

Abstract
Atrial fibrillation (AF) is the most common cardiac arrhythmia diagnosed in clinical practice. Even though hypertension, congestive heart failure, pulmonary disease, and coronary artery disease are the potential risk factors for AF, the underlying molecular pathology is largely unknown. The reversion of the mature cardiomyocytes to fetal phenotype, impaired ketone body metabolism, mitochondrial dysfunction and the cellular effect of reactive oxygen species (ROS) are the major underlying biochemical events associated with the molecular pathology of AF. On this background, the present manuscript sheds light into these biochemical events in regard to the metabolic derangements in cardiomyocyte leading to AF; especially with respect to structural, contractile, and electrophysiological properties. In addition, the article critically reviews the current understanding, potential demerits, and translational strategies in the management of AF.
Keywords: Atrial fibrillation, cardiomyocyte metabolism, cardiomyocyte, cardiac metabolism, arrhythmias
Article Summary:
In this critical review, we discussed critical molecular and cellular events, including fetal metabolic phenotype of glycolytic metabolism over fatty acid metabolism, involvement of ketone body metabolism, mitochondrial dysfunction and the cellular effect of reactive oxygen species (ROS) in the underlying biochemical events associated with the molecular pathology of atrial fibrillation, especially with respect to structural, contractile, and electrophysiological properties. In addition, the article critically reviews the current understanding, potential demerits, and translational strategies in the management of AF.
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia diagnosed in clinical practice and represents a substantial financial burden [1]. AF is characterized by the electrocardiographic (ECG) features consisting of an “irregularly irregular,” non-repetitive R-R interval and non-distinct P waves. Hypertension, congestive heart failure, pulmonary disease, and coronary artery disease are the potential risk factors for AF. Patients are often asymptomatic; however, they may present with symptoms of shortness of breath, heart palpitations, angina, and fatigue. Untreated AF results in reduced cardiac output and thromboembolic events, such as stroke and atrial appendage thrombus formation [2–4]. Initial management consists of strategies that control the rate and/or rhythm including ß-blockers, calcium channel blockers, or antiarrhythmic drugs. In addition, due to the increased risk of thrombus, anti-coagulation is often used. For patients with persistent or refractory AF, radiofrequency catheter ablation has shown higher efficacy rates when compared to antiarrhythmic drugs [5]. Epidemiologically, AF is increasing in the general population, with an additional increase of prevalence in men and with increasing age [6–8]. Generally, the pathophysiology of AF consists of three stages: initiation of the arrhythmia, maintenance of the arrhythmia, and progression of the disease to longer lasting forms [9, 10].
The management of atrial fibrillation depends on the clinical history and evaluation of individual patient and may require either the restoration of normal sinus rhythm or control ventricular rate. The rate and rhythm control can be managed by pharmacological therapies, invasive electrophysiologic interventions, including electrical cardioversion (for rhythm control), catheter-based ablation, and surgery [11]. In the pharmacological therapies, beta blockers or non-dihydropyridine calcium channel blockers are more effective than digoxin to control ventricular rate in patients with paroxysmal, persistent, or permanent atrial fibrillation, whereas amiodarone is the most effective drugs to control abnormal rhythm. Anticoagulants, including both vitamin K antagonist (warfarin) and non-vitamin K antagonists (dabigatran, rivaroxaban and apixaban), and a combination of aspirin and clopidogrel are used in patients with atrial fibrillation and a risk for stroke [11].
Despite the availability of various treatment strategies, the exact molecular mechanisms underlying AF pathology are yet to be identified. Several potential biomarkers have been identified in recurrent AF. These biomarkers include damage associated molecular patterns, heat shock proteins, inflammatory cytokines, non-inflammatory markers, markers of inflammatory cell activity, and markers of collagen deposition and metabolism [12]. However, abnormalities in cardiomyocyte metabolism have been suspected to play a critical role [13].
In order to understand the potential pathologic characteristic in cardiomyocyte, it is important to understand normal cardiomyocyte metabolism. Depending on the physiological situation, the human heart relies on diverse energy substrates for the production of adenosine triphosphate (ATP). Generally, mature cardiomyocytes are adaptable to synthesize ATP from many substrates based on abundance, though cardiomyocytes predominantly utilize fatty acids as substrates [14]. In addition to the abundancy of various substrates, the type of cardiomyocyte metabolism depends on transcriptional regulation and post-translational modification of the crucial proteins in metabolic pathways. However, the cardiomyocyte function is altered due to the pathological nature of AF leading to altered structural, contractile, and electrophysiological properties of cardiomyocytes.
Harada et al. identified four metabolic changes through recent proteomic and metabolic studies that include reversion to the fetal phenotype, increased ketone body metabolism, 5’ AMP-activated protein kinase (AMPK) activation, and mitochondrial dysfunction and reactive oxygen species (ROS) production [13]. In this manuscript we critically reviewed how reversion of fetal phenotype, increased ketone body metabolism, and mitochondrial dysfunction and reactive oxygen species affect cardiomyocyte metabolism leading to AF. Specifically, how cardiomyocytes are altered with respect to structural, contractile, and electrophysiological properties. Understanding how cardiomyocyte metabolism is altered during AF may lead to identifying novel therapeutic targets to develop treatment approaches.
Fetal Phenotype
During cardiac development, cardiomyocytes undergo varying metabolic energy demands that are characteristic to a specific stage in development [15]. While proliferating cardiomyocytes primarily utilize glycolysis for energy demands, the differentiated cardiomyocytes depend primarily on oxidative metabolism [16–18]. In specific stages of AF, differentiated cardiomyocytes switch back to the fetal phenotype of increased glycolytic metabolism and decreased oxidative metabolism [14, 19]. This section addresses the metabolic similarities between the fetal cardiomyocyte metabolic phenotype and the metabolic phenotype that arises from the pathophysiology due to atrial fibrillation.
Normal Cardiomyocyte Metabolic Phenotype Progression
The process of cardiogenesis requires cardiomyocyte proliferation, and the mechanism of fetal cardiomyocyte proliferation has been shown to be different from the metabolism of differentiated, mature cardiomyocytes. Even in the presence of adequate levels of oxygen, proliferating cardiomyocytes utilize an energy metabolism characterized by higher rates of glycolysis and lactate production. This phenotype may be particularly useful for the biosynthesis of cellular amino acids, lipids, nucleotides, and other macromolecules, all of which are important for distribution to daughter cells during mitosis [16]. Cellular growth and proliferation elicit a lower rate of fatty acid β-oxidation and increased rate of lipid synthesis, primarily from glucose-derived carbon [20, 21]. The proliferation of hematopoietic cells is characterized by increased metabolic flux through glycolysis and lactate production, in addition to a decrease in oxygen consumption, which is indicative of a switch from oxidative metabolism to anaerobic metabolism [22]. Also, this metabolic phenotype during cellular proliferation is consistent with the expression of various enzymes that promotes glycolysis and represses fatty acid β-oxidation [21, 22].
For the proper transition from embryonic stem cells to differentiated cardiomyocytes, increase in energy demand channels a switch from glycolytic metabolism to mitochondrial oxidative phosphorylation [17, 18, 23]. The Warburg effect explains that while the mitochondrial oxidative phosphorylation maximizes ATP production, the glycolytic metabolism functions under circumstances when O2 is either present or scarce [16]. Mitochondrial maturation, within the context of cellular proliferation and differentiation, provides insight into circumstances that lead to the transition of glycolytic metabolism to mitochondrial oxidative phosphorylation.
Generally, the pluripotent embryonic cells and undifferentiated stem cells are characterized by small/immature mitochondria. However, in differentiated cells, the mitochondria are characterized by the initiation of oxidative phosphorylation, supported by increased mitochondrial membrane potential, oxygen consumption, and ATP production [17, 18]. In addition, there is a downregulation of glycolytic enzymes coupled with the upregulation of the enzymes involved in the electron transport chain and citric acid cycle. As energy substrate metabolism switches from glycolytic to oxidative phase, there is a loss of pluripotency. However, under hypoxic conditions, embryonic stem cells remain in a proliferative and pluripotent state, suggesting the switch from glycolytic to oxidative metabolism drives the differentiation of embryonic stem cells into mature cardiomyocytes [17, 24, 25]. Furthermore, this suggests that the level of expression of genes involved in glycolytic and oxidative phosphorylation (including their regulators) is important in determining metabolic phenotype during cardiac development.
Metabolic Change to Fetal Phenotype in AF
Cardiomyocyte development mainly utilizes glycolytic metabolism as an energy source, and switches to rely on fatty acid oxidation following the differentiation of cardiomyocytes. Interestingly, since glucose metabolism is more energy efficient, cardiomyocytes undergoing pathological stress switch to the fetal phenotype for metabolic needs (Figure 1) [15]. Such metabolic switch is observed under specific stages of AF. For instance, during a transcriptomic study, patients with permanent AF displayed increased gene expression resembling the fetal metabolic phenotype (glycolytic-related gene expression) compared to patients in sinus rhythm [19].
Figure 1:
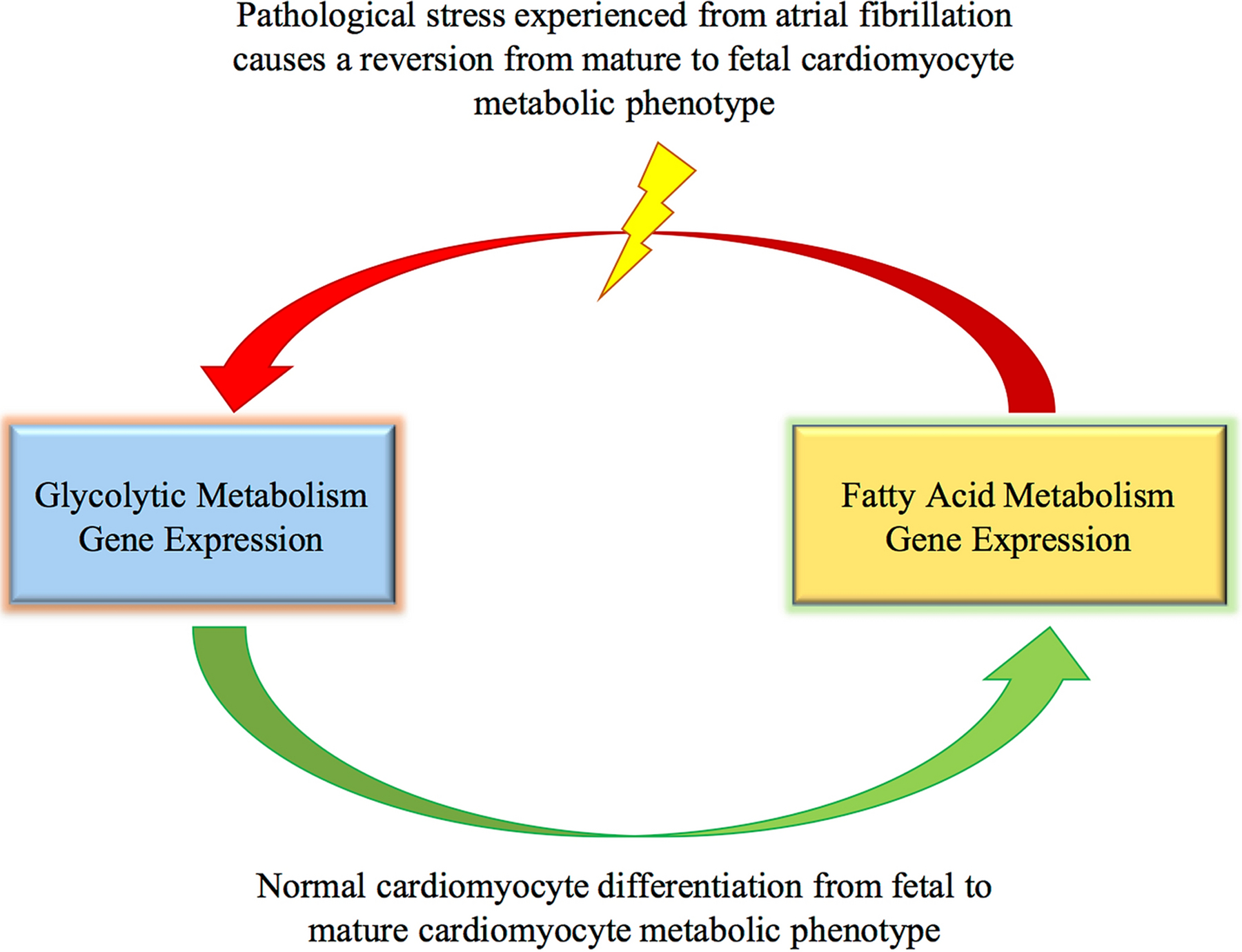
Processes of cardiomyocyte metabolic gene expression, both normally and in AF. The fetal cardiomyocyte metabolic phenotype relies on glycolytic metabolism. As the cardiomyocytes differentiates, they switch to the mature cardiomyocyte metabolic phenotype of fatty acid metabolism. When cardiomyocytes experience pathological stress, they revert back to glycolytic metabolism. This occurs in certain stages of AF.
Structural Remodeling Due to Metabolic Switch in AF
Normal cardiomyocytes in the post-natal period undergo low levels of renewal [26]. Upon encountering growth stimuli, the responses elicited by cardiomyocytes consists of hypertrophy, but not proliferation. Cardiomyocyte hypertrophy occurs in response to increased workload, either pressure and/or volume overload, in order to normalize cardiomyocyte wall stress. It has been shown that when an adult heart hypertrophies, it reverts back to the fetal phenotype of energy metabolism. This reversion consists of increased glycolytic activity and decreased fatty-acid oxidation [27–30]. Cellular hypertrophy along with inflammation, atrial dilation, and fibrosis contributes to AF [31]. In addition, these cellular conditions that initiate AF worsen the severity of AF, showing their critical effect throughout the pathogenesis of AF [32]. In order to appropriately understand the metabolic aspects of the structural changes associated with AF, it is important to identify the actual physiological stages (initiation, maintenance, or progression) associated with the fetal metabolic phenotype. Also, it is critical to establish the associations between the fetal phenotype and specific clinical situations associated with AF. For example, the activity of a glycolytic enzyme (1,6 bisphosphate aldolase) increases during permanent AF [33]. It is possible that the fetal metabolic phenotype is associated with certain stages of AF (either/both physiologically or clinically) and not others, which is important for understanding the pathophysiology of the disease and for possible treatments.
This review explores how transcriptional regulation of hypoxia-inducible factors (HIF-1)α, peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α, peroxisome proliferator-activated receptors (PPAR)α, and PPARγ/δ are involved in glycolytic and fatty acid metabolism, as well as their potential to contribute to AF metabolic pathophysiology.
Transcriptional Regulation/Pathways Involved in Cardiac Metabolic Phenotype: HIF-1α Pathway
Hypoxia-inducible factors (HIFs) are involved in numerous cellular processes, including energy substrate metabolism. HIF-1α regulates the expression of genes involved in a shift that favors the instigation and maintenance of anaerobic glycolysis [34–38]. Therefore, HIF-1α is very important for regulating the metabolic phenotype of the fetal heart, which exists in a low-oxygen environment. HIF-1α is also important for metabolic regulation in an immediate newborn heart, which is very dependent on glycolysis for generating ATP (Table 1) [39, 40]. HIF-1α maintains and promotes glycolysis by upregulating lactate dehydrogenase (LDH)-A, which converts pyruvate to lactate. Furthermore, HIF-1α upregulates pyruvate dehydrogenase kinase (PDK)-1, which restricts the oxidation of pyruvate and keeps it available to be converted into lactate by LDH-A [41]. In short, the regulations and activation imparted by HIF-1α on its target genes show its potential importance on regulating phenotypic metabolic progression from a fetal phenotype (glycolytic) to the postnatal phenotype (oxidative) [13].
Table 1: Potential Involvement of Pathways in Expression of Fetal Metabolic Phenotype in AF.
This table shows the known activity of pathways involving the metabolic regulators HIF-1α, PGC-1α/PPARα, and PGC-1α/ PPARγ/δ. It also provides hypotheses regarding how the expression of these metabolic regulators may be involved in the metabolic switch to the fetal phenotype in AF.
| Fetal Phenotype Pathways | Activity and Function | Possible/Proposed Involvement in AF |
|---|---|---|
| HIF-1α |
|
|
| PGC-1α/PPARα |
|
|
| PGC-1α/PPARγ/δ |
|
|
Transcriptional Regulation/Pathways Involved in Cardiac Metabolic Phenotype: PGC-1α/PPARα axis
Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α and peroxisome proliferator-activated receptors (PPAR)α are transcriptional regulators for fatty acid metabolism, especially in tissues that rely on fatty acid metabolism, such as the heart, kidney, liver, and skeletal muscle [42, 43]. The heart is a tissue with a high mitochondrial density, and therefore has a higher oxidative capacity. PGC-1α has been found to be expressed in tissues with higher amounts of mitochondria and oxidative capacity [44–46]. It has been described as the master regulator of mitochondrial biogenesis and is significant in coordinating and driving energy metabolism, fatty acid oxidation, gluconeogenesis, glucose transport, glycogenolysis, peroxisomal remodeling, oxidative phosphorylation and angiogenesis [47–49]. PPARα expression has been shown to be limited to tissues with higher rate of fatty acid oxidation, such as the heart. The function of PPARα is to enhance fatty acid uptake, β-oxidation, and ketogenesis [50, 51]. Upon formation of a heterodimer with retinoid X receptor (RXR), PPARα regulates the expression of genes involved in fatty acid activation, mitochondrial fatty acid uptake, and fatty acid oxidation. The upregulation of PPARα increases cardiac fatty acid β-oxidation, and its downregulation leads to a decrease in cardiac fatty acid oxidation [52, 53]. PPARα acts by binding to PPAR response elements (PPREs), which are present in the promotor regions of the affected genes. The binding of PGC-1α to the PPARα/RXR heterodimer as a coactivator facilitates the transcriptional regulation [54, 55]. In the heart, PPARα upregulates the transcription of acyl-CoA oxidase, carnitine palmitoyl-transferase 1, carnitine palmitoyl-transferase 2, long-chain acyl-CoA synthetase and medium-chain acyl-CoA dehydrogenase [56]. These transcription factors are crucial for the progression of metabolic phenotype as PPARα and PGC-1α expression increase during the neonatal period, (which requires maximal mitochondrial oxidative capacity) [44, 57, 58].
PGC-1α upregulates the expression of PPAR-α and PPAR-γ/δ. Multiple gene and proteins are associated with both PPAR-α and PPAR-γ/δ. Key genes/proteins involved in fatty acid oxidation are shown in Figure 2. Both PPAR-α and PPAR-γ/δ are altered in both the transcriptomics and proteomics profiles in the atrial tissue of AF patients. The exact role of individual protein/gene in their contribution to the phenotype of AF has not yet been well delineated. However, many of the proteins and genes downstream of PPAR-α and PPAR-γ/δ could serve as potential biomarkers for the disease pathogenesis and progression in AF.
Figure 2:
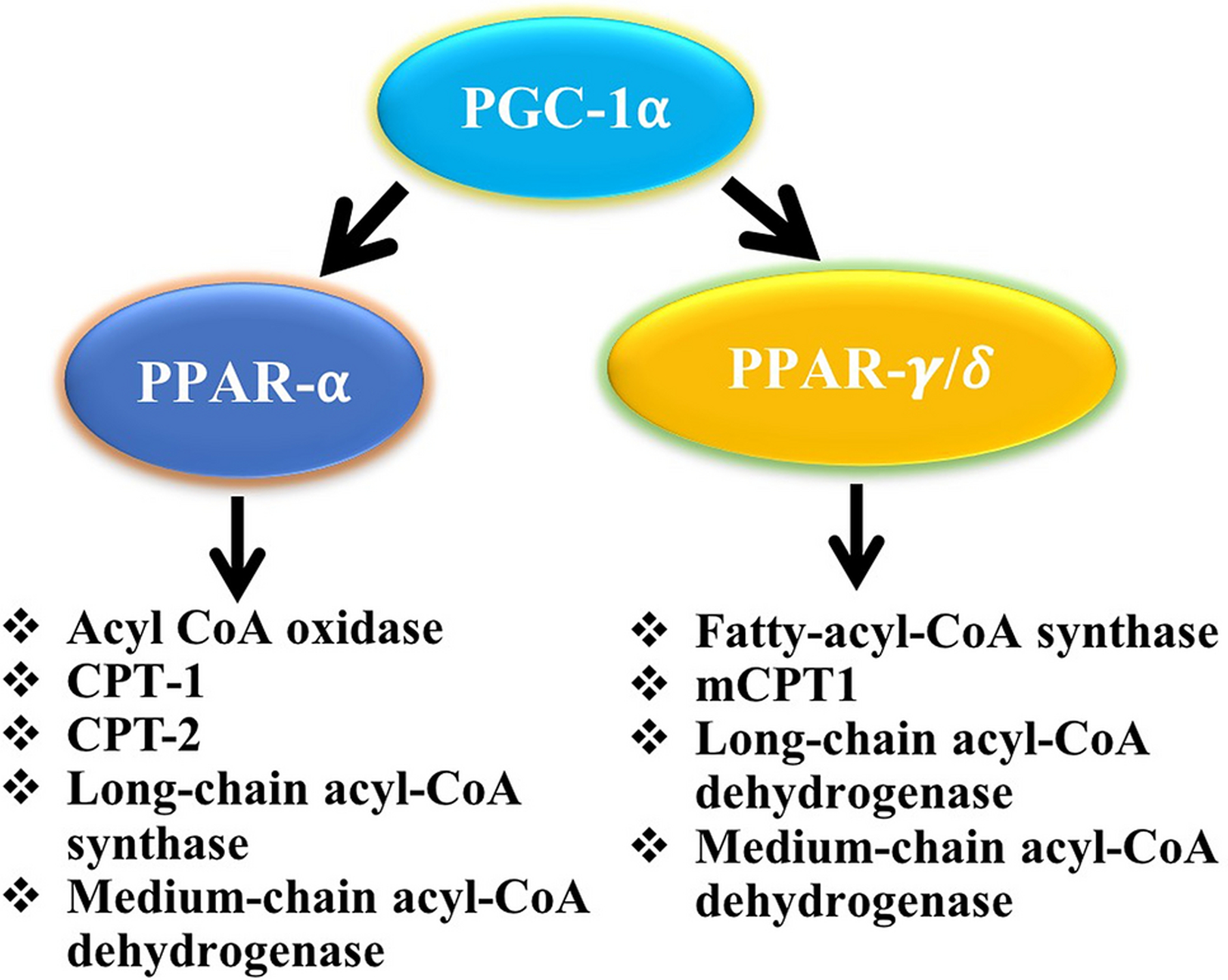
Flow diagram showing the upregulation of PPAR-α and PPARγ/δ expression and downstream fatty acid oxidation-related key proteins/genes in response to the transcriptional activity of PGC-1α. Both PPAR-α and PPARγ/δ are altered in both the transcriptomics and proteomics profiles in the atrial tissue of AF patients.
In vivo, hypoxic conditions are accompanied by a downregulation in genes associated with fatty acid metabolism, including PPARα, malonyl-CoA decarboxylase, and mast cell protease 1 (mCPT1) [59]. In addition, HIF-1α decreases the DNA-binding activity of the PPARα/RXR heterodimer (Figure 3) [59–64] suggesting a dynamic/reciprocal relationship between PPARα and HIF-1α and their respective targets. This reciprocal, interactive relationship promotes the progression from glycolytic metabolism to fatty acid metabolism during cardiomyocyte metabolism, as well as the decrease in fatty acid metabolism and increase in glycolytic metabolism as seen in cardiac hypertrophy and hypoxic conditions.
Figure 3:
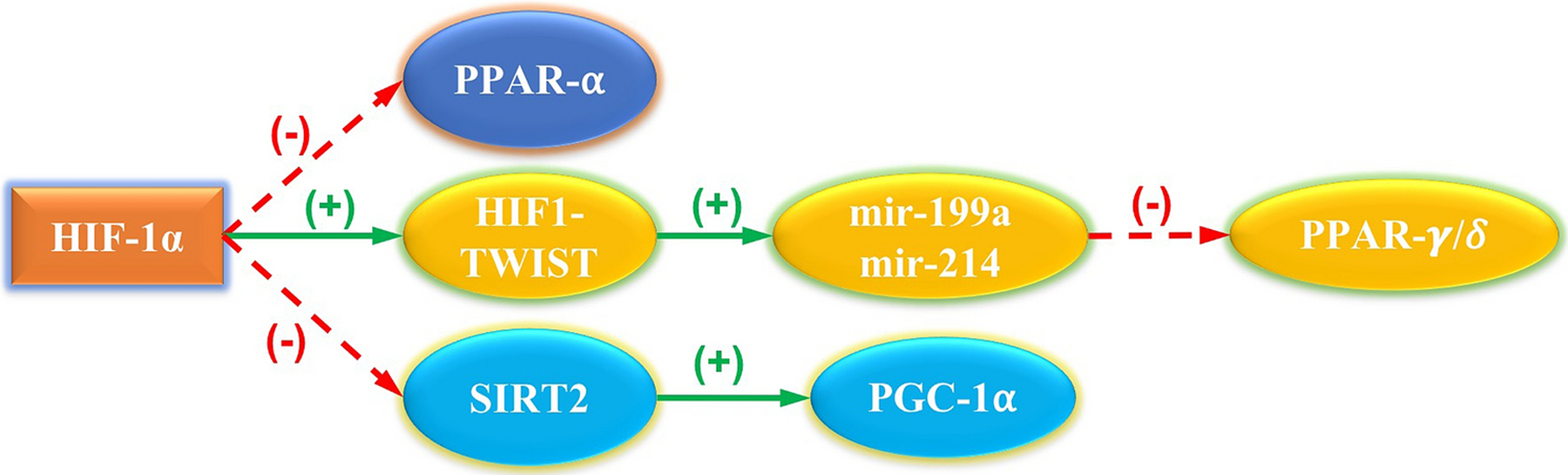
HIF-1 regulation of transcriptomic modulators. HIF-1 has been shown to downregulate gene expression for PPARα [59–61]. HIF-1 directly binds with the TWIST protein, forming the HIF-1/TWIST axis [62]. The HIF-1/TWIST axis activates mir-199a and mir214, which downregulate the expression of PPARγ/δ [63]. HIF-1 represses the transcription of sirtuin 2 (SIRT2), leading to a lack of deacetylation of PGC-1α [64].
Transcriptional Regulation/Pathways Involved in Cardiac Metabolic Phenotype: PGC-1α/PPARβ/δ
PGC-1α is also a coactivator for PPARβ/δ, which is another isoform of PPAR that is present in cardiac muscle. Similar to PPARα, PPARβ/δ has also been shown to upregulate fatty acid metabolism and increase the rate of myocardial fatty acid utilization [65–67]. There seems to be some redundancy in this regulation, as both pathways upregulate the expression of fatty acyl-CoA synthase, mCPT1, long chain acyl CoA dehydrogenase and medium chain acyl-CoA dehydrogenase [65, 68, 69]. However, in comparison to PPARα, PPARβ/δ instigates an increased expression of genes related to myocardial glucose metabolism. In addition, cardiac specific overexpression of PPARβ/δ increases the rate of myocardial glucose oxidation [70]. Moreover, PPARβ/δ regulates the expression of PPARα and PGC-1α and contributes to the hypoxia-induced downregulation of CPT1 expression in cardiomyocytes [60].
Contractile Remodeling Due to Metabolic Switch in AF
The cardiac hypertrophy that occurs in AF results in numerous cellular changes, including alterations in gene expression. In AF patients, there appears to be a myofilament isoform-switch from alpha-myosin heavy chain (α-MHC) to beta-myosin heavy chain (β-MHC) in atrial tissues [71]. This switch is representative of an embryonic phenotype with the purpose of improving the local metabolic economy. α-MHC, the predominant isoform in adult cardiomyocytes, requires more ATP expenditure and oxygen consumption in order to produce a higher-velocity muscle movement in comparison to β-MHC, which is the predominant isoform in embryonic hearts. In patients with permanent AF, atrial tissue shows an increased expression of beta-chain tropomyosin, which is a slow-contracting myofilament with low energy consumption, in addition to increased expression of myosin light-chain embryonic atrial muscle isoform. [33]. The decrease in oxygen consumption that occurs in switching to the β-MHC is similar to the decreased oxygen consumption in glycolytic metabolism compared to fatty acid oxidation.
Electrophysiological Remodeling Due to Metabolic Switch in AF
The conduction system in the heart expresses important electrophysiological proteins in a developmental manner [72]. However, there is no current evidence suggesting the expression pattern of electrophysiological proteins in AF is linked to more energy efficient methods of metabolism, or that compares it to the pattern of expression of electrophysiological proteins during specific developmental stages of cardiomyocytes. However, the reversion to a fetal metabolic phenotype is characterized by a process of dedifferentiation, which includes mitochondrial changes. Just as mitochondria-driven cardiomyocyte energetics strongly affect the regulation of ion channels, the patterns of ionic current impact mitochondrial function [73–79]. It is possible that ionic alterations induce mitochondrial changes that are reminiscent of the fetal metabolic phenotype. Further research is warranted to determine whether a relationship exists between the expression of electrophysiological proteins in AF and energy efficient methods of metabolism or protein expression in the fetal phenotype of cardiomyocyte development.
Ketone Bodies
Ketone bodies serve as energy substrates in both normal and pathophysiological states, such as AF. The information regarding the relationship between ketone body metabolism and the structural, contractile, and electrophysiological changes found in the metabolism AF tissues are limited. Ketone bodies are synthesized in the liver from acetyl-CoA that is mainly derived from fatty acid oxidation. During physiological states of limited carbohydrates and a surplus of fatty acid availability, ketone bodies are transported to extrahepatic tissues for terminal oxidation [80–82]. The major ketone bodies include acetoacetate, β-hydroxybutyrate, and acetone and the physiological states that normally utilize ketone body oxidation include the neonatal period, starvation, post-exercise, and adherence to low-carbohydrate diets [80, 83, 84]. In terms of metabolic processes, hepatic ketogenesis is involved in fatty acid β-oxidation, the tricarboxylic acid (TCA) cycle, and gluconeogenesis. In addition to energy metabolism, ketone bodies serve as substrates to lipogenic and sterol biosynthesis in multiple tissues, including the liver, lactating mammary glands, and the developing brain [85–87].
Structural Remodeling in AF Related to Ketone Bodies
Studies monitoring the levels of metabolic proteins in AF showed a differential expression of 3-oxoacid transferase, a key mitochondrial enzyme utilized in ketolytic energy production [88]. In addition, the findings found an increase in levels of β-hydroxybutyrate and ketogenic amino acids (such as tyrosine and glycine), both of which form acetoacetate and fumarate during catabolism. Furthermore, fumarate levels were elevated in cases of persistent AF, indicating a potential involvement of ketone bodies in persistent AF [88]. It is important to note that ketone bodies contain a higher inherent energy than glucose sources, and data suggests that ketone bodies may present a negative feedback on glucose metabolism in AF [88, 89]. Therefore, it is important to recognize the relationship between ketone body metabolism and glucose metabolism throughout the progression of AF.
Ketone body levels are elevated in patients with dilated and hypertrophic cardiomyopathies [90]. Similarly, hypertrophied and failing hearts showed reduced levels of fatty acid oxidation and upregulation of ketone body oxidation. Interestingly, the ketone bodies and fatty acids are competitive substrates for the normal human heart. It is hypothesized that reduced levels of fatty acids are responsible for the decreased oxidation in most cardiomyopathies including AF. The increased ketone body oxidation occurs due to enhanced delivery of ketone bodies to the failing heart, in addition to increased ketone body synthesis [91–94]. Moreover, the evidence from metabolite profiling and labeled substrate profiling support the shift to utilizing ketone bodies as an alternate fuel source [95].
Contractile Remodeling in AF Related to Ketone Bodies
Despite the evidence suggesting that ketone bodies exerts negative feedback on glucose metabolism in AF, an upregulation of glycolytic enzymes during persistent AF via anaplerosis as a mechanism has been proven [96]. This mechanism, which occurs under normoxic conditions, replaces TCA cycle intermediates by carboxylating pyruvate to oxaloacetate and malate. The anaplerotic reaction is particularly important during prolonged periods of ketone body oxidation, when the Krebs cycle is disrupted due to sequestration of CoA. This sequestration of ACoA inhibits the energy production necessary to sustain normal cardiac function, showing how the metabolic changes in persistent AF contributes to a self-perpetuating state [97].
Electrophysiological Remodeling in AF Related to Ketone Bodies
Patients with AF undergoing cardiac surgery display increased levels of myocardial β-hydroxybutyrate and CoA transferase in comparison to control patients [88]. Even though myocardial ketone body utilization has not yet been associated with cardiomyocytes, it has been shown to affect excitability in murine substantia nigra pars reticulata. It has been reported that β-hydroxybutyrate reduced the firing of GABAergic neurons by opening KATP channels localized to the substantia nigra pars reticulata [98]. It is possible that ketone bodies may affect excitability in the heart, which could contribute to an arrhythmia. Related to cardiomyocytes, L-β-hydroxybutyrate is measured in myocardial extracts, although it does not circulate and likely results from the hydrolysis of L-β-hydroxybutyrate-CoA, a β-oxidation intermediate [99–102]. However, hepatic ketogenesis only produces D-β-hydroxybutyrate, which is the only form compatible as a substrate for oxidation. There is limited research regarding the effect of ketone bodies on the electrophysiological properties of myocardial metabolism, as well as how that may relate to AF. Further research is warranted regarding the potential pathophysiological implications of the L-β-hydroxybutyrate and D-β-hydroxybutyrate stereoisomers on myocardial metabolism in AF [103].
Mitochondrial Dysfunction and ROS Generation
Reactive Oxygen Species (ROS) are reactive and highly unstable molecules containing oxygen that are mostly generated via the mitochondrial electron transport chain (ETC) and oxidative phosphorylation. ROS function as an intermediate in the catalysis of reactions involving metal and to eliminate foreign microbes in the body by the immune system in a process called oxidative/respiratory burst. However, the hyperactivation of ROS lead to cell damage/death mainly by altering membrane and DNA integrity. Interestingly, the ROS hyperactivity is monitored and regulated by the antioxidant defense system which acts by one of three mechanisms, including: scavenging and destroying existing ROS, inhibiting the production of ROS, and reversing the damage done to tissue by ROS. Also, the alterations in the homeostasis between ROS and antioxidants results in pathophysiologic changes in the body. Non-homeostatic levels of ROS have been implicated as a factor associated with AF, with mitochondrial dysfunction closely linked with excess production of ROS.
Structural Remodeling in AF Related to Mitochondrial Dysfunction and ROS Generation
Increased oxidative stress and selective down-regulation of electron transport chain (ETC) activity contributes to the progression of the substrate for AF [104–106]. Though, mitochondrial dysfunction increases oxidative stress, which in turn is arrhythmogenic [105, 107–109]. Patients with AF displayed increased oxidative stress in atrial tissue accompanied by mitochondrial dysfunction with reduced ETC activity that predominantly affected complexes I and II [110]. This phenomenon does not provide evidence of a cause and effect relationship for the development of AF; however, it is a factor that contributes to the progression of the AF substrate. In addition, Cox5b, a ETC enzyme complex responsible for ATP biosynthesis, experiences a decrease in protein expression in the atrial tissue of AF patients compared to sinus-rhythm patients, which links impaired ETC function and energy production with AF [107]. However, further research is warranted to determine the involvement of oxidative stress associated with mitochondrial dysregulation in the cause and effect dynamics of AF.
A recent study that analyzed the relationship between diabetes and atrial fibrillation identified molecular tendencies that involved oxidative stress. First, hyperglycemia and oxidative stress were found to increase atrial fibroblast activation and proliferation, which in turn lead to fibrosis of cardiac tissue and cardiac dysfunction, especially AF. Second, during AF development, excessive ROS production mainly originated from NADPH oxidase (NOX) activity. Third, activation of ROS induced expression of the mitogen-activated protein kinase (MAPK) signaling pathway, which included phospho-c-Jun N-terminal kinase (JNK), p38, phospho-p38, and matrix metalloproteinase 9 (MMP9) [111]. These findings provide insight into the mechanism of AF development in the setting of diabetes and may provide mechanistic implications for AF in other co-morbidities as well.
In addition, mitochondrial DNA (mDNA) deletions are associated with AF. Since aging is also associated with both mDNA deletions and AF, that makes it difficult to discern the relationship between mDNA deletions and AF (mDNA) [112]. Increased ROS induces oxidative damage that accumulates over time, resulting in mDNA deletions [113, 114]. In the most common mDNA deletion, mDNA4977-mut, the mutation deletes a sequence encoding subunits of ATPase and NADH dehydrogenase, which disrupts aerobic metabolism and leads to increased oxidative stress [115, 116]. This deletion is associated with structural remodeling of the left atria in AF. Studies showed that AF patients with the mDNA4977-mut deletion had elevated left atrial size and left ventricle filling pressure, both of which occur as compensation to oxidative stress [117–119]. However, the study indicated that the mDNA4977-mut deletion is more likely to be associated with aging and other factors as opposed to the influence of AF [112]. Alteration of non-mitochondrial gene expression also contributes to the oxidative stress. Case-control studies revealed decreased oxidative phosphorylation and altered gene expression in human atrial biopsies from the patients with chronic AF [71, 120]. In addition, post-operative AF patients were found to have significantly decreased expression of genes involved in oxidation/reduction reactions that contributes to cellular oxidative stress [121].
Contractibility Remodeling in AF Related to Mitochondrial Dysfunction and ROS Generation
Mihm et al. demonstrated that oxidative modification of myofibrillar proteins is increased in atrial myocytes of AF patients due to the loss of fibrillar protein function resulting from oxidative modification [71]. Myofibrillar energetic controllers such as myofibrillar creatine kinase (MM-CK) are proven to be highly vulnerable to oxidative inhibition and the AF patients exhibited a unique impairment of atrial MM-CK activity, while atrial MM-CK content, myosin-ATPase activity, and total CK activity were not altered [122]. Moreover, the MM-CK impairment results in the accumulation of ADP/decreased myofibrillar ATP production [71]. As mentioned in previous sections, myofilament isoform-switching from α-MHC to β-MHC is characteristic of AF which enhances the local metabolic economy of cardiomyocytes. Also, impaired MM-CK activity has been highly correlated with β-MHC expression in AF tissues [71]. Additionally, oxidative stress dependent activities of peroxynitrate led to decreased MM-CK function, and the myosin-heavy-chain nitration (a form of oxidative stress) has been correlated with β-MHC expression [71, 122]. These findings show the relation between oxidative stress and the pathophysiology for contractile changes associated with AF.
Electrophysiological Remodeling in AF Related to Mitochondrial Dysfunction and ROS Generation
Oxidative stress has also been shown to contribute to the electrophysiological remodeling associated with AF. Carnes et al. demonstrated that fibrillating atrial tissue showed increased rates of carbonylation, a pathological outcome of oxidative stress [123]. On a wider scope, Carnes showed involvement of oxidative and nitrosative stress in a rapid atrial pacing model, which provides evidence of increased concentrations of ROS and reactive nitrogen species (RNS) in tachycardias resulting in the impairment of myocardial energetic and electrophysiologic properties [123, 124]. Furthermore, the oxidative stress contributes to changes in atrial electrophysiological properties, including shortened atrial action potential.
A potential electrophysiological manifestation of ROS in AF is mediated by NOX activation. In post-operative AF, oxidative stress from ischemia, reperfusion and mechanical stretch during cardiac surgery may promote electrical remodeling. For example, mechanical stretch induces angiotensin II (Ang II), which acts on multiple ion channels by stimulating Ang II receptor 1. This interaction destabilizes Kv4.3 (potassium channel) mRNA of cardiac myocytes by activating NOX [125, 126]. Ang II is also a potent stimulator of NOX. NOX promotes ROS generation and oxidative modification of protein targets, including activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) [127–129]. CaMKII activation leads to calcium overload via activation of L-type calcium channel and ryanodine receptor 2 hyperphosphorylation. Oxidized CaMKII was increased in atria of AF patients, supporting the possibility of ROS induced CaMKII activation in AF [129, 130]. Moreover, NOX contributes to electrophysiological remodeling through H2O2 production, which has been proven to trigger irregular firing of cardiomyocytes [131].
The presence of mDNA deletions in AF are also associated with more accelerated electro-anatomical remodeling [113–116]. In AF induced hypoxia of the atrium, increased generation of oxidative radicals leading to a deterioration of mitochondrial function (as mDNA damage accumulated) has been observed. The level of ATP in atrial tissue then falls, leading to impaired calcium handling, increased calcium in the cytoplasm, and reduced L-type calcium current [105]. When the atria were electrically remodeled, it led to a cycle in which AF begets AF [32]. However, further investigations are warranted to establish the underlying pathophysiological mechanism of oxidative stress on the contractile changes associated with AF.
Future Directions
Further investigative work is warranted to closely distinguish in what pathophysiological situations (AF initiation, maintenance, and progression) and clinical situations (paroxysmal, persistent, and permanent AF) the fetal phenotype manifests so that potential therapies can be developed to prevent the initiation and progression of AF at different stages of the disease. In tandem with this idea, greater clarification is required regarding manifestation of the fetal phenotype along with structural, contractile, and electrophysiological changes that are associated with different stages of AF. Further research is warranted to analyze how the expression of different PPAR proteins and HIF-1α results in switches between fatty acid oxidation and glycolytic metabolism, including vice versa. Furthermore, the mechanism or pattern of expression regarding the proteins involved in switching back and forth between glycolytic metabolism and fatty acid metabolism, in addition to the identification of potential targets for intervention warrant further investigation.
Limited research exists regarding the involvement of ketone body metabolism in AF. So far, reports indicate that ketone bodies may be involved in persistent AF [88]. Further research is needed to understand the involvement of ketone bodies in various stages of AF. Also, further research is warranted to determine if the L-β-hydroxybutyrate stereoisomer has any pathological effects in human cardiomyocytes [103]. To date, there have been many attempts to correlate the association of ROS with AF, however, the exact underlying molecular mechanism has yet to be determined. Further study is needed to elucidate the role of ROS, specifically, is ROS an initiating factor in the pathogenesis of AF or is the creation of ROS simply a byproduct of a concurrent disease process. Furthermore, the increased expression of MAPK signaling pathway via increased ROS from NOX activation proved insightful for the mechanism of AF development in diabetes, though the implication for this mechanism in non-diabetes settings remains unknown which warrants further investigation. In addition, the mechanism and role casual/contributory factors of AF associated mitochondrial changes in the pathogenesis of AF need to be unveiled. Better understanding of various ROS species and mitochondrial DNA variation on the metabolic nature of AF would pave the ways for the identification of novel therapeutic targets to improve AF treatment and prevention.
Conclusion
The metabolic alterations that occur in AF lend insight to the pathophysiology of AF. AF shows a switch to the fetal metabolic phenotype of glycolytic metabolism over fatty acid metabolism. Numerous metabolic enzymes, including HIF-1α, PGC-1α/PPARα, and the PGC-1α/PPARβ/δ experience changes in expression levels, providing support and potential for further investigation regarding the physiologic changes that occur in AF. Increased levels of ketone bodies, notably β-hydroxybutyrate and ketogenic amino acids, were found in persistent AF. Due to the regulatory nature between glycolytic metabolism and ketone body metabolism, further investigation is warranted to determine their expression status contributing to the progression and persistence of AF. ROS in cardiomyocytes can lead to disruption of ETC, fibrosis of cardiac tissue, and showed relation with mDNA deletions, of which can contribute to the pathogenesis of AF. Further research should place emphasis on associating findings with either the pathophysiological AF classifications (initiation, maintenance, progression) and/or the clinical AF classifications (new onset, paroxysmal, persistent, long standing persistent, permanent).
Funding
This work was supported by research grants R01 HL128063, R01HL144125, and R01HL147662 to DK Agrawal from the National Heart, Lung and Blood Institute, National Institutes of Health, USA. The content of this original research article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Disclosure
The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
Data Availability Statement: All scientific information in this critical review article is gathered from published reports. No new data are presented. Any other information presented in this article will be shared on reasonable request to the corresponding author.
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Wann LS, January CT, Ellenbogen KA, et al. (2011) 2011 ACCF/AHA/HRS Focused Update on the Management of Patients With Atrial Fibrillation (Updating the 2006 Guideline): A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 123:104–123. 10.1161/CIR.0b013e3181fa3cf4 [DOI] [PubMed] [Google Scholar]
- 2.Wood AJJ, Pritchett ELC (1992) Management of Atrial Fibrillation. New England Journal of Medicine 326:1264–1271. 10.1056/NEJM199205073261906 [DOI] [PubMed] [Google Scholar]
- 3.Lip GY, Metcalfe MJ, Rae AP (1993) Management of paroxysmal atrial fibrillation. Q J Med 86:467–472 [DOI] [PubMed] [Google Scholar]
- 4.Disch DL (1994) Managing Chronic Atrial Fibrillation: A Markov Decision Analysis Comparing Warfarin, Quinidine, and Low-Dose Amiodarone. Annals of Internal Medicine 120:449 10.7326/0003-4819-120-6-199403150-00001 [DOI] [PubMed] [Google Scholar]
- 5.Calkins H, Reynolds MR, Spector P, et al. (2009) Treatment of atrial fibrillation with antiarrhythmic drugs or radiofrequency ablation: two systematic literature reviews and meta-analyses. Circ Arrhythm Electrophysiol 2:349–361. 10.1161/CIRCEP.108.824789 [DOI] [PubMed] [Google Scholar]
- 6.Go AS, Hylek EM, Phillips KA, et al. (2001) Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 285:2370–2375. 10.1001/jama.285.18.2370 [DOI] [PubMed] [Google Scholar]
- 7.Heeringa J, van der Kuip DAM, Hofman A, et al. (2006) Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J 27:949–953. 10.1093/eurheartj/ehi825 [DOI] [PubMed] [Google Scholar]
- 8.Chugh SS, Havmoeller R, Narayanan K, et al. (2014) Worldwide Epidemiology of Atrial Fibrillation: A Global Burden of Disease 2010 Study. Circulation 129:837–847. 10.1161/CIRCULATIONAHA.113.005119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wakili R, Voigt N, Kääb S, et al. (2011) Recent advances in the molecular pathophysiology of atrial fibrillation. Journal of Clinical Investigation 121:2955–2968. 10.1172/JCI46315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nattel S, Guasch E, Savelieva I, et al. (2014) Early management of atrial fibrillation to prevent cardiovascular complications. European Heart Journal 35:1448–1456. 10.1093/eurheartj/ehu028 [DOI] [PubMed] [Google Scholar]
- 11.January CT, Wann LS, Alpert JS, et al. (2014) ACC/AHA Task Force Members. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society [erratum in Circulation. 2014;130(23):e270–271]. Circulation. 130(23):2071–2104. doi: 10.1161/CIR.0000000000000040. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg JH, Werner JH, Plitt GD, et al. (2019) Immunopathogenesis and biomarkers of recurrent atrial fibrillation following ablation therapy in patients with preexisting atrial fibrillation. Expert Rev Cardiovasc Therapy 17(3):193–207. doi: 10.1080/14779072.2019.1562902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harada M, Melka J, Sobue Y, Nattel S (2017) Metabolic Considerations in Atrial Fibrillation ― Mechanistic Insights and Therapeutic Opportunities ―. Circulation Journal 81:1749–1757. 10.1253/circj.CJ-17-1058 [DOI] [PubMed] [Google Scholar]
- 14.Kolwicz SC, Purohit S, Tian R (2013) Cardiac Metabolism and its Interactions With Contraction, Growth, and Survival of Cardiomyocytes. Circulation Research 113:603–616. 10.1161/CIRCRESAHA.113.302095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopaschuk GD, Jaswal JS (2010) Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol 56:130–140. 10.1097/FJC.0b013e3181e74a14 [DOI] [PubMed] [Google Scholar]
- 16.Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 324:1029–1033. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung S, Dzeja PP, Faustino RS, et al. (2007) Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med 4 Suppl 1:S60–67. 10.1038/ncpcardio0766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho YM, Kwon S, Pak YK, et al. (2006) Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun 348:1472–1478. 10.1016/j.bbrc.2006.08.020 [DOI] [PubMed] [Google Scholar]
- 19.Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129. 10.1152/physrev.00006.2004 [DOI] [PubMed] [Google Scholar]
- 20.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metabolism 7:11–20. 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 21.DeBerardinis RJ, Lum JJ, Thompson CB (2006) Phosphatidylinositol 3-Kinase-dependent Modulation of Carnitine Palmitoyltransferase 1A Expression Regulates Lipid Metabolism during Hematopoietic Cell Growth. Journal of Biological Chemistry 281:37372–37380. 10.1074/jbc.M608372200 [DOI] [PubMed] [Google Scholar]
- 22.Bauer DE, Harris MH, Plas DR, et al. (2004) Cytokine stimulation of aerobic glycolysis in hematopoietic cells exceeds proliferative demand. The FASEB Journal 18:1303–1305. 10.1096/fj.03-1001fje [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung S, Arrell DK, Faustino RS, et al. (2010) Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. Journal of Molecular and Cellular Cardiology 48:725–734. 10.1016/j.yjmcc.2009.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forristal CE, Wright KL, Hanley NA, et al. (2010) Hypoxia inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. REPRODUCTION 139:85–97. 10.1530/REP-09-0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ezashi T, Das P, Roberts RM (2005) Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA 102:4783–4788. 10.1073/pnas.0501283102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergmann O, Bhardwaj RD, Bernard S, et al. (2009) Evidence for Cardiomyocyte Renewal in Humans. Science 324:98–102. 10.1126/science.1164680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allard MF, Schonekess BO, Henning SL, et al. (1994) Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. American Journal of Physiology-Heart and Circulatory Physiology 267:H742–H750. 10.1152/ajpheart.1994.267.2.H742 [DOI] [PubMed] [Google Scholar]
- 28.Schönekess BO, Allard MF, Henning SL, et al. (1997) Contribution of glycogen and exogenous glucose to glucose metabolism during ischemia in the hypertrophied rat heart. Circ Res 81:540–549 [DOI] [PubMed] [Google Scholar]
- 29.Wambolt RB, Henning SL, English DR, et al. (1999) Glucose utilization and glycogen turnover are accelerated in hypertrophied rat hearts during severe low-flow ischemia. J Mol Cell Cardiol 31:493–502. 10.1006/jmcc.1998.0804 [DOI] [PubMed] [Google Scholar]
- 30.El Alaoui-Talibi Z, Guendouz A, Moravec M, Moravec J (1997) Control of oxidative metabolism in volume-overloaded rat hearts: effect of propionyl-L-carnitine. American Journal of Physiology-Heart and Circulatory Physiology 272:H1615–H1624. 10.1152/ajpheart.1997.272.4.H1615 [DOI] [PubMed] [Google Scholar]
- 31.Allessie MA, Boyden PA, Camm AJ, et al. (2001) Pathophysiology and prevention of atrial fibrillation. Circulation 103:769–777 [DOI] [PubMed] [Google Scholar]
- 32.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA (1995) Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92:1954–1968 [DOI] [PubMed] [Google Scholar]
- 33.Modrego J, Maroto L, Tamargo J, et al. (2010) Comparative Expression of Proteins in Left and Right Atrial Appendages From Patients With Mitral Valve Disease at Sinus Rhythm and Atrial Fibrillation. Journal of Cardiovascular Electrophysiology. 10.1111/j.1540-8167.2010.01718.x [DOI] [PubMed] [Google Scholar]
- 34.Iyer NV, Kotch LE, Agani F, et al. (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12:149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan HE (1998) HIF-1alpha is required for solid tumor formation and embryonic vascularization. The EMBO Journal 17:3005–3015. 10.1093/emboj/17.11.3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wood SM, Wiesener MS, Yeates KM, et al. (1998) Selection and analysis of a mutant cell line defective in the hypoxia-inducible factor-1 alpha-subunit (HIF-1alpha). Characterization of hif-1alpha-dependent and -independent hypoxia-inducible gene expression. J Biol Chem 273:8360–8368 [DOI] [PubMed] [Google Scholar]
- 37.Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185. 10.1016/j.cmet.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 38.Papandreou I, Cairns RA, Fontana L, et al. (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3:187–197. 10.1016/j.cmet.2006.01.012 [DOI] [PubMed] [Google Scholar]
- 39.Makinde AO, Kantor PF, Lopaschuk GD (1998) Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Mol Cell Biochem 188:49–56 [PubMed] [Google Scholar]
- 40.Lopaschuk GD, Collins-Nakai RL, Itoi T (1992) Developmental changes in energy substrate use by the heart. Cardiovasc Res 26:1172–1180 [DOI] [PubMed] [Google Scholar]
- 41.Nau PN, Van Natta T, Ralphe JC, et al. (2002) Metabolic Adaptation of the Fetal and Postnatal Ovine Heart: Regulatory Role of Hypoxia-Inducible Factors and Nuclear Respiratory Factor-1. Pediatric Research 52:269–278. 10.1203/00006450-200208000-00021 [DOI] [PubMed] [Google Scholar]
- 42.Finck BN (2007) The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res 73:269–277. 10.1016/j.cardiores.2006.08.023 [DOI] [PubMed] [Google Scholar]
- 43.Huss JM, Kelly DP (2004) Nuclear Receptor Signaling and Cardiac Energetics. Circulation Research 95:568–578. 10.1161/01.RES.0000141774.29937.e3 [DOI] [PubMed] [Google Scholar]
- 44.Lehman JJ, Barger PM, Kovacs A, et al. (2000) Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. Journal of Clinical Investigation 106:847–856. 10.1172/JCI10268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puigserver P, Wu Z, Park CW, et al. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839 [DOI] [PubMed] [Google Scholar]
- 46.Kelly DP (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes & Development 18:357–368. 10.1101/gad.1177604 [DOI] [PubMed] [Google Scholar]
- 47.Puigserver P (2005) Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-alpha. Int J Obes (Lond) 29 Suppl 1:S5–9. 10.1038/sj.ijo.0802905 [DOI] [PubMed] [Google Scholar]
- 48.Puigserver P, Spiegelman BM (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24:78–90. 10.1210/er.2002-0012 [DOI] [PubMed] [Google Scholar]
- 49.Rowe GC, Jiang A, Arany Z (2010) PGC-1 coactivators in cardiac development and disease. Circulation Research 107:825–838. 10.1161/CIRCRESAHA.110.223818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Escher P, Braissant O, Basu-Modak S, et al. (2001) Rat PPARs: Quantitative Analysis in Adult Rat Tissues and Regulation in Fasting and Refeeding. Endocrinology 142:4195–4202. 10.1210/endo.142.10.8458 [DOI] [PubMed] [Google Scholar]
- 51.Braissant O, Foufelle F, Scotto C, et al. (1996) Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137:354–366. 10.1210/endo.137.1.8536636 [DOI] [PubMed] [Google Scholar]
- 52.Finck BN, Lehman JJ, Leone TC, et al. (2002) The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. Journal of Clinical Investigation 109:121–130. 10.1172/JCI14080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell FM, Kozak R, Wagner A, et al. (2002) A Role for Peroxisome Proliferator-activated Receptor α (PPARα) in the Control of Cardiac Malonyl-CoA Levels: Reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARα are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. Journal of Biological Chemistry 277:4098–4103. 10.1074/jbc.M106054200 [DOI] [PubMed] [Google Scholar]
- 54.Finck BN, Kelly DP (2002) Peroxisome proliferator-activated receptor alpha (PPARalpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol 34:1249–1257 [DOI] [PubMed] [Google Scholar]
- 55.Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20:1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bougarne N, Weyers B, Desmet SJ, et al. (2018) Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocrine Reviews 39:760–802. 10.1210/er.2018-00064 [DOI] [PubMed] [Google Scholar]
- 57.Skárka L, Bardová K, Brauner P, et al. (2003) Expression of mitochondrial uncoupling protein 3 and adenine nucleotide translocase 1 genes in developing rat heart: putative involvement in control of mitochondrial membrane potential. J Mol Cell Cardiol 35:321–330 [DOI] [PubMed] [Google Scholar]
- 58.Panadero M, Herrera E, Bocos C (2000) Peroxisome proliferator-activated receptor-alpha expression in rat liver during postnatal development. Biochimie 82:723–726 [DOI] [PubMed] [Google Scholar]
- 59.Razeghi P, Young ME, Abbasi S, Taegtmeyer H (2001) Hypoxia in Vivo Decreases Peroxisome Proliferator-Activated Receptor α-Regulated Gene Expression in Rat Heart. Biochemical and Biophysical Research Communications 287:5–10. 10.1006/bbrc.2001.5541 [DOI] [PubMed] [Google Scholar]
- 60.Belanger AJ, Luo Z, Vincent KA, et al. (2007) Hypoxia-inducible factor 1 mediates hypoxia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated receptor alpha/retinoid X receptor. Biochem Biophys Res Commun 364:567–572. 10.1016/j.bbrc.2007.10.062 [DOI] [PubMed] [Google Scholar]
- 61.Huss JM, Levy FH, Kelly DP (2001) Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: a mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J Biol Chem 276:27605–27612. 10.1074/jbc.M100277200 [DOI] [PubMed] [Google Scholar]
- 62.Yang M-H, Wu M-Z, Chiou S-H, et al. (2008) Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 10:295–305. 10.1038/ncb1691 [DOI] [PubMed] [Google Scholar]
- 63.el Azzouzi H, Leptidis S, Dirkx E, et al. (2013) The hypoxia-inducible microRNA cluster miR-199a~214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab 18:341–354. 10.1016/j.cmet.2013.08.009 [DOI] [PubMed] [Google Scholar]
- 64.Krishnan J, Danzer C, Simka T, et al. (2012) Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev 26:259–270. 10.1101/gad.180406.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilde AJ, van der Lee KAJM, Willemsen PHM, et al. (2003) Peroxisome Proliferator-Activated Receptor (PPAR) α and PPARβ/δ, but not PPARγ, Modulate the Expression of Genes Involved in Cardiac Lipid Metabolism. Circulation Research 92:518–524. 10.1161/01.RES.0000060700.55247.7C [DOI] [PubMed] [Google Scholar]
- 66.Pellieux C, Montessuit C, Papageorgiou I, Lerch R (2009) Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-α. Cardiovascular Research 82:341–350. 10.1093/cvr/cvp004 [DOI] [PubMed] [Google Scholar]
- 67.Planavila A, Laguna JC, Vázquez-Carrera M (2005) Nuclear factor-kappaB activation leads to down-regulation of fatty acid oxidation during cardiac hypertrophy. J Biol Chem 280:17464–17471. 10.1074/jbc.M414220200 [DOI] [PubMed] [Google Scholar]
- 68.Cheng L, Ding G, Qin Q, et al. (2004) Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med 10:1245–1250. 10.1038/nm1116 [DOI] [PubMed] [Google Scholar]
- 69.Hondares E, Pineda-Torra I, Iglesias R, et al. (2007) PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun 354:1021–1027. 10.1016/j.bbrc.2007.01.092 [DOI] [PubMed] [Google Scholar]
- 70.Burkart EM, Sambandam N, Han X, et al. (2007) Nuclear receptors PPARβ/δ and PPARα direct distinct metabolic regulatory programs in the mouse heart. Journal of Clinical Investigation. 10.1172/JCI32578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mihm MJ, Yu F, Carnes CA, et al. (2001) Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104:174–180 [DOI] [PubMed] [Google Scholar]
- 72.Christoffels VM, Moorman AFM (2009) Development of the Cardiac Conduction System: Why Are Some Regions of the Heart More Arrhythmogenic Than Others? Circulation: Arrhythmia and Electrophysiology 2:195–207. 10.1161/CIRCEP.108.829341 [DOI] [PubMed] [Google Scholar]
- 73.Ozcan C, Holmuhamedov EL, Jahangir A, Terzic A (2001) Diazoxide protects mitochondria from anoxic injury: implications for myopreservation. J Thorac Cardiovasc Surg 121:298–306. 10.1067/mtc.2001.111421 [DOI] [PubMed] [Google Scholar]
- 74.Liu M, Liu H, Dudley SC (2010) Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 107:967–974. 10.1161/CIRCRESAHA.110.220673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Condrescu M, Opuni K, Hantash BM, Reeves JP (2002) Cellular regulation of sodium-calcium exchange. Ann N Y Acad Sci 976:214–223 [DOI] [PubMed] [Google Scholar]
- 76.Halestrap AP, Clarke SJ, Khaliulin I (2007) The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta 1767:1007–1031. 10.1016/j.bbabio.2007.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaasik A, Joubert F, Ventura-Clapier R, Veksler V (2004) A novel mechanism of regulation of cardiac contractility by mitochondrial functional state. FASEB J 18:1219–1227. 10.1096/fj.04-1508com [DOI] [PubMed] [Google Scholar]
- 78.Holmuhamedov EL, Ozcan C, Jahangir A, Terzic A (2001) Restoration of Ca2+-inhibited oxidative phosphorylation in cardiac mitochondria by mitochondrial Ca2+ unloading. Mol Cell Biochem 220:135–140 [DOI] [PubMed] [Google Scholar]
- 79.Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A (2001) Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech Ageing Dev 122:1073–1086 [DOI] [PubMed] [Google Scholar]
- 80.Robinson AM, Williamson DH (1980) Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 60:143–187. 10.1152/physrev.1980.60.1.143 [DOI] [PubMed] [Google Scholar]
- 81.McGarry JD, Foster DW (1980) Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem 49:395–420. 10.1146/annurev.bi.49.070180.002143 [DOI] [PubMed] [Google Scholar]
- 82.Cotter DG, Schugar RC, Crawford PA (2013) Ketone body metabolism and cardiovascular disease. American Journal of Physiology-Heart and Circulatory Physiology 304:H1060–H1076. 10.1152/ajpheart.00646.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cahill GF (2006) Fuel metabolism in starvation. Annu Rev Nutr 26:1–22. 10.1146/annurev.nutr.26.061505.111258 [DOI] [PubMed] [Google Scholar]
- 84.Johnson RH, Walton J, Krebs HA, Williamson DH (1970) Post-exercise ketosis. Lancet 1:195. [PubMed] [Google Scholar]
- 85.Endemann G, Goetz PG, Edmond J, Brunengraber H (1982) Lipogenesis from ketone bodies in the isolated perfused rat liver. Evidence for the cytosolic activation of acetoacetate. J Biol Chem 257:3434–3440 [PubMed] [Google Scholar]
- 86.Freed LE, Endemann G, Tomera JF, et al. (1988) Lipogenesis from ketone bodies in perfused livers from streptozocin-induced diabetic rats. Diabetes 37:50–55 [DOI] [PubMed] [Google Scholar]
- 87.Morris AAM (2005) Cerebral ketone body metabolism. Journal of Inherited Metabolic Disease 28:109–121. 10.1007/s10545-005-5518-0 [DOI] [PubMed] [Google Scholar]
- 88.Mayr M, Yusuf S, Weir G, et al. (2008) Combined Metabolomic and Proteomic Analysis of Human Atrial Fibrillation. Journal of the American College of Cardiology 51:585–594. 10.1016/j.jacc.2007.09.055 [DOI] [PubMed] [Google Scholar]
- 89.Depre C, Vanoverschelde JL, Taegtmeyer H (1999) Glucose for the heart. Circulation 99:578–588 [DOI] [PubMed] [Google Scholar]
- 90.Rudolph W, Schinz A (1973) Studies on myocardial blood flow, oxygen consumption, and myocardial metabolism in patients with cardiomyopathy. Recent Adv Stud Cardiac Struct Metab 2:739–749 [PubMed] [Google Scholar]
- 91.Sack MN, Rader TA, Park S, et al. (1996) Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94:2837–2842 [DOI] [PubMed] [Google Scholar]
- 92.Tian Q, Barger PM (2006) Deranged energy substrate metabolism in the failing heart. Curr Hypertens Rep 8:465–471 [DOI] [PubMed] [Google Scholar]
- 93.Barger PM, Brandt JM, Leone TC, et al. (2000) Deactivation of peroxisome proliferator-activated receptor-α during cardiac hypertrophic growth. Journal of Clinical Investigation 105:1723–1730. 10.1172/JCI9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lahey R, Wang X, Carley AN, Lewandowski ED (2014) Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation 130:1790–1799. 10.1161/CIRCULATIONAHA.114.011687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aubert G, Martin OJ, Horton JL, et al. (2016) The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation CIRCULATIONAHA.115.017355. 10.1161/CIRCULATIONAHA.115.017355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Barth AS, Merk S, Arnoldi E, et al. (2005) Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res 96:1022–1029. 10.1161/01.RES.0000165480.82737.33 [DOI] [PubMed] [Google Scholar]
- 97.Russell RR, Taegtmeyer H (1992) Coenzyme A sequestration in rat hearts oxidizing ketone bodies. J Clin Invest 89:968–973. 10.1172/JCI115679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma W, Berg J, Yellen G (2007) Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci 27:3618–3625. 10.1523/JNEUROSCI.0132-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ito M, Fukui T, Kamokari M, et al. (1984) Purification and characterization of acetoacetyl-CoA synthetase from rat liver. Biochim Biophys Acta 794:183–193 [PubMed] [Google Scholar]
- 100.Lincoln BC, Des Rosiers C, Brunengraber H (1987) Metabolism of S-3-hydroxybutyrate in the perfused rat liver. Arch Biochem Biophys 259:149–156 [DOI] [PubMed] [Google Scholar]
- 101.Reed WD, Ozand PT (1980) Enzymes of L-(+)-3-hydroxybutyrate metabolism in the rat. Arch Biochem Biophys 205:94–103 [DOI] [PubMed] [Google Scholar]
- 102.Scofield RF, Brady PS, Schumann WC, et al. (1982) On the lack of formation of L-(+)-3-hydroxybutyrate by liver. Arch Biochem Biophys 214:268–272 [DOI] [PubMed] [Google Scholar]
- 103.Wang X, Levi AJ, Halestrap AP (1996) Substrate and inhibitor specificities of the monocarboxylate transporters of single rat heart cells. Am J Physiol 270:H476–484. 10.1152/ajpheart.1996.270.2.H476 [DOI] [PubMed] [Google Scholar]
- 104.Akar FG, Aon MA, Tomaselli GF, O’Rourke B (2005) The mitochondrial origin of postischemic arrhythmias. J Clin Invest 115:3527–3535. 10.1172/JCI25371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsuboi M, Hisatome I, Morisaki T, et al. (2001) Mitochondrial DNA deletion associated with the reduction of adenine nucleotides in human atrium and atrial fibrillation. Eur J Clin Invest 31:489–496. 10.1046/j.1365-2362.2001.00844.x [DOI] [PubMed] [Google Scholar]
- 106.Kalifa J, Maixent J-M, Chalvidan T, et al. (2008) Energetic metabolism during acute stretch-related atrial fibrillation. Mol Cell Biochem 317:69–75. 10.1007/s11010-008-9832-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bukowska A, Schild L, Keilhoff G, et al. (2008) Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp Biol Med (Maywood) 233:558–574. 10.3181/0706-RM-155 [DOI] [PubMed] [Google Scholar]
- 108.Chen Y-R, Zweier JL (2014) Cardiac mitochondria and reactive oxygen species generation. Circ Res 114:524–537. 10.1161/CIRCRESAHA.114.300559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van Bragt KA, Nasrallah HM, Kuiper M, et al. (2014) Atrial supply-demand balance in healthy adult pigs: coronary blood flow, oxygen extraction, and lactate production during acute atrial fibrillation. Cardiovasc Res 101:9–19. 10.1093/cvr/cvt239 [DOI] [PubMed] [Google Scholar]
- 110.Emelyanova L, Ashary Z, Cosic M, et al. (2016) Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am J Physiol Heart Circ Physiol 311:H54–63. 10.1152/ajpheart.00699.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liang X, Zhang Q, Wang X, et al. (2018) Reactive oxygen species mediated oxidative stress links diabetes and atrial fibrillation. Mol Med Rep 17:4933–4940. 10.3892/mmr.2018.8472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee JS, Ko Y-G, Shin K-J, et al. (2015) Mitochondrial DNA 4977bp deletion mutation in peripheral blood reflects atrial remodeling in patients with non-valvular atrial fibrillation. Yonsei Med J 56:53–61. 10.3349/ymj.2015.56.1.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hattori K, Tanaka M, Sugiyama S, et al. (1991) Age-dependent increase in deleted mitochondrial DNA in the human heart: possible contributory factor to presbycardia. Am Heart J 121:1735–1742. 10.1016/0002-8703(91)90020-i [DOI] [PubMed] [Google Scholar]
- 114.Linnane AW, Marzuki S, Ozawa T, Tanaka M (1989) Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1:642–645. 10.1016/s0140-6736(89)92145-4 [DOI] [PubMed] [Google Scholar]
- 115.Lin PH, Lee SH, Su CP, Wei YH (2003) Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic Biol Med 35:1310–1318. 10.1016/j.freeradbiomed.2003.07.002 [DOI] [PubMed] [Google Scholar]
- 116.Wei Y-H, Lee H-C (2002) Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood) 227:671–682. 10.1177/153537020222700901 [DOI] [PubMed] [Google Scholar]
- 117.Tsang TSM, Barnes ME, Gersh BJ, et al. (2002) Left atrial volume as a morphophysiologic expression of left ventricular diastolic dysfunction and relation to cardiovascular risk burden. Am J Cardiol 90:1284–1289. 10.1016/s0002-9149(02)02864-3 [DOI] [PubMed] [Google Scholar]
- 118.Tsang TSM, Gersh BJ, Appleton CP, et al. (2002) Left ventricular diastolic dysfunction as a predictor of the first diagnosed nonvalvular atrial fibrillation in 840 elderly men and women. J Am Coll Cardiol 40:1636–1644. 10.1016/s0735-1097(02)02373-2 [DOI] [PubMed] [Google Scholar]
- 119.Pritchett AM, Mahoney DW, Jacobsen SJ, et al. (2005) Diastolic dysfunction and left atrial volume: a population-based study. J Am Coll Cardiol 45:87–92. 10.1016/j.jacc.2004.09.054 [DOI] [PubMed] [Google Scholar]
- 120.Lamirault G, Gaborit N, Le Meur N, et al. (2006) Gene expression profile associated with chronic atrial fibrillation and underlying valvular heart disease in man. J Mol Cell Cardiol 40:173–184. 10.1016/j.yjmcc.2005.09.004 [DOI] [PubMed] [Google Scholar]
- 121.Montaigne D, Marechal X, Lefebvre P, et al. (2013) Mitochondrial dysfunction as an arrhythmogenic substrate: a translational proof-of-concept study in patients with metabolic syndrome in whom post-operative atrial fibrillation develops. J Am Coll Cardiol 62:1466–1473. 10.1016/j.jacc.2013.03.061 [DOI] [PubMed] [Google Scholar]
- 122.Mihm MJ, Coyle CM, Schanbacher BL, et al. (2001) Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc Res 49:798–807. 10.1016/s0008-6363(00)00307-2 [DOI] [PubMed] [Google Scholar]
- 123.Carnes CA, Chung MK, Nakayama T, et al. (2001) Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res 89:E32–38. 10.1161/hh1801.097644 [DOI] [PubMed] [Google Scholar]
- 124.Schild L, Bukowska A, Gardemann A, et al. (2006) Rapid pacing of embryoid bodies impairs mitochondrial ATP synthesis by a calcium-dependent mechanism--a model of in vitro differentiated cardiomyocytes to study molecular effects of tachycardia. Biochim Biophys Acta 1762:608–615. 10.1016/j.bbadis.2006.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou C, Ziegler C, Birder LA, et al. (2006) Angiotensin II and stretch activate NADPH oxidase to destabilize cardiac Kv4.3 channel mRNA. Circ Res 98:1040–1047. 10.1161/01.RES.0000218989.52072.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sridhar A, Nishijima Y, Terentyev D, et al. (2009) Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res 84:227–236. 10.1093/cvr/cvp216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cai H (2005) Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res 68:26–36. 10.1016/j.cardiores.2005.06.021 [DOI] [PubMed] [Google Scholar]
- 128.Cai H, Griendling KK, Harrison DG (2003) The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 24:471–478. 10.1016/S0165-6147(03)00233-5 [DOI] [PubMed] [Google Scholar]
- 129.Swaminathan PD, Purohit A, Soni S, et al. (2011) Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest 121:3277–3288. 10.1172/JCI57833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Erickson JR, He BJ, Grumbach IM, Anderson ME (2011) CaMKII in the cardiovascular system: sensing redox states. Physiol Rev 91:889–915. 10.1152/physrev.00018.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lin Y-K, Lu Y-Y, Chen Y-C, et al. (2010) Nitroprusside modulates pulmonary vein arrhythmogenic activity. J Biomed Sci 17:20 10.1186/1423-0127-17-20 [DOI] [PMC free article] [PubMed] [Google Scholar]