

Abstract
The development of high blood pressure is influenced by genetic and environmental factors, with high salt intake being a known environmental contributor. Humans display a spectrum of sodium-sensitivity, with some individuals displaying a significant blood pressure rise in response to increased sodium intake while others experience almost no change. These differences are, in part, attributable to genetic variation in pathways involved in sodium handling and excretion. The epithelial sodium channel (ENaC) is one of the key transporters responsible for the reabsorption of sodium in the distal nephron. This channel has an important role in the regulation of extracellular fluid volume and consequently blood pressure. Herein we review the role of ENaC in the development of salt-sensitive hypertension, and present mechanistic insights into the regulation of ENaC activity and how it may accelerate sodium-induced damage and dysfunction. We discuss the traditional role of ENaC in renal sodium reabsorption and review work addressing ENaC expression and function in the brain, vasculature and immune cells, and how this has expanded the implications for its role in the initiation and progression of salt-sensitive hypertension.
Keywords: ENaC, salt sensitivity, blood pressure, sodium
Graphical Abstract
Introduction
Blood pressure (BP) is a multifactorial metric, dependent upon complex interactions between environmental, genetic, and demographic factors. One environmental factor known to influence BP is sodium (Na+) intake. Studies have shown increased Na+ raises BP, while a reduction can lower BP in hypertensives1. However, there is a continuum of BP sensitivity to Na+ within the human population. Variability in Na+ sensitivity reflects, in part, gene variances related to the renin-angiotensin-aldosterone system and renal Na+ transporters2. Immune cells3, vascular endothelium and smooth muscle4, and specific regions of the central nervous system (CNS) also contribute to salt-sensing variability. In this review, we discuss how epithelial sodium channels (ENaCs) contribute to the salt-sensitivity of BP, both in the kidney and beyond.
ENaC’s role in the kidney
The kidney is largely responsible for Na+ homeostasis. While the majority of filtered Na+ is reabsorbed in the proximal tubule, the distal nephron, comprised of the distal convoluted tubule, connecting tubule and collecting duct, absorbs 6–10% of filtered Na+, and is where the nephron fine-tunes the amount excreted in urine5. This distal segment is a key site of hormone-regulated Na+ absorption, as principal cells in the distal nephron respond to aldosterone by increasing ENaC apical membrane expression and open probability6. Other hormones such as vasopressin, angiotensin II and insulin also modulate ENaC activity, with their contributions being implicated in Na+ retention in pathophysiological states6. ENaCs are composed of three structurally-related subunits termed α, β, and γ7. While a δ-subunit can replace the α-subunit to form functional channels in non-renal tissues8, it is not expressed in rodents and its functional role is not well understood.
ENaC subunits have large extracellular domains that sense external environmental factors that modulate channel activity7. Each extracellular domain is connected to two transmembrane domains that form the channel pore and gate, and are in turn connected to short intracellular termini7. A number of factors influence channel gating and open probability (summarized in fig. 1A), including α− and γ-subunit proteolytic processing, β− and γ-subunit palmitoylation, intracellular and extracellular Na+, extracellular protons and chloride, acidic phospholipids, and shear stress9. A His-Gly (HG) motif on the cytoplasmic N-termini also influences gating10. In addition to gating and open probability, ENaC is modulated by the number of channels at the membrane. Ubiquitination of cytoplasmic Lys residues on surface-resident channels targets them for internalization and degradation11,12. This process is regulated by the ubiquitin ligase Nedd4–2 that binds to the cytoplasmic C-termini of subunits at a Pro-Tyr (PY) motif12. Recent in vivo work has shown that only the α- and γ-subunits are ubiquitinated, and that the modification favors the cleaved, mature subunits11. A number of protein kinases, including PKA, PKC, ERK1/2, Sgk1, CK2 and MTS3 regulate ENaC and their dysregulation may contribute to hypertension6,13.
Figure 1. Regulation of ENaC in the normal and salt-sensitive kidney.

The top panel, A, illustrates several mechanisms responsible for ENaC regulation in the kidney. ENaC open probability and activity is regulated by shear stress, proteolytic cleavage, palmitoylation (indicated by blue cytoplasmic arrowheads), and extracellular [Na+]. Nedd4–2 facilitates ENaC ubiquitination and endocytosis. Aldosterone activates MR, increasing transcription of αENaC and the regulatory kinase Sgk1. However, in the salt sensitive kidney, panel B, MR is activated by the GTPase Rac1, independent of aldosterone. Rac1 also increases ROS production via NOX, which activates ENaC. Increased proteolytic activation of ENaC has also been reported, however, further work is needed to establish which proteases may be responsible.
While ENaC does not directly couple Na+ movement to other ions, it indirectly influences renal K+ and H+ transport. Na+ reabsorption by ENaC increases the driving force for K+ secretion. These effects explain why the ENaC inhibitors amiloride and triamterene, which work by blocking the channel pore14, also inhibit renal K+ secretion and can lead to hyperkalemia. The activity of other transporters also affects ENaC activity. The Na+-Cl- cotransporter (NCC) is located in the distal convoluted tubule, both upstream of ENaC and in cells where ENaC and NCC are co-expressed. Increased NCC-mediated Na+ absorption reduces Na+ delivery to ENaC15. Direct interaction between the two proteins exists and is increased by aldosterone16. The Cl--HCO3- exchanger pendrin also influences ENaC activity17.
Mutations in genes encoding ENaC subunits (SCNN1A, SCNN1B, and SCNN1G) are associated with monogenic hyper- or hypotensive disorders, underscoring the importance of ENaC in BP regulation. Liddle syndrome is an autosomal dominant disorder associated with ENaC gain-of-function mutations, characterized by early-onset hypertension, hypokalemia and metabolic alkalosis. ENaC mutations associated with Liddle syndrome are found in the genes encoding the β− or γ-subunits12,18. These mutations disrupt the PY motif and impair channel ubiquitination, internalization from the cell surface, and degradation, resulting in increased surface expression and activity6,12. Conversely, pseudohypoaldosteronism type 1 (PHA-1) is an autosomal recessive disorder associated with ENaC loss-of-function mutations, resulting in severe neonatal salt wasting, volume depletion and hyperkalemia. Both frameshift mutations resulting in premature stop codons and missense mutations resulting in a reduction or loss of channel activity have been associated with this disorder, including mutations in the HG motif10,18.
Mutations at other sites affect channel activity as well. Numerous mutations within the α− or γ-subunits modulate activity by changing the Na+ self-inhibition response, where Na+ binds to defined extracellular sites within ENaC and dampens channel open probability9. In this regard, a gain-of-function α-subunit mutation (C479R) was noted in siblings with a mild Liddle syndrome phenotype that exhibited decreased Na+ self-inhibition19,20.
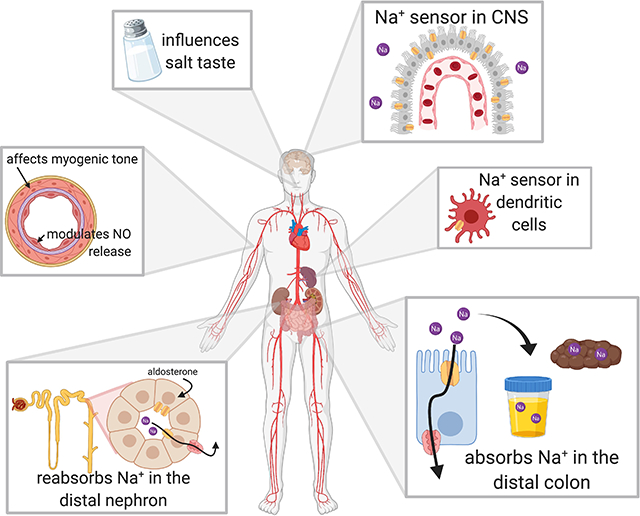
While extreme phenotypes are seen with variants associated with large changes in ENaC activity, the contributions of other channel variants to more subtle BP phenotypes, including salt-sensitive hypertension, is still being unraveled. Inhibitors of renal Na+ transporters are used to treat specific ENaC and NCC gain-of-function disorders. The fact that dietary intervention can partially alleviate specific severe monogenic BP disorders strengthens the idea of a genetic-dietary interaction21. Understanding ENaC’s role in modulating BP sensitivity to environmental factors is critical for identifying individuals who should respond to ENaC-blocking therapies and dietary modulation. A growing body of evidence suggests that ENaC has a role in BP control beyond the kidney, including in other epithelia (lingual epithelium (salt taste) and distal colon (absorption of ingested Na+)), the vasculature, brain and immune system.
Genetic studies linking ENaC to salt-sensitivity and BP
The expanding number of human whole-genome and whole-exome sequences have resulted in identification of thousands of variants within the four genes encoding the ENaC subunits. A few common non-synonymous mutations (e.g., αT663A and αA334T) were identified that alter channel function in heterologous expression systems but generally fail to associate with BP differences in humans22,23. However, the majority of variants are present at low frequency (mean allele frequency <0.05), making analysis of their contribution to specific phenotypes difficult. A β-subunit variant (T549M) in individuals of African ancestry was noted to occur more frequently in hypertensives24, although this is not a consistent finding25. Another variant, βR563Q, was associated with hypertension in black and mixed-ancestry South Africans26. However, its presence in ~20% of the San people who maintain a rural lifestyle with low dietary Na+ intake was not associated with hypertension27. These observations suggest that some ENaC mutations may drive salt-sensitive hypertension, but are tolerated when dietary salt is scarce.
The GenSalt study provided additional evidence of the association of ENaC variants with salt-sensitivity. Carried out in rural northern China, study participants received a 7-day low Na+ intervention (3g of salt/day) followed by a 7-day high Na+ intervention (18g of salt/day) while monitoring BP28. Multiple ENaC single nucleotide polymorphisms were associated with BP variation in response to Na+ manipulation29. Additional non-synonymous single nucleotide variants were identified that increased or decreased ENaC activity in an oocyte expression system, although these functional variants did not significantly correlate with salt-sensitivity in humans30. As these were primarily low frequency variants, a larger population may be necessary to demonstrate significant association. Compensatory mechanisms might also mask the effects of ENaC variants in these individuals. Resequencing of the 300 most salt-sensitive and salt-resistant participants identified carriers of SCNN1A rare variants as having a 0.52 decreased odds of salt-sensitivity as compared to noncarriers, while SCNN1B and SCNN1G rare variants were not associated with salt-sensitivity31.
As outlined above, genetic data support ENaC as a contributor to salt-sensitivity. However, the question remains whether therapeutically targeting ENaC could lower BP in salt-sensitive individuals. ENaC-blockers are rarely prescribed as first-line antihypertensive medications, and relatively few trials have systematically addressed their efficacy32. Amiloride might be most beneficial when administered in a targeted manner. Indeed, when black hypertensives expressing the βT594M variant were taken off their antihypertensive regimen and administered only amiloride, BP was controlled effectively to the same level33. A recent study showed amiloride could blunt the BP response to Na+ loading in a Nigerian population34. Furthermore, an individual with an αC479R variant displaying a modest Liddle phenotype had BP reduction with an ENaC blocker19. Together, these studies provide evidence that ENaC inhibitors lower BP in specific populations and individuals bearing specific ENaC variants.
A number of human gain-of-function variants increase channel activity by suppressing Na+ self-inhibition, and it is likely that additional human gain-of-function variants will be identified9. Future studies will determine whether hypertensive individuals with gain-of-function ENaC variants, identified through genetic and functional screens, respond to ENaC blockers with improved BP.
ENaC’s role in salt-sensitive rodent models
While genetic studies have shown ENaC’s association with salt-sensitive hypertension in certain groups, animal studies and cell culture data provide mechanistic insight into why ENaC may facilitate salt-sensitive hypertension. In a comparison of salt-resistant versus salt-sensitive rat strains, renal ENaC showed dysregulation with high salt diet (HSD). While Na+-loaded salt-resistant strains displayed decreased ENaC in accordance with lower aldosterone, Dahl salt-sensitive rats (Dahl-SS) had a paradoxical increase in ENaC mRNA and protein as well as increased ENaC activity35,36. Dahl-SS rats experienced increased BP within the first week of HS. This was attenuated by amiloride or benzamil, suggesting the increased ENaC expression was, in part, responsible for the salt-induced hypertension35, 36. Additionally, while Dahl-SS rats had low renin levels with HSD, there was a paradoxical activation of the mineralocorticoid receptor (MR), demonstrated by increased expression of the downstream target Sgk137. This hormone-independent activation was shown to be mediated by the small GTPase Rac1, allowing for increased ENaC expression without higher aldosterone38. These observations suggest that ENaC activation through dysregulation of MR signaling contributes to salt-sensitive hypertension in Dahl-SS rats39. Rac1 also serves as a structural unit of NADPH oxidases (NOX), protein complexes responsible for generation of reactive oxygen species (ROS)39. ROS levels have been shown to be elevated in salt-sensitive rats and hypertensive patients, and its production, specifically by NOX4, can positively regulate ENaC activity in the kidney, potentially through mechanisms dependent on PI3-kinase or prostaglandins40 (fig. 1B).
ENaC α- and γ-subunits are activated by cleavage9. The serine protease furin cleaves the α-subunit twice as it traffics through the trans-Golgi network, releasing an inhibitory tract and transitioning channels from a low- to moderate-activity state. The γ-subunit is cleaved once by furin, and cleavage by another protease, such as prostasin, matriptase, kallikrein, plasmin, or elastase, releases a second inhibitory tract, inducing a high-activity state9. While it is still unclear which additional proteases are responsible for cleaving the γ-subunit in the kidney, ENaC proteolysis likely has a role in channel activation in Dahl-SS rats. Dahl-SS rats fed a HSD have enhanced γ-subunit proteolysis, and the serine protease inhibitor camostat mesylate attenuated the rise in BP35,36.
ENaC proteolysis is seen in other models of Na+ retention and hypertension, strengthening the idea this process plays a role in salt-sensitivity. Aldosterone administration is associated with ENaC proteolytic processing, channel activation, and renal Na+ retention41. Proteolytic activation of ENaC is also observed in nephrotic syndrome42. Altogether, while ENaC proteolytic activation is an important channel regulator, further study is needed to understand how it contributes to salt-sensitivity. Other ENaC regulatory mechanisms, such as palmitoylation, phosphorylation and sensitivity to intracellular and extracellular Na+ (discussed above) may also influence salt-sensitivity9. Additionally, while ENaC has a role in the development of salt-sensitive hypertension in specific settings, other transporters and channels involved in Na+ or K+ homeostasis have also been implicated in this phenotype15, as well as signaling pathways ranging from immune activation, nitric oxide (NO) and ROS production, and mTOR-mediated signaling3,4. More work is needed to understand how these signaling pathways implicated in salt-sensitive hypertension may affect ENaC function.
ENaCs contribute to salt-sensitive vascular dysfunction
Salt-mediated dysfunction extends beyond the kidney. Endothelial cells (ECs) are well-known BP regulators, sensing and responding to mechanical stretch and chemical alterations in the blood. ECs translate a mechanical stimulus (i.e., changes in shear stress) to a chemical signal by regulating production of the gaseous vasodilator NO43. Numerous studies have observed that high salt intake or increased aldosterone levels lead to vascular dysfunction, defined by decreased NO production, cellular stiffening, and pressure-independent vascular remodeling44,45. A growing body of evidence suggests endothelial ENaC (EnENaC) has a role in promoting this dysfunction.
EnENaC expression is regulated by aldosterone46. ECs or ex vivo vessel preparations cultured with aldosterone and a high [Na+] developed resistance to mechanical deformation that correlated with increased ENaC surface expression. This was attenuated with the MR antagonist spironolactone or amiloride47,48. The cortical actin cytoskeleton, a meshwork of filaments located within 50–200nm of the cell membrane, appears to mediate this stiffening49. ENaC influences this cytoskeletal architecture by increasing local [Na+], which stabilizes polymerized actin and leads to a denser mesh50. ENaC may also interact with actin directly to stabilize the network51. This stiffening impairs NO production by rendering the cell less sensitive to shear stress49. An increase in the filamentous (F) to globular (G) actin ratio also facilitates interactions between actin filaments and endothelial nitric oxide synthase (eNOS), limiting its ability to convert L-arginine to NO52 (Fig. 2). ENaC inhibition in isolated rat mesenteric vessels led to higher NO production and increased eNOS phosphorylation at Ser1177 (an activating event), providing further proof that this pathway modulates NO availability and vasodilation53.
Figure 2. EnENaC limits NO production in the setting of high [Na+] and aldosterone.

ECs have a thick surface glycocalyx that limits the availability of Na+ for ENaC-dependent Na+ transport. The glycocalyx also enhances the endothelial response to shear stress, inducing an increase in intracellular Ca2+, activation of eNOS, NO production and vasodilation. Increased extracellular [Na+] leads to glycocalyx breakdown and enhanced Na+ entry via ENaC. An increase in the intracellular [Na+] stabilizes F-actin filaments, increasing the density of the cortical cytoskeleton and limiting cellular deformation. This events inhibit eNOS and NO production, leading to vascular dysfunction. Activation of MR by aldosterone in ECs increases ENaC expression, stabilizing the cytoskeleton and reducing deformability.
While renal ENaC is inhibited by increases in intracellular or extracellular [Na+]9, studies in cultured ECs and ex vivo vessel preparations suggest that EnENaC is activated by high extracellular (and presumably intracellular) [Na+]54, a feed-forward activation process. In contrast, in vivo studies where rats were fed a HSD showed a decrease in EnENaC activity over the first 7–14 days but a return to normal after 21 days55. These results suggest that EnENaC regulation varies with acute versus chronic HSD, and that whole animal studies may have circulating regulatory factors not matched in ex vivo studies.
While Na+ may affect ENaC levels directly, increased Na+ may also damage the endothelial glycocalyx, a negatively charged layer of glycoprotein polysaccharides that protects ECs and mediates shear stress responsiveness56. The heparan sulfate component of the glycocalyx was significantly reduced in the presence of high Na+ 57, and removal of this in isolated ECs with heparinase resulted in increased intracellular [Na+] and transendothelial resistance58. These observations suggest increased extracellular Na+ may enhance both EnENaC surface expression and accessibility to extracellular Na+ (Fig. 2).
Long-term administration of low-dose amiloride suggested that EnENaC is involved in high fat diet induced vascular dysfunction in female mice. Amiloride protected against high fat-induced aortic stiffening and small vessel dysfunction, without altering BP59. Studies with endothelial-specific αENaC knockout mice (EC-αENaC KO) support EnENaC’s contribution to vascular stiffening, as these mice had a blunted rise in pulse wave velocity with aldosterone when compared to controls60. In addition, EC-αENaC KO lacked an aldosterone-dependent reduction in acetylcholine-induced vasodilation seen in controls. While EC-αENaC KOs did show a resistance to vascular stiffening consistent with pharmacological studies, discrepancy exists on the effects of pharmacological versus genetic inhibition of ENaC on acetylcholine-induced NO production. Blocking ENaC with benzamil in mesenteric arteries caused a decrease in acetylcholine-induced NO production, whereas genetic deletion of the α-subunit did not affect NO production61. These results were surprising, as previous experiments, discussed above, suggested inhibition of EnENaC should increase NO. Despite this discrepancy, both pharmacological treatment and genetic deletion caused a decrease in flow-mediated vasodilation, suggesting ENaC functions as a mechanosensor in vessels61,62.
ENaCs are also expressed in vascular smooth muscle (VSM) where they participate in regulating the myogenic response, a flow-induced reaction intrinsic to VSM that controls perfusion and blood flow characterized by vasoconstriction in response to increasing perfusion pressure. In isolated, perfused mouse renal arteries, ENaC inhibition through multiple means abolished pressure-induced vasoconstriction63,64. This was also observed in cerebral vessels65. However, there is discrepancy regarding which subunits are expressed across these vessels. In mouse renal interlobar arteries, mRNA and protein expression for only the β- and γ-subunits was observed within freshly dissociated cells63. All three subunits were found in freshly dissociated rat mesenteric VSM cells, and HSD lowered the expression of the α- and γ-subunits without affecting overall expression of the β-subunit66. While some studies have suggested β- and γ-subunits can form functional channels67, an intriguing alternative is that β- and γ-subunits are heteromultimerizing with structurally related polypeptides, such as acid-sensing ion channel (ASIC) subunits. Further work is needed to understand if this occurs, and if these heteromultimeric channels are functional. Overall, while ENaC has a role in the myogenic response, additional studies are needed to define the role of VSM ENaC in the development of salt-sensitive hypertension.
Does ENaC in the brain sense Na+ and contribute to hypertension?
Na+ levels in the CNS influence BP. In rats, BP increases are observed when cerebrospinal fluid (CSF) [Na+] is increased. Furthermore, rats given a HSD experience increases in CSF [Na+] in salt-sensitive strains, but not resistant strains, and this increase precedes BP changes by several days68. Central aldosterone infusion also elicits a BP increase69, and Dahl-SS rats have higher central aldosterone synthesis compared to Sprague-Dawley rats70. Moreover, the increase in BP with a HSD can be prevented via intracerobroventricular (ICV) infusion of spironolactone or benzamil, suggesting the MR and ENaC have roles in Na+-sensing71.
ENaC is expressed in areas of the brain related to fluid and electrolyte balance, including areas that participate in the control of thirst, Na+ excretion, blood volume regulation, and vasopressin secretion72. These sites also express MR72 and proteins that modulate ENaC activity (e.g., Sgk1 and Nedd4–2)73, 74. ICV infusion of Na+-enriched artificial CSF produces elevations in hypothalamic aldosterone levels, attributable to local production75.
Increases in CSF [Na+] by as little as 2mM lead to increased neuronal firing rates, sympathetic activity, and hypertension68. Interestingly, these changes are observed mainly in salt-sensitive versus salt-resistant rats76. In magnocellular neurosecretory cells of the paraventricular and supraoptic nuclei, ENaC appears to mediate a Na+ leak current that affects steady-state cell membrane potential77. Baseline ENaC expression within this region was higher in Dahl-SS as compared to Sprague-Dawley rats and increased further with HSD. Additionally, ENaC appeared in dendrites of these neurons only in Dahl-SS, suggesting ENaC could provide the means for abnormally depolarized local membrane potentials that enhance firing and vasopressin release, contributing to salt-sensitivity78.
Choroid plexus cells uniquely express ENaC on both the basolateral and apical membranes, with the latter expressing channels on small microvilli which extend into the CSF79. In stroke prone versus Wistar Kyoto rats, the choroid plexus displays higher levels of both Sgk1 and ENaC under unstimulated conditions, suggesting baseline MR activation. This increase in expression is enhanced with HSD and presumably allows for greater Na+ transport into CSF, as [Na+] increased only in the stroke prone rats80. Choroid plexus ENaC activity may determine whether increased dietary Na+ affects overall CSF [Na+], influencing whether salt leads to an increase in sympathetic activity.
Studies using ENaC inhibitors provide evidence of ENaC’s role in the central response to HSD, and that the channel can perhaps sense small increases in the extracellular [Na+]. How ENaC affects signaling pathways in response to small increases in CSF [Na+] is unclear. ENaC currents mediated by αβγ channels saturate at a [Na+] well below 140mM81. As ENaCs with non-traditional subunit compositions lack this saturation, it will be important to define the subunit composition and behavior of ENaCs at different CNS sites. Inhibitors can have off-target effects, and rodent models with genetic manipulation of ENaC in specific neural regions are needed to confirm ENaC’s role in CNS Na+-sensing.
ENaCs expressed in immune cells sense Na+ and contribute to inflammation and hypertension
Dendritic cells (DCs) classically process and present antigens within the grooves of major histocompatibility complexes (MHCs) classes I and II. A novel activation of DCs by high salt has recently been described, where Na+ enters DCs through ENaC. An increase in the intracellular [Na+] facilitates calcium influx via the Na+-Ca2+ exchanger, leading to activation of protein kinase C and NADPH oxidase, an increase in superoxide, and protein modification by ketoaldehydes, termed isolevuglandin (isoLG)-protein adducts. These isoLGs incite an autoimmune-like reaction with release of cytokines and an increase in BP82, 83 (Fig. 3).
Figure 3. ENaC-dependent Na+ transport promotes DC activation and inflammation.

ENaC-dependent Na+ transport in DCs triggers an influx of Ca2+ via the Na+-Ca2+ exchanger (NCX), leading to phosphorylation of the p47phox subunit of NOX, facilitating NOX assembly and production of ROS, and creation of IsoLG adducts. DCs with IsoLG-modified surface proteins activate T cells, leading to release of pro-inflammatory cytokines interferon gamma, and IL-1782.
The initial Na+ influx appears to be mediated by ENaC formed by α- and γ-subunits, as β-subunits are not expressed in DCs82, but δ-subunits may contribute to functional channels in human antigen presenting cells. Sgk1 is expressed in DCs and promotes subunit assembly under conditions of high Na+ 84. When DCs primed with high salt are transferred to a naïve animal, hypertension in response to suppressor doses of angiotensin II develops, suggesting that the inflammatory state produced by high salt activation of DCs is sufficient to promote a hypertensive phenotype, and that immune cell ENaC activation has a role in the development of salt-sensitive hypertension.
Exposure of B and T lymphocytes to a high extracellular [Na+] also drives a pro-inflammatory phenotype85, 86, characterized by increased IL-17A that promotes renal and vascular dysfunction87. While ENaC expression has been shown in these cells, the roles of ENaC and other Na+ transporters in mediating Na+ entry have not been determined. Therefore, further work is needed to understand ENaC’s function across different lymphocyte populations.
Conclusions
Guyton’s seminal works described hypertension as a disease of disturbed volume balance and altered pressure-natriuresis response. In this model, ENaC was a perfect culprit for promoting hypertension, given that increased Na+ reabsorption through the channel would lead to increased extracellular volume and ultimately require an increased BP set point to restore volume homeostasis. However, our view of blood pressure control has evolved to include a role for multiple organ systems, in parallel with a growing knowledge of ENaC’s expression in non-epithelial tissues. In addition to its role in the kidney, ENaC acts as a Na+ transporter and a Na+-sensor and/or mechanotransducer in neurons, ECs, VSM and DCs. This positions ENaC as a player in multiple aspects of salt-sensitive hypertension, including inflammatory cytokine release, vascular stiffening, and central activation of sympathetic tone.
Given its involvement in promoting Na+-induced dysfunction across a variety of cell types, ENaC is an attractive pharmacological target for combatting the adverse BP effects of a HSD. While amiloride is an ENaC inhibitor used as a diuretic in the clinical setting, only low doses are needed to reach sufficient concentrations in the lumen of the nephron, preventing therapeutic levels from being achieved in the circulation. Further work is needed to develop ENaC inhibitors that selectively target the channel in non-epithelial cells. The adverse anti-kaliuretic effects of ENaC blockers that limit their therapeutic use must be overcome. While more work is needed to understand the channel’s role in normal and pathophysiologic states in non-epithelial tissues, the combined results of the studies detailed here strongly support ENaC’s role as a mediator of Na+-sensitive hypertension.
Acknowledgments
Figures were created with biorender.com.
Sources of Funding
This work was supported by grants R01 HL147818 (TRK and AK), K01 HL130497 (AK), P30 DK079307 (TRK) and T32 DK007052 (SMM) from the National Institutes of Health. SMM was supported by a postdoctoral fellowship award from Relypsa, Inc.
Footnotes
Disclosure
None
References
- 1.Blumenthal JA, Sherwood A, Smith PJ and Hinderliter A. The Role of Salt Reduction in the Management of Hypertension. J Am Coll Cardiol 2018;71:1597–1598. [DOI] [PubMed] [Google Scholar]
- 2.Kelly TN and He J. Genomic epidemiology of blood pressure salt sensitivity. J Hypertens 2012;30:861–73. [DOI] [PubMed] [Google Scholar]
- 3.Pitzer AL, Van Beusecum JP, Kleyman TR and Kirabo A. ENaC in Salt-Sensitive Hypertension: Kidney and Beyond. Curr Hypertens Rep 2020;22:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mutchler SM and Kleyman TR. New insights regarding epithelial Na+ channel regulation and its role in the kidney, immune system and vasculature. Curr Opin Nephrol Hypertens. 2019;28:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer LG and Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 2015;10:676–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM and Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 2015;10:135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noreng S, Bharadwaj A, Posert R, Yoshioka C and Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife. 2018;7:e39340. doi: 10.7554/eLife.39340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giraldez T, Rojas P, Jou J, Flores C and Alvarez de la Rosa D. The epithelial sodium channel delta-subunit: new notes for an old song. Am J Physiol Renal Physiol 2012;303:F328–38. [DOI] [PubMed] [Google Scholar]
- 9.Kleyman TR and Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol 2020;318:C150–C162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP and Rossier BC. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 1997;16:899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frindt G, Bertog M, Korbmacher C and Palmer LG. Ubiquitination of renal ENaC subunits in vivo. Am J Physiol Renal Physiol 2020;318:F1113–F1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamynina E and Staub O. Concerted action of ENaC, Nedd4–2, and Sgk1 in transepithelial Na(+) transport. Am J Physiol Renal Physiol 2002;283:F377–87. [DOI] [PubMed] [Google Scholar]
- 13.Lu TJ, Kan WC, Yang SS, Jiang ST, Wu SN, Ling P, Bao BY, Lin CY, Yang ZY, Weng YP, Chan CH and Lu TL. MST3 is involved in ENaC-mediated hypertension. Am J Physiol Renal Physiol 2019;317:F30–F42. [DOI] [PubMed] [Google Scholar]
- 14.Kashlan OB, Sheng S and Kleyman TR. On the interaction between amiloride and its putative alpha-subunit epithelial Na+ channel binding site. J Biol Chem 2005;280:26206–15. [DOI] [PubMed] [Google Scholar]
- 15.Hoorn EJ, Gritter M, Cuevas CA and Fenton RA. Regulation of the Renal NaCl Cotransporter and Its Role in Potassium Homeostasis. Physiol Rev 2020;100:321–356. [DOI] [PubMed] [Google Scholar]
- 16.Wynne BM, Mistry AC, Al-Khalili O, Mallick R, Theilig F, Eaton DC and Hoover RS. Aldosterone Modulates the Association between NCC and ENaC. Sci Rep 2017;7:4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wall SM, Verlander JW and Romero CA. The Renal Physiology of Pendrin-Positive Intercalated Cells. Physiol Rev 2020;100:1119–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanukoglu I and Hanukoglu A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579:95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salih M, Gautschi I, van Bemmelen MX, Di Benedetto M, Brooks AS, Lugtenberg D, Schild L and Hoorn EJ. A Missense Mutation in the Extracellular Domain of alphaENaC Causes Liddle Syndrome. J Am Soc Nephrol 2017;28:3291–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Chen J, Shi S, Sheng S and Kleyman TR. Analyses of epithelial Na(+) channel variants reveal that an extracellular beta-ball domain critically regulates ENaC gating. J Biol Chem 2019;294:16765–16775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enslow BT, Stockand JD and Berman JM. Liddle’s syndrome mechanisms, diagnosis and management. Integr Blood Press Control 2019;12:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong Q, Menon AG and Stockand JD. Functional polymorphisms in the alpha-subunit of the human epithelial Na+ channel increase activity. Am J Physiol Renal Physiol 2006;290:F821–7. [DOI] [PubMed] [Google Scholar]
- 23.Wang XF, Lu XM, Lin RY, Wang SZ, Zhang LP, Qian J, Lu DR, Wen H and Jin L. Lack of association of functional variants in alpha-ENaC gene and essential hypertension in two ethnic groups in China. Kidney Blood Press Res 2008;31:268–73. [DOI] [PubMed] [Google Scholar]
- 24.Baker EH, Dong YB, Sagnella GA, Rothwell M, Onipinla AK, Markandu ND, Cappuccio FP, Cook DG, Persu A, Corvol P, Jeunemaitre X, Carter ND and MacGregor GA. Association of hypertension with T594M mutation in beta subunit of epithelial sodium channels in black people resident in London. Lancet. 1998;351:1388–92. [DOI] [PubMed] [Google Scholar]
- 25.Nkeh B, Samani NJ, Badenhorst D, Libhaber E, Sareli P, Norton GR and Woodiwiss AJ. T594M variant of the epithelial sodium channel beta-subunit gene and hypertension in individuals of African ancestry in South Africa. Am J Hypertens 2003;16:847–52. [DOI] [PubMed] [Google Scholar]
- 26.Jones ES, Owen EP, Davidson JS, Van Der Merwe L and Rayner BL. The R563Q mutation of the epithelial sodium channel beta-subunit is associated with hypertension. Cardiovasc J Afr 2011;22:241–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones E and Rayner B. The importance of the epithelial sodium channel in determining salt sensitivity in people of African origin. Pediatr Nephrol 2020:doi: 10.1007/s00467-019-04427-z [DOI] [PubMed] [Google Scholar]
- 28.GenSalt Collaborative Research G. GenSalt: rationale, design, methods and baseline characteristics of study participants. J Hum Hypertens 2007;21:639–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Yang X, Mo X, Huang J, Chen J, Kelly TN, Hixson JE, Rao DC, Gu CC, Shimmin LC, Chen J, Rice TK, Li J, Schwander K, He J, Liu DP and Gu D. Associations of epithelial sodium channel genes with blood pressure: the GenSalt study. J Hum Hypertens 2015;29:224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray EC, Chen J, Kelly TN, He J, Hamm LL, Gu D, Shimmin LC, Hixson JE, Rao DC, Sheng S and Kleyman TR. Human epithelial Na+ channel missense variants identified in the GenSalt study alter channel activity. Am J Physiol Renal Physiol. 2016;311:F908–F914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu X, Gu D, He J, Rao DC, Hixson JE, Chen J, Li J, Huang J, Wu X, Rice TK, Shimmin LC and Kelly TN. Resequencing Epithelial Sodium Channel Genes Identifies Rare Variants Associated With Blood Pressure Salt-Sensitivity: The GenSalt Study. Am J Hypertens 2018;31:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heran BS, Chen JM, Wang JJ and Wright JM. Blood pressure lowering efficacy of potassium-sparing diuretics (that block the epithelial sodium channel) for primary hypertension. Cochrane Database Syst Rev 2012;11:CD008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker EH, Duggal A, Dong Y, Ireson NJ, Wood M, Markandu ND and MacGregor GA. Amiloride, a specific drug for hypertension in black people with T594M variant? Hypertension. 2002;40:13–7. [DOI] [PubMed] [Google Scholar]
- 34.Elias SO, Sofola OA and Jaja SI. Epithelial sodium channel blockade and new beta-ENaC polymorphisms among normotensive and hypertensive adult Nigerians. Clin Exp Hypertens 2019;41:144–151. [DOI] [PubMed] [Google Scholar]
- 35.Pavlov TS, Levchenko V, O’Connor PM, Ilatovskaya DV, Palygin O, Mori T, Mattson DL, Sorokin A, Lombard JH, Cowley AW, Jr. and Staruschenko A. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24:1053–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S and Tomita K. Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens 2009;27:1679–89. [DOI] [PubMed] [Google Scholar]
- 37.Aoi W, Niisato N, Sawabe Y, Miyazaki H, Tokuda S, Nishio K, Yoshikawa T and Marunaka Y. Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt-sensitively hypertensive rats. Cell Biol Int 2007;31:1288–91. [DOI] [PubMed] [Google Scholar]
- 38.Nagase M and Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol 2013;9:86–98. [DOI] [PubMed] [Google Scholar]
- 39.Pavlov TS and Staruschenko A. Involvement of ENaC in the development of salt-sensitive hypertension. Am J Physiol Renal Physiol 2017;313:F135–F140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavlov TS, Palygin O, Isaeva E, Levchenko V, Khedr S, Blass G, Ilatovskaya DV, Cowley AW, Jr. and Staruschenko A. NOX4-dependent regulation of ENaC in hypertension and diabetic kidney disease. FASEB J. 2020;34:13396–13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frindt G and Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol 2015;308:F572–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C and Skott O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 2009;20:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Resnick N, Yahav H, Shay-Salit A, Shushy M, Schubert S, Zilberman LC and Wofovitz E. Fluid shear stress and the vascular endothelium: for better and for worse. Prog Biophys Mol Biol 2003;81:177–99. [DOI] [PubMed] [Google Scholar]
- 44.Fels J, Oberleithner H and Kusche-Vihrog K. Menage a trois: aldosterone, sodium and nitric oxide in vascular endothelium. Biochim Biophys Acta 2010;1802:1193–202. [DOI] [PubMed] [Google Scholar]
- 45.Pimenta E, Gordon RD and Stowasser M. Salt, aldosterone and hypertension. J Hum Hypertens 2013;27:1–6. [DOI] [PubMed] [Google Scholar]
- 46.Kusche-Vihrog K, Sobczak K, Bangel N, Wilhelmi M, Nechyporuk-Zloy V, Schwab A, Schillers H and Oberleithner H. Aldosterone and amiloride alter ENaC abundance in vascular endothelium. Pflugers Arch 2008;455:849–57. [DOI] [PubMed] [Google Scholar]
- 47.Druppel V, Kusche-Vihrog K, Grossmann C, Gekle M, Kasprzak B, Brand E, Pavenstadt H, Oberleithner H and Kliche K. Long-term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J. 2013;27:3652–9. [DOI] [PubMed] [Google Scholar]
- 48.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ and Sowers JR. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ Res 2016;118:935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fels J, Jeggle P, Kusche-Vihrog K and Oberleithner H. Cortical actin nanodynamics determines nitric oxide release in vascular endothelium. PloS one. 2012;7:e41520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oda T, Makino K, Yamashita I, Namba K and Maeda Y. Distinct structural changes detected by X-ray fiber diffraction in stabilization of F-actin by lowering pH and increasing ionic strength. Biophysical J. 2001;80:841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mazzochi C, Benos DJ and Smith PR. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am J Physiol Renal Physiol 2006;291:F1113–22. [DOI] [PubMed] [Google Scholar]
- 52.Su Y, Edwards-Bennett S, Bubb MR and Block ER. Regulation of endothelial nitric oxide synthase by the actin cytoskeleton. Am J Physiol Cell Physiol 2003;284:C1542–9. [DOI] [PubMed] [Google Scholar]
- 53.Perez FR, Venegas F, Gonzalez M, Andres S, Vallejos C, Riquelme G, Sierralta J and Michea L. Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3-kinase/Akt in small-diameter mesenteric arteries. Hypertension. 2009;53:1000–7. [DOI] [PubMed] [Google Scholar]
- 54.Korte S, Strater AS, Druppel V, Oberleithner H, Jeggle P, Grossmann C, Fobker M, Nofer JR, Brand E and Kusche-Vihrog K. Feedforward activation of endothelial ENaC by high sodium. FASEB J. 2014;28:4015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu HB, Zhang J, Sun YY, Li XY, Jiang S, Liu MY, Shi J, Song BL, Zhao D, Ma HP and Zhang ZR. Dietary salt regulates epithelial sodium channels in rat endothelial cells: adaptation of vasculature to salt. Br J Pharmacol 2015;172:5634–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alphonsus CS and Rodseth RN. The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia. 2014;69:777–84. [DOI] [PubMed] [Google Scholar]
- 57.Oberleithner H, Peters W, Kusche-Vihrog K, Korte S, Schillers H, Kliche K and Oberleithner K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch 2011;462:519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korte S, Wiesinger A, Straeter AS, Peters W, Oberleithner H and Kusche-Vihrog K. Firewall function of the endothelial glycocalyx in the regulation of sodium homeostasis. Pflugers Arch 2012;463:269–78. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Lemus LA, Aroor AR, Ramirez-Perez FI, Jia G, Habibi J, DeMarco VG, Barron B, Whaley-Connell A, Nistala R and Sowers JR. Amiloride Improves Endothelial Function and Reduces Vascular Stiffness in Female Mice Fed a Western Diet. Front Physiol 2017;8:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jia G, Habibi J, Aroor AR, Hill MA, Yang Y, Whaley-Connell A, Jaisser F and Sowers JR. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension. 2018;72:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarjus A, Maase M, Jeggle P, Martinez-Martinez E, Fassot C, Loufrani L, Henrion D, Hansen PBL, Kusche-Vihrog K and Jaisser F. The endothelial alphaENaC contributes to vascular endothelial function in vivo. PloS one. 2017;12:e0185319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashley Z, Mugloo S, McDonald FJ and Fronius M. Epithelial Na(+) channel differentially contributes to shear stress-mediated vascular responsiveness in carotid and mesenteric arteries from mice. Am J Physiol Heart Circ Physiol 2018;314:H1022–H1032. [DOI] [PubMed] [Google Scholar]
- 63.Jernigan NL and Drummond HA. Vascular ENaC proteins are required for renal myogenic constriction. Am J Physiol Renal Physiol 2005;289:F891–901. [DOI] [PubMed] [Google Scholar]
- 64.Jernigan NL and Drummond HA. Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous beta and gammaENaC. Am J Physiol Renal Physiol. 2006;291:F1184–91. [DOI] [PubMed] [Google Scholar]
- 65.Kim EC, Ahn DS, Yeon SI, Lim M and Lee YH. Epithelial Na+ channel proteins are mechanotransducers of myogenic constriction in rat posterior cerebral arteries. Exp Physiol 2012;97:544–55. [DOI] [PubMed] [Google Scholar]
- 66.Jernigan NL, LaMarca B, Speed J, Galmiche L, Granger JP and Drummond HA. Dietary salt enhances benzamil-sensitive component of myogenic constriction in mesenteric arteries. Am J Physiol Heart Circ Physiol 2008;294:H409–20. [DOI] [PubMed] [Google Scholar]
- 67.Bonny O, Chraibi A, Loffing J, Jaeger NF, Grunder S, Horisberger JD and Rossier BC. Functional expression of a pseudohypoaldosteronism type I mutated epithelial Na+ channel lacking the pore-forming region of its alpha subunit. J Clin Invest 1999;104:967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang BS, Van Vliet BN and Leenen FH. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am J Physiol Heart Circ Physiol 2004;287:H1160–6. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, Huang BS and Leenen FH. Brain sodium channels and ouabainlike compounds mediate central aldosterone-induced hypertension. Am J Physiol Heart Circ Physiol 2003;285:H2516–23. [DOI] [PubMed] [Google Scholar]
- 70.Gomez-Sanchez EP, Gomez-Sanchez CM, Plonczynski M and Gomez-Sanchez CE. Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol 2010;95:120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang H and Leenen FH. Brain sodium channels and central sodium-induced increases in brain ouabain-like compound and blood pressure. J Hypertens. 2003;21:1519–24. [DOI] [PubMed] [Google Scholar]
- 72.Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS and Leenen FH. Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol 2005;289:R1787–97. [DOI] [PubMed] [Google Scholar]
- 73.Anan T, Nagata Y, Koga H, Honda Y, Yabuki N, Miyamoto C, Kuwano A, Matsuda I, Endo F, Saya H and Nakao M. Human ubiquitin-protein ligase Nedd4: expression, subcellular localization and selective interaction with ubiquitin-conjugating enzymes. Genes Cells. 1998;3:751–63. [DOI] [PubMed] [Google Scholar]
- 74.Imaizumi K, Tsuda M, Wanaka A, Tohyama M and Takagi T. Differential expression of sgk mRNA, a member of the Ser/Thr protein kinase gene family, in rat brain after CNS injury. Brain Res Mol Brain Res 1994;26:189–96. [DOI] [PubMed] [Google Scholar]
- 75.Huang BS, White RA, Ahmad M, Jeng AY and Leenen FH. Central infusion of aldosterone synthase inhibitor prevents sympathetic hyperactivity and hypertension by central Na+ in Wistar rats. Am J Physiol Regul Integr Comp Physiol 2008;295:R166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simchon S, Manger W, Golanov E, Kamen J, Sommer G and Marshall CH. Handling 22NaCl by the blood-brain barrier and kidney: its relevance to salt-induced hypertension in dahl rats. Hypertension. 1999;33:517–23. [DOI] [PubMed] [Google Scholar]
- 77.Teruyama R, Sakuraba M, Wilson LL, Wandrey NE and Armstrong WE. Epithelial Na(+) sodium channels in magnocellular cells of the rat supraoptic and paraventricular nuclei. Am J Physiol Endocrinol Metab 2012;302:E273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mills NJ, Sharma K, Huang K and Teruyama R. Effect of dietary salt intake on epithelial Na(+) channels (ENaCs) in the hypothalamus of Dahl salt-sensitive rats. Physiol Rep 2018;6:e13838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Amin MS, Reza E, Wang H and Leenen FH. Sodium transport in the choroid plexus and salt-sensitive hypertension. Hypertension. 2009;54:860–7. [DOI] [PubMed] [Google Scholar]
- 80.Nakano M, Hirooka Y, Matsukawa R, Ito K and Sunagawa K. Mineralocorticoid receptors/epithelial Na(+) channels in the choroid plexus are involved in hypertensive mechanisms in stroke-prone spontaneously hypertensive rats. Hyperten Res 2013;36:277–84. [DOI] [PubMed] [Google Scholar]
- 81.Sheng S, Carattino MD, Bruns JB, Hughey RP and Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol 2006;290:F1488–96. [DOI] [PubMed] [Google Scholar]
- 82.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG and Kirabo A. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep 2017;21:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J, 2nd and Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 2014;124:4642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, Itani HA, Himmel LE, Harrison DG and Kirabo A. High Salt Activates CD11c(+) Antigen-Presenting Cells via SGK (Serum Glucocorticoid Kinase) 1 to Promote Renal Inflammation and Salt-Sensitive Hypertension. Hypertension. 2019;74:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN and Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, Sun S, Zhang Z, Chen XQ, Qin M, Liu X, Tao J, Jia L, Fan HY, Zhou B, Yu Y, Ying H, Hui L, Liu X, Yi X, Liu X, Zhang L and Duan SZ. High salt primes a specific activation state of macrophages, M(Na). Cell Res. 2015;25:893–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ and Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 2013;97:696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]