

Abstract
Polymicrogyria is a malformation of cortical development characterized by overfolding and abnormal lamination of the cerebral cortex. Manifestations include epilepsy, speech disturbance and motor and cognitive disability. Causes include acquired prenatal insults and inherited and de novo genetic variants. The proportion of patients with polymicrogyria and a causative germline or mosaic variant is not known. The aim of this study was to identify the monogenic causes of polymicrogyria in a heterogeneous cohort of patients reflective of specialized referral services. Patients with polymicrogyria were recruited from two clinical centres in Australia and Belgium. Patients with evidence of congenital cytomegalovirus infection or causative chromosomal copy number variants were excluded. One hundred and twenty-three patients were tested using deep sequencing gene panels including known and candidate genes for malformations of cortical development. Causative and potentially causative variants were identified and correlated with phenotypic features. Pathogenic or likely pathogenic variants were identified in 25/123 (20.3%) patients. A candidate variant was identified for an additional patient but could not be confirmed as de novo, and therefore it was classified as being of uncertain significance with high clinical relevance. Of the 22 dominant variants identified, 5 were mosaic with allele fractions less than 0.33 and the lowest allele fraction 0.09. The most common causative genes were TUBA1A and PIK3R2. The other eleven causative genes were PIK3CA, NEDD4L, COL4A1, COL4A2, GPSM2, GRIN2B, WDR62, TUBB3, TUBB2B, ACTG1 and FH. A genetic cause was more likely to be identified in the presence of an abnormal head size or additional brain malformations suggestive of a tubulinopathy, such as dysmorphic basal ganglia. A gene panel test provides greater sequencing depth and sensitivity for mosaic variants than whole exome or genome sequencing but is limited to the genes included, potentially missing variants in newly discovered genes. The diagnostic yield of 20.3% indicates that polymicrogyria may be associated with genes not yet known to be associated with brain malformations, brain-specific somatic mutations or non-genetic causes.
Keywords: cortical malformation, epilepsy, genetic testing, high-throughput nucleotide sequencing, somatic mutation
Graphical Abstract
Graphical Abstract.
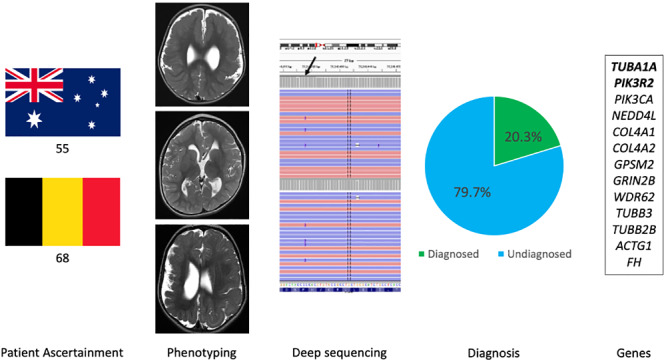
Polymicrogyria is a common cortical malformation, yet genetic causes remain unclear. Stutterd et al. ascertained 123 patients for deep sequencing. Pathogenic variants were identified in 20.3% (25% mosaic). The two most commonly mutated genes were TUBA1A and PIK3R2. A genetic diagnosis found in ∼60% of patients with macrocephaly.
Introduction
Polymicrogyria (PMG) is a malformation of the developing brain characterized by abnormal lamination and excessive folding of the cerebral cortex. It is one of the most common brain malformations and accounts for ∼20% of all malformations of cortical development (Leventer et al., 1999). PMG often occurs in association with other brain malformations and/or multiple congenital anomalies (Jansen and Andermann, 2005). Histologically, PMG is characterized by one or more undulating or festooning neuronal layers replacing the normal six-layered cortical architecture and is frequently associated with thickened leptomeninges (Jansen et al., 2016). In practice, the diagnosis is usually based on neuroimaging, the MRI features of which are an irregular, bumpy appearance of the cortex with apparent cortical thickening and a stippled grey-white matter junction (Leventer et al., 2010). The clinical consequences depend on the extent and regions of the brain affected, associated congenital malformations in the brain or elsewhere, the underlying aetiology and co-morbidities. Common manifestations include epilepsy, intellectual disability, cerebral palsy and oro-motor impairment.
The aetiologies of PMG are heterogeneous and include acquired and genetic causes. It has been proposed that PMG is the common endpoint of many different aetiological processes occurring at specific stages in cortical development (Jansen et al., 2016). Acquired causes include congenital cytomegalovirus infection (Crome and France, 1959) and foetal cerebral ischaemia (Levine et al., 1974). Genetic causes include chromosomal and monogenic variants, the latter of which may be inherited or de novo and can be germline or mosaic. PMG is morphologically heterogeneous, and an identical morphology does not necessarily imply similar aetiology which makes diagnosis of the underlying cause challenging. There are different methods of classification of PMG. It was initially based on the histological appearance (Friede, 1989). Subsequently, it was classified according to imaging patterns (Leventer et al., 2010) and the presumed timing of the aetiology in cortical development (Barkovich et al., 2012). Now that mutations in at least 50 genes have been associated with PMG, these causes can be classified according to the molecular pathways implicated in the aetiology (Stutterd et al., 2018). This recent approach to the classification of cases with suspected genetic aetiology is useful in clinical practice as it can inform genetic testing and reproductive counselling.
Previous genetic studies of PMG have interrogated the contribution of mutations in specific genes or pathways in patient groups with common phenotypic features such as mammalian target of rapamycin (mTOR) pathway mutations in patients with PMG and megalencephaly (Mirzaa et al., 2015) or screening patient cohorts for mutations in tubulin genes (Bahi-Buisson et al., 2014). The proportion of patients with PMG with an identifiable germline or mosaic variant is not known and this clinical question was the motivation for our study. We performed genomic analysis of two phenotypically heterogeneous cohorts from two centres totalling 123 patients. A chromosomal cause and congenital cytomegalovirus (if indicated) were excluded where possible, reflective of clinical practice. We tested for germline and mosaic variants in all genes known to be associated with PMG at the time of the study to establish the diagnostic yield and contribution from mosaic mutations and identify any genotype–phenotype correlations.
Materials and methods
Patient recruitment
Patients were ascertained from the Royal Children’s Hospital (RCH) Melbourne Brain Malformation Research Database and the Center of Medical Genetics, Universitair Ziekenhuis (University Hospital Brussels) Brussel Brain Malformation Database via protocols approved by the RCH Human Research Ethics Committee and the Institutional Review Board at Universitair Ziekenhuis (University Hospital Brussels) Brussels. Patients were included if there was PMG identified on MRI or if they had been referred for genetic testing for the indication of PMG. Patients were excluded if there was evidence of congenital cytomegalovirus infection from neonatal serology or retrospective testing of a dried blood spot sample from their Newborn Screening Card. Patients were excluded if they had a causative copy number variant detected by chromosome microarray. Of the 123 patients included, 55 were recruited from Australia and 68 from Belgium. Results of non-diagnostic chromosome microarray were available for 63/123 patients (55 Australian, 8 Belgium). Congenital cytomegalovirus was excluded for 40/123 patients (34 Australian, 6 Belgium) and testing for this was not clinically indicated for 15/123 (Australian cohort) based on their clinical phenotype.
Clinical and imaging phenotyping
Patient records were reviewed for information regarding sex, family history, prenatal risk factors (twinning, antepartum haemorrhage or infection), head circumference (HC), presence and type of congenital malformation and presence of seizures. All available MRIs (for 112 patients) were reviewed by the paediatric neurologist supervising the project at each site (RJL at RCH and AJ at Universitair Ziekenhuis (University Hospital Brussels) Brussel). The distribution of PMG was recorded as one of the six recurrent topographic patterns as described by Leventer et al. (Leventer et al., 2010): bilateral perisylvian polymicrogyria (BPP), unilateral perisylvian polymicrogyria, bilateral generalized polymicrogyria (BGP), bilateral frontal polymicrogyria, bilateral frontoparietal polymicrogyria, bilateral parasagittal parieto-occipital polymicrogyria or polymicrogyria associated with periventricular nodular heterotopia in addition to patterns consistent with schizencephaly (unilateral or bilateral) or ‘tubulin-related dysgyria’ (TRD). The latter term describes a pattern of cortical malformation seen in association with disordered tubulin function in which there is an abnormal gyral pattern characterized by abnormal sulcal depth or orientation and narrow gyri separated by abnormally deep or shallow sulci (Oegema et al., 2015; Mutch et al., 2016) but is often reported as PMG. The presence and type of additional brain malformations were recorded.
Genomic analysis
Genomic analysis was conducted at two sites using two different gene panels. The Australian cohort was tested at Murdoch Children’s Research Institute (MCRI) using a custom in-house panel targeting 328 genes including confirmed and candidate genes associated with brain malformations. The Belgium cohort was tested at the Center of Medical Genetics, Universitair Ziekenhuis (University Hospital Brussels) Brussel with a custom gene panel targeting 190 genes associated with malformations of cortical development. There were 141 genes common to both gene lists. The panel gene lists and overlapping genes are shown in Supplementary File 1. Of the 50 genes associated with PMG in the GeneReviews Polymicrogyria Overview updated in 2018 (Stutterd et al., 2018), 43 and 47 are included in the Australian and Belgiian panels respectively and 42 in the overlapping gene list (highlighted in bold in each gene list in Supplementary File 1).
Australian cohort: A custom in-house gene panel that targets 328 genes associated with brain malformations was developed using the HaloPlexHS target enrichment system (Agilent Technology). This panel was validated to detect mosaic variants with a minor allele frequency above or equal to 2% (Lee et al., 2019, 2020). Fifty nanograms of genomic DNA from blood or saliva was utilized to prepare the sequencing library according to the manufacturer’s instructions and 2 × 150 bp paired-end sequencing to a median depth of >600× was performed on a NextSeq (Illumina). The raw data files (fastq.gz) were analysed using both SureCall software (Agilent Technology) and Leiden Open Variation Database v.3.0 build 17 software (Leiden University Medical Center). The GRCh37/hg19 version of the human reference genome assembly was used for read alignment. Variants were filtered out based on quality (QUAL < 50), population frequency (>1%, The Exome Aggregation Consortium), region (deep intronic or untranslated regions) and type (synonymous). High-frequency variants within our cohort of cases were used to discard variants likely due to a sequencing artefact. Variant prioritization was performed following two parallel approaches, each of them blind to the other, a clinically driven approach, previously described by Sadedin et al. (2015) and a molecular-driven approach (Supplementary File 2). Variants prioritized by both methods were then further evaluated by a curation team composed of a clinical geneticist, a neurologist and a senior medical scientist. Candidate recessive variants and variants with an uncertain but high chance of clinical significance were segregated to inform pathogenicity.
Belgian cohort: Patients with malformations of cortical development were analysed by the ‘MCD gene panel’, a custom gene panel that targets 190 genes associated with malformations of cortical development. The gene content of the panel is listed in Supplementary File 1. This analysis was performed at the Center of Medical Genetics, Universitair Ziekenhuis (University Hospital Brussels) Brussel in collaboration with the Brussels Interuniversity Genomics High Throughput core according to standard procedures with an average coverage of 500×. In short, 0.5 mg deoxyribonucleic acid (DNA) was fragmented into pieces with an average length of 220 bp. A DNA library was prepared following the manufacturer’s instructions (KAPA Biosystems). Next, five DNA libraries were captured by the use of Roche Nimblegen SeqCap EZ Choice XL enrichment probes. These fragments were amplified on the Illumina cBot machine and sequenced on the Illumina Hiseq1500 instrument or on the Illumina Novaseq 600 machine. Raw data were quality-controlled by use of FastQC (v0.10.1) and mapped to the human reference genome (hg19) with BWA 0.7.10. Mapping qualities were assessed via overall coverage analysis by an in-house designed script. The mapped reads were processed using the GATK 2.7 (Genome Analysis Toolkit) pipeline (IndelRealaginer, BaseRecalibrator, HaplotypeCaller) and the detected variants were annotated by Annovar or Alamut Batch.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.
Results
Clinical features
One hundred and twenty-three patients were included in the study. The sex was known for 121 patients, 60% of whom were male. There were four familial cases in the cohort, all involving two affected siblings, and the remaining 115 cases were sporadic. Four patients were assessed and tested prenatally as foetuses and the remaining 119 patients were children or adults. Eight patients were born from a twin pregnancy, three dizygotic, two monozygotic and three with unknown zygosity. One had a twin sibling who died in utero at four months gestation and seven had an unaffected twin sibling. Ninety-five of 123 patients had a HC documented at the time of genetic testing. Of those with a recorded HC, the majority were normocephalic (60%), 27.3% were microcephalic (<2 standard deviation from mean) and 12.6% were macrocephalic (>2 standard deviation from mean). The presence or absence of congenital malformations outside of the central nervous system was documented in 77 patients and were present in 40 (51.9%), 10 of whom had more than one non-central nervous system malformation. Recurrent malformations were facial dysmorphism, eye anomalies (blepharophimosis, ptosis, strabismus, abnormal palpebral fissures, microphthalmia, cataract and retinal dystrophy), limb malformations (arthrogryposis, talipes), capillary malformation, syndactyly, vocal cord palsy and sensorineural hearing loss. Malformations documented in single cases only were polydactyly, hemihypertrophy, short stature, hypertrichosis and hypospadias. The presence or absence of epilepsy was document for 91 patients, for whom it was present in 59.3%.
PMG distribution
Imaging or information on PMG distribution was available for 112 patients. Forty-four of 112 (39.2%) had bilateral perisylvian polymicrogyria. The next most common distribution patterns were unilateral perisylvian polymicrogyria and BGP seen in 18/112 (16%) and 17/112 (15.2%), respectively. All of the other six distribution patterns were identified amongst the remaining 33 cases. Tubulin-related dysgyria was identified in five patients who had been referred for genetic testing based on an MRI diagnosis of PMG. A review of these MRIs after recruitment to the study led to re-classification of the malformations of cortical development as tubulin-related dysgyria.
Additional brain malformations
Numerous additional brain malformations were identified. The most common were periventricular nodular heterotopia, abnormalities of the corpus callosum (complete or partial absence, thin, thick, dysplastic, elongated), abnormal septum pellucidum (cavum, absence or partial absence), abnormalities of the basal ganglia, brainstem and cerebellum (hypoplasia, dysplasia, asymmetry), calcifications (cortical, subcortical and thalamic), white matter abnormalities, dysplastic ventricles, subcortical heterotopia and porencephalic cyst. Less common abnormalities reported in a single patient only (some in the same patient) were under-rotated hippocampi, hemimegalencephaly, cerebellar cysts, hydrocephalus, fused colliculi, pineal mass, Chiari I malformation, enlarged extra-axial spaces, posterior PNH, optic nerve hypoplasia and choroid plexus cysts.
Genetic causes
A pathogenic or likely pathogenic variant was identified in 25/123 (20.3%) patients and a variant of uncertain significance with high clinical relevance was identified in one patient. Of the 25 pathogenic or likely pathogenic variants, 14 were from the Australian cohort and 11 from the Belgium cohort. One patient from the Belgium cohort was identified to have a variant of uncertain significance with high clinical relevance. The variants identified in these 26 patients involved 13 different genes (Table 1) and included autosomal recessive variants, inherited dominant variants and de novo dominant variants, both germline and mosaic. No X-linked variants were identified. The most commonly implicated genes were TUBA1A (6/26) and PIK3R2 (5/26). Causative variants were also identified in PIK3CA, NEDD4L, COL4A1, COL4A2, GPSM2, GRIN2B, WDR62, TUBB3, TUBB2B, FH and ACTG1. Of the 22 dominant variants, five were likely mosaic variants based on their low allele fractions (AFs) and these were identified in two genes, PIK3R2 and PIK3CA. The lowest AF was 0.09 detected for a PIK3CA variant in a blood-derived DNA sample. Additional genotypic and phenotypic information relating to each variant is provided in Supplementary Table 1. Representative brain MR images from patients with six of the most commonly identified genes are shown in Fig. 1.
Table 1.
Genetic results for patients with causative variant identified
| Patient ID | Gene | Causative variant | Allele fraction a (tissue) | Inheritance | Variant class | PMG distribution | Head size |
|---|---|---|---|---|---|---|---|
| CS34 | ACTG1 | NM_001614.5(ACTG1): c.616C>T; p. Arg206Trp | Het | De novo | LP | BPP | Norm |
| CS53 | COL4A1 | NM_001845.6(COL4A1):c.2842G>C; p. Gly948Arg | Het | De novo | LP | BPP | MIC |
| CS40 | COL4A2 | NM_001846.4(COL4A2):c.2902 + 1G>A | Het | Maternal (sibling of CS41) | LP | UPP | Norm |
| CS41 | COL4A2 | NM_001846.4(COL4A2):c.2902 + 1G>A | Het | Maternal (sibling of CS40) | LP | SCZ | Norm |
| AJ34 | FH | NM_000143.3(FH):c.1169A>G, p. Asn390Ser | Hom | Bi-parental | LP | BGP | NR |
| CS7 | GPSM2 | NM_013296.5(GPSM2):c.1501delA; Ser501Alafs*30 | Hom | Bi-parental | LP | BGP | Norm |
| AJ42 | GRIN2B | NM_000834.4(GRIN2B):c.2437C>G, p. Leu813Val | Het | De novo | LP | BGP | MIC |
| AJ2 | NEDD4L | NM_001144967.3(NEDD4L):c.623G>A, p. Arg208Gln | Het | Maternal | LP | BPP (PNH-PMG) | Norm |
| AJ15 | NEDD4L | NM_001144967.3(NEDD4L):c.623G>A, p. Arg208Gln | Het | De novo | LP | BPP (PNH-PMG) | MAC |
| CS10 | PIK3R2 | NM_005027.3(PIK3R2):c.1117G>A; p. Gly373Arg | 0.14 (blood) | Mosaic | P | BPP | Norm |
| CS27 | PIK3R2 | NM_005027.3(PIK3R2):c.1117G>A; p. Gly373Arg | 0.14 (blood) | Mosaic | P | BPP | MAC |
| CS46 | PIK3R2 | NM_005027.3(PIK3R2):c.1117G>A; p. Gly373Arg | 0.22 (saliva) | Mosaic | P | BGP | MAC |
| CS51 | PIK3R2 | NM_005027.3(PIK3R2):c.1117G>A; p. Gly373Arg | Het | Unknown | P | BFP | MAC |
| AJ57 | PIK3R2 | NM_005027.3(PIK3R2):c.1117G>A; p. Gly373Arg | Het | De novo | P | BGP | MAC |
| AJ52 | PIK3CA | NM_006218.4(PIK3CA):c.1345C>T, p. Pro449Ser |
0.30 (skin) 0.00 (blood) |
Mosaic | LP | BPP | MAC |
| CS3 | PIK3CA | NM_006218.4(PIK3CA):c.2740G>A, p. Gly914Arg |
0.09 (blood) 0.11 (saliva) |
Mosaic | P | BPP | MAC |
| AJ5 | TUBA1A | NM_006009.4(TUBA1A):c.5G>A, p. Arg2His | Het | De novo | P | BPP | MIC |
| CS32 | TUBA1A | NM_006009.4(TUBA1A):c.598T>C, p. Cys200Arg | Het | De novo | LP | BPP | MIC |
| CS37 | TUBA1A | NM_006009.4(TUBA1A):c.746A>G, p. Asn249Ser | Het | De novo | LP | TRD | MIC |
| AJ22 | TUBA1A | NM_006009.4(TUBA1A):c.958C>T, p. Arg320Cys | Het | De novo | P | Unknown | NR |
| AJ58 | TUBA1A | NM_006009.4(TUBA1A):c.1168C>T, p. Arg390Cys | Het | De novo | P | TRD | MIC |
| AJ46 | TUBA1A | NM_006009.4(TUBA1A):c.1182G>C, p. Lys394Asn | Het | Unknown | VUS | TRD | Norm |
| AJ43 | TUBB2B | NM_178012.5(TUBB2B):c.602G>T, p. Cys201Phe | Het | De novo | LP | TRD | NR |
| AJ68 | TUBB3 | NM_006086.4(TUBB3)c.292G>A, p. Gly98Ser | Het | De novo | LP | BPP | Norm |
| CS19 | WDR62 | NM_001083961.2(WDR62):c.836G>A, p. Cys279Tyr | Het | Paternal | LP | BGP | MIC |
| NM_001083961.2(WDR62):c.1480G>A, p. Gly494Arg | Het | Maternal | LP | ||||
| CS20 | WDR62 | NM_001083961.2(WDR62):c.836G>A, p. Cys279Tyr | Het | Paternal | LP | BPP | MIC |
| NM_001083961.2(WDR62):c.1480G>A, p. Gly494Arg | Het | Maternal | LP |
BFP = bilateral frontal polymicrogyria; BG = basal ganglia; BGP = bilateral generalized polymicrogyria; BPP = bilateral perisylvian polymicrogyria; CC = corpus callosum; Comp het = compound heterozygous; CSP = cavum septum pellucidum; Het = heterozygous; Hom = homozygous; LP = likely pathogenic; MAC = macrocephaly; MIC = microcephaly; Norm = normal; NR = No record; P = pathogenic; PIPO = paediatric intestinal pseudo-obstruction; PNH-PMG = polymicrogyria associated with periventricular nodular heterotopia; UPP = unilateral perisylvian polymicrogyria; VUS = variant of uncertain significance.
The AF is provided for mosaic cases only (variants with AF <0.33).
Figure 1.

Representative MRI images for patients with different molecular diagnoses. Images are T2 axial, T1 or T2 parasagittal. TUBA1A (patient CS37, aged 2 years at scan) shows an irregular, overfolded, thickened cortex particularly in the frontal regions. This cortical appearance is now described by some as ‘tubulin-associated dysgyria’. The basal ganglia are dysmorphic, giving the frontal horns a ‘hooked aspect’ (white arrows). The corpus callosum is thin, the cerebellar vermis hypoplastic and brainstem dysmorphic with a flattened pons and bulky medulla. COL4A2(patient CS40, aged 40 years at scan) shows a deep sulcus in the left frontal lobe with PMG branching from its base (white arrow). WDR62(patient CS19, aged 15 months at scan) shows extensive right hemisphere PMG and a dysmorphic right lateral ventricle. NEDD4L(patient AJ15, aged 14 months at scan) shows bilateral PMG centred around the Sylvian fissures (white arrows) and underlying periventricular nodular grey matter heterotopia (black arrows). PIK3R2(patient CS10, aged 23 months at scan) shows generalized PMG with relative sparing of the medial occipital gyri. The Sylvian fissures are abnormally extended and oriented superiorly. PIK3CA(patient CS3, aged five days at scan) shows posterior perisylvian PMG on the right (white arrows). Other slices showed a similar appearance to the left posterior Sylvian fissure.
Tubulinopathies
Eight of the 26 (30.7%) causative or candidate variants were in tubulin-related genes, six in TUBA1A and one each in TUBB2B and TUBB3. Parental testing was available for seven of the eight variants and the variant was shown to be de novo in each case. Parental testing was not available for one variant in TUBA1A, which is a novel missense variant and therefore remains classified as a variant of uncertain significance. As TUBA1A is highly intolerant to missense variation (observed number of variants in population is 0.7 compared with expected number of 260.7; Z score = 5.58) (Lek et al., 2016) and there is a strong phenotype correlation in this case, the variant would be reclassified as ‘likely pathogenic’ if confirmed de novo. None of the TUBA1A variants were recurrent within the cohort and all were missense. Three were previously reported variants and three were novel. The genotype–phenotype correlations identified for this group are discussed below.
mTORopathies
The second most frequently implicated molecular pathway was the mTOR-PI3K-AKT pathway, with causative variants in the PIK3R2 or PIK3CA genes identified in 7 of 26 (26.9%) patients with a causative of candidate variant(s) identified. The recurrent PIK3R2 variant, p. Gly373Arg was identified in five patients and this variant alone constituted 20% of the causative and candidate variants identified in the entire cohort. The AF for the PIK3R2 variants ranged from 0.14 to 0.5. PIK3CA variants were identified in two patients and were mosaic in both cases. For one patient, the variant was detectable only in the patient’s skin sample (AF 0.3) and was not detected in their blood sample (see Table 1 Pt AJ52: p. Pro449Ser). The second PIK3CA variant was detected with a similar AF in the patient’s blood (AF 0.09) and saliva (AF 0.11) samples. This was the lowest AF for a causative variant detected in this study.
Collagenopathies
Three patients from two families were diagnosed with collagenopathies with causative variants identified in COL4A1 and COL4A2. We identified a novel, maternally inherited splice variant in COL4A2 (c.2902 + 1G>A) in two affected sisters, one with intellectual disability and schizencephaly and the other with PMG, epilepsy and normal cognitive function. Family segregation studies showed that the variant was inherited from their mother who had died from small vessel cerebral disease and therefore it was classified as likely pathogenic. We identified a novel, de novo COL4A1 variant (p.Gly948Arg) in a female with microcephaly, cerebral palsy and BPP, porencephaly and multifocal haemorrhagic infarctions. Evidence of cerebrovascular accident had been identified on antenatal ultrasound at 35 weeks’ gestation. This is a novel variant causing substitution of a glycine residue in the triple amino acid repeat sequence of the triple-helical domain which is a common mutational mechanism for collagenopathies.
Other autosomal dominant causes
Two unrelated patients with periventricular nodular heterotopia and syndactyly were identified with a recurrent heterozygous variant in NEDD4L (p.Arg208Gln), inherited from an affected parent in one case and de novo in the other. These patients have been published separately by Stouffs et al. (2020). A de novo variant in GRIN2B (p.Leu813Val) was identified in one patient with microcephaly, epilepsy and BGP with a thin corpus callosum. A novel, de novo variant in ACTG1 (p.Arg206Trp) was identified in a child with BPP without the facial dysmorphism that is typical for Baraitser–Winter syndrome. This finding expands the phenotypic spectrum of ACTG1 to non-syndromic cortical malformation and expands the spectrum of associated cortical malformation to include PMG.
Autosomal recessive causes
Compound heterozygous variants in WDR62 (p.Cys279Tyr and p. Gly494Arg) were identified in two similarly affected sisters with microcephaly, epilepsy and cerebral palsy. A novel, homozygous frameshift variant in GPSM2 (Ser501Alafs*30), was identified in a child born to consanguineous parents with treated hydrocephalus, congenital sensorineural hearing loss and bilateral frontal parasagittal PMG with hypoplastic corpus callosum, consistent with Chudley–McCullough Syndrome. A homozygous variant was identified in the gene encoding fumarate hydratase, FH (p.Asn390Ser) in one patient born to consanguineous parents with BGP and agenesis of the corpus callosum. For this patient, FH activity in fibroblasts and leucocytes was reduced to 25% and MRI findings are consistent with fumarate hydratase deficiency (Kerrigan et al., 2000) providing evidence for likely pathogenicity of this variant for the PMG.
Chromosomal variants of uncertain significance
Two patients included in the study have chromosome copy number variants of uncertain significance that may be relevant to their PMG. A monogenic cause for PMG was not identified in either patient and both patients had congenital cytomegalovirus excluded. One patient (CS55) has BPP and nephronophthisis and a homozygous deletion of NPHP1. This interstitial deletion involves ∼0.12 Mb at 2q13: arr[hg19]2q13(110 859 672–110 982 530)×0. She has normocephaly and no additional brain or other malformations. This homozygous deletion is a known cause for nephronophthisis 1 (MIM#256100) but has never been reported before in association with PMG. At least one other gene associated with Joubert syndrome, AHI1, is known to cause both nephronophthisis and PMG (Dixon-Salazar et al., 2004; Utsch et al., 2006). Therefore, it is possible that the ciliopathy in this case is the cause for the patient’s PMG however, another example of this has not been published. The second patient (CS36) is a male with BPP and a maternally inherited duplication of ∼0.4 Mb at Xq28: arr[hg19]Xq28(154 132 683–154 536 836)×2. He has normocephaly and no additional brain or other malformation. The Xq28 duplication involves seven RefSeq genes including RAB39B. Duplications of this region, and RAB39B specifically, have been associated with intellectual disability but not cortical malformation to date (El-Hattab et al., 2011; Vanmarsenille et al., 2014).
Genotype–phenotype correlations
Family history
The genetic causes identified were not significantly different between male (56%) and female (44%) patients. There were three sets of affected siblings and one set of half-siblings included in the study. A genetic cause was identified in three of these four families, compound heterozygous variants in WDR62 for one family and inherited variants in COL4A2 and NEDD4L for two. Parental consanguinity was reported for four patients, two of whom had homozygous causative variants affecting GPSM2 and FH, respectively.
Prenatal risk factors
Eight patients tested had been born of a twin pregnancy and a genetic cause was identified for only one of these patients. The cause in this dizygous twin was a de novo heterozygous PIK3R2 variant p. Gly373Arg. A genetic cause was not identified in the other seven patients born of a twin pregnancy, two dizygotic, two monozygotic and two of unknown zygotic status.
Head circumference
The highest diagnostic yield was in patients with abnormal head growth, as recorded at the time of genetic testing. A genetic diagnosis was confirmed in 58.3% of patients with macrocephaly. The genes implicated in this group were PIK3R2, PIK3CA and NEDD4L. All patients with an mTORopathy had macrocephaly except one who was mosaic for the recurrent PIK3R2 variant with an AF of 0.14 and had a HC within the normal range. Of the 26 patients in the cohort with microcephaly, a genetic cause was identified in eight, giving a diagnostic yield of 30.8% in this group. The genes implicated in this group were TUBA1A, WDR62, GRIN2B and COL4A1. The proportion of these patients who had an acquired rather than congenital abnormality in head growth is not known. The diagnostic yield according to HC is shown in Fig. 2A. The causative genes implicated according to head size are shown in Supplementary Table 2A.
Figure 2.
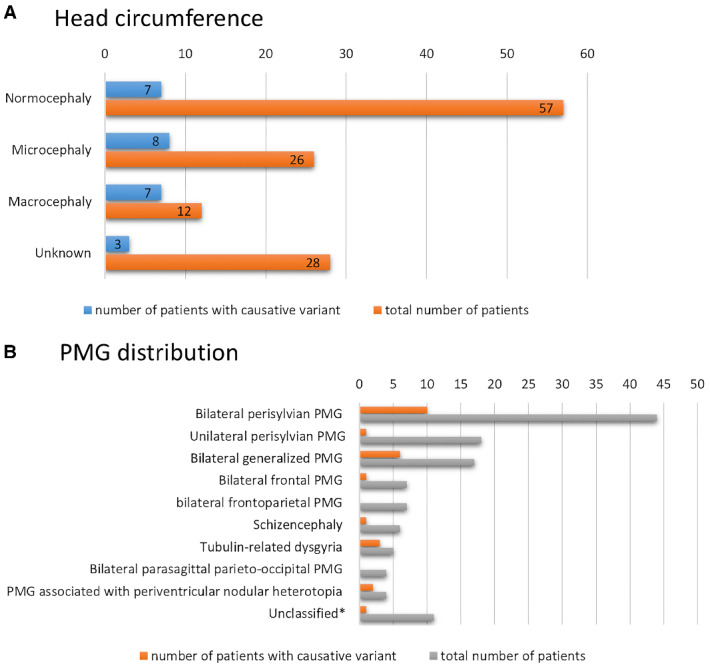
Graphical visualization of the diagnostic yield in patient subgroups. (A) Graphical visualization of the diagnostic yield depending on the HC. (B) Graphical visualization of the diagnostic yield in each morphological subtype of PMG. *Unclassified group refers to patients for whom MR images or detailed data was not available.
Additional congenital anomalies
PMG can be seen as a feature in numerous genetic syndromes (Stutterd and Leventer, 2014). Of the 40 patients with congenital malformation outside the central nervous system, 13 had a genetic cause identified (32.5%). Of the 37 patients with no congenital malformation outside the central nervous system, a genetic diagnosis was identified for 8 (21.6%). This is not significantly different. Of the 6/8 cases of tubulinopathy with records available regarding presence or absence of congenital malformation, five had eye abnormalities. These were documented as abnormal palpebral fissures in four cases, the abnormality of which was ptosis in two cases with blepharophimosis in one case. Two patients had strabismus. The presence of congenital malformations in association with an mTORopathy was variable and not all of these patients had features of the typically associated Megalencephaly-PMG-Polydactyly-Hydrocephalus or Megalencephaly-Capillary malformation and polymicrogyria syndromes. Six of the seven patients had macrocephaly, but additional features were only seen in two; one with capillary malformation and polydactyly and one with capillary malformation only.
PMG distribution
The PMG distribution was not predictive for diagnostic yield except in cases recognized as having the distribution now referred to as ‘tubulin-related dysgyria’ (Oegema et al., 2015), for whom 4/5 patients had a causative or candidate tubulin gene variant identified (TUBA1A or TUBB2B). Outside of tubulin-related dysgyria, the PMG distribution was not predictive for genotype except in one patient who had the rare distribution pattern of frontal parasagittal PMG accompanied by ventriculomegaly and sensorineural hearing loss which is consistent with Chudley–McCullough syndrome and, in this patient, a causative homozygous variant in GPSM2 was identified. The diagnostic yield according to PMG distribution is shown in Fig. 2B. The causative genes implicated according to PMG distribution are shown in Supplementary Table 2B.
Additional brain malformations
The presence or absence of additional brain malformations did not correlate with diagnostic yield in this cohort. However, additional brain malformations were present in all patients diagnosed with a tubulinopathy. The additional brain malformations in these cases included agenesis/hypoplasia or the corpus callosum, dysplastic basal ganglia, brain stem hypoplasia and hypoplasia/dysplasia of the cerebellum/vermis, all of which are recognized malformations in tubulinopathies (Mutch et al., 2016).
Discussion
Diagnostic yield
In this study, 123 individuals with PMG had sequencing on a NGS panel and a pathogenic or likely pathogenic variant was identified in 25 (20.3%), 14/55 patients in the Australian cohort and 11/68 in the Belgium cohort. A variant of uncertain significance with high clinical relevance was identified in one additional patient from the Belgium cohort. Of the 22 autosomal dominant variants identified, five were likely mosaic variants based on their low AF (<0.33) and the lowest AF was 0.09. The most commonly implicated genes were TUBA1A and PIK3R2. Several genotype–phenotype correlations were identified, highlighting the importance of clinical phenotyping for variant prioritization and interpretation. Gene panel analysis of a heterogeneous group of PMG cases was uninformative for 79.7% patients. The causes for the undiagnosed group may include non-genetic factors, brain specific somatic mutations or variants in genes not yet discovered or known to be associated with brain malformations, the latter of which may be detected using a broader genetic test such as whole exome or genome sequencing. Seven of the eight patients born of a twin pregnancy did not have a genetic cause identified, consistent with previous studies that have identified twinning as a risk factor for PMG with the causative mechanism thought to be vascular disruption (Curry et al., 2005).
Monogenic causes
Causative or candidate variants were identified in 13 different genes (Table 1) and included autosomal recessive variants, inherited autosomal dominant variants and de novo autosomal dominant variants, non-mosaic and mosaic. Despite the predominance of males affected and 16 X-linked genes tested by one or both gene panels, no X-linked causes were identified. The majority of genetic causes in this cohort of 123 patients implicated tubulin genes (31%) or genes in the mTOR-PI3K-AKT pathway (27%). The significant contribution of tubulin-related mutations to malformations of cortical development has been recognized in previous studies. Heterogeneous cohorts of patients with malformations of cortical development screened for tubulin mutations have yielded a diagnosis in up to 13.3%. (Bahi-Buisson et al., 2014). Seven of the 26 diagnoses (27%) were in mTOR-PI3K-AKT pathway-related genes, PIK3R2 and PIK3CA. The recurrent PIK3R2 variant p. Gly373Arg constituted 20% of the molecular diagnoses in the entire cohort. Previous studies have identified this variant at similar frequencies, in 10 of 40 (25%) patients with Megalencephaly-PMG-Polydactyly-Hydrocephalus (Riviere et al., 2012) and 19/127 (15%) patients with isolated BPP or Megalencephaly-PMG-Polydactyly-Hydrocephalus (Mirzaa et al., 2015). Beyond these two groups of genes, the causes identified were varied and included isolated and syndromic causes for PMG. Four of the implicated genes are not PMG-associated as published in the GeneReviews Polymicrogyria Overview update in 2018: ACTG1, COL4A1, COL4A2 and FH (Table 1). These four genes had been included in both panels due to a speculative association with PMG or association with a brain malformation other than PMG and these findings highlight the value of broad genetic testing beyond panels that include only the currently known genetic causes.
Novel potential chromosomal causes
Two patients included in the study have chromosome copy number variants of uncertain significance that may be relevant to their PMG. One patient with BPP and nephronophthisis has a homozygous deletion of NPHP1, a known cause for nephronophthisis but never before reported in association with PMG. As PMG is observed in at least one other ciliopathy, that associated with AHI1, it is plausible that the homozygous NPHP1 deletion in this patient explains both their brain and renal phenotype, though there are no other published examples of this. There is one published case of cortical malformation identified in a foetus with compound heterozygous variants in NPHP1, a pathogenic deletion and a missense variant of unknown significance (Reches et al., 2018). The second case with a possible novel chromosomal cause for their PMG is a male with BPP and a maternally inherited duplication at Xq28 involving RAB39B. Duplications of this region, and RAB39B specifically, have been associated with intellectual disability but not cortical malformation to date (El-Hattab et al., 2011; Vanmarsenille et al., 2014).
Genotype–phenotype correlations
Several genotype–phenotype correlations were identified. HC was the most informative clinical feature regarding the likelihood of identifying a genetic cause. A genetic diagnosis was confirmed in 58.3% of patients with macrocephaly and 30.8% in the patients with microcephaly. All patients identified as having a causative or candidate variant in a tubulin gene had a bilateral distribution of PMG and additional brain malformations that are recognized as features of a tubulinopathy (Mutch et al., 2016). Therefore, the presence of this brain phenotype, which includes agenesis/hypoplasia or the corpus callosum, dysplastic basal ganglia, brain stem hypoplasia and hypoplasia/dysplasia of the cerebellum/vermis is highly predictive of a causative variant in a tubulin gene. A causative homozygous variant in GPSM2 was identified in a patient with the typical PMG distribution and syndromic features of Chudley–McCullough syndrome, which is an important genotype to consider in the prenatal setting as the neurodevelopmental outcome is typically good despite extensive CNS malformation. A likely pathogenic variant in ACTG1 was identified in a patient with non-syndromic BPP, expanding the spectrum of cortical malformation associated with this gene which has previously been limited to pachygyria. This finding has informed the need to monitor this patient for comorbidities associated with the Baraitser–Winter Syndrome.
Use of a gene panel
The strength of a gene panel is in its coverage of genes and sensitivity to low-level mosaic variants when compared to exome sequencing. In this study, we detected five causative mosaic variants, three of which were detected with an AF less than 0.15 in blood, a level that would not reliably be detected by clinical exome sequencing. A limitation to the use of a gene panel is that the genomic analysis is limited to the genes included in the panel at the time of its design. This technology prevents future analysis of newly discovered genes and genes not known to be associated with brain development at the time of panel design. Additionally, syndromic causes of PMG may be missed if the causative gene is not typically associated with PMG. In this case, a clinical assessment of the patient’s phenotype and family history may be helpful in determining whether a gene panel or exome sequencing is the more useful test, though access to these tests is also dependent on local availability and resources. A gene panel may be useful to investigate a specific phenotype with suspected mosaic cause. Outside of this, a two-step approach to genome-wide testing is recommended with targeted analysis of exome data focused on genes known to cause PMG and, if negative, an expanded analysis of the clinical exome.
The current rate of gene discovery is high, predominantly due to the application of next-generation sequencing to gene discovery projects. Since the PMG Overview in GeneReviews was updated in 2018 and the panels for these two cohorts were designed, two genes with significant clinical relevance for PMG have been identified. Heterozygous pathogenic missense variants in SCN3A were identified by Smith et al. (2018) in 2018 in six unrelated families with speech and oral motor dysfunction and PMG and Chatron et al. (2019) reported four cases from two unrelated families with a lethal PMG syndrome caused by homozygous ATP1A2 truncating variants in families with a history of migraine in carrier parents. The rate of new gene discovery and the expanding number of molecular pathways implicated in cortical development suggests that diagnoses may have been missed by the gene panel analysis of this cohort as this technology limits the analysis to those genes included in the panel at the time of its design.
Clinical contribution to genomic analysis
A strength of this study is the clinical contribution to patient phenotyping and variant interpretation. This was made possible by a close working relationship between the clinicians and the testing laboratory in both centres. The cohort was refined to those with potential monogenic cause and this was achieved through clinical interpretation of clinical features, family history and chromosomal variants as being contributory or non-contributory. An example of the clinical contribution to variant prioritization included the re-classification of the PMG in tubulin-related dysgyria and subsequent prioritization of tubulinopathy-related genes. Where there was a strong phenotype match with a gene-disease association identified, sufficient to strengthen the evidence for pathogenicity, this led to re-classification of a variant of uncertain significance in some cases to likely pathogenic. The genotype–phenotype correlations identified in this study can assist the clinician in prioritizing candidate genes and variants for curation. These correlations also help to identify patients who are more likely to benefit from genomic testing thereby optimizing the use of resources, especially in resource-poor settings.
Conclusions
Genetic analysis of 123 patients with PMG using deep sequencing panels identified a cause in 20.3%. Genetic testing has the highest diagnostic yield in patients with PMG and abnormal head growth. The highest yield is in those with macrocephaly, in which a mosaic variant may be implicated, and deep sequencing should be considered in these cases. The genetic causes for PMG with microcephaly are unlikely to be mosaic and include recessive and de novo dominant causes. Where non-mosaic causes are being considered, whole exome or genome sequencing is recommended in preference to a gene panel due to the genetic heterogeneity of PMG and the ability to reanalyse the data as new genes are discovered. The causes for the undiagnosed group may include non-genetic factors, brain-specific somatic mutations or variants in genes not yet discovered or known to be associated with brain malformation, the latter of which may be detected by whole-exome or genome sequencing.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
We thank the patients, their families and their referring clinicians for their participation in this study.
Funding
C.A.S. was supported by the Flora Suttie Neurogenetics Fellowship funded by the Suttie family and their supporters, the Thyne Reid Foundation and the Macquarie Foundation, and National Health and Medical Research Council Postgraduate Scholarship (APP1133266). The research at the Murdoch Children’s Research Institute was supported by the Campbell Edwards Trust, the Genet Foundation and the Murdoch Children’s Research Institute Translation Research Fund. P.J.L. was supported by the Vincent Chiodo Foundation. A.J. was supported by a Senior Clinical Investigator Fellowship from the Research Foundation - Flanders. R.J.L. was supported by a Melbourne Children’s Clinician Scientist Fellowship.
Competing interests
The authors report no competing interests.
Glossary
- AF =
allele fraction
- AKT =
protein kinase B
- BGP =
bilateral generalized polymicrogyria
- bp =
base pairs
- BPP =
bilateral perisylvian polymicrogyria
- HC =
head circumference
- hg19 =
human genome assembly 19 (GRCh37) from Genome Reference Consortium
- LOVD =
Leiden Open Variation Database
- Mb =
megabases;
- MCD =
malformation of cortical development
- MIM =
Mendelian inheritance of man
- mTOR =
mammalian target of rapamycin
- PI3K
phosphatidylinositol 3-kinase
- PMG =
polymicrogyria
- PNH-PMG =
polymicrogyria associated with periventricular nodular heterotopia
- RCH =
Royal Children’s Hospital, Melbourne, Australia
- TRD =
tubulin-related dysgyria
- UZ =
Universitair Ziekenhuis (University Hospital Brussels)
References
- Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N,. et al. ; LIS-Tubulinopathies Consortium. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 2014; 137: 1676–700. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB.. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 2012; 135: 1348–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatron N, Cabet S, Alix E, Buenerd A, Cox P, Guibaud L, et al. A novel lethal recognizable polymicrogyric syndrome caused by ATP1A2 homozygous truncating variants. Brain 2019; 142: 3367–74. [DOI] [PubMed] [Google Scholar]
- Crome L, France NE.. Microgyria and cytomegalic inclusion disease in infancy. J Clin Pathol 1959; 12: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry CJ, Lammer EJ, Nelson V, Shaw GM.. Schizencephaly: heterogeneous etiologies in a population of 4 million California births. Am J Med Genet A 2005; 137A: 181–9. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 2004; 75: 979–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Fang P, Jin W, Hughes JR, Gibson JB, Patel GS, et al. Int22h-1/int22h-2-mediated Xq28 rearrangements: intellectual disability associated with duplications and in utero male lethality with deletions. J Med Genet 2011; 48: 840–50. [DOI] [PubMed] [Google Scholar]
- Friede R. Developmental neuropathology. Berlin: Springer; 1989. [Google Scholar]
- Jansen A, Andermann E.. Genetics of the polymicrogyria syndromes. J Med Genet 2005; 42: 369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen AC, Robitaille Y, Honavar M, Mullatti N, Leventer RJ, Andermann E, et al. The histopathology of polymicrogyria: a series of 71 brain autopsy studies. Dev Med Child Neurol 2016; 58: 39–48. [DOI] [PubMed] [Google Scholar]
- Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA.. Fumaric aciduria: clinical and imaging features. Ann Neurol 2000; 47: 583–8. [PubMed] [Google Scholar]
- Lee WS, Stephenson SE, Pope K, Gillies G, Maixner W, Macdonald-Laurs E, et al. Genetic characterisation identifies bottom-of-sulcus dysplasia as an mTORopathy. Neurology 2020; 95: e2542–e2551. [DOI] [PubMed] [Google Scholar]
- Lee WS, Stephenson SEM, Howell KB, Pope K, Gillies G, Wray A, et al. Second-hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann Clin Transl Neurol 2019; 6: 1338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T; Exome Aggregation Consortium, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventer RJ, Jansen A, Pilz DT, Stoodley N, Marini C, Dubeau F, et al. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain 2010; 133: 1415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventer RJ, Phelan EM, Coleman LT, Kean MJ, Jackson GD, Harvey AS.. Clinical and imaging features of cortical malformations in childhood. Neurology 1999; 53: 715–22. [DOI] [PubMed] [Google Scholar]
- Levine DN, Fisher MA, Caviness VS Jr.. Porencephaly with microgyria: a pathologic study. Acta Neuropathol 1974; 29: 99–113. [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Conti V, Timms AE, Smyser CD, Ahmed S, Carter M, et al. Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: a next-generation sequencing study. Lancet Neurol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch CA, Poduri A, Sahin M, Barry B, Walsh CA, Barkovich AJ.. Disorders of Microtubule Function in Neurons: Imaging Correlates. AJNR Am J Neuroradiol 2016; 37: 528–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema R, Cushion TD, Phelps IG, Chung SK, Dempsey JC, Collins S, et al. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum Mol Genet 2015; 24: 5313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reches A, Hiersch L, Simchoni S, Barel D, Greenberg R, Ben Sira L, et al. Whole-exome sequencing in fetuses with central nervous system abnormalities. J Perinatol 2018; 38: 1301–8. [DOI] [PubMed] [Google Scholar]
- Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL,. et al. ; Finding of Rare Disease Genes (FORGE) Canada Consortium. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 2012; 44: 934–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadedin SP, Dashnow H, James PA, Bahlo M, Bauer DC, Lonie A,. et al. ; Melbourne Genomics Health Alliance. Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med 2015; 7: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, et al. Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron 2018; 99: 905–13 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffs K, Verloo P, Brock S, Regal L, Beysen D, Ceulemans B, et al. Recurrent NEDD4L variant in periventricular nodular heterotopia, polymicrogyria and syndactyly. Front Genet 2020; 11: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutterd CA, Dobyns WB, Jansen A, Mirzaa G, Leventer RJ, Polymicrogyria overview In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. GeneReviews (R). Updated 2018 Aug 16 ed. Seattle (WA); 2018. [Google Scholar]
- Stutterd CA, Leventer RJ.. Polymicrogyria: a common and heterogeneous malformation of cortical development. Am J Med Genet 2014; 166: 227–39. [DOI] [PubMed] [Google Scholar]
- Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, Hennies HC, et al. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol 2006; 21: 32–5. [DOI] [PubMed] [Google Scholar]
- Vanmarsenille L, Giannandrea M, Fieremans N, Verbeeck J, Belet S, Raynaud M, et al. Increased dosage of RAB39B affects neuronal development and could explain the cognitive impairment in male patients with distal Xq28 copy number gains. Hum Mutat 2014; 35: 377–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.