

Abstract
Discoveries of adaptive gene knockouts and widespread losses of complete genes have in recent years led to a major rethink of the early view that loss-of-function alleles are almost always deleterious. Today, surveys of population genomic diversity are revealing extensive loss-of-function and gene content variation, yet the adaptive significance of much of this variation remains unknown. Here we examine the evolutionary dynamics of adaptive loss of function through the lens of population genomics and consider the challenges and opportunities of studying adaptive loss-of-function alleles using population genetics models. We discuss how the theoretically expected existence of allelic heterogeneity, defined as multiple functionally analogous mutations at the same locus, has proven consistent with empirical evidence and why this impedes both the detection of selection and causal relationships with phenotypes. We then review technical progress towards new functionally explicit population genomic tools and genotype-phenotype methods to overcome these limitations. More broadly, we discuss how the challenges of studying adaptive loss of function highlight the value of classifying genomic variation in a way consistent with the functional concept of an allele from classical population genetics.
Subject terms: Rare variants, Molecular evolution, Evolutionary biology, Population genetics, Genome evolution
The historical context
Views on loss-of-function mutations—those abolishing a gene’s biomolecular activity—have changed considerably over the last half century. Early theories of molecular evolution that emerged during the 1960’s and 1970’s saw little potential for loss-of-function mutations to contribute to adaptation (Maynard Smith 1970). Except in the case of inactivated gene duplicates, nonfunctional alleles were often assumed to be lethal, with adaptation being generally regarded as a process explained only by the fixation of single, mutationally rare alleles that improved or altered a gene’s function (Orr 2005). Only relatively recently, through discoveries enabled by the availability of molecular sequence data, were alternative views of adaptive loss-of-function alleles formalized, most notably with the “less is more” ideas proposed by Olson (1999). Classical paradigms of molecular evolution had by that time been challenged, for example, by evidence that natural loss-of-function variants of CCR5 lead to reduced HIV susceptibility in humans (Libert et al. 1998). Discoveries during the subsequent two decades have continued to support the idea that loss of function contributes to adaptation (Murray 2020), with cases of adaptive or beneficial loss of function being discovered across diverse organisms, genes, traits, and environments (Fig. 1).
Fig. 1. Examples of genes from different species with adaptive or beneficial loss-of-function alleles.

In each example, multiple independent variants can be combined to constitute the population scale loss-of-function allele state. a Loss of function in SLC30A8 is associated with reduced risk of type 2 diabetes in humans (Flannick et al. 2014; Dwivedi et al. 2019). b Experimental evolution in Pseudomonas aeruginosa resulted repeatedly in loss-of-function mutations in nfxB, conferring antibiotic resistance (Wong et al. 2012). c Experimental evolution in yeast led to consistent disruption of specific signaling pathway genes including MTH1 during adaptation to stable environments (Kvitek and Sherlock 2013). d Populations of Plasmodium falciparum repeatedly evolved loss-of-function alleles in Epac during adaptation to lab culture environments (Claessens et al. 2017). e Natural RDO5 loss-of-function variants in Arabidopsis thaliana occurred at high frequency in northwest Europe and caused reduced seed dormancy, a trait under strong locally adaptive selection (Xiang et al. 2016). f Adaptation to agricultural intensification led to selection for semi-dwarf rice, which is caused by loss-of-function variants in GA20ox2 (Spielmeyer et al. 2002; Sasaki et al. 2002).
Today, reductive genome evolution is viewed as a powerful force of adaptation (Wolf and Koonin 2013) and gene loss is considered an important source of adaptive genetic variation (Albalat and Cañestro 2016; Murray 2020). The flood of -omics data generated in recent years is beginning to reveal the extent of loss of function and gene content variation segregating within species. Pan-genome and pan-transcriptome analyses have found that gene absence variation is pervasive in both prokaryotic and eukaryotic species (Jin et al. 2016; McInerney et al. 2017; Gerdol et al. 2020). And surveys of functional genomic diversity in organisms from Arabidopsis thaliana (Monroe et al. 2018; Xu et al. 2019) to humans (MacArthur et al. 2012; Karczewski et al. 2020) have revealed extensive genetic variation causing predicted loss of function. Yet, the adaptive importance of such variation remains largely unknown.
While the existence of adaptive loss of function is no longer seriously disputed, the assumed maladaptive nature of loss of function from early theories can persist in the language of population genetics such as in the continued use of deleterious as a synonym for loss-of-function (Moyers et al. 2018). Perhaps less visible but more consequential, historical assumptions about loss of function remain implicit in some analyses of DNA sequence variation as many classic tests for evidence of selection or causal relationships with phenotypes are built upon expectations of adaptation only involving hard sweeps of single mutationally rare alleles (Pennings and Hermisson 2006a, b). Contemporary disagreements in population genetics can also reflect differences in views on the functional molecular basis of adaptation. This can be seen for instance in alternative perspectives on the relative importance of soft versus hard selective sweeps, a debate which is inherently connected to the propensity for adaptation to involve recurrent loss-of-function mutations (Messer and Petrov 2013; Jensen 2014).
The aim of this article is to examine theoretical and empirical advances describing the population evolutionary dynamics of beneficial loss-of-function alleles, which remain on one hand a low-hanging fruit when it comes to functionally classifying molecular diversity but on the other, a particularly challenging class of molecular variants to study using common population genetics models. We hope this review will facilitate new considerations of the population genomic diversity now being revealed with the widespread generation of whole genome sequence data (Table 1, Fig. 2). We also hope to shed light on some practical challenges confronting population geneticists (Figs. 3 and 4) and the intriguing dynamics of loss-of-function alleles through the lens of classic models (Fig. 5). We further discuss advances in sequencing technologies and annotation approaches that are facilitating new ways to discover cases of beneficial loss-of-function and more broadly, syntheses between modern genomics and the functional concept of alleles from classic population genetics (Fig. 6).
Table 1.
Recent whole genome re-sequencing projects with functional annotations of variants. Numbers of premature stop, synonymous, and non-synonymous single nucleotide polymorphisms indicate number of variants segregating among the genotypes sequenced (Fig. 2). Sample Size = number of genotypes sequenced.
| Species | Sample size | Premature stops | Synonymous | Non-synonymous | Citation |
|---|---|---|---|---|---|
| Ananas comosus | 89 | 7,084 | 689,019 | 589,484 | (Chen et al. 2019) |
| Arabidopsis thaliana | 1,135 | 27,813 | 803,665 | 1,135,115 | (1001 Genomes Consortium 2016) |
| Bos indicus | 20 | 1,132 | 255,296 | 155,251 | (Iqbal et al. 2019) |
| Bos taurus | 15 | 3,837 | 1,155,244 | 524,103 | (Zhang et al. 2019) |
| Branchiostoma belcheri | 20 | 11,487 | 2,818,189 | 1,467,863 | (Bi et al. 2020) |
| Brassica napus | 991 | 1,413 | 120,926 | 79,018 | (Wu et al. 2019) |
| Caenorhabditis elegans | 330 | 5,084 | 271,950 | 261,538 | (Cook et al. 2017) |
| Cairina moschata | 15 | 285 | 36,517 | 19,817 | (Gu et al. 2020) |
| Canis lupus | 722 | 11,273 | 540,063 | 332,559 | (Plassais et al. 2019) |
| Capsella grandiflora | 15 | 5,209 | 644,326 | 478,238 | (Koenig et al. 2019) |
| Capsella orientalis | 16 | 269 | 11,250 | 13,281 | (Koenig et al. 2019) |
| Capsella rubella | 50 | 2,508 | 194,078 | 171,071 | (Koenig et al. 2019) |
| Cicer arietinum | 16 | 352 | 50,290 | 38,078 | (Thudi et al. 2016) |
| Cucumis melo | 1,175 | 7,030 | 102,687 | 94,426 | (Zhao et al. 2019) |
| Cucurbita pepo | 7 | 864 | 156,828 | 111,687 | (Xanthopoulou et al. 2019) |
| Drosophila melanogaster | 205 | 1,532 | 351,255 | 182,520 | (Huang et al. 2014) |
| Ebola virus | 140 | 3 | 555 | 403 | (Ladner et al. 2015) |
| Echinochloa crus-galli | 328 | 9,264 | 184,746 | 319,816 | (Ye et al. 2019) |
| Felis catus | 54 | 838 | 128,844 | 77,662 | (Buckley et al. 2020) |
| Fraxinus excelsior | 37 | 2,997 | 251,249 | 259,946 | (Sollars et al. 2017) |
| Glycine max | 1,007 | 2,826 | 122,469 | 182,479 | (Torkamaneh et al. 2019) |
| Gossypium spp. | 243 | 6,851 | 101,059 | 128,512 | (Du et al. 2018) |
| Hippotragus niger | 7 | 201 | 11,350 | 11,386 | (Koepfli et al. 2019) |
| Homo sapiens | 141,465 | 133,019 | 2,173,110 | 4,548,307 | (Karczewski et al. 2020) |
| Macaca mulatta | 133 | 2,642 | 148,278 | 126,445 | (Xue et al. 2016) |
| Manihot esculenta | 203 | 4,399 | 265,094 | 299,197 | (Ramu et al. 2017) |
| Mycoplasma pneumoniae | 15 | 88 | 4,382 | 6,891 | (Xiao et al. 2015) |
| Oryza sativa | 3,024 | 198,609 | 2,952,705 | 3,599,083 | Wang et al. 2018) |
| Parastagonospora nodorum | 197 | 2,815 | 226,803 | 160,159 | (Richards et al. 2019) |
| Phaseolus vulgaris | 683 | 1,352 | 112,173 | 97,536 | (Wu et al. 2020) |
| Populus trichocarpa | 1,014 | 8,365 | 231,894 | 333,036 | (Piot et al. 2019) |
| Protobothrops mucrosquamatus | 22 | 883 | 53,023 | 56,553 | (Aird et al. 2017) |
| Puccinia hordei | 5 | 2,629 | 46,763 | 67,526 | (Chen et al. 2019) |
| Rattus norvegicus | 40 | 285 | 42,182 | 26,239 | (Hermsen et al. 2015) |
| SARS-nCoV-2 | 8,053 | 90 | 2,678 | 4,731 | (Rayko and Komissarov 2020) |
| Saccharomyces cerevisiae | 1,011 | 7,207 | 517,729 | 549,300 | (Peter et al. 2018) |
| Solanum melongena | 7 | 438 | 6,645 | 12,828 | (Gramazio et al. 2019) |
| Solanum tuberosum | 201 | 4,962 | 541,208 | 515,492 | (Li et al. 2018) |
| Sorghum bicolor | 44 | 3,114 | 112,255 | 112,108 | (Mace et al. 2013) |
| Trypanosoma evansi | 15 | 1,685 | 30,714 | 53,002 | (Lazaro et al. 2020) |
Fig. 2. Rates of putative loss-of-function variants (stop gained) relative to synonymous single nucleotide polymorphisms reported in recent whole genome re-sequencing projects (Table 1).
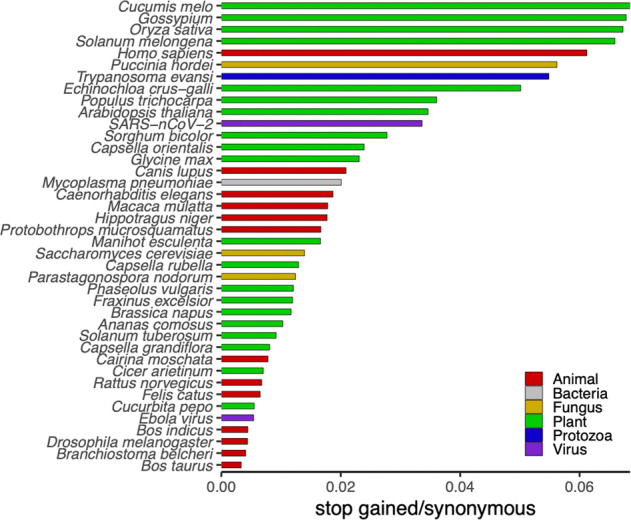
Species exhibit considerable differences in the ratio of stop gained to synonymous single nucleotide polymorphisms, with a 20-fold difference between the greatest (Cucumis melo) and fewest (Bos taurus) observations. The causes for these differences between species and, more generally, the (mal)adaptive nature of this extensive loss-of-function variation remain largely unknown.
Fig. 3. Examples of loss-of-function (LoF) variants.

Shown are those types caused by different kinds of mutations (SNP single nucleotide polymorphism, indel insertions and deletions), which vary in the kind of data needed to detect them (short/long read sequencing) and the predictability of their effect on gene function.
Fig. 4. Theoretical predictions and empirical observations of allelic heterogeneity.

a Predicted values of the number of independent variants of the same allele observed at fixation (k) as a function of mutation rates (u). Equation based on Haldane (1927) and Kimura (1962) and taken from Wilson et al. (2014). Predictions are based on an effective population size (Ne) of 50,000 and selection coefficient (s) of 0.01. Highlighted are frequently observed ranges of empirical estimates of mutation rates from classic population genetics (Muller 1928; Haldane 1933; Rhoades 1941; Stadler 1946, 1948) and sequence-based mutation rates from modern molecular genomics (Lynch et al. 2016). Inset figure illustrates the hypothetical dynamics of multiple independent alleles (each a different color) with positive selection. Collectively the variants increase in frequency, ultimately leading to fixation of adaptive variants (elimination of deleterious ancestral allele), but individually each variant remains at low frequency. b Detected levels of allelic heterogeneity in genes enriched for loss of function in humans (obs > exp) reported by (Karczewski et al. 2020). Highlighted are cases of previously studied genes with evidence of beneficial effects or positive selection CCR5 (Libert et al. 1998), SLC30A8 (Flannick et al. 2014; Dwivedi et al. 2019), TMPRSS11B (Updegraff et al. 2018), TRIM65 (Wang et al. 2016; Wei et al. 2018), PLA2G7 (Song et al. 2012), HDAC10 (Dahiya et al. 2020), CD36 (Fry et al. 2009; Love-Gregory et al. 2011).
Fig. 5. Among genes with high Neutrality Index (NI), those with a high frequency of loss-of-function alleles are enriched for non-synonymous polymorphism (Pn).
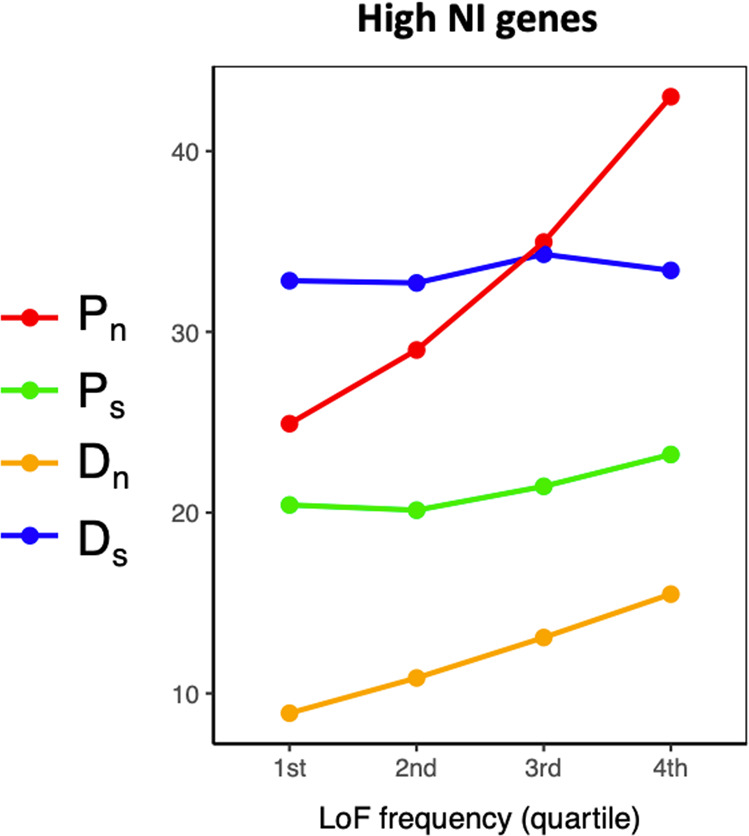
Shown here are mean components of NI in A. thaliana genes with high (top 20%) NI values in relation to loss-of-function (LoF) allele frequencies (binned by quartiles). Loss-of-function calls based on approach from (Monroe et al. 2018; Baggs et al. 2020) and data from (Monroe et al. 2020). Pn = non-synonymous polymorphism, Ps = synonymous polymorphism, Dn = non-synonymous divergence, Ds = synonymous divergence (using A. lyrata as an outgroup).
Fig. 6. Imagined case in which independent loss-of-function (LoF) alleles give rise to adaptive dwarf phenotypes inspired by (Barboza et al. 2013).

In this case, a premature stop codon and a frameshift mutation have arisen in alternative genetic backgrounds distinguished here by a nearby SNP (top). Conventional, functionally agnostic GWAS (bottom left) tests for association between individual variants and the trait of interest, in this case plant height, fail because none of the individual variants capture the functionally definitive variation (indicated by p-values below the significance threshold marked by the dashed line). An alternative approach, functional GWAS, first annotates variants according to predicted functional effects, then defines alleles as functional or non-functional. This corrects for allelic heterogeneity when testing for allele-trait associations and results in a significant allele trait association (bottom right).
What are loss-of-function alleles?
Classifying biological diversity into discrete categories has always been difficult. Descriptions of even the most fundamental biological units such as cell types, populations, and species can be challenging. Yet these classifications provide units of study that help make sense of biological and evolutionary phenomena. This is also true for alleles.
The existence of a category of alleles distinguished by a derived loss of biochemical function has been described by various names: “amorphic” (Muller 1932), “loss-of-function” (Jones 1972), “nonfunctional” (Nei and Roychoudhury 1973), “knockout” (Kulkarni et al. 1999),”null” (Engel et al. 1973), “pseudogene” (Jacq et al. 1977), or simply “gene loss” (Zimmer et al. 1980). Total gene loss is the most obvious case of loss of function. Comparisons of gene content between distantly related species have revealed considerable evidence for adaptation via complete deletion of genes or even entire sets of functionally related genes (Wang et al. 2006; Blomme et al. 2006; Will et al. 2010; McLean et al. 2011; Griesmann et al. 2018; van Velzen et al. 2018; Sharma et al. 2018; Huelsmann et al. 2019; McGowen et al. 2020; Baggs et al. 2020). Pangenome analyses have revealed extensive gene content variation segregating within species. For example, the average Brachypodium distachyon genotype is missing almost half of the genes observed in the species pangenome (Gordon et al. 2017). Yet total gene loss is not the only means by which loss of function can occur. In their review of evolution by gene loss, Albalat and Cañestro (2016) point out that single mutations and many mutation types such as premature stop codons, frameshifts, splice site disruptions, and elimination of regulatory regions required for gene expression can have effects that are functionally indistinguishable from complete gene loss. Here we will discuss how the phenomenon of allelic heterogeneity—that numerous types of mutations can produce the same functionally analogous allele—is important for understanding the evolutionary dynamics and implications of adaptation by loss of function.
First principles and empirical evidence indicate that many types of mutations can have effects that are equivalent to total gene loss, and for the purposes of this review, we employ this definition of complete gene losses being functionally equivalent to other loss-of-function mutations such as premature stop codons. However, there is the practical difficulty that these different types of mutations vary in how easily they can be detected and correctly annotated as loss-of-function alleles (Fig. 3). Insertions and deletions that interrupt the reading frame of a protein coding region (frameshift mutations), for example, might be readily classified as loss-of-function alleles because the downstream amino acid sequence will be severely disrupted. Yet a frameshift mutation at the extreme 3′ end of a coding region affecting only a few amino acids might be functionally distinct from a frameshift mutation at the extreme 5′ end disrupting the entire coding sequence. One simple heuristic to address this ambiguity is a threshold, measured by the portion of the gene affected by functionally disruptive mutations, at which an allele is classified as loss-of-function. This approach can be used to classify premature stop codons, frameshift, splice site disruptions, start loss, and inframe insertions and deletions. In humans (MacArthur et al. 2012; Karczewski et al. 2020) and Arabidopsis thaliana (Monroe et al. 2018; Baggs et al. 2020), loss-of-function mutations affecting only a small fraction (e.g., <10%) of total coding sequence in a gene were ignored when classifying loss-of-function variants. Such cutoffs are supported by the observation that even in genes not thought to be experiencing adaptive loss of function, there is an enrichment for otherwise predicted loss-of-function mutations that affect only small fractions of coding regions (MacArthur et al. 2012; Flowers et al. 2009), suggesting reduced functional impact of such variants.
Other single mutations causing loss of function may be even more difficult to predict. Mutations changing functionally critical amino acids can disable a protein’s molecular function. Indeed, detailed studies of individual genes have uncovered non-synonymous loss-of-function variants (Sasaki et al. 2002; Barboza et al. 2013; Zhang and Jiménez-Gómez 2020; Song et al. 2020, 2014) suggesting the maintenance of extensive cryptic (not easily identified as loss-of-function from standard annotation pipelines) genetic loss-of-function variation within populations (Table 1). But identifying non-synonymous mutations which result in loss-of-function among the numerous non-synonymous polymorphisms is difficult since experimental validation of the functional impacts for every non-synonymous variant is infeasible at genomic scales. Instead, predictions of functional impact must be predicted by more sophisticated methods (Tang and Thomas 2016), such as quantifying changes in the chemical properties of amino acid substitutions (Grantham 1974), sequence homology (Ng and Henikoff 2001), known phenotypic effects (Schwarz et al. 2010), the context of known domains and protein structures, or through integration of multiple methods with tools such as CADD (Kircher et al. 2014; Tang and Thomas 2016). Emerging statistical machine learning approaches, such as unsupervised latent variable models can also detect otherwise cryptic loss of function caused by non-synonymous substitutions (Riesselman et al. 2018). The effect of coding sequence variation on protein folding may also be predicted from deep learning approaches, such as AlphaFold (Senior et al. 2020). Beyond mutations affecting coding sequence, eliminating gene expression could also cause loss of function (Albalat and Cañestro 2016), but identifying such mutations is challenging and validation at genomic scales is currently difficult. However, as with non-synonymous substitutions, advances in machine learning have also led to models that can predict functional consequences of non-coding variants (Zhou and Troyanskaya 2015). These methods can also be used to predict variants causing loss of gene expression. The application of these new tools presents a path forward for a new generation of functionally explicit analyses of genomic diversity. More broadly, a major goal of modern biology is to predict molecular function from genomic sequence data. The study of adaptive loss-of-function alleles could serve as a model class of genetic variation to spearhead this effort.
The accurate prediction of allele function from population genomic data assumes that researchers have complete information about what is functional and about sample sequence diversity. The genomes of reference genotypes used as the basis of comparison for whole genome re-sequencing projects can themselves already harbor loss-of-function alleles, obfuscating definitions of “functional” and therefore loss of function as well. For example, the standard A. thaliana reference is based on the genome of the Col-0 genotype, which is known to harbor an adaptive loss-of-function variant in the vernalization gene FRIGIDA (Johanson et al. 2000). Therefore, to study natural functional variation in this locus, Zhang and Jiménez-Gómez (2020) computationally swapped the reference sequence at this locus with a known functional allele and remapped public short read sequencing data to discover novel loss-of-function variants. Such scenarios at genome-wide scales motivate ongoing efforts to generate and annotate multiple reference genomes for a given species (Michael et al. 2018; Sun et al. 2018; Yang et al. 2019; Jiao and Schneeberger 2020; Zhou et al. 2020; Liu et al. 2020; Michael and VanBuren 2020; Li et al. 2020) to be used as a basis of comparison to describe broader population genetic diversity. Furthermore, most population-scale genome sequencing has been completed using short read sequencing technologies (</= 300 base pairs), which require greater depth to reliably detect small insertions and deletions (compared to single nucleotide polymorphisms) and may be unable to reliably detect large insertions, deletions, and other structural variants altogether (Kishikawa et al. 2019). These unseen variants could be a considerable source of loss-of-function alleles in natural populations, and the difficulty to detect them (Fig. 3) might imply that many adaptive loss-of-function alleles are yet to be discovered. Thus, most assessments of population genetic variation are still limited to only a fraction of functional allelic diversity. Third-generation sequencing technologies are therefore facilitating more complete characterizations of allelic diversity (Alonge et al. 2020; Liu et al. 2020). Precise characterization of alleles at functional molecular resolutions is greater than being a technical challenge for studying sequence variation—it is essential for making sense of genomic sequence data through the lens of classic population genetics theory. We will see how this is exemplified in cases of adaptive loss-of-function alleles, whose high effective mutation rate leads to a breakdown of the assumptions underlying standard approaches used to detect signatures of selection and genotype to phenotype mapping.
Many ways to break a gene: quantifying allelic heterogeneity
A characteristic feature of genes experiencing adaptive loss of function is the existence of multiple functionally equivalent variants. To understand the extent of such variation, we can quantify and predict allelic heterogeneity, the phenomenon where multiple independent molecular variants exist that produce functionally analogous alleles of a given locus (Haldane 1927; Kimura 1962; Wilson Petrov et al. 2014; Ralph and Coop 2015). Assuming a constant effective population size (Ne), mutation rate of an adaptive allele (u), and selection coefficient on that adaptive allele (s), the expected number of mutationally independent alleles of the locus that will be observed in a population at the moment of allele fixation (k), a unit of allelic heterogeneity, is predicted (Wilson Petrov et al. 2014) as:
| 1 |
The expected number of independent alleles at fixation is directly correlated with the mutation rate (Eq. 1, Fig. 4). Early studies of mutation rate quantified the frequency at which mutations gave rise to a particular allelic state, defined by its phenotypic effect. These studies often reported phenotypic mutation rate estimates ranging from 10-4 to 10-6 mutations (change in phenotype) per generation (Muller 1928; Haldane 1933; Rhoades 1941; Stadler 1946, 1948). Estimates of molecular mutation rates at the DNA sequence level are generally orders of magnitude lower: 10−8 to 10−10 mutations (change in sequence) per site per generation (Lynch et al. 2016). A partial explanation for the discrepancy between the range of phenotypic and molecular mutation rate is the obvious fact that many different molecular mutations can give rise to the same phenotypically/functionally effective allele type. Loss-of-function mutations exemplify this reality. Because there are hundreds or thousands of different molecular mutations that can produce a suite of analogous loss-of-function alleles (e.g., any premature stop codon or differently sized deletions along much of the coding region of a gene), the aggregated mutation rate for loss of function is expected to be orders of magnitude greater than the molecular mutation rate. At such high effective mutation rates we should predict, given biologically reasonable population sizes and selection coefficients, the existence of considerable allelic heterogeneity (Fig. 4a), which appears consistent with empirical observations (Figs. 1 and 4b).
Mixed signals in signatures of selection
Early genetics employed a functionally definitive concept of an allele. Alleles were treated as local units of inheritance based on their functional effect, observed at the phenotypic level (e.g., Rhoades 1938). As such, at locus a experiencing adaptive loss of function, the (potentially multiple) variants causing the adaptive trait should act collectively as a single allele, even if due to independent mutational events (Pennings and Hermisson 2006a, b). If, for example, this functionally identical set of variants experiences positive selection, it behaves like a single allele according to predictions of classic population genetic theory (Orr 2005)—increasing in frequency to fixation (Fig. 4a, inset). Indeed, foundational models of population genetics (Haldane 1927) accommodate recurrent mutation and predict that adaptation will often involve multiple independent mutational origins given realistic population sizes, selection coefficients, and mutation rates (Eq. 1, Fig. 4a). But if we encounter such cases through analyses of DNA sequence alone, we may be troubled to find that the sequence variants only exhibit the expected evolutionary dynamics of classical alleles when considered as aggregated functional units, but not when analyzed individually (Remington 2015).
Scenarios like these have been extensively studied in a broad manner, in order to detect signatures of soft sweeps of multiple independent variants. A number of approaches have been developed to study soft sweeps. These generally do not attempt to classify variants into functional allele categories but instead look for evidence of increased frequency of multiple rather than single haplotypes in a functionally agnostic fashion (Schrider and Kern 2016; Hermisson and Pennings 2017; Harris et al. 2018; Mughal and DeGiorgio 2019; Stern et al. 2019; Hartfield and Bataillon 2020; Garud et al. 2020). Nevertheless, it is interesting to note that extensive research into soft sweeps came only after increasing evidence of the potential adaptive value of loss-of-function alleles had been published (Pennings and Hermisson 2006a, b 2006). In contrast, hard sweeps of a single adaptive variant were described during the era predominated by the view that loss-of-function mutations were necessarily deleterious, and adaptation could only proceed through mutationally rare gain-of-function alleles (Maynard Smith and Haigh 1974). Such historical dynamics speak to the interconnectedness, intentional or otherwise, between ideas about the functional molecular basis of adaptation and advances in the development of population genetic models and theories.
Unfortunately, population genetic statistics based on the expectation that adaptive alleles are mutationally rare perform poorly when this assumption is violated. For example, statistics based on the site frequency spectrum such as Tajima’s D do not deviate from neutral expectations in a predictable fashion for adaptive alleles with multiple mutational origins (Pennings and Hermisson 2006a). Similarly, statistics based on linkage disequilibrium around adaptive loci, though they tend to perform better for soft sweeps, also appear neutral if the number of mutational origins of an adaptive allele is high enough (Hermisson and Pennings 2017). For adaptive loss of function, this may often be the case. More generalized methods of detecting soft selective sweeps from independent mutational origins, such as the H12 statistic developed by Garud and colleagues (Garud et al. 2015) might be useful for detecting adaptive loss of function. The reciprocal is also true—known cases of adaptive loss of function could serve as valuable models for testing the limits of test statistics intended to detect soft sweeps.
More functionally explicit statistics of allelic variation are now possible because of the availability of whole genomic sequence data. However, the application of functional test statistics to genes experiencing putatively adaptive loss-of-function can yield surprising results. For example, the Neutrality Index (NI) (McDonald and Kreitman 1991; Rand and Kann 1996) estimates histories of selection by comparing rates of within-species polymorphism and between-species divergence. It is more functionally explicit than many population genetics statistics—comparing putatively functionally impactful (non-synonymous) versus silent (synonymous) variation. Where Pn = non-synonymous polymorphism, Ps = synonymous polymorphism, Dn = non-synonymous divergence, Ds = synonymous divergence
| 2 |
Traditional interpretations of the results are based on the assumption that adaptive variants will become fixed and therefore be observed as diverged (Dn) from related species rather than polymorphic (Pn) within the study species. When genes putatively experiencing adaptive loss of function are investigated, they are often found to have high NI values (Le Corre et al. 2002; Flowers et al. 2009; Will et al. 2010; Rose et al. 2012; Monroe et al. 2016), a pattern that seems paradoxical given that high NI values are commonly interpreted as evidence of purifying selection (Weinreich and Rand 2000). But when considered with the knowledge that non-synonymous variants can themselves cause loss of function, and given the likely independent mutational origins of loss of function, this result is consistent with expectations of an enrichment of non-synonymous polymorphism in genes with both high frequency of loss of function and high NI (Fig. 5).
Increasingly functionally precise statistics such as the sum frequencies of losses of function in a given gene across all variants (Albalat and Cañestro 2016) might better describe loss-of-function alleles than functionally agnostic test statistics or descriptions of individual variants. Accelerations in whole genome sequencing technologies and improved capacity to classify previously cryptic loss-of-function variants may facilitate a new generation of functionally definitive population genetic models and methods. This would not only be valuable for improving the capacity to understand the forces shaping intraspecific loss-of-function, but more generally promote a re-synthesis between studies of molecular sequence variation and the function-based conception of alleles from early population genetic theory.
Functionally explicit genotype-to-phenotype mapping
To identify genes contributing to adaptive phenotypic variation, Genome Wide Association (GWA) scans in natural populations have become a popular alternative to conventional mapping in an experimental population derived from a bi-parental cross. GWA is normally implemented by testing for associations between individual DNA sequence variants in a population and the phenotype (or environmental gradient) of interest. This statistical framework can fail to detect causal loci in the presence of allelic heterogeneity because none of the individual variants are linked to a single causal variant—an assumption of single-locus two-allele population genetic models (Korte and Farlow 2013). This problem is exemplified by loss of function variation in which, with a few notable exceptions (Song et al. 2020), allelic heterogeneity is expected to be the norm (Pennings and Hermisson 2006a, b).
The case of the GA-20 oxidase gene in plants provides a useful illustration of these challenges. This well-studied gene is involved in gibberellin biosynthesis and loss of function produces semi-dwarf phenotypes in wild plants and crop varieties of the Green Revolution (Fig. 1f) (Spielmeyer et al. 2002; Sasaki et al. 2002; Jia et al. 2009; Barboza et al. 2013). While functional experiments have demonstrated that loss of this gene causes considerable reduction in plant height, and investigations of natural molecular variation in A. thaliana identified cases of likely loss-of-function differences between genotypes, a conventional GWA looking for loci explaining variation in plant height failed to detect the GA-20 oxidase locus in A. thaliana (Barboza et al. 2013). However, when all of the genotypes with predicted loss-of-function variants were collapsed into a single allele state and their heights contrasted with the genotypes containing predicted functional variants, the known highly significant effect on plant height was detected (Barboza et al. 2013). Without previous knowledge that this gene plays an important role in plant height, it would have been missed by conventional GWA. This experiment provides a cautionary tale as to how conventional GWA approaches can fail in the presence of allelic heterogeneity at causal loci. It also highlights the power of functionally explicit GWA approaches based on population genetic models that allow for allelic heterogeneity—using predictions about functional effects of individual variants to collapse variants into allele classes (in this case, loss-of-function vs functional) so that a functionally explicit contrast can be made (Fig. 6).
To date, such a framework has been primarily used in the study of rare variants (Wu et al. 2011; Pan and Shen 2011; Zhang et al. 2017) to identify rare deleterious loss-of-function alleles associated with disease phenotypes in humans (Zuk et al. 2014) but it could also be used to find beneficial and adaptive loss of function as well. For example, loss of function in SLC30A8 was found to be strongly associated with decreased risk of type 2 diabetes when all loss-of-function variants were collapsed into a single allele state (Flannick et al. 2014) (Fig. 1a), thus identifying its protein product as a promising therapeutic target to treat diabetes (Dwivedi et al. 2019). With population whole-genome-sequence data becoming available in model and non-model species, this approach can now be readily applied by evolutionary biologists at genome wide scales to discover loss-of-function alleles contributing to phenotypic evolution in populations (Monroe et al. 2020).
A functionally explicit GWA framework may have value beyond scanning genomes for causal loss-of-function alleles. More broadly, it reflects a step toward representing genetic diversity as a matrix of functionally relevant genetic alleles rather than a matrix of DNA sequence variants. While loss of function is currently the easiest allele state to classify, we anticipate that more nuanced and precise allele categories can be identified through analyses of population genomic diversity with advances in sequence annotation. Ideally, these categories would specify the activity of an allele along a scale that reflects Muller’s original categories of amorphic, hypomorphic, hypermorphic, antimorphic, and neomorphic states (Muller 1932). In addition to facilitating discovery of causal loci, functionally explicit methods of population genomics could be useful for predicting quantitative traits (i.e., genomic prediction) and address the problem of missing heritabilities (Manolio et al. 2009) that has frustrated modern geneticists for over a decade (Eichler et al. 2010).
Outlook and concluding remarks
Loss-of-function alleles were once often held up as a paragon of deleterious genetic variation. Today a more nuanced appreciation for their potential role in adaptation has emerged. This new paradigm inspires investigations into deeper questions about the causes and consequences of adaptation by genetic loss of function. For example: Do species or populations differ in their capacity to adapt via loss of function, and if so, why? Does the high effective mutation rate of loss-of-function alleles lead to bias in the probabilities of different evolutionary outcomes? What is the contribution of adaptive loss of function to the phenomena of antagonistic pleiotropy and reproductive isolation? How does adaptation by loss of function affect long term evolutionary trajectories of populations and future evolvability? Ongoing technical breakthroughs promise to scale up the study of loss-of-function alleles experiencing positive selection for population genomic research to address these questions. More broadly, these lines of research provide paths toward advancing tools and concepts that facilitate a continued synthesis between functional molecular genomics and classic population genetic theory.
Acknowledgements
We would like to thank Wim Soppe, Alex Wong, Matthew Rutter, Michael Turelli and Andrew Whitehead for insights and feedback that improved this manuscript. We thank DBCLS (CC BY 4.0), CDC, Mae Melvin (Public Domain), Mogana Das Murtey and Patchamuthu Ramasamy (CC BY 3.0) Aomorikuma (CC BY 4.0) for images used in this manuscript. This work was supported by NSF Awards 1556262 to JKM and 1701918 to JGM, DFG ERA-CAPS 1001 G + to DW, and the Max Planck Society.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Associate editor: Frank Hailer
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1001 Genomes Consortium 1135 genomes reveal the global pattern of polymorphism in arabidopsis thaliana. Cell. 2016;166:481–491. doi: 10.1016/j.cell.2016.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird SD, Arora J, Barua A, Qiu L, Terada K, Mikheyev AS. Population genomic analysis of a pitviper reveals microevolutionary forces underlying venom chemistry. Genome Biol Evol. 2017;9:2640–2649. doi: 10.1093/gbe/evx199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albalat R, Cañestro C. Evolution by gene loss. Nat Rev Genet. 2016;17:379–391. doi: 10.1038/nrg.2016.39. [DOI] [PubMed] [Google Scholar]
- Alonge M, Wang X, Benoit M, Soyk S, Pereira L, Zhang L et al. (2020) Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell [DOI] [PMC free article] [PubMed]
- Baggs E, Monroe JG, Thanki AS, O’Grady R, Schudoma C, Haerty W, et al. (2020) Convergent loss of an EDS1/PAD4 signaling pathway in several plant lineages reveals co-evolved components of plant immunity and drought response. Plant Cell 32:2158–2177 [DOI] [PMC free article] [PubMed]
- Barboza L, Effgen S, Alonso-Blanco C, Kooke R, Keurentjes JJB, Koornneef M, et al. Arabidopsis semidwarfs evolved from independent mutations in GA20ox1, ortholog to green revolution dwarf alleles in rice and barley. Proc Natl Acad Sci USA. 2013;110:15818–15823. doi: 10.1073/pnas.1314979110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi C, Lu N, Han T, Huang Z, Chen J-Y, He C, et al. Whole-genome resequencing of twenty branchiostoma belcheri individuals provides a brand-new variant dataset for branchiostoma. Biomed Res Int. 2020;2020:3697342. doi: 10.1155/2020/3697342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme T, Vandepoele K, De Bodt S, Simillion C, Maere S, Van de Peer Y. The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol. 2006;7:R43. doi: 10.1186/gb-2006-7-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley RM, Davis BW, Brashear WA, Farias FHG (2020). A new domestic cat genome assembly based on long sequence reads empowers feline genomic medicine and identifies a novel gene for dwarfism. bioRxiv [DOI] [PMC free article] [PubMed]
- Chen J, Wu J, Zhang P, Dong C, Upadhyaya NM, Zhou Q, et al. De Novo genome assem comp genomics barley leaf rust pathog puccinia hordei identifies candidates three avirulence genes. G3. 2019;9:3263–3271. doi: 10.1534/g3.119.400450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-Y, VanBuren R, Paris M, Zhou H, Zhang X, Wai CM, et al. The bracteatus pineapple genome and domestication of clonally propagated crops. Nat Genet. 2019;51:1549–1558. doi: 10.1038/s41588-019-0506-8. [DOI] [PubMed] [Google Scholar]
- Claessens A, Affara M, Assefa SA, Kwiatkowski DP, Conway DJ. Culture adaptation of malaria parasites selects for convergent loss-of-function mutants. Sci Rep. 2017;7:41303. doi: 10.1038/srep41303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Zdraljevic S, Roberts JP, Andersen EC. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 2017;45:D650–D657. doi: 10.1093/nar/gkw893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiya S, Beier UH, Wang L, Han R, Jiao J, Akimova T, et al. HDAC10 deletion promotes Foxp3+ T-regulatory cell function. Sci Rep. 2020;10:424. doi: 10.1038/s41598-019-57294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Huang G, He S, Yang Z, Sun G, Ma X, et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat Genet. 2018;50:796–802. doi: 10.1038/s41588-018-0116-x. [DOI] [PubMed] [Google Scholar]
- Dwivedi OP, Lehtovirta M, Hastoy B, Chandra V, Krentz NAJ, Kleiner S, et al. Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat Genet. 2019;51:1596–1606. doi: 10.1038/s41588-019-0513-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel W, Schmidtke J, Vogel W, Wolf U. Genetic polymorphism of lactate dehydrogenase isoenzymes in the carp (Cyprinus carpio) apparently due to a ‘null allele’. Biochem Genet. 1973;8:281–289. doi: 10.1007/BF00486181. [DOI] [PubMed] [Google Scholar]
- Flannick J, Thorleifsson G, Beer NL, Jacobs SBR, Grarup N, Burtt NP, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46:357–363. doi: 10.1038/ng.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers JM, Hanzawa Y, Hall MC, Moore RC, Purugganan MD. Population genomics of the Arabidopsis thaliana flowering time gene network. Mol Biol Evol. 2009;26:2475–2486. doi: 10.1093/molbev/msp161. [DOI] [PubMed] [Google Scholar]
- Fry AE, Ghansa A, Small KS, Palma A, Auburn S, Diakite M, et al. Positive selection of a CD36 nonsense variant in sub-Saharan Africa, but no association with severe malaria phenotypes. Hum Mol Genet. 2009;18:2683–2692. doi: 10.1093/hmg/ddp192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015;11:e1005004. doi: 10.1371/journal.pgen.1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Messer PW, Petrov DA (2020) Detection of hard and soft selective sweeps from Drosophila melanogaster population genomic data. bioRxiv [DOI] [PMC free article] [PubMed]
- Gerdol M, Moreira R, Cruz F, Gómez-Garrido J, Vlasova A, Rosani U et al. (2020) Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol 21:275 [DOI] [PMC free article] [PubMed]
- Gordon SP, Contreras-Moreira B, Woods DP, Des Marais DL, Burgess D, Shu S, et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat Commun. 2017;8:2184. doi: 10.1038/s41467-017-02292-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramazio P, Yan H, Hasing T, Vilanova S, Prohens J, Bombarely A. Whole-genome resequencing of seven eggplant (Solanum melongena) and one wild relative (S. incanum) accessions provides new insights and breeding tools for eggplant enhancement. Front Plant Sci. 2019;10:1220. doi: 10.3389/fpls.2019.01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Griesmann M, Chang Y, Liu X, Song Y, Haberer G, Crook MB et al. (2018) Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 361:eaat1743 [DOI] [PubMed]
- Gu L, Wang F, Lin Z, Xu T, Lin D, Xing M, et al. Genetic characteristics of Jiaji Duck by whole genome re-sequencing. PLoS ONE. 2020;15:e0228964. doi: 10.1371/journal.pone.0228964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. A mathematical theory of natural and artificial selection, Part V: selection and mutation. Math Proc Camb Philos Soc. 1927;23:838–844. doi: 10.1017/S0305004100015644. [DOI] [Google Scholar]
- Haldane JBS. The part played by recurrent mutation in evolution. Am Nat. 1933;67:5–19. doi: 10.1086/280465. [DOI] [Google Scholar]
- Harris AM, Garud NR, DeGiorgio M. Detection and classification of hard and soft sweeps from unphased genotypes by multilocus genotype identity. Genetics. 2018;210:1429–1452. doi: 10.1534/genetics.118.301502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfield M, Bataillon T. Selective sweeps dominance inbreeding. G3. 2020;10:1063–1075. doi: 10.1534/g3.119.400919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. Soft sweeps and beyond: understanding the patterns and probabilities of selection footprints under rapid adaptation (J Kelley, Ed.) Methods Ecol Evol. 2017;8:700–716. doi: 10.1111/2041-210X.12808. [DOI] [Google Scholar]
- Hermsen R, de Ligt J, Spee W, Blokzijl F, Schäfer S, Adami E, et al. Genomic landscape of rat strain and substrain variation. BMC Genomics. 2015;16:357. doi: 10.1186/s12864-015-1594-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Massouras A, Inoue Y, Peiffer J, Ràmia M, Tarone AM, et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 2014;24:1193–1208. doi: 10.1101/gr.171546.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsmann M, Hecker N, Springer MS, Gatesy J, Sharma V, Hiller M. Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Sci Adv. 2019;5:eaaw6671. doi: 10.1126/sciadv.aaw6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal N, Liu X, Yang T, Huang Z, Hanif Q, Asif M, et al. Genomic variants identified from whole-genome resequencing of indicine cattle breeds from Pakistan. PLoS ONE. 2019;14:e0215065. doi: 10.1371/journal.pone.0215065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacq C, Miller JR, Brownlee GG. A pseudogene structure in 5S DNA of Xenopus laevis. Cell. 1977;12:109–120. doi: 10.1016/0092-8674(77)90189-1. [DOI] [PubMed] [Google Scholar]
- Jensen JD. On the unfounded enthusiasm for soft selective sweeps. Nat Commun. 2014;5:5281. doi: 10.1038/ncomms6281. [DOI] [PubMed] [Google Scholar]
- Jiao W-B, Schneeberger K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat Commun. 2020;11:1–10.. doi: 10.1038/s41467-020-14779-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Q, Zhang J, Westcott S, Zhang X-Q, Bellgard M, Lance R, et al. GA-20 oxidase as a candidate for the semidwarf gene sdw1/denso in barley. Funct Integr Genomics. 2009;9:255–262. doi: 10.1007/s10142-009-0120-4. [DOI] [PubMed] [Google Scholar]
- Jin M, Liu H, He C, Fu J, Xiao Y, Wang Y, et al. Maize pan-transcriptome provides novel insights into genome complexity and quantitative trait variation. Sci Rep. 2016;6:18936. doi: 10.1038/srep18936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science. 2000;290:344–347. doi: 10.1126/science.290.5490.344. [DOI] [PubMed] [Google Scholar]
- Jones EW. Fine structure analysis of the ade3 Locus in SACCHAROMYCES CEREVISIAE. Genetics. 1972;70:233–250. doi: 10.1093/genetics/70.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. On the probability of fixation of mutant genes in a population. Genetics. 1962;47:713–719. doi: 10.1093/genetics/47.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of humangenetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishikawa T, Momozawa Y, Ozeki T, Mushiroda T, Inohara H, Kamatani Y, et al. Empirical evaluation of variant calling accuracy using ultra-deepwhole-genome sequencing data. Sci Rep. 2019;9:1784. doi: 10.1038/s41598-018-38346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig D, Hagmann J, Li R, Bemm F, Slotte T, Neuffer B, et al. (2019) Long-term balancing selection drives evolution of immunity genes in Capsella. Elife 8:e43606 [DOI] [PMC free article] [PubMed]
- Koepfli K-P, Tamazian G, Wildt D, Dobrynin P, Kim C, Frandsen PB, et al. Whole genome sequencing and re-sequencing of the sable antelope (Hippotragus niger): a resource for monitoring diversity in ex situ and in situ populations. G3. 2019;9:1785–1793. doi: 10.1534/g3.119.400084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte A, Farlow A. The advantages and limitations of trait analysis with GWAS: a review. Plant methods. 2013;9:1–9. doi: 10.1186/1746-4811-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/S0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 2013;9:e1003972. doi: 10.1371/journal.pgen.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner JT, Wiley MR, Mate S, Dudas G, Prieto K, Lovett S et al. (2015) Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell host & microbe 18:659–669 [DOI] [PMC free article] [PubMed]
- Lazaro JEH, Bascos NAD, Tablizo FA, Abes NS, Paynaganan RID, Miguel MA et al. (2019) Genome-wide Analysis for Variants in Philippine Trypanosoma evansi Isolates with Varying Drug Resistance Profiles. Philippine Journal of Science 148(S1):219–233
- Le Corre V, Roux F, Reboud X. DNA polymorphism at the FRIGIDA gene in Arabidopsis thaliana: extensive nonsynonymous variation is consistent with local selection for flowering time. Mol Biol Evol. 2002;19:1261–1271. doi: 10.1093/oxfordjournals.molbev.a004187. [DOI] [PubMed] [Google Scholar]
- Libert F, Cochaux P, Beckman G, Samson M, Aksenova M, Cao A, et al. The Δccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in northeastern Europe. Hum Mol Genet. 1998;7:399–406. doi: 10.1093/hmg/7.3.399. [DOI] [PubMed] [Google Scholar]
- Li Y, Colleoni C, Zhang J, Liang Q, Hu Y, Ruess H, et al. Genomic analyses yield markers for identifying agronomically important genes in potato. Mol Plant. 2018;11:473–484. doi: 10.1016/j.molp.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Liu J, Seetharam AS, Chougule K, Ou S, Swentowsky KW, Gent JI, et al. Gapless assembly of maize chromosomes using long-read technologies. Genome Biol. 2020;21:121. doi: 10.1186/s13059-020-02029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xiang X, Huang Y, Zhou Y, An D, Dong J, et al. Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nat Commun. 2020;11:17. doi: 10.1038/s41467-019-14023-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love-Gregory L, Sherva R, Schappe T, Qi J-S, McCrea J, Klein S, et al. Common CD36 SNPs reduce protein expression and may contribute to a protective atherogenic profile. Hum Mol Genet. 2011;20:193–201. doi: 10.1093/hmg/ddq449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Ackerman MS, Gout J-F, Long H, Sung W, Thomas WK, et al. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 2016;17:704–714. doi: 10.1038/nrg.2016.104. [DOI] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace ES, Tai S, Gilding EK, Li Y, Prentis PJ, Bian L, et al. Whole-genome sequencing reveals untapped genetic potential in Africa’s indigenous cereal crop sorghum. Nat Commun. 2013;4:2320. doi: 10.1038/ncomms3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McGowen MR, Tsagkogeorga G, Williamson J, Morin PA, Rossiter SJ (2020). Positive selection and inactivation in the vision and hearing genes of cetaceans. Mol Biol Evol 37:2069–2083 [DOI] [PubMed]
- McInerney JO, McNally A, O’Connell MJ. Why prokaryotes have pangenomes. Nat Microbiol. 2017;2:17040. doi: 10.1038/nmicrobiol.2017.40. [DOI] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–219. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer PW, Petrov DA. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol Evol. 2013;28:659–669. doi: 10.1016/j.tree.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael TP, Jupe F, Bemm F, Motley ST, Sandoval JP, Lanz C, et al. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat Commun. 2018;9:541. doi: 10.1038/s41467-018-03016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael TP, VanBuren R. Building near-complete plant genomes. Curr Opin Plant Biol. 2020;54:26–33. doi: 10.1016/j.pbi.2019.12.009. [DOI] [PubMed] [Google Scholar]
- Monroe JG, Arciniegas JP, Moreno JL, Sánchez F, Sierra S, Valdes S et al. (2020) The lowest hanging fruit: Beneficial gene knockouts in past, present, and future crop evolution. Curr Plant Biology 24:100185
- Monroe JG, McGovern C, Lasky JR, Grogan K, Beck J, McKay JK (2016) Adaptation to warmer climates by parallel functional evolution of CBF genes in Arabidopsis thaliana. Molecular ecology 25:3632–3644 [DOI] [PubMed]
- Monroe JG, Powell T, Price N, Mullen JL, Howard A, Evans K et al. (2018) Drought adaptation in Arabidopsis thaliana by extensive genetic loss-of-function. Elife 7 [DOI] [PMC free article] [PubMed]
- Monroe JG, Srikant T, Carbonell-Bejerano P, Exposito-Alonso M, Weng M-L, Rutter MT et al. (2020) Mutation bias shapes gene evolution in Arabidopsis thaliana. BioRxiv
- Moyers BT, Morrell PL, McKay JK. Genetic costs of domestication and improvement. J Hered. 2018;109:103–116. doi: 10.1093/jhered/esx069. [DOI] [PubMed] [Google Scholar]
- Mughal MR, DeGiorgio M. Localizing and classifying adaptive targets with trend filtered regression. Mol Biol Evol. 2019;36:252–270. doi: 10.1093/molbev/msy205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. The measurement of gene mutation rate in drosophila, its high variability, and its dependence upon temperature. Genetics. 1928;13:279–357. doi: 10.1093/genetics/13.4.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. Further studies on the nature and causes of gene mutations. Proc Int Congr Genet. 1932;6:213–255. [Google Scholar]
- Murray AW. Can gene-inactivating mutations lead to evolutionary novelty? Curr Biol. 2020;30:R465–R471. doi: 10.1016/j.cub.2020.03.072. [DOI] [PubMed] [Google Scholar]
- Nei M, Roychoudhury AK. Probability of fixation and mean fixation time of an overdominant mutation. Genetics. 1973;74:371–380. doi: 10.1093/genetics/74.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MV. When less is more: gene loss as an engine of evolutionary change. Am J Hum Genet. 1999;64:18–23. doi: 10.1086/302219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. The genetic theory of adaptation: a brief history. Nature Reviews Genetics. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- Pan W, Shen X. Adaptive tests for association analysis of rare variants. Genet Epidemiol. 2011;35:381–388. doi: 10.1002/gepi.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings PS, Hermisson J. Soft sweeps II—molecular population genetics of adaptation from recurrent mutation or migration. Mol Biol Evol. 2006;23:1076–1084. doi: 10.1093/molbev/msj117. [DOI] [PubMed] [Google Scholar]
- Pennings PS, Hermisson J. Soft sweeps III: the signature of positive selection from recurrent mutation. PLoS Genet. 2006;2:e186. doi: 10.1371/journal.pgen.0020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, et al. Genome evolution across 1011 Saccharomyces cerevisiae isolates. Nature. 2018;556:339–344. doi: 10.1038/s41586-018-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piot A, Prunier J, Isabel N, Klápště J, El-Kassaby YA, Villarreal Aguilar JC, et al. Genomic diversity evaluation of populus trichocarpa germplasm for rare variant genetic association studies. Front Genet. 2019;10:1384. doi: 10.3389/fgene.2019.01384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassais J, Kim J, Davis BW, Karyadi DM, Hogan AN, Harris AC, et al. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat Commun. 2019;10:1489. doi: 10.1038/s41467-019-09373-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph PL, Coop G. Convergent evolution during local adaptation to patchy landscapes. PLoS Genet. 2015;11:e1005630. doi: 10.1371/journal.pgen.1005630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu P, Esuma W, Kawuki R, Rabbi IY, Egesi C, Bredeson JV, et al. Cassava haplotype map highlights fixation of deleterious mutations during clonal propagation. Nat Genet. 2017;49:959–963. doi: 10.1038/ng.3845. [DOI] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Rayko M, Komissarov A (2020) Quality control of low-frequency variants in SARS-CoV-2 genomes. BioRxiv
- Remington DL. Alleles versus mutations: understanding the evolution of genetic architecture requires a molecular perspective on allelic origins. Evolution. 2015;69:3025–3038. doi: 10.1111/evo.12775. [DOI] [PubMed] [Google Scholar]
- Rhoades MM. The genetic control of mutability in maize. Cold Spring Harb Symp Quant Biol. 1941;9:138–144. doi: 10.1101/SQB.1941.009.01.017. [DOI] [Google Scholar]
- Rhoades MM. Effect of the Dt Gene on the Mutability of the a(1) Allele in Maize. Genetics. 1938;23:377–397. doi: 10.1093/genetics/23.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JK, Stukenbrock EH, Carpenter J, Liu Z, Cowger C, Faris JD, et al. Local adaptation drives the diversification of effectors in the fungal wheat pathogen Parastagonospora nodorum in the United States. PLoS Genet. 2019;15:e1008223. doi: 10.1371/journal.pgen.1008223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesselman AJ, Ingraham JB, Marks DS. Deep generative models of genetic variation capture the effects of mutations. Nat Methods. 2018;15:816–822. doi: 10.1038/s41592-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose L, Atwell S, Grant M, Holub EB. Parallel loss-of-function at the RPM1 bacterial resistance locus in Arabidopsis thaliana. Front Plant Sci. 2012;3:287. doi: 10.3389/fpls.2012.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Ashikari M, Ueguchi-Tanaka M, Itoh H, Nishimura A, Swapan D, et al. A mutant gibberellin-synthesis gene in rice. Nature. 2002;416:701–702. doi: 10.1038/416701a. [DOI] [PubMed] [Google Scholar]
- Schrider DR, Kern AD. S/HIC: robust identification of soft and hard sweeps using machine learning. PLoS Genet. 2016;12:e1005928. doi: 10.1371/journal.pgen.1005928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Sharma V, Hecker N, Roscito JG, Foerster L, Langer BE, Hiller M. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat Commun. 2018;9:1215. doi: 10.1038/s41467-018-03667-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J. Natural selection and the concept of a protein space. Nature. 1970;225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23:23–35 [PubMed]
- Sollars ESA, Harper AL, Kelly LJ, Sambles CM, Ramirez-Gonzalez RH, Swarbreck D, et al. Genome sequence and genetic diversity of European ash trees. Nature. 2017;541:212–216. doi: 10.1038/nature20786. [DOI] [PubMed] [Google Scholar]
- Song K, Nelson MR, Aponte J, Manas ES, Bacanu S-A, Yuan X, et al. Sequencing of Lp-PLA2-encoding PLA2G7 gene in 2000 Europeans reveals several rare loss-of-function mutations. Pharmacogenomics J. 2012;12:425–431. doi: 10.1038/tpj.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Li L-S, Arsenault PR, Tan Q, Bigham AW, Heaton-Johnson KJ, et al. Defective Tibetan PHD2 binding to p23 links high altitude adaptionto altered oxygen sensing. J Biol Chem. 2014;289:14656–14665. doi: 10.1074/jbc.M113.541227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Navalsky BE, Guan W, Ingersoll C, Wang T, Loro E, et al. Tibetan PHD2, an allele with loss-of-function properties. Proc Natl Acad Sci USA. 2020;117:12230–12238. doi: 10.1073/pnas.1920546117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmeyer W, Ellis MH, Chandler PM. Semidwarf (sd-1), ‘green revolution’ rice, contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci USA. 2002;99:9043–9048. doi: 10.1073/pnas.132266399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler LJ. Spontaneous mutation at the R Locus in Maize. I. the aleurone-color and plant-color effects. Genetics. 1946;31:377–394. doi: 10.1093/genetics/31.4.377. [DOI] [PubMed] [Google Scholar]
- Stadler LJ. Spontaneous mutation at the R Locus in Maize. II. Race differences in mutation rate. Am Nat. 1948;82:289–314. doi: 10.1086/281585. [DOI] [Google Scholar]
- Stern AJ, Wilton PR, Nielsen R. An approximate full-likelihood method for inferring selection and allele frequency trajectories from DNA sequence data. PLoS Genet. 2019;15:e1008384. doi: 10.1371/journal.pgen.1008384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat Genet. 2018;50:1289–1295. doi: 10.1038/s41588-018-0182-0. [DOI] [PubMed] [Google Scholar]
- Tang H, Thomas PD. Tools for predicting the functional impact of nonsynonymous genetic variation. Genetics. 2016;203:635–647. doi: 10.1534/genetics.116.190033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thudi M, Khan AW, Kumar V, Gaur PM, Katta K, Garg V, et al. Whole genome re-sequencing reveals genome-wide variations among parental lines of 16 mapping populations in chickpea (Cicer arietinum L.) BMC Plant Biol. 2016;16(Suppl 1):10. doi: 10.1186/s12870-015-0690-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamaneh D, Laroche J, Valliyodan B, O’Donoughue L, Cober E, Rajcan I, ... & Belzile F (2019) Soybean haplotype map (GmHapMap): A universal resource for soybean translational and functional genomics. BioRxiv, 534578 [DOI] [PMC free article] [PubMed]
- Updegraff BL, Zhou X, Guo Y, Padanad MS, Chen P-H, Yang C, et al. Transmembrane protease TMPRSS11B promotes lung cancer growth by enhancing lactate export and glycolytic metabolism. Cell Rep. 2018;25:2223–2233.e6. doi: 10.1016/j.celrep.2018.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velzen R, Holmer R, Bu F, Rutten L, van Zeijl A, Liu W, et al. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proc Natl Acad Sci USA. 2018;115:E4700–E4709.. doi: 10.1073/pnas.1721395115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature. 2018;557:43–49. doi: 10.1038/s41586-018-0063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Grus WE, Zhang J. Gene losses during human origins. PLoS Biol. 2006;4:e52. doi: 10.1371/journal.pbio.0040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-L, Shi W-P, Shi H-C, Lu S-C, Wang K, Sun C, et al. Knockdown of TRIM65 inhibits lung cancer cell proliferation, migration and invasion: A therapeutic target in human lung cancer. Oncotarget. 2016;7:81527–81540. doi: 10.18632/oncotarget.13131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wei W-S, Chen X, Guo L-Y, Li X-D, Deng M-H, Yuan G-J, et al. TRIM65 supports bladder urothelial carcinoma cell aggressiveness by promoting ANXA2 ubiquitination and degradation. Cancer Lett. 2018;435:10–22. doi: 10.1016/j.canlet.2018.07.036. [DOI] [PubMed] [Google Scholar]
- Weinreich DM, Rand DM. Contrasting patterns of nonneutral evolution in proteins encoded in nuclear and mitochondrial genomes. Genetics. 2000;156:385–399. doi: 10.1093/genetics/156.1.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will JL, Kim HS, Clarke J, Painter JC, Fay JC, Gasch AP. Incipient balancing selection through adaptive loss of aquaporins in natural Saccharomyces cerevisiae populations. PLoS Genet. 2010;6:e1000893. doi: 10.1371/journal.pgen.1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Petrov DA, Messer PW. Soft selective sweeps in complex demographic scenarios. Genetics. 2014;198:669–684. doi: 10.1534/genetics.114.165571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Koonin EV. Genome reduction as the dominant mode of evolution. Bioessays. 2013;35:829–837. doi: 10.1002/bies.201300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Rodrigue N, Kassen R. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet. 2012;8:e1002928. doi: 10.1371/journal.pgen.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Liang Z, Yan T, Xu Y, Xuan L, Tang J, et al. Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol Plant. 2019;12:30–43. doi: 10.1016/j.molp.2018.11.007. [DOI] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wang L, Fu J, Chen J, Wei S, Zhang S, et al. Resequencing of 683 common bean genotypes identifies yield component trait associations across a north–south cline. Nat Genet. 2020;52:118–125. doi: 10.1038/s41588-019-0546-0. [DOI] [PubMed] [Google Scholar]
- Xanthopoulou A, Montero-Pau J, Mellidou I, Kissoudis C, Blanca J, Picó B, et al. Whole-genome resequencing of Cucurbita pepo morphotypes to discover genomic variants associated with morphology and horticulturally valuable traits. Hortic Res. 2019;6:94. doi: 10.1038/s41438-019-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Song B, Née G, Kramer K, Finkemeier I, Soppe WJJ. Sequence polymorphisms at the REDUCED DORMANCY5 Pseudophosphatase Underlie Natural Variation in Arabidopsis Dormancy. Plant Physiol. 2016;171:2659–2670. doi: 10.1104/pp.16.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Ptacek T, Osborne JD, Crabb DM, Simmons WL, Lefkowitz EJ, et al. Comparative genome analysis of Mycoplasma pneumoniae. BMC Genomics. 2015;16:610. doi: 10.1186/s12864-015-1801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Raveendran M, Harris RA, Fawcett GL, Liu X, White S, et al. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 2016;26:1651–1662. doi: 10.1101/gr.204255.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y-C, Niu X-M, Li X-X, He W, Chen J-F, Zou Y-P, et al. Adaptation and phenotypic diversification in arabidopsis through loss-of-function mutations in protein-coding genes. Plant Cell. 2019;31:1012–1025. doi: 10.1105/tpc.18.00791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Liu J, Gao Q, Gui S, Chen L, Yang L, et al. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet. 2019;51:1052–1059. doi: 10.1038/s41588-019-0427-6. [DOI] [PubMed] [Google Scholar]
- Ye C-Y, Tang W, Wu D, Jia L, Qiu J, Chen M, et al. Genomic evidence of human selection on Vavilovian mimicry. Nat Ecol Evol. 2019;3:1474–1482. doi: 10.1038/s41559-019-0976-1. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhao L, Li B, He Z, Wang GT, Liu DJ, et al. SEQSpark: A Complete Analysis Tool for Large-Scale Rare Variant Association Studies Using Whole-Genome and Exome Sequence Data. Am J Hum Genet. 2017;101:115–122. doi: 10.1016/j.ajhg.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Qu K, Chen N, Hanif Q, Jia Y, Huang Y et al. (2019) Genome-Wide SNPs and InDels characteristics of three chinese cattle breeds. Animals (Basel) 9:596 [DOI] [PMC free article] [PubMed]
- Zhang L, Jiménez-Gómez JM (2020) Functional analysis of FRIGIDA using naturally occurring variation in Arabidopsis thaliana. Plant J 103:154–165 [DOI] [PubMed]
- Zhao G, Lian Q, Zhang Z, Fu Q, He Y, Ma S, et al. A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat Genet. 2019;51:1607–1615. doi: 10.1038/s41588-019-0522-8. [DOI] [PubMed] [Google Scholar]
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Chebotarov D, Kudrna D, Llaca V, Lee S, Rajasekar S, et al. A platinum standard pan-genome resource that represents the population structure of Asian rice. Sci Data. 2020;7:113. doi: 10.1038/s41597-020-0438-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer EA, Martin SL, Beverley SM, Kan YW, Wilson AC. Rapid duplication and loss of genes coding for the alpha chains of hemoglobin. Proc Natl Acad Sci USA. 1980;77:2158–2162. doi: 10.1073/pnas.77.4.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA. 2014;111:E455–E464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]