and coax it against the bottom until it adheres to the dish.NOTE: The best imaging will be achieved for those cases where the gonad itself attaches directly to the cover slip, rather than indirect attachment via overlying tissue. Therefore, as tissue is guided away from the carcass, attempt to have the gonad touch the cover slip first, rather than extraneous tissue.
- Remove as much surrounding tissue from the gonad as possible (Figure 3D) by gently dragging adherent tissue away from the gonad with the needle. Avoid touching the gonad directly, which would damage it (Figure 4E). To ensure the gonad is sufficiently adhered to the dish, move the needle in a gentle circular motion around the gonad—if it moves, touch the needle to the remaining adherent tissue, and direct the gonad toward a fresh region of sticky cover slip. Repeat this process until there is no detectable movement of the gonad.
- Return to the embryo carcass and dissect out the second gonad. Repeat this process on as many embryos as possible but do NOT exceed 25 min. Tissue viability will be compromised in Ringer’s solution after more than ~40–45 min.
-
After dissections are complete, use an indelible marker to add registration marks to the outer rim of the imaging dish to record its orientation during dissection.
- Remove the glue-coated strip from the dish.
-
Gently insert the bottom prong of a pair of forceps underneath the strip, clasp and slowly tilt the strip upwards to free it from the dish with as little sloshing of the Ringer’s solution as possible to minimize disturbance of the adhered gonads.NOTE: The strip should be tilted away from the dissected-out gonads.
-
Gently carry the dish to the imaging microscope in a manner that avoids sloshing of the Ringer’s solution.
Figure 2: Selecting appropriately-aged embryos for gonad dissection.
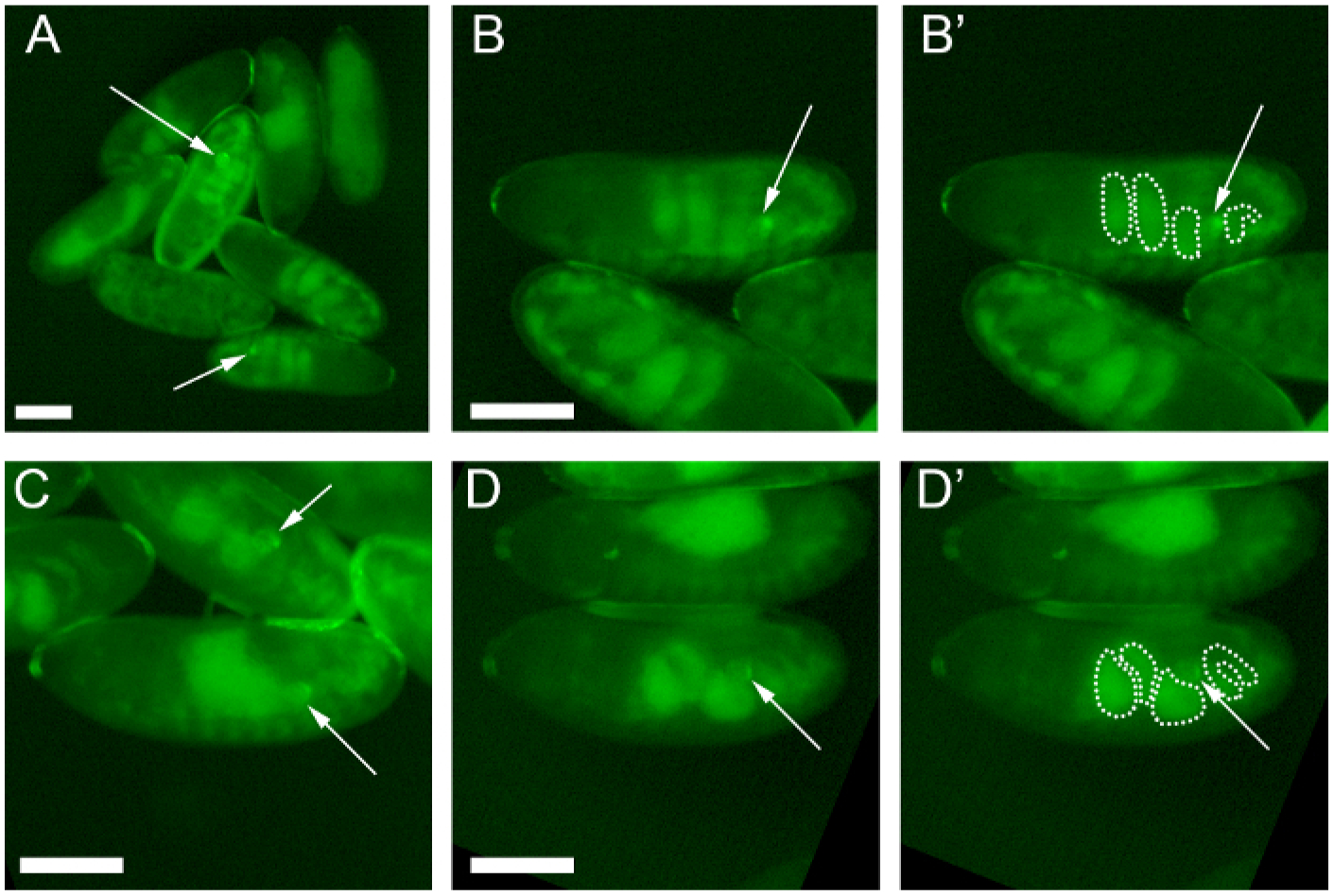
Embryos adhered to a glue-coated coverslip prior to dissection. Embryos express six4-eGFP::moesin (green), which marks the gonad and fat body cells. Note that the gut auto-fluoresces in green. Arrows indicate male gonads (discerned by the presence of brightly fluorescing msSGPs) that are visible in each panel. Scale bars indicate 0.25 mm. (A) Embryos of various stages. (B–D) Embryos of distinct stages in the center of the image, oriented with anterior to the left and dorsal up. (B) An early Stage 16 embryo that is aged appropriately for dissection. (B’) The four stacked gut regions are indicated with dotted white lines. (C) A Stage 15 embryo that is too young to dissect (lower embryo). Note that the fluorescent gut sac just anterior to the gonad does not yet have discrete regions. (D) A late Stage 16 embryo that is difficult to dissect due to developing cuticle. Note that the four gut regions have begun to rotate relative to one another in preparation for gut looping. (D’) The four gut regions are outlined.
Figure 3: The dissection process.
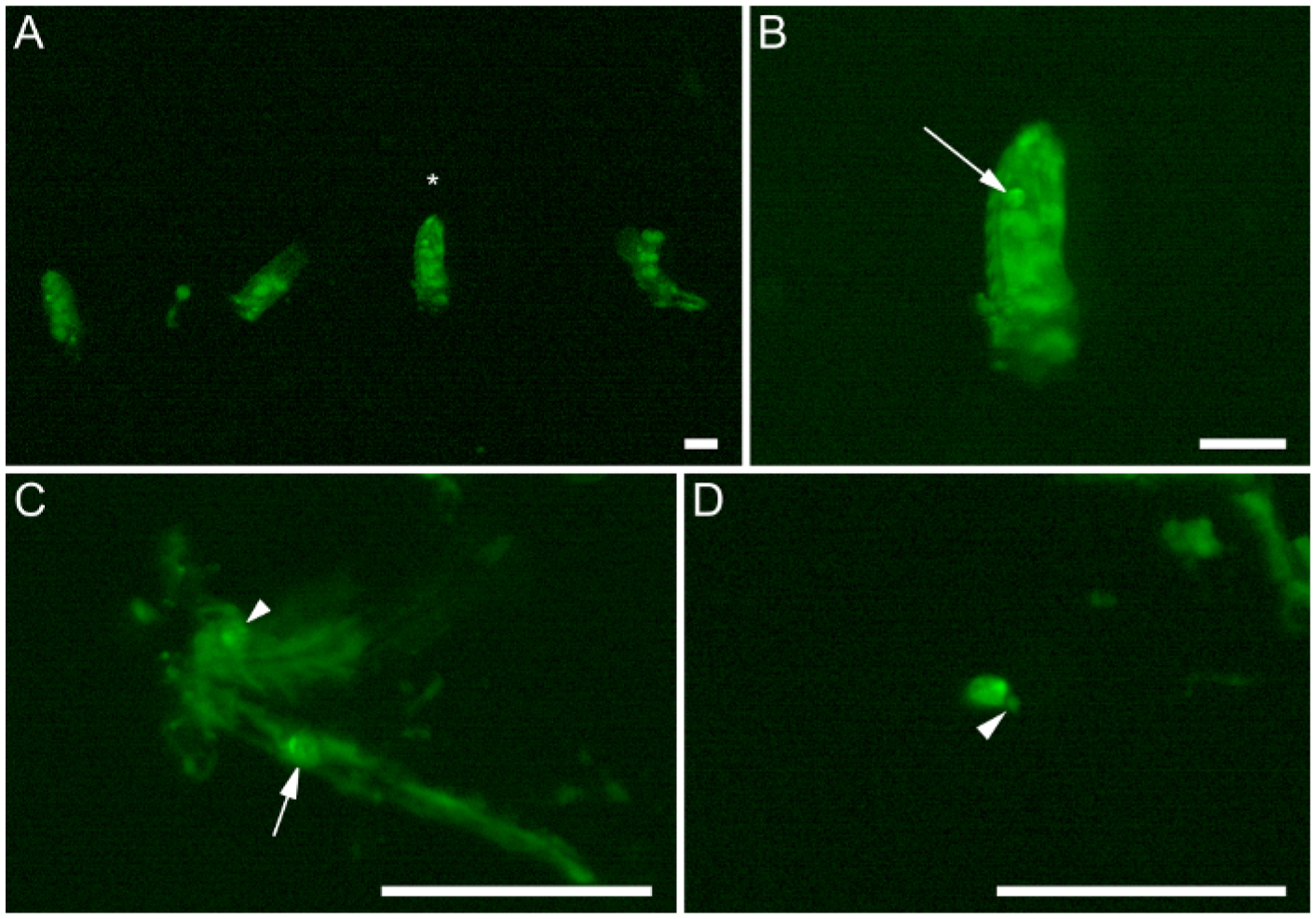
(A–D) Embryos that express six4-eGFP::moesin (green) at sequential stages in the dissection protocol. (A) Four devitellinized embryos that have been transferred to the poly-lysine-coated dissection region of the dish. Note how the embryos are aligned in a neat row to facilitate successive downward dissections in the dish. The small piece of tissue between Embryos 1 and 2 is part of the gut from Embryo 2, extruded during hand devitellinization. (B) Higher magnification view of embryo from (A), indicated by the asterisk. (C) The remaining carcass of an embryo that has been filleted down the midline. The arrowhead indicates an occluded gonad, still heavily embedded in embryonic tissues. (D) A completely dissected gonad. Note only minimal extraneous tissue (arrowhead) remains near the gonad. Arrows point to male gonads. Scale bars are 0.25 mm.
Figure 4: Locating healthy gonads during live-imaging.
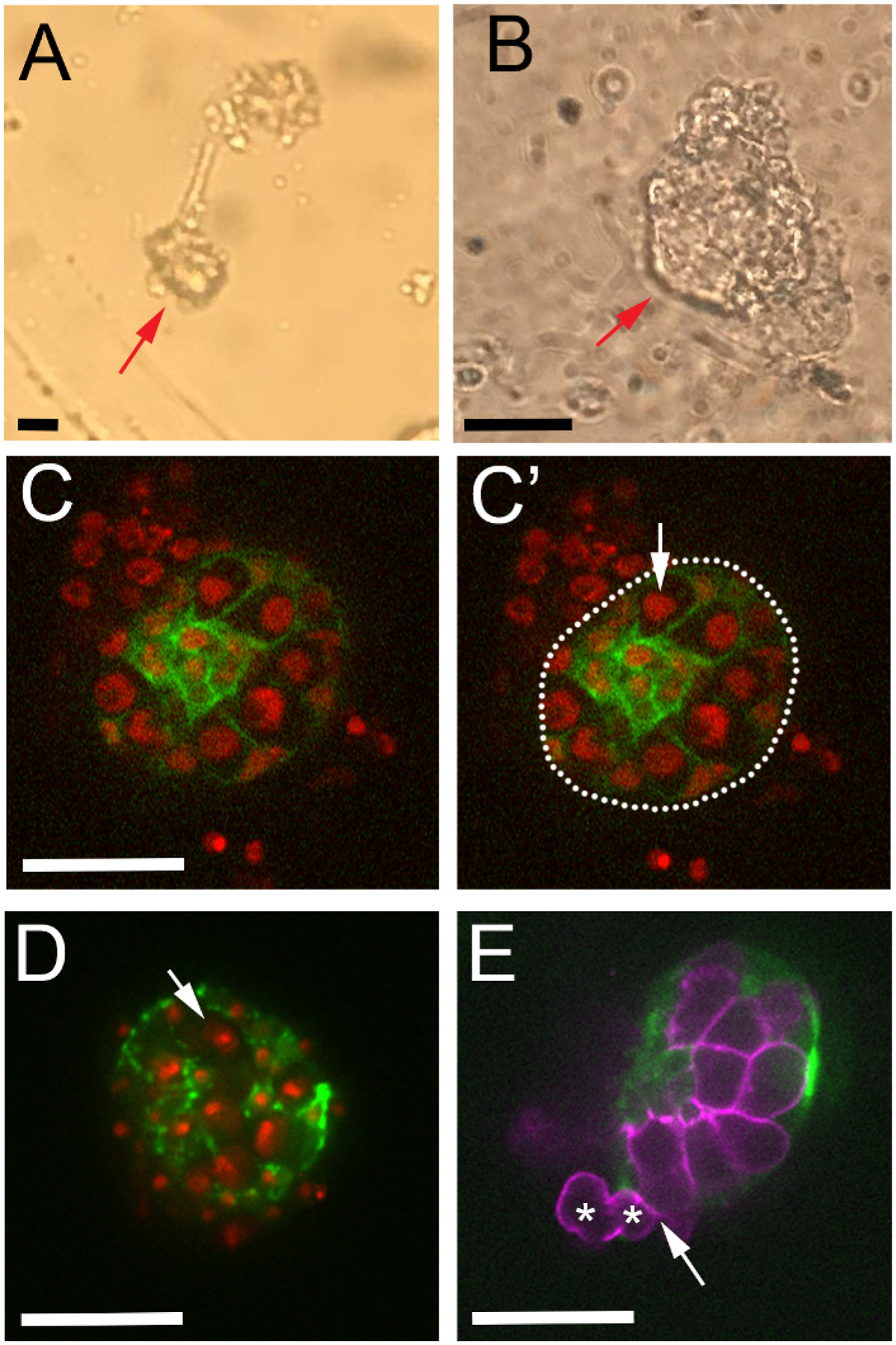
(A) Low and (B) high magnification brightfield views of two dissected gonads adhered to the coverslip. (C–C’) One movie frame from ex vivo imaging of niche compaction in a healthy gonad. Somatic gonadal cells express six4-eGFP::moesin (green), and all cells express His2Av-mRFP (red). (C’) Gonad boundary marked with a white dotted line. His2Av-mRFP visible outside of the gonad boundary is likely fat body that isstill attached to the dissected gonad. Arrow points to a germ cell nucleus with uniform His2Av-mRFP signal, indicating the gonad is healthy. (D–E) Representative negative outcomes of the ex vivo dissection protocol. (D) A frame from an imaging session in which the gonad has dehydrated because of media evaporation. Note pyknotic nuclei as His2Av-mRFP condenses (arrow), and discontinuous spots of six4-eGFP::moesin along the gonad boundary. (E) Ex vivo imaging framein which extracellular matrix has been compromised during dissection. Note that some germ cells (asterisks) are exiting the gonad boundary (arrow). Germ cells are labeled with nos-lifeact::tdtomato (magenta), and somatic gonadal cells with six4-eGFP::moesin (green). Scale bars show 20 μm.
5. Imaging
-
Place the imaging dish in the stage holder, using the registration marks to place the dish in the approximate orientation as during dissection. Using brightfield microscopy and a low power (~10x) objective, identify and bring into focus any piece of tissue that is adhered to the cover slip. Switch the eyepiece settings to reveal fluorescence, and using the binocular eyepieces, systematically scan the dish, marking the position of each gonad within the imaging software (see Table of Materials).
NOTE: Make sure gonads are centered in the field-of-view before marking positions.
Gently remove the entire stage holder assembly, with the imaging dish in its holder, and place the assembly on the work bench.
- Replace the Ringer’s solution with the prepared imaging media containing insulin (see steps 1.2 and 3.1).
- Use a P1000 to remove all Ringer’s solution from the inside upper ledge of the imaging dish (do not pipette Ringer’s from the central cover slip region). Next, switch to a P200, and place its tip just under the surface of the remaining Ringer’s in the central region. Carefully remove 50–100 μL of Ringer’s; do NOT remove the entire solution.
-
Draw up ~200 μL of imaging media, and, again placing the P200 tip just under the surface of the remaining Ringer’s, slowly add this imaging media. Next, add the remaining imaging media (~1,300 μL) to the dish, starting at the outermost rim of the upper ledge. While pipetting, move toward the central dome of the fluid, and eventually merge the two by brushing the pipette tip across them both. Place the lid on the dish.NOTE: Imaging media should cover the entire inside diameter of the dish, with a depth of about 2 mm, to prevent evaporation (Figure 4D).
-
Switch the microscope to a higher power objective (63x, 1.2 NA), apply the proper immersion fluid based on the objective used (immersion fluid type and refractive index required by the objective), and then replace the stage holder assembly. Use brightfield microscopy to focus on tissue adhered to the bottom of the imaging dish.
NOTE: If the stage was not moved, the last gonad should come into view once the objective is focused.
-
Step through each marked gonad position and adjust that position, as necessary. Select which gonads to image based on image clarity, gonad sex, etc. Customize the imaging settings (multi-channel, exposure times, laser intensities, Z-series increments, time-lapse with appropriate intervals, etc.). Begin imaging.
NOTE: Male gonads can be identified by the presence of both a niche, and at the opposite end of the gonad, a cluster of small, highly circular somatic cells called male-specific somatic gonadal precursors (msSGPs). If the six4-eGFP::moesin fluorescent marker is used, the niche cells are the second-brightest cells in the gonad, the brightest being the msSGPs. At this stage, female gonads have neither a niche nor msSGPs.
Representative Results
We illustrate preparation of the imaging dish in Figure 1, as described in “Day-of-dissection preparation.” These methods should ultimately result in well-hydrated embryos adhered to a cover slip strip, which is temporarily fixed to the bottom of the dish and submerged in Ringer’s solution (Figure 1F). A diamond-tipped knife allows one to cleanly slice a 22 × 22 mm cover slip into three to four smaller strips (Figure 1A). While handling these strips with forceps, we use a pipette to transfer enough heptane-glue to coat these strips, which gives them an adhesive, textured surface (Figure 1B). The coated strips are easily stored in an empty cover slip box to keep the adhesive surfaces clean for up to 2 h (Figure 1C). Once these coated strips are made, the goal is to adhere embryos to the strip in an aggregate, near the long edge of the strip (Figure 1E). It is necessary to first transfer a few embryos from the watch glass onto a clean glass slide and dry them (Figure 1D). Then, quickly use forceps to gently touch the coated strip to the embryos so that they adhere (Figure 1E). We then immediately press the strip to the bottom of the dissection dish with embryos facing up, and cover the embryos with Ringer’s solution (Figure 1F). If this goal is met, then viewing embryos under a traditional brightfield stereomicroscope will reveal fully hydrated, healthy embryos within their vitelline membranes (Figure 1G). If, instead, the process of transferring these embryos onto the strip and covering them with solution takes more than approximately 30 s, the embryos will dehydrate and become flaccid (Figure 1G’). Flaccid embryos are not healthy and are incredibly challenging to dissect, so working efficiently during this process is vital.
Viewing an embryo under a stereo-fluorescent microscope allows clear visualization of the gut, which auto-fluoresces in the GFP channel. Gut morphology serves as a proxy for embryonic age when choosing embryos to dissect. Because embryos adhered to the strip of cover slip will vary slightly in age, they will present a diverse array of gut morphologies (Figure 2A). To live-image gonad niche morphogenesis, we dissect early Stage 16 embryos. These embryos present four regionalized gut sections that are stacked in an even row (Figure 2B’, dotted lines). Younger embryos present a sac-like, non-regionalized gut (Figure 2C), and do not yet have sufficient extracellular matrix (ECM) around their gonads to allow for efficient culturing of the intact organ. Older embryos that have already begun niche compaction present four gut regions that are not evenly stacked and are instead shifted relative to one another (Figure 2D’, dotted lines). These embryos have thicker cuticle, which makes the dissection process more challenging.
It is important to dissect embryos that express an indelible fluorescent marker in the gonad, and this marker must be visible under a stereo-fluorescent microscope. Here, we have chosen to use six4-eGFP::moesin20 to mark the gonad (Figure 2 and Figure 3, arrows) for visualization during dissection.
We illustrate the gonad dissection process in Figure 3. The dissection of appropriately-staged embryos on the poly-lysine-coated dish allows for clean isolation of the gonads from these embryos (Figure 3D). Embryos that are devitellinized will adhere to the dish, and can be arranged in a convenient row prior to dissection (Figure 3A). Embryo devitellinization and transfer to the poly-lysine is an imprecise process such that tissue may become extruded from the confines of the embryo body (see piece of tissue between Embryo 1 and Embryo 2, and mangled Embryo 4; Figure 3A). This imprecision is of no importance, as long as the gonads remain unscathed. A standard stereo-fluorescent microscope enables identification of the gonad within the devitellinized embryo body (Figure 3B, arrow). The first few manipulations with a sharp dissection needle should separate the gonads from the embryo carcass, though some auto-fluorescent tissue will remain adhered (Figure 3C). Additional manipulations result in isolated gonads that adhere directly to the coated dish (Figure 3D).
Once the gonads are dissected, one can replace the Ringer’s solution with imaging media, and image cultured gonads directly in the same dish used for dissection. We use a spinning disk confocal microscope. Brightfield visualization of an isolated gonad on a confocal microscope reveals a dark shadow surrounding the gonad periphery (Figure 4A–B, arrows), which is the ECM that maintains integrity of the gonad during imaging. We present an example of a healthy, well-cultured gonad that expresses GFP-labeled F-actin in somatic gonadal cells20 and an RFP-labeled histone marker to visualize all nuclei28 (Figure 4C–C’). Because all cells in this embryo express the RFP histone marker, both gonadal cells, and cells of other adherent tissue are observed in viewing red emission. We discern the boundaries of the gonad using the gonad-specific GFP marker (Figure 4C’, outline). It is clear that this cultured gonad is healthy because the gonad boundary is smooth and round (Figure 4C’, outline), and because gonadal cells have even levels of RFP histone fluorescence throughout nuclei (Figure 4C’, arrow). If, instead, gonads are not sufficiently hydrated during imaging, nuclei and six4-eGFP::moesin fluorescence can become punctate (Figure 4D, arrow) as the gonad tissue shrivels. Further, if the gonad ECM is damaged excessively during dissection, the gonad boundary is compromised, which is evident by the presence of gonad-specific cells (Figure 4E, asterisks) outside of the confines of the gonad (Figure 4E, arrow).
Gonads can be cultured for about 5 h using this ex vivo imaging method, enabling image acquisition of the dynamic morphogenesis events that occur in late embryonic gonad development. In our lab, we have successfully used this protocol to image the compaction of the forming stem cell niche17 (Figure 5). Prior to compaction, the niche is a loose aggregate of somatic cells at the gonad anterior (Figure 5A, green), surrounded by germline cells labeled with nos-lifeact::tdtomato29 (see Table of Materials), the first tier of which will be germline stem cells (Figure 5A, magenta). This niche aggregate initially presents with an irregular boundary (Figure 5A’, dotted line). Throughout the course of imaging, we observe individual niche cells rearranging their positions while the niche aggregate acquires smoother borders. These cellular rearrangements also result in a decrease in niche area (Figure 5C). Our observations of cell rearrangements, and neighboring germ cell divisions that occur concurrently with this phase of development informed our understanding of mechanisms underlying niche compaction17. This protocol thus affords the ability to visualize cell rearrangements, shape changes, divisions, and other cellular events with the resolution required to analyze morphogenetic events in late stage gonads.
Figure 5: An ex vivo cultured gonad undergoing niche compaction.
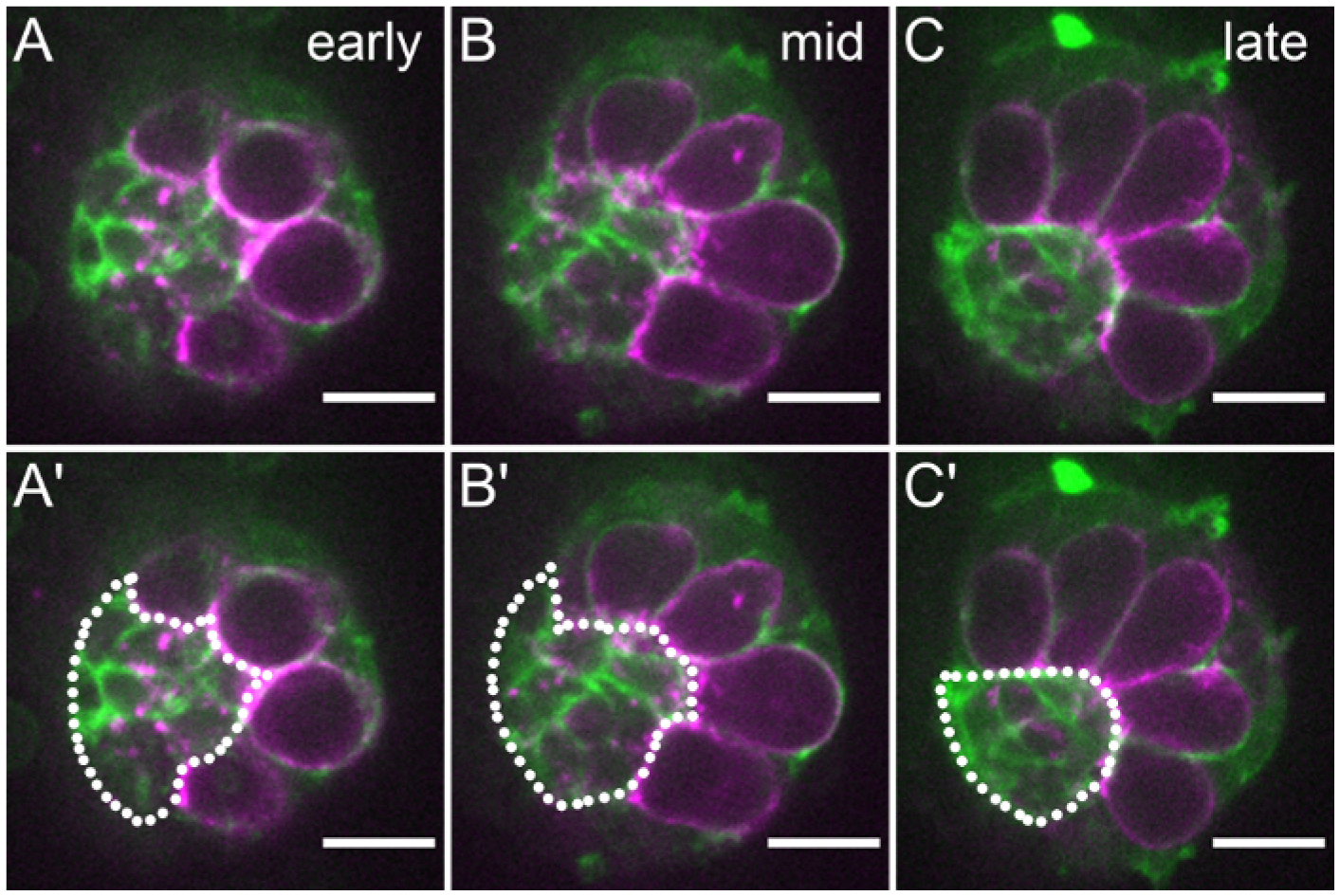
Stills from an imaging series acquired promptly following dissection of gonads from early Stage 16 embryos. (A–C) Germ cells (magenta) are labeled by nos-lifeact::tdtomato and somatic gonadal cells (green) are labeled by six4-eGFP::moesin. The niche (dotted white line) is outlined in A’–C’. Scale bars show 10 μm. Anterior is to the left, and posterior is to the right. (A) At the first timepoint (t = 0 min) in the imaging series, niche cells have finished assembling at the gonad anterior and are at an early stage of compaction. (B) Midway through the imaging series and the compaction process (t = 1 h 48 min), the niche has begun to circularize, but its edge remains irregular. (C) Near the end of the imaging series (t = 4 h 21 min), the niche has a highly smoothened, circularized boundary.
Discussion
During gonadogenesis, the embryonic gonad, and particularly the stem cell niche within the male gonad15, undergoes rapid morphological changes. Developmental mechanisms that underlie these dynamic changes are best understood through live-imaging techniques. However, at embryonic Stage 17, in vivo imaging of the gonad is rendered impossible by the onset of large-scale muscle contractions17. With this protocol, we provide a successful alternative: dissection of the gonads directly onto an imaging dish for ex vivo live-imaging. This protocol presents the only method available to accomplish live-imaging of the late stage embryonic gonad.
The critical steps of the protocol should be executed with an acute focus on dexterity and timing. Prior to dissection, rapid mounting of embryos is paramount to ensure that the embryos do not dehydrate and remain healthy and turgid, which eases devitellinization and dissection. During dissection, it is vital to avoid disruption of the gonadal extracellular matrix, which is accomplished using only delicate and precise manipulations. Use of such dexterity will also ensure that the gonad is in direct contact with the imaging dish, thereby enabling clear imaging directly through the coverslip. After dissection, Ringer’s solution must be switched for imaging media using only gentle suction and expulsion with a pipette to prevent gonad dislodgement. Finally, it is important to limit the overall dissection time to 25 min. This ensures that the amount of time spent in Ringer’s solution during dissection, and location of the tissue at the confocal, is limited to a non-toxic exposure. With increased practice, dissection speed will improve, and personalization of the dissection sequence will occur. In our experience, sufficient dissection may be achieved after about a week of regular practice, with full mastery of the dissection attainable in about 1 month. To ease learning, we recommend practicing individual steps before executing the entire protocol.
There are a number of best practices that ameliorate the challenges associated with this protocol, starting with the genotype of the embryos used for dissection. Incorporation of multiple copies of the transgene used to mark the gonad will make the gonad brighter, and therefore easier to visualize and dissect. The dissection itself works best with a sharp needle, though it should not be overly sharpened, as it will bend against the bottom of the imaging dish and become burdened with extraneous tissue. Schneider’s imaging media auto-fluoresces in the green emission spectrum, which makes it challenging to locate gonads marked with GFP once the media is added. Therefore, it is critical to use the confocal imaging software to mark the location of gonads while they are still in the Ringer’s solution. If locating gonads at the confocal remains difficult, after the next dissection we suggest sketching or taking an image of the relative positioning of gonads in the dish while still at the dissection microscope. Then, mark the outside of the dish to indicate its orientation during dissection, and match this orientation when transitioning to the confocal microscope. Once located at the confocal, if a gonad appears disrupted or mangled, in the next dissection try leaving more adherent tissue surrounding the gonad. On the contrary, if a gonad is present but appears blurry throughout all planes, there is likely tissue between the coverslip and the gonad, and future dissections should strive for better isolation of the gonad from the adherent tissue. In our experience, adopting these practices has facilitated learning and execution of this protocol.
During the formation of this protocol, we identified several alterations that could be applied. For niche compaction studies, we aim to dissect embryos at Stage 16. At this stage, the embryo has not yet developed significant amounts of cuticle, and easily sticks to the poly-lysine-coated dish. However, this technique may be modified to dissect older embryos with cuticle by sticking them to the inner wall of the poly-lysine-coated region for the first incision. Once internal tissues are exposed, the embryo carcass will stick to the poly-lysine, and dissection can proceed as normal. In addition to adjustments for embryo age, we have successfully adjusted this technique to dissect multiple genotypes in the same dish. For example, homozygous mutants can be separated from sibling heterozygotes by the use of an easily discernable marker such as deformed-YFP on the balancer chromosome. After devitellinization, embryos should be transferred to either side of the coverslip based on the presence or absence of the marker. Dissection should proceed downwards in the dish as described in the protocol, with extra care taken to maintain segregation of different genotypes. Also, this protocol could potentially be modified to use culture medium other than Schneider’s, though a superficial investigation of Shields and Sang M3 Insect Media yielded unviable gonads (data not shown). Additionally, this protocol pairs well with pharmacological manipulations by simply combining the drug with the imaging media. Over a 5 h period of imaging, re-addition of drug might be necessary to maintain an effective concentration. Finally, although this protocol was developed with the intention of visualizing niche compaction, a phenomenon specific to male gonads, in theory this protocol could also be implemented to live-image the developing ovary by simply dissecting and imaging gonads that lack a niche and msSGPs.
Limitations associated with this technique relate to the length of culturing time, and the ex vivo nature of the culture. As is the case with mid-stage Drosophila egg chambers30, cultured gonads begin to die after about 5 h of ex vivo live-imaging, evidenced by loss of tissue integrity (nuclei become pyknotic and cell membranes shrivel). Thus, if one wished to explore later-stage events in the male gonad, one would need to dissect gonads from 1st instar or older larvae by making modifications to the protocol presented here, and then image those under conditions similar to those presented here. However, we do not rule out the possibility of ex vivo live-imaging past five hours if improved culture conditions are developed, though there may be a limit if mechanical cues from within the embryo are necessary for gonad development. Perhaps the most severe drawback of the technique is due to its inherent ex vivo nature. When cultured ex vivo, cells can have unnatural potential that is unrepresentative of in vivo biology31. Additionally, subtleties of culturing environments, including media content and matrix stiffness, can have drastic influence over cell behavior32. With this sensitivity in mind, it is vital to ensure that culturing conditions accurately reflect in vivo biology. We have taken measures to attest that in vivo niche development and signaling is recapitulated ex vivo17. Briefly, we ascertained that niche cell fate is maintained and number of niche cells is unchanged using the markers Fasciclin-III and E-cadherin. Also, STAT signaling is present and germline stem cells divide orthogonally, suggesting that niche functionality is maintained. However, we have not verified that the relevant in vivo biology remains intact in other, non-niche tissues within the gonad. Future ex vivo investigations of these other tissues should include a comprehensive analysis to ensure that this is indeed the case.
A future direction one might take with this protocol would include incorporation of a barrier within the imaging dish, such that one dissection session may contain both a control group as well as a drug-treatment group. This enhancement would allow for minimization of error between technical replicates, thereby improving scientific rigor.
There are no other true alternatives to this protocol, as it is the only method available to examine the live events of this organ during late embryonic development. As such, previously unanswerable questions about gonad morphogenesis are now accessible with this advancement. These questions include the intricacies of cell divisions, cytoskeletal changes, and cell intercalation events that govern stem cell niche formation and gonadogenesis. None of these events are detectable in the still images of these processes acquired using a fix-and-stain technique. Overall, this method unlocks the possibility of investigating the dynamics in the late embryonic Drosophila gonad, and such a breakthrough has strong implications for the advancement of a myriad of biological fields, including stem cell-niche biology, piRNA biology, and organogenesis.
Acknowledgments
We would like to thank Lindsey W. Plasschaert and Justin Sui for their substantial contributions to the early development of this protocol. The authors are grateful to the fly community for their generosity with reagents, and particularly to Ruth Lehmann and Benjamin Lin for their gift of the nos5’-Lifeact-tdtomato p2a tdkatushka2 Caax nos3’ line prior to its publication. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. This work was supported by NIH RO1 GM060804, R33AG04791503 and R35GM136270 (S.D) as well as training grants T32GM007229 (B.W.) and F32GM125123 (L.A.).
Footnotes
Disclosures
The authors have nothing to disclose.
References
- 1.Yamashita YM, Jones DL, Fuller MT Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science. 301, 1547–1550 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Inaba M, Yuan H, Salzmann V, Fuller MT, Yamashita YM E-Cadherin is required for centrosome and spindle orientation in Drosophila male germline stem cells. PLoS One. 5, 1–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lenhart KF, DiNardo S Somatic cell encystment promotes abscission in germline stem cells following a regulated block in cytokinesis. Developmental Cell. 34, 192–205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy RW, Tokuyasu KT, Lindsley DL, Garavito M The Germinal Proliferation Center in the Testis of Drosophila melanogaster. Journal of Ultrastructure Research. 69, 180–190 (1979). [DOI] [PubMed] [Google Scholar]
- 5.Fuller MT Spermatogenesis in The Development of Drosophila Melanogaster. (eds. Bate M, Arias AM) Cold Spring Harbor Laboratory Press; 71–147 (1993). [Google Scholar]
- 6.Lin H, Spradling AC A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 124, 2463–2476 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Emilie Q, Amit A, Kai T The piRNA pathway is developmentally regulated during spermatogenesis in Drosophila. RNA. 22, 1044–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marie PP, Ronsseray S, Boivin A From embryo to adult: PiRNA-mediated silencing throughout germline development in Drosophila. G3: Genes, Genomes, Genetics. 7, 505–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michel M, Raabe I, Kupinski AP, Pérez-Palencia R, Bökel C Local BMP receptor activation at adherens junctions in the Drosophila germline stem cell niche. Nature Communications. 2, (2011). [DOI] [PubMed] [Google Scholar]
- 10.Inaba M, Buszczak M, Yamashita YM Nanotubes mediate niche-stem-cell signalling in the Drosophila testis. Nature. 523, 329–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leatherman JL, Dinardo S Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell. 3, 44–54 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leatherman JL, DiNardo S Germline self-renewal requires cyst stem cells, while stat regulates niche adhesion in Drosophila testes. Nature Cell Biology. 12, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tulina N, Matunis E Control of stem cell self-renewal in Drosophila Spermatogenesis by JAK-STAT signaling. Science. 294, 2546–2549 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Gonczy P, Viswanathan S, Dinardo S Probing spermatogenesis in Drosophila with P-element enhancer detectors. Development. 114, 89–98 (1992). [DOI] [PubMed] [Google Scholar]
- 15.Le Bras S, Van Doren M Development of the male germline stem cell niche in Drosophila. Developmental Biology. 294, 92–103 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Cheng J, Hunt AJ Time-lapse live imaging of stem cells in drosophila testis. Current Protocols in Stem Cell Biology. 0331, 1–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anllo L, Plasschaert LW, Sui J, DiNardo S Live imaging reveals hub cell assembly and compaction dynamics during morphogenesis of the Drosophila testis niche. Developmental Biology. 446, 102–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rebecca Sheng X, Matunis E Live imaging of the Drosophila spermatogonial stem cell niche reveals novel mechanisms regulating germline stem cell output. Development. 138, 3367–3376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aboim AN Developpement embryonnaire et post-embryonnaire des gonades normales et agametiques de Drosophila melanogaster. Revue Suisse De Zoologie. 52, 53–154 (1945). [Google Scholar]
- 20.Sano H et al. The Drosophila actin regulator ENABLED regulates cell shape and orientation during gonad morphogenesis. PLoS One. 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark IBN, Jarman AP, Finnegan DJ Live imaging of Drosophila gonad formation reveals roles for Six4 in regulating germline and somatic cell migration. BMC Devlopmental Biology. 7, 1–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheng XR, Brawley CM, Matunis EL Dedifferentiating spermatogonia outcompete somatic stem cells for niche occupancy in the Drosophila testis. Cell Stem Cell. 5, 191–203 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanentzapf G, Devenport D, Godt D, Brown NH Europe PMC Funders Group integrin-dependent anchoring of a stem cell niche. Nature Cell Biology. 9, 1413–1418 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brady J A simple technique for making very fine, durable dissecting needles by sharpening tungsten wire electrolytically a motorized molluscicide dispenser. Bulletin of the World Health Organization. 105–106 (1960). [PMC free article] [PubMed] [Google Scholar]
- 25.Featherstone DE, Chen K, Broadie K Harvesting and preparing Drosophila embryos for electrophysiological recording and other procedures. Journal of Visualized Experiments: JoVE. 3–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Embryo and Larva Harvesting and Preparation. JoVE Science Education Database (2020).
- 27.Ringer’s solution for Drosophila. Cold Spring Harbor Protocols (2011).
- 28.Schuh M, Lehner CF, Heidmann S Incorporation of Drosophila CID/CENP-A and CENP-C into Centromeres during Early Embryonic Anaphase. Current Biology. 17, 237–243 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Lin B, Luo J, Lehmann R Collectively stabilizing and orienting posterior migratory forces disperses cell clusters in vivo. Nature Communications. 11, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasad M, Jang ACC, Starz-Gaiano M, Melani M, Montell DJ A protocol for culturing drosophila melanogaster stage 9 egg chambers for live imaging. Nature Protocols. 2, 2467–2473 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Gabay L, Lowell S, Rubin LL, Anderson DJ Deregulation of dorsoventral patterning by FGF confers trilineage differentiation capacity on CNS stem cells in vitro. Neuron. 40, 485–499 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Engler AJ, Sen S, Sweeney HL, Discher DE Matrix elasticity directs stem cell lineage specification. Cell. 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]