

Abstract
Background & Aims:
Immune checkpoint inhibitors have limited efficacy in many tumors. We investigated mechanisms of tumor resistance to inhibitors of programmed cell death 1 (PDCD1, also called PD1) in mice with gastric cancer, and the role of its ligand, PDL1.
Methods:
Gastrin-deficient mice were given N-methyl-N-nitrosourea (MNU) in drinking water along with Helicobacter felis to induce gastric tumor formation; we also performed studies with H/K-ATPase-hIL1B mice, which develop spontaneous gastric tumors at the antral-corpus junction and have parietal cells that constitutively secrete IL1B. Mice were given injections of an antibody against PD1 or an isotype control before tumors developed, or anti-PD1 and 5-fluorouracil and oxaliplatin, or an antibody against lymphocyte antigen 6 complex locus G (also called Gr-1), which depletes myeloid-derived suppressor cells [MDSCs]), after tumors developed. We generated knockin mice that express PDL1 specifically in the gastric epithelium or myeloid lineage.
Results:
When given to gastrin-deficient mice before tumors grew, anti-PD1 significantly reduced tumor size and increased tumor infiltration by T cells. However, anti-PD1 alone did not have significant effects on established tumors in these mice. Neither early nor late anti-PD1 administration reduced tumor growth in the presence of MDSCs in H/K-ATPase-hIL1B mice. The combination of 5-fluorouracil and oxaliplatin reduced MDSCs, increased numbers of intra-tumor CD8+ T cells, and increased the response of tumors to anti-PD1—however, this resulted in increased tumor expression of PDL1. Expression of PDL1 by tumor or immune cells increased gastric tumorigenesis in mice given MNU. Mice with gastric epithelial cells that expressed PDL1 did not develop spontaneous tumors, but they developed more and larger tumors after administration of MNU and H. felis, with accumulation of MDSCs.
Conclusions:
In mouse models of gastric cancer, 5-fluorouracil and oxaliplatin reduce numbers of MDSCs to increase the effects of anti-PD1, which promotes tumor infiltration by CD8+ T cells. However, these chemotherapeutic agents also induce expression of PDL1 by tumor cells. Expression of PDL1 by gastric epithelial cells increases tumorigenesis in response to MNU and H. felis, and accumulation of MDSCs, which promote tumor progression. The timing and site of PDL1 expression is therefore important in gastric tumorigenesis and should be considered in design of therapeutic regimens.
Keywords: stomach cancer, mouse model, immunosuppression, resistance
Graphical Abstract
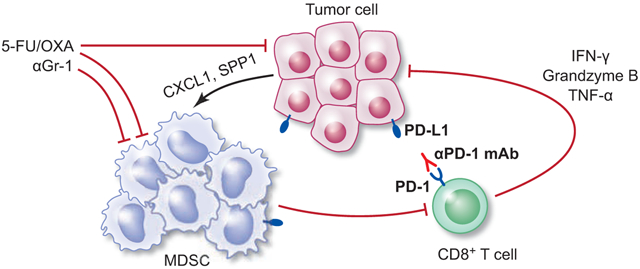
Introduction
Gastric cancer (GC) is the third leading cause of cancer death worldwide1. The prognosis of advanced GC remains poor, with a 5-year survival rate under 30%2. Platinum-fluoropyrimidine combination chemotherapy is considered standard first-line treatment for advanced GC, but chemotherapy has a low complete response rate as well as substantial toxicity3. New therapeutic approaches are urgently needed.
Immune checkpoint blockade (ICB) has yielded important therapeutic benefits against many cancers, yet the clinical activity is limited to a subset of patients4. In 2017, pembrolizumab, a PD-1 inhibitor, was granted accelerated FDA approval for the treatment of recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinomas that express PD-L1, based on a single-armed phase II clinical trial5. PD-L1-expressing tumors had better response rates than PD-L1-negative tumors6, but the objective response rate in PD-L1 expressing tumors was low (11.6%). Similar low objective response rates (11.2%) were seen in the phase 3 ATTRACTION-2 trial of nivolumab in gastro-esophageal cancer patients7. Thus, there is an urgent need to define the roles of PD-1/PD-L1 in immunosuppression.
Patients responsive to ICB often have tumors characterized by pre-existing tumor-infiltrating CD8+ T cells, a higher tumor mutational burden, and/or intratumoral expression of PD-L18. PD-1 ligands are upregulated in many human cancers, and PD-L1/PD-L2 are expressed on human GC9. Amplification of the PD-L1 genes and structural variations disrupting regulatory regions of the PD-L1 gene elevated PD-L1 expression in a subset of GCs10,11. In many malignancies, PD-L1 expression correlates with poor prognosis, suggesting utility as a negative prognostic marker in GC12. However, PD-L1 is expressed in both tumor and tumor-infiltrating immune cells, and clinical responses to PD-1/PD-L1 blockade correlate with both PD-L1 expression in tumor4,13 and in infiltrating immune cells14. Studies of syngeneic mouse models testing the relative role of tumor- versus immune-PD-L1 in mediating anti-PD-1-driven anti-tumor responses yielded mixed results, with several suggesting that expression on tumor cells is critical15-18, whilst other studies pointed to a primary role on non-tumor cells19,20. Of note, these studies all employed implanted, syngeneic models, which have several limitations, most notably the rapid exposure of the host to a plethora of tumor antigens.
Here we evaluate the efficacy of anti-PD-1 in autochthonous mouse models of GC which phenocopy stepwise progression to gastric adenocarcinoma and define the resistance mechanisms to PD-1 blockade. Using conditional PD-L1 knockin mice, we explore the distinct roles of PD-L1 expressed on hematopoietic versus non-hematopoietic cells during gastric carcinogenesis in these physiological models.
Methods
Tumor Models and Treatment
To induce GC, mice received 3-5 cycles of 240 ppm MNU (Gojira Fine Chemicals) in drinking water for a week every other week +/− Helicobacter felis (Hf) inoculation at 6-8 weeks of age21. Treatments used were: anti-PD-1 (mDX400, the murine analog against pembrolizumab; Merck & Co., Inc.) or isotype control at 10 mg/kg by intraperitoneal injection (i.p.) weekly; 5-fluorouracil (5-FU; Sigma) at 30 mg/kg and oxaliplatin (OXA; Tocris) at 5 mg/kg, i.p. biweekly; anti-Gr-1 (RB6-8C5; BioXcell) at 200 μg/mouse, i.p. three times a week.
Additional methods are described in the Supplementary Methods.
Results
Early Anti-PD-1 Treatment Inhibits Tumor Growth in GAS-KO Mice
To study anti-PD-1 therapy against gastric tumors, we induced GC in GAS-KO mice with Hf infection followed by 5 cycles of MNU. MNU-induced GAS-KO mice have microadenocarcinomas in the antrum at 20 weeks post-MNU and later develop large antral tumors that comprise intestinal-type adenocarcinoma21. GAS-KO antral tumors were positive for β-catenin (Figure S1A) and have significantly higher mutation rates than those in WT mice22. Gastric PD-L1 expression was increased as tumors progress, along with PD-1 (Figure 1A). At the protein level, GAS-KO tumors more highly expressed PD-L1 than non-tumor tissues, and the majority of PD-L1-positive cells were immune cells (Figures 1B and S1B-D). Interestingly, a higher density of T cells correlated with high PD-L1 levels in low-grade tumors as opposed to lack of T cells in PD-L1+ high-grade tumors (Figure S1E).
Figure 1. Early anti-PD-1 treatment inhibits tumor growth in GAS-KO mice.

(A) Expression of Pdl1 and Pdcd1 in the antrum following Hf/MNU by qPCR (n = 4/group).
(B) E-cadherin and PD-L1 immunostaining on Hf/MNU/GAS-KO tumors at 30 weeks post-MNU.
(C) Experimental design for early treatment.
(D-E) Gross images (D) and tumor area measured (E) from GAS-KO mice treated with isotype control (n = 8) or anti-PD-1 (n = 6). Dotted lines indicate tumor area.
(F) CD3 immunostaining on treated tumors.
(G-H) The proportion of tumor-infiltrating CD3+ T cells (G) and the subsets (H) among CD45+ cells in treated mice (n = 3-4/group) by flow cytometry.
(I) CD11b immunostaining on treated tumors.
(J-K) The percentage of intratumoral MDSCs (CD11b+Gr-1+) (J), M-MDSCs (CD11b+Ly6ChiLy6G−) and PMN-MDSCs (CD11b+Ly6CloLy6G+) (K) among CD45+ cells from treated mice (n = 3-4/group).
(L) Immunosuppressive activity of PMN-MDSCs isolated from tumors (n = 3/group). Statistically significant differences from No MDSC group.
(M-N) The proportion of macrophages (CD11b+Gr-1−F4/80+) among CD45+ cells (M) and Tregs (CD25+Foxp3+) in CD4+ T cells (N) in treated tumors (n = 3-4/group).
Scale bars, 100 μm (B); 5 mm (D); 50 μm (F and I). Mean ± SEM. one-way ANOVA (A); Student’s t-test (E, G-H and J-N). *P < .05; **P < .01; ***P < .001.
To evaluate early treatment with anti-PD-1, GAS-KO mice were treated before tumors develop starting one week after the last dose of MNU, and subsequently examined at 30 weeks post-MNU (Figure 1C). GAS-KO tumor at 10 weeks was essentially undetectable or else of a microscopic size (< 1 mm2) (Figure S1F) but showed abundant PD-L1 expression compared to controls, in which there were very few PD-L1+ cells (Figure S1G). Early treatment with anti-PD-1 significantly reduced tumor sizes (Figures 1D-E). Anti-PD-1 increased intratumoral T cells, a common outcome with ICB, with significant increases in both CD4+ and CD8+ subsets (Figures 1F-H). Notably, anti-PD-1 markedly decreased tumor-infiltrating myeloid cells, especially CD11b+Gr-1+ cells (Figure 1I-J). In particular, granulocytic cells were significantly decreased, whereas no significant change was seen in the proportion of monocytic cells (Figure 1K). Tumor-infiltrating polymorphonuclear (PMN)-MDSCs had immunosuppressive activity, in that they decreased IFN-γ production by CD8+ T cells (Figure 1L). Additionally, anti-PD-1-treated GAS-KO tumors showed decreased frequency of macrophages as well as regulatory T cells (Tregs) (Figures 1M-N), which have been shown to decline in parallel with decreasing MDSCs23.
To determine whether anti-PD-1 induces systemic anti-tumor immune responses, we analyzed immune cell subsets in peripheral blood and spleen from control and anti-PD-1-treated GAS-KO mice. Although the proportion of circulating and splenic CD3+ T cells was increased in response to anti-PD-1, corresponding changes in T cell subsets were not consistent among blood, spleen and tumors (Figures 1H and S1H). Anti-PD-1 did not affect splenic Tregs (Figure S1I) nor the levels of circulating and splenic myeloid cells (Figure S1J). These data show that early anti-PD-1 treatment of GAS-KO mice has clear local immune effects but limited systemic effects.
Early Anti-PD-1 Treatment of H/K-ATPase-IL-1β Mice Fails to Delay Tumor Growth
Since GAS-KO mice responded to early anti-PD-1 treatment with increased CD8+ T cells and decreased PMN-MDSCs, we further explored MDSCs in immunotherapy of GC using a more MDSC predominant model. We employed H/K-ATPase-IL-1b mice that develop spontaneous gastric tumors at the antral-corpus junction24, with acceleration by Hf/MNU. H/K-ATPase-IL-1β mice via parietal cells constitutively secrete IL-1β, leading to early mobilization and recruitment of MDSCs to the stomach24. Indeed, IL-1β mice showed significantly greater numbers of gastric Gr-1+ cells than GAS-KO mice at the time of early treatment (Figure S2A). Furthermore, IL-1β mice exhibited a high level of Gr-1+ cells even without Hf/MNU while fewer Gr-1+ cells were detected in GAS-KO mice (Figure S2A). There were no differences in tumor burden between GAS-KO and IL-1β mice at the start of early treatment as tumors in IL-1β mice were undetectable or microscopic at 10 weeks (Figure S2B). IL-1β tumors were also positive for β-catenin and PD-L1 and most PD-L1+ cells were immune cells (Figures 2A-B and S2C).
Figure 2. Early anti-PD-1 treatment in H/K-ATPase-IL-1β mice fails to delay tumor growth.

(A) E-cadherin and PD-L1 immunostaining on Hf/MNU/IL-1β tumors at 30 weeks post-MNU.
(B) PD-L1+ cells in EpCAM+ and CD45+ cells isolated from 30-week IL-1β tumors by flow cytometry (n = 3).
(C-D) Gross images (C) and tumor area measured (D) from control (n = 9) or anti-PD-1-treated (n = 14) IL-1β mice. Dotted lines indicate tumor area.
(E) Gr-1 immunostaining on tumors from WT and treated IL-1β mice.
(F-G) Contour plots showing tumor MDSCs (F) and quantification (G) from treated groups (n = 6-8/group).
(H-J) The proportion of MDSC subsets (H), T cells (I-J) in treated tumors (n = 6-8/group).
(K) CD8 immunostaining on treated tumors.
Scale bars, 100 μm (A); 5 mm (C); 50 μm (E and K). Mean ± SEM. Student’s t-test. *P < .05; **P < .01; ****P < .0001.
To test whether increased MDSC density correlated with decreased responsiveness to anti-PD-1, IL-1β mice were given Hf/MNU and treated with anti-PD-1 as described above (Figure 1C). In contrast to GAS-KO mice, anti-PD-1-treated IL-1β mice failed to reduce tumor growth (Figures 2C-D). IL-1β tumors had more abundant Gr-1+ MDSCs than tumors from WT mice (Figure 2E), and in IL-1β tumors, anti-PD-1 did not drive a significant decrease in intratumoral MDSCs due to unchanged PMN-MDSCs (Figures 2E-H). Nevertheless, early anti-PD-1 treatment led to increased intratumoral T cells (Figure 2I). Anti-PD-1 resulted in expansion of CD8+ T cells without a significant increase in CD4+ cells (Figures 2J-K). Together, these results demonstrate that animals with abundant MDSC infiltration do not respond to anti-PD-1, despite increased CD8+ tumor-infiltrating T cells, supporting the notion that MDSCs may mitigate anti-tumor T cell responses promoted by PD-1 blockade.
In IL-1β mice, similar to GAS-KO animals, anti-PD-1 failed to induce systemic effects. Immunophenotyping of blood and splenocytes showed no increase in total T cells or relevant subsets (Figure S2D). No significant changes were observed in splenic Tregs (Figure S2E), nor in circulating of splenic MDSCs (Figure S2F). Overall, these data suggest that the effects of anti-PD-1 in these GC models are primarily local.
Late Treatment with Anti-PD-1 is Effective When Administered in Combination with Chemotherapy
We next performed treatment studies in well-established gastric tumors. We induced tumors in GAS-KO and H/K-ATPase-IL-1β mice with Hf/MNU, and administered anti-PD-1 or chemotherapy (5-FU and OXA), or both starting at 24 weeks post-MNU (Figure 3A). In contrast to the absence of gross tumors or the presence of microscopic tumors at 10 weeks, both mice showed larger tumors at 24 weeks (Figures S1F and S2B). In these established tumors, neither PD-1 monotherapy, nor chemotherapy with a platinum/fluoropyrimidine regimen was effective, but the combination resulted in a significantly decreased tumor burden compared to controls in both GAS-KO and IL-1β mice (Figures 3B-C and S3A-B). Combination treatment was significantly more active than either monotherapy, which in established tumors did not hinder gastric tumor progression. Both control and monotherapy-treated mice developed moderate-to-well differentiated gastric adenocarcinomas, while mice treated with the combination showed dysplasia or almost complete tumor regression (Figures 3D and S3C). Combined treatment decreased expression of the markers of aggressiveness (Ki-67 and nuclear β-catenin), along with enhanced apoptosis induced by chemotherapeutic agents (Figures 3E-G).
Figure 3. Late treatment with anti-PD-1 is effective when administered in combination with chemotherapy.

(A) Experimental scheme for late treatment.
(B-C) Gross images (B) and tumor area measured (C) from GAS-KO mice in indicated treatment groups (n = 12-17/group). Dotted lines indicate tumor area. One-way ANOVA (C).
(D) H&E stains of GAS-KO tumors in different treatment groups. Dashed boxes indicate magnified areas shown at the bottom.
(E-G) Immunostaining for cleaved caspase-3 (E), Ki-67 (F) and β-catenin (G) on treated GAS-KO tumors and quantification (n = 3/group). Arrowheads indicate nuclear β-catenin (G). Student’s t-test.
Scale bars, 5 mm (B); 100 μm (D-G). Mean ± SEM. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Immune Effects of Combination Chemo-Immunotherapy
To elucidate the mechanisms underlying the efficacy of combined treatment, we quantified T cells and MDSCs infiltrated in treated tumors. Combined chemo-immunotherapy significantly increased intratumoral T cells; this increase was driven by an increase in the CD8+ subset (Figures 4A-B and S3D-E). CD8 immunostaining on GAS-KO tumors showed abundant intratumoral infiltrates with combined treatment, as opposed to generally negligible CD8+ T cell infiltration either in control or monotherapy groups (Figure 4B). Importantly, combined treatment led to a significant enrichment of IFN-γ, granzyme B (GzB) and TNF-α-secreting CD8+ T cells (Figure 4C), indicating enhanced effector CD8+ T cell accumulation. Immunostaining also showed that combination treatment decreased infiltration with CD11b+ myeloid cells, of which a fraction were MDSCs (Figures 4D and S3F). Flow cytometric analyses showed that combined chemo-immunotherapy significantly decreased MDSC tumor infiltration (Figures 4E and S3G). In contrast to overall decreases in MDSC subsets in IL-1β mice by combined treatment (Figures S3G), M-MDSCs in GAS-KO mice were neither affected by monotherapy nor by combined treatment (Figure 4E). However, GAS-KO mice receiving combination treatment showed reduced PMN-MDSC infiltration, with a decrease of more than 50%, an effect not mediated by either monotherapy (Figure 4E). Supporting PMN-MDSC depletion as a potential mechanism for the augmentation by chemotherapy, tumor size correlated positively with PMN-MDSC infiltration and negatively with tumor-infiltrating CD8+ T cells (Figure 4F). These data suggest that depleting MDSCs with chemotherapy contributes to the anti-tumor activity of PD-1 blockade in this model.
Figure 4. Immune effects of combination chemo-immunotherapy.

(A) The proportion of intratumoral T cells in treated GAS-KO tumors (n = 8-13/group).
(B) CD8 immunostaining on treated tumors.
(C) Frequency of effector cytokine-secreting CD8+ T cells (n = 3/group).
(D) CD11b immunostaining on treated tumors.
(E) The proportion of MDSCs in treated GAS-KO tumors (n = 8-13/group).
(F) Linear regression analyses between tumor area and the percentage of CD8+ T cells and PMN-MDSCs in GAS-KO tumors (n = 44). r2 and P-values are shown.
(G) MFI (mean fluorescence intensity) of PD-L1 expressed on EpCAM+ cells isolated from GAS-KO tumors treated as indicated (n = 3-5/group).
Scale bars, 50 μm. Mean ± SEM. Student’s t-test. *P < .05; **P < .01; ***P < .001.
To further explore whether the increased MDSCs in these mice drive tumor resistance to anti-PD-1, we targeted MDSCs with anti-Gr-1. GAS-KO tumors were given anti-Gr-1 alone or combined with anti-PD-1 (Figure S4A). Combination treatment significantly decreased gastric tumor growth whereas anti-Gr-1 monotherapy did not affect growth (Figures S4B-C). Anti-Gr-1 depleted CD11b+Gr-1+ cells by approximately 50%, with a marked reduction in PMN-MDSCs (Figures S4D-E). As anticipated, anti-PD-1 increased CD8+ T cell tumor infiltration (Figure S4F). These data show that abundant MDSCs limit the adaptive anti-tumor activity of PD-1 blockade and that even partially depleting MDSCs augments the response rate of GC to ICB.
Generation of R26-LSL-PD-L1-EGFP Mice
Although chemotherapy augmented the activity of anti-PD-1, likely by depleting MDSCs, we observed increased PD-L1 expression on tumor cells in both groups receiving chemotherapy (that is, chemotherapy alone and combined with anti-PD-1) (Figure 4G). In the anti-PD-1-treated groups, PD-L1 upregulation might be expected to occur via a JAK/STAT-dependent mechanism driven by IFN-γ secretion from infiltrating CD8+ T cells8, but it was surprising to observe this with chemotherapy alone. Thus, these data suggested a potentially unique role for tumor cell-expressed PD-L1 in response to therapy in these GC models.
To explore the relative role of tumor cell-expressed PD-L1, we generated knockin mice which conditionally express the mouse Pdl1 gene. To create this mouse line, a loxP-stop-loxP-Pdl1-IRES-EGFP construct was inserted into the Rosa26 (referred to as R26) locus (Figure 5A). To determine the contribution of PD-L1 from tumor epithelial cells versus immune cells in the development of GC, we crossed R26-LSL-Pdl1-EGFP mice with Tff2-Cre25 and LysM-Cre26 mice to target PD-L1 expression specifically to the gastric epithelium and myeloid lineage, respectively. GFP expression marked Pdl1-expressing cells (Figures 5B and S5A) and Cre-expressing R26-PD-L1 mice exhibited high levels of Pdl1 expression in the targeted tissues (Figures S5B-C).
Figure 5. PD-L1 overexpression in gastric epithelial cells promotes gastric tumorigenesis.

(A) Gene construct of R26-LSL-Pdl1-IRES-EGFP mice.
(B) Endogenous GFP expression in the stomach. The targeted tissues by Cre-expressing mice are gastric epithelial cells (Tff2-Cre) and immune/myeloid cells (LysM-Cre).
(C-D) Gross images (C) and tumor area measured (D) from R26-PD-L1 and Tff2-Cre; R26-PD-L1 mice at 36 weeks post-MNU +/− Hf. Dotted lines indicate tumor area (n = 6-11/group).
(E) H&E stains of gastric tumors. Dashed lines indicate intramucosal well-differentiated adenocarcinoma. An arrowhead indicates submucosal immune cell infiltrates.
(F) Ki-67 and β-catenin staining on gastric tissues and quantification (n = 3/group).
(G-H) The proportion of MDSCs by flow cytometry (G) and Gr-1 immunostaining (H) on tumors (n = 5-6/group).
(I) The percentage of intratumoral MDSC subsets (n = 5-6/group).
(J-K) mRNA expression of chemokines in tumors (J, n = 4/group) and isolated EpCAM+ cells from tumors (K, n = 3/group) by qPCR.
(L) The percentage of tumor-infiltrating T cells (n = 5-6/group).
(M) The percentage of cytokine secreting-intratumoral CD8+ T cells (n = 3/group).
Scale bars, 100 μm (B and E-F); 5 mm (C); 50 μm (H). Mean ± SEM. One-way ANOVA (D); Student’s t-test (F, G and I-M). *P < .05; **P < .01; ***P < .001; n.s., not significant.
To assess whether PD-L1 overexpression alone (i.e. in the absence of carcinogen) in gastric epithelium induces histological changes, we examined stomach tissues from R26-PD-L1 and Tff2-Cre; R26-PD-L1 mice at 56 weeks of age. PD-L1-overexpressing animals appeared grossly normal, with no weight loss or other macroscopic signs. Histologically, malignancy was not observed in gastric tissue from Tff2-Cre; PD-L1 mice, although there appeared to be relative hyperplasia. Mild gastric hyperplasia was also observed in R26-PD-L1 and LysM-Cre; R26-PD-L1 mice (Figure S5D). Additionally, few immune cells were detected on gastric tissue from R26-PD-L1 and Tff2-Cre; R26-PD-L1 mice without treatment at baseline. No differences were seen in gastric myeloid and T cells between the groups (Figures S5E-G). Similarly, LysM-Cre; R26-PD-L1 mice did not display changes in gastric immune infiltrates relative to controls (Figure S5E). Together, these data show that, in the absence of other factors, PD-L1 overexpression in gastric epithelium alone is not sufficient to drive tumorigenesis.
Overexpression of PD-L1 in Gastric Epithelial Cells Promotes Inflammation-Driven Gastric Tumorigenesis
Helicobacter infection induces chronic inflammatory immune responses in the gastric mucosa that can progress to the development of GC27. To test for a potential tumor-promoting role of chronic inflammation in PD-L1-overexpressing mice, control and PD-L1-overexpressing mice received MNU +/− Hf and were examined at 36 weeks (Figure S6A). MNU-induced tumors were located mainly in the antrum and appeared as well-differentiated adenocarcinomas (Figures 5C and E). Control mice had small tumors, with the tumor cells confined to the limited area of gastric mucosa (Figures 5C-E). PD-L1 overexpression in gastric epithelial cells promoted tumor growth induced by MNU (Figures 5C-D). In Tff2-Cre; R26-PD-L1 mice, gastric adenocarcinomas were found throughout the mucosa, were rich in stromal cells, with dense immune cell infiltration in the submucosa (Figure 5E). Tff2-Cre; R26-PD-L1 tumors showed higher Ki-67 expressions and increased cytosolic/nuclear β-catenin levels than controls (Figure 5F). In controls, Helicobacter infection accelerated MNU-induced tumor growth (Figures 5C-D). Notably, PD-L1 expression by gastric epithelial cells further potentiated gastric tumorigenesis, even in the setting of Hf/MNU (Figures 5C-D).
PD-L1-Overexpressing Tumors Accumulate MDSCs
Tumors in Tff2-Cre; R26-PD-L1 mice showed significantly increased MDSCs compared to R26-PD-L1 mice (Figures 5G-H). PMN-MDSCs were remarkably increased in PD-L1-overexpressing tumors, accompanied by a trend of increased M-MDSCs (Figure 5I) but the proportion of macrophages was lower than controls (Figure S6B). To address possible mechanisms for the accumulated PMN-MDSCs, we analyzed the expression of chemokines/cytokines involved in MDSC migration and differentiation. Chemokines primarily attracting granulocytes, including Cxcl1 and Cxcl2, and their receptor Cxcr228, were significantly upregulated (Figure 5J), whereas chemokines mediating monocyte migration such as Ccl2 and Ccl5 were not significantly changed (Figure S6C). Furthermore, growth factors and cytokines responsible for granulocyte expansion and differentiation, including Csf2, Csf3, Il1b, and Il6, were dramatically increased (Figure S6D). To determine the source of chemokines/cytokines associated with MDSCs, we sorted EpCAM+ cells from dissociated tumors in Tff2-Cre; R26-PD-L1 and R26-PD-L1 mice and performed qPCR. Compared to controls, PD-L1-overexpressing tumor cells expressed higher levels of Cxcl1, the key chemokine for PMN-MDSC recruitment, as well as Spp1 which has been known to affect recruitment and immunosuppressive activity of MDSCs (Figure 5K). Together, these data suggest that selective upregulation of chemokines mediating granulocyte recruitment may increase PMN-MDSC tumor infiltration in Tff2-Cre; R26-PD-L1 mice.
In contrast to myeloid cell expansion, Tff2-Cre; R26-PD-L1 mice did not show altered frequency of intratumoral T cells (Figure 5L). To determine whether PD-L1-overexpressing tumor cells inhibit T cell responses, we tested T cell function by performing intracellular cytokine staining on isolated tumor-infiltrating CD8+ T cells. Compared with controls, CD8+ T cells from Tff2-Cre; R26-PD-L1 tumors showed decreased expression of IFN-γ and TNF-α as well as a reduced level of GzB (Figure 5M), a cytotoxic granule reflective of CD8+ T cell killing activity. Moreover, in contrast to the higher proportion of PD-L1+ tumor cells, CD11b+PD-L1+ myeloid cells were significantly less in Tff2-Cre; R26-PD-L1 tumors than controls (Figure S6E). To further support that tumor-derived PD-L1 restricts T cell functionality, MNU-induced Tff2-Cre; R26-PD-L1 mice at 24 weeks were treated with anti-PD-1 weekly for 6 weeks, and T cell function assessed by flow cytometry. Anti-PD-1 led to reduction in tumor growth (Figures S6F-G) and partially restored T cell function. Notably, anti-PD-1-treated animals showed increased expression of GzB, LAMP-1 and perforin in CD8+ T cells (Figure S6H), effector molecules for CD8+ T cell cytotoxicity. Together, these data demonstrate that PD-L1-expressing tumor cells restrain functionally active T cells within tumors, highlighting the role of tumor PD-L1 in evasion of immune surveillance.
PD-L1-Overexpressing Myeloid Cells Facilitate Gastric Tumorigenesis by Suppressing Tumor-Infiltrating CD8+ T cells
Targeting PD-L1 expression to the myeloid lineage by crossing R26-PD-L1 mice to LysM-Cre mice also markedly enhanced MNU-induced gastric tumorigenesis (Figures 6A-B). Similar to Tff2-Cre; R26-PD-L1 mice, MNU-induced LysM-Cre; R26-PD-L1 mice had higher levels of tumor MDSCs than controls, with significantly increased PMN-MDSCs and a trend of increased M-MDSCs (Figure 6C). There was indeed a significant correlation between tumor PD-L1 expression scored by CPS and myeloid cells infiltrated in human GCs, and tumors with higher PD-L1 expression appeared highly infiltrated with myeloid cells in PD-L1-positive tumors (Figures S7A-B).
Figure 6. PD-L1-overexpressing myeloid cells facilitate gastric tumorigenesis.

(A-B) Gross images (A) and tumor area measured (B) from R26-PD-L1 and LysM-Cre; R26-PD-L1 mice at 36 weeks post-MNU +/− Hf (n = 5-12/group). Dotted lines indicate tumor area. Scale bars, 5 mm (A).
(C-D) The percentage of tumor-infiltrating MDSCs and T cells (n = 4-9/group).
(E-F) The percentage of Ki-67+ (E), effector cytokine-positive cells (F) in CD8+ T cells isolated from MNU-induced R26-PD-L1 and LysM-Cre; R26-PD-L1 tumors (n = 3/group).
Mean ± SEM. one-way ANOVA (B); Student’s t-test (C-F). *P < .05; **P < .01; ***P < .001; n.s., not significant.
Notably, LysM-Cre; R26-PD-L1 mice showed decreased intratumoral T cells relative to controls; this decrease was driven by a reduction in the CD8+ subset (Figure 6D). Furthermore, there was a marked decrease in Ki-67-positive intratumoral CD8+ T cells (Figure 6E), with reduced CD8+ T cell activation and attenuated anti-tumor immunity. PD-L1-expressing myeloid cells also caused significant decreases in the expression of IFN-γ, TNF-α and IL-2 but not GzB, LAMP-1 and perforin in CD8+ T cells (Figure 6F). In the CD4+ subset, a decrease was seen in only IFN-γ-expressing cells (Figure S8A). These data suggest that the primary effect of host immune cell-PD-L1-mediated inhibition of CD8+ T cells involves blocking their cytokine secretion. Collectively, PD-L1 overexpression on myeloid cells impaired the proliferative activity of intratumoral CD8+ T cells as well as their production of effector cytokines.
Interestingly, forced PD-L1 expression in myeloid cells did not potentiate MNU-induced gastric tumorigenesis under Helicobacter infection (Figures 6A-B). Helicobacter infection combined with MNU increased tumor MDSCs compared to uninfected controls (Figure 6C). The total MDSCs and the subsets from Hf/MNU-induced R26-PD-L1 tumors were comparable to those from MNU-induced LysM-Cre; R26-PD-L1 tumors (Figure 6C). Furthermore, no difference was observed in the proportion of intratumoral T cells between R26-PD-L1 and LysM-Cre; PD-L1 mice in the setting of Hf/MNU (Figure 6D). Helicobacter-infected R26-PD-L1 mice under MNU treatment showed markedly reduced CD8+ T cell infiltration compared to uninfected controls (Figure 6D). Importantly, Hf/MNU-induced R26-PD-L1 tumors had more abundant PD-L1+ cells than the MNU group (Figure S8B), and flow cytometric analyses confirmed a higher level of PD-L1+ cells in tumor-infiltrating myeloid cells in Helicobacter-infected animals (Figure S8C). Collectively, these data show that immunosuppression and tumor promotion by Helicobacter-induced chronic inflammation are comparable in several respects to the effects of PD-L1 overexpression on myeloid cells, and upregulation of PD-L1+ myeloid cells by Helicobacter infection may contribute to gastrointestinal carcinogenesis.
Epithelial-Derived PD-L1 Increases Susceptibility to Chemical Carcinogenesis
We explored whether gastric epithelial PD-L1 expression can directly promote tumorigenesis early in cancer development. We induced epithelial-PD-L1 overexpression in Tff2-Cre; R26-PD-L1 mice and treated them with 3 cycles of MNU, fewer cycles than is needed to optimally initiate tumors, and assessed tumor formation at 24 weeks (Figure 7A). Surprisingly, 10 out of 11 Tff2-Cre; R26-PD-L1 mice (> 90%) developed tumors whereas only 20% of controls exhibited tumors (Figures 7B-C). Tumor sizes were variable within the Tff2-Cre; R26-PD-L1 group, but overall were significantly larger than in controls (Figure 7D), suggesting a more aggressive phenotype. However, LysM-Cre; R26-PD-L1 mice failed to accelerate tumor formation with just 3 cycles of MNU (Figures 7E-F).
Figure 7. Epithelial-derived PD-L1 increases susceptibility to gastric carcinogenesis.

(A) Experimental design to assess susceptibility to MNU-induced gastric tumorigenesis.
(B-D) Gross images (B), tumor incidence rates (C), and tumor area measured (D) in R26-PD-L1 and Tff2-Cre; R26-PD-L1 mice (n = 10-11/group) at 24 weeks post-MNU.
(E-F) Gross images (E) and tumor area measured (F) from R26-PD-L1 and LysM-Cre; R26-PD-L1 mice at 24 weeks (n = 5/group).
(G) Pdl1 expression in antral tissues at 10 weeks by qPCR (n = 3/group).
(H-I) The proportion of T cells (H) and MDSCs (I) in antral tissues from R26-PD-L1 and Tff2-Cre; R26-PD-L1 mice at various time points following MNU induction (n = 3-5/group).
Scale bars, 5 mm (B and E). Mean ± SEM. Student’s t-test. *P < .05; **P < .01; ****p < .0001.
To examine whether the level of PD-L1 may affect differences in the early stage of tumorigenesis, we compared gastric Pdl1 expression among the groups at 10 weeks. Tff2-Cre; R26-PD-L1 mice had a higher level of gastric PD-L1 than LysM-Cre; R26-PD-L1 and R26-PD-L1 mice (Figure 7G). LysM-Cre; R26-PD-L1 mice did not show significantly increased PD-L1 expression relative to R26-PD-L1 mice (Figure 7G) as there are much fewer gastric immune infiltrates following 3 cycles of MNU and less myeloid cells are recruited at a very early time point. To further determine whether tumor initiation and growth in Tff2-Cre; R26-PD-L1 mice involved the early creation of an immunosuppressive microenvironment, we performed immunophenotyping during various stages of tumor progression. While R26-PD-L1 mice showed increased gastric CD8+ T cells over time, Tff2-Cre; R26-PD-L1 mice had much less CD8+ infiltrates, particularly at 10 and 18 weeks (Figure 7H), supporting the notion that PD-L1+ precancerous cells can evade immune surveillance. However, we did not see significantly increased MDSCs until 24 weeks when macroscopic tumors develop (Figure 7I). These findings suggest that epithelial-derived PD-L1 is important in tumorigenesis by enhancing immune escape. In summary, these data demonstrate that PD-L1 expression in epithelial cells render them more susceptible to chemical-induced carcinogenesis, subsequently promoting tumor initiation and progression. PD-L1 on myeloid cells may also play a key role in later tumor progression by modulating the tumor microenvironment, with a lesser role in early tumor development.
Discussion
In this study, we tested the efficacy of anti-PD-1 in autochthonous mouse models of GC, which better recapitulate the complexity of host-tumor interactions with disease progression. While poorly understood, resistance to ICB appears to stem from an immunosuppressive tissue-specific microenvironment that is progressively shaped during tumor development29 and likely contributes to low response rates with ICB6,7,30. KEYNOTE-059, which first tested ICB in GC, was a phase II non-randomized, single arm study of pembrolizumab + 5-FU + cisplatin31, which resulted in an overall response rate of 60% versus the historical ~45% response with 5-FU and cisplatin alone32. The phase 2 ATTRACTION-4 study showed a 76.5% response rates with nivolumab + capecitabine + oxaliplatin33. The phase III KEYNOTE-062 study showed that pembrolizumab alone was noninferior to chemotherapy as first line treatment for advanced/metastatic GC, nor was the combination superior to chemotherapy alone34. Recent industry announcements from the large KEYNOTE-590 and CheckMate-649 Phase 3 clinical trials have reported significant improvements with ICB/chemo combinations in overall survival and progression free survival, consistent with our findings.
Here, we found that early anti-PD-1 treatment potently inhibited gastric tumor growth in GAS-KO mice, while anti-PD-1 at later stages failed to control established tumors. Indeed, recent studies in melanoma and lung cancer have shown better activity for anti-PD-1 given early in the disease course35,36 and neoadjuvant PD-1 blockade was effective by initiating CD8+ T cell immune response earlier in cancer development37. Since GAS-KO tumors have a higher mutation rate in response to MNU22, the responsiveness to ICB may be attributed in part to a larger mutational burden. MNU induces mutagenesis but also modifies histone proteins leading to chromatin remodeling in human GC21. We chose Hf/MNU/GAS-KO model because of the high mutational load and PD-L1 expression, and mimics in some ways the MSI GC subtype, with antral localization, intestinal-type features and hypermutated DNA.
Early anti-PD-1 treatment in GAS-KO mice elicited robust anti-tumor immunity, as demonstrated by enhanced T cell tumor infiltration and markedly reduced PMN-MDSCs. Treatment of late-stage tumors in the same model was ineffective, with robust infiltration of PMN-MDSCs that correlated with a lack of response to anti-PD-1. MDSCs suppress CD8+ T cell immunity by increasing expression of ROS, NO, arginase-1 and PGE-2 and through PD-L1/PD-1 interaction38, and accumulate in tumor-bearing mice and cancer patients39,40. High levels of MDSCs in patients correlated with more advanced GC stage with reduced survival41. These data support the hypothesis that MDSCs may be one important mediator of anti-PD-1 resistance. IL-1β mice, characterized by early recruitment of MDSCs to tumors, were unresponsive to anti-PD-1, further supporting MDSCs as a likely limiting factor for immune-oncology therapy. Anti-PD-1 combined with standard chemotherapeutic drugs depleted MDSCs, while concomitantly enhancing CD8+ T cell expansion and effector cytokine secretion, resulting in delayed cancer progression. MDSC depletion with anti-Gr-1 sensitized gastric tumors to anti-PD-1, similar to chemotherapy, supporting the conclusion that MDSCs contribute to anti-PD-1 resistance, in agreement with recent studies42,43. Our data do not exclude the possible role by other immunosuppressive cells in ICB resistance.
PD-L1 expression is induced by inflammatory cytokines, particularly IFN-γ, although it is also regulated by tumor cell-intrinsic mechanisms8. Indeed, our findings showed prevalent PD-L1 expression with a higher density of T cells in low-grade tumors, reflective of IFN-γ-induced adaptive regulation. However, such a positive correlation of PD-L1 expression and T cell infiltration was not shown in PD-L1+ higher-grade tumors, supporting the notion that PD-L1 expression impairs T cell-mediated anti-tumor immunity and that PD-L1 status along with CD8+ infiltrates could better predict response to immunotherapy. PD-L1 is highly expressed in GC patients and higher levels of tumor PD-L1 significantly correlated with a more advanced tumor stage, bigger tumor size, greater tumor invasion, and distant metastasis44. We found that PD-L1-overexpressing tumors showed aggressive phenotypes and larger adenocarcinoma lesions. Blocking PD-1/PD-L1 axis by anti-PD-1 treatment immediately after MNU exposure significantly reduced tumor lesions, corroborating PD-L1 contribution to early malignant transformation. Further studies are needed to define involvement of PD-L1 in tumor invasion and metastasis.
The relative importance of PD-L1 on hematopoietic versus non-hematopoietic cells in tumors has been debated. While some studies showed that PD-L1 on tumor cells is sufficient for immune evasion15,18, others suggested that myeloid-derived PD-L1 is more important19,20. Our data demonstrate that overexpression of PD-L1 in either immune or non-immune cells can inhibit anti-tumor immunity and promote tumorigenesis. Indeed, we found that tumor-derived PD-L1 potently attenuated markers of cytotoxic function in CD8+ T cells as well as cytokine production, supporting PD-L1 as a “molecular shield” on tumor cells, preventing cytolysis by T cells45,46. Myeloid-derived PD-L1 strongly suppressed T cell proliferation and production of IFN-γ, TNF-α, and IL-2, suggesting that infiltrating myeloid cells can respond extremely rapidly, preempting CD8+ effector T cell responses. Recent studies have shown that PD-L1 knockdown in human GC cell lines inhibited tumor growth and increased sensitivity to T cell-mediated killing47, augmenting T cell activity in myeloid cells48. Thus, the sum total of PD-L1 expression in the two cell compartments may determine the timing and level of immunosuppression.
Although myeloid-expressed PD-L1 failed to enhance much the tumor-promoting effects of chronic inflammation, tumor-derived PD-L1 further accelerated tumor progression, suggesting a potentially non-redundant role of PD-L1 expressed on tumor and host immune cells16,17. The importance of epithelial-PD-L1 expression was further supported by our observation that PD-L1-expressing gastric epithelial cells were more susceptible to low-dose chemical-induced carcinogenesis, leading to early onset of tumorigenesis and subsequent MDSC accumulation. PD-L1+ precancerous cells evaded immune surveillance during the early phase of tumor establishment, suggesting a more direct and earlier role for PD-L1 in cancer development. Evidence suggests PD-L1 expression confers intrinsic signals to some types of cancer cells, favoring tumor progression by promoting cancer stemness, cell proliferation and invasion, epithelial-to-mesenchymal transition, and chemoresistance49. Emerging tumors may benefit from PD-L1 expression in order to overcome one or more of the roadblocks to cancer initiation. At later stages, tumor-derived PD-L1 may limit further anti-tumor immunity and promote tumorigenesis.
In summary, our findings showed that PD-L1 expression on either tumor or immune cells enhanced carcinogen-induced gastric tumorigenesis. Since PD-1 blockade alone is often insufficient to generate effective anti-tumor responses resulting in low clinical response rates, combining PD-1 blockade with MDSC targeting may succeed in overcoming resistance to ICB. A better understanding of the distinct roles of PD-L1 expressed on tumor and host immune cells will be essential for the future design of effective cancer therapy.
Supplementary Material
What you need to know:
Background and Context: Immune checkpoint inhibitors have limited efficacy in many tumors. This study investigated mechanisms of tumor resistance to inhibitors of programmed cell death 1 (PDCD1, also called PD1) in mice with gastric cancer, and the role of its ligand, PDL1.
New Findings: In mouse models of gastric cancer, 5-fluorouracil and oxaliplatin reduced numbers of MDSCs to increase the effects of anti-PD1, which promoted tumor infiltration by CD8+ T cells. However, these chemotherapeutic agents also induced expression of PDL1 by tumor cells. Expression of PDL1 by gastric epithelial cells increased tumorigenesis in response to MNU and H. felis, and accumulation of MDSCs, which promote tumor progression.
Limitations: This study was performed in mice; further studies are needed in humans.
Impact: The timing and site of PDL1 expression is therefore important in gastric tumorigenesis and should be considered in design of therapeutic regimens.
Lay Summary: This study identified mechanisms by which gastric tumors in mice escape the effects of a class of anti-tumor drugs called immune checkpoint inhibitors. The study identified cell types that can be depleted from tumors to increase the efficacy of these drugs.
Acknowledgments:
We thank Bryana R. Belin for assistance in maintenance of mouse colonies; Chyuan-Sheng Lin for pronuclear DNA injection; Merck & Co., Inc. for providing mDX400.
Funding: This research was funded in part through the NIH/NCI Cancer Center Support Grant P30CA013696 and used the resources of the Herbert Irving Comprehensive Cancer Center Flow Cytometry Shard Resources and the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University. T.C.W. received grants from the NIH (R35CA210088) and the Merck Foundation. H.J.N. was supported by a German Research Foundation grant (NI 1810/1-1). Y.H. was supported by JSPS, P-CREATE, and PRIME grants.
Abbreviations:
- GC
gastric cancer
- ICB
immune checkpoint blockade
- PD-1
programmed cell death-1
- PD-L
programmed cell death-ligand
- GAS-KO
gastrin-deficient
- MDSC
myeloid-derived suppressor cell
- Hf
Helicobacter felis
- MNU
N-methyl-N-nitrosourea
- Treg
regulatory T cell
- 5-FU
5-fluorouracil
- OXA
oxaliplatin
- WT
wild-type
- IFN-γ
interferon-gamma
- GzB
granzyme B
- TNF-α
tumor necrosis factor-alpha
- LAMP-1
lysosomal-associated membrane protein 1
- IL-2
interleukin-2
Footnotes
Disclosures:
The authors disclose no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2016, National Cancer Institute; Bethesda, MD, https://seer.cancer.gov/csr/1975_2016/, based on November 2018 SEER data submission, posted to the SEER web site, April 2019. 2019. [Google Scholar]
- 3.Smyth EC, Verheij M, Allum W, et al. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016;27:v38–v49. [DOI] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fashoyin-Aje L, Donoghue M, Chen H, et al. FDA Approval Summary: Pembrolizumab for Recurrent Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Adenocarcinoma Expressing PD-L1. Oncologist 2019;24:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs CS, Doi T, Jang RW, et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol 2018;4:e180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:2461–2471. [DOI] [PubMed] [Google Scholar]
- 8.Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018;48:434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Teng F, Kong L, et al. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther 2016;9:5023–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature 2016;534:402–6. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiang X, Yu PC, Long D, et al. Prognostic value of PD -L1 expression in patients with primary solid tumors. Oncotarget 2018;9:5058–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juneja VR, McGuire KA, Manguso RT, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleinovink JW, Marijt KA, Schoonderwoerd MJA, et al. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology 2017;6:e1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau J, Cheung J, Navarro A, et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun 2017;8:14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noguchi T, Ward JP, Gubin MM, et al. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res 2017;5:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Wei S, Hurt EM, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 2018;128:805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang H, Liang Y, Anders RA, et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest 2018;128:580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomita H, Takaishi S, Menheniott TR, et al. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology 2011;140:879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang W, Wang H, Kim W, et al. Hormonal Suppression of Stem Cells Inhibits Symmetric Cell Division and Gastric Tumorigenesis. Cell Stem Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Takemoto Y, Deng H, et al. Histidine decarboxylase (HDC)-expressing granulocytic myeloid cells induce and recruit Foxp3(+) regulatory T cells in murine colon cancer. Oncoimmunology 2017;6:e1290034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14:408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubeykovskaya Z, Si Y, Chen X, et al. Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nat Commun 2016;7:10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clausen BE, Burkhardt C, Reith W, et al. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999;8:265–77. [DOI] [PubMed] [Google Scholar]
- 27.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest 2007;117:60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Sun H, Wei J, et al. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res 2017;77:3655–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 2016;16:121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shitara K, Ozguroglu M, Bang YJ, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 2018;392:123–133. [DOI] [PubMed] [Google Scholar]
- 31.Bang YJ MK, Fuchs CS, et al. KEYNOTE-059 cohort 2: Safety and efficacy of pembrolizumab (pembro) plus 5-fluorouracil (5-FU) and cisplatin for first-line (1L) treatment of advanced gastric cancer. J Clin Oncol 2017;35:4012–4012.28934000 [Google Scholar]
- 32.Al-Batran SE, Hartmann JT, Probst S, et al. Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: a study of the Arbeitsgemeinschaft Internistische Onkologie. J Clin Oncol 2008;26:1435–42. [DOI] [PubMed] [Google Scholar]
- 33.Boku N, Ryu MH, Kato K, et al. Safety and efficacy of nivolumab in combination with S-1/capecitabine plus oxaliplatin in patients with previously untreated, unresectable, advanced, or recurrent gastric/gastroesophageal junction cancer: interim results of a randomized, phase II trial (ATTRACTION-4). Ann Oncol 2019;30:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shitara K, Van Cutsem E, Bang YJ, et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribas A, Hamid O, Daud A, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016;315:1600–9. [DOI] [PubMed] [Google Scholar]
- 36.Markowitz GJ, Havel LS, Crowley MJ, et al. Immune reprogramming via PD-1 inhibition enhances early-stage lung cancer survival. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forde PM, Chaft JE, Smith KN, et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med 2018;378:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Youn JI, Nagaraj S, Collazo M, et al. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008;181:5791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elliott LA, Doherty GA, Sheahan K, et al. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front Immunol 2017;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Chang EW, Wong SC, et al. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol 2013;190:794–804. [DOI] [PubMed] [Google Scholar]
- 42.Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med 2014;6:237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu X, Horner JW, Paul E, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017;543:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Zhang X-H, Bei S-H, et al. PD-1/PD-L1 antagonists in gastric cancer: Current studies and perspectives. World Journal of Meta-Analysis 2019;7:101–109. [Google Scholar]
- 45.Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azuma T, Yao S, Zhu G, et al. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008;111:3635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Chen L, Xiong Y, et al. Knockdown of PD-L1 in Human Gastric Cancer Cells Inhibits Tumor Progression and Improves the Cytotoxic Sensitivity to CIK Therapy. Cell Physiol Biochem 2017;41:907–920. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Chen W, Xu ZP, et al. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front Immunol 2019;10:2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong P, Xiong Y, Yue J, et al. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front Oncol 2018;8:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.