

Abstract
Coronary artery no-reflow is a complex problem in the area of reperfusion therapy, and the molecular mechanisms underlying coronary artery no-reflow injury have not been fully elucidated. In the present study, we explored whether oxidative stress caused damage to coronary endothelial cells by inducing mitochondrial fission and activating the JNK pathway. The hypoxia/reoxygenation (H/R) model was induced in vitro to mimic coronary endothelial no-reflow injury, and mitochondrial fission, mitochondrial function, and endothelial cell viability were analyzed using western blotting, quantitative polymerase chain reaction (qPCR), enzyme-linked immunosorbent assay (ELISA), and immunofluorescence. Our data indicated that reactive oxygen species (ROS) were significantly induced upon H/R injury, and this was followed by decreased endothelial cell viability. Mitochondrial fission was induced and mitochondrial bioenergetics were impaired in cardiac endothelial cells after H/R injury. Neutralization of ROS reduced mitochondrial fission and protected mitochondrial function against H/R injury. Our results also demonstrated that ROS stimulated mitochondrial fission via JNK-mediated Drp1 phosphorylation. These findings indicate that the ROS-JNK-Drp1 signaling pathway may be one of the molecular mechanisms underlying endothelial cell damage during H/R injury. Novel treatments for coronary no-reflow injury may involve targeting mitochondrial fission and the JNK-Drp1 signaling pathway.
1. Introduction
Primary percutaneous coronary intervention (PPCI) is the gold standard for the treatment of ST segment elevation myocardial infarction (STEMI) because it results in the timely opening of the occluded coronary arteries [1]. Fresh blood flow is restored in >90% of STEMI patients after PPCI. However, there is still a small number of patients who develop severe microvascular obstruction (MVO) even though epicardial arteries are successfully opened following PPCI. This phenomenon is called coronary no-reflow injury [2]. The early clinical manifestation of patients with coronary no-reflow injury includes recurring angina pectoris, malignant arrhythmias (e.g., ventricular tachycardia, ventricular fibrillation, and atrioventricular block), acute left heart failure, cardiogenic shock, and cardiac rupture or other malignant complications [3]. Although the incidence of coronary no-reflow varies between studies because of differences in evaluation methods and types of no-reflow injuries used [4], it is estimated that the incidence of no-reflow injury is ~26% in patients with anterior wall STEMI [5].
It is believed that the initial cause of coronary no-reflow injury is related to microvascular dysfunction [6]. The myocardial blood supply is composed of the subepicardial coronary artery and the capillaries that penetrate the myocardium [7], and microvessels form the main resistance to myocardial perfusion blood flow [8]. Further, coronary blood flow is inversely correlated with microvascular resistance. Several mechanisms, including endothelial cell death, inflammation response, oxidative stress, and coronary spasm, have been introduced to explain microvascular damage after PPCI [9–12]. It was reported that the microvessel wall was thickened, the lumen became smaller, and the endothelial cells were swelling in patients undergoing myocardial biopsy [13]. It has also been reported that endothelial cell damage is primarily caused by oxidative stress; however, the detailed molecular mechanisms underlying this damage have not been fully elucidated [14].
Although previous studies have reported that excessive oxidative stress is associated with mitochondrial dysfunction [15] (as evidenced by decreased mitochondrial membrane potential and increased mitochondrial proapoptotic factor release), the mechanisms underlying ROS-induced mitochondrial damage in endothelial cells following coronary no-reflow injury remain unknown. Recent investigations have demonstrated that mitochondrial dynamics disorder may be an upstream regulator of mitochondrial function [16–18]. Excessive mitochondrial fission divides mitochondria into several fragmentations that contain damaged mitochondrial DNA [19]. Since these fragmented mitochondria cannot synthesize the normal mitochondrial respiration complex, the physiological mitochondrial membrane potential cannot be generated [20]. Additionally, a decreased mitochondrial membrane potential is associated with increased mitochondrial membrane hyperpermeability, which results in the leakage of proapoptotic proteins into the cytoplasm [21]. Interestingly, the relationship between ROS and mitochondrial dynamics disorders has not been fully elucidated [22]. A recent study illustrated that mitochondrial fission was mainly regulated by Drp1, which was activated through posttranscriptional phosphorylation following activation of the JNK pathway [23]. In the present study, we investigated whether ROS mediated endothelial cell damage by inducing mitochondrial fission via the JNK pathway. Specifically, the purpose of this study was to investigate the role of ROS-modified mitochondrial fission through the JNK-Drp1 pathway in cultured endothelial cells using a hypoxia/reoxygenation (H/R) injury model.
2. Materials and Methods
2.1. Cardiac Endothelial Cells (CECs)
Murine CECs were purchased from CELLutions Biosystems Inc. (Ontario, Canada) and cultured according to the supplier's instructions [24]. CECs were maintained in Dulbecco's Modified Eagle's Medium (glucose [4500 mg/L], L-glutamine [0.584 g/L], sodium bicarbonate [3.7 g/L], without sodium pyruvate [Sigma-Aldrich]) that was supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL; both from GIBCO), 10 mmol/L HEPES (HyClone, Thermo Fisher Scientific), and 5% fetal bovine serum (FBS; Sigma-Aldrich). When confluent (2-3 days after plating), the CECs were harvested using a 0.25% trypsin/EDTA solution (GIBCO), subcultured, or used for functional assays and RNA purification [25].
2.2. ELISA
The contents of SOD, GSH, GPX, and MDA in the culture media were detected using ELISA kits following the manufacturer's instructions (Shanghai Hengyuan Biological Technology, China) [26]. The culture media was added to a microdroplet plate that was coated with purified SOD, GSH, GPX, and MDA primary antibodies and incubated at 37°C for 2 hours [27]. After incubation, the plate was washed and HRP-labeled secondary antibodies were added. After incubation with the secondary antibodies, the plate was washed thoroughly, and a solution of 3,3′,5,5′-tetramethylbenzidine substrate was added. A sulfuric acid solution was added to stop the reaction, and the color was analyzed using spectrophotometry at a wavelength of 450 nm [28]. The concentrations of SOD, GPX, GSH, and MDA in the culture media were then calculated according to the standard curve [29].
2.3. Immunofluorescence Analysis
The protocol for the immunofluorescent staining of the heart tissues was performed as described below. Samples were fixed using 4% paraformaldehyde and mounted on microscope slides [30]. The slides were subsequently treated with primary antibodies, and the relative immunofluorescence intensity was calculated as previously described [31].
2.4. Oxygen-Glucose Deprivation (OGD) Followed by Reoxygenation
Cells were incubated in 6-well plates (4 × 105 cell/well) for 24 h, cultured in glucose-free DMEM, and incubated in an anaerobic chamber containing 95% N2 and 5% CO2 at 37°C for 3 h to mimic ischemic-like conditions in vitro [32]. After incubation, the CECs were returned to the atmosphere with 95% air and 5% CO2 and cultured in glucose-containing DMEM for 3 h for reoxygenation. The subsequent experiment was performed 24 h after reoxygenation [33].
2.5. RT-qPCR
Total RNA was extracted using TRIzol reagent kits (Invitrogen, CA, USA). After extraction, 1 μg of total RNA was reverse transcribed to cDNA using the PrimeScript RT reagent kits (Takara) [34]. RT-qPCR was performed using a SYBR Premix Ex Taq II (TaKaRa) and the ABI 7500 real-time PCR system (Applied Biosystems). Relative RNA expression was normalized to that of GAPDH and was determined using the 2−ΔΔCt method [35].
2.6. CCK-8 Assay
Transfected cells were seeded into 96-well plates and cultured for 24 h. Next, 10 μL of the CCK-8 solution was added to each well, and the cells were incubated for 2 h. After incubation, absorbance was measured at 450 nm using a microplate reader (Bio-Rad, USA) [36].
2.7. Western Blot
Western blot was conducted as previously reported. The primary antibodies were purchased from Abcam (Cambridge, MA, USA), and HRP-conjugated secondary antibodies were obtained from Sangon (Shanghai, China) [37].
2.8. Mitochondrial Respiratory Chain Complex Activity Analysis
The activity of the mitochondrial respiratory chain was determined using the Mitochondrial Respiratory Chain Complex Activity Assay Kit (Solarbio, Beijing, China) according to the manufacturer's instructions [38]. Briefly, the mitochondrial complex was extracted from the ovaries using the suitable reagents, and 10 μL of the extract was added to each well of a 96-well plate. Then, the detection reagents were added to the wells, mixed gently, and incubated at 37°C for 2 min. The corresponding absorbance was evaluated before and after the reaction using a microplate reader (BioTek, Vermont, VT), and the difference in absorbance was calculated [39]. Finally, the enzymatic activity of the complex was calculated using the corresponding formula that was provided in the kit manual [40].
2.9. Measurement of ATP Levels
ATP production was detected using the luminometric ATP Assay Kit (AAT Bioquest, Sunnyvale, CA) according to the manufacturer's instructions [41]. Briefly, 50 oocytes were seeded into each well of a 96-well white plate, and 200 μL of the ATP assay solution was added to the oocyte cultures. After gently mixing and incubating for 20 min at room temperature [42], the luminescence intensity was measured using the luminometer mode on a plate reader (Tecan, Zurich, Switzerland). The readings were normalized to the total protein content [43].
2.10. Measurement of ROS Production
Cells were cultured in clear bottom 6-well black plates (Corning, Corning, NY, USA), incubated in the presence or absence of compounds for 48 h at 37°C, washed with PBS (pH 7.4), and incubated with 2′,7′-dichlorofluorescin diacetate (H2DCFDA) for 18 h [44]. Following incubation, the cells were washed twice with PBS (pH 7.4), and the fluorescence of 2′,7′-dichlorofluorescin (DCF) was then measured at excitation/emission (Ex/Em) wavelengths of 485/530 nm [45].
2.11. Statistical Analysis
Data shown are mean ± SEM of the number of independent experiments indicated (n) and represent experiments performed on at least three separate occasions with similar outcomes. Data were analyzed using one-way or two-way ANOVAs, and comparisons between groups were performed using a protected Tukey's test. Statistical significance was defined as p < 0.05.
3. Results
3.1. H/R Injury Mediates Endothelial Cell Oxidative Stress
In the present study, an H/R model was used in cardiac endothelial cells (CECs) to mimic coronary microvascular no-reflow injury in vitro. Cell viability was measured using the Cell Counting Kit-8 (CCK-8) assay. As shown in Figure 1(a), cell viability was significantly suppressed in H/R-treated CECs as compared with control CECs. Recent studies have reported that oxidative stress is the primary pathological factor that induces endothelial cell damage during coronary no-reflow injury. To confirm this finding, an enzyme-linked immunosorbent assay (ELISA) was used to analyze alterations in the activities of antioxidants in CECs following H/R injury. As shown in Figures 1(b)–1(d), the activities of glutathione (GSH), superoxide dismutase (SOD), and glutathione peroxidase (GPX) were significantly increased in the H/R-treated cells as compared with the control group. This suggests that there was a decline in the antioxidative capacity of CECs following H/R injury. Additionally, the levels of malondialdehyde (MDA) were increased, and this may have been due to increased ROS production in the cytoplasm (Figure 1(e)). To further observe the oxidative stress in CECs, an ROS probe was used to stain the intracellular ROS. As shown in Figures 1(f) and 1(g), the levels of cytoplasmic ROS and mitochondrial ROS were significantly elevated in the H/R-treated CECs as compared with the control group. These results indicate that H/R is followed by oxidative stress in CECs under H/R injury.
Figure 1.
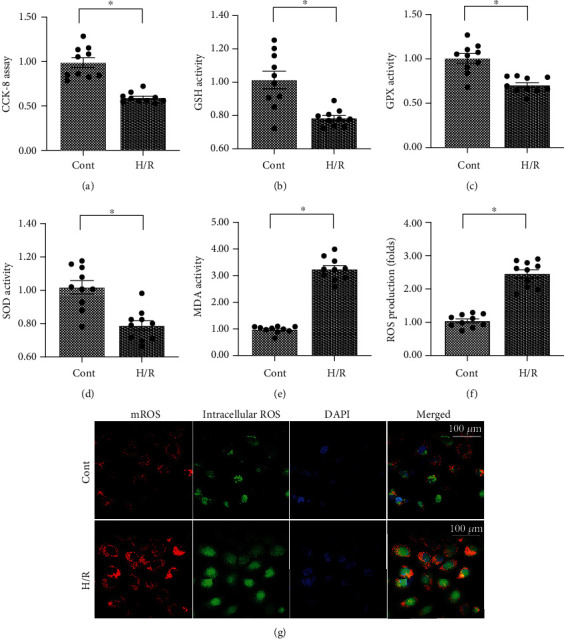
Hypoxia/reoxygenation injury mediates endothelial cell oxidative stress. (a) Cell viability was measured using the CCK-8 assay. Cardiac endothelial cells (CECs) underwent hypoxia/reoxygenation (H/R) injury. (b–e) The content of antioxidative factors was measured using an enzyme-linked immunosorbent assay (ELISA). (f, g) Intracellular ROS and mitochondrial ROS (mROS) were detected using DCFHDA and MitoSOX red mitochondrial superoxide indicator, respectively. ∗p < 0.05.
3.2. Mitochondrial ROS Promotes Drp1 Phosphorylation and Mitochondrial Fission in Endothelial Cells following H/R Injury
Previous studies have identified that mitochondrial ROS induces endothelial cell dysfunction through the disruption of mitochondrial homeostasis. Further investigations demonstrated that mitochondrial performance (especially mitochondrial fission) in endothelial cells is drastically regulated by mitochondrial dynamics. Therefore, we investigated whether mitochondrial ROS affected mitochondrial dysfunction in endothelial cells under H/R injury via activating mitochondrial fission. Mitochondrial morphology was observed through immunofluorescence. The data shown in Figures 2(a) and 2(b) demonstrated that the morphology of mitochondria was converted into a fragmented shape after exposure to H/R injury. To determine if mitochondrial ROS play a causal role in inducing mitochondrial fission, Mito-Q, a mitochondria-specific antioxidant, was incubated with CECs before H/R injury, and mitochondrial morphology was observed again after H/R injury. As shown in Figures 2(a) and 2(b), the elongated mitochondrial morphology was sustained in CECs that underwent Mito-Q treatment before H/R injury as compared with CECs that underwent H/R injury but were not treated with Mito-Q. To further support the role of mitochondrial ROS-induced mitochondrial fission, RNA transcriptions of mitochondrial fission-related factors were conducted. The transcriptions of Drp1, Mff, and Fis1 were significantly elevated in CECs that underwent H/R injury as compared with those in control CECs (Figures 2(c)–2(e)). Conversely, treatment with Mito-Q prevented the increase of these profission factors (Figures 2(c)–2(e)), suggesting that mitochondrial ROS plays a contributory role in initiating mitochondrial fission in endothelial cells.
Figure 2.
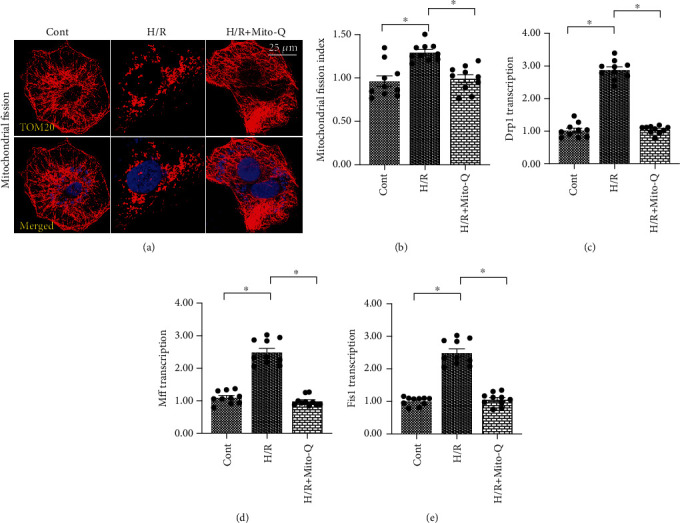
Mitochondrial ROS promotes Drp1 phosphorylation and mitochondrial fission in endothelial cells under hypoxia/reoxygenation injury. (a, b) Mitochondrial fission was measured using immunofluorescence. Cardiac endothelial cells (CECs) underwent hypoxia/reoxygenation (H/R) injury. Mito-Q, a mitochondrial antioxidant, was added to the CEC media to neutralize mROS. (c–e) qPCR was performed to analyze the transcription of Drp1, Mff, and Fis1 in CECs under H/R injury. ∗p < 0.05.
3.3. Mitochondrial Dysfunction Is Induced by Mitochondrial ROS
Although we reported that mitochondrial fission could be activated by mitochondrial ROS in endothelial cells under H/R injury, it remains unclear whether mitochondrial function is disturbed by mitochondrial ROS in H/R-treated CECs. To address this question, mitochondrial function was assessed in response to Mito-Q treatment. As shown in Figure 3(a), cellular adenosine triphosphate (ATP) production, which is primarily generated by mitochondria, was downregulated in H/R-treated CECs as compared with control CECs. Additionally, ATP production was reversed to near-normal levels with Mito-Q administration. We also found that the activities of the mitochondrial respiration complex were reduced in H/R-treated CECs (Figures 3(b)–3(d)), and this alteration was improved by Mito-Q treatment. This finding suggests that mitochondrial ROS regulate mitochondria-mediated bioenergetics.
Figure 3.
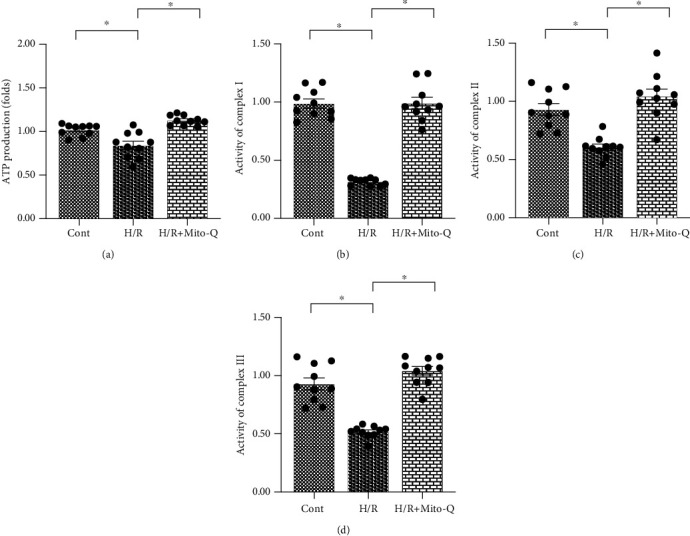
Mitochondrial dysfunction is induced by mitochondrial ROS. (a) Adenosine triphosphate (ATP) production was measured using an enzyme-linked immunosorbent assay (ELISA). Cardiac endothelial cells (CECs) underwent hypoxia/reoxygenation (H/R) injury. Mito-Q, a mitochondrial antioxidant, was added to the CEC media to neutralize mROS. (b–d) The activity of the mitochondrial respiration complex was determined using an ELISA. ∗p < 0.05.
3.4. Inhibition of Mitochondrial Fission Also Sustains Mitochondrial Function in Endothelial Cells
To understand whether mitochondrial fission is also involved in the regulation of mitochondrial function in endothelial cells under H/R injury, Mdivi-1, an inhibitor of mitochondrial fission, was added to the endothelial cell culture media, and mitochondrial function was evaluated again [46]. As shown in Figure 4(a), cellular ATP production was significantly reduced in H/R-treated endothelial cells as compared with control endothelial cells. Interestingly, administration of Mdivi-1 drastically improved ATP content in endothelial cells, and this was similar to the results obtained from the Mito-Q-treated endothelial cells. Our data also demonstrated that the activities of the mitochondrial respiration complex were inhibited in H/R-treated CECs (Figures 4(b)–4(d)). Interestingly, Mdivi-1 supplementation enhanced the activities of the mitochondrial respiration complex (Figures 4(b)–4(d)). These results suggest that mitochondria-related bioenergetics are normalized by mitochondrial fission inhibition.
Figure 4.
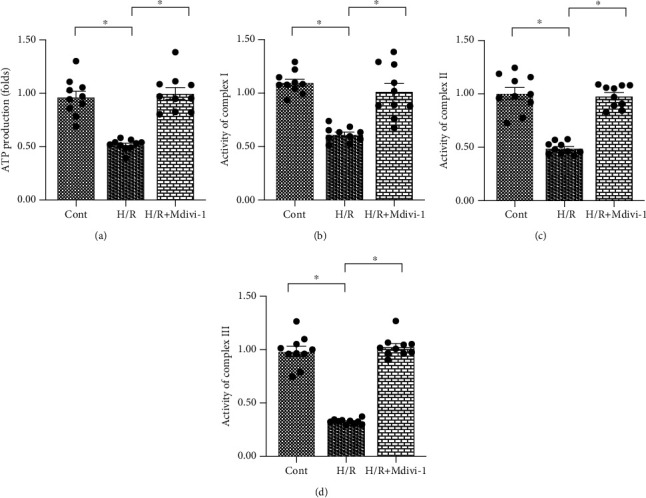
Inhibition of mitochondrial fission also sustains mitochondrial function in endothelial cells. (a) Adenosine triphosphate (ATP) production was measured using an enzyme-linked immunosorbent assay (ELISA). Cardiac endothelial cells (CECs) underwent hypoxia/reoxygenation (H/R) injury. Mdivi-1, a mitochondrial fission inhibitor, was added to the CEC culture media to repress mitochondrial fission. (b–d) The activity of the mitochondrial respiration complex was determined using an ELISA. ∗p < 0.05.
3.5. ROS Causes Drp1-Related Mitochondrial Fission through the JNK Pathway
Lastly, we evaluated the molecular mechanism underlying ROS-induced mitochondrial fission. Previous studies have demonstrated that mitochondrial fission is mainly regulated by Drp1 phosphorylation, and Drp1 posttranscriptional phosphorylation can be modified by the JNK pathway [47]. Thus, we investigated if ROS-mediated mitochondrial fission could be achieved through JNK-induced Drp1 phosphorylation. Results from western blotting demonstrated that the JNK pathway was significantly activated by H/R injury in CECs as evidenced by the presence of phosphorylated JNK (Figures 5(a)–5(c)). We also observed an increase in Drp1 phosphorylation after H/R injury. Interestingly, supplementation of Mito-Q repressed JNK phosphorylation and inhibited Drp1 phosphorylation (Figures 5(a)–5(c)), suggesting that mitochondrial ROS promote JNK and Drp1 phosphorylation (p-JNK and p-Drp1) in CECs. This finding was also supported by our immunofluorescent findings. As shown in Figures 5(d)–5(f), the expressions of p-Drp1 and p-JNK were significantly increased in H/R-treated cells as compared with control cells. Interestingly, Mito-Q treatment repressed the increase in p-Drp1 and p-JNK following H/R injury (Figures 5(d)–5(f)). These findings suggest that mitochondrial fission in CECs is controlled by the ROS-Drp1-JNK signaling pathway.
Figure 5.
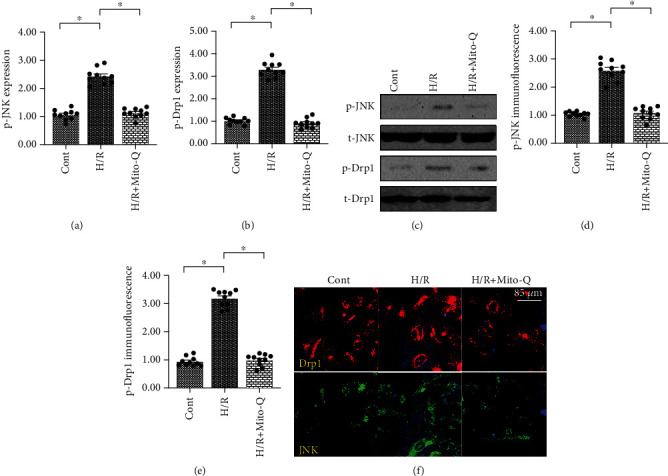
ROS causes Drp1-related mitochondrial fission through the JNK pathway. (a–c) Proteins were isolated from hypoxia/reoxygenation- (H/R-) treated CECs. Western blotting was performed to analyze the expression of JNK and Drp1. Mito-Q, a mitochondrial antioxidant, was added to the CEC culture media to neutralize mROS. (d–f) Immunofluorescence was performed to verify the expressions of JNK and Drp1. Mito-Q, a mitochondrial antioxidant, was added to the CEC culture media to neutralize mROS. ∗p < 0.05.
4. Discussion
Coronary artery no-reflow is a complex problem in the area of reperfusion therapy [48]. Although there has been an advancement in drugs and interventional therapies in recent years, breakthrough progress has not been achieved in the prevention and treatment of coronary no-reflow injury following reperfusion therapy [2, 49]. Since infarction size, cardiac remodeling, myocardial function, and all-cause death are also determined by coronary no-reflow injury; understanding the molecular mechanisms underlying coronary no-reflow will promote specific diagnoses and therapies in clinical practice [11, 50]. In the present study, we used an H/R injury model to mimic coronary artery no-reflow injury in vitro. Our findings demonstrated that CEC damage was associated with mitochondrial dysfunction that was caused by increased mitochondrial fission. Further, we reported that oxidative stress was the primary inducer of mitochondrial fission via the activation of the JNK-Drp1 signaling pathway. ROS promoted JNK phosphorylation, which augmented Drp1 phosphorylation and resulted in mitochondrial fission in CECs. Excessive mitochondrial fission impaired mitochondrial bioenergetics, as evidenced by decreased ATP production and blunted mitochondrial respiration complex, which ultimately contributed to endothelial cell death. Overall, these findings demonstrate the molecular basis underlying ROS-induced endothelial cell damage and also identify the ROS-JNK-Drp1 pathway as the potential therapeutic target for the treatment of coronary no-reflow.
At the stage of myocardial ischemia, mitochondrial respiration is significantly impaired and glycolysis is augmented because of insufficient oxygen supply and interrupted blood flow, resulting in the accumulation of lactic acid [51, 52]. Since carbon dioxide in the blood stream cannot be quickly removed, the pH in endothelial cells significantly decreases [53]. As the pH decreases, massive hydrogen ions within endothelial cells promote the opening of Na+/H+ channels, which in turn promotes sodium translocation into the cytoplasm [54]. Subsequently, abnormal sodium stimulates sodium-calcium exchange channels that trigger intracellular calcium overload, which leads to coronary spasm and blunted coronary artery relaxation [55]. These alterations promote slow blood flow or terminal blood flow under hypoxic conditions. Although the recovery of blood flow will bring sufficient oxygen to the ischemic zone during the reperfusion stage [56], the damage to mitochondrial respiration that is induced by ischemic stress cannot be quickly restored in a short time, and this results in excessive ROS production in coronary endothelial cells [57]. In addition to oxidative stress, the inflammatory response is also initiated at the reperfusion stage. An uncontrolled inflammation response will aggravate myocardial edema [58] and ultimately compress the coronary artery and limit the diastolic function of the coronary artery. More importantly, inflammatory responses will also activate platelets to induce the formation of coronary thrombi [59, 60], leading to microcirculation embolism in the reperfusion phase. These alterations promote the development of coronary no-reflow injury even though the occluded epicardial arteries have been opened.
In the present study, we found that ROS are induced by H/R injury in CECs and correlate with the survival of CECs by mediating mitochondrial bioenergetics. Similarly, it was previously reported that ischemia/reperfusion-mediated senescence and vascular dysfunction of endothelial cells were attenuated by oxidants [61]. Endothelial cell necroptosis is also induced by ROS-mediated opening of the mitochondrial permeability transition pore (mPTP) [62]. Endothelial nitric oxide (NO) is a key regulator of vascular tone, and increased NO is associated with coronary relaxation. However, excessive oxidative stress impairs the bioactivity of NO and ultimately represses NO-mediated vascular relaxation [63]. Mitochondrial fragmentation is induced by superoxide anion production in coronary endothelial cells in diabetic mice [64], suggesting a possible relationship between mitochondrial fission and oxidative stress. In a mouse model of cardiac ischemia/reperfusion injury, mitochondrial fission in coronary endothelial cells was regulated by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (Nox-2) signaling pathway and ROS production [65]. This finding confirms that oxidative stress plays a causal role in mediating mitochondrial fission in endothelial cells. Our data further identified that the JNK-Drp1 signaling pathway was the primary mechanism responsible for ROS-modified mitochondrial fission in coronary endothelial cells. This finding will help us to better understand the relationship between oxidative stress and mitochondrial dynamics in coronary endothelial cells during coronary no-reflow injury.
Mitochondrial dynamics are mitochondrial morphological mechanisms that include mitochondrial fission and fusion. Mitochondrial fission is activated by hypoxia and/or reoxygenation, and multiple studies have confirmed that mitochondrial fission plays a pathological role in exacerbating myocardial damage during reperfusion therapy. For example, it was demonstrated that cardiac function was improved by the inhibition of mitochondrial fission at the reperfusion stage [66]. Another study revealed that reperfusion-mediated energetic crises were attenuated by mitochondrial fission suppression in a mouse model of cardiac ischemia/reperfusion injury [67]. It was also reported that intimal thickening after coronary damage was enhanced by mitochondrial fission due to dysregulated macrophage function [68, 69]. Further, it was demonstrated that inflammation and apoptosis of coronary endothelial cells were reduced via inhibition of mitochondrial fission or activation of mitophagy [70]. Decreased mitochondrial fission significantly promotes the production of NO in coronary endothelial cells, and this effect is followed by increased endothelium-dependent vasodilation [71]. Similarly, our data revealed that mitochondrial fission caused endothelial cell dysfunction as evidenced by decreased cell viability, impaired mitochondrial bioenergetics, and reduced ATP production. Based on these findings, mitochondrial fission may be a potential therapeutic target that sustains endothelial cell function and viability.
Overall, our results illustrate that coronary no-reflow injury is associated with endothelial cell damage that is caused by excessive oxidative stress. The ROS-JNK-Drp1 signaling pathway is activated at the stage of cardiac ischemia/reperfusion injury and contributes to mitochondrial fission, resulting in abnormal mitochondrial function and decreased endothelial cell viability. However, there are several limitations in the present study. First, our results are primarily based on cellular experiments, and animal studies are necessary to support our findings. Second, we used chemical inhibitors to determine the role of the ROS-JNK-Drp1 signaling pathway in regulating endothelial cell viability during coronary no-reflow. Additional studies using genetic ablation mice are required to confirm our findings.
Data Availability
The analyzed datasets that were generated during the study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Y.C. contributed to the study concepts, study design, data acquisition, and manuscript preparation; Y.C. and C.L. contributed to the study concepts, study design, literature research, data acquisition, manuscript preparation, and editing; P.Z., J.L., X.Z., and Y.W. were the guarantors of integrity of the entire study and contributed to the definition of intellectual content, statistical analysis, and manuscript review; R.C. was the guarantor of integrity of the entire study and contributed to the data analysis and manuscript review; L.S. and H.Z. contributed to the clinical studies and data acquisition; and L.C. and H.Y. contributed to the experimental studies and data acquisition. The final version of the manuscript has been read and approved by all authors.
References
- 1.Reyes-Retana J. A., Duque-Ossa L. C. Acute myocardial infarction biosensor: a review from bottom up. Current Problems in Cardiology. 2020;(article 100739) doi: 10.1016/j.cpcardiol.2020.100739. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz B. G., Kloner R. A. Coronary no reflow. Journal of Molecular and Cellular Cardiology. 2012;52(4):873–882. doi: 10.1016/j.yjmcc.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Eeckhout E., Kern M. J. The coronary no-reflow phenomenon: a review of mechanisms and therapies. European Heart Journal. 2001;22(9):729–739. doi: 10.1053/euhj.2000.2172. [DOI] [PubMed] [Google Scholar]
- 4.Rezkalla S. H., Kloner R. A. Coronary no-reflow phenomenon: from the experimental laboratory to the cardiac catheterization laboratory. Catheterization and Cardiovascular Interventions. 2008;72(7):950–957. doi: 10.1002/ccd.21715. [DOI] [PubMed] [Google Scholar]
- 5.Scarpone M., Cenko E., Manfrini O. Coronary no-reflow phenomenon in clinical practice. Current Pharmaceutical Design. 2018;24(25):2927–2933. doi: 10.2174/1381612824666180702112536. [DOI] [PubMed] [Google Scholar]
- 6.Zhou H., Toan S. Pathological roles of mitochondrial oxidative stress and mitochondrial dynamics in cardiac microvascular ischemia/reperfusion injury. Biomolecules. 2020;10(1):p. 85. doi: 10.3390/biom10010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsunari I. Microvascular function measurement in mice: From large to small. Journal of Nuclear Cardiology. 2020 doi: 10.1007/s12350-020-02097-1. [DOI] [PubMed] [Google Scholar]
- 8.Altara R., Booz G. W. Editorial: cardiac microvascular endothelium contribution to cardiac myocyte growth, structure, and contractile function. Frontiers in Cardiovascular Medicine. 2019;6:p. 130. doi: 10.3389/fcvm.2019.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong J., Ouyang H., Sun M., et al. Tanshinone IIA attenuates cardiac microvascular ischemia-reperfusion injury via regulating the SIRT1-PGC1α-mitochondrial apoptosis pathway. Cell Stress & Chaperones. 2019;24(5):991–1003. doi: 10.1007/s12192-019-01027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heusch G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Research in Cardiology. 2019;114(6):p. ???. doi: 10.1007/s00395-019-0756-8. [DOI] [PubMed] [Google Scholar]
- 11.Cuijpers I., Simmonds S. J., van Bilsen M., et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Research in Cardiology. 2020;115(4):p. 39. doi: 10.1007/s00395-020-0798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao X. M., Su Y., Moore S., et al. Relaxin mitigates microvascular damage and inflammation following cardiac ischemia-reperfusion. Basic Research in Cardiology. 2019;114(4):p. 30. doi: 10.1007/s00395-019-0739-9. [DOI] [PubMed] [Google Scholar]
- 13.Kumar J., O’Connor C. T., Kumar R., Arnous S. K., Kiernan T. J. Coronary no-reflow in the modern era: a review of advances in diagnostic techniques and contemporary management. Expert Review of Cardiovascular Therapy. 2019;17(8):605–623. doi: 10.1080/14779072.2019.1653187. [DOI] [PubMed] [Google Scholar]
- 14.Zhu H., Toan S., Mui D., Zhou H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiologica. 2020;(article e13590) doi: 10.1111/apha.13590. [DOI] [PubMed] [Google Scholar]
- 15.Fan H., He Z., Huang H., et al. Mitochondrial quality control in cardiomyocytes: a critical role in the progression of cardiovascular diseases. Frontiers in Physiology. 2020;11:p. 252. doi: 10.3389/fphys.2020.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maneechote C., Palee S., Kerdphoo S., Jaiwongkam T., Chattipakorn S. C., Chattipakorn N. Pharmacological inhibition of mitochondrial fission attenuates cardiac ischemia-reperfusion injury in pre-diabetic rats. Biochemical Pharmacology. 2020;182:p. 114295. doi: 10.1016/j.bcp.2020.114295. [DOI] [PubMed] [Google Scholar]
- 17.Zhou H., Toan S., Zhu P., Wang J., Ren J., Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Research in Cardiology. 2020;115(2):p. 11. doi: 10.1007/s00395-019-0773-7. [DOI] [PubMed] [Google Scholar]
- 18.Wang J., Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharmaceutica Sinica B. 2020;10(10):1866–1879. doi: 10.1016/j.apsb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng J., Lu C. Oxidized LDL causes endothelial apoptosis by inhibiting mitochondrial fusion and mitochondria autophagy. Frontiers in Cell and Development Biology. 2020;8:p. 600950. doi: 10.3389/fcell.2020.600950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Huang M., Wei R., Wang Y., Su T., Li P., Chen X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biology. 2018;16:303–313. doi: 10.1016/j.redox.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Q., Zhang M., Torres G., et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2016;66(1):193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallert M., Ziegler M., Wang X., et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biology. 2019;26, article 101292 doi: 10.1016/j.redox.2019.101292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin R., Lin D., Zhang L., Xiao F., Guo L. Mst1 deletion reduces hyperglycemia-mediated vascular dysfunction via attenuating mitochondrial fission and modulating the JNK signaling pathway. Journal of Cellular Physiology. 2019;235(1):294–303. doi: 10.1002/jcp.28969. [DOI] [PubMed] [Google Scholar]
- 24.Adapala R. K., Kanugula A. K., Paruchuri S., Chilian W. M., Thodeti C. K. TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Research in Cardiology. 2020;115(2):p. 14. doi: 10.1007/s00395-020-0775-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bausch D., Fritz S., Bolm L., et al. Hedgehog signaling promotes angiogenesis directly and indirectly in pancreatic cancer. Angiogenesis. 2020;23(3):479–492. doi: 10.1007/s10456-020-09725-x. [DOI] [PubMed] [Google Scholar]
- 26.Afşar E., Kırımlıoglu E., Çeker T., Yılmaz Ç., Demir N., Aslan M. Effect of ER stress on sphingolipid levels and apoptotic pathways in retinal pigment epithelial cells. Redox Biology. 2020;30:p. 101430. doi: 10.1016/j.redox.2020.101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Y., Zhong M., Tang Y., et al. The role and underlying mechanism of exosomal CA1 in chemotherapy resistance in diffuse large B cell lymphoma. Molecular Therapy - Nucleic Acids. 2020;21:452–463. doi: 10.1016/j.omtn.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Che L., Alavattam K. G., Stambrook P. J., Namekawa S. H., Du C. BRUCE preserves genomic stability in the male germline of mice. Cell Death and Differentiation. 2020;27(8):2402–2416. doi: 10.1038/s41418-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan X., Li K., Zhu L., et al. Prolonged therapeutic effects of photoactivated adipose-derived stem cells following ischaemic injury. Acta Physiologica. 2020;230(1, article e13475) doi: 10.1111/apha.13475. [DOI] [PubMed] [Google Scholar]
- 30.Hausenloy D. J., Ntsekhe M., Yellon D. M. A future for remote ischaemic conditioning in high-risk patients. Basic Research in Cardiology. 2020;115(3):1–4. doi: 10.1007/s00395-020-0794-2. [DOI] [PubMed] [Google Scholar]
- 31.Domingues A., Boisson-Vidal C., Marquet de Rouge P., et al. Targeting endothelial thioredoxin-interacting protein (TXNIP) protects from metabolic disorder-related impairment of vascular function and post-ischemic revascularisation. Angiogenesis. 2020;23(2):249–264. doi: 10.1007/s10456-019-09704-x. [DOI] [PubMed] [Google Scholar]
- 32.Wang J., Zhu P., Li R., Ren J., Zhang Y., Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020;10(1):384–397. doi: 10.7150/thno.40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou H., Zhu P., Wang J., Toan S., Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduction and Targeted Therapy. 2019;4(1):p. 56. doi: 10.1038/s41392-019-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seano G., Jain R. K. Vessel co-option in glioblastoma: emerging insights and opportunities. Angiogenesis. 2020;23(1):9–16. doi: 10.1007/s10456-019-09691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steffen E., Mayer von Wittgenstein W. B. E., Hennig M., et al. Murine sca1/flk1-positive cells are not endothelial progenitor cells, but B2 lymphocytes. Basic Research in Cardiology. 2020;115(2):p. 18. doi: 10.1007/s00395-020-0774-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janus P., Toma-Jonik A., Vydra N., et al. Pro-death signaling of cytoprotective heat shock factor 1: upregulation of NOXA leading to apoptosis in heat-sensitive cells. Cell Death and Differentiation. 2020;27(7):2280–2292. doi: 10.1038/s41418-020-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song Y., Wang B., Zhu X., et al. Human umbilical cord blood–derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biology Toxicology. 2020 doi: 10.1007/s10565-020-09530-8. [DOI] [PubMed] [Google Scholar]
- 38.Li K., Liu Y., Xu Z., et al. Prevention of avian retrovirus infection in chickens using CRISPR-Cas9 delivered by Marek's disease virus. Molecular Therapy - Nucleic Acids. 2020;21:343–353. doi: 10.1016/j.omtn.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh E., Redgrave R. E., Phillips H. M., Arthur H. M. Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis. 2020;23(4):559–566. doi: 10.1007/s10456-020-09731-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szulcek R., Sanchez-Duffhues G., Rol N., et al. Exacerbated inflammatory signaling underlies aberrant response to BMP9 in pulmonary arterial hypertension lung endothelial cells. Angiogenesis. 2020;23(4):699–714. doi: 10.1007/s10456-020-09741-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson S. A., Dendorfer A., Thum T., Perbellini F. A practical guide for investigating cardiac physiology using living myocardial slices. Basic Research in Cardiology. 2020;115(6):p. 61. doi: 10.1007/s00395-020-00822-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nawaz M. I., Rezzola S., Tobia C., et al. D-Peptide analogues of Boc-Phe-Leu-Phe-Leu-Phe-COOH induce neovascularization via endothelial N-formyl peptide receptor 3. Angiogenesis. 2020;23(3):357–369. doi: 10.1007/s10456-020-09714-0. [DOI] [PubMed] [Google Scholar]
- 43.Kammoun M., Piquereau J., Nadal-Desbarats L., et al. Novel role ofTieg1in muscle metabolism and mitochondrial oxidative capacities. Acta Physiologica. 2019;228(3, article e13394) doi: 10.1111/apha.13394. [DOI] [PubMed] [Google Scholar]
- 44.Wang J., Zhu P., Toan S., Li R., Ren J., Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biology and Toxicology. 2020;36(4):365–378. doi: 10.1007/s10565-020-09513-9. [DOI] [PubMed] [Google Scholar]
- 45.Vasseur A., Cabel L., Tredan O., et al. Prognostic value of CEC count in HER2-negative metastatic breast cancer patients treated with bevacizumab and chemotherapy: a prospective validation study (UCBG COMET) Angiogenesis. 2020;23(2):193–202. doi: 10.1007/s10456-019-09697-7. [DOI] [PubMed] [Google Scholar]
- 46.Wang J., Toan S., Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;23(3):299–314. doi: 10.1007/s10456-020-09720-2. [DOI] [PubMed] [Google Scholar]
- 47.Wang J., Zhu P., Li R., Ren J., Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biology. 2020;30:p. 101415. doi: 10.1016/j.redox.2019.101415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caiazzo G., Musci R. L., Frediani L., et al. State of the art: no-reflow phenomenon. Cardiology Clinics. 2020;38(4):563–573. doi: 10.1016/j.ccl.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 49.Hughes W. E., Beyer A. M., Gutterman D. D. Vascular autophagy in health and disease. Basic Research in Cardiology. 2020;115(4):p. 41. doi: 10.1007/s00395-020-0802-6. [DOI] [PubMed] [Google Scholar]
- 50.Sawashita Y., Hirata N., Yoshikawa Y., Terada H., Tokinaga Y., Yamakage M. Remote ischemic preconditioning reduces myocardial ischemia-reperfusion injury through unacylated ghrelin-induced activation of the JAK/STAT pathway. Basic Research in Cardiology. 2020;115(4):p. 50. doi: 10.1007/s00395-020-0809-z. [DOI] [PubMed] [Google Scholar]
- 51.Maldonado C., Nguyen M. D., Bauer P., et al. Rapid lipid modification of endothelial cell membranes in cardiac ischemia/reperfusion injury: a novel therapeutic strategy to reduce infarct size. Cardiovascular Drugs and Therapy. 2021;35(1):113–123. doi: 10.1007/s10557-020-07101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuo Y. H., Liu Y. B., Cheng C. S., et al. Isovaleroylbinankadsurin A ameliorates cardiac ischemia/reperfusion injury through activating GR dependent RISK signaling. Pharmacological Research. 2020;158:p. 104897. doi: 10.1016/j.phrs.2020.104897. [DOI] [PubMed] [Google Scholar]
- 53.Daiber A., Münzel T. Interplay of the red blood cell and vascular endothelial nitric oxide synthase system to combat cardiac complications of anemia. Basic Research in Cardiology. 2020;115(4):p. 44. doi: 10.1007/s00395-020-0801-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haushalter K. J., Schilling J. M., Song Y., et al. Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIα. American Journal of Physiology. Heart and Circulatory Physiology. 2019;317(6):H1231–h1242. doi: 10.1152/ajpheart.00237.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuznetsov A. V., Javadov, Margreiter, Grimm, Hagenbuchner, Ausserlechner The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants. 2019;8(10):p. 454. doi: 10.3390/antiox8100454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X., Luo S., Zhang J., et al. lncRNA H19 alleviated myocardial I/RI via suppressing miR-877-3p/Bcl-2-mediated mitochondrial apoptosis. Molecular Therapy - Nucleic Acids. 2019;17:297–309. doi: 10.1016/j.omtn.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sedighi M., Sewell R. D. E., Nazari A., et al. A review on the most important medicinal plants effective in cardiac ischemia-reperfusion injury. Current Pharmaceutical Design. 2019;25(3):352–358. doi: 10.2174/1381612825666190329144016. [DOI] [PubMed] [Google Scholar]
- 58.Kohlhauer M., Pell V. R., Burger N., et al. Protection against cardiac ischemia-reperfusion injury by hypothermia and by inhibition of succinate accumulation and oxidation is additive. Basic Research in Cardiology. 2019;114(3):p. 18. doi: 10.1007/s00395-019-0727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bradic J., Zivkovic V., Srejovic I., et al. Protective effects of Galium verum L. extract against cardiac ischemia/reperfusion injury in spontaneously hypertensive rats. Oxidative Medicine and Cellular Longevity. 2019;2019:11. doi: 10.1155/2019/4235405.4235405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lubos N., van der Gaag S., Gerçek M., Kant S., Leube R. E., Krusche C. A. Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy. Basic Research in Cardiology. 2020;115(4):p. 42. doi: 10.1007/s00395-020-0803-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bidault G., Garcia M., Capeau J., Morichon R., Vigouroux C., Béréziat V. Progerin expression induces inflammation, oxidative stress and senescence in human coronary endothelial cells. Cells. 2020;9(5):p. 1201. doi: 10.3390/cells9051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu P., Hu S., Jin Q., et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biology. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grossini E., Gramaglia C., Farruggio S., et al. Asenapine increases nitric oxide release and protects porcine coronary artery endothelial cells against peroxidation. Vascular Pharmacology. 2014;60(3):127–141. doi: 10.1016/j.vph.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Makino A., Scott B. T., Dillmann W. H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53(8):1783–1794. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou H., Shi C., Hu S., Zhu H., Ren J., Chen Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. 2018;21(3):599–615. doi: 10.1007/s10456-018-9611-z. [DOI] [PubMed] [Google Scholar]
- 66.Zhao Y., Guo R., Li L., et al. Tongmai formula improves cardiac function via regulating mitochondrial quality control in the myocardium with ischemia/reperfusion injury. Biomedicine & Pharmacotherapy. 2020;132:p. 110897. doi: 10.1016/j.biopha.2020.110897. [DOI] [PubMed] [Google Scholar]
- 67.Chen L., Chen X. Y., Wang Q. L., et al. Astragaloside IV derivative (LS-102) alleviated myocardial ischemia reperfusion injury by inhibiting Drp1Ser616 phosphorylation-mediated mitochondrial fission. Frontiers in Pharmacology. 2020;11:p. 1083. doi: 10.3389/fphar.2020.01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Umezu R., Koga J. I., Matoba T., et al. Macrophage (Drp1) dynamin-related protein 1 accelerates intimal thickening after vascular injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(7):e214–e226. doi: 10.1161/atvbaha.120.314383. [DOI] [PubMed] [Google Scholar]
- 69.Qiao K., Liu Y., Xu Z., et al. RNA m6A methylation promotes the formation of vasculogenic mimicry in hepatocellular carcinoma via Hippo pathway. Angiogenesis. 2020 doi: 10.1007/s10456-020-09744-8. [DOI] [PubMed] [Google Scholar]
- 70.Zhou H., Wang J., Zhu P., et al. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Research in Cardiology. 2018;113(4):p. 23. doi: 10.1007/s00395-018-0682-1. [DOI] [PubMed] [Google Scholar]
- 71.Giedt R. J., Yang C., Zweier J. L., Matzavinos A., Alevriadou B. R. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free Radical Biology & Medicine. 2012;52(2):348–356. doi: 10.1016/j.freeradbiomed.2011.10.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed datasets that were generated during the study are available from the corresponding author upon reasonable request.