

Abstract
Cystic fibrosis (CF), an autosomal recessive genetic disease, is recognized as one of the most prevalent diseases in Caucasian populations. Epidemiological data show that the incidence of CF varies between countries and ethnic groups in the same region. CF occurs due to pathogenic variants in the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR), located on chromosome 7q31.2. To date, more than 2,000 variants have been registered in the CFTR database. The study of these variants leads to the diagnosis and the possibility of a specific treatment for each patient through precision medicine. In this study, complete screening of CFTR was performed through next-generation sequencing (NGS) to gain insight into the variants circulating in the population of Rio de Janeiro and to provide patient access to treatment through genotype-specific therapies. Samples from 93 patients with an inconclusive molecular diagnosis were subjected to full-length screening of CFTR using an Illumina NGS HiSeq platform. Among these patients, 46 had two pathogenic variants, whereas 12 had only one CFTR variant. Twenty-four variants were not part of our routine screening. Of these 24 variants, V938Gfs∗37 had not been described in the CF databases previously. This research achieved a molecular diagnosis of the patients with CF and identification of possible molecular candidates for genotype-specific treatments.
1. Introduction
The cystic fibrosis transmembrane conductance regulator gene (CFTR; OMIM #602421) encodes a chloride channel that is located in the apical membrane of epithelial cells [1]. Variants in this gene cause a reduction or complete absence of channel activity, leading to the development of a life-threatening illness known as cystic fibrosis (CF; OMIM #219700) or mucoviscidosis [2]. CF is characterized as a multisystem disease with an autosomal recessive inheritance pattern. Patients exhibit progressive manifestations of obstructive pulmonary disease, pancreatic insufficiency, and high concentrations of chloride in the sweat [3–5].
With the identification of CFTR in 1989 [6], genetic analysis to identify disease-causing variants in this gene began, improving the diagnosis of CF and identification of pathogenic variant carriers. The most prevalent pathogenic variant was discovered 30 years ago, having a deletion of a phenylalanine at position 508 of the protein (F508del; c.1521_1523delCTT; p.Phe508del), present in one or both alleles in approximately 90% of cases in some populations [7, 8]. Additionally, genetic studies helped clarify the correlation between CFTR dysfunction and the clinical characteristics, revealing that defects in CFTR can create other phenotypes besides CF [9–11].
Currently, more than 2,000 variants have been described over all 27 exons of CFTR (http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html), although only some of them are pathogenic [12]. Pathogenic variants are grouped into six classes according to their primary biological defects [13]. Understanding the process of CFTR synthesis up to its targeting to the plasma membrane is essential for the development of specific treatments. These treatments could be used to correct defective CFTRs according to the pathogenic variant of each patient. The genotype-specific therapeutic approach focuses on the detection of small modulatory molecules capable of correcting deficient subcellular trafficking of CFTR (“correctors”) or on the defective gating (“potentiators”) [14, 15]. The identification of pathogenic variants is important for early diagnosis, allowing a more effective treatment and a longer life expectancy for the patients [12, 16].
The heterogeneous distribution of CFTR variants worldwide and the size of the gene represent major challenges for the molecular diagnosis of CF. Thus, establishing population-specific mutation panels is extremely important [17, 18]. With new sequencing technologies becoming easily available, it is possible to rapidly generate a large amount of sequencing data, expanding the analysis of CFTR and uncovering population-specific mutation panels, increasing the sensitivity and specificity of available diagnostic strategies for various populations or ethnic groups [19, 20].
The aim of this study was to perform a complete screening of CFTR through next-generation sequencing (NGS) to investigate the variants prevalent in the population in Rio de Janeiro, Brazil.
2. Materials and Methods
2.1. Ethics Statement
The study protocol was approved by the Ethics and Research Committee of the Oswaldo Cruz Foundation (CAEE: 55095316.4.0000.5248/Protocol No: 2.010.565/17). All participants provided written informed consent prior to their inclusion in this study.
2.2. Patients and Samples
Patients from three specialized centers, namely, the Pulmonology Sector of the National Institute of Women and Adolescent Health Fernandes Figueira/FIOCRUZ, the Pedro Ernesto University Hospital/UERJ, and the Carioca Association of Assistance to Mucoviscidosis, were received in the Human Genetics Laboratory of the Oswaldo Cruz Institute for molecular diagnosis of CF. All individuals were undergoing treatment and were recruited by specialized clinicians. Patients with clinical manifestations suggestive of CF, i.e., positive sweat test (>60 mEq), positive newborn screening test, or suggestive clinical features following the most recent diagnosis of CF guidelines [21], were invited to participate in this study. In total, 217 patients agreed to participate in the study (Figure 1). Most of these patients (198 individuals) were initially screened for 27 known CF variants (Table 1) through our routine molecular panel test, and both mutated alleles were found in 124 patients. In the remaining 74 patients, only one mutated allele (36 patients) or no mutated alleles (38 patients) were found. Nineteen patients were not tested in this initial panel. To search for the missing pathogenic variants in these 93 patients, we sequenced the entire CFTR (exons and introns) through NGS.
Figure 1.
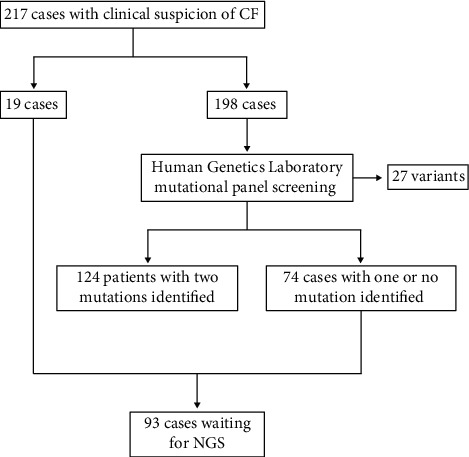
Flowchart of the studied sample. Of 198 probands with clinical suspicion of cystic fibrosis (CF), 74 candidates for next-generation sequencing (NGS) were part of an initial genetic screening of 27 variants. After the first stage, these patients were found to have one or no mutated CFTR allele. Additionally, 19 patients had not undergone any initial screening, totaling 93 patients.
Table 1.
Mutation panel used in routine testing in the Human Genetics Laboratory of the Oswaldo Cruz Institute.
| No | Legacy name | cDNA HGVS name | Protein HGVS name | dbSNP ID | Method1 |
|---|---|---|---|---|---|
| 1 | F508del | c.1521_1523delCTT | p.Phe508del | rs113993960 | Sanger sequencing |
| 2 | 3120+1G>A | c.2988+1G>A | No protein | rs75096551 | Real-time PCR |
| 3 | G542X | c.1624G>T | p.Gly542X | rs113993959 | Real-time PCR |
| 4 | G85E | c.254G>A | p.Gly85Glu | rs75961395 | Real-time PCR |
| 5 | R334W | c.1000C>T | p.Arg334Trp | rs121909011 | Real-time PCR |
| 6 | 3849+10KBC>T | c.3717+12191C>T | — | rs75039782 | Real-time PCR |
| 7 | S4X | c.11C>A | p.Ser4X | rs397508173 | Sanger sequencing |
| 8 | R1162X | c.3484C>T | p.Arg1162X | rs74767530 | Sanger sequencing |
| 9 | N1303K | c.3909C>G | p.Asn1303Lys | rs80034486 | Real-time PCR |
| 10 | P205S | c.613C>T | p.Pro205Ser | rs121908803 | Real-time PCR |
| 11 | S549R | c.1547T>G | p.Ser549Arg | rs121909005 | Sanger sequencing |
| 12 | Y1092X | c.3276C>A | p.Tyr10292X | rs121908761 | Real-time PCR |
| 13 | R347P | c.1040G>C | p.Arg347Pro | rs77932196 | Sanger sequencing |
| 14 | 232del18 | c.100_117delTTGTCAGACATATACCAA | p.Leu34_Gln39del | rs397508141 | Sanger sequencing |
| 15 | R553X | c.1657C>T | p.Arg553X | rs74597325 | Real-time PCR |
| 16 | W1282X | c.3846G>A | p.Trp1282X | rs77010898 | Real-time PCR |
| 17 | 2183del AA>G | c.2051_2052delAAinsG | rs121908799 | Sanger sequencing | |
| 18 | G551D | c.1652G>A | p.Gly551Asp | rs75527207 | Sanger sequencing |
| 19 | 2184delA | c.2052delA | p.Lys684AsnfsX38 | rs121908746 | Sanger sequencing |
| 20 | S549N-I | c.1646G>A or c.1646G>T | p.Ser549Asn or p.Ser549Ile | rs121908755 | Sanger sequencing |
| 21 | 120del23 | — | — | — | Sanger sequencing |
| 22 | S168L | c.635C>T | p.Ser168Leu | rs869249241 | Sanger sequencing |
| 23 | S1255X | c.3764C>A | p.Ser1255X | rs76649725 | Real-time PCR |
| 24 | G1244E | c.3731G>A | p.Gly1244Glu | rs267606723 | Sanger sequencing |
| 25 | CFTRdup2-3 | — | — | — | MLPA2 |
| 26 | Del25-26 | — | — | — | MLPA |
| 27 | del25-27CORTBP2 | — | — | — | MLPA |
1Methodology previously used to screening mutations at the Human Genetics Laboratory of the Oswaldo Cruz Institute. 2Multiplex ligation-dependent probe amplification.
The 93 unrelated patients comprised 44 males and 49 females (age range, 4–47 years). In addition, two control subjects were included: a healthy control and a carrier control with the 3849+10KBC>T (c.3717+12191C>T) intronic variant. Additionally, a reaction control from the NGS kit was used.
2.3. DNA Extraction
Genomic DNA was extracted from peripheral blood leukocytes using a PureLink Genomic DNA Kit (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions.
2.4. NGS
For sequencing, we used an Illumina HighSeq System (Illumina, USA) from the IOC High-Performance Sequencing Platform. Targeting of the region of interest from the entire sequence of CFTR, containing exons and introns, was achieved by designing a custom enrichment kit using the Illumina Design Studio tool. A total of 990 amplicons were generated to cover 158,462 nucleotides from the entire CFTR, from nucleotide chr7:117,119,917 to chr7:117,308,801 (188,884 bases), resulting in 83.89% of coverage. CFTR exon coordinates were obtained from the human genome assembly Hg19 (Genome Reference Consortium).
Each sample was quantified using a Qubit® 2.0 fluorometer (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA). For library preparation and enrichment of the targeted regions, we utilized the TruSeq® Custom Amplicon kit (Illumina Inc., San Diego, CA, USA), following the standard protocol. The final enriched library was sequenced via HiSeq 2500 (Illumina Inc., San Diego, CA, USA) using 150 bp paired-end reads.
Data were processed as follows: reads were trimmed using Trimmomatic (v.0.35) [22] for the removal of Truseq adapters and low-quality bases. Afterward, the reads were mapped against the reference sequence of the 990 targeted amplicons using the BWA-MEM (Burrows-Wheeler Aligner) version 0.7.17 algorithm [23]. The sequence alignment map (SAM) file was sorted and converted using samtools-1.3 [24]. Variants were called using freebayes (v.1.1.0) [25]. At the end of the process, variants were sorted and annotated using SnpSift (v.4.3) [26] and SnpEff (v.4.3) [27] and compared with the available databases dbSNP (https://www.ncbi.nlm.nih.gov/SNP/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Cystic Fibrosis Mutation Database (CFMDB; http://www.genet.sickkids.on.ca/app).
2.5. Sanger Sequencing
Twenty-four variants detected through NGS were confirmed by Sanger sequencing. Fifteen variants were previously observed in our diagnostic routine panel either by Sanger sequencing performed previously or through restriction fragment length polymorphism (RFLP). As a result, all variants observed through NGS were confirmed by at least one additional method. Sanger sequencing was performed using a Big Dye Terminator V3.1 kit (Applied Biosystems, Austin, TX, USA) in an ABI PRISM 3130xl DNA analyzer (Applied Biosystems). The CFTR DNA amplification was achieved with the set of primers listed in Supplementary Table 1. PCR products were visualized on 1.5% agarose gels and purified using a Sweep Clean up kit (Applied Biosystems, Vilnius, Lithuania). The obtained sequences were aligned with the reference sequence of CFTR in Ensembl (ENST00000003084.10). Sequence analysis was performed with Chromas Lite 2.0 software (Technelysium) and BioEdit Sequence Alignment Editor v6.0.6 (Ibis Therapeutics).
3. Results
Of the 309,361,463 pairs of reads generated in our experiment, 97.52% passed the quality control, of which 99.73% were assigned individually to each of the 95 subjects, with an average of 6,364,353 reads per individual. The lowest number and highest number of reads in the samples were 1,593,382 and 17,301,187, respectively. On an average, 88.73% of the reads were successfully mapped with the reference, indicating the quality of the data. The mean reading coverage over the region of interest was 4,000 times, with a standard deviation of 2,445, indicating a satisfactory depth. The minimum base quality parameters of 20 and a minimum mapping quality of 30 were used for detecting the variants. These results indicate high resolution and high capacity for variant identification.
Analysis of the NGS data allowed us to identify 39 variants (Table 2); 24 were not part of our routine screening. Of these, 22 were exonic and 2 were intronic; 14 already had their confirmed pathogenicity in the CFTR2 database (https://www.cftr2.org/). The 24 variants were found in exons 3, 4, 6, 8, 10, 12, 13, 14, 15, 17, 19, 20, and 22 and in introns 5 and 19, totaling 13 missense, 6 nonsense, 2 frameshift, 1 deletion, and 2 splicing variants.
Table 2.
CFTR variants identified through next-generation sequencing.
| No | Legacy name | cDNA HGVS name | Protein HGVS name | Exon/intron | dbSNP ID | Classification1 | Mutation panel2 | Alleles (in 108 alleles) | Mutation type |
|---|---|---|---|---|---|---|---|---|---|
| 01 | R75Q4 | c.224G>A | p.Arg75Gln | Exon 3 | rs1800076 | Non-CF-causing | No | 1 | Missense |
| 02 | Q98X4 | c.292C>T | p.Gln98X | Exon 4 | rs397508461 | CF-causing | No | 1 | Nonsense |
| 03 | R117H4 | c.350G>A | p.Arg117His | Exon 4 | rs78655421 | Varying clinical consequence | No | 1 | Missense |
| 04 | I148T4 | c.443T>C | p.Ile148Thr | Exon 4 | rs35516286 | Non-CF-causing | No | 1 | Missense |
| 05 | 711+1G>T4 | c.579+1G>T | — | Intron 5 | rs77188391 | CF-causing | Yes | 1 | Intronic |
| 06 | L206W4 | c.617T>G | p.Leu206Trp | Exon 6 | rs121908752 | CF-causing | No | 2 | Missense |
| 07 | R347H4 | c.1040G>A | p.Arg347His | Exon 8 | rs77932196 | CF-causing | No | 1 | Missense |
| 08 | S434X4 | c.1301C>A | p.Ser434X | Exon 10 | rs367934560 | CF-causing | No | 1 | Nonsense |
| 09 | A559T4 | c.1675G>A | p.Ala559Thr | Exon 12 | rs75549581 | CF-causing | No | 2 | Missense |
| 10 | A561E4 | c.1682C>A | p.Ala561Glu | Exon 13 | rs121909047 | CF-causing | No | 1 | Missense |
| 11 | G576A4 | c.1727G>C | p.Gly576Ala | Exon 13 | rs1800098 | Non-CF-causing | No | 2 | Missense |
| 12 | S589N4 | c.1766G>A | p.Ser589Asn | Exon 13 | rs397508300 | — | No | 2 | Missense |
| 13 | R764X4 | c.2290C>T | p.Arg764X | Exon 14 | rs121908810 | CF-causing | No | 1 | Nonsense |
| 14 | E831X4 | c.2491G>T | p.Glu831X | Exon 15 | rs397508387 | CF-causing | No | 1 | Nonsense |
| 15 | Y913X4 | c.2739T>A | p.Tyr913X | Exon 17 | rs149790377 | CF-causing | No | 2 | Nonsense |
| 16 | V938Gfs∗374,5 | c.2812_2813insG | p.Val938GlyfsX37 | Exon 17 | Novel | — | No | 1 | Frameshift |
| 17 | L997F4 | c.2991G>C | p.Leu997Phe | Exon 19 | rs1800111 | Non-CF-causing | No | 1 | Missense |
| 18 | 3199dell4 | c.3067_3072delATAGTG | — | Exon 19 | rs121908767 | — | No | 1 | Deletion |
| 19 | 3272-26A>G4 | c.3140-26A>G | — | Intron 19 | rs76151804 | CF-causing | No | 2 | Intronic |
| 20 | R1066C4 | c.3196C>T | p.Arg1066Cys | Exon 20 | rs78194216 | CF-causing | No | 4 | Missense |
| 21 | W1089X4 | c.3266G>A | p.Trp1089X | Exon 20 | rs78802634 | CF-causing | No | 2 | Nonsense |
| 22 | W1098C4 | c.3294G>T | p.Trp1098Cys | Exon 20 | rs397508533 | CF-causing | No | 1 | Frameshift |
| 23 | Q1100P4 | c.3299A>C | p.Gln1100Pro | Exon 20 | rs397508535 | — | No | 1 | Missense |
| 24 | I1234V4 | c.3700A>G | p.Ile1234Val | Exon 22 | rs75389940 | CF-causing | No | 1 | Missense |
| 25 | 232del18 | c.100_117delTTGTCAGACATATACCAA | p.Leu34_Gln39del | Exon 2 | rs397508141 | — | No | 1 | Deletion |
| 26 | G85E | c.254G>A | p.Gly85Glu | Exon 3 | rs75961395 | CF-causing | Yes | 7 | Missense |
| 27 | S168L | c.635C>T | p.Ser168Leu | Exon 5 | rs869249241 | — | No | 1 | Missense |
| 28 | P205S | c.613C>T | p.Pro205Ser | Exon 6 | rs121908803 | CF-causing | No | 1 | Missense |
| 29 | R334W | c.1000C>T | p.Arg334Trp | Exon 8 | rs121909011 | CF-causing | Yes | 2 | Missense |
| 30 | F508del | c.1521_1523delCTT | p.Phe508del | Exon 11 | rs113993960 | CF-causing | Yes | 29 | Deletion |
| 31 | S549R | c.1547T>G | p.Ser549Arg | Exon 12 | rs121909005 | CF-causing | No | 1 | Missense |
| 32 | G542X | c.1624G>T | p.Gly542X | Exon 12 | rs113993959 | CF-causing | Yes | 9 | Nonsense |
| 33 | R668C | c.2002C>T | p.Arg668Cys | Exon 14 | rs1800100 | Non-CF-causing | No | 3 | Missense |
| 34 | 2183delAA>G | c.2051_2052delAAinsG | p.Lys684SerfsX38 | Exon 14 | rs121908799 | CF-causing | No | 2 | Frameshift |
| 35 | 3120+1G>A | c.2988+1G>A | — | Intron 18 | rs75096551 | CF-causing | Yes | 12 | Intronic |
| 36 | Y1092X | c.3276C>A | p.Tyr10292X | Exon 20 | rs121908761 | CF-causing | No | 2 | Nonsense |
| 37 | R1162X | c.3484C>T | p.Arg1162X | Exon 22 | rs74767530 | CF-causing | Yes | 1 | Nonsense |
| 38 | 3849+10KBC>T | c.3717+12191C>T | — | Intron 22 | rs75039782 | CF-causing | Yes | 2 | Intronic |
| 39 | N1303K | c.3909C>G | p.Asn1303Lys | Exon 24 | rs80034486 | CF-causing | Yes | 1 | Missense |
1Classification based on CFTR2 database (clinical and functional translation of CFTR). 223 ACMG/ACOG mutation panel—American College of Medical Genetics and the American College of Obstetricians and Gynecologists. 3Number of CFTR alleles found in this study. 4Twenty-four mutations found in the next-generation sequencing performed in this study. 5Novel mutation found in the next-generation sequencing performed in this study.
A new frameshift variant, V938Gfs∗37 (c.2812_2813insG; p.Val938GlyfsX37), which occurs in exon 17 of CFTR, where guanine is inserted at position 2812 of the cDNA, resulting in a stop codon 37 bases after the amino acid change, was reported. This variant was identified in heterozygosity with the F508del mutation in a 27-year-old male patient with a positive sweat test (>60 mEq) and typical CF respiratory manifestations. In silico analyses by MutationTaster showed that V938Gfs∗37 is predicted as pathogenic and affects the structure of CFTR. The variant was submitted to the CFMDB.
Based on these results, both mutated alleles were identified in 46 individuals, 12 individuals presented only one CF variant, and 35 presented no genetic variant related to CF. Among the patients with two variants identified, 37 had the genotype CF-causing/CF-causing, 9 patients presented the combination CF-causing/unknown clinical significance, and 1 had CF-causing/novel variant, according to the CFTR2 database.
4. Discussion
The identification of new variants causing CF continues to occur even after almost 30 years of CFTR identification. Currently, more than 2,000 variants have been registered in the CFMDB; however, only 442 are annotated in the CFTR2 database, of which 360 are considered pathogenic. These variants vary in frequency and distribution in different populations. Historically, CF has been regarded as a disease limited to people of European descent. However, research has shown that CF is not ancestry linked. Therefore, in order to obtain a high detection rate, diagnosis through population-specific mutation panels should consider the molecular heterogeneity of the population and the variants to be included [11, 19]. For example, panels used in European populations to diagnose African descent patients eventually lead to inconclusive results [28]. In countries with heterogeneous populations, such as those in Latin America, the use of these panels also leads to misdiagnosis, which can compromise the patient's health and treatment [29–32]. In Brazil, with its highly mixed population, the choice of mutation panels designed for other populations has become ineffective for the diagnosis, leading to a low detection rate [33]. This shows the importance of NGS for diagnostics in these populations.
Genetic testing of CF in Brazil is not performed with uniformity, since there are no epidemiological studies or a comprehensive neonatal screening to estimate the incidence of the disease in different regions of the country. Raskin [34] estimated that only 10% of the patients are diagnosed, leading to a false impression of low incidence in the Brazilian population. According to the latest report of the Brazilian Registry of Cystic Fibrosis (REBRAFC), a large increase was observed in the percentage of patients with genotype investigation. In 2013, 40.6% of the patients in a total of 2,942 individuals had their genotyping performed, and in 2017, the number of patients genotyped reached almost 80% of the 5,128 individuals analyzed. This improvement is due to advances in molecular diagnostic techniques [35].
Here, we used NGS in a cohort of 93 patients to conclude their molecular diagnosis and search for new or rare CF pathogenic variants in the Brazilian population. Thus, 74 patients from a sample of 198 individuals were tested. These individuals had already been screened for 27 common CF pathogenic variants. This means that patients with both alleles identified from this panel were not used in this study, causing the frequency of these common pathogenic variants to be underestimated by our NGS results.
The four most frequent pathogenic variants observed in our sample of 217 patients were F508del in 42% alleles, wherein 72 patients were heterozygous, and 55 were homozygous for this pathogenic variant. The 3120+1G>A, G542X, and G85E variants were observed in 5.8%, 4.1%, and 3.2% alleles, respectively. All four variants were part of the routine testing for CF molecular diagnosis performed in our laboratory. Nunes et al. [36] published the first Brazilian study using a NGS methodology using Ion Torrent PGM (Life Technologies), with pediatric patients from the Children's Institute at Hospital das Clínicas of the University of São Paulo Medical School (HCFMUSP). The three most frequent pathogenic variants described in their study were F508del (59.1%), G542X (7.3%), and 3120+1G>A (5.3%). Our findings corroborate the observations presented by Nunes et al. [36], in which they justify the high frequency of the G542X variant as a result of the migration flow of Spanish, Portuguese, and Italians to Brazil between the 19th and 20th centuries.
The Brazilian population has a significant genetic heterogeneity, mainly resulting from a trihybrid ethnic mixture of Europeans, Africans, and indigenous populations, which varies proportionally between the different Brazilian regions [37]. Thus, it is clear that CF genetic tests preestablished for populations defined as Caucasian may present limitations when employed in a scenario as heterogeneous as the Brazilian one. For example, if we had used a panel of 23 variants recommended by the American College of Medical Genetics and the American College of Obstetricians and Gynecologists [38] that detected 88% of non-Hispanic Caucasians, we would have reduced our rate of pathogenic variant recognition. Furthermore, of the 39 variants found in our study, only 10 would be part of this panel. In addition, five patients would remain without any identified CFTR variant. Thus, genetic diagnosis for the 23 patients would not have been completed with variants identified in both alleles. Rispoli et al. proposed a panel of 11 variants as a complement to the screening of the F508del variant performed by the Brazilian Public Health System in Rio Grande do Sul, Brazil [39]. Despite being a panel developed for a Brazilian region, it does not include all the variants observed in our sample.
A total of 46 out of 93 patients who participated in the NGS had two pathogenic variants. Among these variants, we detected a new one, i.e., V938Gfs∗37. Predictive analysis of the possible effect of this insertion in the MutationTaster program was positive for pathogenicity. As a class I mutation, it leads to complete or near-complete loss of CFTR activity [12, 40]. Even with complete sequencing of CFTR, 12 patients were identified with only one CFTR variant. Among these, seven patients presented a defined pathogenic allele (F508del/unknown, G542X/unknown, 3120+1G>A/unknown, R334W/unknown, and 2183delAA/unknown). Notably, our designed method of sequencing is not capable of detecting large exonic and intronic deletions and duplications or copy number variations, a type of variant that is known to cause CF in some cases. We believe that such pathogenic variants may be responsible for some of these cases with only one defined allele.
Five individuals had one CFTR variant classified as “non-CF causing” according to CFTR2: R668C (c.2002C>T; p.Arg668Cys), G576A (c.1727G>C; p.Gly576Ala), R75Q (c.224G>A; p.Arg75Gln), and L997F (c.2991G>C; p.Leu997Phe). Two of the five patients presented the R668C and G576A variants. In 1992, Fanen et al. [41] considered the R668C variant as a polymorphism, and in 2003, Pagani et al. [42] described G576A as a variant that likely induced the skipping of exon 12 in splicing, leading to reduced levels of normal CFTR transcripts [43]. In a study by Ziętkiewicz et al. [44], the R668C variant was considered pathogenic and G576A a compound allele element. Based on previous studies, these variants when combined with a CF pathogenic variant are associated with a moderate phenotype (CFTR-related disorders; CFTR-RDs), in particular with congenital bilateral absence of the vas deferens (CBAVD). However, we cannot affirm that both variants R668C and G576A form a complex allele in our patients, since a segregation study could not be performed. According to El-Seedy et al., the variants G576A and R668C affect the chloride channel activity [45]. The R75Q variant leads to the exchange of arginine to glutamine. Zielenski et al. [46] initially reported R75Q as a neutral variant that was not involved in CF. Gené et al. [47] evaluated the impact of this variant on the functioning of the CFTR channel and found a pattern of glycosylation and subcellular distribution similar to that of wild-type CFTR. The variant L997F was initially considered as a polymorphism and was subsequently reported to cause CFTR-RDs, such as lung diseases, disseminated bronchiectasis, idiopathic pancreatitis, CBAVD, and neonatal hypertrypsinemia with normal sweat test [48].
It is now known that the severity of CF is influenced not only by CFTR variants but also by modifier genes, intragenic polymorphisms, environmental factors, and lifestyle, which explains individuals with the same variant having different clinical manifestations [19]. To this end, great efforts have been made to develop therapeutics for correcting the consequences of CFTR variants on the function of the protein [49], some of which are already available for the treatment of patients with certain genotypes [15].
5. Conclusions
Through the NGS-based study of CFTR, we expanded our knowledge of the variants that circulate in the population of Rio de Janeiro, allowing us to offer genetic support for patients seeking specific treatments. In addition, NGS made it possible to increase our previous panel of variants from 27 to 51 (41 CFTR pathogenic variants). This study highlights the importance of considering the distribution of pathogenic variants specific for admixed populations for choosing the right molecular diagnostic method. The use of NGS for the entire gene has an advantage over the mutation-specific panels available, allowing the discovery of disease-causing variants that are population specific. Moreover, this method provides an opportunity for patients from countries with heterogeneous populations, which are not well covered by commercial diagnostic panels, to have a molecular diagnosis for receiving genotype-specific therapy and creates the scope for providing genetic counseling to the family.
Acknowledgments
We thank all CF patients from Rio de Janeiro and their parents for agreeing to participate in this research. We also thank the IOC High-Performance Sequencing Platform and DNA Sequencing Platform (RPT01A), Brazil. We acknowledge the important clinical additions made to this work from Mônica de Cássia Firmida, MD, PhD, Pedro Ernesto University Hospital (HUPE), Tania Wrobel Folescu, MD, PhD, National Institute of Women and Adolescent Health Fernandes Figueira (IFF), and Renata Zlot, MD, MSc, Carioca Association of Assistance to Mucoviscidosis. This study was financially supported by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (grant number E-26/110.288/2014).
Data Availability
The data generated by the next-generation sequencing are available upon request.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Supplementary Materials
Supplementary Table 1: primers used for Sanger sequencing.
References
- 1.O’sullivan B. P., Freedman S. D. Cystic fibrosis. The Lancet. 2009;373(9678):1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 2.Cholon D. M., Gentzsch M. Recent progress in translational cystic fibrosis research using precision medicine strategies. Journal of Cystic Fibrosis. 2018;17(2):S52–S60. doi: 10.1016/j.jcf.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis P. B. Cystic fibrosis since 1938. American Journal of Respiratory and Critical Care Medicine. 2006;173(5):475–482. doi: 10.1164/rccm.200505-840OE. [DOI] [PubMed] [Google Scholar]
- 4.Cutting G. R. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Annals of the New York Academy of Sciences. 2010;1214(1):57–69. doi: 10.1111/j.1749-6632.2010.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goetz D., Ren C. L. Review of cystic fibrosis. Pediatric Annals. 2019;48(4):e154–e161. doi: 10.3928/19382359-20190327-01. [DOI] [PubMed] [Google Scholar]
- 6.Kerem B., Rommens J., Buchanan J., et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 7.Riordan J. R. CFTR function and prospects for therapy. Annual Review of Biochemistry. 2008;77(1):701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 8.Flume P. A., Liou T. G., Borowitz D. S., et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142(3):718–724. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cutting G. R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nature Reviews Genetics. 2015;16(1):45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucarelli M., Bruno S. M., Pierandrei S., et al. A genotypic-oriented view of CFTR genetics highlights specific mutational patterns underlying clinical macrocategories of cystic fibrosis. Molecular Medicine. 2015;21(1):257–275. doi: 10.2119/molmed.2014.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov M., Matsvay A., Glazova O., et al. Targeted sequencing reveals complex, phenotype-correlated genotypes in cystic fibrosis. BMC Medical Genomics. 2018;11(13):21–32. doi: 10.1186/s12920-018-0328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sosnay P. R., Siklosi K. R., Van Goor F., et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nature Genetics. 2013;45(10):1160–1167. doi: 10.1038/ng.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callebaut I., Hoffmann B., Lehn P., Mornon J. P. Molecular modelling and molecular dynamics of CFTR. Cellular and Molecular Life Sciences. 2017;74(1):3–22. doi: 10.1007/s00018-016-2385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreindler J. L. Cystic fibrosis: exploiting its genetic basis in the hunt for new therapies. Pharmacology and Therapeutics. 2010;125(2):219–229. doi: 10.1016/j.pharmthera.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castellani C., Assael B. M. Cystic fibrosis: a clinical view. Cellular and Molecular Life Sciences. 2017;74(1):129–140. doi: 10.1007/s00018-016-2393-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farinha M. C. CFTR and cystic fibrosis. In: Farinha M. C., editor. CFTR and Cystic Fibrosis: From Structure to Function. Springer Briefs in Molecular Science; 2018. pp. 1–56. [Google Scholar]
- 17.Loukas Y. L., Thodi G., Molou E., Georgiou V., Dotsikas Y., Schulpis K. H. Clinical diagnostic next-generation sequencing: the case of CFTR carrier screening. Scandinavian Journal of Clinical and Laboratory Investigation. 2015;75(5):374–381. doi: 10.3109/00365513.2015.1031689. [DOI] [PubMed] [Google Scholar]
- 18.Lucarelli M., Bruno S. M., Pierandrei S., et al. The impact on genetic testing of mutational patterns of CFTR gene in different clinical macrocategories of cystic fibrosis. Journal of Molecular Diagnostics. 2016;18(4):554–565. doi: 10.1016/j.jmoldx.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Castellani C., Cuppens H., Macek M., Jr., et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. Journal of Cystic Fibrosis. 2008;7(3):179–196. doi: 10.1016/j.jcf.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucarelli M., Porcaro L., Biffignandi A., et al. A new targeted CFTR mutation panel based on next-generation sequencing technology. Journal of Molecular Diagnostics. 2017;19(5):788–800. doi: 10.1016/j.jmoldx.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Farrell P. M., White T. B., Ren C. L., et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. The Journal of Pediatrics. 2017;181:S4–S15. doi: 10.1016/j.jpeds.2016.09.064. [DOI] [PubMed] [Google Scholar]
- 22.Bolger A. M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Handsaker B., Wysoker A., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. 2012. https://arxiv.org/abs/1207.3907.
- 26.Ruden D. M., Cingolani P., Patel V. M., et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Frontiers in Genetics. 2012;3(35):1–9. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cingolani P., Platts A., Wang L. L., et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2014;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart C., Pepper M. S. Cystic fibrosis in the African diaspora. Annals of the American Thoracic Society. 2017;14(1):1–7. doi: 10.1513/AnnalsATS.201606-481FR. [DOI] [PubMed] [Google Scholar]
- 29.Pérez M. M., Luna M. C., Pivetta O. H., Keyeux G. CFTR gene analysis in Latin American CF patients: heterogeneous origin and distribution of mutations across the continent. Journal of Cystic Fibrosis. 2007;6(3):194–208. doi: 10.1016/j.jcf.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Pepermans X., Mellado S., Chialina S., et al. Identification and frequencies of cystic fibrosis mutations in central Argentina. Clinical Biochemistry. 2016;49(1–2):154–160. doi: 10.1016/j.clinbiochem.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Sánchez K., de Mendonca E., Matute X., Chaustre I., Villalón M., Takiff H. Analysis of the CFTR gene in Venezuelan cystic fibrosis patients, identification of six novel cystic fibrosis-causing genetic variants. The Application of Clinical Genetics. 2016;9:33–38. doi: 10.2147/TACG.S78241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz-Cabezas J. C., Barros F., Sobrino B., et al. Mutational analysis of CFTR in the Ecuadorian population using next-generation sequencing. Gene. 2019;696:28–32. doi: 10.1016/j.gene.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 33.Faucz F. R., Souza D. A., Olandoski M., Raskin S. CFTR allelic heterogeneity in Brazil: historical and geographical perspectives and implications for screening and counseling for cystic fibrosis in this country. Journal of Human Genetics. 2010;55(2):71–76. doi: 10.1038/jhg.2009.123. [DOI] [PubMed] [Google Scholar]
- 34.Raskin S. Estudo multicêntrico das bases da genética molecular e da epidemiologia da fibrose cística em populações brasileiras. Curitiba (PR): Universidade Federal do Paraná; 2001. [Google Scholar]
- 35.GBEFC – Grupo Brasileiro de Estudos de Fibrose Cística. Registro Brasileiro de Fibrose Cística. São Paulo: GBEFC. 2016. September 2020, http://portalgbefc.org.br/ckfinder/userfiles/files/REBRAFC_2017_EN.pdf.
- 36.Nunes L. M., Ribeiro R., Niewiadonski V. D. T., et al. A new insight into CFTR allele frequency in Brazil through next generation sequencing. Pediatric Pulmonology. 2017;52(10):1300–1305. doi: 10.1002/ppul.23774. [DOI] [PubMed] [Google Scholar]
- 37.Cabello G. M., Cabello P. H., Llerena J. C., Jr., Fernandes O. Polymorphic markers suggest a gene flow of CFTR gene from sub-Saharan/Arabian and Mediterranean to Brazilian population. Journal of Heredity. 2006;97(4):313–317. doi: 10.1093/jhered/esl016. [DOI] [PubMed] [Google Scholar]
- 38.Watson M. S., Cutting G. R., Desnick R. J., et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genetics in Medicine. 2004;6(5):387–391. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rispoli T., Martins de Castro S., Grandi T., et al. A low-cost and simple genetic screening for cystic fibrosis provided by the Brazilian public health system. The Journal of Pediatrics. 2018;199:272–277.e3. doi: 10.1016/j.jpeds.2018.03.047. [DOI] [PubMed] [Google Scholar]
- 40.Behar D. M., Inbar O., Shteinberg M., et al. Nationwide genetic analysis for molecularly unresolved cystic fibrosis patients in a multiethnic society: implications for preconception carrier screening. Molecular Genetics and Genomic Medicine. 2017;5(3):223–236. doi: 10.1002/mgg3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fanen P., Ghanem N., Vidaud M., et al. Molecular characterization of cystic fibrosis: 16 novel mutations identified by analysis of the whole cystic fibrosis conductance transmembrane regulator (CFTR) coding regions and splice site junctions. Genomics. 1992;13(3):770–776. doi: 10.1016/0888-7543(92)90152-I. [DOI] [PubMed] [Google Scholar]
- 42.Pagani F., Stuani C., Tzetis M., et al. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Human Molecular Genetics. 2003;12(10):1111–1120. doi: 10.1093/hmg/ddg131. [DOI] [PubMed] [Google Scholar]
- 43.Polizzi A., Tesse R., Santostasi T., et al. Genotype-phenotype correlation in cystic fibrosis patients bearing [H939R;H949L] allele. Genetics and Molecular Biology. 2011;34(3):416–420. doi: 10.1590/S1415-47572011000300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziętkiewicz E., Rutkiewicz E., Pogorzelski A., Klimek B., Voelkel K., Witt M. CFTR mutations spectrum and the efficiency of molecular diagnostics in Polish cystic fibrosis patients. PLoS One. 2014;9(2, article e89094) doi: 10.1371/journal.pone.0089094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El-Seedy A., Girodon E., Norez C., et al. CFTR mutation combinations producing frequent complex alleles with different clinical and functional outcomes. Human Mutation. 2012;33(11):1557–1565. doi: 10.1002/humu.22129. [DOI] [PubMed] [Google Scholar]
- 46.Zielenski J., Bozon D., Kerem B., et al. Identification of mutations in exons 1 through 8 of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10(1):229–235. doi: 10.1016/0888-7543(91)90504-8. [DOI] [PubMed] [Google Scholar]
- 47.Gené G. G., Llobet A., Larriba S., et al. N-terminal CFTR missense variants severely affect the behavior of the CFTR chloride channel. Human Mutation. 2008;29(5):738–749. doi: 10.1002/humu.20721. [DOI] [PubMed] [Google Scholar]
- 48.Lucarelli M., Narzi L., Pierandrei S., et al. A new complex allele of the CFTR gene partially explains the variable phenotype of the L997F mutation. Genetics in Medicine. 2010;12(9):548–555. doi: 10.1097/GIM.0b013e3181ead634. [DOI] [PubMed] [Google Scholar]
- 49.Clancy J. P., Jain M. Personalized medicine in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2012;186(7):593–597. doi: 10.1164/rccm.201204-0785PP. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: primers used for Sanger sequencing.
Data Availability Statement
The data generated by the next-generation sequencing are available upon request.