

Abstract
Emerging studies indicate that DNA damage in cancer cells triggers antitumor immunity, but its intrinsic regulatory mechanism in breast cancer cells remains poorly understood. Here, we show that ZMYND8 is upregulated and inhibits micronucleus formation and DNA damage in breast cancer cells. Loss of ZMYND8 triggered activation of the DNA sensor cyclic guanosine monophosphate-adenosine monophosphate synthase in micronuclei, leading to further activation of the downstream signaling effectors stimulator of interferon genes and NF-κB, but not TANK-binding kinase 1 and interferon regulatory factor 3, thereby inducing the expression of interferon-β and interferon-stimulated genes (ISGs) in breast cancer cells in vitro and tumors in vivo. ZMYND8 knockout (KO) in breast cancer cells promoted infiltration of CD4+ and CD8+ T cells leading to tumor inhibition in syngeneic mouse models, which was significantly attenuated by treatment of anti-CD4/CD8 depleting antibodies or anti-IFNAR1 antibody and in immunodeficient Rag1 KO mice. In human breast tumors, ZMYND8 was negatively correlated with ISGs, CD4, CD8A, CD8B, and the tumor-lymphocyte infiltration phenotype. Collectively, these findings demonstrate that maintenance of genome stability by ZMYND8 causes breast cancer cells to evade cytotoxic T-lymphocyte surveillance which leads to tumor growth.
Keywords: DNA damage, micronucleus, innate immune response, T cells, immunosurveillance
Introduction
Breast cancer is the most commonly diagnosed cancer and the second leading cause of cancer death among women. Emerging studies support a critical role of the immune system in the treatment of several types of human cancers, including melanoma, non-small cell lung cancer and colon carcinoma (1–3). Although a PD-L1 antibody has been approved by the US Food and Drug Administration for the treatment of triple-negative breast cancer, the response rate to the single immune checkpoint inhibitor is still very low (~5%) in overall breast cancer patients (4). Low tumor mutational burden is one of the mechanisms of immune evasion in breast tumors (5). Understanding how to increase tumor mutational burden in breast tumors would lead to the development of innovative approaches for a better treatment of this human disease.
Genome instability is a hallmark of human cancers (6). Recent clinical studies have revealed that tumor mutational burden correlates with lymphocyte infiltration and a better response to immune checkpoint inhibitors in human cancers (7–9). DNA damage triggers the immune response to increase lymphocyte infiltration in tumors, leading to tumor regression (10,11). Cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS) is a primary DNA sensor in the mammalian innate immune system (12). Previous studies reported that cGAS locates at micronuclei in the cytosol, where it binds to double-stranded DNA and then is activated (13). Activated cGAS catalyzes GTP and ATP to produce 2’3’-cGAMP, which acts as a secondary messenger that binds and induces the structural change of the downstream factor stimulator of interferon genes (STING) for its activation (12). STING then recruits and activates the TANK-binding kinase 1 (TBK1)/interferon regulatory factor 3 (IRF3) and NF-κB signaling pathways to induce the expression of inflammatory genes including type I interferon (IFN) and interferon-stimulated genes (ISGs), thereby initiating antitumor immunity in human cancers (12,14). This antitumor signaling cascade, initially discovered in immune cells, was also observed in cancer cells (15,16). However, how this intrinsic cGAS/STING signaling mechanism is regulated in cancer cells remains incompletely understood.
Human ZMYND8 gene was cloned from the hippocampus cDNA library screen in 2000. It encodes a ~180 kDa histone reader ZMYND8 (zinc finger MYND-type containing 8) protein containing PHD-BRD-PWWP domains at its N-terminus and a MYND domain at its C-terminus (17,18). Its N-terminal PHD-BRD-PWWP domains recognize several acetyl and methyl lysine residues on histones in vitro, including acetyl lysine 14 of histone H3 (H3K14ac), H4K16ac, and di- and tri-methyl lysine 36 of histone H3 (18,19). Upon DNA damage stimuli, ZMYND8 is recruited to sites of DNA damage and facilitates homologous recombination (18,20). Our group found that ZMYND8 is highly expressed in human breast tumors and promotes breast cancer progression (21). Whether or not ZMYND8-mediated DNA damage response inhibits antitumor immunity leading to breast cancer progression remains unknown.
Here, we showed that loss of ZMYND8 increased micronucleus formation and activated the cGAS-STING-NF-κB signaling cascade to induce IFN-β and ISGs in breast cancer cells and tumors. Furthermore, ZMYND8 knockout (KO) in breast cancer cells promoted infiltration of CD4+ and CD8+ T cells leading to tumor inhibition in mice, which was significantly attenuated by treatment of anti-CD4/CD8 depleting antibodies or anti-IFNAR1 antibody and in immunodeficient Rag1 KO mice. ZMYND8 is highly expressed in human breast tumors and negatively correlated with ISGs, CD8A, CD8B, CD4, and tumor-lymphocyte infiltration in human breast tumors. Collectively, these findings uncover that ZMYND8 inhibits an intrinsic antitumor immunity that causes breast cancer cells to evade immunosurveillance for their growth.
Materials and methods
Plasmid constructs
Mouse ZMYND8, cGAS, STING, p65, and scrambled control (SC) single-guide RNAs (sgRNAs) listed in Supplementary Table 1 were cloned into lentiCRISPR-V2. All plasmids were verified by DNA sanger sequencing. Other plasmid constructs have been described previously (21).
Cell culture and stable KO cell lines
MDA-MB-231 (a gift from Brekken Lab in 2015), MCF-7 (ATCC in 2014), and 4T1 (Fu Lab in 2018) cells were cultured in DMEM or RPMI1640 with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a 5% CO2/95% air incubator. PY8119 cells were obtained from ATCC in 2018 and cultured in Ham’s F12 containing 1.5 g/L NaHCO3 and 5% FBS at 37°C in a 5% CO2/95% air incubator. ZMYND8, cGAS, STING, and p65 KO cells were selected with puromycin (3–4 μg/mL) after transient transfection using PolyJet DNA transfection reagent (SignaGen). No Cas9 expression was confirmed in these KO lines. Three to six single KO cells were mixed for this study. ZMYND8-rescued MDA-MB-231 cells were generated by lentiviral transduction of ZMYND8 cDNA in ZMYND8 KO cells. Cells within twenty passages were used for experiments. MDA-MB-231 and MCF-7 cells have been authenticated by STR DNA profiling analysis in 2016. All cell lines were mycoplasma-free.
In vitro cell growth assay
MDA-MB-231 (2.5 × 105 cells/well), PY8119 (3.0 × 105 cells/well), and 4T1 (2.0 × 105 cells/well) cells were seeded onto a 6-well plate and cultured for 24, 36, 48, and 60 hours. The cell number at each time point was determined by trypan blue assay.
Immunoblot assay
Cells were lyzed with NETN lysis buffer (150 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0, 0.5% NP-40, 1 mM Na3VO4, 10 mM NaF, and protease inhibitor cocktail), followed by sonication for 15 seconds. After centrifugation, supernatant was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The following antibodies were used: anti-ZMYND8 antibody (A302–089A, Bethyl Laboratories), anti-cGAS antibody (31659S, Cell Signaling Technology), anti-STING antibody (13647S, Cell Signaling Technology), anti-phospho-TBK1 antibody (5483S, Cell Signaling Technology), anti-TBK1 antibody (3013S, Cell Signaling Technology), anti-phospho-IRF3 (Ser396) antibody (29047S, Cell Signaling Technology), anti-IRF3 antibody (4302S, Cell Signaling Technology), anti-phospho-p65 antibody (3033S, Cell Signaling Technology), anti-p65 antibody (8242S, Cell Signaling Technology), anti-phospho-Chk1 (Ser296) antibody (2349S, Cell Signaling Technology), anti-Chk1 antibody (sc-8408, Santa Cruz Biotechnology), anti-γH2A.X antibody (05–636, Sigma), anti-actin antibody (A2066, Sigma), and anti-H2A.X antibody (10856–1-AP, Proteintech). Proteins were visualized by chemiluminescence with ECL prime (GE Healthcare).
Immunostaining assay
Cells were seeded with 10% confluence onto glass coverslips placed in a 12-well plate and cultured for 36 hours. After washing with phosphate-buffered saline (PBS) once, cells were fixed for 20 min with methanol at room temperature, permeabilized for 15 min with 0.1% Triton X-100 in PBS, and blocked for 60 min with 5% BSA in PBS. Cells then were incubated overnight with anti-cGAS and/or anti-γH2A.X antibody (1:200 dilution in PBS with 1% BSA) in a 12-well plate at 4 °C, washed with PBST (PBS with 0.1% Tween-20) for 3 times, incubated for 60 min with Alexa Fluo® 488 goat anti-rabbit IgG and/or Cy™3 donkey anti-mouse IgG (1:1000 dilution in PBST with 1% BSA) in dark, washed again with PBST for 3 times, and incubated for 5 min with DAPI (1:1000 dilution in PBS) in dark. After washing 3 times, cells were mounted with anti-fade mounting medium. Mounted slides were observed with a Zeiss Axio Observer Z1 fluorescence microscope. Cells with at least five or more γH2A.X foci in the nucleus were considered as positive. All quantifications were performed under blinded conditions.
Chromosome aberration assay
Cells with 70% confluence were treated for 2 hours with 1 μg/mL colcemid, harvested, and resuspended for 30 min in 1 mL of 75 mM KCl at 37 °C. After centrifugation, cells were fixed with cold methanol/acetic acid (3:1) buffer and incubated for 15 min at room temperature. Metaphase spreads were made by dropping cells onto the slide, air-dried, stained with DAPI, and visualized under a Zeiss Axio Observer Z1 microscope.
Quantitative reverse transcription-polymerase chain reaction (RT–qPCR) assay
Total RNA was isolated with Trizol and treated with DNase (Invitrogen). First-strand cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Real-time qPCR was performed as described previously (22) and data were normalized to 18S rRNA or actin. qPCR primers used in this study are shown in Supplementary Table 2.
Chromatin immunoprecipitation (ChIP)-qPCR assay
Cells were fixed in 10 mL of culture media containing 1% formaldehyde for 10 minutes at room temperature and quenched by addition of 1 mL of 2 M glycine. After washing with TBSE (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA), cells were lysed in lysis buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10 mM EDTA, 0.25% Triton X-100, and protease inhibitor cocktail) and centrifuged. The nuclei pellet was collected and lysed in lysis buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1% SDS, and protease inhibitor cocktail) to extract chromatin. The chromatin DNA was fragmented by sonication and then centrifuged at 15000 rpm for 30 min at 4 ℃. The supernatant was diluted in ChIP immunoprecipitation buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail) and then subjected to immunoprecipitation overnight in the presence of Dynabeads (ThermoFisher) with anti-phospho-p65 (Ser536) antibody (3033S, Cell Signaling Technology) or normal rabbit IgG (2729S, Cell Signaling Technology) at 4 °C. Beads were washed, eluted, and reverse-crosslinked at 65°C for 4 hours, followed by treatment with proteinase K at 45°C for 1 hour. The precipitated DNA was purified with phenol/chloroform/isoamyl alcohol (25:24:1, v/v) and quantified by real-time qPCR assay. The primers used for ChIP-qPCR are listed in Supplementary Table 2. Fold enrichment was calculated based on Ct as 2−Δ(ΔCt), where ΔCt = CtIP - CtInput and Δ(ΔCt) = ΔCtantibody - ΔCtIgG.
2’3’-cGAMP ELISA assay
3 × 105 cells were seeded onto a 6-well plate and lyzed after 36-hour culture. The cellular 2’3’-cGAMP levels were measured using a 2’3’-cGAMP ELISA kit (Cayman) in accordance with the manufacturer’s instructions.
IFN-β ELISA assay
3 × 105 cells were seeded onto a 6-well plate with 1 mL of culture medium. After 48-hour culture, media were collected for IFN-β protein measurement with a mouse IFN-β ELISA kit (PBL Assay Science) in accordance with the manufacturer’s instructions. The concentration of IFN-β protein was normalized to cell numbers.
Senescence assay
Cells were seeded onto a 6-well plate. After 24 hours, cells were treated with vehicle (DMSO) or etoposide (12.5 μM) for 24 hours and cultured with fresh media for an additional 3 days. Senescence staining was performed with a Senescence β-Galactosidase Staining Kit (9860S, Cell Signaling Technology) according to the manufacturer’s instructions.
Animal studies
Animal studies were approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee. 1–2 × 105 cells suspended in PBS/Matrigel (1:1, Corning) were implanted into the second left mammary fat pad of 6–8-week old female C57BL/6J, Balb/c, or Rag1 KO mice (Jackson Laboratory). Tumors were measured as described (21). For CD4+ and CD8+ T-cell depletion experiment, 200 μg anti-CD4 and anti-CD8 antibodies (BioXCell) or control IgG were injected (i.p.) into mice every 3 days for total 4 times starting 1 day before tumor cell implantation (23). For IFN-β blockade experiment, 200 μg anti-IFNAR1 antibody (BE0241, BioXCell) was injected (i.p.) into mice every 3 days starting day 0 until the tumor volume reached about 200 mm3, and then 100 μg anti-IFNAR1 antibody was intratumorally injected every 3 days (23).
Immunohistochemistry (IHC) assay
IHC assay was performed on a Dako Autostainer Link 48 system as described previously (21). The anti-CD4 (1:100, 25229S) and anti-CD8 (1: 400, 98941S) antibodies were purchased from Cell Signaling Technology.
Flow cytometry assay
Tumors were harvested from Balb/c mice and cut into small pieces in PBS with 5% FBS. After centrifugation at 4 °C, tumor pieces were resuspended in 4 mL of digestion buffer (1 mg/mL collagenase A and 0.05 mg/mL DNase I in PBS with 5% FBS) and incubated for 30 min at 37 °C by gently shaking. Cell suspension was filtered using a 70 μm-strainer. After centrifugation, single cells were resuspended in 4 mL of PBS with 5% FBS and counted. 1 × 107 cells in 100 μL of PBS with 5% FBS were incubated for 10 min with anti-CD16/32 antibody (2.4G2, BioXCell) on ice to block Fc receptors. After centrifugation, cells were incubated for 30 min with anti-CD45.2 (109827, BioLegend), CD4 (100548, BioLegend), CD8a (100730, BioLegend), CD3e (564379, BD Biosciences), eFluor 506 (65086614, Invitrogen), and B220 (103226, BioLegend) antibodies diluted in 100 μL of PBS with 5% FBS at 4 °C in dark, washed, and resuspended in 300 μL of PBS with 5% FBS. Data were collected on a CytoFLEX flow cytometer (Beckman Coulter) and analyzed by FlowJo software.
Statistical analysis
Statistical analysis was performed by 2-tailed Student’s t test between 2 groups, and 1- or 2-way ANOVA with multiple testing correction within multiple groups. Gene correlation was analyzed by Pearson’s correlation method. Data represent mean ± SEM from three independent experiments. p < 0.05 is considered significant.
Results
ZMYND8 inhibits micronucleus formation and DNA damage in breast cancer cells
To study whether ZMYND8 regulates genome stability in breast cancer cells, we established SC and ZMYND8 KO MDA-MB-231 cells (Fig. 1A). ZMYND8-rescued MDA-MB-231 cells were also generated by lentiviral transduction of ZMYND8 cDNA in KO cells (Fig. 1A). The number of micronuclei-positive cells was significantly increased by ZMYND8 KO in MDA-MB-231 (Fig. 1B and C). Re-expression of ZMYND8 in KO cells reversed micronucleus formation (Fig. 1B and C). Similarly, we found that ZMYND8 KO increased micronucleus formation by 1.5- to 6-fold in another human breast cancer cell line MCF-7 and mouse mammary cancer cell lines 4T1 and PY8119 (Fig. 1D–F, Supplementary Fig. S1A–F). Micronucleus formation is caused by chromosome mis-segregation and indicates DNA damage in the cell (24). To confirm that ZMYND8 inhibits DNA damage in breast cancer cells, we first analyzed the levels of a DNA damage marker γH2A.X by immunoblot assay. γH2A.X protein levels were increased in ZMYND8 KO cells as compared with SC cells, which was reversed by expression of exogenous ZMYND8 (Fig. 1A). Consistently, immunostaining assay showed that ZMYND8 KO significantly increased the numbers of γH2A.X-positive micronuclei/nuclei and γH2A.X foci in MDA-MB-231 and PY8119 cells (Fig. 1B, E, and G–L). Re-expression of ZMYND8 attenuated γH2A.X staining in MDA-MB-231 cells (Fig. 1B and G–I). Next, we analyzed chromosome aberrations in ZMYND8 KO breast cancer cells. As shown in Fig. 1M and N, chromosome aberrations including breaks, dicentrics, and gaps were significantly increased in two independent ZMYND8 KO MDA-MB-231 cell lines as compared with their parental cells. Collectively, these findings indicate that ZMYND8 inhibits micronucleus formation and increases genome stability in breast cancer cells.
Figure 1. ZMYND8 inhibits micronucleus formation and DNA damage in breast cancer cells.
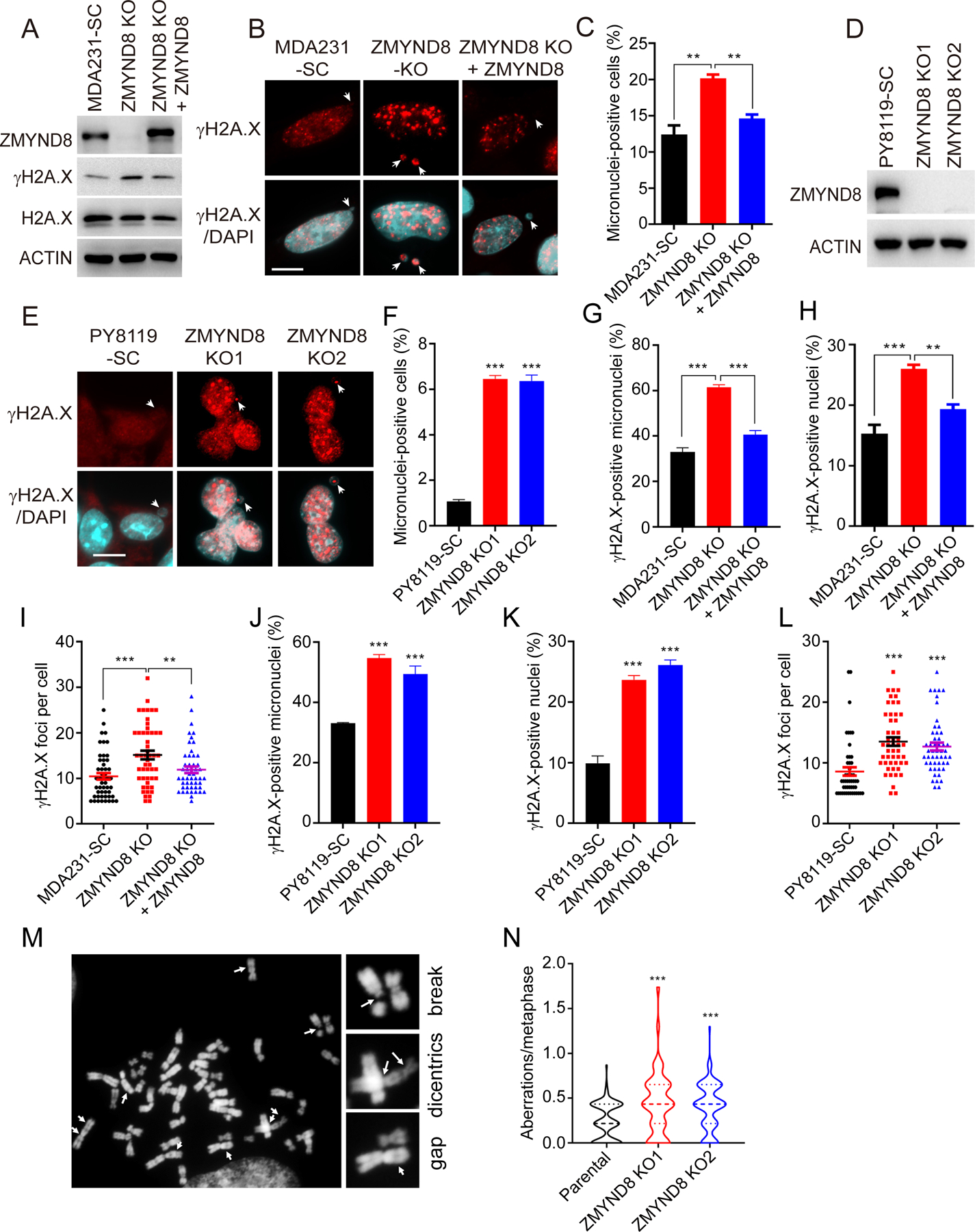
(A) Immunoblot analysis of indicated proteins in SC, ZMYND8 KO, and ZMYND8-rescued MDA-MB-231 (MDA231) cells. (B) Representative immunostaining of γH2A.X and micronuclei (indicated by white arrows) in SC, ZMYND8 KO, and ZMYND8-rescued MDA-MB-231 cells. Scale bar, 10 μm. (C) Quantification of micronuclei-positive MDA-MB-231 cells (n = 500, mean ± SEM). **p < 0.01 by one-way ANOVA with Dunnett’s test. (D) Immunoblot analysis of ZMYND8 and actin in SC and ZMYND8 KO PY8119 cells. (E) Representative immunostaining of γH2A.X and micronuclei (indicated by white arrows) in SC and ZMYND8 KO PY8119 cells. Scale bar, 10 μm. (F) Quantification of micronuclei-positive PY8119 cells (n = 500, mean ± SEM). ***p < 0.001 vs. SC by one-way ANOVA with Dunnett’s test. (G-L) Quantification of γH2A.X-positive micronuclei (n = 50, G and J), γH2A.X-positive nuclei (n = 500, H and K), and γH2A.X foci per cell (n = 50, I and L). Data are shown in mean ± SEM. **p < 0.01; ***p < 0.001 by one-way ANOVA with Dunnett’s test. (M) Representative images of chromosome aberrations (indicated by white arrows) in ZMYND8 KO MDA-MB-231 cells. (N) Quantification of chromosome aberrations in parental and ZMYND8 KO MDA-MB-231 cells (n = 100, mean ± SEM). ***p < 0.001 by one-way ANOVA with Dunnett’s test.
ZMYND8 suppresses the intrinsic innate immune response in breast cancer cells
DNA damage triggers the innate immune response by inducing expression of IFN-β (11). To find out whether ZMYND8 KO upregulates IFN-β and immune response in breast cancer cells, we analyzed ZMYND8-dependent transcriptome in parental and ZMYND8 KO MDA-MB-231 cells by RNA-sequencing (RNA-seq, GSE108833). Loss of ZMYND8 led to upregulation of 940 genes but downregulation of 1393 genes (FDR < 0.05, LogCPM > 0, fold change > 1.5) (Fig. 2A). The gene ontology analysis of ZMYND8-downregulated genes identified multiple immune and inflammatory responses-related biological processes, which ranked after biological adhesion and cell adhesion (Fig. 2B; Supplementary Fig. S2A). Reactome pathway analysis also revealed interferon α/β signaling as the second top pathway as well as several other immune-related pathways (Supplementary Fig. S2B). The effect of ZMYND8 on cell adhesion and extracellular matrix will be studied in the future. To validate the RNA-seq data, RT-qPCR assay was performed in ZMYND8 KO and rescued MDA-MB-231 cells. ZMYND8 KO significantly increased the mRNA expression of IFN-β but not the non-target gene RPL13A (Fig. 2C). Similar results were observed in PY8119 cells (Fig. 2D). IFN-β protein levels were also significantly higher in culture media from ZMYND8 KO PY8119 cells than SC cells (Fig. 2E), indicating that ZMYND8 KO increases IFN-β release from breast cancer cells. IFN-α and IFN-γ were not detectable in parental and ZMYND8 KO cells (Fig. 2D). In line with IFN-β induction, ZMYND8 KO significantly enhanced the mRNA expression of ISGs including ISG15, OASL, OAS1, and OAS2 in MDA-MB-231 cells (Fig. 2C). Additional ISGs including Oasl1, Oasl2, Oas1a, Oas3, Mx1, and Cxcl10 mRNAs were also upregulated by ZMYND8 KO in PY8119 and 4T1 cell lines (Fig. 2D and Supplementary Fig. S2C). Re-expression of ZMYND8 reversed upregulation of IFN-β and ISGs conferred by ZMYND8 KO in MDA-MB-231 cells (Fig. 2C), suggesting the specific regulation of IFN-β and ISGs by ZMYND8. Together, these data indicate that ZMYND8 suppresses the expression of IFN-β and ISGs in breast cancer cells.
Figure 2. ZMYND8 suppresses the expression of IFN-β and ISGs in breast cancer cells.
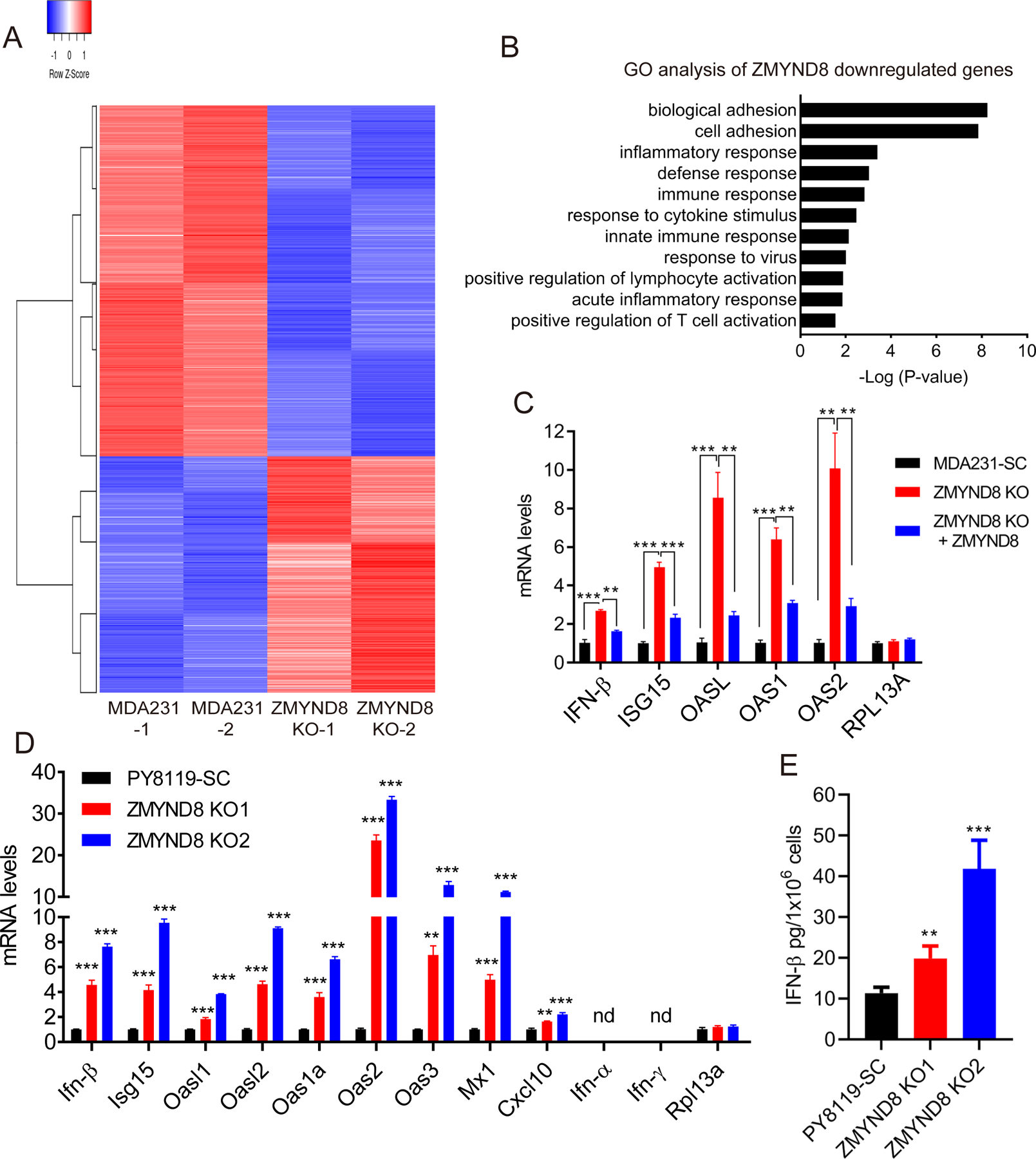
(A) Heatmap of ZMYND8-up- and -down-regulated genes in MDA-MB-231 (MDA231) cells. (B) Gene ontology (GO) analysis of ZMYND8-downregulated genes in MDA-MB-231 cells by the DAVID. (C and D) RT-qPCR analysis of IFN-β and ISGs mRNAs in MDA-MB-231 (C) and PY8119 (D) cells (n = 3, mean ± SEM). **p < 0.01; ***p < 0.001 by one-way ANOVA with Dunnett’s test. nd, not detected. (E) ELISA analysis of IFN-β protein release from SC and ZMYND8 KO PY8119 cells (n = 4, mean ± SEM). **p < 0.01; ***p < 0.001 by one-way ANOVA with Dunnett’s test.
DNA damage often induces the cell-cycle checkpoint response (25). We found that ZMYND8 KO did not induce Chk1 phosphorylation but slightly reduced the levels of total Chk1 in MDA-MB-231 and PY8119 cells (Supplementary Fig. S3A and B). The positive control MNNG effectively triggered the cell-cycle checkpoint response in both parental and ZMYND8 KO cells (Supplementary Fig. S3A and B). These results indicate that ZMYND8 loss does not induce DNA damage to activate the cell-cycle checkpoint response in breast cancer cells.
Activated cGAS-STING axis by ZMYND8 loss is required for the innate immune response in breast cancer cells
To determine if the DNA sensor cGAS is required for IFN-β and ISGs expression in ZMYND8 KO cells, we first examined the cellular distribution of cGAS in ZMYND8 KO cells. cGAS was localized at the nucleus and cytosol of PY8119-SC cells (Fig. 3A and B). ZMYND8 KO increased cGAS translocation into the micronucleus, where cGAS was colocalized with γH2A.X (Fig. 3A and B), suggesting that ZMYND8 loss causes cGAS recruitment to sites of DNA damage in breast cancer cells. To determine if cGAS is activated by ZMYND8 KO, we measured the intracellular 2’3’-cGAMP levels in PY8119 cells using ELISA and found that the 2’3’-cGAMP levels were significantly increased by about 4–5-fold in ZMYND8 KO cells (Fig. 3C). cGAS protein levels were not affected by ZMYND8 KO (Fig. 3D), which rules out a role of cGAS protein expression changes in ZMYND8 loss-induced cGAS activation. Next, we generated two independent ZMYND8/cGAS double KO (DKO) PY8119 cell lines by transfecting cGAS sgRNAs in ZMYND8 KO cells (Fig. 3D). RT-qPCR results showed that ISGs mRNAs were upregulated in ZMYND8 KO PY8119 cells, which was completely abolished by cGAS KO (Fig. 3E). In contrast, the non-target gene Rpl13a mRNA was not affected by ZMYND8 KO alone or ZMYND8/cGAS DKO (Fig. 3E). These data indicate that cGAS is required for ZMYND8 loss-induced ISGs upregulation in breast cancer cells.
Figure 3. cGAS-STING-NF-κB activation is required for ZMYND8 KO-induced ISGs expression in breast cancer cells.
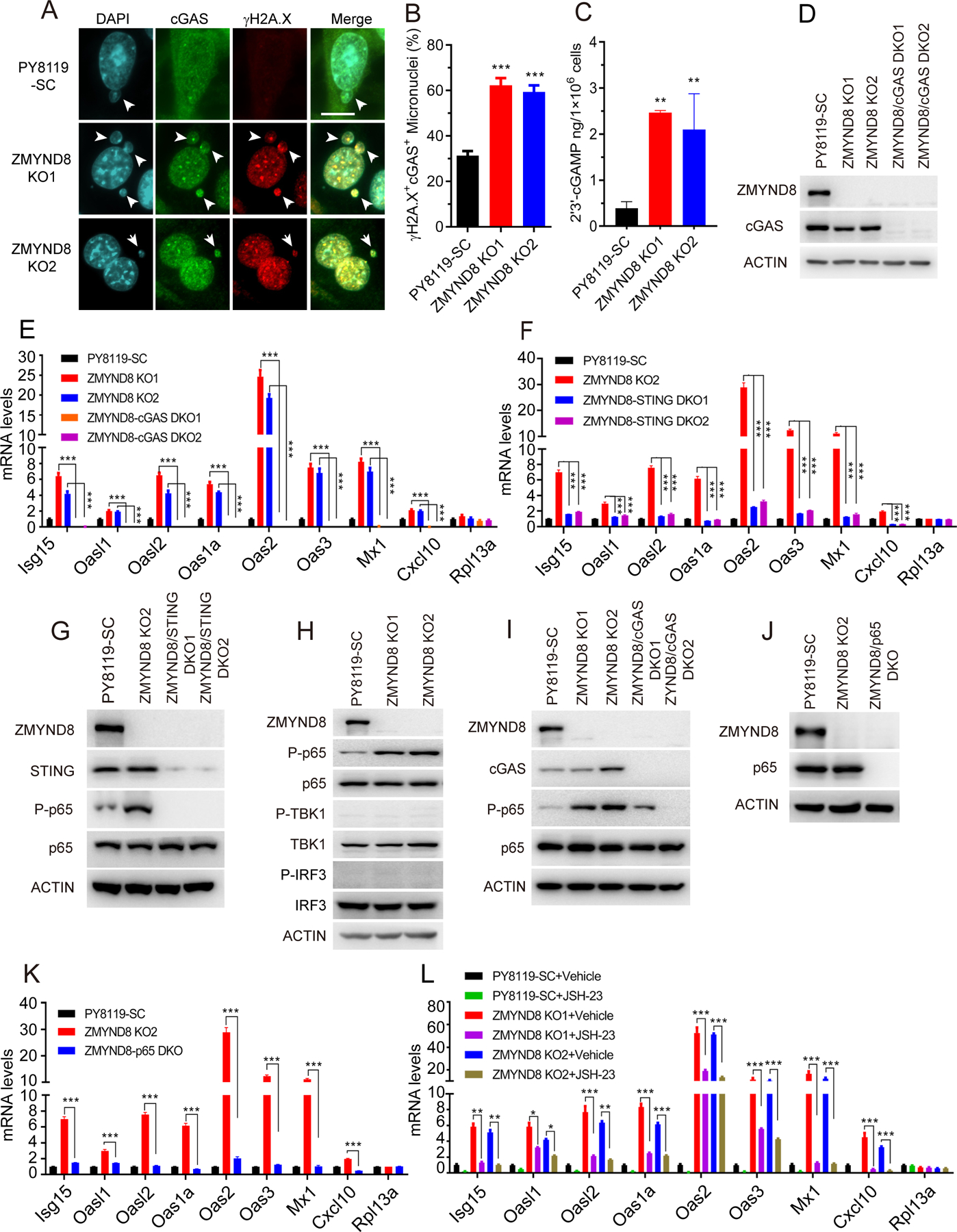
(A) Representative immunostaining of cGAS and γH2A.X in SC, ZMYND8 KO1, and ZMYND8 KO2 PY8119 cells. Scale bar, 10 μm. (B) Quantification of cGAS- and γH2A.X-positive (+) micronuclei in SC, ZMYND8 KO1, and ZMYND8 KO2 PY8119 cells (n = 150, mean ± SEM). **p < 0.01 by one-way ANOVA with Dunnett’s test. (C) ELISA analysis of cellular 2’3’-cGAMP levels in SC and ZMYND8 KO PY8119 cells (n = 3, mean ± SEM). **p < 0.01 by one-way ANOVA with Dunnett’s test. (D) Immunoblot analysis of indicated proteins in SC, ZMYND8 KO, and ZMYND8/cGAS DKO PY8119 cells. (E and F) RT-qPCR analysis of ISGs and Rpl13a mRNAs in SC, ZMYND8 KO, and ZMYND8/cGAS DKO (E) or ZMYND8/STING DKO (F) PY8119 cells (n = 3, mean ± SEM). ***p < 0.001 by two-way ANOVA with Tukey’s test. (G) Immunoblot analysis of indicated proteins in SC, ZMYND8 KO, and ZMYND8/STING DKO PY8119 cells. P, phosphorylation. (H) Immunoblot analysis of indicated proteins in SC and ZMYND8 KO PY8119 cells. (I) Immunoblot analysis of indicated proteins in SC, ZMYND8 KO, and ZMYND8/cGAS DKO PY8119 cells. (J) Immunoblot analysis in SC, ZMYND8 KO, and ZMYND8/p65 DKO PY8119 cells. (K) RT-qPCR analysis of ISGs and Rpl13a mRNAs in SC, ZMYND8 KO, and ZMYND8/p65 DKO PY8119 cells (n = 3, mean ± SEM). ***p < 0.001 by two-way ANOVA with Tukey’s test. (L) RT-qPCR analysis of ISGs mRNAs in SC and ZMYND8 KO PY8119 cells treated with a NF-κB inhibitor JSH-23 or vehicle (n = 3, mean ± SEM). *p < 0.05; **p < 0.01; ***p < 0.001 by two-way ANOVA with Tukey’s test.
2’3’-cGAMP produced by cGAS binds to STING to initiate the signaling cascade (12). To determine whether STING is also involved in upregulation of ISGs in ZMYND8 KO PY8119 cells, we generated ZMYND8/STING DKO PY8119 cells using the CRISPR technique and found by RT-qPCR assay that upregulation of ISGs mRNAs by ZMYND8 KO was reversed when STING was knocked out in ZMYND8 KO PY8119 cells (Fig. 3F and G), which was consistent with that in ZMYND8/cGAS DKO PY8119 cells (Fig. 3E). KO of cGAS or STING did not change micronucleus formation in ZMYND8 KO PY8119 cells (Supplementary Fig. S4A and B), confirming that downregulation of ISGs in these DKO cells is attributed to inactivation of the cGAS-STING axis. Taken together, these findings indicate that the cGAS-STING axis is activated by ZMYND8 loss and required for ISGs upregulation in ZMYND8 KO breast cancer cells.
Previous studies showed that cGAS activation triggers senescence (26–28). To study whether ZMYND8 loss regulates senescence via activation of cGAS, we performed senescence-associated β-galactosidase assay in SC and ZMYND8 KO cells. β-galactosidase-positive cells were barely detected in either SC or ZMYND8 KO MDA-MB-231, PY8119, and 4T1 cells (Supplementary Fig. S5A–F). Treatment of the known senescence inducer etoposide (12.5 μM) induced a robust senescence phenotype in both SC and ZMYND8 KO cells, suggesting that the senescence machinery is intact in these cells (Supplementary Fig. S5A–F). We further analyzed the expression of senescence-associated genes from our RNA-seq experiment and found that IL1A, IL1B, and CDKN1A mRNAs were upregulated by ZMYND8 loss, while CCL2 mRNA was downregulated by ZMYND8 loss (Supplementary Fig. S5G). IL6 and IL8 mRNAs were not significantly affected by ZMYND8 loss (Supplementary Fig. S5G). These results indicate that ZMYND8 loss has a mixed effect on the expression of senescence-associated genes and fails to induce senescence in breast cancer cells.
Activated NF-κB by ZMYND8 loss is required for the innate immune response in breast cancer cells and tumors
Next, we investigated which transcription factor downstream of cGAS/STING is responsible for ZMYND8 loss-induced ISGs expression in breast cancer cells. TBK1/IRF3 and NF-κB are phosphorylated and activated following activation of cGAS/STING to induce ISGs expression (12,14). Immunoblot assay showed that no phospho-TBK1 and phospho-IRF3 were detected in SC and ZMYND8 KO PY8119 cells (Fig. 3H). In contrast, levels of phospho-p65, also known as RELA that is a key subunit of NF-κB heterodimer, were remarkably increased in ZMYND8 KO PY8119 cells as compared with SC cells (Fig. 3H). Total p65 protein levels were not affected by ZMYND8 KO in PY8119 cells (Fig. 3H). Moreover, KO of cGAS or STING abolished phosphorylation of p65 in ZMYND8 KO PY8119 cells (Fig. 3G and I). These results indicate that NF-κB, but not TBK1/IRF3, is activated by cGAS/STING in ZMYND8 KO breast cancer cells.
We next studied whether NF-κB is required for ZMYND8 loss-induced ISGs expression in breast cancer cells. To this end, we generated ZMYND8/p65 DKO PY8119 cells (Fig. 3J) and performed RT-qPCR assay. As expected, ZMYND8 KO significantly increased the expression of ISGs in PY8119 cells, which was completely abolished by p65 KO (Fig. 3K). We further treated SC and ZMYND8 KO PY8119 cells with a NF-κB inhibitor JSH-23 (10 μM). In line with genetic KO of p65 (Fig. 3K), pharmacological treatment of JSH-23 significantly inhibited ZMYND8 KO-induced mRNA expression of ISGs but not Rpl13a in PY8119 cells (Fig. 3L). We further studied whether ZMYND8 also inhibits NF-κB activity and IFN-β and ISGs expression in breast tumors in vivo. To this end, we orthotopically implanted SC and ZMYND8 KO1 or KO2 PY8119 cells into the mammary fat pad of female C57BL/6J mice, respectively. Both ZMYND8 KO1 and KO2 inhibited mammary tumor growth in mice (Fig. 4A–C), which was consistent with our recent findings in immunodeficient mice (21). ZMYND8 KO was validated in primary tumors (Fig. 4D). Phospho-p65 levels were increased in ZMYND8 KO1 and KO2 tumors (Fig. 4D), similar to that in cultured breast cancer cells (Fig. 3H). Total p65, cGAS, and STING protein levels were not affected by ZMYND8 KO in tumors (Fig. 4D). Furthermore, RT-qPCR results showed that IFN-β and ISGs were significantly upregulated in ZMYND8 KO1 and KO2 tumors as compared with SC tumors (Fig. 4E), which is consistent with our in vitro results (Fig. 2D). The non-target gene Rpl13a expression was not affected by ZMYND8 KO in tumors (Fig. 4E). Finally, we studied whether NF-κB directly binds to the Ifnb1 gene to regulate its transcription in ZMYND8 KO cells. ChIP-qPCR assay showed that ZMYND8 KO significantly increased phospho-p65 enrichment at the promoter of the Ifnb1 gene in PY8119 cells (Fig. 4F). In contrast, it had no significant effect on phospho-p65 occupancy at ISGs genes including Isg15, Oas2, Oas3, and Mx1 in PY8119 cells (Fig. 4F), suggesting that ISGs are induced by ZMYND8 KO indirectly through IFN-β in breast cancer cells. Collectively, these findings indicate that ZMYND8 specifically inhibits the cGAS-STING-NF-κB signaling cascade leading to suppression of IFN-β in breast cancer cells.
Figure 4. ZMYND8 inhibits NF-κB activation and expression of IFN-β and ISGs in breast tumors.
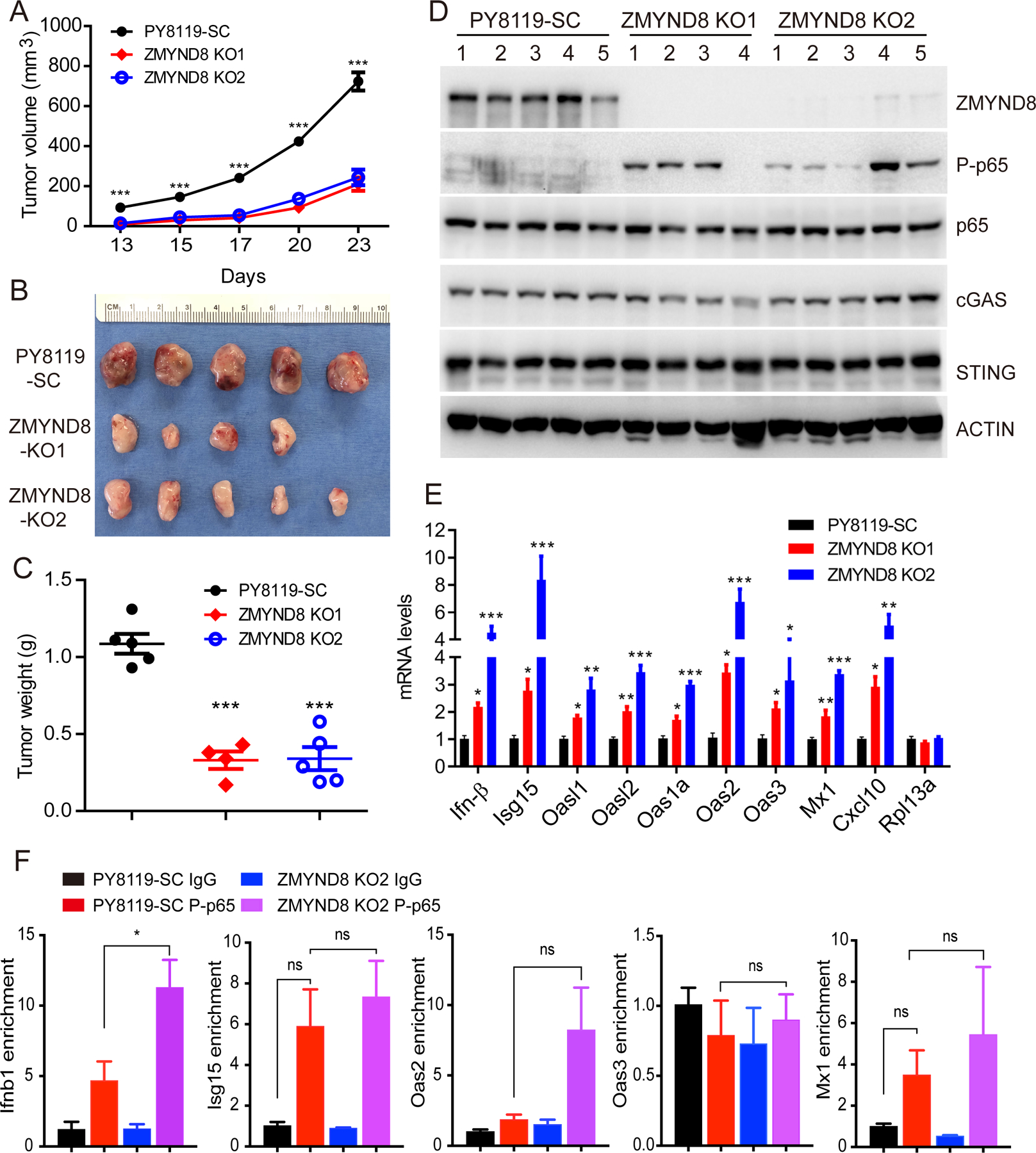
(A-C) SC and ZMYND8 KO PY8119 cells were orthotopically implanted into the mammary fat pad of female C57BL/6J mice, respectively. Tumor growth curve is shown in A (n = 4–5, mean ± SEM). Tumor image on day 23 is shown in B. Tumor weight on day 23 is shown in C (n = 4–5, mean ± SEM). ***p < 0.001 by one-way ANOVA with Dunnett’s test. (D) Immunoblot analysis of indicated proteins in SC and ZMYND8 KO PY8119 tumors. (E) RT-qPCR analysis of IFN-β, ISGs and Rpl13a mRNAs in SC and ZMYND8 KO PY8119 tumors (n = 4–5, mean ± SEM). *p < 0.05; **p < 0.01; ***p < 0.001 vs. SC by one-way ANOVA with Dunnett’s test. (F) ChIP-qPCR analysis of phospho-p65 (P-p65) enrichment at Ifnb1, Isg15, Oas2, Oas3, Mx1 genes in SC and ZMYND8 KO2 PY8119 cells (n = 3, mean ± SEM). *p < 0.05 by two-way ANOVA with Tukey’s test. ns, not significant.
Previous studies showed that STAT1 and STAT3 regulate ISGs expression (29,30). We treated SC and ZMYND8 KO PY8119 cells with a STAT1 inhibitor fludarabine (20 μM) and found that fludarabine treatment had no effect on ZMYND8 KO-induced IFN-β and ISGs expression in PY8119 cells (Supplementary Fig. S6A). Treatment of a STAT3 inhibitor C188–9 (20 μM) increased IFN-β expression and selectively inhibited ZMYND8 KO-induced mRNA expression of Oas1a and Oasl2 but not other ISGs in PY8119 cells (Supplementary Fig. S6B). These data suggest that both STAT1 and STAT3 are unlikely involved in ZMYND8 loss-induced IFN-β and ISGs expression in breast cancer cells.
ZMYND8 inhibits cytotoxic T cells-mediated antitumor immunity to promote breast tumorigenesis
To study whether ZMYND8 inhibits the innate immune response to promote tumor growth, we orthotopically implanted the equal number of parental and ZMYND8 KO 4T1 cells respectively into the mammary fat pad of both female immunocompetent Balb/c and immunocompromised Rag1 KO mice. ZMYND8 KO in 4T1 cells significantly reduced primary tumor growth in both Balb/c and Rag1 KO mice (Fig. 5A and B). This was not due to reduced cancer cell proliferation as ZMYND8 KO had no effect on proliferation of breast cancer cells in vitro (Supplementary Fig. S7A–C). However, ZMYND8 KO tumors grew faster in Rag1 KO mice than Balb/c mice (Fig. 5A and B), suggesting a critical role of the immune system in ZMYND8 KO tumor growth. Flow cytometry analysis showed that CD8+ and CD4+ T cells were remarkably increased in ZMYND8 KO tumors harvested from Balb/c mice (Fig. 5C and D), which was confirmed by IHC analysis (Fig. 5E–G), indicating that ZMYND8 expression in breast cancer cells inhibits infiltration of cytotoxic CD8+ and CD4+ T cells to promote breast tumor growth. To further confirm the antitumor role of CD8+ and CD4+ T cells in ZMYND8 KO tumors, we treated parental or ZMYND8 KO 4T1 tumor-bearing Balb/c mice intraperitoneally with anti-CD4 and CD8 antibodies to deplete CD4+ and CD8+ T cells. As expected, ZMYND8 KO significantly inhibited 4T1 tumor growth in mice treated with control IgG, which was significantly reversed by anti-CD4 and CD8 depleting antibody treatment (Fig. 5H and I). ZMYND8 KO in primary tumors was validated by immunoblot assay (Fig. 5J). Together, these findings indicate that ZMYND8 inhibits CD8+ and CD4+ T cells-mediated antitumor immunity to promote breast tumor growth in mice.
Figure 5. ZMYND8 expression in breast cancer cells inhibits infiltration of CD4+ and CD8+ T cells to promote tumor growth in mice.
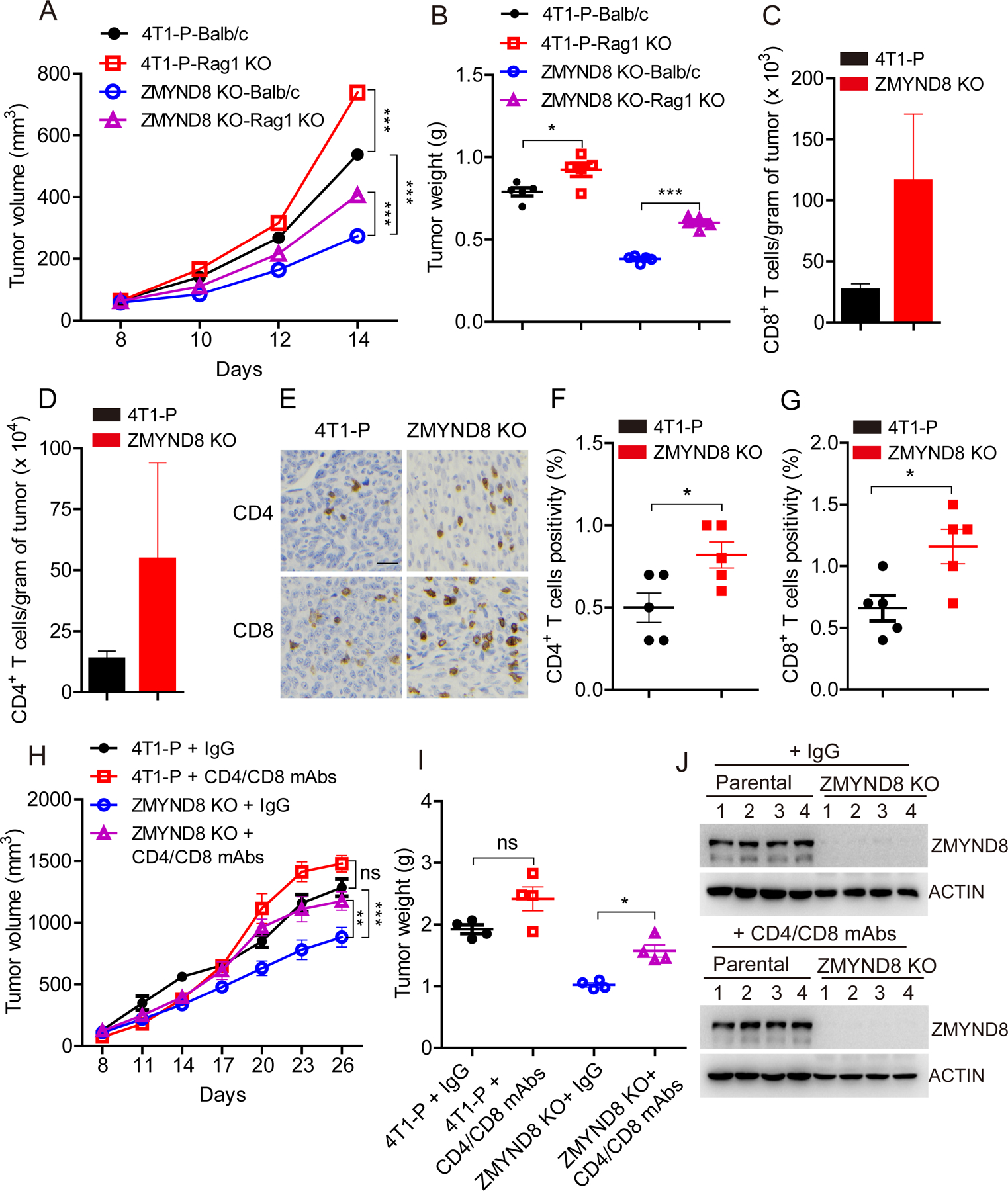
(A and B) Parental (P) and ZMYND8 KO 4T1 cells were orthotopically implanted into the mammary fat pad of female Balb/c and Rag1 KO mice, respectively. Tumor growth curve is shown in A (n = 5, mean ± SEM). Tumor weight on day 14 is shown in B (n = 5, mean ± SEM). *p < 0.05; ***p < 0.001 by two-way ANOVA with Tukey’s test. (C and D) Flow cytometry quantification of CD8+ (C) and CD4+ (D) T cells in parental and ZMYND8 KO 4T1 tumors harvested from Balb/c mice on day 14 (n = 5, mean ± SEM). (E-G) IHC analysis of CD8+ and CD4+ T cells in parental and ZMYND8 KO 4T1 tumors harvested from Balb/c mice on day 14 (n = 5, mean ± SEM). *p < 0.05 by Student’s t test. (H and I) Parental and ZMYND8 KO 4T1 cells were orthotopically implanted into the mammary fat pad of female Balb/c mice, respectively. Mice were treated with monoclonal anti-CD4/CD8 depleting antibodies (mAbs) or control IgG. Tumor growth curve is shown in H (n = 4, mean ± SEM). Tumor weight on day 26 is shown in I (n = 4, mean ± SEM). *p < 0.05; **p < 0.01, ***p < 0.001 by two-way ANOVA with Tukey’s test. ns, not significant. (J) Immunoblot analysis of ZMYND8 and actin in parental and ZMYND8 KO 4T1 tumors treated with anti-CD4/CD8 depleting mAbs or control IgG.
To study the involvement of IFN-β in ZMYND8 loss-caused antitumor immunity, we blocked IFN-β signaling using anti-IFNAR1 antibody in SC and ZMYND8 KO PY8119 xenograft tumor models. Intratumoral administration of anti-IFNAR1 antibody significantly restored ZMYND8 KO tumor growth in C57BL/6J mice (Fig. 6A–C). ZMYND8 KO in tumors was validated by immunoblot assay (Fig. 6D). These results indicate that IFN-β induced by ZMYND8 loss is required for antitumor effect in vivo.
Figure 6. IFN-β is required for ZMYND8 loss-mediated antitumor effect.
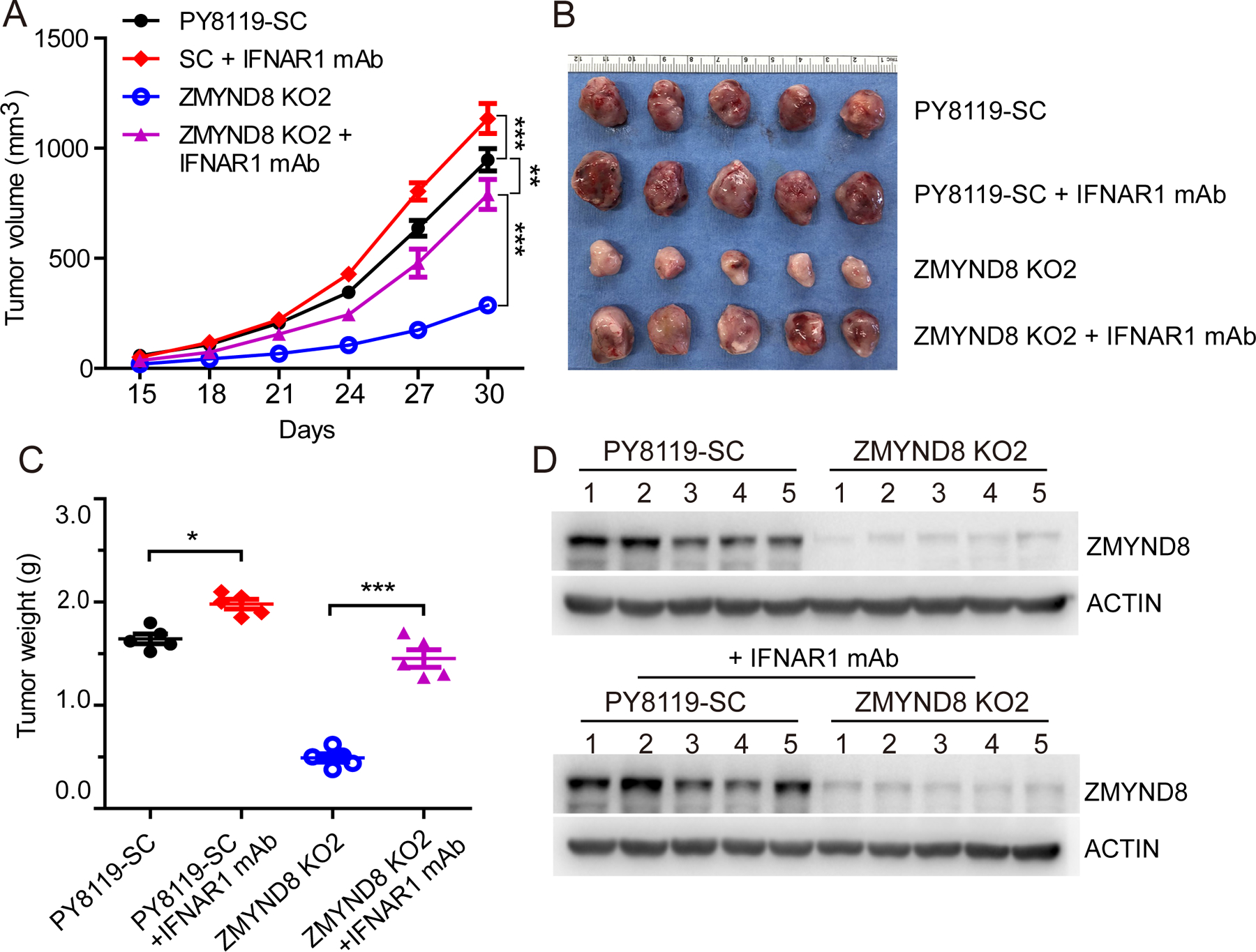
SC and ZMYND8 KO2 PY8119 cells were orthotopically implanted into the mammary fat pad of female C57BL/6J mice, respectively. 5 mice each group were administrated with monoclonal anti-IFNAR1 depleting antibody (mAb). (A) Tumor growth curve (n = 5, mean ± SEM). (B) Tumor image on day 30. (C) Tumor weight on day 30 (n = 5, mean ± SEM). (D) Immunoblot analysis of ZMYND8 and actin in tumors. *p < 0.05; **p < 0.01, ***p < 0.001 by two-way ANOVA with Tukey’s test.
ZMYND8 is upregulated and negatively correlated with the tumor-lymphocyte infiltration phenotype in human breast tumors
Lastly, we studied the clinical significance of ZMYND8-induced immunosuppression in human breast tumors. By analyzing a cohort of 994 breast cancer patients in The Cancer Genome Atlas (TCGA), we found that ZMYND8 gene is amplified and/or overexpressed in breast tumors from about 10% of patients (Fig. 7A and B), which was consistent with our recent findings of ZMYND8 protein upregulation in breast tumors (21). ZMYND8 amplification/overexpression was observed in different types of invasive breast tumors with the highest frequency in invasive mixed mucinous breast carcinoma (Fig. 7C). Gene correlation analysis across the TCGA cohort of breast cancer patients revealed a significant negative correlation of ZMYND8 with ISGs including CXCL10, MX1, and ISG15 (Fig. 7D–F), consistent with gene expression data in breast cancer cells and xenograft tumors (Fig. 2C, 2D, and 4E). Importantly, we found that ZMYND8 was negatively correlated with the expression of CD8A, CD8B, and CD4 in human breast tumors (Fig. 7G–I), which strongly supports our findings on increased infiltration of CD8+ and CD4+ T cells in mouse ZMYND8 KO tumor models (Fig. 5C–G). Furthermore, ZMYND8 was negatively associated with the 17-gene transcriptional signature of tumor-infiltrating lymphocytes in human breast tumors (Fig. 7J) (31). Similar results were also found in other two tumor-lymphocyte infiltration gene signatures (Fig. 7K and L) (32,33). Together, these findings reveal that ZMYND8 is highly expressed and negatively correlated with the tumor-lymphocyte infiltration phenotype in human breast tumors.
Figure 7. ZMYND8 is amplified and negatively correlated with the tumor-lymphocyte infiltration phenotype in human breast tumors.
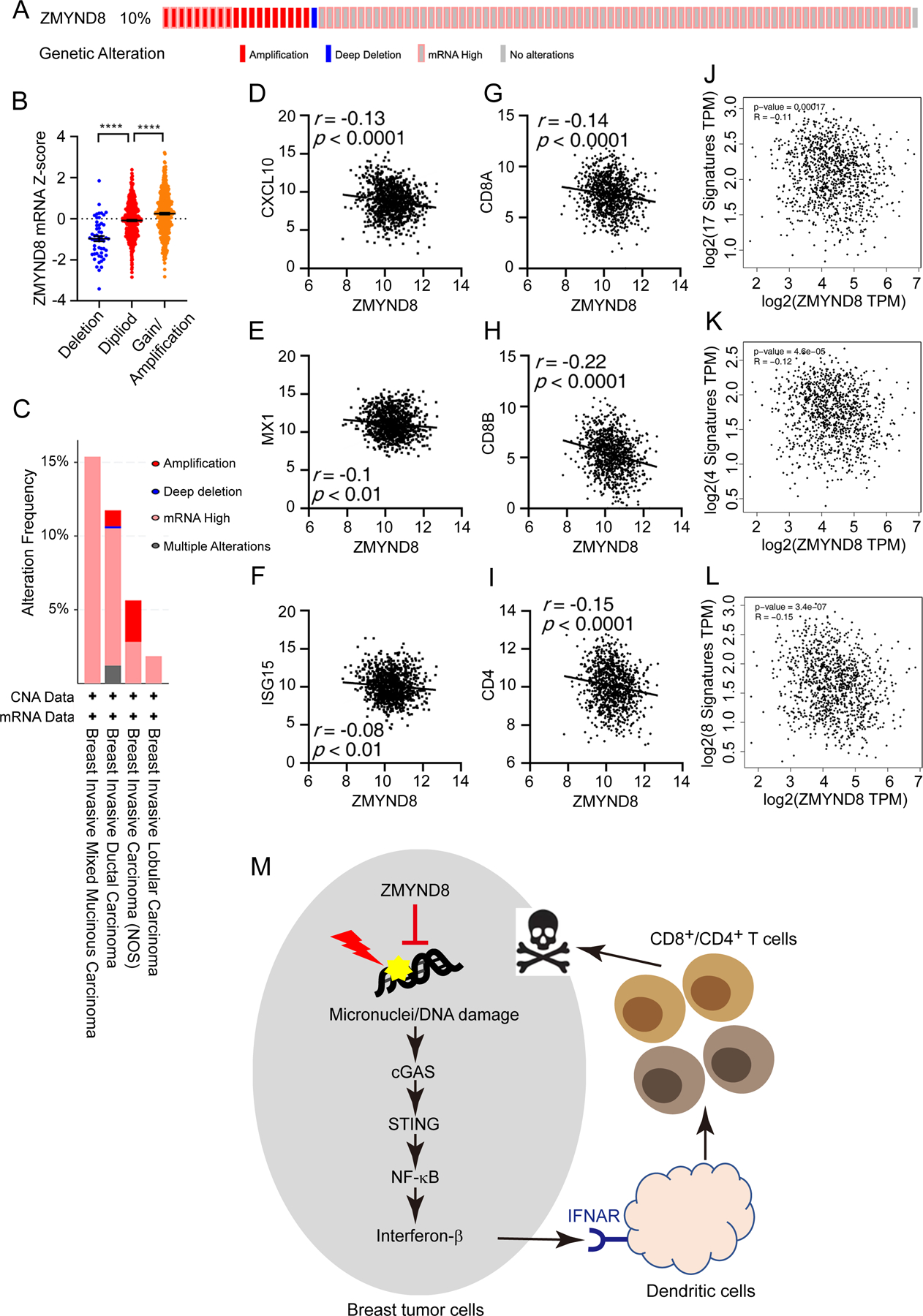
(A) Oncoprint summary of genetic alteration of ZMYND8 in human breast tumors. Data were retrieved from the TCGA PanCancer Atlas in cBioPortal. (B) Association of ZMYND8 copy number alteration with its mRNA expression in the TCGA cohort of breast cancer patients (n = 994). ****p < 0.0001 by one-way ANOVA with Dunnett’s test. (C) Frequency of ZMYND8 genetic alterations in different types of human breast tumors. Data were retrieved from the TCGA PanCancer Atlas in cBioPortal. (D-I) Expression correlation of ZMYND8 mRNA with CXCL10 (D), MX1 (E), ISG15 (F), CD8A (G), CD8B (H) and CD4 (I) mRNAs in human TCGA breast tumors (n = 1048). (J-L) ZMYND8 is negatively correlated to the 17-gene (J), 4-gene (K), and 8-gene (L) transcriptional signatures of tumor-infiltrating lymphocytes in human TCGA breast tumors. Data were generated by GEPIA2. (M) A proposed model for ZMYND8 suppressing antitumor immunity to promote breast tumor growth. ZMYND8 is highly expressed and inhibits micronucleus formation and DNA damage in breast cancer cells, leading to inactivation of the cGAS-STING-NF-κB signaling pathway and subsequent suppression of IFN-β release from breast cancer cells, thereby inhibiting infiltration of CD4+ and CD8+ T cells to facilitate breast tumor growth.
Discussion
In the present study, we identified an inhibitory role of ZMYND8 in cytotoxic T-cell-mediated antitumor immunity. We showed that ZMYND8 is highly expressed and reduces micronucleus formation and DNA damage in breast cancer cells, leading to inactivation of the cGAS-STING-NF-κB signaling pathway and subsequent suppression of IFN-β release from breast cancer cells, thereby inhibiting infiltration of CD4+ and CD8+ T cells to promote breast tumor growth in mice (Fig. 7M). Our results highlight that ZMYND8 is an important “safety guard” for genome stability that causes breast cancer cells to evade immunosurveillance for their growth.
ZMYND8 is known to be recruited to sites of DNA damage upon laser microirradiation to promote homologous recombination DNA repair (18,19). In this regard, ZMYND8 functions as an adaptor to recruit the NuRD complex leading to repression of gene transcription, thereby facilitating homologous recombination. In the present study, we showed that ZMYND8 KO increases micronucleus formation and DNA damage in breast cancer cells, which has been supported by another recent study (34), indicating that ZMYND8 is a critical regulator of micronucleus formation. Micronucleus formation is caused by chromosome mis-segregation and represents a type of DNA damage that occurs during cell mitosis (24). We indeed found that chromosome aberrations are significantly increased in ZMYND8 KO breast cancer cells. p53 is highly mutated in breast cancer and involved in chromosome mis-segregation and genome instability (35). We observed consistent micronucleus formation and DNA damage response by ZMYND8 loss in multiple breast cancer cell lines with distinct p53 genotypes, suggesting that ZMYND8 loss-caused micronucleus formation and DNA damage is p53-independent. Future investigation is needed to address how ZMYND8 regulates chromosome mis-segregation in breast cancer cells. Nevertheless, our present work uncovered another layer of the cellular mechanism of ZMYND8-mediated maintenance of genome stability in breast cancer.
Here, we characterized the signaling pathway by which ZMYND8 maintains genome stability to suppress the intrinsic innate immune response in breast cancer cells (Fig. 7M). We observed the increased recruitment of the DNA sensor cGAS to sites of DNA damage in micronuclei of ZMYND8 KO cells, which triggers activation of cGAS and its downstream signaling pathway leading to IFN-β production in breast cancer cells. Interestingly, cGAS/STING prefer to activate NF-κB but not TBK1 and IRF3 in ZMYND8 KO breast cancer cells. Numerous studies have revealed that TBK1/IRF3 are the canonical cGAS/STING downstream signaling effectors (12,14). However, a recent study reported that etoposide-induced DNA damage causes cGAS-independent STING activation, leading to predominant activation of NF-κB (36). Thus, our results along with others suggest that selective activation of cGAS/STING downstream signaling effector NF-κB or TBK1/IRF3 might depend on the type of DNA damage or additional unknown signaling regulator. cGAS is activated by cytosolic double-stranded DNA from nucleus, mitochondria, and micronuclei under conditions of genome instability or stress (12,13,37). Our findings uncover ZMYND8 as a new negative and intrinsic regulator of the cGAS-dependent innate immune response in breast tumor cells, which strongly suggests that ZMYND8 is a possible target for antitumor immunotherapy and that targeting ZMYND8 may render breast tumor cells to be more immunogenic. Future research into the identification of the small molecule inhibitor of ZMYND8 would have a significant clinical relevance in breast cancer as emerging studies suggest that turning tumor cells into antigen presenting cells may improve immunotherapy (38).
Previous studies showed that ZMYND8 is involved in gene repression by interacting with gene repressors KDM5C and CHD4 (18,39). Our current work identified a distinct ZMYND8-mediated gene repression mechanism, which depends on inactivation of the cGAS-STING-NF-κB signaling cascade (Fig. 7M). Whether or not ZMYND8 represses IFN-β through the corepressors remains to be investigated in the future. It was reported that gene repression by the ZMYND8-NuRD complex axis facilitates DNA damage repair (18,20). However, we found that ZMYND8-mediated downregulation of IFN-β and ISGs does not have the similar role in DNA damage response. ZMYND8 is enriched at the active promoters and enhancers and also involved in gene activation besides gene repression (20,21,39,40). ZMYND8 binds to hypoxia-inducible factor (HIF)-1α and HIF-2α to coactivate HIF target gene expression in breast cancer cells under hypoxia (21). It also regulates the recruitment of the P-TEFb complex to control gene activation (40). Therefore, ZMYND8 has the dual gene repression and activation functions via multiple distinct mechanisms.
Although the majority of breast tumors have very low mutational burden as compared with melanoma and non-small cell lung cancer, which is associated with reduced tumor-lymphocyte infiltration leading to tumor cell survival, triple-negative breast cancer has relatively high mutational burden (5,41,42). Recent studies showed that increasing genome instability by manipulation of DNA mismatch repair proteins can lead to high tumor mutational burden and tumor-lymphocyte infiltration in colorectal cancer (3,10). We presented evidence that ZMYND8 inhibits micronucleus formation and DNA damage in breast cancer cells, leading to suppression of IFN-β production and CD8+ and CD4+ T cell infiltration in triple-negative breast tumors. The clinical data from TCGA breast cancer patients also reveal a negative correlation of ZMYND8 with the tumor-lymphocyte infiltration phenotype, strongly supporting our preclinical findings and also unraveling translational potential of targeting ZMYND8 with its small molecule inhibitors in patients with triple-negative breast cancer. We also showed that ZMYND8 loss causes micronucleus formation in estrogen receptor-positive breast cancer MCF-7 cells, but its antitumor immunity role in estrogen receptor-positive breast cancer remains to be confirmed in the future. It has been known that IFN-β promotes dendritic cell maturation to increase priming and activation of cytotoxic T-lymphocytes (43,44). Thus, ZMYND8 expression in breast cancer cells has a negative impact on cytotoxic T-cell-mediated antitumor immunity in breast tumors. We found that treatment of anti-CD4/8 depleting antibodies partially restores ZMYND8 KO tumor growth in Balb/c mice. In contrast, anti-IFNAR1 antibody treatment almost completely prevents ZMYND8 KO tumor regression in syngeneic mice, suggesting that other types of immune cells besides T cells might also contribute to ZMYND8 loss-triggered antitumor immunity. It was reported that IFN-β can upregulate PD-L1 and PD-L2 in cancer cells (45,46). PD-L1/2 expression in turn induces an immunosuppressive microenvironment and attenuates the killing activity of CD8+ T cells (47). Our RNA-seq data (GSE108833) showed that ZMYND8 KO increases PD-L2 expression in MDA-MB-231 cells. Whether or not PD-L2 impairs ZMYND8 loss-induced antitumor immunity remains to be investigated in the future. Similar to ZMYND8, ZMYND8-interacting proteins the NuRD complex are also shown to regulate DNA damage and antitumor immunity and their expression is inversely correlated with T cell infiltration in tumors (48,49). However, it remains unknown whether ZMYND8 and the NuRD complex share the same mechanism of antitumor immunity. Nonetheless, we have previously shown that ZMYND8 enhances HIF-dependent transcriptional programs to promote breast tumor progression in vitro and in mice (21). Past and current research uncovered that ZMYND8 not only potentiates hypoxic responses in the tumor microenvironment, but also inhibits an intrinsic antitumor immunity that causes breast cancer cells to evade immunosurveillance, thereby promoting tumor growth. Therefore, ZMYND8 is a critical regulator that shapes the tumor microenvironment and may be a potential target for the treatment of breast cancer.
Supplementary Material
Significance:
Findings show that ZMYND8 is a new negative and intrinsic regulator of the innate immune response in breast tumor cells, and ZMYND8 may be a possible target for antitumor immunotherapy.
Acknowledgments
We thank the UTSW Cancer Center Tissue Resource for assistance with IHC, which is supported by NCI Cancer Center grant P30CA142543. This work was supported by grants from the Mary Kay Foundation (08-19), NIH (R01CA222393), the CPRIT (RR140036, RP190358), the Susan G. Komen® Foundation (CCR16376227), and the Welch Foundation (I-1903-20190330) to W.L., and the NIH (R35GM124693 and R01AG066166), the CPRIT (RP170671), and the Welch Foundation (I-1939-20160319) to Y.W.. W.L. is a CPRIT Scholar in Cancer Research.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med 2018;378:1976–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. J Natl Compr Canc Netw 2020;18:479–89 [DOI] [PubMed] [Google Scholar]
- 5.Thomas A, Routh ED, Pullikuth A, Jin G, Su J, Chou JW, et al. Tumor mutational burden is a determinant of immune-mediated survival in breast cancer. Oncoimmunology 2018;7:e1490854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 7.Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov 2017;7:675–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parikh AR, He Y, Hong TS, Corcoran RB, Clark JW, Ryan DP, et al. Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: implications for therapy. Oncologist 2019;24:1340–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017;552:116–20 [DOI] [PubMed] [Google Scholar]
- 11.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 2019;9:646–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339:786–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548:461–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, Kranzusch PJ. Modular architecture of the STING C-terminal tail allows interferon and NF-κB signaling adaptation. Cell Rep 2019;27:1165–75.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014;41:843–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep 2019;29:1236–48.e7 [DOI] [PubMed] [Google Scholar]
- 17.Fossey SC, Kuroda S, Price JA, Pendleton JK, Freedman BI, Bowden DW. Identification and characterization of PRKCBP1, a candidate RACK-like protein. Mamm Genome 2000;11:919–25 [DOI] [PubMed] [Google Scholar]
- 18.Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev 2015;29:197–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savitsky P, Krojer T, Fujisawa T, Lambert JP, Picaud S, Wang CY, et al. Multivalent histone and DNA engagement by a PHD/BRD/PWWP triple reader cassette recruits ZMYND8 to K14ac-rich chromatin. Cell Rep 2016;17:2724–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spruijt CG, Luijsterburg MS, Menafra R, Lindeboom RG, Jansen PW, Edupuganti RR, et al. ZMYND8 co-localizes with NuRD on target genes and regulates poly(ADP-ribose)-dependent recruitment of GATAD2A/NuRD to sites of DNA damage. Cell Rep 2016;17:783–98 [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Zhang B, Bao L, Jin L, Yang M, Peng Y, et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J Clin Invest 2018;128:1937–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A 2012;109:E3367–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, et al. Non-canonical NF-κB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 2018;49:490–503.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature 2015;522:179–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanz MC, Dibitetto D, Smolka MB. DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J 2019;38:e101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017;550:402–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 2017;19:1061–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 2017;114:E4612–E20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WB, Levy DE, Lee CK. STAT3 negatively regulates type I IFN-mediated antiviral response. J Immunol 2011;187:2578–85 [DOI] [PubMed] [Google Scholar]
- 30.Hartman SE, Bertone P, Nath AK, Royce TE, Gerstein M, Weissman S, et al. Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev 2005;19:2953–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang B, Chou J, Tao Y, Wu D, Wu X, Li X, et al. An assessment of prognostic immunity markers in breast cancer. NPJ Breast Cancer 2018;4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res 2011;13:R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Criscitiello C, Bayar MA, Curigliano G, Symmans FW, Desmedt C, Bonnefoi H, et al. A gene signature to predict high tumor-infiltrating lymphocytes after neoadjuvant chemotherapy and outcome in patients with triple-negative breast cancer. Ann Oncol 2018;29:162–9 [DOI] [PubMed] [Google Scholar]
- 34.Kim JJ, Lee SY, Gong F, Battenhouse AM, Boutz DR, Bashyal A, et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev 2019;33:1751–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cazzola A, Schlegel C, Jansen I, Bochtler T, Jauch A, Kramer A. TP53 deficiency permits chromosome abnormalities and karyotype heterogeneity in acute myeloid leukemia. Leukemia 2019;33:2619–27 [DOI] [PubMed] [Google Scholar]
- 36.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell 2018;71:745–60.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014;159:1549–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Charette M, Marabelle A, Houot R. Turning tumour cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134–47 [DOI] [PubMed] [Google Scholar]
- 39.Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, et al. Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell 2016;165:331–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh K, Tang M, Kumari N, Nandy A, Basu S, Mall DP, et al. Positive regulation of transcription by human ZMYND8 through its association with P-TEFb complex. Cell Rep 2018;24:2141–54.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narang P, Chen M, Sharma AA, Anderson KS, Wilson MA. The neoepitope landscape of breast cancer: implications for immunotherapy. BMC Cancer 2019;19:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu S, Yu Y, Zhang M, Wang W, Cao X. The involvement of TNF-α-related apoptosis-inducing ligand in the enhanced cytotoxicity of IFN-β-stimulated human dendritic cells to tumor cells. J Immunol 2001;166:5407–15 [DOI] [PubMed] [Google Scholar]
- 44.Renneson J, Salio M, Mazouz N, Goldman M, Marchant A, Cerundolo V. Mature dendritic cells differentiated in the presence of interferon-β and interleukin-3 prime functional antigen-specific CD8 T cells. Clin Exp Immunol 2005;139:468–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 2017;19:1189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morimoto Y, Kishida T, Kotani SI, Takayama K, Mazda O. Interferon-β signal may up-regulate PD-L1 expression through IRF9-dependent and independent pathways in lung cancer cells. Biochem Biophys Res Commun 2018;507:330–6 [DOI] [PubMed] [Google Scholar]
- 47.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget 2016;7:7390–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shao S, Cao H, Wang Z, Zhou D, Wu C, Wang S, et al. CHD4/NuRD complex regulates complement gene expression and correlates with CD8 T cell infiltration in human hepatocellular carcinoma. Clin Epigenetics 2020;12:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.