

Abstract
Heart failure (HF) is a global pandemic with a poor prognosis after hospitalization. Despite HF syndrome complexities, evidence of significant sympathetic overactivity in the manifestation and progression of HF is universally accepted. Confirmation of this dogma is observed in guideline-directed use of neurohormonal pharmacotherapies as a standard of care in HF. Despite reductions in morbidity and mortality, a growing patient population is resistant to these medications, while off-target side effects lead to dismal patient adherence to lifelong drug regimens. Novel therapeutic strategies, devoid of these limitations, are necessary to attenuate the progression of HF pathophysiology while continuing to reduce morbidity and mortality. Renal denervation is an endovascular procedure, whereby the ablation of renal nerves results in reduced renal afferent and efferent sympathetic nerve activity in the kidney and globally. In this review, we discuss the current state of preclinical and clinical research related to renal sympathetic denervation to treat HF.
Keywords: sympathetic nerve system, heart failure, cardiorenal syndrome, renal denervation, hear failure with preserved ejection fraction
INTRODUCTION
Heart failure (HF) is a global pandemic, affecting greater than 26 million people worldwide (1) and 6.2 million people in the United States (2). The prevalence of HF is projected to increase 46% by the year 2030 and cost $70 billion in the United States alone (3), while deaths attributable to HF from 2007 to 2017 increased by 42% (2). HF is a complex syndrome, resulting from a variety of pathophysiological conditions that lead to adverse cardiac remodeling and dysfunction. The manifestation of HF is observed as dyspnea upon exertion, fatigue, exercise intolerance, and congestion. It is well established that sympathetic nervous system (SNS) activation is a critical compensatory response to cardiovascular insults, such as acute myocardial infarction, resulting in reduced cardiac function and inability to maintain cardiac output (4). Initially, activation of the SNS is a beneficial adaptation that serves to maintain blood pressure and perfusion of organs. However, chronic activation of the SNS results in profound pathological consequences that impact the heart and circulation (4). Attenuation of the downstream consequences of pathological overactivation of the SNS has been a therapeutic target for cardiovascular diseases, including hypertension and HF. A number of drugs have been developed to counteract the SNS, including beta-blockers, angiotensin-converting enzyme inhibitors (ACEi), angiotensin II receptor blockers (ARBs), and mineralocorticoid receptor antagonists. Although these agents are effective, their benefits are limited by side effects and poor patient compliance. Furthermore, these agents fail to inhibit the SNS in a significant manner and do not attenuate proximal SNS signals but rather downstream consequences.
Chronic neurohormonal activation may aid the heart in maintaining cardiac output in cardiovascular disease, but it eventually leads to deleterious remodeling and pathological consequences and cellular signaling in the heart, vasculature, and kidneys. The kidneys are a critical regulator of central SNS outflow (5) and contribute to neurohormonal mediator production via the reninangiotensin-aldosterone system (RAAS) (6). Deranged renal function is well characterized in the progression of cardiovascular disease, including essential hypertension and HF (7, 8). Hypertension is the most common comorbidity associated with cardiovascular mortality worldwide (9). This relationship necessitates the development of novel therapeutics to provide alternative strategies to treat these patients for which pharmacological therapies fail to achieve acceptable reductions in blood pressure.
One attempt to address this treatment gap in hypertension involves the use of catheter-based approaches for renal sympathetic denervation (RDN) as a therapeutic strategy to downregulate both afferent and efferent sympathetic nerve activity (10). This technique is a one-time, minimally invasive, catheter-based procedure that ablates renal sympathetic nerves in a bilateral manner using radiofrequency energy, ultrasound energy, or chemical ablation. RDN has been shown to exert clinically relevant blood pressure–lowering effects in patients with hypertension (11–13). While initial clinical trials were positive, these trials were not blinded appropriately and lacked sham controls (14, 15). A subsequent trial (16) involving a sham treatment arm and appropriate blinding of study subjects failed to demonstrate a significant reduction in blood pressure in hypertensive patients. More recently, newer RDN catheters have been developed for clinical use, users have gained more experience with experimental RDN devices, and clinical trial designs have been modified and optimized to involve the most responsive patient populations. It has now become clear that RDN does significantly reduce blood pressure in the setting of hypertension. Despite the focus of catheter-based RDN therapy for resistant hypertension, basic research investigating this modality in the context of HF has come to the forefront (17, 18). This review summarizes the current state of preclinical and clinical research related to the use of RDN and alternative autonomic modulatory approaches to treat HF.
REGULATION OF SYMPATHETIC NERVOUS SYSTEM IN HEART FAILURE
HF is characterized by the inability of heart to maintain cardiac output and appropriate organ perfusion (19). Changes in cardiac output and blood pressure are detected by both mechano-and chemoreceptors within the circulation that relay this information to cardiovascular centers in the central nervous system (20). Information provided by the chemo- and pressure receptors is integrated to regulate cardiac, renal, and vascular function maintaining blood pressure within the normal, physiological range via the level of activation of the parasympathetic and sympathetic nervous systems. In the setting of HF (Figure 1), the SNS is activated maximally and inappropriately as a result of a significant reduction in cardiac output and blood pressures resulting in under perfusion of organs throughout the body. In this case, sustained activation of cardiac sympathetic efferent nerves results in pathological cardiac hypertrophy, fibrosis, and arrhythmias. The vasculature (macro- and microcirculations) is subject to endothelial cell dysfunction, smooth muscle cell hypertrophy, and vasoconstriction following activation of the sympathetic nerves innervating the vasculature. Finally, increased renal sympathetic activity augments sodium reabsorption, renal release of renin and activation of the RAAS, and renal injury. Pathological effects in the kidneys following renal SNS activation ultimately increase circulating blood volume, tissue edema, and systemic vasoconstriction via angiotensin II to significantly exacerbate HF.
Figure 1.
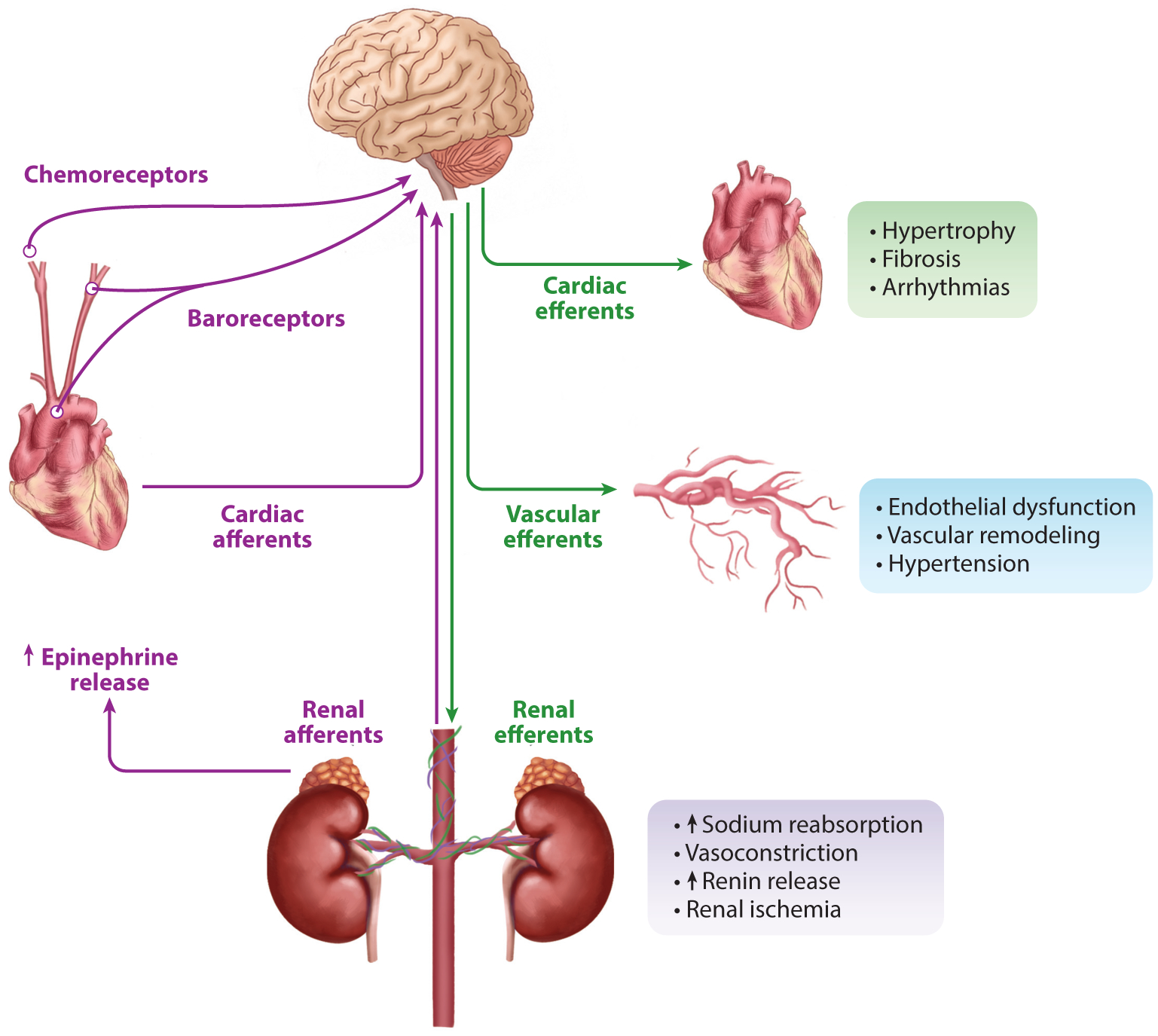
Role of the sympathetic nervous system in heart failure. After a cardiovascular event, such as an acute myocardial infarction, the heart may lack the ability to maintain cardiac output and proper tissue perfusion. Mechanosensitive baroreceptors, which sense changes in blood pressure, and chemoreceptors, which sense a rise in CO2 in the carotid arteries, aortic arch, and peripherally in the kidneys, integrate afferent nerve signaling into autonomic regulatory centers within the central nervous system. Through hypothalamic and other autonomic centers, sympathetic efferent nerve activity can provide initial compensation. However, chronic sympathetic nerve system overactivation, from a variety of etiologies, leads to maladaptive remodeling in target areas such as the heart, vasculature, and kidneys, which can initiate heart failure and exacerbate progression and severity of symptoms.
A HISTORICAL PERSPECTIVE ON RENAL DENERVATION
In 1924, the first surgical renal denervation procedure was performed by Papin & Ambard (21) as an intervention for nephralgia and limited hydronephrosis. In four of six cases of surgical renal denervation, pain was relieved; however, in all cases here, hydronephrosis was not controlled(21). A decade later, in 1934, prior to the era of antihypertensive drugs, the first case of surgical renal denervation was performed in a patient with uncontrolled hypertension and nephritis (22). Surgical denervation in this patient did not lead to a drop in blood pressure and had no effect on renal function (22). A subsequent follow-up study, in 1935, demonstrated a drop in blood pressure over weeks and months; however, blood pressure–lowering effects were not permanent (23). The authors concluded in four of the five cases, renal denervation (a) attenuated proteinuria, (b) preserved renal efficiency in clearance on urea, and (c) despite initial blood pressure–lowering effects, had no major effect on blood pressure over time (23). In 1953, a clinical research study of 1,266 patients suffering from essential hypertension that underwent splanchnicectomy were assessed for blood pressure–lowering effects and subsequent mortality (24). The results demonstrated a 45% reduction in arterial blood pressure at the 5-year postoperative follow-up; however, the authors emphasized a hesitation in choosing surgical methods over drug therapy due to a lack of superiority(24). Furthermore, surgical patients suffered from complications, including orthostatic hypotension, impotence, and incontinence. Consequentially, sympatholytic surgery became obsolete with the development of more tolerable and efficacious antihypertensive pharmacotherapies (24).
CATHETER-BASED RENAL DENERVATION IN THE ERA OF INTERVENTIONAL CARDIOLOGY
The first attempts to modulate autonomic nervous system activity using a percutaneous endovascular catheter approach were performed in a preclinical model of atrial fibrillation (25). The study authors sought to control heart rate through interruption of the parasympathetic nervous system. Schauerte et al. (25) demonstrated that atrial radiofrequency ablation abolished vagal-driven atrial fibrillation. This work provided proof-of-concept results for the concept of implementing radiofrequency energy-based catheter devices through an interventional approach. The first adoption of a radiofrequency energy ablation catheter to denervate the kidneys was by Ardian, Inc., a US-based company acquired by Medtronic, Inc. The vascular safety was first demonstrated by preclinical studies in a porcine model (26). The Symplicity® catheter was utilized in a safety and proof-of-concept clinical trial published in 2009 (27). This catheter system and clinical trial were the cornerstones for the development of several other systems over the next decade, including five radiofrequency-based catheters: Symplicity Flex® and Spyral® (Ardian-Medtronic); EnligHTN® (previously St. Jude Medical, now Abbott); V2 (Vessix Vascular®; Boston Scientific); and RENLANE® (Cordis Corporation); one ultrasound-based catheter, Paradise® (ReCor Medical); and one chemical ablations catheter, Peregrine System® (Ablative Solutions). As the catheters were developed, the focus became on designing catheters with multielectrodes for more complete circumferential denervation and catheter maneuverability for potential tortuous vasculature. New catheter designs and modifications resulted in more reliable and efficacious RDN in hypertensive patients. Despite initial success in treating hypertension, the development of RDN was not without its misfortunes and failures. Over the last decade, obstacles regarding the technology, clinical trial design, patient selection, and appropriate study end points have severely hampered the clinical translation of this promising technology.
Following the failure of the Symplicity HTN-3 study, several rigorous follow-up clinical trials demonstrated efficacy. These have sparked renewed interest in the therapeutic potential of RDN for hypertension. Some of these trials include the SPYRAL HTN-OFF (13) (NCT02439749) and ON MED (NCT02439775) trials (12), and the RADIANCE-HTN SOLO/TRIO trial (NCT02649426) (11). These trials all have similar designs, with rigorous inclusion/exclusion criteria (28) and established procedures that would permit the maximum effect of RDN to be observed on lowering blood pressure. These more recent clinical trials have demonstrated significant and clinically relevant reductions in blood pressure compared to sham controls (11–13). Careful design and well-executed trials have reestablished RDN’s ability to provide clinically meaningful blood pressure–lowering effects. This opened the door for alternative denervation systems, like the Peregrine catheter, whereby alcohol ablation of the renal interstitium is performed. Ongoing clinical trials in the United States and Europe are currently evaluating the safety and efficacy of this device (NCT02910414 and NCT03503773). Moreover, pivotal trials are now underway, such as SPYRAL PIVOTAL– SPYRAL HTN-OFF MED (NCT02439749) trial and RADIANCE II Pivotal Study (NCT03614260), with completion dates expected in 2022 and 2024, respectively.
RENAL SYMPATHETIC DENERVATION IN HEART FAILURE WITH REDUCED EJECTION FRACTION
HF with a reduced ejection fraction (HFrEF) [left ventricular ejection fraction (LVEF) <40%] (29) is primarily characterized by a significant deficit in systolic function of the heart, with or without congestion depending on the progression of the HF syndrome. Previous research (30) has demonstrated a substantial increase in renal norepinephrine spillover in HF patients. When compared to individuals with essential hypertension (31), the levels of norepinephrine spillover from the kidneys in HF patients are significantly higher (30), suggesting increased SNS activity. Increases in muscle sympathetic nerve activity were positively correlated to increased HF severity and demonstrated a tenfold increase in peripheral muscle sympathetic nerve activity (32). The effectiveness of neurohormonal modulators, such as beta-blockers, ACEi, ARBs, aldosterone antagonists, diuretics, and neprilysin inhibition, as standards of care to manage HFrEF, is a testament to the significant role the SNS plays in exacerbating HF severity (9, 33–35). Despite the success of these pharmacotherapies in reducing morbidity and early mortality, resistance to pharmacotherapies, off-target side effects, and the lack of patient adherence to drug regimens (36, 37) still lead to worsened HF symptoms over time. It should also be noted that the current available drugs to treat HFrEF do delay the progression of the disease, but they do not halt or reverse this condition. Therefore, a clinical unmet need for adjunctive or alternative therapeutic strategies to combat HFrEF still exists.
Preclinical studies have investigated the potential beneficial effects of RDN in the setting of myocardial ischemia-reperfusion and asymptomatic HFrEF (17, 18, 38). In a spontaneously hypertensive rat model, Polhemus et al. (38) investigated the role of RDN on SNS modulation in ischemia-reperfusion injury to evaluate the potential acute cardioprotective effects of renal sympathetic denervation. This study elucidated the novel remote preconditioning cardioprotective mechanisms of RDN in the setting of ischemia-reperfusion in conjunction with chronic hypertension. The results demonstrate a significant reduction in myocardial infarct size and preservation of cardiac function 7 days postreperfusion following RDN pretreatment as compared to a sham control (38). RDN treatment in spontaneously hypertensive rats resulted in marked attenuation of oxidative stress, reduced G protein–coupled receptor kinase 2 (GRK2) pathological signaling in the heart, and increased myocardial nitric oxide bioavailability (38).
Subsequent studies involved investigations into the effects of RDN in HFrEF following acute myocardial infarction. Polhemus et al. (17) report on the beneficial effects of delayed radiofrequency RDN to improve outcomes in the setting of HFrEF in rats subjected to myocardial ischemia and reperfusion. This study was performed in both spontaneously hypertensive and normotensive Wistar Kyoto rats who underwent 45 min of myocardial ischemia followed by 12 weeks of reperfusion. At 4 weeks, following myocardial infarction, the rats were randomized to sham RDN or radiofrequency RDN. The radiofrequency RDN-treated animals displayed significant improvement in left ventricular function, vascular reactivity, and reduced cardiac fibrosis. Furthermore, these investigators observed greater than twofold increases in plasma levels of natriuretic peptides [NPs; B-type (BNP), atrial (ANP), and C-type (CNP)] that were mediated via inhibition of renal neprilysin activity (17).
To evaluate the clinical potential of radiofrequency RDN in the setting of chronic HFrEF, we recently embarked on experiments utilizing a swine model of HF secondary to acute myocardial infarction (18). These studies utilized the St. Jude EnligHTN radiofrequency RDN system to perform circumferential RDN in both the proximal and distal regions of the renal arteries following the onset of HFrEF at a time when LVEF was significantly reduced. Normotensive Yucatan mini-swine underwent 75 min of myocardial ischemia and subsequent reperfusion for 18 weeks. After manifestation of reduced LVEF (<40%), animals were randomized in a blinded manner to receive sham RDN or radiofrequency RDN treatment and 12 weeks of follow-up. There was a significant reduction in renal norepinephrine, a biomarker of renal sympathetic nerve activity, and highly significant reductions in both circulating angiotensin I and II following radiofrequency RDN treatment (18). These data confirm robust inhibition of renal sympathetic nerve activity coupled with attenuation of renal renin-angiotensin system activation as a key beneficial effect of RDN in HF. This swine study confirmed earlier results obtained in the rat study (17) as evidence by increased NP levels and improved left ventricular structure and function after radiofrequency RDN when compared to sham RDN. We also observed improved coronary artery vasorelaxation to endothelial-dependent and -independent vasodilators. Similar findings have been reported by others in both small (39–41) and large (42, 43) animal HFrEF models. Figure 2 summarizes our findings on the protective actions of radiofrequency RDN in rodent and swine studies of HFrEF secondary to acute myocardial infarction.
Figure 2.
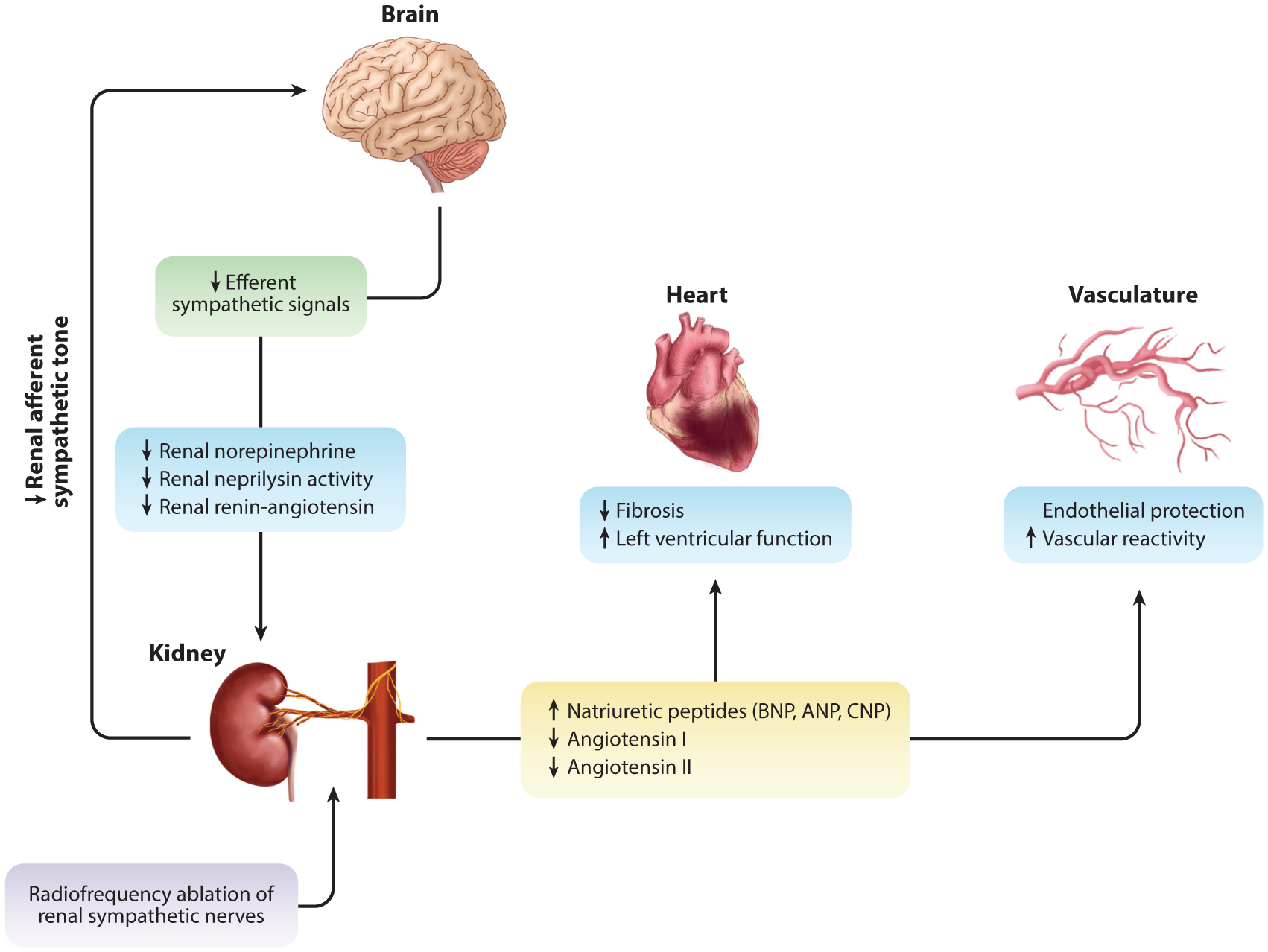
The effects of renal sympathetic nerve ablation in HFrEF. With chronic overactivation observed in heart failure, renal afferent input into the central nervous system plays an integral role in efferent output. Through renal sympathetic nerve ablation (purple box), preclinical data suggest a reduction in efferent signaling to the kidney (green box) through a reduction in renal norepinephrine and concurrent reduction in renal neprilysin activity and attenuation renin-angiotensin system activation (blue boxes). This leads to reduced renal fibrosis and improved function (17, 18). Neprilysin inhibition promotes sustained exposure to natriuretic peptides in circulation, and reduced efferent sympathetic signal inhibits renin production and downstream circulating levels of angiotensin I and II (yellow box). Improved natriuretic peptide exposure and reduced angiotensin I and II lead to reduced cardiac fibrosis and improved function. Further cardiovascular protection is afforded to the vasculature, whereby there is improved vascular reactivity to classical vasodilators [i.e., bradykinin, substance P (endothelial-dependent), and sodium nitroprusside (endothelial-independent)] (blue boxes). Abbreviations: ANP, atrial natriuretic peptide; BNP, B-type natriuretic peptide; C-type natriuretic peptide; HRrEF, heart failure with a reduced ejection fraction.
One of the unexpected findings from studies of RDN in HFrEF is the effect of RDN to inhibit neprilysin in the kidney during HF to augment the bioavailability of NPs and improve myocardial and vascular function. NPs are a well-established biomarker of HF severity (44–46). It was discovered four decades ago that the heart was an endocrine organ (47), producing and secreting a family of hormones known as NPs (48–50). NPs’ primary mechanism of action is the reduction of cardiac preload and afterload to ultimately reduce myocardial wall stress that occurs as HF progresses in severity (51). Dilatation of the left ventricular chamber and wall thinning promote increased stretch that is sensed by ventricular myocytes, stimulating NP secretion. NPs signal to the vasculature, which induces smooth muscle relaxation-mediated vasodilation to reduce peripheral vasculature resistance. In the kidney, it promotes natriuresis and diuresis to reduce blood volume, subsequently reducing arterial pressure in an attempt to correct pathological myocardial wall tension (52). In addition, the autocrine effects of NPs elicit cardioprotective signaling within the heart. NPs signal through NP receptors A, B, and C (53, 54). Two of the receptors (A and B) are G protein coupled, activating guanylyl cyclase generation of cyclic guanosine monophosphate (53, 55). This signaling cascade leads to activation of protein kinase G (56), which has been shown to inhibit hypertrophic remodeling and improve cardiac contractility (57, 58). NPs can also attenuate transforming growth factor beta activation of fibroblasts within the myocardium, leading to attenuation of fibrosis (59). Figure 3 summarizes the powerful cardioprotective effects that can occur with increased circulating NPs (52).
Figure 3.
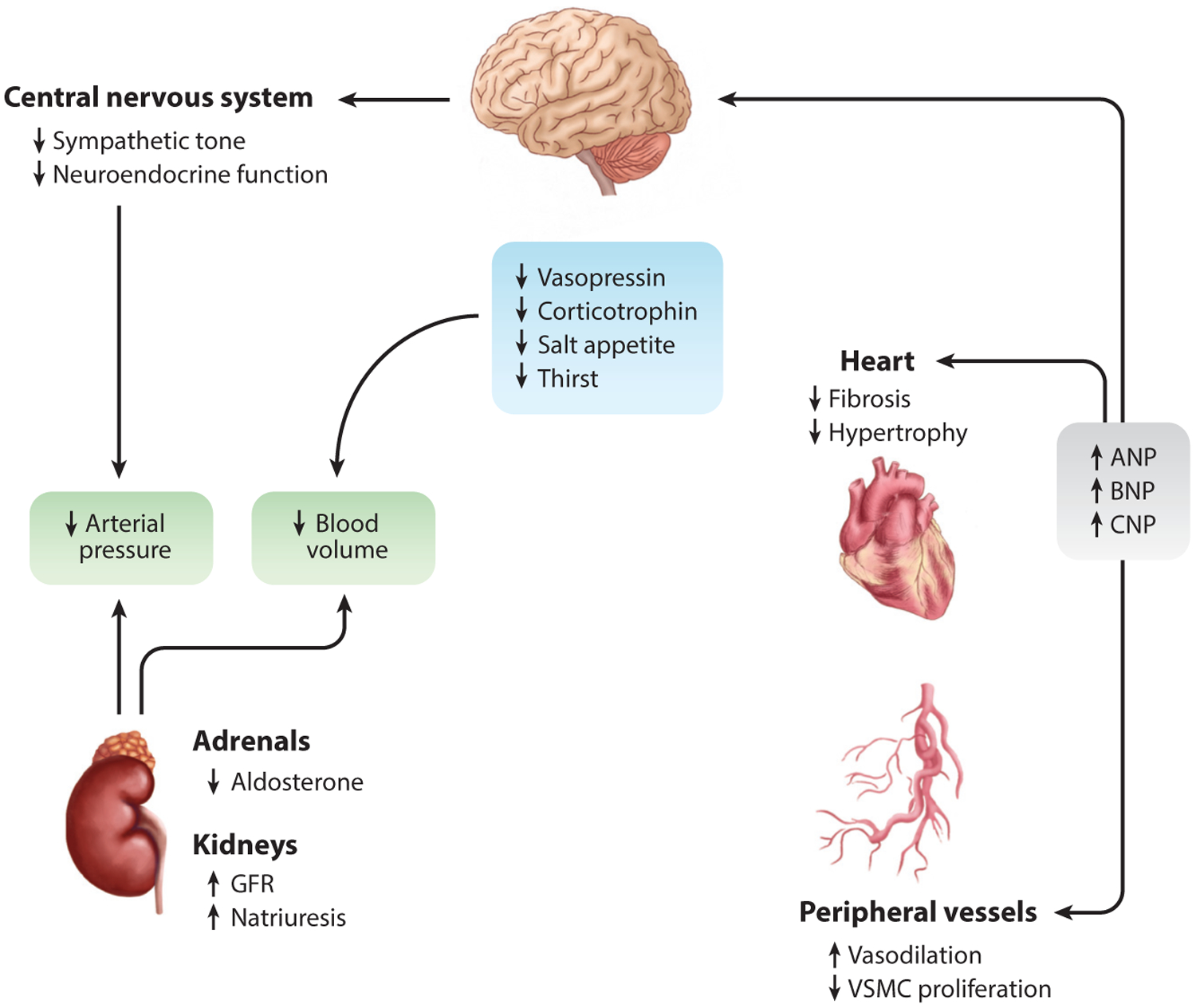
Cardioprotection by NPs in heart failure. Circulating NPs have cardioprotective effects in relation to cardiac, vasculature, central nervous system, and renal physiology. Increased circulating NPs have paracrine effects on peripheral organs (i.e., brain, adrenal glands, kidneys, and vasculature) and autocrine effects on the heart. Circulating NPs reduce central nervous system pathways that modulate natriuresis and diuresis, also providing reduced sympathetic tone. This offers several direct and indirect mechanisms culminating in reduced arterial blood pressure. Furthermore, NPs act in an autocrine manner on cardiomyocytes through inhibition of hypertrophy and other maladaptive signaling cascades. These local and remote mechanisms provide cardioprotection in the setting of heart failure. Abbreviations: ANP, atrial natriuretic peptide; BNP, B-type natriuretic peptide; CNP, C-type natriuretic peptide; GFR, glomerular filtration rate; NP, natriuretic peptide; VSMC, vascular smooth muscle cell. Figure adapted with permission from Reference 52; copyright 2013, American Physiological Society.
Following RDN in the setting of HFrEF we found that the primary enzyme responsible for NP degradation, neprilysin, was significantly inhibited (17, 18). Neprilysin is a membrane metallo-endopeptidase found primarily in the kidney and lungs that cleaves hormone peptides (60). In Figure 4, our hypothesis suggests that the SNS plays a critical role in renal neprilysin activity, which subsequently affects NPs as described. Under conditions of chronic SNS overactivation, it is our belief that renal β1-adrenergic signaling increases neprilysin activity, through posttranslational modifications that have yet to be defined. This leads to an increased degradation of NPs and a loss of cardioprotective effects permitting further adverse cardiovascular remodeling and worsening HF. Neprilysin inhibition is at the forefront of current HF treatment strategies(61). Sacubitril, a neprilysin inhibitor combined with the ARB valsartan, has been shown to be highly effective in HFrEF to reduce cardiovascular mortality. In the PARADIGM-HF study (62), sacubitril/valsartan reduced combine primary end points of cardiovascular death and hospital admission by 20% when compared to enalapril in 8,442 patients with HFrEF. Interestingly, radiofrequency RDN not only inhibits neprilysin activity but very significantly halts activation of the renin-angiotensin system, resulting in low circulating levels of angiotensin I and II (17, 18). These data suggest that RDN acts in a very similar manner to the combination therapy of neprilysin inhibition and angiotensin receptor blockade (i.e., LCZ696, sacubitril/valsartan). One potential advantage of RDN over LCZ696 is that RDN would only need to be performed once as opposed to daily drug dosing over many years.
Figure 4.
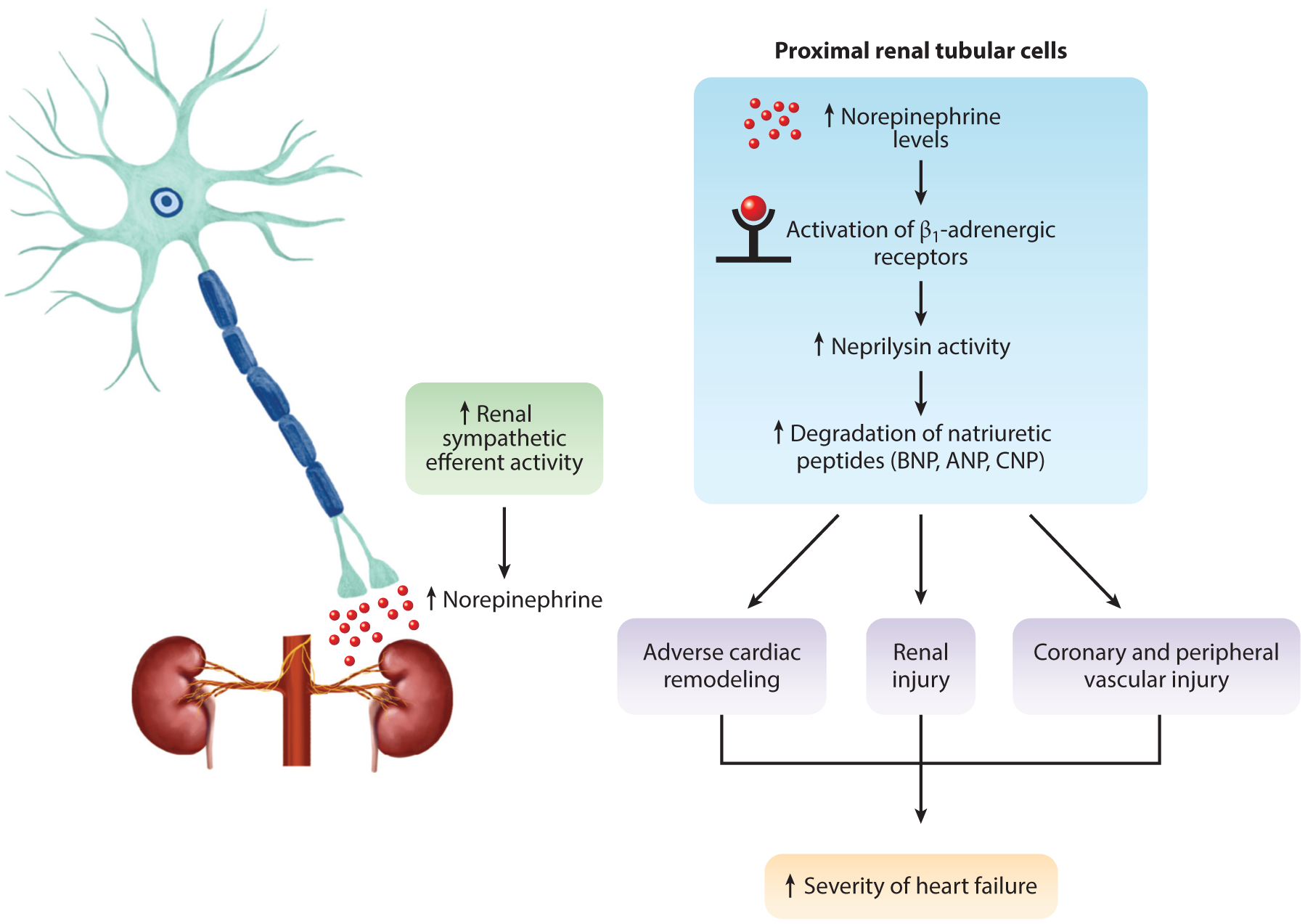
Proposed regulation of renal neprilysin activity in the kidney in relation to heart failure. In the presence of increased sympathetic nerve activity to the kidney in heart failure, Polhemus et al. (17) and Sharp et al. (18) propose that overactive catecholamine β1-adrenergic signaling in the proximal renal tubular cells drives downstream posttranslational modification, which increases neprilysin activity. With increased activity, there is a significant increase in natriuretic peptide degradation, resulting in further adverse cardiac remodeling, potential renal injury, and vascular dysfunction synergizing to exacerbate heart failure severity. The application of a novel therapeutic intervention, such as renal denervation, inhibits norepinephrine signaling and attenuates neprilysin activity and natriuretic peptide degradation, providing remote cardioprotection. Abbreviations: ANP, atrial natriuretic peptide; BNP, B-type natriuretic peptide; CNP, C-type natriuretic peptide.
Cardiac arrhythmias are a result of complex autonomic dysregulation of the electrophysiological circuitry within the heart under pathophysiological conditions. Many cardiac arrhythmias are associated with an imbalanced increase in SNS activity and withdrawal of parasympathetic regulation (63). The desynchrony within the heart can lead to adverse cardiac remodeling, perpetuating a sustained and chronic arrhythmogenic state, such as recurrent ventricular tachycardia or atrial fibrillation. The incidence of ventricular tachycardia is associated with ischemic and nonischemic insults, whereby disruption in the cardiac conduction pathways occur due to myocardial tissue loss and increased fibrosis seen with ischemia or hypertrophic remodeling as occurs in heart failure (64–66). Since RDN can potentially modulate global sympathetic tone without side effects of hemodynamic disarrangement, this procedure may be beneficial in this context. In a proof-of-principle murine model of myocardial ischemia-induced HF, chemical RDN with phenol resulted in a reduced susceptibility of ventricular arrhythmias (39). The translational application of this work to a porcine model of myocardial ischemia validated the use of RDN to significantly suppress ventricular tachycardia when compared to sham-operated animals (67). In cases where cardiac sympathetic denervation failed and persistent refractory ventricular tachycardia is present, interventionists are performing adjunctive RDN to cardiac sympathetic denervation procedures. In 10 patients with cardiomyopathies (20% ischemic) and HFrEF, Bradfield et al. (68) demonstrated the potential benefit of cardiac sympathetic denervation with adjunctive RDN when compared to cardiac denervation alone, whereby a significant reduction in cardioverter-defibrillator shocks was seen in patients 6 months post-RDN. The authors also concluded that RDN following cardiac denervation is indicative of poor prognosis (68). Additional case reports and clinical studies have been performed aiming to understand the efficacy of RDN as a primary or adjunctive therapy in patients suffering from dilated and hypertrophic cardiomyopathy as well as refractory ventricular tachycardia after myocardial ischemia (69, 70). Large-scaled prospective studies are necessary to validated RDN in this context.
A unique and potentially important aspect of RDN compared to current pharmacological therapies is that RDN’s site of action to inhibit renal norepinephrine is upstream of pharmacotherapy used today in HF (e.g., ACEi, ARBs, beta-blockers). Current drug therapies work to disrupt intermediate-level signaling and end-organ receptors, and this approach fails to address critical and proximal pathological signals that drive the pathology of HF and disease progression. In contrast, RDN modulates renal afferent signaling to the central nervous system and inhibition of efferent signaling to the kidney and potentially other organs. Reduction of norepinephrine levels in target tissues and spillover into the circulation inhibit maladaptive sympathetic signaling and its downstream signaling pathways (β1-adrenergic signaling, RAAS activation, increased neprilysin activity, reduced circulating NPs), providing beneficial effects on HF progression.
Despite the overwhelming positive preclinical data in HFrEF and clinical data in hypertension, little has been done clinically to evaluate the safety and efficacy of RDN in HFrEF patients. Davies et al. (71) published the first-in-man clinical trial evaluating RDN in chronic systolic HF (REACH Pilot study; NCT01584700). In 7 patients, there was improved HF symptoms and exercise capacity, as measured by a 6-min walk test at 6 months post-RDN with no adverse events (71). This trial preceded the results of the Symplicity HTN-3 study. In 2013, the PRESERVE clinical trial (NCT01954160) was initiated to study the safety, efficacy, and effect of RDN on renal sodium excretion in HFrEF patients but was abruptly terminated by the Data and Safety Monitoring Board when the Symplicity HTN-3 results were published. Since then, there has been a paucity of data in alternative indications like HFrEF.
RENAL SYMPATHETIC DENERVATION IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
A recent reevaluation of the dichotomy within the HF population has shed light on the ever growing number of patients that present with classical HF symptoms (dyspnea, fatigue, lack of functional reserve) yet maintain a preserved ejection fraction (HFpEF) (LVEF >50%) (29). HFpEF is a systemic syndrome consisting of multiorgan pathophysiological abnormalities and driving end-organ damage, including cardiovascular remodeling and exertional intolerance (72). Of importance, exclusion of other causes of HFpEF or dyspnea must be ruled out, including but not limited to valvular disease, pericardial disease, and infiltrative myocarditis (73). HFpEF accounts for more than half of all HF diagnoses, and treatments for this rapidly developing disease are extremely limited, as no US Food and Drug Administration (FDA)-approved drug has been shown to be effective in the setting of HFpEF (74). Neurohormonal therapies such as ACEi (75), ARBs (76, 77), and aldosterone antagonist (78), as well as combination therapy, are efficacious in HFrEF (79) but have uniformly failed to provide meaningful benefit in HFpEF patients (80). Furthermore, nitric oxide donors (81) and soluble guanylate cyclase activators (82) have been tested in clinical trials and were found to be neutral compared to optimal medical therapy. It is becoming increasingly clear that HFpEF is driven, to some degree, by a distinct subset of pathophysiology mechanisms (74). SNS activity has been shown to be elevated in HFpEF patients(83), but what cannot be discerned is which process is the cause and/or result of the other.
HFpEF is recognized as a systemic syndrome that involves an inflammatory response propagated by comorbidities, including hypertension, diabetes mellitus, obesity, and kidney disease, which ultimately lead to HF characterized by diastolic dysfunction. Dependent upon the predominating organ system affected, the clinical presentation of HFpEF can vary widely. Phenomapping, through computer-based algorithms (84), has revealed the dominant phenotypes that are presented clinically; however, no two patients are the same, and with a lack of therapeutic interventions comes a need to develop new strategies and preclinical models for testing safety and efficacy. Herein, we discuss the potential mechanisms by which RDN might be able to impact the most predominant etiologies of HFpEF pathobiology.
With the ever evolving clinical knowledge base about HFpEF, it has been a struggle for basic researchers to come to a consensus on proper models to elucidate the pathology of this complex disease and for the testing of novel therapeutic strategies. Small (85) and large (86) animal models have been proposed to date, yet each has noteworthy limitations, with little agreement on the interpretation of results (87, 88). An understanding of the target phenotype within HFpEF should be taken into consideration in model development; for instance, a model of cardiometabolic HFpEF. Furthermore, clinical translation must be at the forefront of model development and study design. Although proof-of-concept work in rodent models may elucidate mechanisms, it rarely translates to humans with respect to pathophysiological and therapeutic responses (89). We believe that furthering our understanding of the underlying causes, along with developing and refining rigorous animal models, will begin to allow for some agreement among basic and clinical researchers alike. Figure 5 illustrates the potential beneficial effects that RDN might exert in HFpEF, leading to improved HF symptoms. Sympathetic tone is strongly associated with HFpEF along with all the chronic comorbidities, including metabolic syndrome, hypertension, chronic kidney disease, pulmonary hypertension, and atrial fibrillation (90–94). End-organ dysfunction at the level of the heart, lungs, vasculature, kidneys, and skeletal muscle could be attenuated with RDN modulation of SNS activity. Future preclinical and potentially clinical studies are required to test the efficacy of RDN in the setting of HFpEF, and given the diverse and highly complex nature of the HFpEF patient population, it is critical to determine which HFpEF patients suffer from SNS overactivation and might be responsive to RDN therapy.
Figure 5.
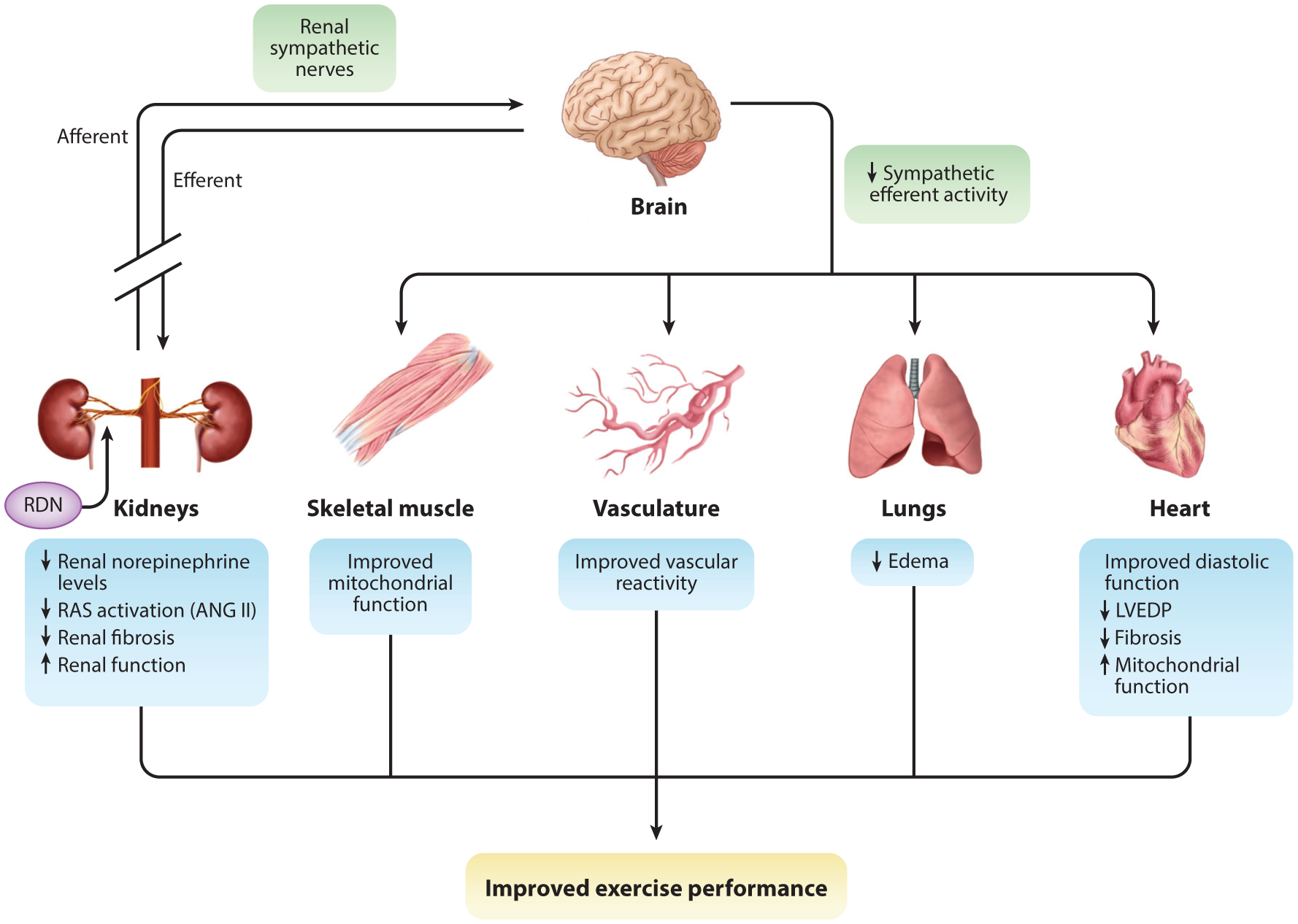
Proposed effects of renal nerve ablation in HFpEF. Renal nerve ablation reduces sympathetic afferent and efferent signaling to the kidney, followed by a coordinated disruption in global sympathetic tone and neurohormonal signaling pathways (green boxes). In the kidney, there is reduced norepinephrine levels, which dampens renin-angiotensin system (RAS) activation. This could also provide therapeutic benefit toward renal disease and the concurrent inflammation, thereby reducing renal fibrosis and improving function. Remotely, the global alteration in circulating norepinephrine, angiotensin II (ANG II), and sympathetic tone could lead to improvements in a variety of systems, including skeletal muscle, vasculature, lungs, and heart. Sympathetic drive is a systemic regulator of energy production and consumption. Chronic sympathetic overactivation leads to mitochondrial dysfunction and an imbalance in the energy supply and demand. Upon exertion, the body is unable to cope with the demand so that an energy production deficit arises, which can manifest itself as skeletal muscle weakness and exercise intolerance. We propose that renal sympathetic denervation (RDN) can provide remote mito-protection and reestablish proper energy production and consumption. The ability to reduce RAS activity, which directly affects vascular function, would provide an improvement of the body’s ability to distribute blood and proper oxygenation to organ systems, further improving the systemic pathophysiology observed in HFpEF. By improving renal and vascular function through RDN, attenuation of improper natriuresis and diuresis can occur; this would afford a reduction in blood volume and blood pressure. In reducing the load with which the heart must work against, it may provide congestion relief that usually manifests itself in the pulmonary circulation. These remote effects provide significant benefit to cardiac structure and function. By reducing the load, this should improve diastolic function and reduce both elevated cardiac pressures and myocardial stress, which leads to inflammation and fibrosis. Improvement in the supply and demand of energy in the heart should also occur. We propose that these effects (blue boxes) synergize to provide greater functional capacity and reserve, leading to improved exercise capacity and quality of life for patients with HFpEF. Other abbreviations: HFpEF, heart failure with a preserved ejection fraction; LVEDP, left ventricular end-diastolic pressure.
In the setting of HFpEF, increasing NPs through RDN-induced neprilysin inhibition may be an effective way to increase protein kinase G signaling and improve outcomes via the impact on a number of organs, including the heart. Clinical trials have targeted neprilysin inhibition using a combination of sacubitril/valsartan (LCZ696) as compared to valsartan in HFpEF patients. The PARAGON-HF trial (NCT01920711) (79) enrolled 4,822 patients randomized to a combination of valsartan-only therapy. The study demonstrated no significant difference in the rate of hospitalization or cardiovascular-related mortality between treatment groups (79). Despite neutral results, this mechanism of action may still be a viable target if RDN is applied. We stress the concept that RDN works more proximally on the underlying issue of SNS overactivation by inhibiting renal afferent and efferent signaling, which can provide remote cardioprotective effects in conjunction with neprilysin inhibition.
Small retrospective analyses and observational clinical research have been performed to examine RDN in patients with HFpEF. In a randomized clinical trial of resistant hypertension, a quarter of the patients were clinically diagnosed with HFpEF and received RDN (95). Brandt et al. (95) reported that at 6 months after RDN, the RDN-treated patients had a reduced left ventricular mass, improved systolic function, and indexes of diastolic function (mitral E deceleration time, E/e′ ratio) as compared to optimal medical therapy. In a recent multicenter cardiac magnetic resonance (CMR) imaging study (96), 16 patients with resistant hypertension and diagnosed HFpEF underwent RDN. There was a significant improvement in global longitudinal strain, suggesting improved diastolic function in these patients. While CMR imaging is a more reliable imaging modality, with less interobserver error, large clinical trials with long-term follow-up are truly necessary for more concrete conclusions to be made. The RDT-PEF randomized clinical trial (NCT01840059) (97) was going to take on this task; however, due to difficulties in recruitment the study was unpowered to demonstrate improvement in quality of life, exercise, and biomarker and cardiac remodeling. However, in those patients enrolled, the procedure was safe (97). As the field of catheter-based denervation garners regulatory approval for its primary indications, the flood gates might soon open on alternative indications, such as HFpEF. Only then will we begin to get answers on clinical efficacy in an ever-growing HF population.
ALTERNATIVE STRATEGIES FOR AUTONOMIC MODULATION IN HEART FAILURE
Renal integration into the SNS has a major impact on the autonomic nervous system in health and disease, and RDN is poised to become an effective procedure to combat sympathetic overactivity observed in a myriad of disease conditions. Notwithstanding, RDN is not the only technology under development to attenuate sympathetic overactivity to treat hypertension or cardiovascular diseases. Other technologies are being developed to target more centrally located autonomic integration centers, including the baroreceptor reflex (98–100), as well as vagal parasympathetic innervation (101, 102).
The MobiusHD®, by Vascular Dynamics, Inc. is a proprietary-designed device that is placed through an endovascular procedure in the internal carotid artery to create a geometric alteration on the carotid bulb, leading to changes in the carotid sinus nerve input to the central nervous system. The device is termed an endovascular baroreflex amplification technology. The baroreceptor reflex, over time, is lost or “reset” in patients with essential hypertension (103). This device, when implanted, changes the pulsatile strain of the carotid bulb, thereby inducing a reduction in blood pressure. The first patient procedure was performed in 2015 and reported a blood pressure–lowering effect in a single patient with resistant hypertension (100). A first-in-man, multicenter, prospective, open-labeled trial (CALM-FIM_EUR; NCT01900897) reported a significant blood pressure–lowering effect in 30 patients with resistant hypertension (104). The study demonstrated that the MobiusHD device also had an acceptable safety profile; however, it did have adverse events in four patients (104) (i.e., hypotension, worsening hypertension, intermediate claudication, and one wound infection). There are four active/nonrecruiting studies, one of which is the CALM-2 pivotal study (NCT03179800), which is estimated to enroll 300 patients and measure changes in 24-h systolic arterial blood pressure from baseline to 180 days. The MobiusHD is not the only device that targets the baroreflex.
CVRx, Inc. developed a technology that activates the baroreflex through an implantable pulse generator and lead system called Rheos® (98). The technology was initially conceived as an alternative therapeutic intervention for resistant hypertension (105). The device is implanted subcutaneously and sends pulse waves to the lead implanted near the carotid bodies to rectify autonomic imbalance in patients. To understand the implications of baroreflex activation in the pathogenesis of HF, the device is being tested in current clinical trials (NCT00957073, NCT01471860, and NCT01720160). Initial reports (99, 106) demonstrate an improved quality of life, 6-min walk test, and LVEF and a reduction in HF hospitalization in NYHA Class III HF patients. Others have taken a different approach to understanding the autonomic imbalance by directing effort to stimulate the vagal nerve and induce parasympathetic activation. While several clinical trials have been performed (101, 102), data would suggest that safe, favorable, or neutral outcomes were observed in patients over long-term follow-up. Results demonstrate an improvement in HF symptoms but no difference in mortality (101, 102).
Other devices are taking an indirect approach at providing autonomic modulation. A hallmark of HF, observed clinically, is congestion, both pulmonary and/or peripheral (19, 29). Cardiac shunt devices are being implemented in hope of providing congestion relief in HF patients. Although they do not directly affect the SNS, strategically placed shunts may elicit changes in arteriovenous hemodynamics and shift blood volume. By centrally unloading the left atria, it may permit the left ventricle to work more efficiently. These alterations are sensed by the autonomic sensory afferent nerve, which becomes integrated in the central nervous system, thereby affecting SNS efferent signaling to the cardiopulmonary and cardiovascular systems.
Regardless whether a patient suffers from HFrEF or HFpEF, increased left atrial pressure and pulmonary congestion are precipitating symptoms of symptomatic and acute decompensated HF; therefore, these shunting devices should be beneficial for both forms of HF. Several studies (107–109) have demonstrated the utility of performing a permanent interatrial shunt. Providing left atrial decompression relieves the pulmonary vasculature from edema and results in better gas exchange and, ultimately, a relief of dyspnea at rest and upon exertion. While most reports have focused on the implementation of this device in HFpEF (107, 108), owing to the lack of efficacious therapeutics, there is a limited data set in the HFrEF population (109). The V-wave clinical study followed HF patients (n = 38) with interatrial shunts for 1 year (109). This first-in-man clinical study deployed a proprietary designed interatrial V-shaped shunt in HF patients with a NYHA class of III or IV; 30 of the patients were diagnosed with HFrEF, while 8 suffered from HFpEF (109). The results demonstrated an improvement in NYHA class, quality of life, and 6-min walk test with no changes in left- or right-sided cardiac function (109). The improvement in the 6-min walk test, which is a metric of cardiovascular reserve, provides evidence that the shunt not only results in cardiac unloading but also reduces dyspnea. This sustained unloading over time elicits changes systemically (potentially to sympathetic tone), which provides remote mechanism(s) of cardiovascular protection. In this study (109), the major limitation was shunt closure over time; at 3 months, all devices were patent; however, at 12-months, ~50% were occluded or became stenotic. Patients with patent shunts exhibited sustained cardiovascular benefits through the 12-month study duration (109). Second-generation shunts are currently under development, which will hopefully help to overcome this obstacle; furthermore, large-scale, randomized clinical trials with long-term follow-up are necessary. It is important to investigate all viable options with respect to adjunctive therapeutic strategies to optimal medical therapy to combat the development, manifestation, and severity of HF.
CONCLUSION
HF, a clinical syndrome with several manifestations, has a complex and incompletely understood pathophysiology at play that results in significant morbidity and mortality. The prevalence of HF and the resultant economic burden this disease imposes will continue to rise worldwide, and no guideline-based optimal medical therapy has proven to halt or reverse disease progression for those who suffer from HFrEF. Approved HFrEF therapies only treat HF symptoms but fail to reverse this devastating disease. The current state of affairs is even more bleak for HFpEF, with a complete lack of effective agents to treat this form of HF. There is an abundance of basic and clinical research demonstrating the powerful compensatory and subsequent maladaptive response of the autonomic nervous system to cardiovascular injury and HF. To that end, currently, drug-based neurohormonal modulation is our best therapeutic intervention. Still, there are several limitations to these agents, including the lack of effect in some patients, pharmacotherapy resistance, untoward side effects, and a lack of patient adherence to the drug regimen. With this landscape, it is imperative to develop and test novel therapeutic strategies in the setting of HF.
The kidneys play a critical role in SNS activity and modulation. Therapeutically targeting renal afferent and efferent sympathetic nerves, through RDN, has provided an alternative strategy that is performed remotely but delivers central modulation of the autonomic nervous system and cardioprotective and vasculoprotective effects in HF. Through simultaneous inhibition of the renal afferent and efferent activity, RDN may reset central nervous system integration and therefore global efferent output. Furthermore, by dampening sympathetic efferent signaling to remote targets and the kidney, RDN modulates global catecholamine signaling pathways, improves global physiology, and halts the maladaptive activation of the renal neurohormonal axes at its origin. Therapeutic mechanisms by which RDN improves outcomes in preclinical models of HFrEF have yet to be fully elucidated and are the subject of ongoing research efforts.
SUMMARY POINTS.
The heart’s inability to maintain cardiac output and tissue perfusion results in compensatory activation of the SNS and the renin-angiotensin system.
A series of coordinated responses throughout the body collectively work to reestablish homeostasis; however, chronic activation is a maladaptive response that can drive the progression of HF.
While antagonism of the neurohormonal system forms the basis of current pharmacotherapy for HF, untoward side effects and complicated drug regimens lead to patient nonadherence.
Renal denervation represents a one-time procedure and highly effective therapeutic strategy to downregulate sympathetic activity and antagonize neurohormonal activation in the setting of HF.
Preclinical research suggests that renal denervation reduces norepinephrine release in the kidney, providing inhibition of the renin-angiotensin system to improve outcomes in HFrEF.
Renal denervation reduces neprilysin activity, promoting elevated levels of cardioprotective natriuretic peptides in the setting of HFrEF.
We proposed that the observed cardioprotective mechanisms afforded by renal denervation in HFrEF models can translate to both HFrEF and HFpEF patients.
Footnotes
DISCLOSURE STATEMENT
D.J.L. has a provisional patent on the use of renal denervation for the treatment of myocardial infarction and heart failure. T.E.S. is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Savarese G, Lund LH. 2017. Global public health burden of heart failure. Card. Fail. Rev 3:7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, et al. 2020. Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation 141:e139–596 [DOI] [PubMed] [Google Scholar]
- 3.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, et al. 2013. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ. Heart Fail 6:606–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartupee J, Mann DL. 2017. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol 14:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Converse RL Jr., Jacobsen TN, Toto RD, Jost CM, Cosentino F, et al. 1992. Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med 327:1912–18 [DOI] [PubMed] [Google Scholar]
- 6.DiBona GF, Kopp UC. 1997. Neural control of renal function. Physiol. Rev 77:75–197 [DOI] [PubMed] [Google Scholar]
- 7.Smith GL, Lichtman JH, Bracken MB, Shlipak MG, Phillips CO, et al. 2006. Renal impairment and outcomes in heart failure: systematic review and meta-analysis. J. Am. Coll. Cardiol 47:1987–96 [DOI] [PubMed] [Google Scholar]
- 8.Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Bohm M, Krum H. 2011. Sympatho-renal axis in chronic disease. Clin. Res. Cardiol 100:1049–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bozkurt B, Aguilar D, Deswal A, Dunbar SB, Francis GS, et al. 2016. Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American Heart Association. Circulation 134:e535–78 [DOI] [PubMed] [Google Scholar]
- 10.Leong KT, Walton A, Krum H. 2014. Renal sympathetic denervation for the treatment of refractory hypertension. Annu. Rev. Med 65:349–65 [DOI] [PubMed] [Google Scholar]
- 11.Azizi M, Schmieder RE, Mahfoud F, Weber MA, Daemen J, et al. 2018. Endovascular ultrasound renal denervation to treat hypertension (RADIANCE-HTN SOLO): a multicentre, international, single-blind, randomised, sham-controlled trial. Lancet 391:2335–45 [DOI] [PubMed] [Google Scholar]
- 12.Kandzari DE, Böhm M, Mahfoud F, Townsend RR, Weber MA, et al. 2018. Effect of renal denervation on blood pressure in the presence of antihypertensive drugs: 6-month efficacy and safety results from the SPYRAL HTN-ON MED proof-of-concept randomised trial. Lancet 391:2346–55 [DOI] [PubMed] [Google Scholar]
- 13.Townsend RR, Mahfoud F, Kandzari DE, Kario K, Pocock S, et al. 2017. Catheter-based renal denervation in patients with uncontrolled hypertension in the absence of antihypertensive medications (SPYRAL HTN-OFF MED): a randomised, sham-controlled, proof-of-concept trial. Lancet 390:2160–70 [DOI] [PubMed] [Google Scholar]
- 14.Krum H, Schlaich MP, Sobotka PA, Böhm M, Mahfoud F, et al. 2014. Percutaneous renal denervation in patients with treatment-resistant hypertension: final 3-year report of the Symplicity HTN-1 study. Lancet 383:622–29 [DOI] [PubMed] [Google Scholar]
- 15.Symplicity HTN-2 Investigators. 2010. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 376:1903–9 [DOI] [PubMed] [Google Scholar]
- 16.Bhatt DL, Kandzari DE, O’Neill WW, D’Agostino R, Flack JM, et al. 2014. A controlled trial of renal denervation for resistant hypertension. N. Engl. J. Med 370:1393–401 [DOI] [PubMed] [Google Scholar]
- 17.Polhemus DJ, Trivedi RK, Gao J, Li Z, Scarborough AL, et al. 2017. Renal sympathetic denervation protects the failing heart via inhibition of neprilysin activity in the kidney. J. Am. Coll. Cardiol 70:2139–53 [DOI] [PubMed] [Google Scholar]
- 18.Sharp TE, Polhemus DJ, Li Z, Spaletra P, Jenkins JS, et al. 2018. Renal denervation prevents heart failure progression via inhibition of the renin-angiotensin system. J. Am. Coll. Cardiol 72:2609–21 [DOI] [PubMed] [Google Scholar]
- 19.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, et al. 2016. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. J. Heart Fail 18:891–975 [DOI] [PubMed] [Google Scholar]
- 20.Singh JP, Kandala J, Camm AJ. 2013. Non-pharmacological modulation of the autonomic tone to treat heart failure. Eur. Heart J 35:77–85 [DOI] [PubMed] [Google Scholar]
- 21.Papin E, Ambard L. 1924. Resection of the nerves of the kidney for nephralgia and small hydronephroses. J. Urol 11:337–48 [Google Scholar]
- 22.Page IH, Heuer GJ. 1935. The effect of renal denervation on the level of arterial blood pressure and renal function in essential hypertension. J. Clin. Investig 14:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Page IH, Heuer GJ. 1935. The effect of renal denervation on patients suffering from nephritis. J. Clin. Investig 14:443–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smithwick RH, Thompson JE. 1953. Splanchnicectomy for essential hypertension; results in 1,266 cases. J. Am. Med. Assoc 152:1501–4 [DOI] [PubMed] [Google Scholar]
- 25.Schauerte P, Scherlag BJ, Scherlag MA, Goli S, Jackman WM, Lazzara R. 1999. Ventricular rate control during atrial fibrillation by cardiac parasympathetic nerve stimulation: a transvenous approach. J. Am. Coll. Cardiol 34:2043–50 [DOI] [PubMed] [Google Scholar]
- 26.Rippy MK, Zarins D, Barman NC, Wu A, Duncan KL, Zarins CK. 2011. Catheter-based renal sympathetic denervation: chronic preclinical evidence for renal artery safety. Clin. Res. Cardiol 100:1095–101 [DOI] [PubMed] [Google Scholar]
- 27.Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, et al. 2009. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 373:1275–81 [DOI] [PubMed] [Google Scholar]
- 28.Papademetriou V, Stavropoulos K, Doumas M, Tsioufis K. 2019. Now that renal denervation works, how do we proceed? Circ. Res 124:693–95 [DOI] [PubMed] [Google Scholar]
- 29.Metra M, Teerlink JR. 2017. Heart failure. Lancet 390:1981–95 [DOI] [PubMed] [Google Scholar]
- 30.Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. 1986. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 73:615–21 [DOI] [PubMed] [Google Scholar]
- 31.DiBona GF, Esler M. 2010. Translational medicine: the antihypertensive effect of renal denervation. Am. J. Physiol. Regul. Integr. Comp. Physiol 298:R245–53 [DOI] [PubMed] [Google Scholar]
- 32.Ferguson DW, Berg WJ, Sanders JS, Kempf JS. 1990. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: evidence from direct micronenrographic recordings. J. Am. Coll. Cardiol 16:1125–34 [DOI] [PubMed] [Google Scholar]
- 33.Remme WJ. 1995. Neurohormonal modulation in heart failure: ACE inhibition and beyond. Eur. HeartJ 16:73–78 [DOI] [PubMed] [Google Scholar]
- 34.Braunwald E 2015. The path to an angiotensin receptor antagonist-neprilysin inhibitor in the treatment of heart failure. J. Am. Coll. Cardiol 65:1029–41 [DOI] [PubMed] [Google Scholar]
- 35.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, et al. 1999. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. New Engl. J. Med 341:709–17 [DOI] [PubMed] [Google Scholar]
- 36.Rossignol P, Dobre D, McMurray JJ, Swedberg K, Krum H, et al. 2014. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ. Heart Fail 7:51–58 [DOI] [PubMed] [Google Scholar]
- 37.Fitzgerald AA, Powers JD, Ho PM, Maddox TM, Peterson PN, et al. Impact of medication nonadherence on hospitalizations and mortality in heart failure. J. Card. Fail 17:664–69 [DOI] [PubMed] [Google Scholar]
- 38.Polhemus DJ, Gao J, Scarborough AL, Trivedi R, McDonough KH, et al. 2016. Radiofrequency renal denervation protects the ischemic heart via inhibition of GRK2 and increased nitric oxide signaling. Circ. Res 119:470–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang S-N, Chang S-H, Yu C-C, Wu C-K, Lai L-P, et al. 2017. Renal denervation decreases susceptibility to arrhythmogenic cardiac alternans and ventricular arrhythmia in a rat model of post-myocardial infarction heart failure. JACC Basic Transl. Sci 2:184–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang HJ, Wang W, Cornish KG, Rozanski GJ, Zucker IH. 2014. Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 64:745–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng H, Liu X, Sharma NM, Patel KP. 2016. Renal denervation improves cardiac function in rats with chronic heart failure: effects on expression of β-adrenoceptors. Am. J. Physiol. Heart Circ. Physiol 311:H337–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao SY, Zhen Z, Liu Y, Au KW, Lai WH, et al. 2017. Improvement of myocardial function following catheter-based renal denervation in heart failure. JACC Basic Transl. Sci 2:270–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Song L, Li C, Feng Q, Xu M, et al. 2018. Renal denervation improves cardiac function by attenuating myocardiocyte apoptosis in dogs after myocardial infarction. BMC Cardiovasc. Disord 18:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. 2000. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 355:1126–30 [DOI] [PubMed] [Google Scholar]
- 45.Vodovar N, Seronde MF, Laribi S, Gayat E, Lassus J, et al. 2014. Post-translational modifications enhance NT-proBNP and BNP production in acute decompensated heart failure. Eur. Heart J 35:3434–41 [DOI] [PubMed] [Google Scholar]
- 46.Zile MR, Claggett BL, Prescott MF, McMurray JJ, Packer M, et al. 2016. Prognostic implications of changes in N-terminal pro-b-type natriuretic peptide in patients with heart failure. J. Am. Coll. Cardiol 68:2425–36 [DOI] [PubMed] [Google Scholar]
- 47.de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. 1981. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 28:89–94 [DOI] [PubMed] [Google Scholar]
- 48.Sudoh T, Kangawa K, Minamino N, Matsuo H. 1988. A new natriuretic peptide in porcine brain. Nature 332:78–81 [DOI] [PubMed] [Google Scholar]
- 49.Sudoh T, Minamino N, Kangawa K, Matsuo H. 1990. C-type natriuretic peptide (CNP): a new member of natriuretic peptide family identified in porcine brain. Biochem. Biophys. Res. Commun 168:863–70 [DOI] [PubMed] [Google Scholar]
- 50.Yandle TG. 1994. Biochemistry of natriuretic peptides. J. Intern. Med 235:561–76 [DOI] [PubMed] [Google Scholar]
- 51.Nishikimi T, Maeda N, Matsuoka H. 2006. The role of natriuretic peptides in cardioprotection. Cardiovasc. Res 69:318–28 [DOI] [PubMed] [Google Scholar]
- 52.Moro C, Lafontan M. 2013. Natriuretic peptides and cGMP signaling control of energy homeostasis. Am. J. Physiol. Heart Circ. Physiol 304:H358–68 [DOI] [PubMed] [Google Scholar]
- 53.Koller KJ, Goeddel DV. 1992. Molecular biology of the natriuretic peptides and their receptors. Circulation 86:1081–88 [DOI] [PubMed] [Google Scholar]
- 54.King L, Wilkins MR. 2002. Natriuretic peptide receptors and the heart. Heart 87:314–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chinkers M, Singh S, Garbers DL. 1991. Adenine nucleotides are required for activation of rat atrial natriuretic peptide receptor/guanylyl cyclase expressed in a baculovirus system. J. Biol. Chem 266:4088–93 [PubMed] [Google Scholar]
- 56.Airhart N, Yang Y-F, Roberts CT, Silberbach M. 2003. Atrial natriuretic peptide induces natriuretic peptide receptor-cGMP-dependent protein kinase interaction. J. Biol. Chem 278:38693–98 [DOI] [PubMed] [Google Scholar]
- 57.Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, et al. 1997. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. PNAS 94:14730–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Booz GW. 2005. Putting the brakes on cardiac hypertrophy: exploiting the NO-cGMP counter-regulatory system. Hypertension 45:341–46 [DOI] [PubMed] [Google Scholar]
- 59.Kapoun AM, Liang F, O’Young G, Damm DL, Quon D, et al. 2004. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-β in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ. Res 94:453–61 [DOI] [PubMed] [Google Scholar]
- 60.Bayes-Genis A, Barallat J, Richards AM. 2016. A test in context: neprilysin: function, inhibition, and biomarker. J. Am. Coll. Cardiol 68:639–53 [DOI] [PubMed] [Google Scholar]
- 61.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, et al. 2013. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure trial (PARADIGM-HF). Eur. J. Heart Fail 15:1062–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, et al. 2014. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med 371:993–1004 [DOI] [PubMed] [Google Scholar]
- 63.Shen MJ, Zipes DP. 2014. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ. Res 114:1004–21 [DOI] [PubMed] [Google Scholar]
- 64.Opthof T, Misier AR, Coronel R, Vermeulen JT, Verberne HJ, et al. 1991. Dispersion of refractoriness in canine ventricular myocardium. Effects of sympathetic stimulation. Circ. Res 68:1204–15 [DOI] [PubMed] [Google Scholar]
- 65.Tomaselli GF, Zipes DP. 2004. What causes sudden death in heart failure? Circ. Res 95:754–63 [DOI] [PubMed] [Google Scholar]
- 66.Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ. 2003. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J. Am. Coll. Cardiol 42:873–79 [DOI] [PubMed] [Google Scholar]
- 67.Jackson N, Gizurarson S, Azam MA, King B, Ramadeen A, et al. 2017. Effects of renal artery denervation on ventricular arrhythmias in a postinfarct model. Circ. Cardiovasc. Interv 10:e004172. [DOI] [PubMed] [Google Scholar]
- 68.Bradfield JS, Hayase J, Liu K, Moriarty J, Kee ST, et al. 2020. Renal denervation as adjunctive therapy to cardiac sympathetic denervation for ablation refractory ventricular tachycardia. Heart Rhythm 17:220–27 [DOI] [PubMed] [Google Scholar]
- 69.Ukena C, Bauer A, Mahfoud F, Schreieck J, Neuberger HR, et al. 2012. Renal sympathetic denervation for treatment of electrical storm: first-in-man experience. Clin. Res. Cardiol 101:63–67 [DOI] [PubMed] [Google Scholar]
- 70.Hoffmann BA, Steven D, Willems S, Sydow K. 2013. Renal sympathetic denervation as an adjunct to catheter ablation for the treatment of ventricular electrical storm in the setting of acute myocardial infarction. J. Cardiovasc. Electrophysiol 24:1175–78 [DOI] [PubMed] [Google Scholar]
- 71.Davies JE, Manisty CH, Petraco R, Barron AJ, Unsworth B, et al. 2013. First-in-man safety evaluation of renal denervation for chronic systolic heart failure: primary outcome from REACH-Pilot study. Int.J. Cardiol 162:189–92 [DOI] [PubMed] [Google Scholar]
- 72.Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, et al. 2020. Research priorities for heart failure with preserved ejection fraction. Circulation 141:1001–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reddy YNV, Borlaug BA. 2016. Heart failure with preserved ejection fraction. Curr. Probl. Cardiol 41:145–88 [DOI] [PubMed] [Google Scholar]
- 74.Patel RB, Shah SJ. 2019. Drug targets for heart failure with preserved ejection fraction: a mechanistic approach and review of contemporary clinical trials. Annu. Rev. Pharmacol. Toxicol 59:41–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kitzman DW, Hundley WG, Brubaker PH, Morgan TM, Moore JB, et al. 2010. A randomized double-blind trial of enalapril in older patients with heart failure and preserved ejection fraction: effects on exercise tolerance and arterial distensibility. Circ. Heart Fail 3:477–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, et al. 2008. Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med 359:2456–67 [DOI] [PubMed] [Google Scholar]
- 77.Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, et al. 2003. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 362:777–81 [DOI] [PubMed] [Google Scholar]
- 78.Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, et al. 2014. Spironolactone for heart failure with preserved ejection fraction. New Engl. J. Med 370:1383–92 [DOI] [PubMed] [Google Scholar]
- 79.Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, et al. 2019. Angiotensin–neprilysin inhibition in heart failure with preserved ejection fraction. New Engl. J. Med 381:1609–20 [DOI] [PubMed] [Google Scholar]
- 80.Tschöpe C, Lam CSP. 2012. Diastolic heart failure: what we still don’t know. Herz 37:875–79 [DOI] [PubMed] [Google Scholar]
- 81.Borlaug BA, Anstrom KJ, Lewis GD, Shah SJ, Levine JA, et al. 2018. Effect of inorganic nitrite versus placebo on exercise capacity among patients with heart failure with preserved ejection fraction: the INDIE-HFpEF randomized clinical trial. JAMA 320:1764–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, et al. 2017. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur. Heart J 38:1119–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Verloop WL, Beeftink MMA, Santema BT, Bots ML, Blankestijn PJ, et al. 2015. A systematic review concerning the relation between the sympathetic nervous system and heart failure with preserved left ventricular ejection fraction. PLOS ONE 10:e0117332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, et al. 2015. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 131:269–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alex L, Russo I, Holoborodko V, Frangogiannis NG. 2018. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol 315:H934–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schwarzl M, Hamdani N, Seiler S, Alogna A, Manninger M, et al. 2015. A porcine model of hypertensive cardiomyopathy: implications for heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol 309:H1407–18 [DOI] [PubMed] [Google Scholar]
- 87.Valero-Muñoz M, Backman W, Sam F. 2017. Murine models of heart failure with preserved ejection fraction: a “fishing expedition.” JACC. Basic Transl. Sci 2:770–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Riehle C, Bauersachs J. 2019. Small animal models of heart failure. Cardiovasc. Res 115:1838–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, et al. 2012. Animal models of heart failure. Circ. Res 111:131–50 [DOI] [PubMed] [Google Scholar]
- 90.Esler M, Kaye D. 2000. Sympathetic nervous system activation in essential hypertension, cardiac failure and psychosomatic heart disease. J. Cardiovasc. Pharmacol 35:S1–7 [DOI] [PubMed] [Google Scholar]
- 91.Landsberg L, Troisi R, Parker D, Young JB, Weiss ST. 1991. Obesity, blood pressure, and the sympathetic nervous system. Ann. Epidemiol 1:295–303 [DOI] [PubMed] [Google Scholar]
- 92.Kaur J, Young BE, Fadel PJ. 2017. Sympathetic overactivity in chronic kidney disease: consequences and mechanisms. Int. J. Mol. Sci 18:1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaillancourt M, Chia P, Sarji S, Nguyen J, Hoftman N, et al. 2017. Autonomic nervous system involvement in pulmonary arterial hypertension. Respir. Res 18:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen P-S, Chen LS, Fishbein MC, Lin S-F, Nattel S. 2014. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ. Res 114:1500–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brandt MC, Mahfoud F, Reda S, Schirmer SH, Erdmann E, et al. 2012. Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J. Am. Coll. Cardiol 59:901–9 [DOI] [PubMed] [Google Scholar]
- 96.Mahfoud F, Urban D, Teller D, Linz D, Stawowy P, et al. 2014. Effect of renal denervation on left ventricular mass and function in patients with resistant hypertension: data from a multi-centre cardiovascular magnetic resonance imaging trial. Eur. Heart J 35:2224–31 [DOI] [PubMed] [Google Scholar]
- 97.Patel HC, Rosen SD, Hayward C, Vassiliou V, Smith GC, et al. 2016. Renal denervation in heart failure with preserved ejection fraction (RDT-PEF): a randomized controlled trial. Eur. J. Heart Fail 18:703–12 [DOI] [PubMed] [Google Scholar]
- 98.Georgakopoulos D, Little WC, Abraham WT, Weaver FA, Zile MR. 2011. Chronic baroreflex activation: a potential therapeutic approach to heart failure with preserved ejection fraction. J. Card. Fail 17:167–78 [DOI] [PubMed] [Google Scholar]
- 99.Zile MR, Abraham WT, Weaver FA, Butter C, Ducharme A, et al. 2015. Baroreflex activation therapy for the treatment of heart failure with a reduced ejection fraction: safety and efficacy in patients with and without cardiac resynchronization therapy. Eur. J. Heart Fail 17:1066–74 [DOI] [PubMed] [Google Scholar]
- 100.Habib N, Mahmoodi BK, Bos WJ, Tromp SC, Suttorp MJ, et al. 2015. Initial experience with therapeutic geometric modification of the carotid bulb for true resistant hypertension. EuroIntervention 11:117–20 [DOI] [PubMed] [Google Scholar]
- 101.Gold MR, Van Veldhuisen DJ, Hauptman PJ, Borggrefe M, Kubo SH, et al. 2016. Vagus nerve stimulation for the treatment of heart failure: The INOVATE-HF Trial. J. Am. Coll. Cardiol 68:149–58 [DOI] [PubMed] [Google Scholar]
- 102.De Ferrari GM, Stolen C, Tuinenburg AE, Wright DJ, Brugada J, et al. 2017. Long-term vagal stimulation for heart failure: eighteen month results from the NEural Cardiac TherApy foR Heart Failure (NECTAR-HF) trial. Int. J. Cardiol 244:229–34 [DOI] [PubMed] [Google Scholar]
- 103.Victor RG. 2015. Carotid baroreflex activation therapy for resistant hypertension. Nat. Rev. Cardiol 12:451–63 [DOI] [PubMed] [Google Scholar]
- 104.Spiering W, Williams B, Van der Heyden J, van Kleef M, Lo R, et al. 2017. Endovascular baroreflex amplification for resistant hypertension: a safety and proof-of-principle clinical study. Lancet 390:2655–61 [DOI] [PubMed] [Google Scholar]
- 105.Lohmeier TE, Barrett AM, Irwin ED. 2005. Prolonged activation of the baroreflex: A viable approach for the treatment of hypertension? Curr. Hypertens. Rep 7:193–98 [DOI] [PubMed] [Google Scholar]
- 106.Abraham WT, Zile MR, Weaver FA, Butter C, Ducharme A, et al. 2015. Baroreflex activation therapy for the treatment of heart failure with a reduced ejection fraction. JACC Heart Fail. 3:487–96 [DOI] [PubMed] [Google Scholar]
- 107.Søndergaard L, Reddy V, Kaye D, Malek F, Walton A, et al. 2014. Transcatheter treatment of heart failure with preserved or mildly reduced ejection fraction using a novel interatrial implant to lower left atrial pressure. Eur. J. Heart Fail 16:796–801 [DOI] [PubMed] [Google Scholar]
- 108.Hasenfuß G, Hayward C, Burkhoff D, Silvestry FE, McKenzie S, et al. 2016. A transcatheter intracardiac shunt device for heart failure with preserved ejection fraction (REDUCE LAP-HF): a multicentre, open-label, single-arm, phase 1 trial. Lancet 387:1298–304 [DOI] [PubMed] [Google Scholar]
- 109.Rodes-Cabau J, Bernier M, Amat-Santos IJ, Ben Gal T, Nombela-Franco L, et al. 2018. Interatrial shunting for heart failure: early and late results from the first-in-human experience with the V-wave system. JACC Cardiovasc. Interv 11:2300–10 [DOI] [PubMed] [Google Scholar]